 Boyuan Huang
Boyuan Huang Xuesong Li2†
Xuesong Li2† Yuntao Li
Yuntao Li Hongbo Zhang
Hongbo Zhang- 1Department of Neurosurgery, Beijing Electric Power Hospital, Beijing, China
- 2Department of Neurosurgery, Huizhou Third People’s Hospital, Guangzhou Medical University, Huizhou, China
- 3Department of Neurosurgery, Zhujiang Hospital, Southern Medical University, The National Key Clinical Specialty, The Engineering Technology Research Center of Education Ministry of China, Guangdong Provincial Key Laboratory on Brain Function Repair and Regeneration, Guangzhou, China
- 4Department of Neurosurgery, The First Affiliated Hospital, Jinan University, Guangzhou, China
- 5Department of Neurosurgery, Jiujiang Hospital of Traditional Chinese Medicine, Jiujiang, China
Glioblastoma multiforme (GBM) is the most common and aggressive malignant tumor found in the central nervous system. Currently, standard treatments in the clinic include maximal safe surgical resection, radiation, and chemotherapy and are mostly limited by low therapeutic efficiency correlated with poor prognosis. Immunotherapy, which predominantly focuses on peptide vaccines, dendritic cell vaccines, chimeric antigen receptor T cells, checkpoint inhibitor therapy, and oncolytic virotherapy, have achieved some promising results in both preclinical and clinical trials. The future of immune therapy for GBM requires an integrated effort with rational combinations of vaccine therapy, cell therapy, and radio- and chemotherapy as well as molecule therapy targeting the tumor microenvironment.
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive primary malignant tumor in the central nervous system (CNS) in adults (1). It is mainly classified into two groups: isocitrate dehydrogenase (IDH)-wildtype GBM, which has been previously referred to as primary GBM and represents about 90% of cases, and IDH-mutant GBM, which is developed from a lower-grade diffuse glioma and represents about 10% of cases. The current standard treatments for GBM include a combination of surgical resection, radiation, and chemotherapy. At present, there are only two drugs approved by the FDA to treat GBM via systematical administration: temozolomide (TMZ) for the treatment of newly diagnosed GBM (ndGBM) and bevacizumab for the treatment of recurrent GBM (rGBM) (2, 3). Unfortunately, current therapeutic approaches have very limited impact on improving the prognosis of GBM patients, showing 15 months of median survival and less than 5% of a 5-year survival rate (1). Thus, opportunities and challenges remain in finding more efficient treatments against GBM.
Immunotherapy, which manipulates the immune system to attack tumor cells with minimal adverse effects and prevents tumor remission, has drawn extensive attention (4). However, there are still challenges that need to be overcome in the development of immunotherapy for GBM. The CNS is considered to be an “immune-privileged” organ, attributed to the lack of lymphatic involvement and the selectivity of the blood–brain barrier (BBB) to immune cells (5). Antigens in the brain can still drain into the cervical lymph nodes through lymphatic vessels in the dura and meninges (6). Moreover, microglia, as the brain’s resident immune cells, can function as potential antigen presentation cells (APCs), and T cells are activated in the cervical lymph nodes entering the brain parenchyma through the cerebrospinal fluid (CSF) (7). These observations suggest that the brain is immune privileged to a certain degree, and blood-derived immune cells are not completely precluded from the brain (8, 9). Moreover, GBM cells can exert local immunosuppressive effects in many ways. On the one hand, GBM cells themselves can secrete various protumor cytokines and/or chemokines, which can influence macrophage polarization, promote regulatory T cell (Treg) recruitment, and inhibit dendritic cell (DC) maturation and natural killer (NK) cell function. On the other hand, GBM cells can express immunosuppressive molecules, such as programmed cell death protein 1 ligand (PD-L1), which can prevent T cell proliferation and activation (10). In spite of these challenges, immunotherapy for GBM still obtains considerable achievements, which have given rise to a number of clinical trial investigations. Increasing immunotherapeutic approaches for GBM treatments have also been established. In this review, we present an overview of the current immunotherapy for GBM, including peptide vaccines, DC vaccines, chimeric T-cell receptors, checkpoint inhibitors, and oncolytic virotherapy.
Peptide Vaccines
Peptide vaccines are about 8–30 amino acids in length. They are designed to encompass tumor-specific antigens (TSA), which derive from mutations only expressed in tumor cells but are absent in normal cells, or tumor-associated antigens (TAA), which derive from overexpressed normal proteins that are present in both tumoral and normal tissue. Unlike other solid tumors, GBM is notorious for possessing a relatively low level of mutation, resulting in only a minority of mutations used as TSA (11). At present, the peptide vaccines under investigation in GBM include rindopepimut (12), IMA950, and isocitrate dehydrogenase 1 (IDH1). The epidermal growth factor receptor variant III (EGFRvIII), with a mutated deletion in 20%–30% of tumors, is the most relevant and uncontroversial TSA for GBM. Thus, targeting EGFRvIII as a primary example of TSA-based peptide vaccines has been extensively investigated in the immunotherapy against GBM. In a phase II clinical trial, 65 patients with EGFRvIII-positive GBM were administrated with rindopepimut as well as with standard adjuvant TMZ (13) (Table 1). As a result, a progression-free survival (PFS) at 5.5 months of 66% and a median overall survival (OS) of 21.8 months were observed (13). In another phase II clinical trial, bevacizumab plus rindopepimut or a placebo were tested in rGBM patients, indicating that PFS at 6 months was 27%, and the median OS was 12 months, which is significantly improved compared with the control group of a PSF at 6 months of 11% and a median OS of 8.8 months (14). Following these achievements, a large, randomized, double-blind, placebo-controlled phase III clinical trial, enrolling 745 patients with ndGBM was terminated early after showing no significant improvement in the median OS. However, the data demonstrate patients with decent humoral immune responses (15) (Table 1). Notably, lost expression of EGFRvIII (antigen escape) was observed in the control arm to a similar degree as that of the treatment arm, which challenges the notion that therapies targeting EGFRvIII should be responsible for the outgrowth of EGFRvIII-deficient GBM cells (16, 17). In addition, this study also highlights that targeting a single tumor antigen may not be sufficient enough to induce durable antitumor responses.
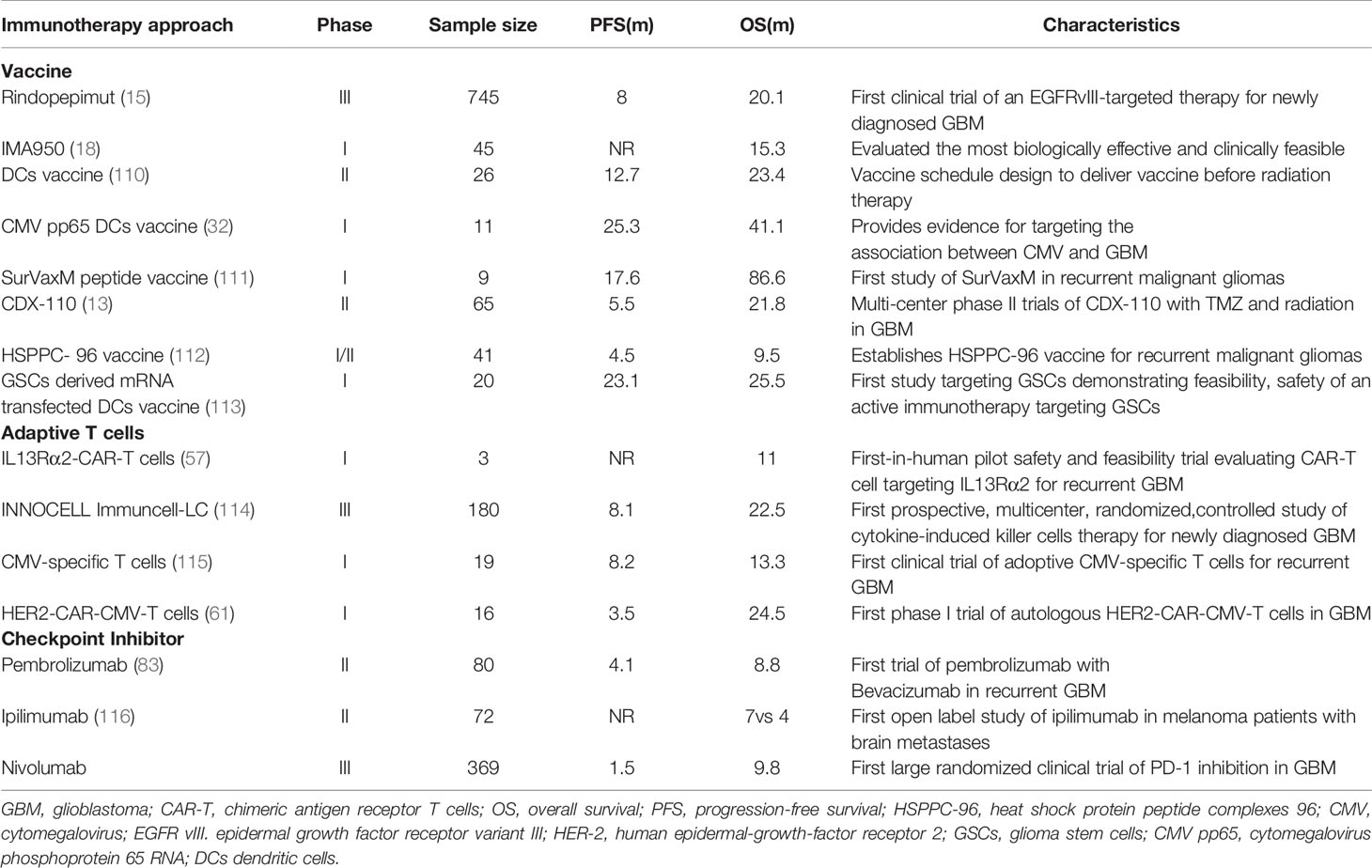
Table 1 Completed representative clinical trials of immunotherapy.
IMA950 is a novel therapeutic vaccine that includes nine synthetic tumor-associated HLA-A2-restricted peptides (TUMAP), two MHC class II–binding peptides, and one HLA-A2-restricted HBV-derived peptide, and the last one was also used as a marker of vaccine immunogenicity. IMA950 can trigger the stimulation of TUMAP-specific cytotoxic T cells, leading to the destruction of malignant tumor cells. In a phase I trial, patients diagnosed with ndGBM after tumor resection were injected intradermally with IMA950 either prior to or just after the initiation of chemoradiotherapy. The majority of patients were found to respond well with a PFS at 6 months of 74% and a median OS of 15.3 months (18) (Table 1). In a recently completed phase I/II trial, IMA950 with vaccine adjuvant poly-ICLC in combination with TMZ were tested in 19 patients (16 with GBM and three with anaplastic astrocytoma). Patients from the overall cohort showed a median OS of 21 months from the date of surgery, compared with the GBM-only cohort of 19 months. PFS of patients from the overall cohort were 93% and 56% at 6 and 9 months, respectively (19). As for rGBM, however, IMA950 has no benefit in any preclinical trial. In a previous clinic trial, patients with recurrent high-grade gliomas who were administrated bevacizumab with the IMA950/poly-ICLC peptide vaccine did not show improved OS and PFS compared to nonvaccinated patients (20).
IDH1 mutations can be found in nearly 90% of low-grade gliomas, and more than 90% of IDH1 mutations contain an arginine-to-histidine switch at position 132 (IDH1R132H). In GBM, IDH1 mutations can predict whether the tumors are secondarily developed from lower-grade gliomas because IDH1 mutations are rarely found in primary GBM. This high-frequency neoantigen is expressed in more than 70% of rGBMs, which can induce the formation of the oncometabolite 2-hydroxyglutarate and the inhibition of NADPH production (21, 22). Preclinical studies suggest peptide vaccines spanning the IDH1 mutation, may elicit IDH1R132H-reactive CD4+ and CD8+ responses for antitumor (23). A phase I clinical trial at Duke University is ongoing in which the intradermal IDH1 peptide vaccine is tested in IDH1-positive grade II primary brain tumors (ClinicalTrials.gov identifier: NCT02193347). In another phase I trial, the safety of the IDH1 peptide vaccine for high-grade gliomas was also being evaluated. This study was completed in 2019 (ClinicalTrials.gov identifier: NCT02454634). Data collection is ongoing, and the therapeutic efficiency of IDH1 vaccines will be further estimated.
To date, several peptide vaccine strategies are shown to have safe and efficient profiles in phase I and II clinical trials, and some vaccines have significantly improved patient survival compared with historical controls. However, supportive data from phase III trials are still lacking. Although a phase III clinical trial on the EGFRvIII-based vaccine has failed in ndGBM patients, this vaccine could still induce decent humoral immune responses (15). Accordingly, more phase III trials on the peptide vaccine are required to support the therapeutic potential of peptide vaccines in GBM treatment. In addition, the single-antigen targeted strategy may lead to antigen escape due to high heterogeneity in the tumor. Therefore, alternative vaccine approaches are needed to target multiple tumor neoantigens. Heat shock protein (HSP) peptide complexes 96 (HSPPC-96) is one solution to handling this problem. HSPPC-96 is a primary resident chaperone of the endoplasmic reticulum, which can be internalized into APCs for efficient class I and II MHC-mediated presentation of tumor peptides (24). In a phase I clinical trial, an HSPPC-96 vaccination induced a tumor-specific peripheral immune response in 11 of 12 high-grade glioma patients (25). A subsequent open-label phase II multicenter clinical trial in surgically resectable rGBM patients treated with HSPPC-96-loaded antigens, which were extracted from patient-derived glioma tissue, showed an impressive median OS of 42.6 weeks and a 6-month OS of 29.3%, respectively (26). These results have sparked multiple ongoing clinical trials: NCT00905060, a completed phase II trial exploring the application of autologous HSPPC-96 following tumor resection and adjuvant RT and TMZ in ndGBM, and NCT01814813, a multi-institutional trial investigating the safety, tolerability, and efficacy of HSPPC-96 combined with bevacizumab in rGBM patients.
DC Vaccines
DCs are able to present tumor antigens to CD4+ and CD8+ T cells to stimulate an immune response. Therefore, vaccines based on DCs represent another immunotherapeutic approach. This type of vaccine is typically produced through the ex vivo generation of DCs harvested from patients. The isolated DCs are stimulated by either tumor antigens or mRNA-expressing MHC molecules before administration (27, 28). Currently, there are strategies for DC vaccines exposed to either single specific antigens or multiple tumor antigens. In a phase I trial, seven patients with high-grade gliomas were administered Wilms’ tumor 1 (WT1)-pulsed autologous DCs. Five patients showed stable clinical responses, and the OS was 12.3 months in the cohort after the first DC vaccination (29). Cytomegalovirus phosphoprotein 65 RNA (CMV pp65) is also incorporated into DC vaccines because CMV nucleic acids and proteins are found in both primary and recurrent GBM (30). In another phase I trial, patients with ndGBM were administered pp65-specific DCs in combination with preconditioning using tetanus-diphtheria toxoid (Td). It achieved a promising PFS of 15.4–47.3 months and OS of 20.6–47.3 months (31). Batich et al. applied dose-intensified TMZ followed by a CMV pp65 DC vaccine to treat 11 ndGBM patients in a phase I trial. Both median PFS and OS are longer than predicted ones (32) (Table 1). Currently, a randomized phase II trial involving a CMV pp65 DC vaccine is recruiting ndGBM patients (ClinicalTrials.gov identifier: NCT02465268) (Table 2). Another similar clinical trial on IDH1 DC vaccine for glioma treatments is also under investigation in China (ClinicalTrials.gov identifier: NCT02771301).
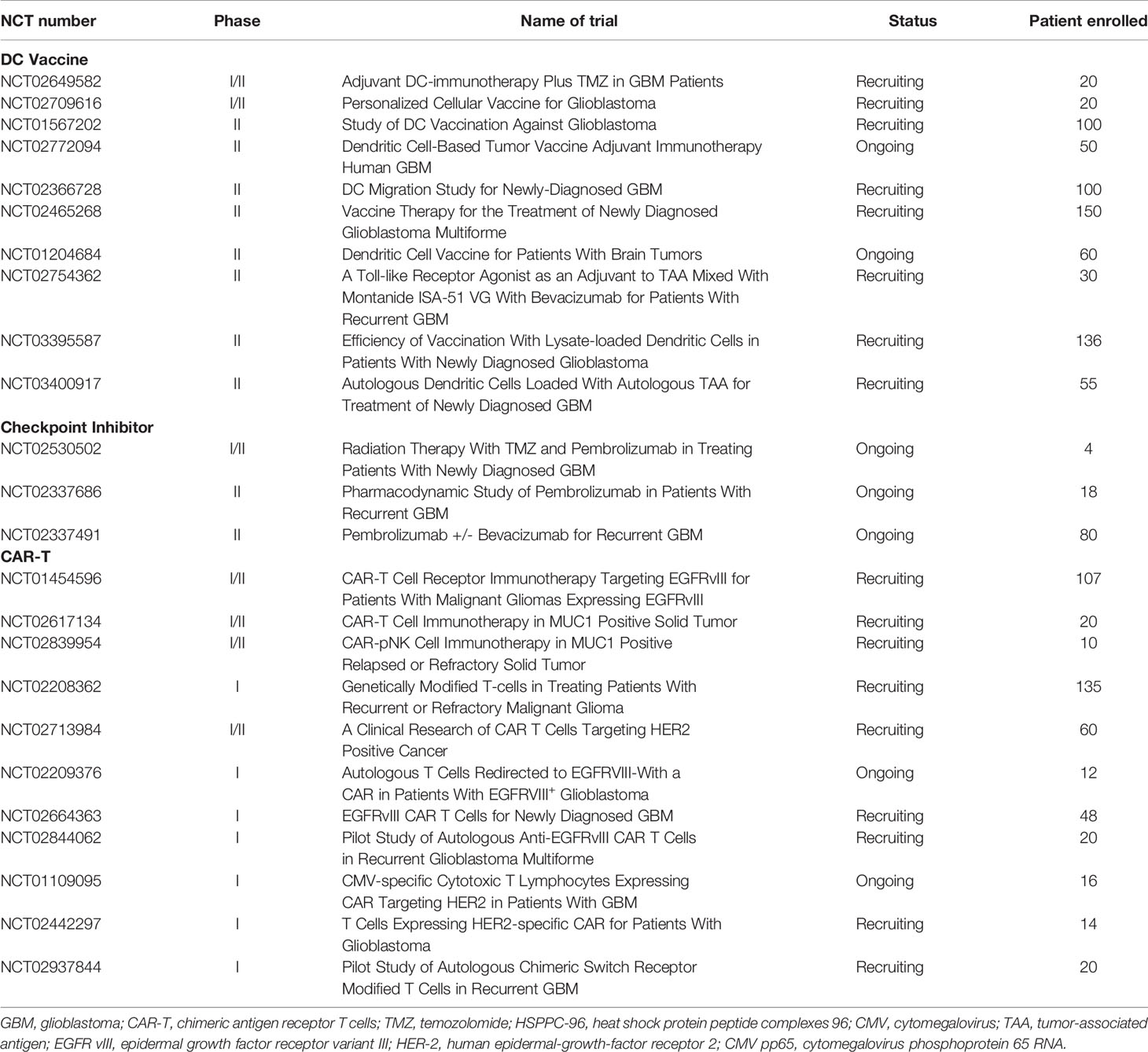
Table 2 Ongoing clinical trials involving DC vaccine, checkpoint inhibitor and CAR-T.
In addition, there are also DC vaccines exposed to multiple tumor antigens to induce a more robust immune response. In a phase I clinical study, an autologous DC vaccine pulsed with class I peptides from TAA highly expressed on gliomas and a cancer stem cell population (ICT-107) were administered to 15 ndGBM patients. Median PFS was 16.9 months and median OS was 38.4 months. It is also worth noting that six patients showed no evidence of tumor recurrence in a follow-up of 40.1 months (33). In another phase I/II trial, patients with recurrent glioma were administered α-type 1 polarized DCs loaded with EphA2, IL13Rα2, YKL-40, and gp100 and combined with poly-ICLC. It was observed that nine of 22 patients achieved PFS lasting at least 12 months, and one rGBM patient exhibited a sustained complete response (34). Recently, a novel DC vaccine, called DCVax-L, has been prepared from tumor lysate. In a phase I/II clinical trial, a DC vaccine was prepared with the patient’s own tumor cells prior to administration to the patients. Sixteen GBM patients were enrolled in this trial. The data show that median and 5-year survival were 525 days and 18.8%, respectively (35). A randomized phase II trial on DCVax-L and nivolumab in rGBM patients is ongoing (ClinicalTrials.gov identifier: NCT03014804). Another randomized phase III trial on DCVax-L is currently underway in 348 GBM patients (ClinicalTrials.gov identifier: NCT00045968). Given that DC cocultured with tumor lysate for the generation of DCVax-L, this kind of vaccine should be more efficient in the elimination of tumor cells because it is able to target more tumor-related antigens. However, theoretically there is also a high risk that it may cause an autoimmune response. Therefore, it still remains a challenge for researchers to choose a suitable tumor lysate for the generation of DC vaccines regarding the high heterogeneity of GBM. There is still much work that needs to be done to understand the influences of tumor genotypes and microenvironments on DC vaccine production to prevent the undesired autoimmune response during administration.
Adoptive T Cell Therapy
The functional advantage of adoptive T cell therapy lies in its ability to harvest, train, and expand autologous T cells which are then transfered back into patients (36). The primary forms of adoptive T cell therapy can be generally classified as tumor-infiltrating lymphocytes (TILs), T-cell receptor (TCR) treatment, and chimeric antigen receptor T (CAR-T) cells. The application of TILs requires highly accessible and immunogenic tumor cells; however, only melanoma can meet sufficient expansion of TILs from their respective tumor samples (37). In a prospective pilot study including six rGBM, locally infused autologous TILs did not show powerful cytotoxicity against the autologous tumor (38). Apart from the desire for improvement in expansion of brain tumor–derived TILs, this study also implied the significance of maintaining autologous TIL activation within the brain TME. TCR treatment was the first successful application of adoptive T cell therapy that utilized autologous T cells transduced with human TCR recognizing a melanoma antigen recognized by T cells 1 (MART-1) to treat patients with metastatic melanoma (39). As far as gliomas are concerned, however, no clinical trials based on TCR-T cell therapies have been initiated. The little progress made in TILs and TCR against gliomas force researchers to seek other ways, and the efforts to overcome MHC restriction result in the development of CAR-T cell therapy.
Recently, genetically engineered T cells expressing chimeric antigen receptors (CARs) to recognize specific tumor antigens have brought in a new era of cancer immunotherapy. CARs are artificial fusion proteins that incorporate an intracellular T-cell signaling domain that consists of one or more single-chain variable fragments (scFv) and an extracellular antigen-recognition domain to target specific neoplastic cells. These complex domains include CD28, CD3ζ, 4-1BB, or OX40 derived from the same part of CD28/CD8 or a corresponding domain of T-cell receptors (TCRs) (40, 41) (Figure 1). In addition to being endowed with a specific affinity to TSAs or targets of interest, CAR-T cells can be stimulated without MHC involvement and prevent the challenges associated with adoptive T-cell transfer (42, 43). Currently, CD19-specific CAR-T cells have induced sustained and durable antitumor immune responses in patients with multiple myeloma, acute and chronic lymphocytic leukemia, and refractory diffuse large B-cell lymphoma (DLBCL) (44–49). These encouraging results have prompted FDA approvals of two therapies: CTL019, a treatment for patients younger than 25 with relapsed or refractory B-cell precursor acute lymphoblastic leukemia, and another CD19-targeted CAR T-cell treatment, axicabtagene ciloleucel, for patients with failed DLBCL for at least two prior therapies (50, 51). Inspired by the success in blood tumors, increasing interest has focused on the treatments of CAR-T cells against GBM. These CAR-T cells mainly target the following antigens: EGFRvIII, IL-13Rα2, and HER2. EGFRvIII is abundantly expressed in approximately 30% of GBM to enhance glioma cell proliferation, angiogenesis, and invasiveness (52). In preclinical studies, CAR-T cells targeting EGFRvIII could effectively infiltrate to tumor sites and suppress the growth of glioma xenografts in murine models (53). In a human clinical trial, EGFRvIII-targeting CAR-T cells showed feasibility and safety in the treatment for 10 rGBM patients without toxicity or cytokine release syndromes (54). It demonstrates that transient expansion of EGFRvIII-targeting CAR-T cells could be detected in peripheral blood of all patients. The median OS was approximately 8 months, and one patient experienced residual stable disease at 18 months (54). The promising clinical trials are still ongoing to assess the efficiency of this approach (Figure 1).
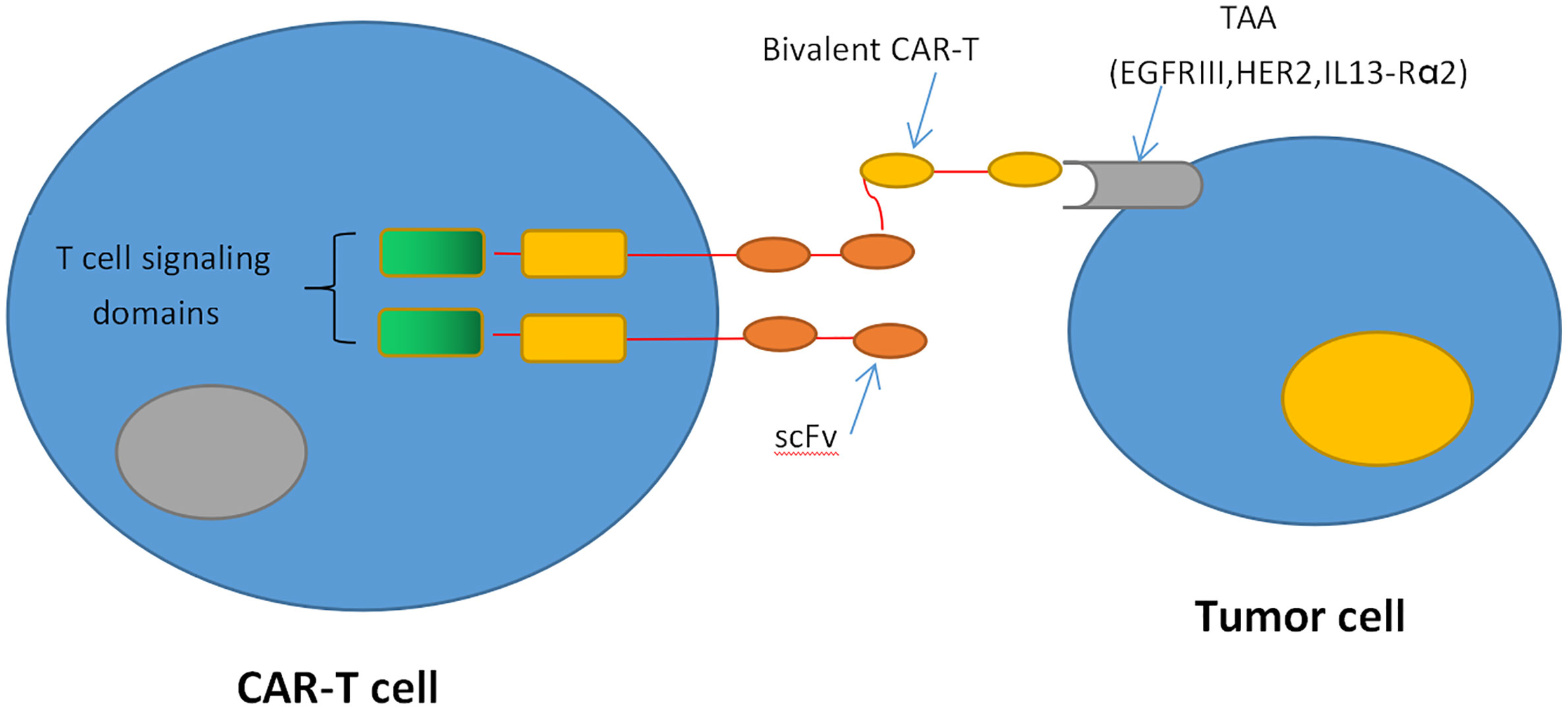
Figure 1 CAR T-cell therapy. CAR-T cells produce an artificial T cell receptor that has big tumor-specific surface antigens.
Another target of CAR-T cell treatment for GBM patients is IL-13Rα2, which presents in more than 75% of GBM tumors associated with tumor invasiveness and poor prognosis (55, 56). As the first CAR-T targeting IL-13Rα2 therapy, the feasibility and safety of IL13-zetakine CD8+ CTL against rGBM have been evaluated by Brown et al. In this trial, intracranial delivery of the IL13-zetakine+ cytotoxic T lymphocytes (CTL) into the resection cavity was well tolerated in three patients. A transient antiglioma response was observed in two patients (57) (Table 1). In a following report, CAR T-cells targeting IL-13Rα2 incorporated with costimulatory immunoreceptor CD137 were initially delivered into the resection cavity of grade 3 or higher GBM. Consequently, regression of all intracranial and spinal tumors was observed without any toxic effects. Moreover, a robust increase of inflammatory cytokines and chemokines in the CSF with limited CAR T-cell accumulation and expansion was also found. Eventually, this clinical response lasted for 7.5 months after the initiation of CAR T-cell therapy (58).
Human epidermal growth factor receptor 2 (HER2) is a transmembrane tyrosine kinase receptor overexpressed in 80% of GBM. It is identified as an independent unfavorable prognostic indicator for GBM patients (59, 60). Considering that HER2 is also expressed in normal tissues, there is a theoretical high risk of off-target toxicity resulting from HER2-targeting CAR-T cells. Intriguingly, a phase I clinical trial demonstrated the feasibility and safety of HER2-targeting CAR-T cells, which were well-tolerated in 17 patients with progressive HER2-positive GBM without any dose-limiting toxic effects (61) (Table 1). The median OS was 11.1 months (95% CI, 4.1-27.2 months) from the first T-cell infusion and 24.5 months (95% CI, 17.2-34.6 months) from diagnosis. Three patients had no progression between 24 to 29 months (61).
Although the results from these studies are encouraging, CAR-T cells targeting a single antigen may still inevitably lead to antigen escape. To deal with this intractable dilemma, CAR-T cells targeting multiple tumor antigens have been established to overcome the heterogeneity of GBM. Hegde et al. created CAR-T cells expressing a HER2-binding scFv and an IL-13Rα2-binding IL-13 mutein, which could efficiently recognize and kill either HER2 or IL-13Rα2 positive tumor cells (62). These bispecific CAR-T cells are more sustainable and capable of improving the survival in GBM murine models and mitigating antigen escape (62). Taking this approach one step further, the same research group generated trivalent CAR-T cells targeting HER2, IL-13Rα2, and EphA2, which could overcome the interpatient variability and capture nearly 100% of tumor cells. In a murine model, the trivalent CAR-T cells exhibited superior antitumor efficacy. It significantly inhibited tumor growth and improved animal survival compared with biCAR-T cells or single CAR T-cells (63).
CAR T-cell therapy in GBM has just begun. Preliminary results demonstrate its feasibility and safety, and bi- or tri-CAR-T cells may be a promising strategy for the intractable dilemma of antigen loss. However, several problems and challenges in solving CAR-T treatment still exist.. First, T-cell proliferation and persistence is still a limitation for solid tumor treatment because the peripheral blood is not the therapeutic site. It also raises a related issue regarding whether precondition of lymphodepleting, which has been approved as a standard in CAR-T treatment of hematologic malignancies, is able to improve CAR T-cell expansion and efficacy in GBM (64, 65). Although it has not been reported to use lymphodepleting preconditioning in ndGBM (54, 57, 58), rGBM patients often accept “lymphodepletion” before CAR-T treatment due to the effects of standard radiation and TMZ (66). Another issue that needs to be addressed is the immunosuppressive TME. The TME of GBM can present many obstacles to CAR-T cells, including immunosuppressive immune cells, tumor-derived soluble factors and cytokines, and physical and metabolic barriers (67, 68). Therefore, intensive investigations are urgently needed to improve the efficacy of CAR-T treatment in GBM patients.
Checkpoint Inhibitors
Immune checkpoints are the coinhibitory molecules that could attenuate the intensity and duration of T-cell-mediated immune responses to maintain self-tolerance and prevent uncontrolled inflammatory responses. Currently, the most well-studied coinhibitory molecules in hematologic and solid tumors include cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1) and its ligand PD-L1, T-cell immunoglobulin and mucin domain 3 (TIM-3), and indoleamine 2,3-dioxygenase-1 (IDO1).
CTLA-4 is one of the most extensively studied immune checkpoint inhibitors, and it suppresses T-cell stimulation by competing with the costimulatory molecule CD28 for binding its ligands CD80 and CD86 (69, 70) (Figure 2). Ipilimumab (trade name Yervoy) was the first FDA-approved checkpoint for immunotherapy targeting CTLA-4 applied in metastatic melanoma and now approved for several solid tumors. In murine glioma models, blockade of CTLA-4 could induce tumor regression and promote long-term survival without eliciting experimental allergic encephalomyelitis (71). For GBM, combinatorial blockade of CTLA-4 and PD-1 were demonstrated to cure 75% of immunocompetent murine GBM models even against advanced, later-stage tumors (72). Until now, blockade of CTLA-4 could lead to robust antitumor immunity only at the preclinical stage. Although there has been no published data on CTLA-4 inhibitors solely treating GBM yet, some clinical trials are currently ongoing to evaluate CTLA-4 inhibitors in GBM combined with other therapeutic agents, such as VEGF inhibitors, checkpoint inhibitors, tumor treating fields, and radiation therapy (73).
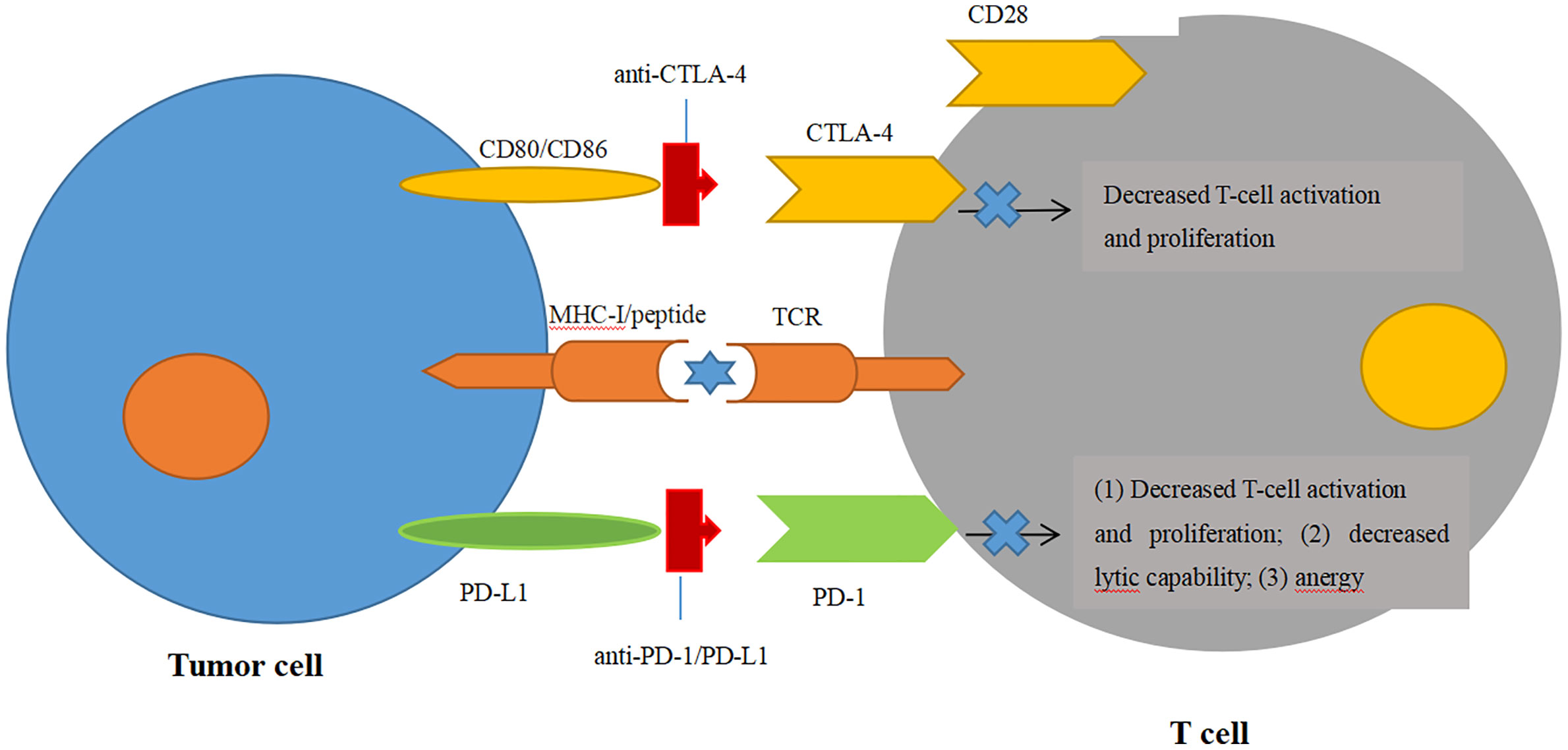
Figure 2 Blockade of immune checkpoint inhibitors. Engagement of CTLA-4 with its ligands CD80/CD86 can prevent the ligands binding to the T-cell activation and proliferation. Engagement of PD-1 with one of its ligands, PD-L1, can decrease the T-cell tumor lytic capacity and induces T-cell anergy.
PD-1, an immunoglobulin receptor belonging to the extended CD28/CTLA-4 family of T-cell regulators, is expressed on activated T, B, myeloid, and NK cells. It binds to the ligands PD-L1 and PD-L2 (74). The PD-1/PD-L1 axis is proven to be the major negative regulation of CTL in the TME, whose protumor function, including suppression of T-cell activation and infiltration, is inhibiting the secretion of pro-inflammatory factors and inactivation of TCR signaling (74, 75) (Figure 2). Owing to the success of antibodies targeting the PD-1/PD-L1 axis in the clinical trials against advanced melanoma, monoclonal PD-1 antibodies (Pembrolizumab and Nivolumb) were approved by the FDA for the treatment of melanoma, non-small cell lung cancer (NSLC), and other solid tumors (76–80). For GBM, the therapeutic effects of PD-1/PD-L1 antibodies remain largely elusive. In a preclinical study, the combination of PD-1 antibody and radiotherapy achieved a twofold increase in median survival in GL261 glioma mouse models, and 15%–40% of mice gained long-term survival compared with a single treatment (81). In another preclinical trial, the combination of a DC vaccine and PD-1 antibody achieved long-term survival in intracranial glioma tumor-bearing mice that were solely dependent on CD8+ T cells (82). Moreover, this combination of a DC vaccine and PD-1 antibody also resulted in the upregulation of homing integrin and immunologic memory markers on TILs (82). These encouraging preclinical studies prompted the first large phase III clinical trial of PD-1 checkpoint blockade in rGBM through the comparison of nivolumab monotherapy with standard care using bevacizumab (NCT02017717). Although the median OS was comparable between nivolumab and bevacizumab among the overall enrolled patients, this trial was still closed in 2017 on account of failing to meet the primary OS endpoint (83) (Table 1). Another phase III randomized trial, CheckMate 548, was processed to evaluate the effects of nivolumab with or without radiation therapy and TMZ in O6-methylguanine-DNA methyltransferase (MGMT)-methylated ndGBM patients. This study has also failed to achieve the endpoint for the inability of nivolumab concomitant with radiation therapy and TMZ to improve the median OS (84). Another similar phase III trial, CheckMate 498, for patients with MGMT-unmethylated tumors also declared that nivolumab combined with TMZ failed to improve patients’ median OS. Although nivolumab has not yet shown efficiency in clinical trials, other antibody therapies targeting the PD-1/PD-L1 axis have emerged in clinical trials. Pembrolizumab, another PD-1 antibody, was tested as neoadjuvant or adjuvant-only therapy in 35 surgically resectable rGBM patients in a single-arm phase II clinical trial. Patients accepting pembrolizumab showed a statistically significant increase in OS with a median value of 417 days compared with those in the adjuvant group with 228.5 days. PFS in the neoadjuvant group was also significantly increased over the adjuvant group (99.5 days vs. 72.5 days). The study also found that neoadjuvant anti-PD-1 blockade was related to an upregulation of the IFN-γ responsive gene signature and a declined cell cycle–related gene signature in the tumor (85). In a single-arm phase I trial, pembrolizumab accompanied by hypofractionated stereotactic irradiation and bevacizumab were well tolerated in 23 rGBM patients. More than half of the patients achieved durable objective responses, and 64% of the patients were still alive within 12 months (86). Another phase I trial on combinatorial pembrolizumab with bevacizumab (NCT02337491) in rGBM patients showed a median OS of 8.8 months and PFS of 4.1 months (https://clinicaltrials.gov/ct2/show/results/NCT02337491) (Table 2). Additionally, durvalumab (MEDI4736), a humanized PD-Ll monoclonal antibody, is currently being tested in a multicenter phase II trial combined with radiotherapy and bevacizumab in GBM patients (NCT02336165) (87). It is striking that one patient obtained a long-period OS of 86 weeks (87). In contrast to the monotherapy by PD-1/PD-L1 inhibitors with few successes, combinatorial therapy of PD-1/PD-L1 antibodies with radiation therapy and/or chemotherapy seem more promising in the clinical trials against GBM.
In addition to CTLA-4 and PD-1/PD-L1 therapy, another two checkpoint targets have received researcher interest. TIM-3, an immunosuppressive receptor expressed on T cells, Tregs, DCs, NK cells, and macrophages, can promote T-cell exhaustion similar to PD-1 (88, 89). There are ongoing clinical trials testing TIM-3-targeted antibodies in solid tumors (NCT02608268, NCT02817633) and hematological malignancies (NCT03066648). IDO1 is a cytoplasmic enzyme promoting tryptophan catabolism through the kynurenine pathway. It is demonstrated that depletion of IDO1 can suppress T-cell function and elevate expression of IDO1 in a tumor, which is correlated with poor prognosis in GBM patients (90, 91). So far, there are some clinical trials evaluating IDO1 inhibitors in melanoma (92) and breast cancer (NCT01792050) but none showing a survival benefit. There is a phase I clinical trial including various treatments, such as IDO1 inhibitor therapy, chemotherapy, and radiation therapy in pediatric brain tumors. Twenty-nine patients enrolled in this trial showing a median PFS of 6.2 months, and the median time to regimen failure is 11.7 months (NCT02502708). There are also other ongoing clinical trials testing the IDO1 inhibitor combined with other therapies in malignant brain tumors (93) and rGBM (NCT03707457). Results from these trials are still pending. Furthermore, the efficacy and safety of these agents need to be evaluated in GBM patients.
Despite the great advances in treating hematological malignancies and solid tumors as well as promising results from preclinical and early-phase trials in GBM, immune checkpoint inhibitors have not yet demonstrated efficacy in GBM through large phase III clinical trials as a monotherapy or combination therapy with other treatments. The BBB should first be taken into account as it may block the antibody penetration into the CNS. Moreover, a tumor mutational burden that predicts the efficacy of immune checkpoint inhibitors across multiple solid tumors is actually associated with poor prognosis in glioma patients (94). Last but not least, the immunosuppressive TME and dynamic responses to tumorigenesis of GBM may also contribute to the obstacles faced by the immune checkpoint inhibitors. Thus, further investigations on the optimal combinations of multiple therapies as well as tumor genomic and immune characteristics are urgently required to clarify the role of checkpoint inhibitors in GBM in the future.
Oncolytic Virotherapy
Oncolytic virotherapy (OV) employs naturally occurring or artificially engineered viruses, which are typically delivered intratumorally or postsurgically into the resection cavity to infect and lyse tumor cells, simultaneously triggering inflammation and immune responses to tumor cells and the virus (95). Multifarious virus species have been studied as oncolytic virus platforms for cancer therapy, such as herpes simplex virus (HSV), adenovirus, vaccinia virus, measles virus, poliovirus, and reovirus. In 2015, talimogene laherparepvec (T-VEC), a genetically modified HSV, was approved by the FDA for advanced melanoma as the first OV therapeutic in the United States (96). GBM virotherapy clinical trials started in 1991; Martuza et al. first reported engineered HSV for their capability of selective replication and killing of GBM cells (97). Since then, multifarious OVs have been tested in gliomas; however, they seldomly demonstrate efficacy in improving median OS in randomized trials (98). Here, we present evidence that OVs have recently been advanced to phase I/II trials in glioma patients, demonstrating remarkable efficacy in subsets of patients.
DNX-2401 (Ad5-Delta-24-RGD;tasadenoturev) is a replication-competent adenovirus with enhanced infectivity, high tumor selectivity, and a specific mutation to restrict viral replication. This virus can target integrins on GBM cells with a glycine/aspartic/arginine acid motif, which can increase infective specificity for tumor cells (99, 100). In a phase I trial of DNX-2401 (NCT00805376), 37 rGBM patients received a single intratumoral injection of DNX-2401 through the biopsy needle (cohort 1) or a permanently implanted catheter followed by tumor resection (cohort 2). In cohort 1, 20% of patients survived more than 3 years after treatment, and 3 patients showed more than 3 years of PFS with dramatic tumor reduction (95% or more, CR). Immunohistochemical analysis of post-treatment surgical specimens from cohort 2 revealed that DNX-2401 replicated and spread within the tumor and induced CD8+ and T-bet+ cell infiltration. No dose-limiting toxicities were observed, and adverse effects were reported in 15% of patients with no serious virus-related events of grade 3 or higher noted (101). Thus, this clinical trial, for the first time, showed direct oncolytic effects in GBM and provided evidence for elicitation of anti-GBM immune responses. In another phase I/II clinical trial that was initiated in 2010 for rGBM patients (NCT01582516), DNX-2401 was administered by catheters targeting the tumor mass and the surrounding infiltrated brain. Analysis of CSF from patients showed an elevated level of some cytokines that can increase the levels of CD64, a marker of M1-polarization, implying that DNX-2401 therapy can promote a macrophage phenotype shift from M2 to M1 (102). Currently, the combination of DNX-2401 treatment with pembrolizumab is under investigation in a phase II trial for rGBM patients (CAPTIVE/KEYNOTE-192, NCT02798406). Interim results were reported at the SNO 2018 annual meeting, including that the combinatorial therapy was well tolerated, and 100% 9-month survival for the first seven patients treated was noted (103). Publication of longer follow-up data is eagerly awaited.
The polio-rhinovirus chimera (PVS-RIPO) is a replication-competent, live attenuated poliovirus vaccine/human rhinovirus chimera that is engineered with a foreign (rhinovirus) ribosome entry site to ablate neurovirulence. PVS-RIPO can target the poliovirus receptor CD155 that is expressed on APC or overexpressed on tumor cells. In a phase I trial (NCT01491893), 61 patients with recurrent supratentorial grade IV malignant glioma received PVS-RIPO intratumorally by convection-enhanced delivery via a catheter. The patients who received PVS-RIPO had an OS rate of 21% at 24 and 36 months with two patients obtaining complete response and remaining alive for more than 70.4 months (104). A randomized phase II trial of PVS-RIPO solely or combined with lomustine in patients with recurrent grade IV malignant glioma (NCT02986178) is ongoing.
Other OVs, such as ParvOryx (oncolytic H-1 parvovirus), Toca 511 (a retroviral replication-competent vector), Reovirus, and HSV type 1 have also been tested in a phase I/II trial for GBM patients and obtained promising results (105–108). Although these early phase clinical trials demonstrate a survival benefit that OV has brought, these benefits were only appreciated by some subsets of patients with glioma. Recently, a comprehensive analysis of virotherapy trials for rGBM revealed that virotherapy can improve the 2- and 3-year survival rates compared with non-virotherapy clinical trials (2-year survival: 15% vs. 12%; 3-year survival rate: 9% vs. 6%) (109). Thus, further investigations and more large randomized controlled phase II/III trials need to be done to evaluate the benefit of OV.
Conclusion
Current clinical trials of immunotherapy predominantly focus on the investigation of peptide vaccines, DC vaccines, CAR-T cells, checkpoint inhibitors, and OV. Many promising clinical outcomes have been achieved (110–116) however, immunotherapeutic successes in GBM are still lacking. Multiple factors challenge immunotherapy in GBM, including the immunosuppressive TME, tumor heterogeneity, tumor genomic characteristics, persistence and delivery of the vaccines, and efficiency of drug penetration through the BBB. Moreover, there remains a need for appropriate pre- and post-therapeutic biomarkers that may facilitate the establishment of a valid and standardized assessment for clinical efficacy in GBM. Immunotherapy for GBM requires integrated efforts with rational combinations of vaccine therapy, cell therapy, and radio- and chemotherapy, as well as molecule therapy targeting TME. These contributions promote the development of an optimal personalized therapeutic strategy for GBM patients.
Author Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by BH, XL, YL, ZZ, and HZ. The first draft of the manuscript was written by BH and JZ, and all authors commented on previous versions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This program was funded by major science and technology special general projects of Jiangxi Province (20203BBGL73178).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Davis ME. Epidemiology and Overview of Gliomas. Semin Oncol Nurs (2018) 34(5):420–9. doi: 10.1016/j.soncn.2018.10.001
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
3. Cohen MH, Shen YL, Keegan P, Pazdur R. FDA Drug Approval Summary: Bevacizumab (Avastin®) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist (2012) 17:1482–2. doi: 10.1634/theoncologist.2009-0121erratum
4. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell (2019) 176:677. doi: 10.1016/j.cell.2019.01.008
5. Sampson JH, Maus MV, June CH. Immunotherapy for brain tumors. J Clin Oncol (2017) 35:2450–6. doi: 10.1200/JCO.2017.72.8089
6. McGranahan T, Li G, Nagpal S. History and current state of immunotherapy in glioma and brain metastasis. Ther Adv Med Oncol (2017) 9:347–68. doi: 10.1177/1758834017693750
7. Sampson JH, Maus MV, June CH. Immunotherapy for brain tumors. J Clin Oncol (2017) 35:2450–6. doi: 10.1200/JCO.2017.72.8089
8. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity (2018) 48:380–395.e6. doi: 10.1016/j.immuni.2018.01.011
9. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome infuences prenatal and adult microglia in a sex-specific manner. Cell (2018) 172:500–16.e16. doi: 10.1016/j.cell.2017.11.042
10. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol (2015) 17 Suppl 7:vii9–vii14. doi: 10.1093/neuonc/nov151
11. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol (2017) 19:1047–57. doi: 10.1093/neuonc/nox026
12. Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res (2003) 9(11):4247–54.
13. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol (2015) 17:854–61. doi:10.1093/neuonc/nou348
14. Reardon DA, Schuster J, Tran DD, Fink KL, Nabors LB, Li G, et al. ReACT: overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J Clin Oncol (2015) 33(suppl):2009. doi:10.1200/jco.2015.33.15_suppl.2009
15. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII expressing glioblastoma (ACT IV): a randomized, double-blind, international phase 3 trial. Lancet Oncol (2017) 18:1373–785. doi: 10.1016/S1470-2045(17)30517-X
16. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28(31):4722–9. doi: 10.1200/JCO.2010.28.6963
17. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol (2011) 13(3):324–33. doi: 10.1093/neuonc/noq157
18. Rampling R, Peoples S, Mulholland PJ, James A, Al-Salihi O, Twelves CJ, et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin Cancer Res (2016) 22(19):4776–85. doi: 10.1158/1078-0432.CCR-16-050
19. Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, Widmer V, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol (2019) 21(7):923–33. doi: 10.1093/neuonc/noz040
20. Boydell E, Marinari E, Migliorini D, Dietrich PY, Patrikidou A, Dutoit V. Exploratory Study of the Effect of IMA950/Poly-ICLC Vaccination on Response to Bevacizumab in Relapsing High-Grade Glioma Patients. Cancers (Basel) (2019) 11(4):E464. doi: 10.3390/cancers11040464
21. Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res (2009) 15(19):6002–7. doi: 10.1158/1078-0432.CCR-09-0715
22. Cohen A, Holmen S, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neuro Neurosci Rep (2013) 13:345. doi: 10.1007/s11910-013-0345-4
23. Schumacher T, Bunse L, Pusch S, Sahm F, Wielster B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature (2014) 512(7514):324–7. doi: 10.1038/nature13387
24. Amato RJ. Heat-shock protein-peptide complex-96 for the treatment of cancer. Expert Opin Biol Ther (2007) 7(8):1267–73. doi: 10.1517/14712598.7.8.1267
25. Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specifc immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res (2013) 19:205–14. doi: 10.1158/1078-0432.CCR-11-3358
26. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-Oncology (2014) 16:274–9. doi: 10.1093/neuonc/not203
27. Filley AC, Dey M. Dendritic cell based vaccination strategy: an evolving paradigm. J Neurooncol (2017) 133:223–35. doi: 10.1007/s11060-017-2446-4
28. Lynes J, Sanchez V, Dominah G, Nwankwo A, Nduom E. Current Options and Future Directions in Immune Therapy for Glioblastoma. Front Oncol (2018) 8:578. doi: 10.3389/fonc.2018.00578
29. Sakai K, Shimodaira S, Maejima S, Udagawa N, Sano K, Higuchi Y, et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J Neurosurg (2015) 123(4):989–97. doi: 10.3171/2015.1.JNS141554
30. Nair SK, Sampson JH, Mitchell DA. Immunological targeting of cytomegalovirus for glioblastoma therapy. OncoImmunology (2014) 3:e29289. doi: 10.4161/onci.29289
31. Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature (2015) 519(7543):366–9. doi: 10.1038/nature14320
32. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res (2017) 23(8):1898–909. doi: 10.1158/1078-0432.CCR-16-2057
33. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuño MA, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother (2013) 62(1):125–35. doi: 10.1007/s00262-012-1319-0
34. Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol (2011) 29(3):330–6. doi: 10.1200/JCO.2010.30.7744
35. Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, Kubota T, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci (2011) 18(8):1048–54. doi: 10.1016/j.jocn.2010.11.034
36. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348:62–8. doi: 10.1126/science.aaa4967
37. Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science (2020) 367:446–53. doi: 10.1126/science.aay5967
38. Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. Clin Trial J Neurooncol (1999) 45(2):141–57. doi: 10.1023/A:1006293606710
39. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314:5. doi: 10.1126/science.1129003
40. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
41. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
42. Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol (2015) 36(8):494–502. doi: 10.1016/j.it.2015.06.004
43. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discovery (2015) 14(7):499–509. doi: 10.1038/nrd4597
44. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med (2015) 373:1040–7. doi: 10.1056/NEJMoa1504542
45. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
46. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
47. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
48. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR t cell therapy in refractory aggressive lymphoma. Mol Ther (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
49. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
50. Buechner J, Grupp SA, Maude SL, Boyer M, Bittencourt H, Laetsch TW, et al. Global reigstration trial of effcacy and safety of CTL019 in pediatric and young adult patients with relapsed/refractory (R/R) acute lymphoblastic leukemia (ALL): update to the interim analysis. Haematologica (2017) 102(2):178. doi: 10.1016/j.clml.2017.07.030
51. Neelapu SS, Locke FL, Bartlett NL, Lekakis LG, Miklos D, Jacobson CA, et al. Axicabtagene ciloleucel (axicel; kte-c19) in patients with refractory aggressive non-Hodgkin lymphomas (NHL): primary results of the pivotal trial ZUMA-1. Hematol Oncol (2017) 35(Suppl 2):28. doi: 10.1002/hon.2437_7
52. Yang J, Yan J, Liu B. Targeting EGFRvIII for glioblastoma multiforme. Cancer Lett (2017) 403:224–30. doi: 10.1016/j.canlet.2017.06.024
53. Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PloS One (2014) 9:e94281. doi: 10.1371/journal.pone.0094281
54. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
55. Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res (1999) 5:985–90.
56. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PloS One (2013) 8(10):e77769. doi: 10.1371/journal.pone.0077769
57. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res (2015) 21(18):4062–72. doi: 10.1158/1078-0432.CCR-15-0428
58. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
59. Mineo JF, Bordron A, Baroncini M, Maurage CA, Ramirez C, Siminski RM, et al. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J Neurooncol (2007) 85:281–7. doi: 10.1007/s11060-007-9424-1
60. Koka V, Potti A, Forseen SE, Pervez H, Fraiman GN, Koch M, et al. Role of Her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am J Clin Oncol (2003) 26:332–5. doi: 10.1097/01.COC.0000020922.66984.E7
61. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specifc chimeric antigen receptormodifed virus-specifc T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
62. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ra2 mitigate tumor antigen escape. J Clin Invest (2016) 126:3036–52. doi: 10.1172/JCI83416
63. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol (2018) 20:506–18. doi: 10.1093/neuonc/nox182
64. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics (2016) 3:16006. doi: 10.1038/mto.2016.6
65. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7(303):303ra139. doi: 10.1126/scitranslmed.aac5415
66. Yovino S, Kleinberg L, Grossman SA, Narayanan M, Ford E. The etiology of treatment-related lymphopenia in patients with malignant gliomas: modeling radiation dose to circulating lymphocytes explains clinical observations and suggests methods of modifying the impact of radiation on immune cells. Cancer Invest (2013) 31(2):140–4. doi: 10.3109/07357907.2012.762780
67. Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med (2017) 68:139–52. doi: 10.1146/annurev-med-062315-120245
68. Mirzaei R, Sarkar S, Yong VW. T cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol (2017) 38(2):104–15. doi: 10.1016/j.it.2016.11.005
69. Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex afnity. Cancer Cell (2013) 23(4):516–26. doi: 10.1016/j.ccr.2013.03.018
70. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
71. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 Blockade Ameliorates Glioma-Induced Changes to the CD4+ T Cell Compartment Without Affecting Regulatory T-cell Function. Clin Cancer Res (2007) 13(7):2158–67. doi: 10.1158/1078-0432.CCR-06-2070
72. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res (2016) 4(2):124–35. doi: 10.1158/2326-6066.CIR-15-0151
73. Desai K, Hubben A, Ahluwalia M. The Role of Checkpoint Inhibitors in Glioblastoma. Target Oncol (2019) 14(4):375–94. doi: 10.1007/s11523-019-00655-3
74. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18:153–67. doi: 10.1038/nri.2017.108
75. Wintterle S, Schreiner B, Mitsdoerfer M, Schneider D, Chen L, Meyermann R, et al. Expression of the B7-related molecule B7–H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res (2003) 63(21):7462–7.
76. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s Lymphoma. N Engl J Med (2015) 372(4):311–9. doi: 10.1056/NEJMoa1411087
77. Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee J-L, Fong L, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med (2017) 376(11):1015–26. doi: 10.1056/NEJMoa1613683
78. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
79. Motzer RJ, Tannir NM, McDermott DF, Frontera OA, Melichar B, Choueiri TK, et al. Nivolumab Plus Ipilimumab Versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
80. Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol (2018) 36(8):773–9. doi: 10.1200/JCO.2017.76.9901
81. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 Blockade and Stereotactic Radiation Produce Long-Term Survival in Mice With Intracranial Gliomas. Int J Radiat Oncol Biol Phys (2013) 86(2):343–9. doi: 10.1016/j.ijrobp.2012.12.025
82. Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, et al. PD-1 Blockade Enhances the Vaccination-Induced Immune Response in Glioma. JCI Insight (2016) 1(10):e87059. doi: 10.1172/jci.insight.87059
83. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol (2020). doi: 10.1001/jamaoncol.2020.1024
84. Bristol-Myers Squibb Provides Update on Phase 3 Opdivo. (nivolumab) CheckMate -548 Trial in Patients with Newly Diagnosed MGMT-Methylated Glioblastoma Multiforme. Available at: https://news.bms.com/category/press-release-category/%5Bcatpath-raw%5D.
85. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat Med (2019) 25(3):470–6. doi: 10.1038/s41591-018-0339-5
86. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn GP, Lim M, et al. Phase 2 study to evaluate safety and efcacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. J Clin Oncol (2017) 35(15_Suppl):2042. doi: 10.1200/JCO.2017.35.15_suppl.2042
87. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn GP, Lim M, et al. Phase 2 study to evaluate safety and efcacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. J Clin Oncol (2017) 35(15_Suppl):2042. doi: 10.1200/JCO.2017.35.15_suppl.2042
88. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specifc cell surface protein TIM-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
89. Kanzaki M, Wada J, Sugiyama K, Nakatsuka A, Teshigawara S, Murakami K, et al. Galectin-9 and T cell immunoglobulin mucin-3 pathway is a therapeutic target for type 1 diabetes. Endocrinology (2012) 153(2):612–20. doi: 10.1210/en.2011-1579
90. Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res (2017) 77(24):6795–811. doi: 10.1158/0008-5472.CAN-17-2285
91. Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res (2017) 23(21):6650–60. doi: 10.1158/1078-0432.CCR-17-0120
92. Soliman HH, Minton SE, Ismail-Khan R, Han HS, Vahanian NN, Ramsey WJ, et al. A phase 2 study of docetaxel in combination with indoximod in metastatic breast cancer. J Clin Oncol (2014) 32(15_Suppl):TPS3124. doi: 10.1200/jco.2014.32.15_suppl.tps3124
93. Zakharia Y, Johnson TS, Colman H, Vahanian NN, Link CJ, Kennedy E, et al. A phase I/II study of the combination of indoximod and temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors. J Clin Oncol (2014) 32(15_Suppl):TPS2107. doi: 10.1200/jco.2014.32.15_suppl.tps2107
94. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet (2019) 51(2):202–6. doi: 10.1038/s41588-018-0312-8
95. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol (2012) 30(7):658–70. doi: 10.1038/nbt.2287
96. Andtbacka RH II, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol (2015) 33(25):2780–8. doi: 10.1200/JCO.2014.58.3377
97. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science (1991) 252(5007):854–6. doi: 10.1126/science.1851332
98. Eissa IR, ustos-Villalobos I, Ichinose T, Matsumura S, Naoe Y, Miyajima N, et al. The Current Status and Future Prospects of Oncolytic Viruses in Clinical Trials against Melanoma, Glioma, ancreatic, and Breast Cancers. Cancers (Basel) (2018) 10(10):356. doi: 10.3390/cancers10100356
99. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol (2018) 36:1419–27. doi: 10.1200/JCO.2017.75.8219
100. Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst (2003) 95:652–60. doi: 10.1093/jnci/95.9.652
101. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol (2018) 36(14):1419–27. doi: 10.1200/JCO.2017.75.8219
102. van Den Bossche WBL, Kleijn A, Teunissen CE, Voerman JSA, Teodosio C, Noske DP, et al. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro Oncol (2018) Oct 920(11):1494–504. doi: 10.1093/neuonc/noy082
103. Gea Z. Abstract – interim results of a phase II multicenter study of the conditionally replicative oncolytic adenovirus DNX-2401 with pembrolizumab(Keytruda) for recurrent glioblastoma; CAPTIVE STUDY (LEYNOTE-192). Neuro-Oncology (2018) 20(suppl_6):vi6. doi: 10.1093/neuonc/noy148.019
104. Desjardins A, Gromeier M, Herndon JE2, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med (2018) 379(2):150–61. doi: 10.1056/NEJMoa1716435
105. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ, et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol Ther (2017) 25(12):2620–34. doi: 10.1016/j.ymthe.2017.08.016
106. Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther (2009) 17(1):199–207. doi: 10.1038/mt.2008.228
107. Cloughesy TF, Landolfi J, Hogan DJ, Bloomfield S, Carter B, Chen CC, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med (2016) 8(341):341ra75. doi: 10.1126/scitranslmed.aad9784
108. Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther (2014) 22(5):1056–62. doi: 10.1038/mt.2014.21
109. Antonio Chiocca E, Nassiri F, Wang J, Peruzzi P, Zadeh G. Viral and other therapies for recurrent glioblastoma: is a 24-month durable response unusual? Neuro Oncol (2019) 21(1):14–25. doi: 10.1093/neuonc/noy170
110. Inogés S, Tejada S, de Cerio ALD, Pérez–Larraya 6 JG, Espinós J, Idoate MG, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med (2017). doi: 10.1186/s12967-017-1202-z
111. Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother (2016). doi: 10.1007/s00262-016-1890-x
112. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro Oncol (2014) 16(2):274–9. doi: 10.1093/neuonc/not203
113. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due–Tønnesen P, Suso EMI, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother (2013). doi: 10.1007/s00262-013-1453-3
114. Kong D-S, Nam D-H, Kang S-H, Lee JW, Chang J-H, Kim J-H, et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in korea. Oncotarget (2015). doi: 10.18632/oncotarget.12273
115. Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous t-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res (2014) 74(13):3466–76. doi: 10.1158/0008-5472.CAN-14-0296
Keywords: immunotherapy, glioblastoma multiforme, glioma, vaccines, checkpoint inhibitors
Citation: Huang B, Li X, Li Y, Zhang J, Zong Z and Zhang H (2021) Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 11:603911. doi: 10.3389/fimmu.2020.603911
Received: 08 September 2020; Accepted: 29 December 2020;
Published: 09 March 2021.
Edited by:
Xiaoxing Xiong, Renmin Hospital of Wuhan University, ChinaReviewed by:
Yu Hao, Peking University First Hospital, ChinaSoldano Ferrone, Massachusetts General Hospital and Harvard Medical School, United States
Hailong Liu, Capital Medical University, China
Copyright © 2021 Huang, Li, Li, Zhang, Zong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhitao Zong, eGlhbmdmYW56aGJAMTYzLmNvbQ==; Hongbo Zhang, aG9uZ2Jvemhhbmc5OUBzbXUuZWR1LmNu
†These authors have contributed equally to this work