 Stéphanie Jacquet
Stéphanie Jacquet Dominique Pontier
Dominique Pontier Lucie Etienne
Lucie Etienne
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 18 December 2020
Sec. Comparative Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.605270
This article is part of the Research Topic Nucleic Acid-Associated Inflammation View all 22 articles
Studying the evolutionary diversification of mammalian antiviral defenses is of main importance to better understand our innate immune repertoire. The small HERC proteins are part of a multigene family, including HERC5 and HERC6, which have probably diversified through complex evolutionary history in mammals. Here, we performed mammalian-wide phylogenetic and genomic analyses of HERC5 and HERC6, using 83 orthologous sequences from bats, rodents, primates, artiodactyls, and carnivores—the top five representative groups of mammalian evolution. We found that HERC5 has been under weak and differential positive selection in mammals, with only primate HERC5 showing evidences of pathogen-driven selection. In contrast, HERC6 has been under strong and recurrent adaptive evolution in mammals, suggesting past and widespread genetic arms-races with viral pathogens. Importantly, the rapid evolution of mammalian HERC6 spacer domain suggests that it might be a host-pathogen interface, targeting viral proteins and/or being the target of virus antagonists. Finally, we identified a HERC5/6 chimeric gene that arose from independent duplication in rodent and bat lineages and encodes for a conserved HERC5 N-terminal domain and divergent HERC6 spacer and HECT domains. This duplicated chimeric gene highlights adaptations that potentially contribute to rodent and bat immunity. Our findings open new research avenues on the functions of HERC6 and HERC5/6 in mammals, and on their implication in antiviral innate immunity.
As a result of sustained exposure to viral infections, mammals have evolved a sophisticated and diversified immune repertoire against viruses. A hallmark of mammalian antiviral immunity is the induction of type I interferon (IFN) upon viral infection. This cytokine upregulates the transcription of hundreds of interferon-stimulated genes (ISGs) in viral infected cells (1). Many of these ISGs encode proteins with antiviral functions, named restriction factors, which are critical players in the first line of the innate immune defense inhibiting different steps of the viral replication cycle (2).
Viruses have adapted to circumvent, subvert, or antagonize these host restriction factors (3). Reciprocally, restriction factors have rapidly and repeatedly evolved to maintain defenses against evolving viral pathogens, leading to virus-host evolutionary arms-races (3, 4). These dynamics of reciprocal adaptations can leave genetic signatures in the host restriction factors. Significant accumulations of non-synonymous changes over synonymous substitutions—designated as positive selection, as well as codon deletions or insertions that may alter the virus-host interface—are common genetic signatures of such long-term evolutionary arms-races (3–6). At the genomic level, gene duplication and recombination are among the most important mechanisms that can diversify the antiviral immune repertoire. In particular, gene duplication can generate adaptive molecular novelty allowing hosts to escape viral antagonism, evolve new immune functions, or increase the depth of antiviral response (3, 7, 8). The weight of such evolutionary mechanisms in mammalian immunity is highlighted by the extent of multigene families, which encode important ISG-encoded proteins, such as the Tripartite Motif-containing (TRIM) (9–12), Apolipoprotein B Editing Complex (APOBEC3) (13–15), Interferon-induced Protein with Tetratricopeptide Repeats (IFIT) (16–18), Interferon induced Transmembrane protein (IFITM) (19) families. For example, the APOBEC3 family has expanded in a lineage-specific manner in primates (20), artiodactyls (21), and bats (15), generating variability in mammalian antiviral response (14). However, the evolutionary and functional diversification of many antiviral families remains poorly characterized in mammals. Deciphering the evolutionary trajectories of multigene family members can provide insights into the genetic mechanisms underlying the diversification of antiviral responses and may allow identifying novel antiviral proteins.
The HECT and RLD domain containing E3-ubiquitin protein ligases, known as HERC proteins, are encoded by a multigene superfamily that is poorly studied in mammals. With six gene members, the HERC family is divided into two subfamilies, the large (HERC1 and HERC2) and the small (HERC3–6) HERCs (22). The small HERC proteins are structurally characterized by a N-terminal RCC1-like domain (RLD), a spacer region, and a C-terminal HECT (Homologous E6-AP Carboxyl Terminus) ubiquitin E3-ligase domain, while the large HERCs possess at least two RLD domains in addition to a HECT domain (23). This structural difference between large and small HERCs reflects their independent evolutionary history (24). In the antiviral immune context, much attention has been devoted to the small HERCs, in particular to HERC5 - an ISG-encoded antiviral effector - and HERC6 its closest relative (24, 25). In humans, HERC5 acts as a HECT ubiquitin and E3-ligase (26–28). HERC5 notably conjugates the ubiquitin-like protein ISG15 to different protein targets, a process termed ISGylation (29–31). The protein targets may be non-specific newly synthetized viral proteins, specific viral proteins, or specific host proteins (29–31). ISGylated proteins are modified, functionally disrupted, or altered in their localization within the cells. Through this ISGylation activity, HERC5 has an antiviral function against highly divergent viruses, including retrovirus (Human and Simian immunodeficiency viruses, HIV and SIV), papillomavirus, and influenza virus (25, 31–33). For example, HERC5 targets the early stage of HIV assembly by catalyzing the ISGylation of the viral Gag protein (30), while it reduces influenza A viral replication through the conjugation of ISG15 to the viral NS1 protein (31). Besides, HERC5 appears to further interfere with HIV replication in an ISGylation-independent manner by impacting the nuclear export of Rev/RRE-dependent viral RNA, most likely through determinants in the RLD domain (33). In contrast, although HERC6 is the most closely-related protein of HERC5, little is known about its functional implication in mammalian antiviral immunity (25). The antiviral role of HERC6 has mainly been described in mouse, in which the HERC5 gene has been lost and functionally substituted by HERC6, the main murine E3-ligase of ISG15 (28, 34, 35). In humans, although the HERC6 protein possesses a HECT E3-ligase domain, it is devoid of ISGylation function (25).
These evolutionary and functional differences between mammals suggest lineage-specific adaptive changes in HERC5 and HERC6. Two previous studies showed that HERC5 and HERC6 genes have evolved under positive selection during vertebrate evolution (25, 33). They further showed that the RLD domain plays an important role in the antiviral activity of HERC5 and HERC6 proteins. While these studies have provided important insights into the diversification of HERCs, the evolutionary analyses have certain limitations: (i) the scarcity of species analyzed (only 12 species, versus 81 species with at least 10 species per order in this current study), (ii) the overrepresentation of primates compared to other mammalian species (seven primates, two to three carnivores, two artiodactyls, and one perissodactyl), (iii) the integration of highly divergent species, which may bias the genetic inferences by increasing the number of false positives (36). Moreover, a recent study in primates have shown differences in HERC5 and HERC6 selective pressures (37). Therefore, how HERC5 and HERC6 genes have evolved within mammalian orders has not been fully deciphered. Nor is the evolutionary dynamic of HERC5 and HERC6 expansions and contractions across mammals.
Here, we decipher the evolutionary history of mammalian HERC5 and HERC6 via mammalian-wide and lineage-specific phylogenetic and genomic analyses. We analyzed the orthologous sequences of HERC5 and HERC6 from bats, rodents, primates, artiodactyls, and carnivores the top five mammalian orders in terms of zoonotic viral diversity they host (38, 39). First, we show that HERC6—and to a much lesser extent HERC5—has been under strong positive selection. Second, we stressed the HERC6 spacer region as a potential pathogen-mammal interface, targeting viral proteins and/or being the target of virus antagonists. Finally, we identified independent gene duplications through recombination between HERC5 and HERC6 in some bat and rodent lineages, which have led to the fixation of a chimeric HERC5/6 gene in both mammalian orders. Taken together, our results suggest that HERC6 may be an important antiviral protein in mammals and identified a novel chimeric HERC member in bats and rodents that may contribute to unique antiviral responses in these species.
Full-length HERC5 and HERC6 coding sequences were analyzed in bats, rodents, primates, artiodactyls, and carnivores. HERC5 and HERC6 coding sequences from each group were obtained using the Little Brown bat (Myotis lucifigus), mouse (Mus musculus), human (Homo sapiens), cow (Bos taurus), and dog (Canis lupus familiaris) Refseq proteins as queries, respectively, through tBLASTn searches of the “Nucleotide” database in GenBank (40, 41). The species and accession numbers are presented in Supplementary Table 1.
The genomic locus of HERC5 and HERC6 genes in Little Brown bat, mouse, human, dog, and cow were obtained from Ensembl (http://www.ensembl.org/index.html), and GenBank Refseq genome database (41). Their coding sequences were used as queries for BLASTn searches against a total of 110 whole genome assemblies from bats, rodents, primates, carnivores, and artiodactyls (Supplementary Table 1) available in GenBank database (41). We analyzed eight additional mammalian genomes from Proboscidea, Lagomorpha, Scandentia, Perissodactyla, Sirenia, Soricomorpha, Eulipotyphla, and Tubulidentata orders (Orycteropus afer, Loxodonta Africana, Trichechus manatus, Tupaia chinensis, Condylura cristata, Ceratotherium simum, Equus przewalskii, and Sorex araneus, respectively) (Supplementary Table 1). The synteny of HERC5 and HERC6 genes was analyzed and visualized through BLAST searches against the 110 annotated genomes in GenBank database (41). Newly identified HERC-like paralogs (see Results) were confirmed by blasting and aligning their whole sequence (intron and exon regions) to the genomic region containing HERC5 and HERC6 genes in three bat species (Myotis lucifigus, Rhinolophus ferrumequinum, and Phyllostomus discolor), and three rodent species (Chinchilla lanigera, Mastomys coucha, Cavia porcellus), which are representative genomes with good assembling quality (based on the N50, the number of scaffolds, and sequencing coverage).
HERC5 and HERC6 orthologs as well as HERC-like paralogous sequences were aligned for each mammalian order separately using the program MACSE (42), and the alignments were manually curated. A phylogenetic tree was then built for each gene and mammalian order, and for a combined dataset of HERC5 and HERC6 genes (rooted with HERC3 as an outgroup, which is the most closely related gene to HERC5 and HERC6), using the maximum likelihood method implemented in the ATGC-PhyML Web server (43). Each phylogenetic tree was based on the best substitution model (GTR+G+I), as determined by the Smart Model Selection (SMS) program in PhyML (44) and node statistical support was computed through 1,000 bootstrap replicates.
To test whether recombination has occurred in HERC5, HERC6, and HERC-like genes, we ran the GARD (Genetic Algorithm for Recombination Detection) method (45) implemented in the HyPhy package (46, 47), using a general discrete site-to-site rate variation with three rate classes. The program uses a genetic algorithm to screen multiple-sequence alignment for putative recombination breakpoints and provides the probability of support for each breakpoint. GARD analyses were run for each mammalian order and each gene separately, including the newly identified HERC-like paralog.
To determine whether HERC5 and HERC6 have been targets of natural selection during mammalian evolution, we carried out positive selection analyses on orthologous coding sequences from bats (n = 10 and 13, respectively), rodents (n = 11 and 16), primates (n = 19 and 20), carnivores (n = 20 and 23), and artiodactyls (n = 21 and 11). As combining highly divergent sequences for positive selection analyses can lead to misleading results, we performed positive selection analyses on separate dataset for each mammalian order. For Artiodactyls, three different datasets were analyzed: the first dataset included all the available species, the second was restricted to cetacean species, and the third excluded the cetaceans. Analyzing each mammalian order separately allowed us to qualitatively compare the evolutionary profile of both genes in each mammalian order. We first tested for positive selection at the gene level using two different methods available in the Codeml program, which is implemented in the PAML package (48). This program allows both gene- and site-specific detection of positive selection by comparing constrained models that disallow positive selection (models M1 and M7) to unconstrained models allowing for positive selection (M2 and M8). We ran the different models with the codon frequencies of F61 and F3x4 with a starting omega ω (dN/dS ratio) of 0.4. Likelihood ratio tests were computed to compare the models (M1 vs M2 and M7 vs M8), and codons evolving under significant positive selection (dN/dS >1) were identified using the Bayes Empirical Bayes (BEB) with a posterior probability ≥0.95. The residues under positive selection were further assessed using two other methods, the Fast, Unconstrained Bayesian AppRoximation for Inferring Selection (FUBAR) (49) and the Mixed Effects Model of Evolution (MEME) (50), both implemented in the HYPHY package. To increase the specificity of our results, we only kept the sites that were identified as being under significant positive selection by at least two of the four methods used. When significant recombination breakpoints were detected, positive selection analyses were carried out for each fragment identified by GARD. Similarly, we sought for signatures of adaptive selection in the HERC-like paralogs of rodents (see Results), for which five orthologous coding sequences were available. Furthermore, we tested if the three HERC domains (RLD, spacer region, and HECT) have similarly been subjects of positive selection, by analyzing each domain separately.
Finally, to determine if HERC5 and HERC6 have experienced episodic selection within mammalian orders, we carried out the branch-specific analysis aBSREL (51, 52), implemented in the HYPHY package. This program allows testing the significance of positive selection and quantifying the dN/dS ratio for each branch independently.
To determine the genomic evolutionary history of HERC5 and HERC6 in mammals, we performed a complete synteny analysis for 14 chiropteran, 25 rodent, 25 primate, 23 artiodactyl, and 23 carnivore species, and eight more species from different orders. We first found that HERC5 and HERC6 synteny is mostly conserved throughout eutherian mammals (Figure 1, Supplementary Figure 1).
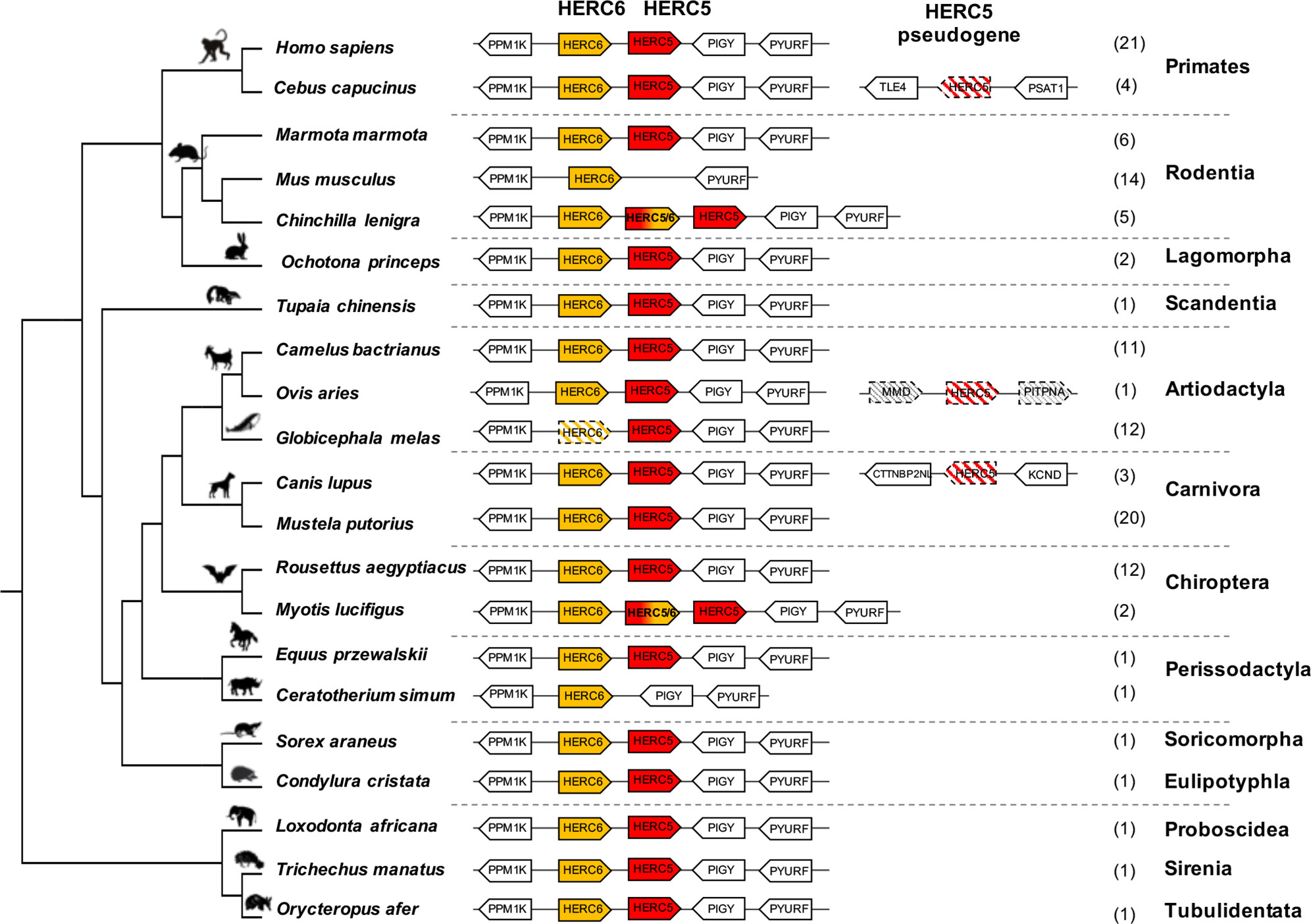
Figure 1 Evolutionary dynamics of mammalian HERC5 and HERC6 gene loci. Representation of the HERC5 and HERC6 gene loci from mammalian genomes. Plain colored arrows represent intact HERC5 and HERC6 genes, striped colored arrows indicate HERC5 or HERC6 pseudogenes, and white arrows are adjacent syntenic genes. The newly identified chimeric HERC5/6 genes are bicolored. The numbers in brackets indicate the total number of genomes analyzed which contain the corresponding genomic organization in each mammalian order. In primates and carnivores, the HERC5 and HERC6 genes are well conserved. In the cetacean and rhinoceros’ species, the HERC6 has been pseudogenized and lost, respectively, while rodent HERC5 has been lost in the Muridae and Cricetidae families. A duplicated HERC5/6 fused gene is independently fixed in several rodent species and in the Myotis genus in bats. Duplication followed by pseudogenization of HERC5 in primates, carnivores and artiodactyls is presented.
However, we also detected that gene erosion has repeatedly shaped the evolution of HERC5 and HERC6 in mammals. While primate and carnivore genomes encode both genes, the artiodactyls and the rodents have experienced gene loss or pseudogenization, generating HERC gene copy number variation in each group. Indeed, although all the studied artiodactyl species encode an intact HERC5 gene, the cetacean genomes showed HERC6 pseudogenization, through nucleotide deletions that impacted frameshift, as well as early stop codons (Figure 1, Supplementary Figure 2). Moreover, we confirmed the erosion of HERC5 in rodents (25), and we further showed that this loss has most probably occurred in the common ancestor of the Cricetidae and Muridae. Interestingly, we also did not find the HERC5 gene in the rhinoceros genome, suggesting at least two independent losses of HERC5 in mammals, specifically in the Rodent and Perissodactyl orders (Figure 1).
Finally, in addition to these independent losses of HERC5 or HERC6 in mammals, we found multiple evidences of HERC5 duplication followed by pseudogenization in primate (Sapajus apella, Cebus capucinus, Aotus nancymaae, and Saimiri boliviensis), carnivore (Canis lupus, Vulpes Vulpes, leptonychotes weddellii), and artiodactyl (Ovis aries) species, highlighting a strong dynamic of gene gain and loss in mammalian HERC5 and HERC6.
While most mammalian species possess one or both HERC genes, we found independent duplications of HERC5 in the chiropteran Myotis genus and the rodent Hystricognathi infra-order (Figure 1, Supplementary Figure 3). These duplicated genes were identified in three bat Myotis species (M. lucifigus, M. brandtii, and M. davidii) and five rodent species (Fukomys damarensis, Heterocephalus glaber, Cavia porcellus, Chinchilla lanigera, and Octodon degus) (Table S1). This dates these duplication events to at least 30 MYA (million years ago) and 44 MYA, respectively.
Surprisingly, alignments of HERC5, HERC-like, and HERC6 proteins in each mammalian group revealed 96–99% amino acid identity between the HERC5 and HERC-like N-terminals, and 74–84% amino acid identity between the HERC-like and HERC6 C-terminals (Figure 2A, Supplementary Figure 4). To test whether this may be reminiscent of recombination in rodents and bats, we used the GARD program. We identified a significant recombination breakpoint located upstream from the spacer region at 1,103 bp in bats and 1,118 bp in rodents (Figures 2A, B). The phylogenetic analyses of the resulting fragments (identified by GARD) confirmed the recombination in both bats and rodents. Specifically, we found that the HERC-like 5’-fragment clustered with the HERC5 gene (clade supported by a significant bootstrap), while the 3’-fragment grouped with the HERC6 gene (Figures 2C–F). Taken together, our findings reveal that a similar ancient mechanism has independently led to the fixation of a HERC-like gene, which in fact is a chimeric HERC5/6 gene containing the HERC5 RLD domain and the HERC6 spacer and HECT domains in bats and rodents.
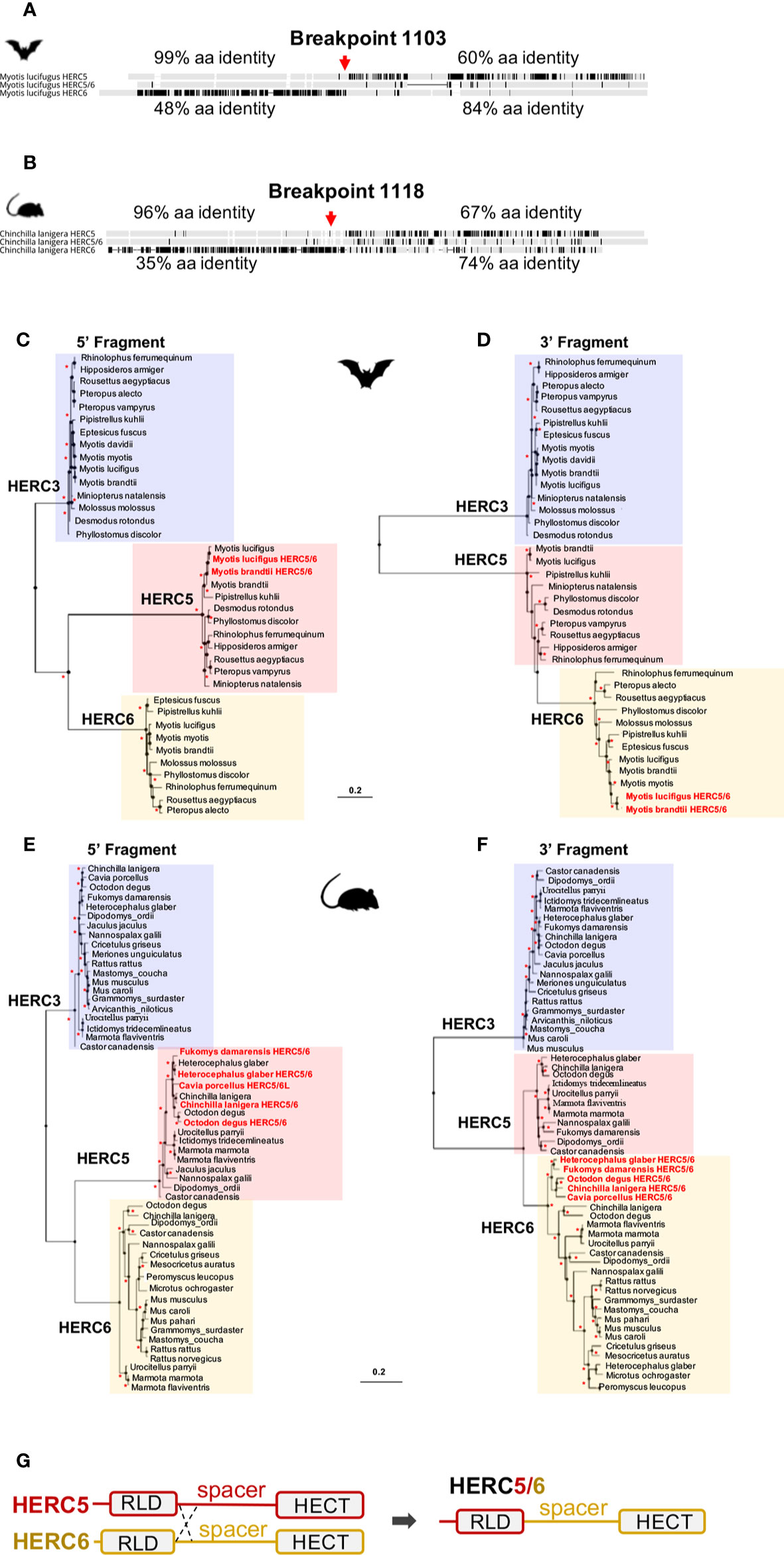
Figure 2 Independent duplication of a chimeric HERC5/6 gene through recombination in bats and rodents (A, B). Alignment of the protein sequence of HERC5, HERC5/6, and HERC6 genes from bats and rodents, respectively. Additional sequence alignments are shown in Supplementary Figure 4. The percentages of pairwise amino acid identities between the N-terminals of HERC5/6 and HERC5 or HERC6, as well as the C-terminals of HERC5/6 and HERC5 or HERC6 are indicated. The significant recombination breakpoints (red arrows, p-value <0.05) assigned by the GARD program are shown for the rodent and bat HERC5/6 gene (C–F). Maximum likelihood phylogenetic tree generated with the 5’ (at the left) and 3’ (at the right) of the HERC5, HERC6, HERC5/6, and HERC3 (as an outgroup) nucleotide gene alignment based on GARD recombination results (corresponding to the breakpoint 1103 in Myotis lucifigus in bats, and 1118 in Chinchilla lanigera in rodents). The duplicated chimeric HERC5/6 genes are shown in red. Asterisks indicate bootstrap values greater than 80%. The scale bar represents genetic variation of a 0.2 (20%) for the length of the scale (G). A linear representation of HERC5 and HERC6 structures showing a chromosomal crossover between the 5’ regions, upstream of the spacer region. This mechanism has led to a duplicated recombined HERC5/6 gene containing the HERC5 RLD domain and HERC6 spacer region and HECT domain.
Moreover, by analyzing the phylogenetic trees of HERCs in bats and rodents (Figures 2C–F), we found that the 5’ fragment of HERC5/6 was genetically closer to HERC5 from the same species. This was not the case for the 3’ end, where all 3’ fragments of the HERC5/6 genes significantly grouped together. Combined with several GARD analyses, this supports that recent recombinations further occurred between the RLD domains of HERC5 and HERC5/6 genes (Figure 2G).
We next investigated whether HERC5 and HERC6 have been under selective pressure in mammals.
Our results revealed some signatures of positive selection in artiodactyl, primate, and bat HERC5 (p-values <10−3), but none in rodent and carnivore species (p-value >0.5) (Table 1). At the codon level, the signal was overall very weak, with less than three positively selected codons assigned per order (posterior probability threshold fixed at 0.95, and p-value <0.05) (Table 2). In primates, two significant positively selected sites were identified in the RLD domain (Figure 3). Similarly, less than two codons evolved under positive selection in bats and artiodactyls (except for the MEME method which identified multiple positively selected sites in the artiodactyls), and none of the codons were common between methods (Table 2). Interestingly, the separate analysis of cetacean species revealed stronger signatures of positive selection at the gene (p-value = 6.10−6) and the codon levels (five sites identified by at least two methods), suggesting a lineage-specific adaptation.
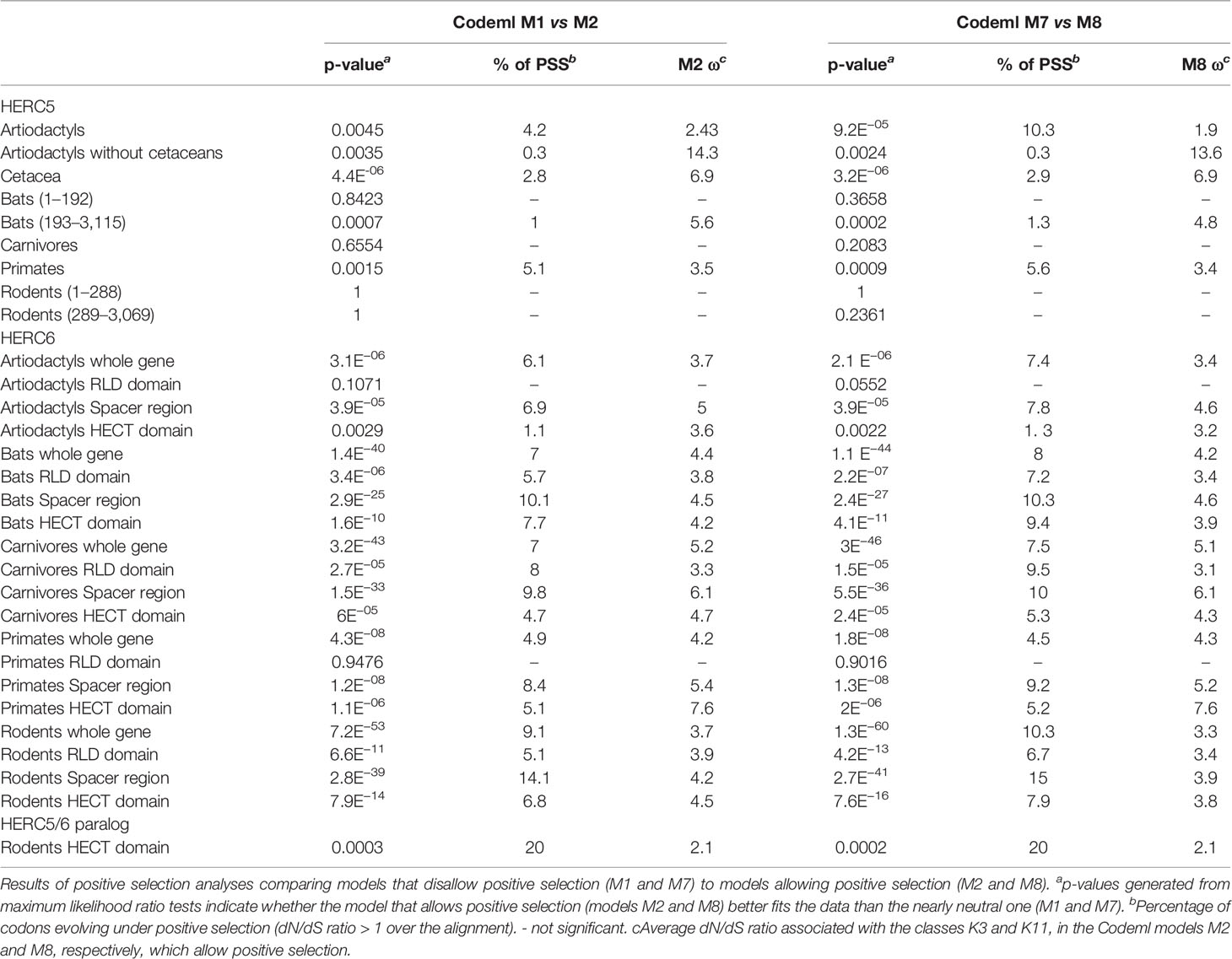
Table 1 Positive selection analyses of mammalian HERC5 and HERC6 genes.
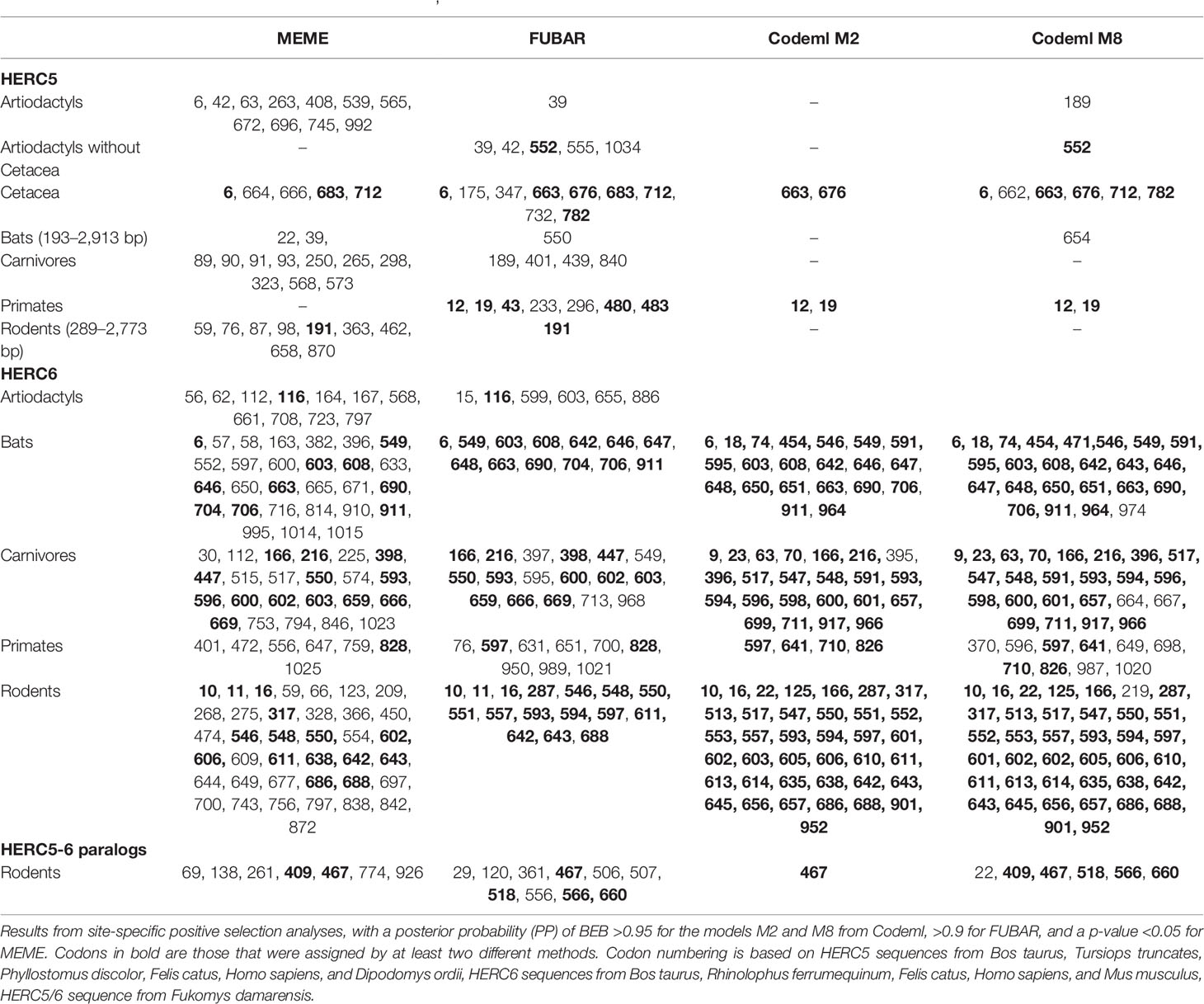
Table 2 Positively selected codons in mammalian HERC5, HERC6 and HERC5/6 genes.
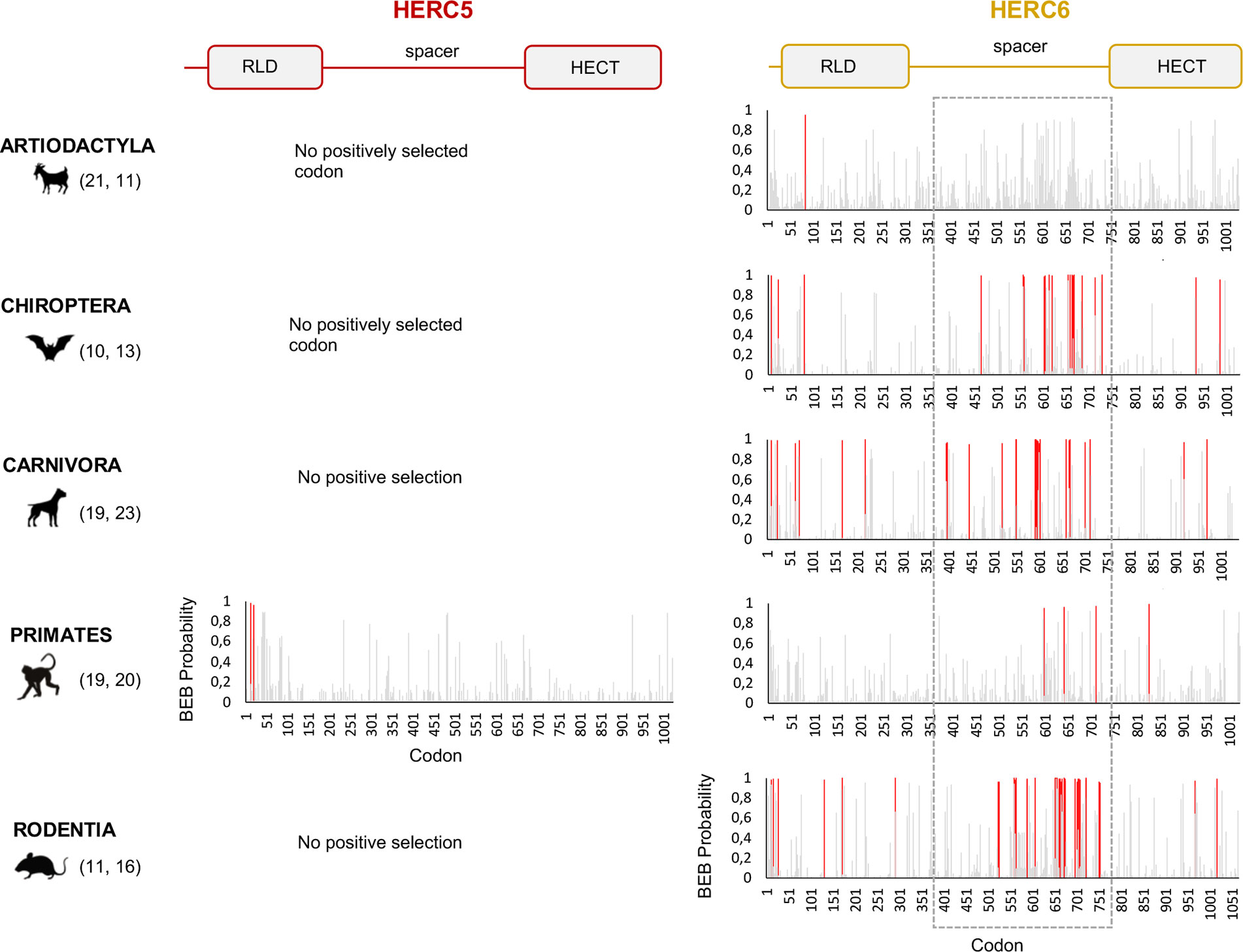
Figure 3 HERC6, and not HERC5, has experienced strong and mammalian-wide positive selection. Graphic panels represent the posterior probabilities of positive selection (Bayes empirical Bayes, BEB) (y axis) in the M2 Codeml model (allowing for positive selection, ω >1) for each codon (x axis) in HERC5 (left) and HERC6 (right) alignments. Red bars indicate the sites identified by both models, M2 and M8, with a BEB posterior probability greater than 0.95. Numbers in brackets are total species analyzed in each mammalian order for each gene. Site numbering is based on HERC5 protein sequences from Homo sapiens, HERC6 sequences from Bos taurus, Rhinolophus ferrumequinum, Felis catus, Homo sapiens, and Mus musculus. Above is a linear representation of HERC5 and HERC6 showing the structural domains, the RLD, spacer region, and HECT domains.
In contrast, we found very strong signatures of ancient and recurrent positive selection in HERC6, at both the gene and the codon levels. All five mammalian orders exhibited a significant excess of non-synonymous rate along the HERC6 coding sequences, specifically in bats, carnivores, and rodents (p-value <10−43 in bats, carnivores, and rodents; and p-value <10−6 in artiodactyls and primates). Positive selection was observed in each of the three domains of HERC6—the RLD domain, the spacer region, and the HECT domain—in bat, carnivore, and rodent species, while many of the signatures were concentrated in the spacer region and the HECT domains in primates and artiodactyls (Table 1, Figure 3). Remarkably, the fastest-evolving codons mapped into the HERC6 spacer region of all five mammalian orders (p-value <10−26 in bats, carnivores, and rodents, p-value <10−5 in artiodactyls and primates). More than 14 sites were identified by at least two methods in bats, carnivores, and rodents, thereby constituting a hotspot of highly variable sites in mammalian HERC6 (Table 2 and Figure 3). In line with this finding, alignment of the spacer region revealed that it is an extremely divergent domain, characterized by multiple amino acid changes and indels between and within groups, in particular in rodents and bats (Figure 4).

Figure 4 Rapid evolution of mammalian HERC6 spacer region is characterized by multiple amino acid changes and major indels. Multiple alignment and comparison of the HERC6 spacer region between and within mammalian orders. Left, cladogram with the number of sequences used for each clade (n = 6 to 9). Right, colors indicate site variations between the sequences as compared to the consensus sequence with a threshold of 25% (Geneious, Biomatters; blue/red, hydrophilic/hydrophobic residues), while gray represents similarity with the consensus. The average pairwise percentage of identity is graphically represented above in gray. The codon numbers are based on human HERC6 sequence.
Therefore, although HERC5 presents low evidence of positive selection in mammals, HERC6 has experienced very strong adaptive evolution. Both genes showed differential evolutionary profiles across/between mammals, with lineage-specific and domain-specific adaptations.
We then addressed whether the newly identified chimeric HERC5/6 gene, which contains the HERC5 RLD domain and the HERC6 spacer and HECT domains, has also experienced positive selection. As coding sequences from five rodent species were available, we assessed the inter-species evolutionary history of the chimeric HERC5/6 gene within this group. Of note, there were insufficient bat sequences/species to perform the corresponding analyses. The likelihood ratio tests revealed significant positive selection in rodent HERC5/6 (p-value <0.0003, Table 2). Importantly, all the positively selected codons mapped in the spacer region, and were concentrated between the amino acids 409 and 660 (Figure 3), suggesting that the spacer domain has been the target of strong positive selection as observed in HERC6.
Finally, we tested whether positive selection has differentially impacted the evolution of HERC5 and HERC6 across mammals. We found that the chiropteran and rodent HERC6 genes have experienced intensive episodic positive selection compared to the other groups (Figure 5). In particular, five chiropteran lineages distributed along bat phylogeny (Rhinolophus ferrumequinum, Pteropus ancestral branch, Phylllostomus discolor, Molossus molossus, and Pipistrellus kuhlii) and five rodent branches (two ancestral branches of mouse related clade, Cricetulus griseus, Mesocricetus auratus, and Urocitellus parryii) have undergone a significant excess of amino acid changes, with a ratio ω >3.8. Differential selective pressure in HERC5 was also evidenced across mammalian lineages, but to a lesser extent: only two branches were found under significant positive selection, in bats (Hipposideros armiger, ω = 189) and carnivores (Ailuoropoda melanoleuca, ω = 78).
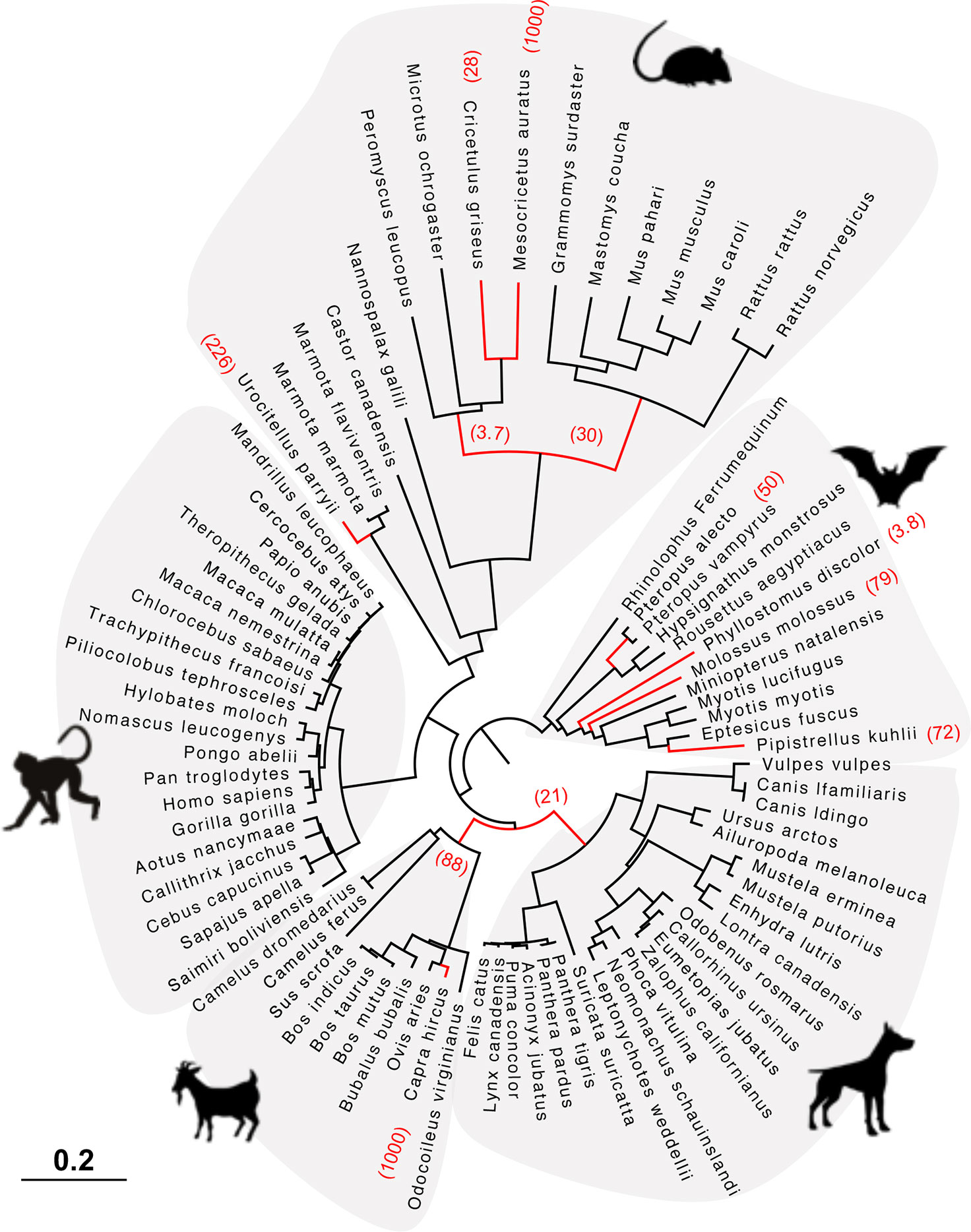
Figure 5 HERC6 has been under strong positive selection during rodent and bat evolution. Maximum likelihood phylogenetic tree of mammalian HERC6 gene showing the branches under significant positive selection (p-value <0.05, in red) assigned by aBSREL from the HYPHY package. The numbers in brackets indicate the estimated values of the ω at the branch. The scale bar indicates the proportion of genetic variation.
Deciphering the evolutionary and functional diversification of the antiviral innate immunity in mammals is of primary importance to better understand modern viral pathogens, virus-host interfaces, and identify novel antiviral strategies. The functional significance of HERC5 and HERC6 is underlined by their ancient origin and conserved expression in vertebrates (25). However, their evolutionary history in mammals has remained unclear. Here, we have carried out in-depth phylogenetic and genomic analyses to address how mammalian HERC5 and HERC6 have evolved over millions of years of divergence.
Although mammalian HERC5 was previously reported as a rapidly evolving gene (33), we only found strong evidences of recurrent positive selection in primates. In particular, two codons in primate HERC5 have rapidly evolved in the RLD domain, possibly reminiscent of pathogen-exerted pressure (3, 53–55). In line with this, blade 1 of the primate RLD domain was recently reported to be an important functional region for HERC5 anti-HIV antiviral activity (25). Thus, retroviruses may have played a role in the diversification of HERC5 during primate evolution. Such patterns have been reported in many primate restriction factors, including BST2 (56–58), TRIM5 (10, 59–61), and APOBEC3 (14, 62–64). Because the role of HERC5 is not limited to host defense against retroviruses, its evolution in primates may also reflect past selection against other viral pathogens such as influenza viruses and papillomaviruses.
In contrast, positive selection was solely evident at the gene level for artiodactyl and bat HERC5, and absent in rodents and carnivores. This pattern may be a result of lineage-specific selective drivers: differential viral exposure history, distinct mechanisms of viral antagonism, and/or may reflect overall pressure to maintain efficient cellular functions of HERC5. For example, apart from its antiviral role, some evidences suggest that HERC5 might be functionally involved in other pathways, such as spermatogenesis and cell cycle (22), as well as cancer (65). HERC5 may have thus evolved to maintain effective cellular functions rather than to escape viral antagonisms or to target viral pathogens in bats and artiodactyls. In mammals not exhibiting positive selection, viruses may have evolved indirect mechanisms of antagonism to counteract HERC5 function. In line with this, many viruses encode proteins that interfere with the ISGylation activity of HERC5, through direct interaction with the ISG15 protein (66–69). For example, the NS1B protein encoded by influenza virus antagonizes ISG15 conjugation through direct interaction (66).
HERC6 is the only gene from the small HERC family exhibiting such high levels of adaptive changes with an extremely divergent spacer region in all mammals, except artiodactyls. Such rapid evolution of HERC6, with accumulated mutations replacing the amino acids and multiple amino acid insertions/deletions, most likely mirrors pathogen-driven adaptations as a result of past evolutionary arms-races (3, 53–55). This highlights a fundamental antiviral role for HERC6 in mammals. Previous functional evidences support that HERC6 is involved in mammalian antiviral immune response (28, 34, 35, 66). However, available studies have only focused on HERC6 anti-retroviral activity and ISGylation function. For example, HERC6 has been shown to be an IFN-inducible E3-ligase of ISG15 conjugation in mouse (28, 34, 35). In contrast, human HERC6 lacks the ISGylation activity, but it potently inhibits the primate lentivirus SIVmac viral production (25).
Specifically, we identified the spacer region as a potential pathogen—HERC6 interface, involved in the recognition of viral proteins and/or being the target of viral antagonists. Up to now, most studies have been devoted to the functional characterization of the RLD and HECT domains of HERC proteins, as they belong to the well-characterized protein families, RCC1 and E3-ligases, respectively. In contrast, the structural characteristic of the spacer region has not been related to any known protein, which hinders its description and functional role. However, its propensity for amino acid insertions/deletions and accumulated non-synonymous mutations, including changes with strong chemicophysical differences highlights a strong evolutionary plasticity. Such a hotspot of variability in unstructured regions has been reported for several restriction factors, such as MX1, in which the highly variable L4 loop has led to differential virus-host interfaces and plasticity (70, 71).
It is noteworthy that the RLD and HECT domains of HERC6 were also subjected to positive selection in bats, carnivores, and rodents. These signatures may reflect different virus-host interfaces. This pattern was reported in the primate PKR protein, in which signatures of pathogen-driven selection are scattered along the protein as a result of interactions with multiple viral antagonists (72, 73). However, it is also possible that all positively selected sites cluster in a same spatial region of the protein. A 3D structural analysis of the HERC6 protein would help assessing how the rapidly evolving sites are located in the protein, and allow determining whether HERC6 presents multiple or unique host-pathogen interface(s). Up to now, the 3D structure has only been solved for the HECT domain and RLD domain separately [e.g (74, 75)]. Therefore, how the spacer region connects the HECT and RLD domains in a 3D structural dimension is currently unknown. Further studies on small HERC protein structure would help to better understand how the high variability in HERC6 impacts its structure and function.
We found differential genetic profiles of HERC6 across mammalian orders. The strongest signal was found in bats, carnivores, and rodents, suggesting that different strength/intensity of selective pressures have shaped the evolution of HERC6 in mammals. This was confirmed by the branch-specific analyses, in which rodents and bats exhibit significant lineage-specific adaptive changes. Rodents and bats are the two most diverse mammalian orders, and host the highest viral richness among mammals (38). Both orders have thus been exposed to a greater viral diversity, compared to primates and artiodactyls. This may have increased the strength and extent of selective pressure exerted on the HERC6 protein.
Lineage-specific expansions of multigene families have shaped and complexified the mammalian antiviral repertoire over million years of evolution. Consequently, many unrecognized genes encoding for antiviral proteins are yet to be discovered. Here, we have identified duplications of a HERC paralog in the rodent Hystricognathi infra-order and the chiropteran Myotis genus, which has occurred around 30 MYA (76, 77) and 44 MYA (78–80), respectively.
Interestingly, these paralogs are chimeric HERC5/6 genes coding for the HERC5 RLD fused to the HERC6 spacer region and HECT domain. This suggests that an independent duplication has occurred through a similar mechanism in bats and rodents. Gene duplication can occur by different modes, mainly including unequal crossing over, retroposition, or chromosomal duplication (81, 82). In the former case, duplicated genes are physically linked in the chromosome, and can contain a fragment of a gene, a whole gene, or several genes (81). In contrast, retroposition engenders a retrotranscribed complementary DNA incorporated into the genome, which generally lacks intronic regions and regulatory sequences (81). Given that the HERC5/6 are located in the canonical locus of HERC5 and HERC6 and contain the parental intronic regions, they have most likely resulted from an unequal crossing over between HERC5 and HERC6.
This hypothesis is supported by the recombination analyses, which detected a significant breakpoint upstream of the spacer region in both mammalian orders. The fact that the recombination occurred at the same genetic location can be explained by two different, but not necessarily mutually exclusive, hypotheses. First, the recombination event can only occur at this location because of genomic structural constraints (i.e. genomic homology between paralogs). Second, the HERC6 spacer region and HECT domains are required for functional HERC5/6 proteins. The best example of such tandem duplication with domain fusion is the lineage-specific expansion of the APOBEC3 family in mammals. Originating from an ancestral APOBEC3 gene, tandem duplications as well as retrocopying events have radically expanded the repertoire of mammalian APOBEC3 genes (14, 20). In primates, several of the APOBEC3 genes have most likely resulted from the fusion of A3 domains, while the murine genome encodes a unique APOBEC3Z2-APOBEC3Z3 fused gene (83), highlighting lineage specific functional adaptations.
Such gene duplications accompanied with gene fusion are major genetic innovations that functionally diversify the antiviral arsenal (3, 4, 7). For example, the expansion of the APOBEC3 family has functionally diversified the antiviral activities and specificity of targeted viruses in primates (20, 62, 84). Given the antiviral role of HERC5 and the potential implication of HERC6 in antiviral immunity, we hypothesize that the HERC5/6 paralog provides a functional advantage against pathogenic viruses. This is supported by both the extremely rapid episodic evolution of HERC6 in rodents and bats, and the signatures of positive selection in rodent HERC5/6. The fused HERC5/6 gene may have evolved combined functional features of HERC5 and HERC6. Alternatively, it may have retained the functional implication of the HERC5 RLD domain, but has functionally diverged from HERC6.
This latter hypothesis is more likely as we found that the RLD domain of HERC5 and of HERC5/6 are highly similar and cluster together within species (more than 95% pairwise amino acid identity). This pattern may reflect ongoing gene conversions between the RLD domains of HERC5 and HERC5/6, thereby maintaining a N-terminal similar to the parental HERC5 protein.
Whether the independent fixation of the HERC5/6 paralog in rodents and bats is a functional/phenotypic convergent evolutionary event has to be investigated. Moreover, it raises the question why both lineages have undergone such genetic innovation. While this duplication could be hazardous, it is possible that the rodent Hystricognathi infra-order and the bat Myotis genus share a common selective pressure, such as a viral pathogen family. Other forces such as ecological and/or environmental factors may have played a role, such as life-history traits or biodiversity changes.
Studying the genetic adaptations of host innate immunity can provide insights into the evolutionary and functional determinants of host antiviral response. This approach has been a powerful tool for assessing the functional diversification of virus-host interfaces in many systems [e.g (59, 62, 85, 86)]. In this present study, we identified HERC6 and HERC5/6 as potential unrecognized restriction factors in mammals. Further functional investigations are now required to (i) decipher the antiviral function of HERC6 and HERC5/6, (ii) determine how the accumulated variability in the spacer domain may impact their structure, function, and stability, (iii) unravel HERC6 binding interface with potential viral antagonists or targeted viral proteins, and (iv) determine on the other side which pathogens are targeted by those proteins. It will also be interesting to investigate whether and how the evolution of HERCs may impact the cellular partners involved in the ISGylation functions. For example, although we found that ISG15 is mostly conserved in mammals, it bears important variations at the C-terminal (data not shown). This could reflect host protein-protein co-evolutions or indirect viral antagonism of HERC restriction. Moreover, because the HERCs have been mostly studied in primates and rodents, it is possible that other proteins than ISG15 are involved in the ISGylation function in other mammals. In addition, deciphering whether and how the HERC5/6 paralogs afford a selective advantage against pathogens in rodent and bat lineages is of main interest in virology and immunology fields, as both orders are important reservoirs of zoonotic pathogens. Finally, studying the functional implications of HERC6 adaptation in mammals will not only allow to better understand how pathogens have shaped host immunity, but will provide important insights into the overlooked role of small HERC proteins in mammalian antiviral response.
Altogether, our results represent avenues for future functional studies of importance in mammalian innate immunity.
Publicly available datasets were analyzed in this study. This data can be found here: NCBI (https://www.ncbi.nlm.nih.gov/) and accession numbers are available in Supplementary Table 1.
SJ carried out the analyses of the data. SJ, LE, and DP performed the investigations. SJ, LE, and DP wrote the original draft of the paper. LE and DP acquired the funding, and coordinated the study. All authors contributed to the article and approved the submitted version.
This work is funded by the ANR LABEX ECOFECT (ANR-11-LABX-0048 of the Université de Lyon, within the program Investissements d’Avenir [ANR-11-IDEX-0007] operated by the French National Research Agency). LE is further supported by the CNRS and by grants from amfAR (Mathilde Krim Phase II Fellowship no. 109140-58-RKHF), the Fondation pour la Recherche Médicale (FRM Projet Innovant no. ING20160435028), the FINOVI (“recently settled scientist” grant), the ANRS (no. ECTZ19143 and ECTZ118944), and a JORISS incubating grant. DP is supported by the CNRS, the European Regional Development Fund (ERDF) 2018-4512410/convention P-2020-BAFE-9 and 2018-4729510/convention P-2020-BAFE-23, and the ANR EBOFAC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are particularly grateful to Adil El Filali (UMR5558) for his help on genomic analyses, and Laurent Duret (UMR5558) for his comments on the study. We thank Andrea Cimarelli, head of the Host-Pathogen Interaction during Lentiviral Infection team at the CIRI Lyon, as well as the members of the “Intercontinental Retrovirus Zoomposium” for helpful discussion. We also thank all the contributors of the LBBE (UMR5558) bioinformatic server and the publicly available genome sequences. We would also like to thank the reviewers for their comments and suggestions on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.605270/full#supplementary-material
Supplementary Figure 1 | Maximum likelihood phylogenetic tree generated with the whole coding sequences of HERC5, HERC6, and HERC3 nucleotide alignment from artiodactyl, carnivore, rodent, bat, and primate species. Asterisks indicate bootstrap values greater than 80%. The scale bar represents the proportion of genetic variation (0.2 for the scale), and is indicated at the bottom. Sequences are collapsed in each order for better readability.
Supplementary Figure 2 | Pseudogenization of cetacean HERC6 (A). Multiple amino acid alignment of six cetacean HERC6 sequences showing multiple substitutions, insertions, and deletions (B). Multiple nucleotide alignment with corresponding amino acids of cetacean, rodent, bat, ruminant, primate, and carnivore HERC6, highlighting a conserved stop codon in the cetacean species (codon 174 in Balaenoptera acutorostrata). Nucleotide and amino acid sequences are shown using Geneious.
Supplementary Figure 3 | Maximum likelihood phylogenetic tree generated with the whole coding sequences of HERC5, HERC6, HERC5/6, and HERC3 nucleotide alignment in bats (left) and rodents (right). The chimeric duplicated HERC5/6 genes are shown in red. Asterisks indicate bootstrap values greater than 80%. The scale bar at 0.2 is indicated below.
Supplementary Figure 4 | Alignment of the protein sequence of HERC5, HERC5/6, and HERC6 from bats and rodents. The percentages of pairwise amino acid identities between the N-terminals of HERC5/6 and HERC5 or HERC6, as well as the C-terminals of HERC5/6 and HERC5 or HERC6 are indicated. The significant recombination breakpoints (red arrows, p-value <0.05) assigned by GARD program are shown for bat and rodent HERC5/6 gene. Because the coding sequence of HERC6 gene from Heterocephalus glaber was incomplete with many missing data, it was not included in the figure. Likewise, some portions of the N-terminal of HERC5, as well as the C-terminals, from HERC5/6 and HERC6 are missing in the protein alignment of the chiropteran species, Myotis brandtii (top).
Supplementary Table 1 | Information on publicly available datasets analyzed in this study. Accession numbers are available in NCBI (https://www.ncbi.nlm.nih.gov/).
1. Hubel P, Urban C, Bergant V, Schneider WM, Knauer B, Stukalov A, et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat Immunol (2019) 20:493–502. doi: 10.1038/s41590-019-0323-3
2. Kluge SF, Sauter D, Kirchhoff F. SnapShot: Antiviral Restriction Factors. Cell (2015) 163:774–774e1. doi: 10.1016/j.cell.2015.10.019
3. Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol (2012) 12:687–95. doi: 10.1038/nri3295
4. Daugherty MD, Malik HS. Rules of Engagement: Molecular Insights from Host-Virus Arms Races. Annu Rev Genet (2012) 46:677–700. doi: 10.1146/annurev-genet-110711-155522
5. Sironi M, Cagliani R, Forni D, Clerici M. Evolutionary insights into host–pathogen interactions from mammalian sequence data. Nat Rev Genet (2015) 16:224–36. doi: 10.1038/nrg3905
6. Hölzer M, Schoen A, Wulle J, Müller MA, Drosten C, Marz M, et al. Virus- and Interferon Alpha-Induced Transcriptomes of Cells from the Microbat Myotis daubentonii. iScience (2019) 19:647–61. doi: 10.1016/j.isci.2019.08.016
7. Daugherty MD, Zanders SE. Gene conversion generates evolutionary novelty that fuels genetic conflicts. Curr Opin Genet Dev (2019) 58–59:49–54. doi: 10.1016/j.gde.2019.07.011
8. Kondrashov FA. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc R Soc B Biol Sci (2012) 279:5048–57. doi: 10.1098/rspb.2012.1108
9. Tareen SU, Sawyer SL, Malik HS, Emerman M. An expanded clade of rodent Trim5 genes. Virology (2009) 385:473–83. doi: 10.1016/j.virol.2008.12.018
10. Sawyer SL, Emerman M, Malik HS. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PloS Pathog (2007) 3:1918–29. doi: 10.1371/journal.ppat.0030197
11. Boso G, Shaffer E, Liu Q, Cavanna K, Buckler-White A, Kozak CA. Evolution of the rodent Trim5 cluster is marked by divergent paralogous expansions and independent acquisitions of TrimCyp fusions. Sci Rep (2019) 9:1–14. doi: 10.1038/s41598-019-47720-5
12. Malfavon-Borja R, Sawyer SL, Wu LI, Emerman M, Malik HS. An Evolutionary Screen Highlights Canonical and Noncanonical Candidate Antiviral Genes within the Primate TRIM Gene Family. Genome Biol Evol (2013) 5:2141–54. doi: 10.1093/gbe/evt163
13. Münk C, Willemsen A, Bravo IG. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol Biol (2012) 12:71. doi: 10.1186/1471-2148-12-71
14. Ito J, Gifford RJ, Sato K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc Natl Acad Sci U S A (2020) 117:610–8. doi: 10.1073/pnas.1914183116
15. Hayward JA, Tachedjian M, Cui J, Cheng AZ, Johnson A, Baker ML, et al. Differential evolution of antiretroviral restriction factors in pteropid bats as revealed by APOBEC3 gene complexity. Mol Biol Evol (2018) 35:1626–37. doi: 10.1093/molbev/msy048
16. Liu Y, Zhang YB, Liu TK, Gui JF. Lineage-Specific Expansion of IFIT Gene Family: An Insight into Coevolution with IFN Gene Family. PloS One (2013) 8(6):e66859. doi: 10.1371/journal.pone.0066859
17. Daugherty MD, Schaller AM, Geballe AP, Malik HS. Evolution-guided functional analyses reveal diverse antiviral specificities encoded by ifit1 genes in mammals. eLife (2016) 5:1–22. doi: 10.7554/eLife.14228
18. Zhou X, Michal JJ, Zhang L, Ding B, Lunney JK, Liu B, et al. Interferon induced IFIT family genes in host antiviral defense. Int J Biol Sci (2013) 9:200–8. doi: 10.7150/ijbs.5613
19. Hickford D, Frankenberg S, Shaw G, Renfree MB. Evolution of vertebrate interferon inducible transmembrane proteins. BMC Genomics (2012) 13. doi: 10.1186/1471-2164-13-155
20. Yang L, Emerman M, Malik HS, McLaughlin Jnr RN. Retrocopying expands the functional repertoire of APOBEC3 antiviral proteins in primates. eLife (2020) 9:1–18. doi: 10.7554/eLife.58436
21. LaRue RS, Jónsson SR, Silverstein KAT, Lajoie M, Bertrand D, El-Mabrouk N, et al. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol (2008) 9:1–20. doi: 10.1186/1471-2199-9-104
22. Garcia-Gonzalo FR, Rosa JL. The HERC proteins: Functional and evolutionary insights. Cell Mol Life Sci (2005) 62:1826–38. doi: 10.1007/s00018-005-5119-y
23. Hochrainer K, Mayer H, Baranyi U, Binder BR, Lipp J, Kroismayr R. The human HERC family of ubiquitin ligases: Novel members, genomic organization, expression profiling, and evolutionary aspects. Genomics (2005) 85:153–64. doi: 10.1016/j.ygeno.2004.10.006
24. Marin I. Animal HECT ubiquitin ligases: Evolution and functional implications. BMC Evol Biol (2010) 10:1–12. doi: 10.1186/1471-2148-10-56
25. Paparisto E, Woods MW, Coleman MD, Moghadasi SA, Kochar DS, Tom SK, et al. Evolution-Guided Structural and Functional Analyses of the HERC Family Reveal an Ancient Marine Origin and Determinants of Antiviral Activity. J Virol (2018) 92:e00528–18. doi: 10.1128/jvi.00528-18
26. Wong JJY, Pung YF, Sze NSK, Chin KC. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc Natl Acad Sci U S A (2006) 103:10735–40. doi: 10.1073/pnas.0600397103
27. Dastur A, Beaudenon S, Kelley M, Krug RM, Huibregtse JM. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J Biol Chem (2006) 281:4334–8. doi: 10.1074/jbc.M512830200
28. Ketscher L, Basters A, Prinz M, Knobeloch KP. MHERC6 is the essential ISG15 E3 ligase in the murine system. Biochem Biophys Res Commun (2012) 417:135–40. doi: 10.1016/j.bbrc.2011.11.071
29. Villarroya-Beltri C, Guerra S, Sánchez-Madrid F. ISGylation - a key to lock the cell gates for preventing the spread of threats. J Cell Sci (2017) 130:2961–9. doi: 10.1242/jcs.205468
30. Woods MW, Kelly JN, Hattlmann CJ, Tong JGK, Xu LS, Coleman MD, et al. Human HERC5 restricts an early stage of HIV-1 assembly by a mechanism correlating with the ISGylation of Gag. Retrovirology (2011) 8:95. doi: 10.1186/1742-4690-8-95
31. Zhao C, Hsiang TY, Kuo RL, Krug RM. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc Natl Acad Sci U S A (2010) 107:2253–8. doi: 10.1073/pnas.0909144107
32. Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol Cell (2010) 38:722–32. doi: 10.1016/j.molcel.2010.05.002
33. Woods MW, Tong JG, Tom SK, Szabo PA, Cavanagh PC, Dikeakos JD, et al. Interferon-induced HERC5 is evolving under positive selection and inhibits HIV-1 particle production by a novel mechanism targeting Rev/RRE-dependent RNA nuclear export. Retrovirology (2014) 11:1–16. doi: 10.1186/1742-4690-11-27
34. Oudshoorn D, van Boheemen S, Sánchez-Aparicio MT, Rajsbaum R, García-Sastre A, Versteeg GA. HERC6 is the main E3 ligase for global ISG15 conjugation in mouse cells. PloS One (2012) 7(1):e29870. doi: 10.1371/journal.pone.0029870
35. Arimoto KI, Hishiki T, Kiyonari H, Abe T, Cheng C, Yan M, et al. Murine HERC6 plays a critical role in protein ISGylation in vivo and has an ISGylation-independent function in seminal vesicles. J Interferon Cytokine Res (2015) 35:351–8. doi: 10.1089/jir.2014.0113
36. McBee RM, Rozmiarek SA, Meyerson NR, Rowley PA, Sawyer SL. The effect of species representation on the detection of positive selection in primate gene data sets. Mol Biol Evol (2015) 32:1091–6. doi: 10.1093/molbev/msu399
37. Picard L, Ganivet Q, Allatif O, Cimarelli A, Gueguen L, Etienne L. DGINN, an automated and highly-flexible pipeline for the Detection of Genetic INNovations on protein-coding genes. Nucleic Acids Res (2020) 48(18):e103. doi: 10.1093/nar/gkaa680
38. Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. Host and viral traits predict zoonotic spillover from mammals. Nature (2017) 546:646–50. doi: 10.1038/nature22975
39. Mollentze N, Streicker DG. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc Natl Acad Sci U S A (2020) 117:9423–30. doi: 10.1073/pnas.1919176117
40. NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res (2016) 44:D7–19. doi: 10.1093/nar/gkv1290
41. Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I. GenBank. Nucleic Acids Res (2018) 47:D94–9. doi: 10.1093/nar/gky989
42. Ranwez V, Douzery EJP, Cambon C, Chantret N, Delsuc F. MACSE v2: Toolkit for the Alignment of Coding Sequences Accounting for Frameshifts and Stop Codons. Mol Biol Evol (2018) 35:2582–4. doi: 10.1093/molbev/msy159
43. Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst Biol (2010) 59:307–21. doi: 10.1093/sysbio/syq010
44. Lefort V, Longueville J-E, Gascuel O. SMS: Smart Model Selection in PhyML. Mol Biol Evol (2017) 34:2422–4. doi: 10.1093/molbev/msx149
45. Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW. GARD: a genetic algorithm for recombination detection. Bioinformatics (Oxford England) (2006) 22:3096–8. doi: 10.1093/bioinformatics/btl474
46. Pond SLK, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics (2004) 21:676–9. doi: 10.1093/bioinformatics/bti079
47. Kosakovsky Pond SL, Poon AFY, Velazquez R, Weaver S, Hepler NL, Murrell B, et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol Biol Evol (2019) 37:295–9. doi: 10.1093/molbev/msz197
48. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol (2007) 24:1586–91. doi: 10.1093/molbev/msm088
49. Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol (2013) 30:1196–205. doi: 10.1093/molbev/mst030
50. Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting Individual Sites Subject to Episodic Diversifying Selection. PloS Genet (2012) 8:e1002764. doi: 10.1371/journal.pgen.1002764
51. Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Mol Biol Evol (2015) 32:1342–53. doi: 10.1093/molbev/msv022
52. Kosakovsky Pond SL, Murrell B, Fourment M, Frost SDW, Delport W, Scheffler K. A Random Effects Branch-Site Model for Detecting Episodic Diversifying Selection. Mol Biol Evol (2011) 28:3033–43. doi: 10.1093/molbev/msr125
53. McLaughlin RN, Malik HS. Genetic conflicts: the usual suspects and beyond. J Exp Biol (2017) 220:6–17. doi: 10.1242/jeb.148148
54. Meyerson NR, Sawyer SL. Two-stepping through time: Mammals and viruses. Trends Microbiol (2011) 19:286–94. doi: 10.1016/j.tim.2011.03.006
55. Sauter D, Kirchhoff F. Key Viral Adaptations Preceding the AIDS Pandemic. Cell Host Microbe (2019) 25:27–38. doi: 10.1016/j.chom.2018.12.002
56. Blanco-Melo D, Venkatesh S, Bieniasz PD. Origins and Evolution of tetherin, an Orphan Antiviral Gene. Cell Host Microbe (2016) 20:189–201. doi: 10.1016/j.chom.2016.06.007
57. Heusinger E, Kluge SF, Kirchhoff F, Sauter D. Early Vertebrate Evolution of the Host Restriction Factor Tetherin. J Virol (2015) 89:12154–65. doi: 10.1128/jvi.02149-15
58. Lim ES, Malik HS, Emerman M. Ancient Adaptive Evolution of Tetherin Shaped the Functions of Vpu and Nef in Human Immunodeficiency Virus and Primate Lentiviruses. J Virol (2010) 84:7124–34. doi: 10.1128/jvi.00468-10
59. Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A (2005) 102:2832–7. doi: 10.1073/pnas.0409853102
60. McCarthy KR, Kirmaier A, Autissier P, Johnson WE. Evolutionary and Functional Analysis of Old World Primate TRIM5 Reveals the Ancient Emergence of Primate Lentiviruses and Convergent Evolution Targeting a Conserved Capsid Interface. PloS Pathog (2015) 11:1–26. doi: 10.1371/journal.ppat.1005085
61. Wu F, Kirmaier A, Goeken R, Ourmanov I, Hall L, Morgan JS, et al. TRIM5 alpha Drives SIVsmm Evolution in Rhesus Macaques. PloS Pathog (2013) 9(8):e1003577. doi: 10.1371/journal.ppat.1003577
62. Sawyer SL, Emerman M, Malik HS. Ancient Adaptive Evolution of the Primate Antiviral DNA-Editing Enzyme APOBEC3G. PloS Biol (2004) 2:e275. doi: 10.1371/journal.pbio.0020275
63. Zhang Z, Gu Q, de Manuel Montero M, Bravo IG, Marques-Bonet T, Häussinger D, et al. Stably expressed APOBEC3H forms a barrier for cross-species transmission of simian immunodeficiency virus of chimpanzee to humans. PloS Pathog (2017) 13:1–25. doi: 10.1371/journal.ppat.1006746
64. Compton AA, Hirsch VM, Emerman M. The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe (2012) 11:91–8. doi: 10.1016/j.chom.2011.11.010
65. Xue F, Higgs BW, Huang J, Morehouse C, Zhu W, Yao X, et al. HERC5 is a prognostic biomarker for post-liver transplant recurrent human hepatocellular carcinoma. J Trans Med (2015) 13:379. doi: 10.1186/s12967-015-0743-2
66. Versteeg GA, Hale BG, van Boheemen S, Wolff T, Lenschow DJ, García-Sastre A. Species-Specific Antagonism of Host ISGylation by the Influenza B Virus NS1 Protein. J Virol (2010) 84:5423–30. doi: 10.1128/jvi.02395-09
67. Eduardo-Correia B, Martínez-Romero C, García-Sastre A, Guerra S. ISG15 is counteracted by vaccinia virus E3 protein and controls the proinflammatory response against viral infection. J Virol (2014) 88:2312–8. doi: 10.1128/JVI.03293-13
68. Swatek KN, Aumayr M, Pruneda JN, Visser LJ, Berryman S, Kueck AF, et al. Irreversible inactivation of ISG15 by a viral leader protease enables alternative infection detection strategies. Proc Natl Acad Sci (2018) 115:2371–76. doi: 10.1073/pnas.1710617115
69. Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J Virol (2012) 86:3839–50. doi: 10.1128/JVI.06466-11
70. Mitchell PS, Patzina C, Emerman M, Haller O, Malik HS, Kochs G. Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe (2012) 12:598–604. doi: 10.1016/j.chom.2012.09.005
71. Mitchell PS, Emerman M, Malik HS. An evolutionary perspective on the broad antiviral specificity of MxA. Curr Opin Microbiol (2013) 16:493–9. doi: 10.1016/j.mib.2013.04.005
72. Elde NC, Child SJ, Geballe AP, Malik HS. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature (2009) 457:485–9. doi: 10.1038/nature07529
73. Rothenburg S, Seo EJ, Gibbs JS, Dever TE, Dittmar K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat Struct Mol Biol (2009) 16:63–70. doi: 10.1038/nsmb.1529
74. Renault L, Nassar N, Vetter I, Becker J, Klebe C, Roth M, et al. The 1.7 Å crystal structure of the regulator of chromosome condensation (RCC1) reveals a seven-bladed propeller. Nature (1998) 392:97–101. doi: 10.1038/32204
75. Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, et al. Structure of an E6AP-UbcH7 Complex: Insights into Ubiquitination by the E2-E3 Enzyme Cascade. Science (1999) 286:1321–6. doi: 10.1126/science.286.5443.1321
76. Teeling EC, Springer MS, Madsen O, Bates P, O’Brien SJ, Murphy WJ. A molecular phylogeny for bats illuminates biogeography and the fossil record. Supporting online material. Science (2005) 307:580–4. doi: 10.1126/science.1105113
77. Stadelmann B, Lin LK, Kunz TH, Ruedi M. Molecular phylogeny of New World Myotis (Chiroptera, Vespertilionidae) inferred from mitochondrial and nuclear DNA genes. Mol Phylogenet Evol (2007) 43:32–48. doi: 10.1016/j.ympev.2006.06.019
78. Opazo JC. A molecular timescale for caviomorph rodents (Mammalia, Hystricognathi). Mol Phylogenet Evol (2005) 37:932–7. doi: 10.1016/j.ympev.2005.05.002
79. Fabre PH, Hautier L, Dimitrov D, P Douzery EJ. A glimpse on the pattern of rodent diversification: A phylogenetic approach. BMC Evol Biol (2012) 12:88. doi: 10.1186/1471-2148-12-88
80. Voloch CM, Vilela JF, Loss-Oliveira L, Schrago CG. Phylogeny and chronology of the major lineages of New World hystricognath rodents: Insights on the biogeography of the Eocene/Oligocene arrival of mammals in South America. BMC Res Notes (2013) 6:1–9. doi: 10.1186/1756-0500-6-160
81. Zhang J. Evolution by gene duplication: An update. Trends Ecol Evol (2003) 18:292–8. doi: 10.1016/S0169-5347(03)00033-8
82. Hurles M. Gene Duplication: The Genomic Trade in Spare Parts. PloS Biol (2004) 2:e206. doi: 10.1371/journal.pbio.0020206
83. Conticello SG, Thomas CJF, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC Family of Polynucleotide (Deoxy)cytidine Deaminases. Mol Biol Evol (2005) 22:367–77. doi: 10.1093/molbev/msi026
84. Etienne L, Bibollet-Ruche F, Sudmant PH, Wu LI, Hahn BH, Emerman M. The Role of the Antiviral APOBEC3 Gene Family in Protecting Chimpanzees against Lentiviruses from Monkeys. PloS Pathog (2015) 11:e1005149. doi: 10.1371/journal.ppat.1005149
85. Laguette N, Rahm N, Sobhian B, Chable-Bessia C, Münch J, Snoeck J, et al. Evolutionary and functional analyses of the interaction between the myeloid restriction factor SAMHD1 and the lentiviral Vpx protein. Cell Host Microbe (2012) 11:205–17. doi: 10.1016/j.chom.2012.01.007
Keywords: HERC, restriction factor, antiviral immunity, gene duplication, genetic conflicts, positive selection, HERC5, HERC6
Citation: Jacquet S, Pontier D and Etienne L (2020) Rapid Evolution of HERC6 and Duplication of a Chimeric HERC5/6 Gene in Rodents and Bats Suggest an Overlooked Role of HERCs in Mammalian Immunity. Front. Immunol. 11:605270. doi: 10.3389/fimmu.2020.605270
Received: 14 September 2020; Accepted: 10 November 2020;
Published: 18 December 2020.
Edited by:
Christelle Langevin, Institut National de Recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Stephen Barr, Western University, CanadaCopyright © 2020 Jacquet, Pontier and Etienne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stéphanie Jacquet, c3RlcGhhbmllLmphY3F1ZXRAdW5pdi1seW9uMS5mcg==; Dominique Pontier, ZG9taW5pcXVlLnBvbnRpZXJAdW5pdi1seW9uMS5mcg==; Lucie Etienne, bHVjaWUuZXRpZW5uZUBlbnMtbHlvbi5mcg==
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.