 Xiangyun Li
Xiangyun Li Nathanial K. Berg
Nathanial K. Berg Tingting Mills
Tingting Mills Kaiying Zhang1
Kaiying Zhang1 Holger K. Eltzschig
Holger K. Eltzschig Xiaoyi Yuan
Xiaoyi Yuan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 January 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.604944
This article is part of the Research Topic Purinergic Signaling and Inflammation View all 10 articles
Hypoxia and inflammation often coincide in pathogenic conditions such as acute respiratory distress syndrome (ARDS) and chronic lung diseases, which are significant contributors to morbidity and mortality for the general population. For example, the recent global outbreak of Coronavirus disease 2019 (COVID-19) has placed viral infection-induced ARDS under the spotlight. Moreover, chronic lung disease ranks the third leading cause of death in the United States. Hypoxia signaling plays a diverse role in both acute and chronic lung inflammation, which could partially be explained by the divergent function of downstream target pathways such as adenosine signaling. Particularly, hypoxia signaling activates adenosine signaling to inhibit the inflammatory response in ARDS, while in chronic lung diseases, it promotes inflammation and tissue injury. In this review, we discuss the role of adenosine at the interphase of hypoxia and inflammation in ARDS and chronic lung diseases, as well as the current strategy for therapeutic targeting of the adenosine signaling pathway.
Acute respiratory distress syndrome (ARDS) is common in critically ill patients, characterized by respiratory failure, pulmonary edema independent of left heart failure, as well as high morbidity and mortality (1). The mortality rate was 30–40% in the most recent studies despite the latest improvement in clinical management (2). Pathological characters of ARDS in the acute “exudative” phase (~7 days) include alveolar epithelial and endothelial injury, resulting in interstitial and alveolar edema, hyaline membrane formation, and alveolar hemorrhage, as well as the accumulation of immune cells (1, 3). The main causes for ARDS include pneumonia, aspiration of gastric contents, severe trauma as well as sepsis (1, 4, 5). The recent global outbreak of Coronavirus disease 2019 (COVID-19) has placed viral infection-induced ARDS under the spotlight. COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and has resulted in a worldwide pandemic rapidly because of its high transmissibility and pathogenicity (6). ARDS is one of the most common organ dysfunctions for severe COVID-19, which accounts for the cause of death in 70% of fatal cases (7, 8). There are several emerging viruses in the past 20 years, which can induce ARDS-related mortality, such as influenza H1N1 2009, influenza H5N1 and H7N9 viruses, the severe acute respiratory syndrome coronavirus (SARS), and Middle East respiratory syndrome coronavirus (MERS) (9). It is reported that about 30–40% of the hospitalized patients infected with influenza virus progress to pneumonia, and influenza A shows a higher predisposition to ARDS in adults (10). Compared to SARS (10%) and MERS (35%), COVID-19 shows lower mortality rates of approximately 5.2%, but higher infectiousness (9, 11). As of September 6th, 2020, the pandemic of COVID-19 had affected over 26 million individuals around the world and caused more than 800,000 deaths worldwide. Therefore, the search for effective therapeutic approaches for the preventing and treatment of COVID-19 associated ARDS has become an urgency. Currently, although there are certain improvements in the management of ARDS, the treatment for ARDS is in urgent need. Therefore, the fundamental pathogenesis and effective treatments for ARDS are still under intensive investigation.
Persistent pulmonary inflammation and tissue remodeling result in the gradual decline in pulmonary function in patients suffering from chronic lung diseases including chronic obstructive pulmonary disease (COPD), asthma, and idiopathic pulmonary fibrosis (IPF) (3, 12–16). Chronic lung disease ranks the third leading cause of death in the United States. The risk factors of chronic lung diseases included genes, environmental factors, and aging (3, 12, 13, 17). However, one of the common characteristics among these diseases is dysregulated recruitment or activation of immune cells, such as neutrophils, macrophages, dendritic cells, and other effector cells, such as fibroblasts, myofibroblasts, and airway epithelial cells (AECs), which accelerates pulmonary remodeling and inflammatory response (3, 4). The therapeutic approaches for chronic lung diseases focus on providing symptomatic relief, but pharmacologic compounds are still lacking to reverse the profound tissue remodeling and restore lung function in these patients.
Adenosine was first isolated from the heart muscle and identified as an “adenine compound” that could change cardiac rhythm when injected in guinea pigs in 1927 (18). Besides its function in cardiac rhythm, adenosine also modulates inflammatory responses during hypoxic conditions (19–21). In this review, we will discuss the interaction between hypoxia and adenosine signaling, including adenosine, adenosine receptors, and adenosine metabolism, in acute lung inflammation and chronic lung diseases. We will also focus on the currently available approaches for therapeutic targeting of the hypoxia-adenosine axis in these disease conditions.
Adenosine, along with ATP and ADP, is considered the main purinergic signaling molecules (Figure 1). The release of ATP from intracellular to the extracellular environment contributes to the formation of adenosine especially when the tissue is in inflammatory, ischemic, and hypoxic conditions (23, 24). ATP/ADP in the extracellular space can be converted to adenosine monophosphate (AMP) by ectonucleoside triphosphate diphosphohydrolase-1 (CD39) (25, 26). Then AMP is further converted by ecto-5’-nucleotidase (CD73) to extracellular adenosine (25, 26). Mice with CD39 or CD73 deficiency are viable, which indicates that nucleotide phosphohydrolysis regulated by ectoenzymes is not vital in regular physiologic conditions (27). However, ectonucleotidases still have a crucial role in disease conditions. For example, the upregulation of adenosine generation and CD39 and CD73 expression is one of the protective mechanisms to reduce apoptosis, and alleviate inflammation in kidney ischemia/reperfusion (I/R) injury models (28). The deletion of CD39 in mice leads to increased level of ATP/ADP, and reduced adenosine levels, along with elevated risk of dysregulated inflammation and tissue injury (29, 30). Similarly, genetic deletion of CD73 results in higher mortality and delayed acute lung injury resolution when compared with WT mice because of the dampened generation of adenosine in regulatory T cells (Tregs) (31). Therefore, the conversion of ATP/ADP to adenosine is considered beneficial in many ischemic and inflammatory disorders.
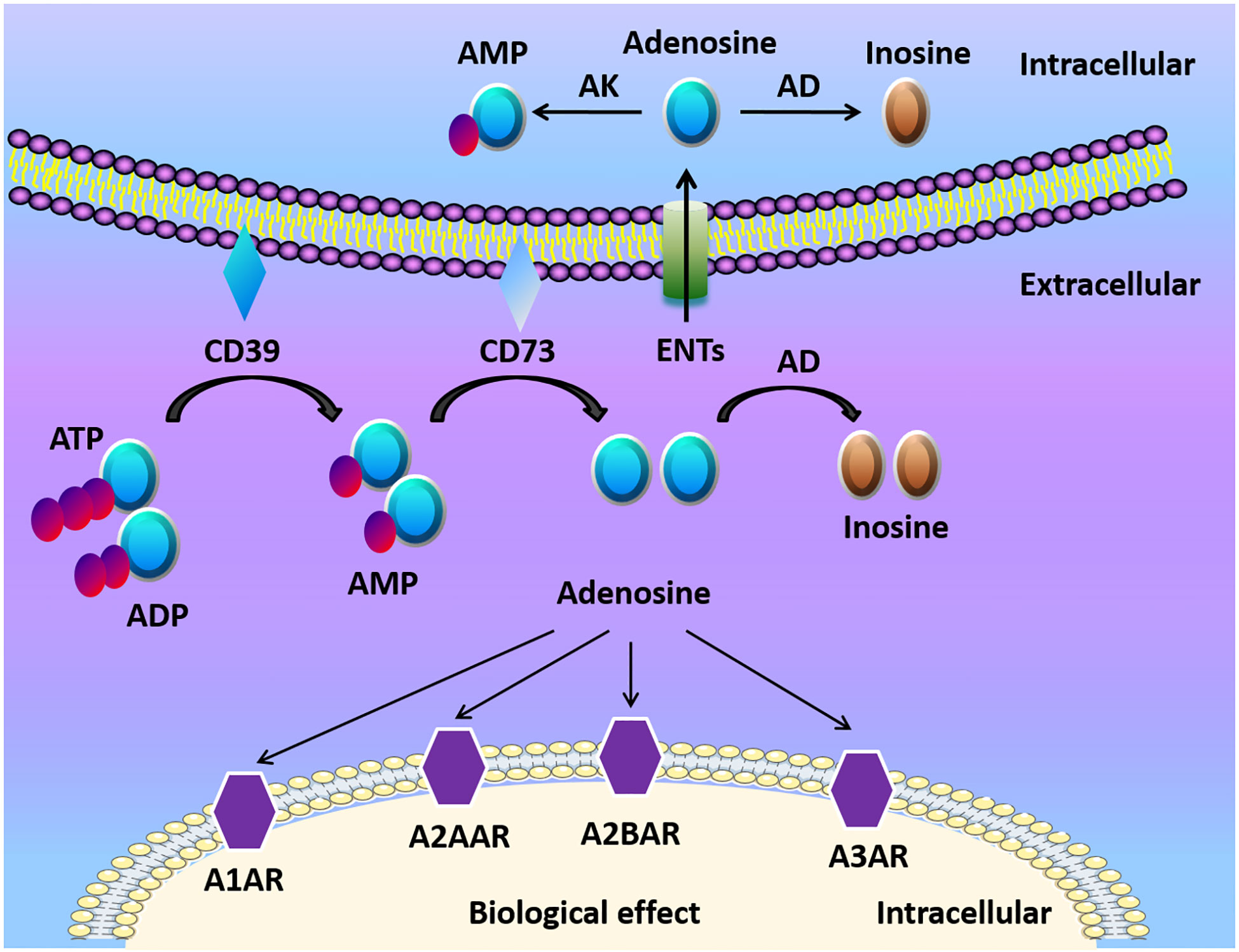
Figure 1 Adenosine biogenesis and signaling. ATP and ADP are the main resources of extracellular adenosine. ATP and ADP are dephosphorylated to AMP on the cell surface by Ecto-nucleotide triphosphate diphosphohydrolase 1 (CD39) and ecto-5’-nucleotidase (CD73) dephosphorylates AMP to adenosine. Adenosine activates adenosine receptors (A1AR, A2AAR, A2BAR, A3AR) and plays a crucial role in different cells and organs. Adenosine can be transported into the cell by equilibrative nucleoside transports (ENTs), or be transformed to inosine via CD26-bound adenosine deaminase (ADA) at the cell surface. Under normoxic conditions, adenosine has a high affinity with adenosine receptors and ENTs. Under hypoxia conditions, the release of extracellular ATP/ADP increased. Finally, HIFs enhanced the release of extracellular adenosine and adenosine receptors, which modulates tissue barriers and inflammatory response.
Adenosine receptors, which include four distinct G-protein coupled seven membrane-spanning cell surface receptors: the adenosine A1 receptor (A1AR), the adenosine A2A receptor (A2AAR), the adenosine A2B receptor (A2BAR), and the adenosine A3 receptor (A3AR), are crucial for adenosine mediated responses (3, 19, 21, 27). Both A2AAR and A2BAR are linked to Gs protein involving activation of adenylate cyclase, to stimulate cAMP production followed by PKA activation (32–35). A1AR and A3AR, on the other hand, bear a distinct signal transduction pathway. For example, A1AR activation inhibits cAMP accumulations in Chinese hamster ovary cells (36). The coupling of A1AR to the Gi/o protein pathway attenuates cAMP signal transduction in hepatic stellate cells (33). Furthermore, A3AR has been indicated to attenuate adenosine-induced increase of cAMP in rat vascular smooth muscle cells in vitro (37) and A3AR knockout mice show an increased level of cAMP in the cardiovascular system (38). Functionally, Dr. Michail Sitkovsky’s laboratory identified that A2AAR is crucial for limiting inflammatory responses as mice with A2AAR deficiency showed profound tissue damage in inflammation and endotoxin-induced septic shock (21). The expression of adenosine receptor subtypes is different in various cell types. For example, neutrophils and lymphocytes have higher expression levels of A2AAR, while vascular endothelial cells have higher levels of A2BAR (39–41). It has been elucidated that adenosine receptors have important functions in pathologic conditions. For instance, adenosine has a selective role in reducing the heart rate via A1AR, which would be a potential therapeutic method for superventricular tachycardia in mice (42). Adenosine signaling via A2AAR or A2BAR has a beneficial effect via shifting proinflammatory immune response to anti-inflammatory immune response as well as promoting barrier protection in different animal models (43–48). A3AR is related to the aqueous humor production in the eye in a preclinical study (49), and its agonist showed efficacy in treating dry eye syndrome in a clinical study (50).
The termination of adenosine signaling is mediated by the transportation of adenosine from the extracellular to the intracellular space (Figure 1) (27, 51). ENTs and concentrative nucleoside transporters (CNTs) are nucleoside transporters found on various cell types (52, 53). According to the concentration gradient, adenosine moves freely across these channels because of its diffusion-limited character (53). Adenosine signaling can be diminished by the transportation of adenosine into the cell and then metabolized to inosine via adenosine deaminase (ADA) (54). Additionally, adenosine kinase can convert adenosine to AMP (55). The activation of mucosal A2B signaling combined with the repression or deletion of epithelial ENT2 dampens mucosal inflammation (56). Another study also showed that elevations of adenosine protect from liver injury after the genetic deletion or inhibition of Ent1 via A2B signaling in liver ischemia and reperfusion models (57).
Hypoxia and inflammation frequently occur in pathogenic conditions such as cancer, inflammatory bowel diseases, ischemia/reperfusion injury, and inflammatory lung diseases (58). Hypoxia-inducible factors (HIFs) are crucial in the responses mediating the crosstalk between hypoxia and inflammation. Hypoxia-inducible factors (HIFs) have a central role in regulating tissue adaptation to low oxygen conditions. HIFs belongs to αβ-heterodimeric transcription factors that include HIF-1α, HIF-2α, and HIF-1β/ARNT subunits. When oxygen is abundant, HIF-1α or HIF-2α binds to the von Hippel-Lindau (VHL) gene product, a part of the E3 ubiquitin ligase complex, and result in proteasomal degradation (59–61). HIFα and VHL binding are related to the hydroxylation of HIFα proline residues, which rely on prolyl hydroxylases (PHDs) and factor-inhibiting HIF (FIH) (60, 61). Under hypoxia, HIFα subunits can not be hydroxylated as efficiently due to the lack of oxygen as a substrate for PHDs, which results in the stabilization of HIF-1α and HIF-2α. Once stabilized, HIFα translocates to the nucleus and binds to HIF-1β to form a complex, and in turn bind to hypoxia-responsive elements (HRE) of the promoter region in the target genes for start transcriptional regulation (46, 62, 63). Most of the HIFs target genes are related to metabolism, proliferation, oxygen transport, and other processes important for hypoxia adaptation (64). HIF stabilization is demonstrated in inflammatory conditions and diseases, such as lung injury, inflammatory bowel disease, and ischemia-reperfusion injury through various mechanisms (Figure 2) (3, 58, 65). Tissue metabolism in inflammatory disease has higher local oxygen demand, which induces tissue hypoxia. Additionally, the supply of oxygen decreased due to the shortage of tissue blood supply in trauma, ischemia, and vascular occlusion disease, which aggravate tissue inflammation (46, 66). Moreover, cytokines (e.g., IL-1, IFN-β, TNF-α) released during inflammation have an impact on HIF-1α expression (67, 68). The hypoxic environment in solid tumors and during ischemia/reperfusion activates NF-κB, which is a crucial transcription factor regulating inflammation and immune response (69–71). Therefore, hypoxia and inflammation usually occur simultaneously during pathogenic conditions, and they are closely linked to each other.
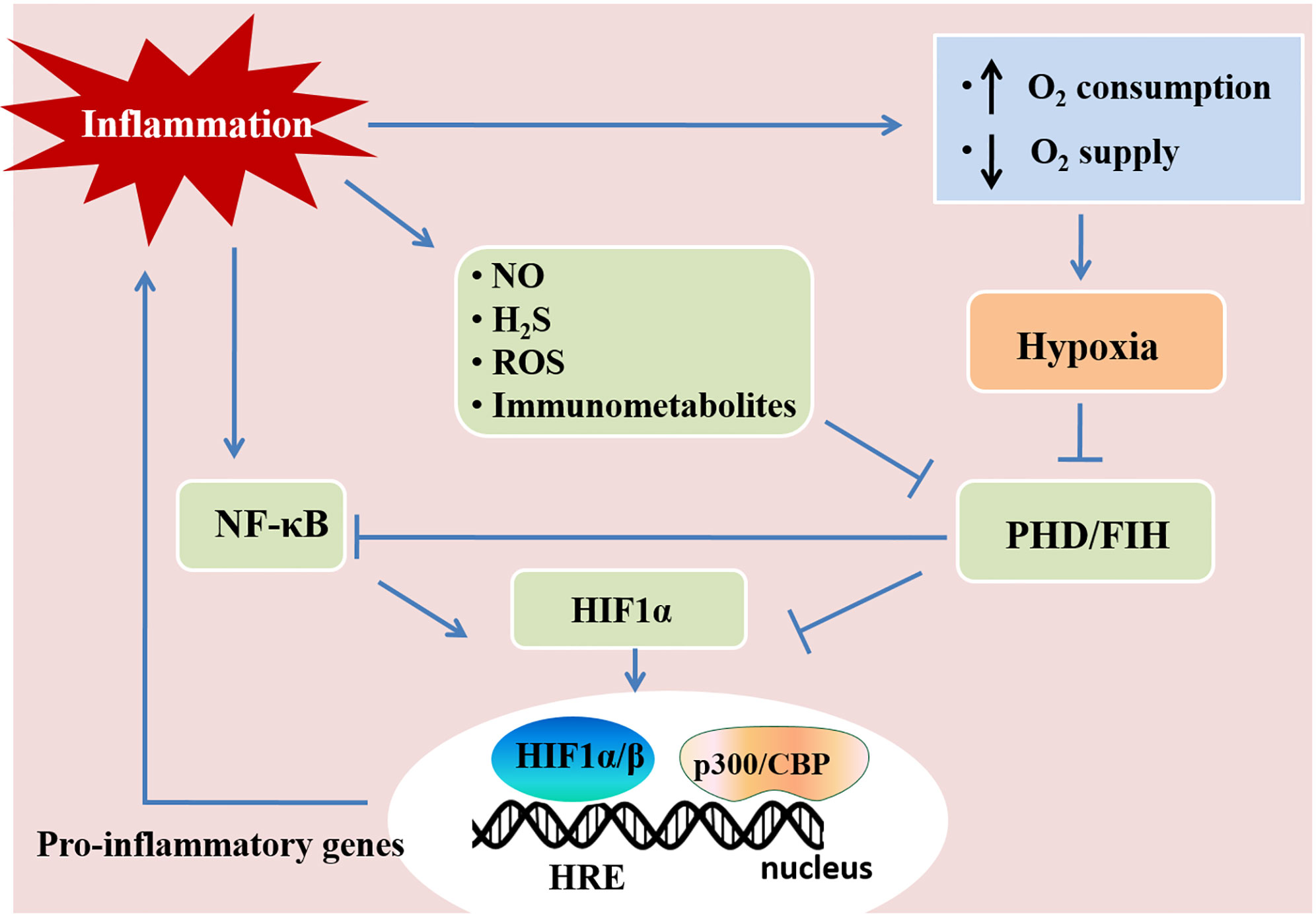
Figure 2 Hypoxia and inflammation. Inflammation and hypoxia are co-incidental events in several pathological conditions. Inflammatory stimuli, such as cytokines, bacterial products, and hypoxia, activate the nuclear factor-κB (NF-κB) pathway. The activation of NF-κB enhances the transcription of HIF-1α mRNA and promotes HIF activity. Inflammatory mediators, such as nitric oxide (NO), hydrogen sulfide (H2S), reactive oxygen species (ROS), and immunometabolites also control HIF activity in immune cells, which regulates immunity and inflammation. Activated HIF-1α translocates to the nucleus and promotes the transcription of pro-inflammatory genes by associating with HIF-1β and the cofactor p300/CBP. This figure is adapted from Regulation of immunity and inflammation by hypoxia in immunological niches; Cormac T. Taylor and Sean P. Colgan, Nature Reviews Immunology; 17, pages 774–785(2017) (22).
Recently, increasing research effort has provided convincing evidence of the link between hypoxia and inflammation in ARDS (72–75). For example, HIF-1α is stabilized under normoxic conditions by mechanical stretch of alveolar epithelial cells in vitro and in ventilation-induced lung injury (VILI) in mice in vivo (72). The normoxic stabilization of HIF-1a by mechanical stretch could be explained by the inhibition of succinate dehydrogenase (SDH). Functionally, HIF-1α stabilization dampens lung inflammation through the regulation of glucose metabolism in alveolar epithelial cells, because only mice with alveolar epithelial cell-specific deletion of HIF-1α show profoundly increased lung inflammation and pulmonary edema (72). The protective effect of HIF-1α in alveolar epithelial cells has also been demonstrated in acute cobalt-induced lung injury models as more neutrophilic infiltration and Th1 cytokines were observed in alveolar epithelial-specific HIF-1α-deficient mice (76). Additionally, HIF-2α activation improved endothelial adherens junction integrity in endotoxin-mediated injury through increasing its target gene vascular endothelial protein tyrosine phosphatase (VE-PTP) (77). Furthermore, the pharmacologic activator of HIF, dimethyloxalylglycine (DMOG), protects the lung alveolar epithelium during murine VILI and LPS induced acute lung injury via enhancement of glycolysis (72, 78). Another study showed that DMOG treatment attenuates Fas Ligand (FasL)-induced apoptosis in MLE-12 cells in vitro and dampens lung inflammation, and histopathological changes intratracheal FasL induced lung injury in mice in vivo (79). These studies suggests that pharmacological HIF activator could offer lung protection during ARDS via maintaining alveolar epithelial and endothelial functions during injury.
Viral infection-induced ARDS has been the center of attention because of the recent pandemic of COVID-19. Influenza virus infection is one of the most studied models for viral pneumonia (80–83). Several studies have shown a close relationship between hypoxia and inflammation in viral infection-induced ARDS. For example, respiratory syncytial virus infection in mice results in the stabilization of HIF-1α in an oxygen-independent manner (84). Besides, earlier studies indicated that influenza A (H1N1) virus infection could induce HIF-1α nuclear translocation but did not change its expression levels in A549 cells in vitro (85). A recent study indicated that H1N1 infection stabilizes HIF-1α under normoxic conditions in A549 cells in vitro and in murine models of H1N1 mediated viral pneumonia in vivo (86). The normoxic stabilization of HIF-1α is dependent on the inhibition of proteasome function and decreasing the expression of factor inhibiting HIF-1 (FIH-1) (86). Moreover, influenza A virus (IAV) infection-induced acute lung injury (ALI) also results in hypoxia, and further contribute to the stabilization of HIF-1α in mouse lung tissue (87). Functionally, alveolar epithelial type II cell-specific deficient Hif1afl/fl SPCCre mice showed increased lung inflammation and mortality during IAV infection in vivo (87). Mechanistically, HIF-1α deficiency promotes influenza A virus replication in A549 cells in vitro via reducing glycolysis and enhancing autophagy (87). The functional role of HIF in SARS-CoV-2 infection associated ARDS needs to be further investigated.
IPF is one of the most common and severe forms of interstitial lung disease (88). IPF patients suffer from an impaired pulmonary gas exchange and chronic arterial hypoxemia (89). The important role of hypoxia and HIFs on fibroblast proliferation and differentiation has been studied extensively (90–92). Besides the direct impact on fibroblasts, hypoxia is regarded as one of the potent stimuli for the production of proinflammatory cytokines. For example, protein kinase C (PKC) activation promotes the expression of TNF-α and IL-1β in the pulmonary artery under hypoxic conditions (93). Additionally, vascular endothelial growth factor (VEGF), a known target gene of HIF, is an angiogenesis factor with proinflammatory, permeability-inducing roles in murine bleomycin-induced pulmonary fibrosis (94). Furthermore, HIF-1α stabilization has been observed in alternatively activated macrophages in a murine model of bleomycin-induced pulmonary fibrosis and HIF-1α inhibition in macrophages inhibits the expression of profibrotic mediators including IL-7 and CXCL1 (95). However, the involvement of hypoxia signaling in other subtypes of immune cells during IPF has yet to be elucidated.
Inflammation and hypoxia are also tightly linked in COPD, including chronic bronchitis and emphysema. For example, cigarette smoking significantly increases inflammation mediators expression, such as IL-6, IL-8, and TNF-α (96). These factors contribute to the activation of hypoxia response genes (including HIFs, NF-κB) and promote the development of COPD in rats (97). HIFs are overexpressed in the lung tissue of COPD patients (98) and HIF-1α level is positively correlated with the severity of COPD in patients (99). HIF-2α, on the other hand, has been shown to be decreased in lung tissue from emphysema patients compared to healthy control (100). Furthermore, endothelial cell-specific deletion of HIF-2α in mice results in emphysematous changes in the lung, which was exaggerated by the treatment of SU5416, a vascular endothelial growth factor receptor 2 (VEGFR2) inhibitor (100). On the other hand, endothelial-specific overexpression of HIF-2α in mice was protected from emphysema (100), suggesting therapeutic activation of HIF-2 α as a treatment for emphysema.
Hypoxia is frequently encountered in patients suffering from severe asthma or acute exacerbation (101). How hypoxia and HIFs influence allergic airway inflammation has been studied extensively. An earlier study suggested that HIF-1α is stabilized in lung tissues from asthmatic patients and in a murine model of allergic airway inflammation induced by ovalbumin sensitization (102). This study also demonstrated that deficiency in HIF-1β significantly dampens allergic airway inflammation and reduced ovalbumin-specific antibodies in mice (102). Consistently, HIF-1α antagonist YC-1 reduced airway hyperresponsiveness and lung inflammation in a murine model of asthma (103, 104). Besides its global impact on allergic airway inflammation, the functional role of HIF-1α in different subsets of immune cells has also been investigated in asthma. For instance, myeloid-specific deletion of HIF-1α in mice results in reduced airway hyperresponsiveness (AHR), and HIF-1α deficient eosinophils show reduced chemotaxis (104). Furthermore, a recent study indicates that exposure to 3% oxygen leads to increased T helper type 2 cells (Th2) cytokine expression in CD8+ T cells and adoptive transfer of these cells exaggerated AHR and lung inflammation in the ovalbumin model of murine allergic airway disease (105). Additionally, HIF-1α inhibition reduced Th2 cytokines expression in CD8+ T cells upon hypoxia exposure, and the adoptive transfer of HIF-1α deficient CD8+ T cells underwent hypoxia attenuates AHR and airway inflammation in mice (105). In summary, HIF-1α is important for the development of AHR and airway inflammation by modulating immune cell chemotaxis and function. However, the detailed mechanism, such as the identification of HIF target genes in specific immune cells during asthma, needs to be further investigated.
In the past decades, studies have provided ample evidence that hypoxia signaling is tightly linked with adenosine signaling (46, 58, 106–112). Previous studies showed that hypoxic condition or inflammation contributes to the accumulation of extracellular ATP/ADP due to the damage in the cell membrane (3, 23, 27, 113–115). The increased level of extracellular ATP and ADP is essential for the generation of extracellular adenosine, which is a key mediator of inflammatory responses (116, 117). It has been demonstrated that HRE in the promoter of CD73 gene is crucial for HIF-1α mediated expression in epithelial cells under hypoxic conditions, and the inhibition of HIF-1α decreases the hypoxia-inducible CD73 expression (118). Besides HIF-1α, transcription factor Sp1 is also involved in the transcription of CD39 under hypoxia conditions, and its protective effect has been demonstrated during cardiac and hepatic ischemia (29, 46, 119). Moreover, A2AAR has been identified as a target gene of HIF-2α in human lung endothelial cells (120), while A2BAR has been identified as a target gene of HIF-1α (121, 122). The links between HIF and adenosine are not only through regulation of ectonucleotidases and adenosine receptors but also via equilibrative nucleoside transporters (ENTs) and its G-protein-coupled receptors. For example, HIFs are implicated in the repression of ENT1 and ENT2 (53, 123) and abolish the conversion of adenosine to AMP by adenosine kinase in cells (46, 55). The close relationship between hypoxia and adenosine signaling in acute and chronic lung injury have been established during the past decades (Figure 3).
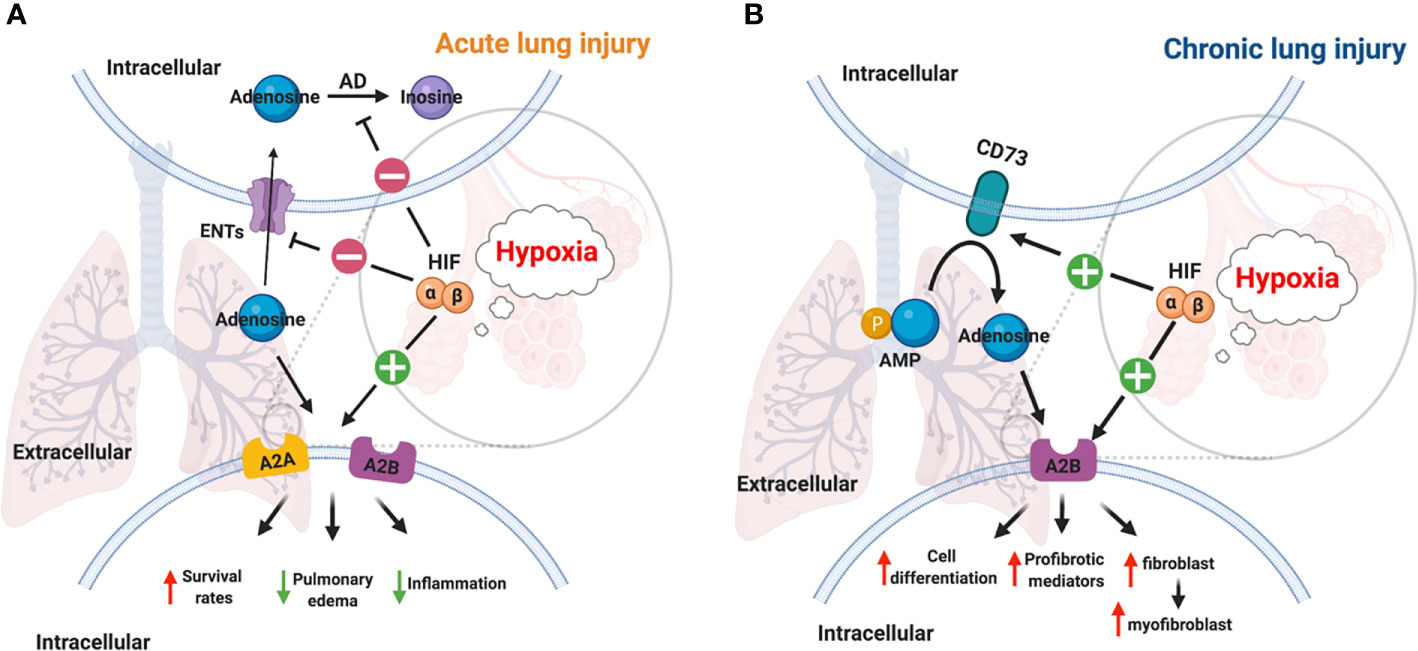
Figure 3 Links between HIF and adenosine signaling in acute/chronic lung injury. (A) Inflammation and infection results in the stabilization of HIFs in acute lung injury (112). HIF-1a dependent inhibition of ENT1, ENT2, and adenosine kinase contributes to the accumulation of adenosine (55, 123). A2AAR and A2BAR are two adenosine receptors that are regulated by HIF-2α and HIF-1α respectively in lung tissue (109, 120, 121). Therefore, the higher level of adenosine, and the activation of its receptors reduced mortality, pulmonary edema, inflammation in acute lung injury. (B) The activation of the hypoxic-adenosinergic system has been investigated in chronic lung injury. CD73 and A2BAR are two hypoxia-inducible genes in patients with idiopathic pulmonary fibrosis and pulmonary hypertension (124). The upregulation of A2BAR enhances cell differentiation, produces profibrotic mediators, and promotes fibroblast to myofibroblast in chronic lung injury (95, 125).
Several studies suggest that adenosine level increases following hypoxia exposure in animal studies (126) and in human studies (127, 128). In vitro cell culture experiments and in vivo animal studies indicated that endogenous adenosine generation inhibits neutrophil accumulation during hypoxia (129). Particularly, CD39 deficient mice show an increased level of MPO in colon, lung, kidney, and liver after 4 h of exposure to hypoxia (8% O2) compared to wild-type mice. Pharmacological inhibition or genetic deletion of CD73 in mice leads to a similar phenotype as CD39 deficient mice, suggesting the importance of extracellular adenosine generation in hypoxia-induced inflammation. Moreover, short term exposure to hypoxia increases plasma levels of adenosine, attenuates pro-inflammatory cytokine release, and results in an elevated level of IL-10 during experimental endotoxemia models in humans (130). Extracellular adenosine levels increase after the mechanical ventilation in mice or stretched pulmonary epithelial cells (106, 112). Pharmacological inhibition or genetic deletion of CD39 or CD73 in mice leads to severe lung inflammation with mechanical ventilation, suggesting the protective effect of adenosine (106). The relationship between HIF and adenosine in ARDS during viral pneumonia has not been clearly demonstrated yet. Nucleotide ATP and adenosine in BALF have been shown to be increased after the infection of influenza A virus in mice (131, 132). However, adenosine levels and pathogenesis of ALI did not show any difference between WT and CD73-knockout mice after the infection of influenza A virus. Therefore, CD73 is not considered as one of the crucial factors for the development of influenza-induced ALI (133).
Cellular stress and damage induce the generation of adenosine in lung tissue of patients with chronic lung disease. For example, the hypoxic-adenosinergic pathway is activated in IPF patients with pulmonary hypertension (PH), as marked by increased expression of HIF-1α, adenosine, adenosine A2B receptor, CD73, and ENT1 (124). Other studies showed that adenosine levels are increased in the serum, lymphocytes, and erythrocytes in healthy smokers compared to healthy non-smokers and continue to increase with the severity in COPD patients (134). The same study also demonstrated that patients with higher levels of adenosine tend to have reduced forced expiratory volume in one second (FEV1), suggesting a potential functional link (134). Furthermore, adenosine signaling is significantly enhanced in COPD as represented by increased CD73 activity and adenosine receptor levels in lung tissue from patients with COPD or in murine model of emphysema (135, 136). Adenosine signaling is also enhanced in asthma, and consequently, a high level of adenosine induces airway hyperresponsiveness and bronchoconstriction and promotes human mast cells to release allergen-induced mediators (137).
A1AR has diverse roles in lung injury. For example, A1AR deficient mice have increased susceptibility to LPS-induced acute lung injury with increased PMN recruitment and microvascular permeability (138). The same study indicated that pretreatment of A1AR agonist, 2’Me-2-chloro-N6-cyclopentyladenosine, attenuates PMN recruitment and microvascular permeability. On the other hand, post-infection treatment of a combination of A1AR antagonist L-97-1 and ciprofloxacin improves the outcome of Y. pestis infection in rats, indicating a protective effect of A1AR (139). Furthermore, A1AR knockout mice show significantly increased macrophage and neutrophil infiltration in the airway after influenza A/WSN/33 (H1N1) infection when compared to wild-type counterparts and daily treatment of A1AR antagonist 8-cyclopentyl-1,3-dipropylxanthine results in improved outcome (140). Besides acute lung injury and infection, the activation of A1AR has also been found on bronchial epithelial cells, and inflammatory cells, which enhanced the asthma phenotype (141). Mice with ADA deficiency experience lung injury and inflammation (142). A1AR deletion in mice exaggerated the pulmonary inflammation marked by increased expression of IL-4 and IL-13, as well as matrix metalloproteinases, suggesting a protective role of A1AR in chronic lung injury (142).
Exposure to hypoxia (10% O2) attenuates lung inflammation during LPS-induced lung injury in mice and A2AAR is indispensable for hypoxia-mediated lung protection (143). A2AAR expression in myeloid cells is crucial for the control of neutrophil recruitment to the lung injury and an A2AAR specific agonist ATL202 offers lung protection in mice during LPS-induced lung injury (144). The lung protective effect of A2AAR has also been implicated in cardiopulmonary bypass-mediated lung injury. Pretreatment of A2AAR agonist CGS21680 in juvenile rats dampens inflammatory cytokines and myeloperioxidase levels in the serum as well as pulmonary edema and lung injury score during cardiopulmonary bypass−induced organ injury. Another study demonstrated that A2AAR activation induces the expression of peroxisome proliferator-activated receptors γ (PPARγ) via cAMP and PKA pathways in murine macrophages (145). Combining PPARγ agonist ROSI and A2AAR agonist CGS21680 significantly reduces lung pathology and the production of inflammatory cytokines in the lung during murine model of LPS-induced ALI (145). Moreover, treatment of CGS21680 after the onset of trauma/hemorrhagic shock-induced lung injury attenuates pulmonary edema and MPO levels in Sprague-Dawley rats (146). Interestingly, A2AAR has been identified as a HIF-2α target in pulmonary endothelial cells in vitro, implicating the crosstalk between adenosine signaling and hypoxia signaling (120). Furthermore, treatment of A2AAR agonist CGS21680 reduces inflammatory cell infiltration to the airway in murine models of asthma (147). A2AAR deficient mice experience exaggerated lung inflammation and airway hyperactivity, suggesting the protective role of A2AAR in allergic airway diseases.
A2BAR is an important link between hypoxia and adenosine signaling in acute lung injury. HIF-1α has been shown to transcriptionally induce the expression of A2BAR in murine VILI model (109, 121). For instance, genetic silence or pharmaceutical inhibition of HIF-1α dampens the expression of A2BAR in mice during VILI or alveolar epithelial cells exposed to cyclic stretch (109). A2BAR-dependent adenosine signaling offers lung protection during endotoxin-induced ALI in mice by potentiating the regulatory T cell population (148). Furthermore, hypoxia-induced vascular leakage also exaggerates in siRNA-mediated knockdown of A2BAR or A2BAR deficient mice (149). Furthermore, HIF-1α-dependent induction of netrin-1 attenuated neutrophil transmigration and dampens inflammation through A2BAR at pulmonary and colon mucosal surface (150), suggesting another layer of complexity in the crosstalk between HIF and adenosine signaling.
In chronic lung injury, hypoxia potentiates the function of adenosine and promotes the production of IL-6, and induce the differentiation of fibroblasts to myofibroblasts by increasing adenosine A2B receptor expression in human fibroblasts (125). Furthermore, adenosine deaminase-deficient mice have higher expression of A2BAR and exhibit progressive pulmonary fibrosis and respiratory distress (151). The crosstalk between hypoxia and adenosine signaling has been established in the murine model of IPF (95). For example, HIF-1α inhibition via the treatment of 17-DMAG results in reduced pulmonary fibrosis and A2BAR expression in the late stages of murine bleomycin-induced lung fibrosis in vivo (95). Additionally, HIF-1α inhibition along with A2BAR deletion or pharmacological inhibition result in disruption of alternatively activated macrophages differentiation and IL-6 production in vitro (95).
Of note, while A2AAR and A2BAR reduce mortality, pulmonary edema, and inflammation in acute lung injury, A2BAR enhances cell differentiation, produces profibrotic mediators, and promotes fibroblast to myofibroblast differentiation in chronic lung injury. The differential role of A2BAR could possibly be stemming from the different impacts of downstream signaling in acute or chronic lung injury. As mentioned above, A2AAR and A2BAR activation lead to the activation of cAMP and PKA pathway (32–35). The cAMP-CREB axis is important for the maintenance of endothelial integrity and the attenuation of lung inflammation during endotoxin-induced lung injury in mice (152). The protective effect of cAMP in LPS-induced endothelial permeability is mediated through PKA (153). cAMP synthesis and PKA activity are inhibited in oleic acid-induced lung injury, and the treatment of hydroxysafflor yellow A enhances the cAMP/PKA pathway and dampened lung inflammation in mice (154). Furthermore, pretreatment of phosphodiesterase antagonist PTX enhances cAMP signaling and results in the attenuation of lung injury during cecal ligation and puncture in mice (155). These studies suggest a protective role of cAMP and PKA during acute lung injury. In chronic lung injury, cAMP and PKA regulate hypercontractility in human airway smooth muscle cells (156) and phosphodiesterase inhibitors, which prevents the breakdown of cAMP, are currently being studied as a treatment for asthma (157). In addition, dibutyryl-cAMP treatment increases endogenous cAMP levels, enhances PKA signaling in vitro, and blocked myofibroblast differentiation in vivo (158). Other cAMP elevating agents also inhibits the proliferation and collagen production in pulmonary fibroblasts (159). Thus, the divergent function of A2BAR in acute and chronic lung injury might not be based on the downstream activation of the cAMP and PKA signaling pathway. Other factors could contribute to the response to cAMP activation as lung fibroblasts from pulmonary fibrosis patients has a deficiency in the phosphorylation of cAMP response element-binding protein (160). Future studies are needed to elucidate the signaling mechanism of A2BAR mediated responses in pulmonary injuries.
A3AR is also expressed in the lung and several previous studies have indicated the functional role of A3AR in lung injury. The protective role of A3AR in lung ischemia/reperfusion injury has been demonstrated by an early study in which the pretreatment of A3AR agonist IB-MECA attenuated alveolar injury and apoptosis during lung ischemia and reperfusion injury of isolated cat lung ex vivo (161). The protective role of A3AR is further supported as pretreatment of IB-MECA offers lung protection during lung ischemia/reperfusion injury in cat in vivo (162). In addition, A3AR agonist CI-IB-MECA pretreatment alleviates lung ischemia/reperfusion injury in mice, and the protective effect is abolished in mice with genetic deletion of A3AR (163). Besides lung ischemia/reperfusion injury, the protective role of A3AR has also been indicated in LPS-induced lung injury. Indeed, A3AR deficient mice showed exaggerated PMN infiltration after LPS inhalation and pretreatment of CI-IB-MECA attenuates the inflammatory responses and injury (164). Furthermore, A3AR activation is associated with mast cell degranulation and airway hyperreactivity. For example, selective activation of A3AR via IB-MECA results in the release of histamine in mast cells in vitro and nebulizer treatment of IB-MECA in mice results in mast cell degranulation in the lung in wild type mice but not in A3AR deficient mice (165). Adenosine administration results in airway responsiveness in mice and mice with A3AR deficiency show attenuated responses marked by reduced mast cell degranulation and neutrophil infiltration (166). Other studies also demonstrate the contribution of A3AR in chronic airway inflammation (167, 168).
Besides the impact on adenosine receptors, HIF-1α dependent repression of ENT1 and ENT2 decreases adenosine uptake and increases extracellular adenosine, which dampens neutrophil accumulation and protects vascular barrier during hypoxia in endothelia and epithelia (123). HIF-1α-dependent repression of adenosine kinase leading to increased extracellular adenosine attenuates hypoxia-induced vascular leak in murine models of sepsis or ALI (169). In addition, adenosine deaminase activity, ADA2 in particular, is significantly reduced in serum from COPD patients and smokers when compared to non-smokers (134), which could further explain the increased level of adenosine in COPD patients.
Direct therapeutic targeting of the hypoxia signaling pathway could profoundly modulate the adenosine signaling pathway. Pharmacologic compounds have been developed for normoxic stabilization of HIFs by functioning as inhibitors of PHDs. Several preclinical studies show that these compounds can be given to animals that are kept under normoxic conditions, and result in robust stabilization of HIFs (170, 171). In line with this concept, preclinical studies have shown that pretreatment with the HIF activator dimethyloxalylglycine (DMOG) is associated with attenuated organ injury in the heart, lungs, or kidneys (72, 172, 173). Moreover, recently, several pharmaceutical companies have developed HIF activators as orally available compounds and several ongoing clinical trials have used them in patients for the treatment of anemia associated with chronic kidney disease. These compounds include roxadustat (FG-4592, sponsored by FibroGen, Astellas, & AstraZeneca), vadadustat (AKB-6548, sponsored by Akebia), and daprodustat (GSK-1278863, sponsored by GlaxoSmithKline). Based on phase 3 clinical trials showing efficiency in increasing hemoglobin levels in patients with anemia associated with renal insufficiency (174, 175), roxadustat has been approved for treating chronic kidney disease-related anemia in China and is currently in phase 3 clinical trials in the United States. In the meantime, several phase 2 clinical trials indicated that oral vadadustat is safe and effective as a treatment for anemia in patients with non-dialysis-dependent chronic kidney diseases (176, 177), and in patients receiving hemodialysis previously received erythropoiesis-stimulating agents (178). Currently, vadadustat is evaluated by a randomized, double-blinded and placebo-controlled phase 2 clinical trial as a treatment of COVID-19 associated ARDS (Table 1) (180). These oral available HIF activators could potentially be efficient for enhancing adenosine signaling pathways in patients for the prevention of acute lung injury. On the other hand, HIF inhibitors could potentially inhibit adenosine signaling as a therapeutic approach for chronic lung diseases. Currently, HIF-2α inhibitors such as PT2385 and PT2977 have been investigated by clinical trials mainly as novel therapeutic approaches for renal cell carcinoma (181). However, HIF-1α specific inhibitor has yet to be developed for clinical use, which will be crucial for the inhibition of adenosine signaling in chronic lung diseases.
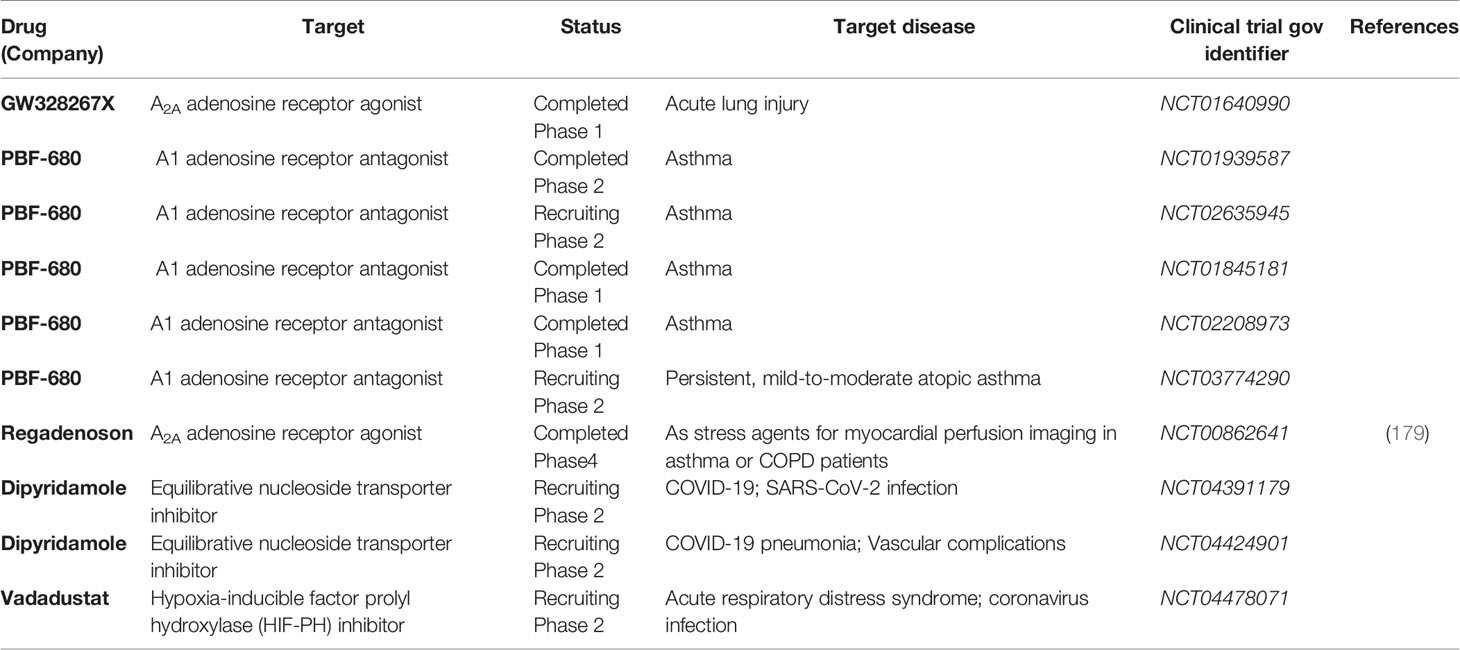
Table 1 Clinical trials targeting adenosine signaling in lung diseases.
Adenosine signaling could potentially be targeted for lung protection during acute lung inflammation via direct administration of adenosine or utilizing specific adenosine receptor agonists in both preclinical and clinical settings (182, 183). Several preclinical studies have indicated that direct administration of adenosine attenuates lung injury (184, 185). The safety of adenosine administration has also been supported by previous clinical studies (186, 187). However, due to the short half-life of adenosine in vivo, adenosine analogs might be a more feasible option. Adenosine receptor agonists have been developed for preclinical and clinical use (188). For example, pretreatment of A2AAR agonist ATL202 inhibits LPS-induced PMN recruitment, reduced the release of inflammatory cytokines in the lung, and reduced vascular leakage in mice (144). A2AAR agonist GW328267C improves lung function in three models of ALI (HCl instillation 1 h, LPS instillation 16 h, and live Escherichia coli instillation) in rats (189). The delivery of A2BAR-specific agonist BAY 60-6583 attenuate pulmonary edema, inhibits lung inflammation, and improves histologic lung injury in murine ALI (73, 107). Furthermore, mice treated with BAY60-6583 show attenuated oleic acid (OA)-induced ALI by inhibiting alveolar epithelial cell apoptosis (190). However, only A1AR, A2AAR, and A3AR agonists have been evaluated in the clinical setting while the safety and efficacy ofA2BAR agonists have yet to be established by clinical studies (188). The usage of adenosine receptor agonists in clinical trials related to lung injury is summarized in Table 1.
Adenosine receptor antagonists have been developed as treatment of chronic lung diseases in both preclinical and clinical settings. For instance, LASSBio-897 (3-thienylidene-3, 4-methylenedioxybenzoylhydrazide) can block the activity of A2AAR agonist and has anti-inflammatory and anti-fibrotic role in a mouse model of silicosis (191). Additionally, the treatment of A2BAR antagonist CVT-6883 dampens lung inflammation, reduces fibrosis, and attenuates alveolar airspace enlargement in ADA-deficient mice (192). Similarly, CVT-6883 treatment reduced inflammation and lung fibrosis in murine bleomycin-induced lung injury (192). Finally, A1AR antagonist PBF-680 has been and is currently being evaluated by several phase 1 and phase 2 clinical trials as a treatment of asthma (Table 1). Although adenosine receptor antagonists have been investigated for inflammatory conditions, neurodegenerative diseases, and mood disorders (193), their potential impact on chronic lung diseases needs to be further evaluated.
Adenosine signaling could also be targeted via modification of adenosine metabolism. For instance, inhibition or deletion of ENT1/2 elevates extracellular adenosine levels in lung tissue and improves pulmonary function by activating A2AAR and A2BAR receptor and preventing NLRP3 inflammasome activation in Pseudomonas aeruginosa infection-induced acute lung injury in mice (194). Moreover, ENT inhibitor dipyridamole treatment decreases adenosine uptake, and in turn improves vascular barrier and reduces neutrophil accumulation in acute pulmonary inflammation in preclinical studies (108, 123, 195). Along the same line, dipyridamole is currently investigated by several clinical trials as a treatment for COVID-19 and associated vascular manifestation (NCT04391179, NCT04424901, Table 1). Besides targeting ENTs, ADA administration reduced lung pathology in IL-13 transgenic mice, which spontaneously develop lung inflammation, alveolar destruction, and fibrosis (196). Furthermore, PEGylated adenosine deaminase is currently employed as an enzyme replacement therapy for patients suffering adenosine deaminase severe combined immunodeficiency (197) and has lately been shown to alleviate fibrosis and inflammation in a murine model of systemic sclerosis (198). PEGylated adenosine deaminase should be further investigated as a therapeutic approach for chronic lung diseases.
Adenosine signaling is one of the most crucial mediators in the cross-talk between hypoxia and inflammation. In this review, many studies suggest that targeting hypoxia and adenosine signaling could be a promising therapeutic approach for ARDS and chronic lung diseases. However, further investigation is needed to address the knowledge gaps in the mechanism of how HIF-adenosine contributes to different disease conditions and how to target this pathway in patients. For instance, the functional link between HIF and adenosine pathway in viral pneumonia induced ARDS needs to be established, especially for COVID-19 associated ARDS. Furthermore, the functional role of the HIF-adenosine pathway needs to be demonstrated in COPD and asthma for the development of novel therapies targeting this pathway. Pharmacological agents to modulate adenosine signalings, such as adenosine receptor antagonists and PEGylated adenosine deaminase, have been investigated in several disease conditions. However, its potential use for chronic lung diseases needs to be further evaluated. Taken together, a detailed understanding of the functional role of the HIF-adenosine axis is needed for the development of efficient and safe therapy in pulmonary diseases.
XL drafted the manuscript. NB assisted with the literature search. TM revised the manuscript. KZ assisted with the figures. HE revised the manuscript and provided critical advice on the structure and content of the manuscript. XY drafted and finalized the manuscript and provided critical advice on the structure and content of the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by R01 DK097075, POI-HL114457, R01-HL109233, R01-DK109574, R01-HL119837, R01-DK082509, R01-HL154720, and R01-HL133900 to HE, the American Thoracic Society Unrestricted Grant, American Heart Association Career Development Award (19CDA34660279), American Lung Association Catalyst Award (CA-622265), the Center for Clinical and Translational Sciences, McGovern Medical School Pilot Award (1UL1TR003167–01), the Parker B. Francis Fellowship to XY, and the China Scholarship Council State Scholarship Fund to XL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers (2019) 5:18. doi: 10.1038/s41572-019-0069-0
2. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA (2012) 307:2526–33. doi: 10.1001/jama.2012.5669
3. Le TT, Berg NK, Harting MT, Li X, Eltzschig HK, Yuan X. Purinergic Signaling in Pulmonary Inflammation. Front Immunol (2019) 10:1633. doi: 10.3389/fimmu.2019.01633
4. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet (1967) 2:319–23. doi: 10.1016/S0140-6736(67)90168-7
5. Yu J, Shi J, Wang D, Dong S, Zhang Y, Wang M, et al. Heme Oxygenase-1/Carbon Monoxide-regulated Mitochondrial Dynamic Equilibrium Contributes to the Attenuation of Endotoxin-induced Acute Lung Injury in Rats and in Lipopolysaccharide-activated Macrophages. Anesthesiology (2016) 125:1190–201. doi: 10.1097/ALN.0000000000001333
6. Zhang J, Xie B, Hashimoto K. Current status of potential therapeutic candidates for the COVID-19 crisis. Brain Behav Immun (2020) 87:59–73. doi: 10.1016/j.bbi.2020.04.046
7. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
8. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol (2020) 28:1–12. doi: 10.1038/s41577-020-0311-8
9. Gralinski LE, Baric RS. Molecular pathology of emerging coronavirus infections. J Pathol (2015) 235:185–95. doi: 10.1002/path.4454
10. Kalil AC, Thomas PG. Influenza virus-related critical illness: pathophysiology and epidemiology. Crit Care (2019) 23:258. doi: 10.1186/s13054-019-2539-x
11. Phua J, Weng L, Ling L, Egi M, Lim CM, Divatia JV, et al. Intensive care management of coronavirus disease 2019 (COVID-19): challenges and recommendations. Lancet Respir Med (2020) 8(5):506–17. doi: 10.1016/S2213-2600(20)30161-2
12. Zhou Y, Schneider DJ, Blackburn MR. Adenosine signaling and the regulation of chronic lung disease. Pharmacol Ther (2009) 123:105–16. doi: 10.1016/j.pharmthera.2009.04.003
13. Thannickal VJ, Toews GB, White ES, Lynch JP 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med (2004) 55:395–417. doi: 10.1146/annurev.med.55.091902.103810
14. Yuan X, Shan M, You R, Frazier MV, Hong MJ, Wetsel RA, et al. Activation of C3a receptor is required in cigarette smoke-mediated emphysema. Mucosal Immunol (2015) 8:874–85. doi: 10.1038/mi.2014.118
15. Millien VO, Lu W, Mak G, Yuan X, Knight JM, Porter P, et al. Airway fibrinogenolysis and the initiation of allergic inflammation. Ann Am Thorac Soc (2014) 11(Suppl 5):S277–83. doi: 10.1513/AnnalsATS.201403-105AW
16. Knight JM, Mak G, Shaw J, Porter P, McDermott C, Roberts L, et al. Long-Acting Beta Agonists Enhance Allergic Airway Disease. PLoS One (2015) 10:e0142212. doi: 10.1371/journal.pone.0142212
17. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet (2017) 389:1941–52. doi: 10.1016/S0140-6736(17)30866-8
18. Drury AN, Szent-Gyorgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol (1929) 68:213–37. doi: 10.1113/jphysiol.1929.sp002608
19. Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol (2004) 22:657–82. doi: 10.1146/annurev.immunol.22.012703.104731
20. Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol (2005) 5:712–21. doi: 10.1038/nri1685
21. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414:916–20. doi: 10.1038/414916a
22. Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol (2017) 17:774–85. doi: 10.1038/nri.2017.103
23. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509:310–7. doi: 10.1038/nature13085
24. Eltzschig HK. Extracellular adenosine signaling in molecular medicine. J Mol Med (Berl) (2013) 91:141–6. doi: 10.1007/s00109-013-0999-z
25. Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med (2013) 19:355–67. doi: 10.1016/j.molmed.2013.03.005
26. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev (2017) 276:121–44. doi: 10.1111/imr.12528
27. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
28. Zhou Q, Gong X, Kuang G, Jiang R, Xie T, Tie H, et al. Ferulic Acid Protected from Kidney Ischemia Reperfusion Injury in Mice: Possible Mechanism Through Increasing Adenosine Generation via HIF-1alpha. Inflammation (2018) 41:2068–78. doi: 10.1007/s10753-018-0850-3
29. Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood (2009) 113:224–32. doi: 10.1182/blood-2008-06-165746
30. Kohler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, et al. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation (2007) 116:1784–94. doi: 10.1161/CIRCULATIONAHA.107.690180
31. Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, et al. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J (2013) 27:2207–19. doi: 10.1096/fj.12-225201
32. Iwamoto T, Umemura S, Toya Y, Uchibori T, Kogi K, Takagi N, et al. Identification of adenosine A2 receptor-cAMP system in human aortic endothelial cells. Biochem Biophys Res Commun (1994) 199:905–10. doi: 10.1006/bbrc.1994.1314
33. Yang Y, Wang H, Lv X, Wang Q, Zhao H, Yang F, et al. Involvement of cAMP-PKA pathway in adenosine A1 and A2A receptor-mediated regulation of acetaldehyde-induced activation of HSCs. Biochimie (2015) 115:59–70. doi: 10.1016/j.biochi.2015.04.019
34. Strohmeier GR, Reppert SM, Lencer WI, Madara JL. The A2b adenosine receptor mediates cAMP responses to adenosine receptor agonists in human intestinal epithelia. J Biol Chem (1995) 270:2387–94. doi: 10.1074/jbc.270.5.2387
35. Sun Y, Huang P. Adenosine A2B Receptor: From Cell Biology to Human Diseases. Front Chem (2016) 4:37. doi: 10.3389/fchem.2016.00037
36. Akbar M, Okajima F, Tomura H, Shimegi S, Kondo Y. A single species of A1 adenosine receptor expressed in Chinese hamster ovary cells not only inhibits cAMP accumulation but also stimulates phospholipase C and arachidonate release. Mol Pharmacol (1994) 45:1036–42.
37. Zhao Z, Francis CE, Ravid K. An A3-subtype adenosine receptor is highly expressed in rat vascular smooth muscle cells: its role in attenuating adenosine-induced increase in cAMP. Microvasc Res (1997) 54:243–52. doi: 10.1006/mvre.1997.2044
38. Zhao Z, Makaritsis K, Francis CE, Gavras H, Ravid K. A role for the A3 adenosine receptor in determining tissue levels of cAMP and blood pressure: studies in knock-out mice. Biochim Biophys Acta (2000) 1500:280–90. doi: 10.1016/S0925-4439(99)00111-8
39. Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest (1990) 85:1150–7. doi: 10.1172/JCI114547
40. Wallace KL, Linden J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood (2010) 116:5010–20. doi: 10.1182/blood-2010-06-290643
41. Johnston-Cox HA, Koupenova M, Ravid K. A2 adenosine receptors and vascular pathologies. Arterioscler Thromb Vasc Biol (2012) 32:870–8. doi: 10.1161/ATVBAHA.112.246181
42. Koeppen M, Eckle T, Eltzschig HK. Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS One (2009) 4:e6784. doi: 10.1371/journal.pone.0006784
43. Yang Z, Day YJ, Toufektsian MC, Ramos SI, Marshall M, Wang XQ, et al. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation (2005) 111:2190–7. doi: 10.1161/01.CIR.0000163586.62253.A5
44. Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD, Linden J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. Am J Physiol Gastrointest Liver Physiol (2004) 286:G285–93. doi: 10.1152/ajpgi.00348.2003
45. Aherne CM, Saeedi B, Collins CB, Masterson JC, McNamee EN, Perrenoud L, et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal Immunol (2015) 8:1324–38. doi: 10.1038/mi.2015.22
46. Bowser JL, Lee JW, Yuan X, Eltzschig HK. The hypoxia-adenosine link during inflammation. J Appl Physiol (1985) (2017) 123:1303–20. doi: 10.1152/japplphysiol.00101.2017
47. Poth JM, Brodsky K, Ehrentraut H, Grenz A, Eltzschig HK. Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. J Mol Med (Berl) (2013) 91:183–93. doi: 10.1007/s00109-012-0988-7
48. Eltzschig HK, Rivera-Nieves J, Colgan SP. Targeting the A2B adenosine receptor during gastrointestinal ischemia and inflammation. Expert Opin Ther Targets (2009) 13:1267–77. doi: 10.1517/14728220903241666
49. Mitchell CH, Peterson-Yantorno K, Carre DA, McGlinn AM, Coca-Prados M, Stone RA, et al. A3 adenosine receptors regulate Cl- channels of nonpigmented ciliary epithelial cells. Am J Physiol (1999) 276:C659–66. doi: 10.1152/ajpcell.1999.276.3.C659
50. Avni I, Garzozi HJ, Barequet IS, Segev F, Varssano D, Sartani G, et al. Treatment of dry eye syndrome with orally administered CF101: data from a phase 2 clinical trial. Ophthalmology (2010) 117:1287–93. doi: 10.1016/j.ophtha.2009.11.029
51. Eltzschig HK, Weissmuller T, Mager A, Eckle T. Nucleotide metabolism and cell-cell interactions. Methods Mol Biol (2006) 341:73–87. doi: 10.1385/1-59745-113-4:73
52. Loffler M, Morote-Garcia JC, Eltzschig SA, Coe IR, Eltzschig HK. Physiological roles of vascular nucleoside transporters. Arterioscler Thromb Vasc Biol (2007) 27:1004–13. doi: 10.1161/ATVBAHA.106.126714
53. Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology (2009) 136:607–18. doi: 10.1053/j.gastro.2008.10.037
54. Eltzschig HK, Faigle M, Knapp S, Karhausen J, Ibla J, Rosenberger P, et al. Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood (2006) 108:1602–10. doi: 10.1182/blood-2006-02-001016
55. Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood (2008) 111:5571–80. doi: 10.1182/blood-2007-11-126763
56. Aherne CM, Collins CB, Rapp CR, Olli KE, Perrenoud L, Jedlicka P, et al. Coordination of ENT2-dependent adenosine transport and signaling dampens mucosal inflammation. JCI Insight (2018) 3 3(20):e121521. doi: 10.1172/jci.insight.121521
57. Zimmerman MA, Tak E, Ehrentraut SF, Kaplan M, Giebler A, Weng T, et al. Equilibrative nucleoside transporter (ENT)-1-dependent elevation of extracellular adenosine protects the liver during ischemia and reperfusion. Hepatology (2013) 58:1766–78. doi: 10.1002/hep.26505
58. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med (2011) 364:656–65. doi: 10.1056/NEJMra0910283
59. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell (2001) 107:43–54. doi: 10.1016/S0092-8674(01)00507-4
60. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi: 10.1126/science.1059796
61. Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol (2000) 2:423–7. doi: 10.1038/35017054
62. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci USA (1991) 88:5680–4. doi: 10.1073/pnas.88.13.5680
63. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol (1992) 12:5447–54. doi: 10.1128/MCB.12.12.5447
64. Bogdanovski DA, DiFazio LT, Bogdanovski AK, Csoka B, Jordan GB, Paul ER, et al. Hypoxia-inducible-factor-1 in trauma and critical care. J Crit Care (2017) 42:207–12. doi: 10.1016/j.jcrc.2017.07.029
65. Yuan X, Lee JW, Bowser JL, Neudecker V, Sridhar S, Eltzschig HK. Targeting Hypoxia Signaling for Perioperative Organ Injury. Anesth Analg (2018) 126:308–21. doi: 10.1213/ANE.0000000000002288
66. Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discovery (2014) 13:852–69. doi: 10.1038/nrd4422
67. Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry WL Jr, Reichner JS. HIF-1 expression in healing wounds: HIF-1alpha induction in primary inflammatory cells by TNF-alpha. Am J Physiol Cell Physiol (2001) 281:C1971–7. doi: 10.1152/ajpcell.2001.281.6.C1971
68. Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest (2004) 114:1098–106. doi: 10.1172/JCI200421086
69. Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA (2006) 103:18154–9. doi: 10.1073/pnas.0602235103
70. Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology (2010) 139:2093–101. doi: 10.1053/j.gastro.2010.06.068
71. Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med (2003) 9:575–81. doi: 10.1038/nm849
72. Eckle T, Brodsky K, Bonney M, Packard T, Han J, Borchers CH, et al. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol (2013) 11:e1001665. doi: 10.1371/journal.pbio.1001665
73. Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar Epithelial A2B Adenosine Receptors in Pulmonary Protection during Acute Lung Injury. J Immunol (2015) 195:1815–24. doi: 10.4049/jimmunol.1401957
74. McClendon J, Jansing NL, Redente EF, Gandjeva A, Ito Y, Colgan SP, et al. Hypoxia-Inducible Factor 1alpha Signaling Promotes Repair of the Alveolar Epithelium after Acute Lung Injury. Am J Pathol (2017) 187:1772–86. doi: 10.1016/j.ajpath.2017.04.012
75. Wu G, Xu G, Chen DW, Gao WX, Xiong JQ, Shen HY, et al. Hypoxia Exacerbates Inflammatory Acute Lung Injury via the Toll-Like Receptor 4 Signaling Pathway. Front Immunol (2018) 9:1667. doi: 10.3389/fimmu.2018.01667
76. Saini Y, Greenwood KK, Merrill C, Kim KY, Patial S, Parameswaran N, et al. Acute cobalt-induced lung injury and the role of hypoxia-inducible factor 1alpha in modulating inflammation. Toxicol Sci (2010) 116:673–81. doi: 10.1093/toxsci/kfq155
77. Gong H, Rehman J, Tang H, Wary K, Mittal M, Chatturvedi P, et al. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest (2015) 125:652–64. doi: 10.1172/JCI77701
78. Tojo K, Tamada N, Nagamine Y, Yazawa T, Ota S, Goto T. Enhancement of glycolysis by inhibition of oxygen-sensing prolyl hydroxylases protects alveolar epithelial cells from acute lung injury. FASEB J (2018) 32:2258–68. doi: 10.1096/fj.201700888R
79. Nagamine Y, Tojo K, Yazawa T, Takaki S, Baba Y, Goto T, et al. Inhibition of Prolyl Hydroxylase Attenuates Fas Ligand-Induced Apoptosis and Lung Injury in Mice. Am J Respir Cell Mol Biol (2016) 55:878–88. doi: 10.1165/rcmb.2015-0266OC
80. Wu F, Huang JH, Yuan XY, Huang WS, Chen YH. Characterization of immunity induced by M2e of influenza virus. Vaccine (2007) 25:8868–73. doi: 10.1016/j.vaccine.2007.09.056
81. Wu F, Yuan XY, Li J, Chen YH. The co-administration of CpG-ODN influenced protective activity of influenza M2e vaccine. Vaccine (2009) 27:4320–4. doi: 10.1016/j.vaccine.2009.04.075
82. Wu F, Yuan XY, Huang WS, Chen YH. Heterosubtypic protection conferred by combined vaccination with M2e peptide and split influenza vaccine. Vaccine (2009) 27:6095–101. doi: 10.1016/j.vaccine.2008.11.037
83. Hong MJ, Gu BH, Madison MC, Landers C, Tung HY, Kim M, et al. Protective role of gammadelta T cells in cigarette smoke and influenza infection. Mucosal Immunol (2018) 11:894–908. doi: 10.1038/mi.2017.93
84. Haeberle HA, Durrstein C, Rosenberger P, Hosakote YM, Kuhlicke J, Kempf VA, et al. Oxygen-independent stabilization of hypoxia inducible factor (HIF)-1 during RSV infection. PLoS One (2008) 3:e3352. doi: 10.1371/journal.pone.0003352
85. Guo X, Zhu Z, Zhang W, Meng X, Zhu Y, Han P, et al. Nuclear translocation of HIF-1alpha induced by influenza A (H1N1) infection is critical to the production of proinflammatory cytokines. Emerg Microbes Infect (2017) 6:e39. doi: 10.1038/emi.2017.21
86. Ren L, Zhang W, Han P, Zhang J, Zhu Y, Meng X, et al. Influenza A virus (H1N1) triggers a hypoxic response by stabilizing hypoxia-inducible factor-1alpha via inhibition of proteasome. Virology (2019) 530:51–8. doi: 10.1016/j.virol.2019.02.010
87. Zhao C, Chen J, Cheng L, Xu K, Yang Y, Su X. Deficiency of HIF-1alpha enhances influenza A virus replication by promoting autophagy in alveolar type II epithelial cells. Emerg Microbes Infect (2020) 9:691–706. doi: 10.1080/22221751.2020.1742585
88. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med (2018) 379:797–8. doi: 10.1056/NEJMc1807508
89. Plantier L, Cazes A, Dinh-Xuan AT, Bancal C, Marchand-Adam S, Crestani B. Physiology of the lung in idiopathic pulmonary fibrosis. Eur Respir Rev (2018) 27(147):170062. doi: 10.1183/16000617.0062-2017
90. Senavirathna LK, Huang C, Yang X, Munteanu MC, Sathiaseelan R, Xu D, et al. Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci Rep (2018) 8:2709. doi: 10.1038/s41598-018-21073-x
91. Goodwin J, Choi H, Hsieh MH, Neugent ML, Ahn JM, Hayenga HN, et al. Targeting Hypoxia-Inducible Factor-1alpha/Pyruvate Dehydrogenase Kinase 1 Axis by Dichloroacetate Suppresses Bleomycin-induced Pulmonary Fibrosis. Am J Respir Cell Mol Biol (2018) 58:216–31. doi: 10.1165/rcmb.2016-0186OC
92. Aquino-Galvez A, Gonzalez-Avila G, Jimenez-Sanchez LL, Maldonado-Martinez HA, Cisneros J, Toscano-Marquez F, et al. Dysregulated expression of hypoxia-inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir Res (2019) 20:130. doi: 10.1186/s12931-019-1100-4
93. Tsai BM, Wang M, Pitcher JM, Meldrum KK, Meldrum DR. Hypoxic pulmonary vasoconstriction and pulmonary artery tissue cytokine expression are mediated by protein kinase C. Am J Physiol Lung Cell Mol Physiol (2004) 287:L1215–9. doi: 10.1152/ajplung.00179.2004
94. Hamada N, Kuwano K, Yamada M, Hagimoto N, Hiasa K, Egashira K, et al. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J Immunol (2005) 175:1224–31. doi: 10.4049/jimmunol.175.2.1224
95. Philip K, Mills TW, Davies J, Chen NY, Karmouty-Quintana H, Luo F, et al. HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J (2017) 31:4745–58. doi: 10.1096/fj.201700219R
96. MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc (2005) 2:258–66; discussion 290-1. doi: 10.1513/pats.200504-045SR
97. Jiang H, Zhu Y, Xu H, Sun Y, Li Q. Activation of hypoxia-inducible factor-1alpha via nuclear factor-kappa B in rats with chronic obstructive pulmonary disease. Acta Biochim Biophys Sin (Shanghai) (2010) 42:483–8. doi: 10.1093/abbs/gmq041
98. Zhang HX, Yang JJ, Zhang SA, Zhang SM, Wang JX, Xu ZY, et al. HIF-1alpha promotes inflammatory response of chronic obstructive pulmonary disease by activating EGFR/PI3K/AKT pathway. Eur Rev Med Pharmacol Sci (2018) 22:6077–84. doi: 10.26355/eurrev_201809_15946
99. Fu X, Zhang F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med (2018) 16:4553–61. doi: 10.3892/etm.2018.6785
100. Pasupneti S, Tian W, Tu AB, Dahms P, Granucci E, Gandjeva A, et al. Endothelial HIF-2alpha as a Key Endogenous Mediator Preventing Emphysema. Am J Respir Crit Care Med (2020) 202(7):983–95. doi: 10.1164/rccm.202001-0078OC
101. Hypoxemia and hypocapnia in asthma. N Engl J Med (1968) 278(19):1068. doi: 10.1056/NEJM196805092781912
102. Huerta-Yepez S, Baay-Guzman GJ, Bebenek IG, Hernandez-Pando R, Vega MI, Chi L, et al. Hypoxia inducible factor promotes murine allergic airway inflammation and is increased in asthma and rhinitis. Allergy (2011) 66:909–18. doi: 10.1111/j.1398-9995.2011.02594.x
103. Dewitz C, McEachern E, Shin S, Akong K, Nagle DG, Broide DH, et al. Hypoxia-inducible factor-1alpha inhibition modulates airway hyperresponsiveness and nitric oxide levels in a BALB/c mouse model of asthma. Clin Immunol (2017) 176:94–9. doi: 10.1016/j.clim.2017.01.002
104. Crotty Alexander LE, Akong-Moore K, Feldstein S, Johansson P, Nguyen A, McEachern EK, et al. Myeloid cell HIF-1alpha regulates asthma airway resistance and eosinophil function. J Mol Med (Berl) (2013) 91:637–44. doi: 10.1007/s00109-012-0986-9
105. Ning F, Takeda K, Schedel M, Domenico J, Joetham A, Gelfand EW. Hypoxia enhances CD8(+) TC2 cell-dependent airway hyperresponsiveness and inflammation through hypoxia-inducible factor 1alpha. J Allergy Clin Immunol (2019) 143:2026–37 e7. doi: 10.1016/j.jaci.2018.11.049
106. Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol (2007) 178:8127–37. doi: 10.4049/jimmunol.178.12.8127
107. Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest (2008) 118:3301–15. doi: 10.1172/JCI34203
108. Eckle T, Hughes K, Ehrentraut H, Brodsky KS, Rosenberger P, Choi DS, et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J (2013) 27:3078–89. doi: 10.1096/fj.13-228551
109. Eckle T, Kewley EM, Brodsky KS, Tak E, Bonney S, Gobel M, et al. Identification of hypoxia-inducible factor HIF-1A as transcriptional regulator of the A2B adenosine receptor during acute lung injury. J Immunol (2014) 192:1249–56. doi: 10.4049/jimmunol.1100593
110. Eckle T, Koeppen M, Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiol (Bethesda) (2009) 24:298–306. doi: 10.1152/physiol.00022.2009
111. Schingnitz U, Hartmann K, Macmanus CF, Eckle T, Zug S, Colgan SP, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol (2010) 184:5271–9. doi: 10.4049/jimmunol.0903035
112. Vohwinkel CU, Hoegl S, Eltzschig HK. Hypoxia signaling during acute lung injury. J Appl Physiol (1985) (2015) 119:1157–63. doi: 10.1152/japplphysiol.00226.2015
113. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5:304. doi: 10.3389/fimmu.2014.00304
114. Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, et al. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol (1997) 159:1451–8.
115. Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med (2007) 13:913–9. doi: 10.1038/nm1617
116. Hasko G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol (2013) 4:85. doi: 10.3389/fimmu.2013.00085
117. Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol (2004) 25:33–9. doi: 10.1016/j.it.2003.11.003
118. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110:993–1002. doi: 10.1172/JCI0215337
119. Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol (2010) 184:4017–24. doi: 10.4049/jimmunol.0901851
120. Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo X, et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci USA (2009) 106:10684–9. doi: 10.1073/pnas.0901326106
121. Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J (2006) 20:2242–50. doi: 10.1096/fj.06-6419com
122. Eltzschig HK, Bonney SK, Eckle T. Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol Med (2013) 19:345–54. doi: 10.1016/j.molmed.2013.02.005
123. Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schonfeld C, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med (2005) 202:1493–505. doi: 10.1084/jem.20050177
124. Garcia-Morales LJ, Chen NY, Weng T, Luo F, Davies J, Philip K, et al. Altered Hypoxic-Adenosine Axis and Metabolism in Group III Pulmonary Hypertension. Am J Respir Cell Mol Biol (2016) 54:574–83. doi: 10.1165/rcmb.2015-0145OC
125. Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol (2005) 32:2–8. doi: 10.1165/rcmb.2004-0103OC
126. Wang T, Sodhi J, Mentzer R, Van Wylen DG. Changes in interstitial adenosine during hypoxia: relationship to oxygen supply:demand imbalance, and effects of adenosine deaminase. Cardiovasc Res (1994) 28:1320–5. doi: 10.1093/cvr/28.9.1320
127. Saito H, Nishimura M, Shinano H, Makita H, Tsujino I, Shibuya E, et al. Plasma concentration of adenosine during normoxia and moderate hypoxia in humans. Am J Respir Crit Care Med (1999) 159:1014–8. doi: 10.1164/ajrccm.159.3.9803100
128. Song A, Zhang Y, Han L, Yegutkin GG, Liu H, Sun K, et al. Erythrocytes retain hypoxic adenosine response for faster acclimatization upon re-ascent. Nat Commun (2017) 8:14108. doi: 10.1038/ncomms14108
129. Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood (2004) 104:3986–92. doi: 10.1182/blood-2004-06-2066
130. Kiers D, Wielockx B, Peters E, van Eijk LT, Gerretsen J, John A, et al. Short-Term Hypoxia Dampens Inflammation in vivo via Enhanced Adenosine Release and Adenosine 2B Receptor Stimulation. EBioMedicine (2018) 33:144–56. doi: 10.1016/j.ebiom.2018.06.021
131. Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, et al. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med (2008) 178:969–76. doi: 10.1164/rccm.200803-455OC
132. Aeffner F, Bratasz A, Flano E, Powell KA, Davis IC. Postinfection A77-1726 treatment improves cardiopulmonary function in H1N1 influenza-infected mice. Am J Respir Cell Mol Biol (2012) 47:543–51. doi: 10.1165/rcmb.2012-0112OC
133. Aeffner F, Woods PS, Davis IC. Ecto-5’-nucleotidase CD73 modulates the innate immune response to influenza infection but is not required for development of influenza-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol (2015) 309:L1313–22. doi: 10.1152/ajplung.00130.2015
134. Singh Patidar B, Meena A, Kumar M, Menon B, Rohil V, Kumar Bansal S. Adenosine Metabolism in COPD: A Study on Adenosine Levels, 5’-Nucleotidase, Adenosine Deaminase and Its Isoenzymes Activity in Serum, Lymphocytes and Erythrocytes. COPD (2018) 15:559–71. doi: 10.1080/15412555.2018.1537365
135. Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One (2010) 5:e9224. doi: 10.1371/journal.pone.0009224
136. Varani K, Caramori G, Vincenzi F, Adcock I, Casolari P, Leung E, et al. Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2006) 173:398–406. doi: 10.1164/rccm.200506-869OC
137. Fozard JR. The case for a role for adenosine in asthma: almost convincing? Curr Opin Pharmacol (2003) 3:264–9. doi: 10.1016/S1471-4892(03)00039-0
138. Ngamsri KC, Wagner R, Vollmer I, Stark S, Reutershan J. Adenosine receptor A1 regulates polymorphonuclear cell trafficking and microvascular permeability in lipopolysaccharide-induced lung injury. J Immunol (2010) 185:4374–84. doi: 10.4049/jimmunol.1000433
139. Wilson CN, Vance CO, Doyle TM, Brink DS, Matuschak GM, Lechner AJ. A novel post-exposure medical countermeasure L-97-1 improves survival and acute lung injury following intratracheal infection with Yersinia pestis. Innate Immun (2012) 18:373–89. doi: 10.1177/1753425911411595
140. Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol (2014) 88:10214–27. doi: 10.1128/JVI.01068-14
141. Wilson CN. Adenosine receptors and asthma in humans. Br J Pharmacol (2008) 155:475–86. doi: 10.1038/bjp.2008.361
142. Sun CX, Young HW, Molina JG, Volmer JB, Schnermann J, Blackburn MR. A protective role for the A1 adenosine receptor in adenosine-dependent pulmonary injury. J Clin Invest (2005) 115:35–43. doi: 10.1172/JCI22656
143. Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, et al. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol (2005) 3:e174. doi: 10.1371/journal.pbio.0030174
144. Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol (2007) 179:1254–63. doi: 10.4049/jimmunol.179.2.1254
145. He X, Hu JL, Li J, Zhao L, Zhang Y, Zeng YJ, et al. A feedback loop in PPARgamma-adenosine A2A receptor signaling inhibits inflammation and attenuates lung damages in a mouse model of LPS-induced acute lung injury. Cell Signal (2013) 25:1913–23. doi: 10.1016/j.cellsig.2013.05.024
146. Hasko G, Xu DZ, Lu Q, Nemeth ZH, Jabush J, Berezina TL, et al. Adenosine A2A receptor activation reduces lung injury in trauma/hemorrhagic shock. Crit Care Med (2006) 34:1119–25. doi: 10.1097/01.CCM.0000206467.19509.C6
147. Bonneau O, Wyss D, Ferretti S, Blaydon C, Stevenson CS, Trifilieff A. Effect of adenosine A2A receptor activation in murine models of respiratory disorders. Am J Physiol Lung Cell Mol Physiol (2006) 290:L1036–43. doi: 10.1152/ajplung.00422.2005
148. Ehrentraut H, Westrich JA, Eltzschig HK, Clambey ET. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PLoS One (2012) 7:e32416. doi: 10.1371/journal.pone.0032416
149. Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood (2008) 111:2024–35. doi: 10.1182/blood-2007-10-117044
150. Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, et al. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol (2009) 10:195–202. doi: 10.1038/ni.1683
151. Chunn JL, Mohsenin A, Young HW, Lee CG, Elias JA, Kellems RE, et al. Partially adenosine deaminase-deficient mice develop pulmonary fibrosis in association with adenosine elevations. Am J Physiol Lung Cell Mol Physiol (2006) 290:L579–87. doi: 10.1152/ajplung.00258.2005
152. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, et al. IL-1beta suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest (2020) 130:3684–98. doi: 10.1172/JCI136908
153. Bogatcheva NV, Zemskova MA, Kovalenkov Y, Poirier C, Verin AD. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J Cell Physiol (2009) 221:750–9. doi: 10.1002/jcp.21913
154. Wang C, Huang Q, Wang C, Zhu X, Duan Y, Yuan S, et al. Hydroxysafflor yellow A suppresses oleic acid-induced acute lung injury via protein kinase A. Toxicol Appl Pharmacol (2013) 272:895–904. doi: 10.1016/j.taap.2013.07.021
155. Li Q, Hu X, Sun R, Tu Y, Gong F, Ni Y. Resolution acute respiratory distress syndrome through reversing the imbalance of Treg/Th17 by targeting the cAMP signaling pathway. Mol Med Rep (2016) 14:343–8. doi: 10.3892/mmr.2016.5222
156. Baarsma HA, Han B, Poppinga WJ, Driessen S, Elzinga CRS, Halayko AJ, et al. Disruption of AKAP-PKA Interaction Induces Hypercontractility With Concomitant Increase in Proliferation Markers in Human Airway Smooth Muscle. Front Cell Dev Biol (2020) 8:165. doi: 10.3389/fcell.2020.00165
157. Matera MG, Page C, Cazzola M. PDE inhibitors currently in early clinical trials for the treatment of asthma. Expert Opin Invest Drugs (2014) 23:1267–75. doi: 10.1517/13543784.2014.921157
158. Liu Y, Xu H, Geng Y, Xu D, Zhang L, Yang Y, et al. Dibutyryl-cAMP attenuates pulmonary fibrosis by blocking myofibroblast differentiation via PKA/CREB/CBP signaling in rats with silicosis. Respir Res (2017) 18:38. doi: 10.1186/s12931-017-0523-z
159. Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol (2004) 286:C1089–99. doi: 10.1152/ajpcell.00461.2003
160. Liu X, Sun SQ, Ostrom RS. Fibrotic lung fibroblasts show blunted inhibition by cAMP due to deficient cAMP response element-binding protein phosphorylation. J Pharmacol Exp Ther (2005) 315:678–87. doi: 10.1124/jpet.105.090324
161. Rivo J, Zeira E, Galun E, Matot I. Activation of A3 adenosine receptors attenuates lung injury after in vivo reperfusion. Anesthesiology (2004) 101:1153–9. doi: 10.1097/00000542-200411000-00015
162. Rivo J, Zeira E, Galun E, Matot I. Activation of A3 adenosine receptor provides lung protection against ischemia-reperfusion injury associated with reduction in apoptosis. Am J Transplant (2004) 4:1941–8. doi: 10.1111/j.1600-6143.2004.00620.x
163. Mulloy DP, Sharma AK, Fernandez LG, Zhao Y, Lau CL, Kron IL, et al. Adenosine A3 receptor activation attenuates lung ischemia-reperfusion injury. Ann Thorac Surg (2013) 95:1762–7. doi: 10.1016/j.athoracsur.2013.01.059
164. Wagner R, Ngamsri KC, Stark S, Vollmer I, Reutershan J. Adenosine receptor A3 is a critical mediator in LPS-induced pulmonary inflammation. Am J Physiol Lung Cell Mol Physiol (2010) 299:L502–12. doi: 10.1152/ajplung.00083.2010
165. Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol (2003) 171:338–45. doi: 10.4049/jimmunol.171.1.338
166. Tilley SL, Tsai M, Williams CM, Wang ZS, Erikson CJ, Galli SJ, et al. Identification of A3 receptor- and mast cell-dependent and -independent components of adenosine-mediated airway responsiveness in mice. J Immunol (2003) 171:331–7. doi: 10.4049/jimmunol.171.1.331
167. Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan LN, et al. A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J Immunol (2004) 173:1380–9. doi: 10.4049/jimmunol.173.2.1380
168. Young HW, Sun CX, Evans CM, Dickey BF, Blackburn MR. A3 adenosine receptor signaling contributes to airway mucin secretion after allergen challenge. Am J Respir Cell Mol Biol (2006) 35:549–58. doi: 10.1165/rcmb.2006-0060OC
169. Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1–dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood (2008) 111:5571–80. doi: 10.1182/blood-2007-11-126763
170. Hill P, Shukla D, Tran MG, Aragones J, Cook HT, Carmeliet P, et al. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol (2008) 19:39–46. doi: 10.1681/ASN.2006090998
171. Ahn JM, You SJ, Lee YM, Oh SW, Ahn SY, Kim S, et al. Hypoxia-inducible factor activation protects the kidney from gentamicin-induced acute injury. PLoS One (2012) 7:e48952. doi: 10.1371/journal.pone.0048952
172. Ockaili R, Natarajan R, Salloum F, Fisher BJ, Jones D, Fowler AA 3rd, et al. HIF-1 activation attenuates postischemic myocardial injury: role for heme oxygenase-1 in modulating microvascular chemokine generation. Am J Physiol Heart Circ Physiol (2005) 289:H542–8. doi: 10.1152/ajpheart.00089.2005
173. Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation (2008) 118:166–75. doi: 10.1161/CIRCULATIONAHA.107.758516
174. Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N Engl J Med (2019) 381:1011–22. doi: 10.1056/NEJMoa1901713
175. Chen N, Hao C, Peng X, Lin H, Yin A, Hao L, et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N Engl J Med (2019) 381:1001–10. doi: 10.1056/NEJMoa1813599
176. Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM. Clinical Trial of Vadadustat in Patients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am J Nephrol (2017) 45:380–8. doi: 10.1159/000464476
177. Pergola PE, Spinowitz BS, Hartman CS, Maroni BJ, Haase VH. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int (2016) 90:1115–22. doi: 10.1016/j.kint.2016.07.019
178. Haase VH, Chertow GM, Block GA, Pergola PE, deGoma EM, Khawaja Z, et al. Effects of vadadustat on hemoglobin concentrations in patients receiving hemodialysis previously treated with erythropoiesis-stimulating agents. Nephrol Dial Transplant (2019) 34:90–9. doi: 10.1093/ndt/gfy055
179. Prenner BM, Bukofzer S, Behm S, Feaheny K, McNutt BE. A randomized, double-blind, placebo-controlled study assessing the safety and tolerability of regadenoson in subjects with asthma or chronic obstructive pulmonary disease. J Nucl Cardiol (2012) 19:681–92. doi: 10.1007/s12350-012-9547-4
180. Akebia Therapeutics. Akebia Announces Initiation of Investigator-Sponsored Study Evaluating Vadadustat for Prevention and Treatment of ARDS in Patients Hospitalized with COVID-19. (2020). Available at: https://ir.akebia.com/node/11061/pdf.
181. Fallah J, Rini BI. HIF Inhibitors: Status of Current Clinical Development. Curr Oncol Rep (2019) 21:6. doi: 10.1007/s11912-019-0752-z
182. Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discovery (2008) 7:759–70. doi: 10.1038/nrd2638
183. Antonioli L, Blandizzi C, Pacher P, Hasko G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases. Pharmacol Rev (2019) 71:345–82. doi: 10.1124/pr.117.014878
184. Lu Q, Harrington EO, Newton J, Casserly B, Radin G, Warburton R, et al. Adenosine protected against pulmonary edema through transporter- and receptor A2-mediated endothelial barrier enhancement. Am J Physiol Lung Cell Mol Physiol (2010) 298:12. doi: 10.1152/ajplung.00330.2009
185. Gonzales JN, Gorshkov B, Varn MN, Zemskova MA, Zemskov EA, Sridhar S, et al. Protective effect of adenosine receptors against lipopolysaccharide-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol (2014) 306(6):L497–507. doi: 10.1152/ajplung.00086.2013
186. Garcia-Dorado D, Garcia-del-Blanco B, Otaegui I, Rodriguez-Palomares J, Pineda V, Gimeno F, et al. Intracoronary injection of adenosine before reperfusion in patients with ST-segment elevation myocardial infarction: a randomized controlled clinical trial. Int J Cardiol (2014) 177:935–41. doi: 10.1016/j.ijcard.2014.09.203
187. Jin Z, Duan W, Chen M, Yu S, Zhang H, Feng G, et al. The myocardial protective effects of adenosine pretreatment in children undergoing cardiac surgery: a randomized controlled clinical trial. Eur J Cardiothorac Surg (2011) 39:e90–6. doi: 10.1016/j.ejcts.2010.12.052
188. Jacobson KA, Tosh DK, Jain S, Gao ZG. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front Cell Neurosci (2019) 13:124. doi: 10.3389/fncel.2019.00124
189. Folkesson HG, Kuzenko SR, Lipson DA, Matthay MA, Simmons MA. The adenosine 2A receptor agonist GW328267C improves lung function after acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol (2012) 303:L259–71. doi: 10.1152/ajplung.00395.2011
190. Xu X, Zhu Q, Niu F, Zhang R, Wang Y, Wang W, et al. A2BAR activation attenuates acute lung injury by inhibiting alveolar epithelial cell apoptosis both in vivo and in vitro. Am J Physiol Cell Physiol (2018) 315:C558–70. doi: 10.1152/ajpcell.00294.2017
191. Carvalho VF, Ferreira TPT, de Arantes ACS, Noël F, Tesch R, Sant’Anna CMR, et al. LASSBio-897 Reduces Lung Injury Induced by Silica Particles in Mice: Potential Interaction with the A. Front Pharmacol (2017) 8:778. doi: 10.3389/fphar.2017.00778
192. Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, et al. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest (2006) 116:2173–82. doi: 10.1172/JCI27303
193. Lopez-Cruz L, Salamone JD, Correa M. Caffeine and Selective Adenosine Receptor Antagonists as New Therapeutic Tools for the Motivational Symptoms of Depression. Front Pharmacol (2018) 9:526. doi: 10.3389/fphar.2018.00526
194. Chambers ED, White A, Vang A, Wang Z, Ayala A, Weng T, et al. Blockade of equilibrative nucleoside transporter 1/2 protects against Pseudomonas aeruginosa-induced acute lung injury and NLRP3 inflammasome activation. FASEB J (2020) 34:1516–31. doi: 10.1096/fj.201902286R
195. Morote-Garcia JC, Kohler D, Roth JM, Mirakaj V, Eldh T, Eltzschig HK, et al. Repression of the equilibrative nucleoside transporters dampens inflammatory lung injury. Am J Respir Cell Mol Biol (2013) 49:296–305. doi: 10.1165/rcmb.2012-0457OC
196. Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, et al. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest (2003) 112:332–44. doi: 10.1172/JCI200316815
197. Onuora S. Targeting adenosine in SSc. Nat Rev Rheumatol (2020) 16:298. doi: 10.1038/s41584-020-0435-z
Keywords: adenosine, inflammation, hypoxia, hypoxia-inducible factor, acute lung injury, chronic lung injury
Citation: Li X, Berg NK, Mills T, Zhang K, Eltzschig HK and Yuan X (2021) Adenosine at the Interphase of Hypoxia and Inflammation in Lung Injury. Front. Immunol. 11:604944. doi: 10.3389/fimmu.2020.604944
Received: 10 September 2020; Accepted: 25 November 2020;
Published: 14 January 2021.
Edited by:
Detlef Neumann, Hannover Medical School, GermanyReviewed by:
Tomoyuki Nishizaki, Innovative Bioinformation Research Organization, JapanCopyright © 2021 Li, Berg, Mills, Zhang, Eltzschig and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyi Yuan, WGlhb3lpLnl1YW5AdXRoLnRtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.