 Jia Li1†
Jia Li1† Liwei Li
Liwei Li Rufang Zhang
Rufang Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 14 January 2021
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.603437
This article is part of the Research Topic Immune Evasion Mechanisms by RNA Viruses View all 32 articles
Viral myocarditis is caused by a viral infection and characterized by the inflammation of the myocardium. Coxsackievirus B3 (CVB3) infection is one of the most common among the infections caused by this virus. The host’s early innate immune response to CVB3 infection particularly depends on the functions of type I interferons (IFNs). In this study, we report that a host microRNA, miR-30a, was upregulated by CVB3 to facilitate its replication. We demonstrated that miR-30a was a potent negative regulator of IFN-I signaling by targeting tripartite motif protein 25 (TRIM25). In addition, we found that TRIM25 overexpression significantly suppressed CVB3 replication, whereas TRIM25 knockdown increased viral titer and VP1 protein expression. MiR-30a inhibits the expression of TRIM25 and TRIM25-mediated retinoic acid-inducible gene (RIG)-I ubiquitination to suppress IFN-β activation and production, thereby resulting in the enhancement of CVB3 replication. These results indicate the proviral role of miR-30a in modulating CVB3 infection for the first time. This not only provides a new strategy followed by CVB3 in order to modulate IFN-I–mediated antiviral immune responses by engaging host miR-30a but also improves our understanding of its pathogenesis.
Viral myocarditis (VMC) is an inflammatory disease, which appears to be a major cause of sudden cardiac death in children and young adults (1). Coxsackievirus B3 (CVB3) has been considered as the commonest pathogen for VMC (2). Acute VMC is the consequence of myocyte damage induced by early direct virus via apoptosis and necrosis, followed by host inflammatory and immune responses (3). Although excessive activation of immune response triggered by virus infection may be a significant cause of tissue injuries, the virus itself is vital to VMC progression via direct attack on cardiomyocytes (4, 5). To date, no effective therapies or vaccines for VMC are available currently, although type I interferon (IFN) may show certain efficacy (2, 6). Therefore, studying of the fundamental mechanisms involved in CVB3 infection may provide new clues for researching and developing new therapeutic options against CVB3-induced diseases.
MicroRNAs (miRNAs) are noncoding RNAs that function as gene regulators by repressing translation or degrading target mRNAs (7). The seed region (2–8 nucleotides at the 5′ end) of an miRNA is identified as the key to exerting its silencing function, most commonly by targeting the 5′ or 3′ untranslated region (5′- or 3′-UTR) (8, 9). There is a growing body of evidence that host miRNA-mediated RNA interference is critical in numerous viral infections as negative or positive factors of antiviral immune response (10–12). For example, hepatitis C virus–induced miR-93-5p up-regulation inhibits IFN signaling pathway by targeting interferon alpha receptor 1 (13). MiR-194 facilitated influenza A virus replication by negatively regulating type I IFN production (14). MiR-3570 negatively regulates the retinoic acid-inducible gene (RIG)-I–dependent innate immune response to rhabdovirus by targeting mitochondrial antiviral signaling protein (15). Previous studies suggested that miR-30c regulated macrophage-mediated inflammation and pro-atherosclerosis pathways (16), and miR-30b was identified to be a negative regulator of immune responses by binding Notch1 (17). However, the ability of miR-30 family to modulate CVB3 infection has not been reported.
As an E3 ubiquitin ligase, the tripartite motif protein 25 (TRIM25) is involved in regulating the antiviral immune response (18). Pathogen-associated molecular patterns are immediately recognized by host pattern recognition receptors (PRRs) after virus infection (19). RIG-I is the main cytoplasmic PRR of RNA viruses (20). Then, the caspase recruitment domains (CARDs) of RIG-I are modified by ubiquitin. TRIM25 mediates this progress, which is essential for the activation of type I and III IFNs, mediating viral clearance, and inhibiting viral replication and spread (21, 22). TRIM25 inhibits various viruses, e.g., herpesvirus and influenza virus (23, 24).
We show that host miR-30a could be upregulated upon CVB3 infection. Overexpression of miR-30a exhibits an important role in facilitating CVB3 replication through targeting TRIM25. Mir-30a suppresses TRIM25 expression and TRIM25-mediated RIG-I ubiquitination to inhibit IFN-β activation and production. Together, these findings suggest a novel mechanism through which CVB3 infection suppresses host antiviral immunity by utilizing cellular miRNA and provide insight into the regulatory mechanism between CVB3 and the host.
HeLa cells and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Waltham, MA). Cells were maintained at 37°C with 5% CO2. Primary cardiomyocytes were isolated from one-day-old BALB/c mice (Slac Animal Inc., Shanghai, China) and cultured as described previously (25). The CVB3 (Nancy strain) was stored in our laboratory. CVB3 was maintained by passage through HeLa cells, and cultured in DMEM containing 10% FBS. Virus titer was measured by a 50% tissue culture infectious dose (TCID50) assay in HeLa cells.
miR-30 family mimics (30a, 30b, 30c, 30d, and 30e) were synthesized by GenePharma (Shanghai, China). The sense sequences are listed in Table 1. The negative-control (NC) mimic sequence was 5′-uucuccgaacgugucacgutt-3′. The sequences of NC inhibitor (NC-inhi) and miR-30a inhibitor (30a-inhi) are also listed in Table 1.

Table 1 Sequences of microRNA (miRNA) mimics and inhibitors used in this study.
RNAs were extracted using the miRNeasy MiniKit or RNeasy MiniKit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For detection of endogenous miRNAs, an miRcute miRNA First-Strand cDNA Synthesis (TIANGEN BIOTECH, Beijing, China) was used for polyadenylation and reverse transcription. A commercial miRcute miRNA qPCR detection kit was used for measuring miRNA abundance. The U6 small nuclear RNA (TIANGEN BIOTECH) was used for normalization. To detect the relative mRNA levels, a PrimeScript RT Master Mix kit (Takara, Dalian, China) was used for reverse transcription. SYBR Premix Ex Taq (Takara) was used to quantify levels of IFN-α, IFN-β, interferon-stimulated gene 15 (ISG15), and MX1 mRNA. The relative expression levels of these genes were analyzed using the ΔΔCt method (26), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used as a control. Primers are listed in Table 2. All PCR experiments were performed in triplicate.
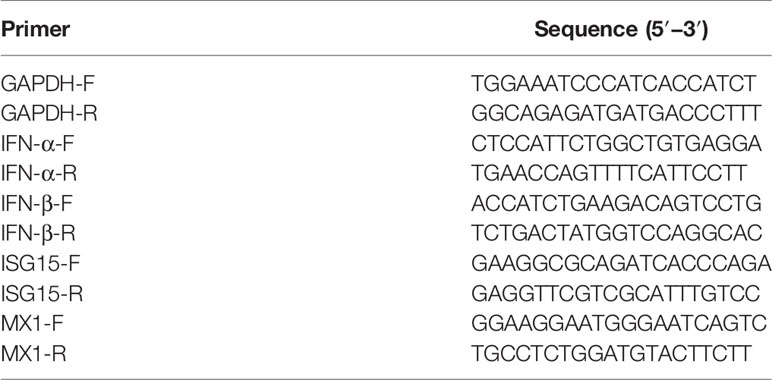
Table 2 Sequence of oligonucleotide primers used in this study.
The pGL3 luciferase reporter plasmids containing the 3′UTR sequence of TRIM25 were cloned downstream of the luciferase open reading frame to test the binding regions in TRIM25. To verify the target gene of miR-30a, 293T cells were co-transfected with 100 ng/well of pRL-TK and 500 ng/well of the indicated reporter plasmid along with the indicated amount of miR-30a or NC mimics. Cells were lysed 24 h post-transfection (hpt) to determine the luciferase activities using a dual-luciferase reporter assay kit (Promega). Data are presented as the relative luciferase activities. To generate the miR-30a target-mutated reporter plasmid (pGL3-TRIM25-mut), mutations at positions corresponding to the miR-30a seed region were introduced. DNA sequencing confirmed mutant plasmid (pGL3-TRIM25-mut) and the primer sequences are available upon request. Luciferase activity in 293T cells were determined. Human IFN-β promoter, NF-κB promoter, and primary miR-30a promoter [a fragment ∼3 kb upstream from the primary miR-30a (pri-miR-30a) coding sequences] plasmids were generated by cloning sequences into the upstream of firefly luciferase gene in the pGL3-Basic vector. To test the promoter activities, HeLa or 293T cells seeded in 24-well plates were co-transfected with the indicated expression plasmids or miRNA mimics along with 100 ng/well of the indicated luciferase reporter plasmid and 20 ng/well of pRL-TK. Twenty-four hpt, luciferase activities were measured and the results are expressed as means ± standard deviation (SD) from three independent experiments.
Full-length TRIM25- and RIG-I-encoding sequences were cloned into pCAGGS vector to generate pCAGGS-TRIM25-Myc, pCAGGS-RIG-I-HA, pCAGGS-RIG-I-Flag, or pCAGGS-2CARD-Flag. All plasmids were constructed by homologous recombination using the NEBuilder® HiFi DNA assembly master mix (New England Biolabs; Ipswich, MA). Sequences of all primers will be made available upon request.
MiRNA or NC mimics were transfected into HeLa cells at a concentration of 80 nM (except for dosage experiments) using X-tremeGENE siRNA Transfection Reagent (Roche). To investigate the effect of TRIM25 on CVB3 replication, HeLa cells were transfected with 2 μg of pCAGGS-TRIM25-Myc using X-treme GENE HP DNA reagent (Roche). Next, 24 hpt, cells were infected with CVB3 (multiplicity of infection, MOI = 0.1). The supernatants were harvested at 12, 24, and 36 h post-infection (hpi), and the cells were lysed using RIPA lysis buffer (Thermo Fisher Scientific). Viral titers were determined and the amount of viral VP1 was detected by western blotting (WB). WB assays were performed as described previously (27), except using an anti-VP1 antibody (1:1,000) purchased from Leica Biosystems Newcastle Ltd.
Small interfering RNAs (siRNAs) against TRIM25 were synthesized by GenePharma (Shanghai, China). The siRNA molecule sequence was CCU GGA GUA UUA CGU UAA ATT (si-TRIM25). The control siRNA (si-con) sequence was UUC UCC GAA CGU GUC ACG UTT. HeLa cells were transfected with 50 nM si-TRIM25 or si-con using X-tremeGENE siRNA Transfection Reagent (Roche). The cells were lysed 24 hpt, and WB detected the effects of siRNAs using an anti-TRIM25 monoclonal antibody (cat. no. 13773; Cell Signaling Technology; Danvers, MA; 1:1,000). Twenty-four hpt, the cells were infected with CVB3 at an MOI of 0.1. The supernatant and cells were harvested 12 and 24 hpi, and were later analyzed based on virus titers and by WB.
An RISC immunoprecipitation assay was performed as previously described (28). 293T cells were transfected with NC or miR-30a mimics using X-tremeGENE siRNA Transfection Reagent (Roche). Cells were harvested 24 hpt. The anti-Ago2 antibody (cat. no. 2897; Cell Signaling Technology) was used for the immunoprecipitation of RISC and the detection of Ago2.
Statistical analysis was conducted using GraphPad Prism 6 (GraphPad, La Jolla, CA). The statistical significance was analyzed using Student’s t-test, with p < 0.05 considered statistically significant.
To investigate whether miR-30 family can be regulated by CVB3, we performed a time-course assay to analyze the expression of the miR-30 family members in HeLa cells infected with CVB3 (MOI = 1) or mock infected. The miR-30 family contains five members (miR-30a–e). Results indicated that miR-30a was significantly increased at 12 hpi and peaked at 24 hpi (∼6.6-fold induction) and the expression of other miR-30 family members showed slight alteration (Figure 1A). To confirm that the target cells of CVB3 function equivalently, primary cardiomyocytes were cultured from newborn mice and infected with CVB3 (MOI = 1). Results showed that CVB3 infection enhanced the relative expression of miR-30a at 12 and 24 hpi, thereby demonstrating that miR-30a expression is upregulated by CVB3 infection, which may be involved in regulating CVB3 infection (Figure 1B). Then, Hela cells were infected with CVB3 or heat-inactivated CVB3 (HI-CVB3; 70°C for 60 min) at MOI of 0.1. The expression of miR-30a in the cells inoculated with HI-CVB3 was not altered (Figure 1C), suggesting that the increased miR-30a depends on CVB3 replication.
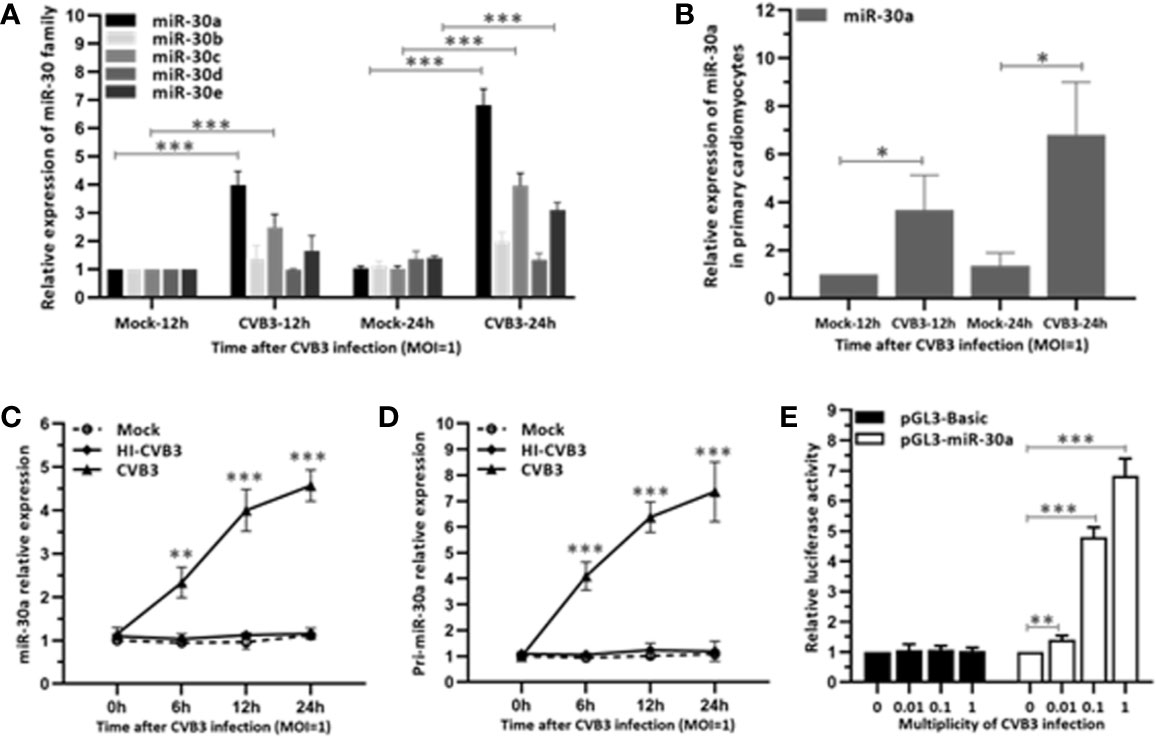
Figure 1 miR-30a is up-regulated in response to CVB3 infection in vitro. (A) qRT-PCR analysis of miR-30 family was performed in HeLa cells inoculated with CVB3 at an MOI of 1 for the indicated times. (B) Primary cardiomyocytes of mice were mock infected or infected by CVB3 (MOI = 1) for 12 and 24 h. The relative level of miR-30a was analyzed by qRT-PCR analysis. (C) qRT-PCR analysis of miR-30a was performed in HeLa cells inoculated with HI-CVB3 (MOI = 1) at 0, 6, 12, and 24 hpi. (D) qRT-PCR analysis of pri-miR-30 was performed in HeLa cells inoculated with CVB3 (MOI = 1) at 24 hpi. (E) After co-transfection of 100 ng pGL3-basic or pGL3-miR-30a promoter along with pRL-TK (20 ng) into HeLa cells for 12 h, cells were infected with CVB3 at an MOI of 0.01, 0.1, or 1. At 24 hpi, luciferase activities were analyzed. Data represent the mean ± SD of three independent experiments. Statistical significance was analyzed using t tests. *P < 0.05; **P < 0.01; ***P < 0.001.
To verify the induction of miR-30a by CVB3 infection, the expression of its primary transcript pri-miR-30a was analyzed. We demonstrated that pri-miR-30a was significantly upregulated and reached a significant increment to ∼7.2-fold at 24 hpi (Figure 1D). To corroborate the findings further, pri-miR-30a promoter was transfected into HeLa cells, followed by CVB3 infection. CVB3 infection enhanced the activity of the miR-30a promoter with increasing MOIs, and it increased ∼4.7- and ∼6.8-fold at MOIs of 0.1 and 1, respectively (Figure 1E).
To verify whether miR-30a can affect CVB3 replication, the expression of CVB3 capsid protein VP1 and viral titers in HeLa cells at 24 hpi were detected. Both CVB3 growth and the amount of VP1 mRNA level were upregulated as a function of the dose of miR-30a mimics (Figures 2A, B). Consistent with this, transfecting the miR-30a mimics induced the accumulation of VP1 (Figure 2C). Results above suggest that viral replication and progeny release in HeLa cells were significantly increased by miR-30a. To illustrate the proviral effect of miR-30a on CVB3 replication, we analyzed viral infection in the presence of the miR-30a inhibitor. MiR-30a inhibitor significantly reduced virus titer to that of NC inhibitor at 12, and 24 hpi, respectively (Figure 2D). This indicated that miR-30a promoted CVB3 replication in HeLa cells.
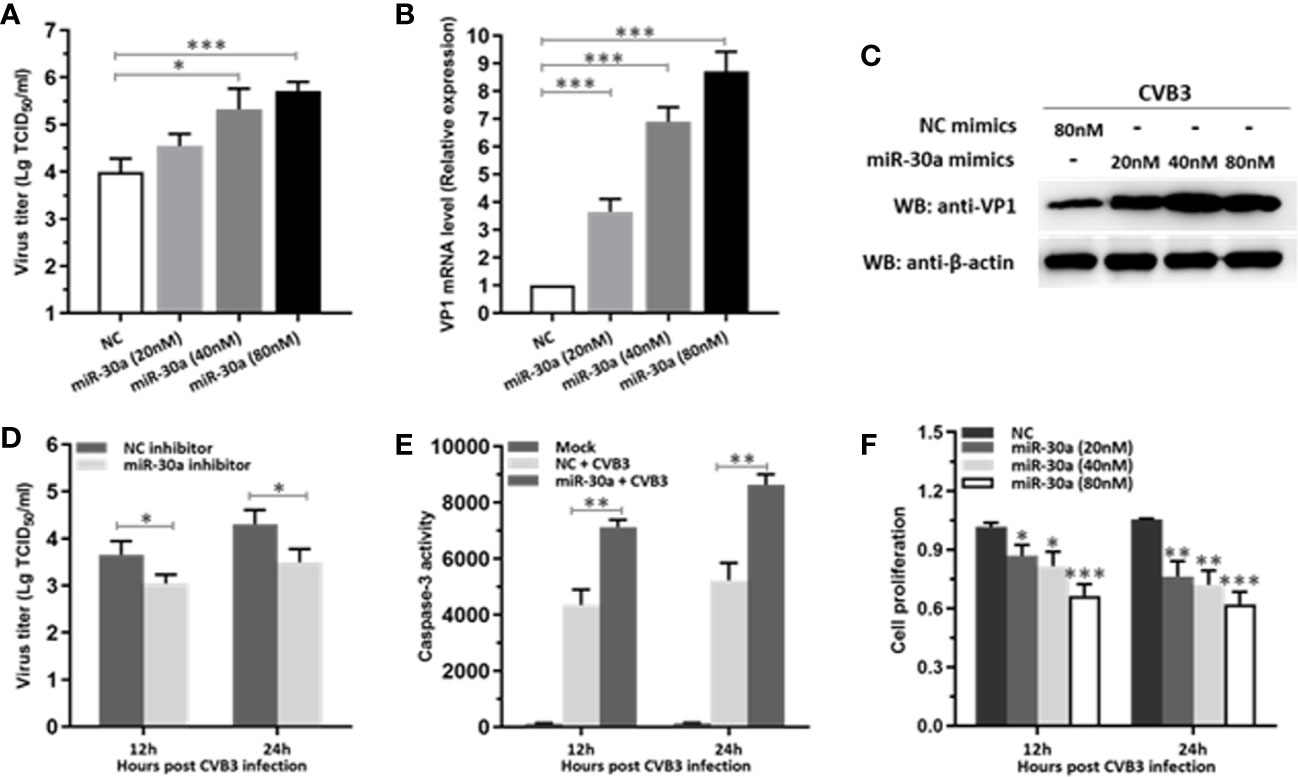
Figure 2 miR-30a significantly facilitates CVB3 replication. (A) HeLa cells were transfected with miR-30a or NC mimics at the indicated doses (20–80 nM), followed by CVB3 infection (MOI = 0.1). The supernatants were collected at the indicated times for viral titer determination. (B) qRT-PCR analysis of VP1 mRNA levels in HeLa cells transfected with miR-30a or NC mimics at the indicated doses (20–80 nM), followed by CVB3 infection (MOI = 0.1). (C) The experiments were performed as described for panel (B). Cells were collected at 24 hpi for WB analysis of VP1 expression. β-actin expression was analyzed as a loading control. (D) HeLa cells were transfected with miR-30a inhibitor or NC inhibitor (80 nM), followed by CVB3 infection (MOI = 0.1). Supernatants were collected at 12 and 24 hpi for viral titer determination. (E) HeLa cells were with miR-30a or NC mimics, followed by CVB3 infection (MOI = 0.1). The caspase-3 activity was detected at 12 and 24 hpi. (F) Cell viability assay was performed at 12 and 24 hpi with the indicated dose of miR-30a or NC mimics. The viability was tested by MTT assay. Data represent the mean ± SD of three independent experiments. Statistical significance was analyzed using t tests. *P < 0.05; **P < 0.01; ***P < 0.001.
We next sought to investigate the effect of miR-30a on apoptosis post CVB3 infection. As shown in Figure 2E, CVB3 infection caused a dramatic increase in the caspase-3 activity in miR-30a-transfected cells than that in NC-transfected cells, suggesting a potential promoting role of miR-30a for CVB3-induced apoptosis in infected cells. Indeed, miR-30a significantly promoted the proliferation in CVB3-infected cells in a dose- and time-dependent manner (Figure 2F), indicating that miR-30a also aggravated CVB3‐induced apoptosis. Taken together, these results suggest that miR-30a promoted CVB3 replication and aggravated CVB3‐induced apoptosis in HeLa cells.
Next, to investigate the potential mechanisms for miR-30a to enhance CVB3 infection, we tested whether miR-30a can alter NF-κB and IFN-I activation. We performed a series of assays to monitor the activation of NF-κB promoter and IFN-β promoter after stimulating with poly(I:C). Our data indicated that miR-30a significantly repressed the activation of IFN-β under poly(I:C) stimulation (Figure 3A), whereas it had no effect on the activation of NF-κB promoter (Figure 3B). However, miR-30a mutant with mutated seed sequences (GUAAACA to GAUUUGA) lost its ability to inhibit the activation of IFN-β (Figure 3C). Results above suggest that miR-30a regulates type I IFN activation. The inhibition of IFN-β activation by miR-30a occurred in a dose-dependent manner (Figure 3D), whereas miR-30a inhibitor enhanced IFN-β activation (Figure 3E). To confirm these results, we found that the relative mRNA levels of IFN-α/β expression decreased in miR-30a-transfected cells, whereas miR-30a inhibitors had the opposite effect (Figure 3F). These results demonstrated the role of miR-30a as a negative regulator of type I IFN activation and production.
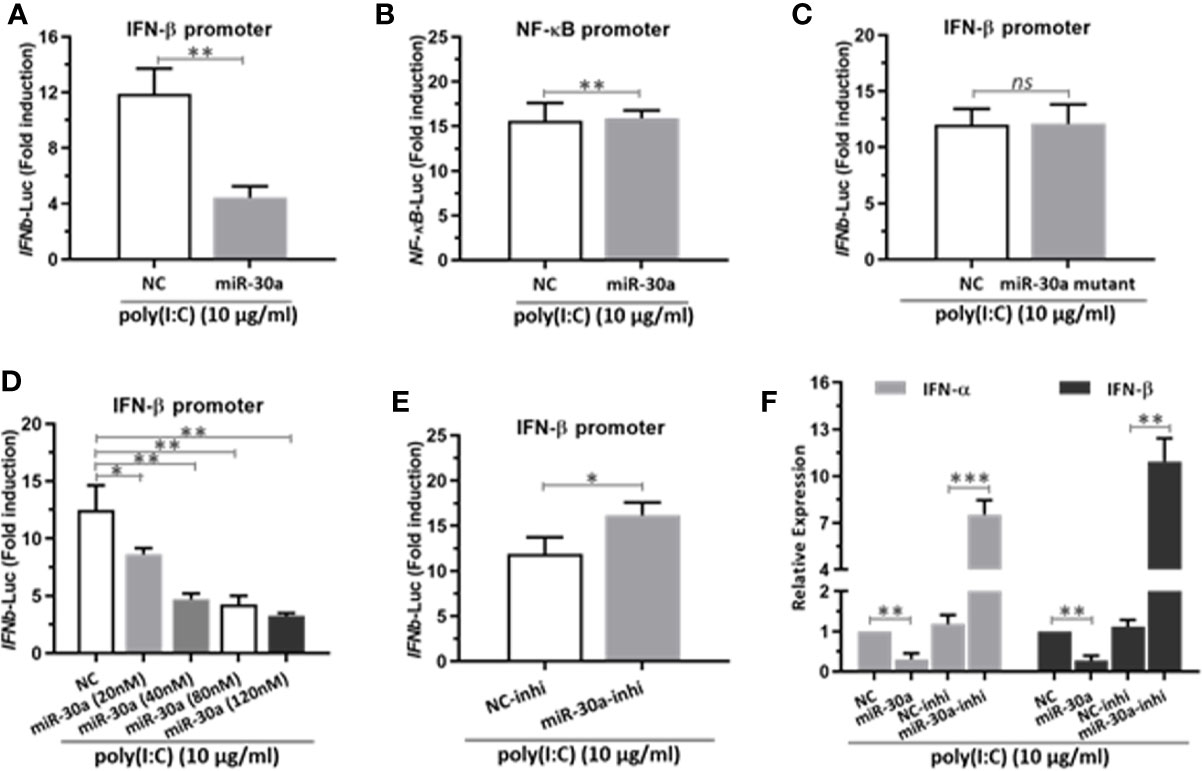
Figure 3 miR-30a represses IFN-α/β activation and production. (A) IFN-β or (B) NF-κB promoter vector along with pRL-TK were co-transfected with 80 nM of the miR-30a or NC mimics into 293T cells for 24 h, followed by stimulation with poly(I:C) (10 μg/ml) for 12 h. Then, 293T cells were harvested for luciferase assay. (C–E) The experiments were performed as described for panel (A), except that miR-30a mutant, the indicated doses (20–120 nM) of miR-30a mimics, or miR-30a inhibitor were used. (F) qRT-PCR analysis of type I IFN α/β mRNA expression in HeLa cells transfected with NC mimics, miR-30a mimics, NC inhibitor, or miR-30a inhibitor for 24 h, followed by stimulation with poly(I:C) (10 μg/ml) for 12 h. Data were normalized to GAPDH expression and are the mean ± SD of three independent experiments. Statistical significance was analyzed using t tests. *P < 0.05; **P < 0.01; ***P < 0.001.
To investigate the major target of miR-30a that is involved in modulating IFN-I responses, we used TargetScan program (http://www.targetscan.org) to predict the target genes. Analysis showed that miR-30a could target TRIM25 through a site in the 3′UTR region (Figure 4A). To validate whether TRIM25 is a direct target of miR-30a, the cDNA fragments containing the predicted target site were constructed into a firefly luciferase reporter vector pGL3-Control; meanwhile, a mutant vector was constructed to eliminate the recognition by replacing five seed nucleotides (UGUUUAC to UCAAAUC). In the presence of miR-30a, the luciferase activity of TRIM25 3′UTR was reduced to ∼29% relative to NC. However, the effects produced by miR-30a were eliminated in the vector bearing the mutant target site (Figure 4B). These results confirmed that the miR-30a target site is harbored in the 3′UTR of TRIM25. To clarify that miR-30a targets TRIM25 mRNA directly by binding to its 3′UTR region, the cells were transfected with miR-30a for 24 h and then lysed and harvested for the pull-down of miR-30a and TRIM25 mRNA by anti-Ago2 monoclonal antibody. Quantitative real-time PCR (qRT-PCR) data showed that the transfection of miR-30a increased the fraction of TRIM25 mRNA bound by Ago2, and the enrichment efficiency was about 1.8-fold (Figure 4C). At the same time, miR-30a associated with Ago2 was significantly enriched by about 7.6-fold in the miR-30a–overexpressed sample compared to NC (Figure 4D). As Ago2 had a similar input among different treatments (Figure 4E), we concluded that miR-30a physically bound TRIM25 mRNA along with Ago2 in RISC. The results suggest that miR-30a physically binds to the 3′UTR region of TRIM25 mRNA. To further verify TRIM25 as a target of miR-30a, its expression was tested in 293T cells transfected with miR-30a mimics or inhibitors. The mRNA levels of TRIM25 were decreased when miR-30a was overexpressed but were increased when the miR-30a inhibitor was applied (Figure 4F). MiR-30a mimics significantly inhibited the level of TRIM25 protein in a dose-dependent manner (Figure 4G). Taken together, results above demonstrate that TRIM25 is a target of miR-30a.
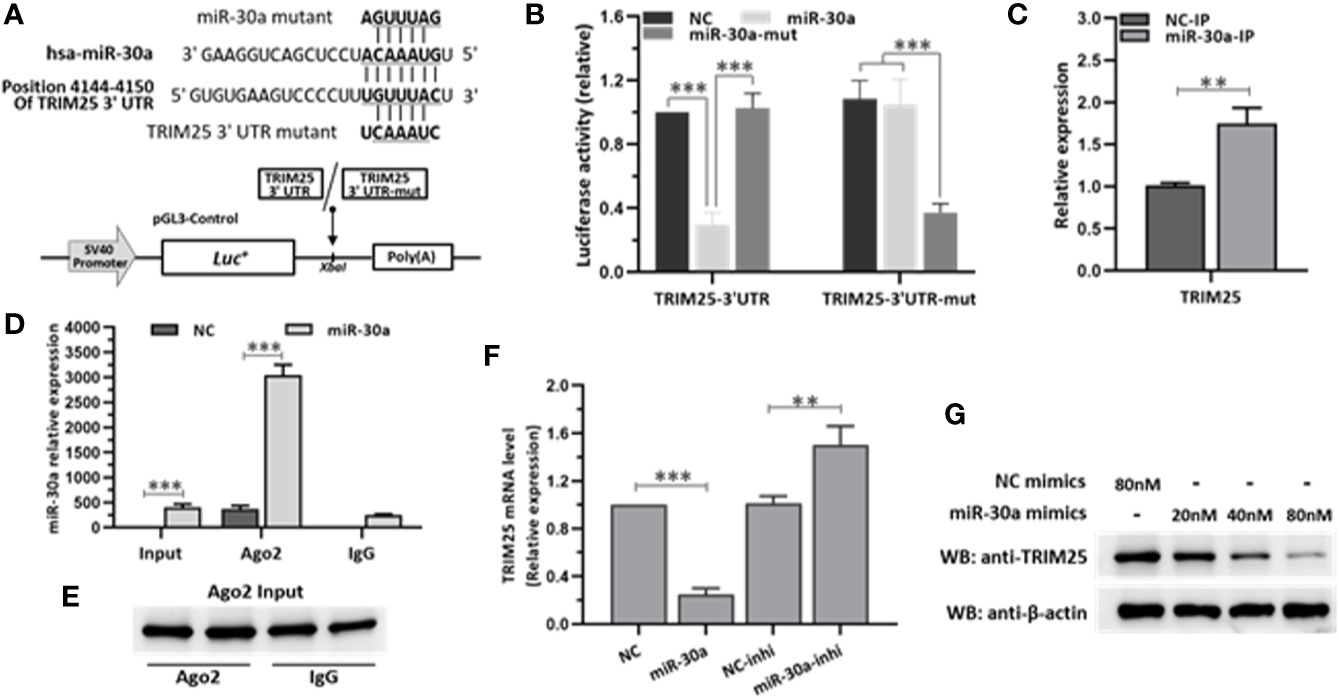
Figure 4 miR-30a inhibits the expression of TRIM25 by targeting the 3′UTR of TRIM25. (A) Computational prediction of potential miR-30a target sites in the 3′UTR of TRIM25. The predicted target sites of miR-30a are underlined and were mutated as indicated. The fragments were inserted in XbaI site. (B) Luciferase activity in lysates of 293T cells co-transfected with TRIM25-3′UTR or TRIM25-3′UTR-mutant luciferase reporter vector and miR-30a mimics or NC mimics. (C–E) 293T cells were transfected with miR-30a or NC mimics (80 nM) for 24 h. Cells were analyzed by RISC-IP followed by qRT-PCR to analyze TRIM25 (normalized to GADPH) (C), or miR-30a compared to RNAs extracted from total samples of lysates from 293T cells (normalized to U6) (D) or WB to analyze Ago2 expression from input (E). (F) qRT-PCR analysis of TRIM25 mRNA levels in 293T cells transfected with miR-30a mimics or miR-30a inhibitor (80 nM). Data are presented as mean ± SD. Statistical significance was analyzed using t-tests. **P < 0.01; ***P < 0.001. (G) WB analysis of TRIM25 expression levels in cells transfected with miR-30a mimics at the indicated doses (20–80 nM). β-actin expression was analyzed as a loading control.
To investigate the role of TRIM25 on CVB3 replication, HeLa cells were transfected with a plasmid expressing TRIM25 or vector alone and then infected with CVB3 at MOI of 0.1. The efficiency of overexpression of TRIM25 was confirmed by WB (Figure 5A). The supernatants were subjected to TCID50 assay to determine the role of TRIM25 on viral progeny release. As shown in Figure 5B, TRIM25 overexpression significantly reduced the virus particle release. Furthermore, the protein level of VP1 was significantly inhibited by TRIM25 overexpression (Figure 5C). To verify the antiviral effect of TRIM25, we designed and screened a specific siRNA targeting the open reading frame of TRIM25, which led to a 70% reduction in the overall levels of the TRIM25 protein (Figure 5D). Knockdown of TRIM25 significantly increased CVB3 progeny production (Figure 5E) and viral capsid protein VP1 expression (Figure 5F), compared to the effect of control siRNA. Collectively, the results confirmed that TRIM25 significantly restricted CVB3 replication in vitro. To confirm IFN-β promoting role of TRIM25, HeLa cells were co-transfected with TRIM25 vector and IFN-β promoter-luciferase plasmid. As demonstrated in Figure 5G, overexpression of TRIM25 enhanced the activity of IFN-β promoter in a dose-dependent manner after CVB3 infection. Furthermore, TRIM25 significantly increased the IFN-β, ISG15, and MX1 mRNA levels upon CVB3 infection (Figure 5H). Thus, our data suggested that TRIM25 suppresses CVB3 replication by upregulating the activation of IFN signaling pathway.

Figure 5 TRIM25 restricts CVB3 replication by upregulating IFN-β activation and production. (A) HeLa cells were transiently transfected with pCAGGS-TRIM25-Myc or pCAGGS. 24 hpt, cells were subjected to WB for detecting TRIM25 expression levels. (B, C) HeLa cells were transiently transfected with pCAGGS-TRIM25-Myc or pCAGGS. 24 hpt, cells were infected with CVB3 at an MOI of 0.1. Supernatants were collected and subjected to TCID50 (B) and WB analyzed VP1 levels (C). (D) HeLa cells were transfected with control siRNA (si-con) or TRIM25 siRNA (si-TRIM25) and the knockdown efficiency was analyzed by WB at 24 hpt. (E, F) Cells were transfected with si-con or si-TRIM25 (50 nM) for 24 h and infected with CVB3 (MOI = 0.1). Cells and supernatants were harvested at the indicated times. (E) TCID50 assay and (F) WB analysis evaluated CVB3 replication. (G) IFN-β promoter vector along with pRL-TK were co-transfected with pCAGGS-TRIM25-Myc or pCAGGS into cells for 24 h, followed by stimulation with poly(I:C) (10 μg/ml) for 12 h. Then, cells were harvested for luciferase assay. (H) qRT-PCR analysis of IFN-α, IFN-β, MX1, and ISG15 expression in cells transfected with pCAGGS-TRIM25-Myc or pCAGGS for 24 h and then infected with CVB3 for 24 h at an MOI of 0.1. Data were normalized to GAPDH expression and are the mean ± SD of three independent experiments. Statistical significance was analyzed using t tests. *P < 0.05; **P < 0.01; ***P < 0.001.
TRIM25-mediated RIG-I ubiquitination is essential for the activation of type I and III IFNs. In the current study, we demonstrated that miR-30a significantly suppressed the expression of TRIM25. Whether miR-30a can suppress TRIM25-mediated RIG-I activation was unknown. We first verified the effect of miR-30a overexpression on the interaction between RIG-I and TRIM25. When RIG-I-HA and TRIM25-Myc were co-transfected with miR-30a mimics, the interaction between TRIM25 and RIG-I was markedly diminished. Meanwhile, when TRIM25-Myc and RIG-I-HA were co-transfected with NC mimics, the interaction between TRIM25 and RIG-I was not affected (Figure 6A). To investigate whether TRIM25-mediated RIG-I ubiquitination is regulated by miR-30a, 293T cells were co-transfected with pCAGGS-Flag-RIG-I (0.5 μg per well) and HA-ubiquitin (0.5 μg per well), and the indicated dose of miR-30a mimics. The experiment revealed that TRIM25-mediated RIG-I ubiquitination was potentiated under poly(I:C) stimulation but was substantially suppressed by increasing miR-30a in a dose-dependent manner (Figure 6B). To examine regulation of RIG-I activity by miR-30a, a luciferase reporter under the control of the IFN-β promoter was used to quantify promoter activation. Consistent with the inhibition of RIG-I ubiquitination by miR-30a, IFN-β promoter activation induced by RIG-I or RIG-I CARD domain overexpression was significantly inhibited by miR-30a in a dose-dependent manner (Figures 6C, D). However, co-expression of TRIM25 with miR-30a counteracted this inhibitory effect mediated by miR-30a (Figure 6E). In addition, suppression of RIG-I ubiquitination by miR-30a was partially rescued by TRIM25 overexpression (Figure 6F). These observations indicated that the miR-30a inhibits TRIM25-mediated RIG-I ubiquitination to suppress IFN-β activation and production.
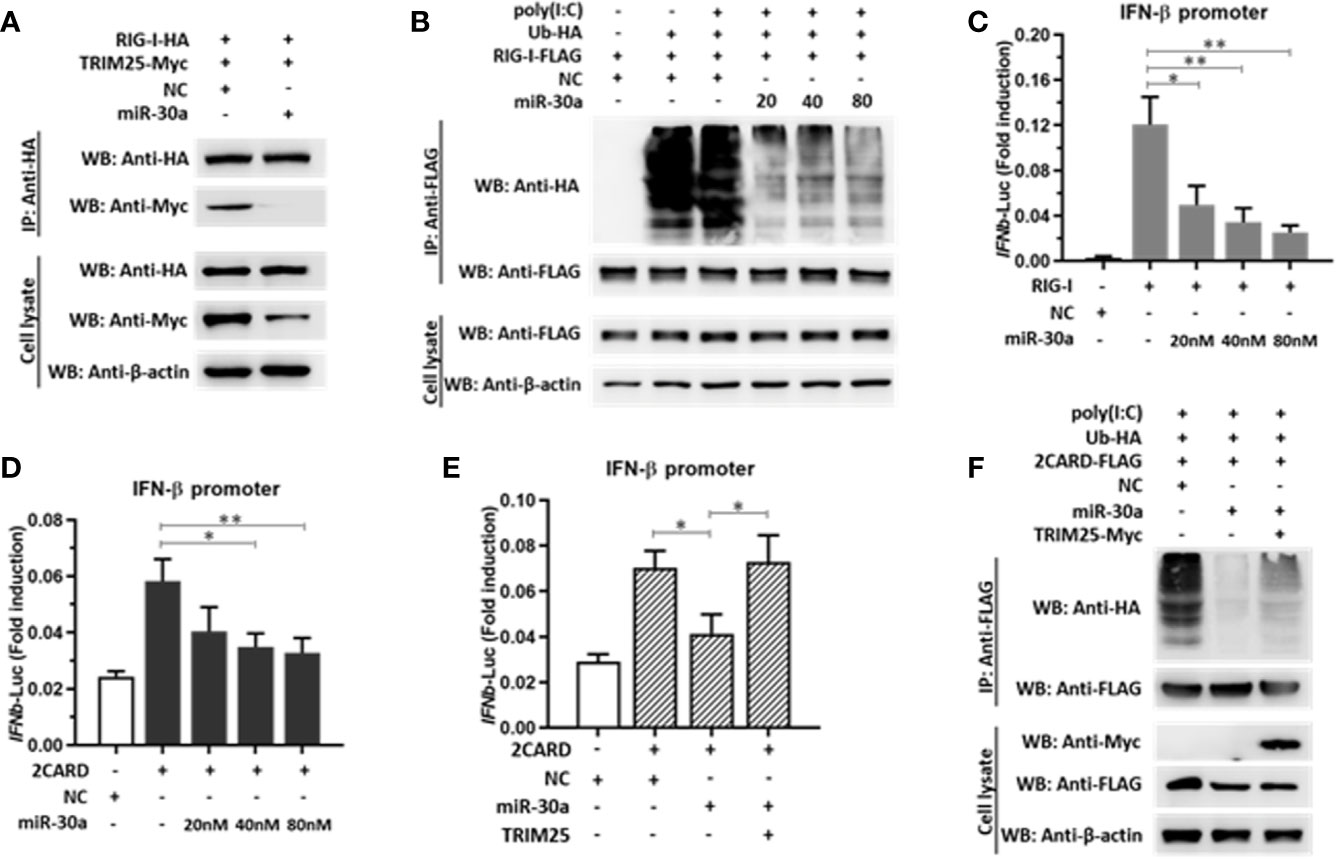
Figure 6 miR-30a suppresses TRIM25-mediated RIG-I activation and IFN production. (A) pCAGGS-RIG-I-HA and pCAGGS-TRIM25-Myc were co-transfected with or without miR-30a. The interaction was probed by immunoprecipitation. (B) 293T cells were co-transfected with the indicated plasmids and miR-30a or NC mimics for 36 h, and were treated with or without poly(I:C) (10 μg/ml) for 12 h. Anti-Flag immunoprecipitates prepared from the cell extracts were analyzed by WB using the indicated antibodies. (C, D) 293T cells seeded in 24-well plates were co-transfected using the IFN-β-Luc firefly luciferase reporter plasmid and the Renilla luciferase control reporter plasmid pRL-TK. For the experiment, pCAGGS-RIG-I-Flag or pCAGGS-2CARD-Flag were co-transfected with or without miR-30a. (E) pCAGGS-2CARD-Flag (0.25 μg) and pCAGGS-TRIM25-Myc (0.5 μg) plasmids with or without miR-30a were co-transfected. The luciferase activity in cell lysates was measured. The data are presented as the mean ± SD from three experiments. Statistical significance was analyzed using t tests. *P < 0.05; **P < 0.01. (F) Ubiquitin-HA (0.5 μg), pCAGGS-2CARD-Flag (0.5 μg), or pCAGGS-TRIM25-Myc (1.0 μg) were co-transfected with or without miR-30a. 24 hpt, cells were treated with poly(I:C) (10 μg/ml) for 12 h, cell lysates were analyzed by immunoprecipitation using the indicated antibodies to detect the ubiquitination of RIG-I-2CARD.
Recently, host miRNAs emerged as key regulators and potential drug targets in CVB3 infection. miR‐221 and miR‐155 have been reported to participate in a CVB3‐induced inflammatory response (29, 30). miR‐34a aggravates CVB3‐induced apoptosis of cardiomyocytes through the SIRT1‐p53 pathway (31). MiR-223/Pknox1 axis protects mice from CVB3-induced VMC by modulating macrophage polarization (32). miRNA-21 shows protective role against CVB3 infection by targeting the MAP2K3/P38 MAPK signaling pathway (33). In this study, we demonstrated for the first time that miR‐30a participates in the pathogenesis during CVB3 infection in vitro.
Previous studies report that miR-30c facilitated porcine reproductive and respiratory syndrome virus infection by targeting JAK1 and IFN-alpha/beta receptor beta chain (34, 35). However, influenza A virus restrained host type I IFN-mediated antiviral immune responses by decreasing the expression of miR-30 family members via targeting SOCS1 and SOCS3 expression (36).
In this study, we showed that miR-30a functions by facilitating CVB3 replication, and that it could act as a host-derived promoter of the virus (Figure 2). Furthermore, miR-30a facilitates CVB3 infection by repressing IFN-I signaling and by targeting TRIM25 (Figures 3 and 4). As an E3 ubiquitin ligase, TRIM25 can regulate the antiviral immune response (22). TRIM25-mediated ubiquitination of the cytosolic PRR RIG-I is an essential step for the initiation of an intracellular antiviral response (37). Data presented (Figure 5) revealed that upon TRIM25 overexpression, CVB3 replication was significantly suppressed by upregulating type I IFNs and interferon-stimulated genes (ISGs) production. However, in the course of natural infection, CVB3 can complete the replication cycle and efficiently spread. Hence, CVB3 has evolved several general strategies to suppress host innate immunity therefore does not cause robust IFN responses and ISG expressions. In the current study, miR-30a was significantly up-regulated in response to CVB3 infection in HeLa cells and cardiomyocytes, thereby suggesting that miR-30a may be involved in regulating of CVB3 infection and the progression of VMC or other CVB3-induced diseases (Figure 1). Mir-30a suppressed TRIM25 expression and TRIM25-mediated RIG-I ubiquitination to suppress IFN-β activation and production (Figure 6). This might be the mechanism adopted by CVB3 to antagonize the antiviral response of host.
In the present study, we confirmed that miR-30a modulates type I IFN responses to facilitate CVB3 replication via targeting TRIM25. Furthermore, CVB3 can antagonize the antiviral activity of TRIM25 by increasing miR-30a level in vitro. These results improve the understanding of the effect of miR-30a on CVB3 replication and the mechanism by which CVB3 evades the TRIM25-mediated innate immune response. Hence, the current study not only offers a novel mechanism through which miR-30a inhibits host innate immunity but also provides improved understanding of the mutual regulatory mechanism between CVB3 and the host innate immune responses.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
This study was conceived and designed by RZ. All authors participated in the experiments. JL wrote the main manuscript text and prepared the figures. LL and YX performed the experiments. XL, LS, and JG prepared the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Shanghai Science and Technology Innovation Action Plan (17411969000) and the Foundation of Key Laboratory of Veterinary Biotechnology (No. shklab202004), Shanghai, P.R. China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J (2013) 34(33):2636–48, 2648a-2648d. doi: 10.1093/eurheartj/eht210
2. Esfandiarei M, McManus BM. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol (2008) 3:127–55. doi: 10.1146/annurev.pathmechdis.3.121806.151534
3. Fairweather D, Stafford KA, Sung YK. Update on coxsackievirus B3 myocarditis. Curr Opin Rheumatol (2012) 24(4):401–7. doi: 10.1097/BOR.0b013e328353372d
4. Liu PP, Mason JW. Advances in the understanding of myocarditis. Circulation (2001) 104(9):1076–82. doi: 10.1161/hc3401.095198
5. Huber SA, Gauntt CJ, Sakkinen P. Enteroviruses and myocarditis: viral pathogenesis through replication, cytokine induction, and immunopathogenicity. Adv Virus Res (1998) 51:35–80. doi: 10.1016/s0065-3527(08)60783-6
6. Pollack A, Kontorovich AR, Fuster V, Dec GW. Viral myocarditis–diagnosis, treatment options, and current controversies. Nat Rev Cardiol (2015) 12(11):670–80. doi: 10.1038/nrcardio.2015.108
7. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5
8. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136(2):215–33. doi: 10.1016/j.cell.2009.01.002
9. Gottwein E, Cullen BR. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe (2008) 3(6):375–87. doi: 10.1016/j.chom.2008.05.002
10. Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, et al. A cellular microRNA mediates antiviral defense in human cells. Science (2005) 308(5721):557–60. doi: 10.1126/science.1108784
11. Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science (2007) 315(5818):1579–82. doi: 10.1126/science.1136319
12. Nathans R, Chu CY, Serquina AK, Lu CC, Cao H, Rana TM. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell (2009) 34(6):696–709. doi: 10.1016/j.molcel.2009.06.003
13. He CL, Liu M, Tan ZX, Hu YJ, Zhang QY, Kuang XM, et al. Hepatitis C virus core protein-induced miR-93-5p up-regulation inhibits interferon signaling pathway by targeting IFNAR1. World J Gastroenterol (2018) 24(2):226–36. doi: 10.3748/wjg.v24.i2.226
14. Wang K, Lai C, Gu H, Zhao L, Xia M, Yang P, et al. miR-194 Inhibits Innate Antiviral Immunity by Targeting FGF2 in Influenza H1N1 Virus Infection. Front Microbiol (2017) 8:2187. doi: 10.3389/fmicb.2017.02187
15. Xu T, Chu Q, Cui J, Bi D. Inducible MicroRNA-3570 Feedback Inhibits the RIG-I-Dependent Innate Immune Response to Rhabdovirus in Teleost Fish by Targeting MAVS/IPS-1. J Virol (2018) 92(2):e01594–17. doi: 10.1128/JVI.01594-17
16. Ceolotto G, Giannella A, Albiero M, Kuppusamy M, Radu C, Simioni P, et al. miR-30c-5p regulates macrophage-mediated inflammation and pro-atherosclerosis pathways. Cardiovasc Res (2017) 113(13):1627–38. doi: 10.1093/cvr/cvx157
17. Su X, Qian C, Zhang Q, Hou J, Gu Y, Han Y, et al. miRNomes of haematopoietic stem cells and dendritic cells identify miR-30b as a regulator of Notch1. Nat Commun (2013) 4:2903. doi: 10.1038/ncomms3903
18. Heikel G, Choudhury NR, Michlewski G. The role of Trim25 in development, disease and RNA metabolism. Biochem Soc Trans (2016) 44(4):1045–50. doi: 10.1042/BST20160077
19. Sparrer KM, Gack MU. Intracellular detection of viral nucleic acids. Curr Opin Microbiol (2015) 26:1–9. doi: 10.1016/j.mib.2015.03.001
20. Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol (2016) 14(6):360. doi: 10.1038/nrmicro.2016.45
21. Gack MU, Albrecht RA, Urano T, Inn K-S, Huang I-C, Carnero E, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe (2009) 5(5):439–49. doi: 10.1016/j.chom.2009.04.006
22. Martin-Vicente M, Medrano LM, Resino S, Garcia-Sastre A, Martinez I. TRIM25 in the Regulation of the Antiviral Innate Immunity. Front Immunol (2017) 8:1187. doi: 10.3389/fimmu.2017.01187
23. Meyerson NR, Zhou L, Guo YR, Zhao C, Tao YJ, Krug RM, et al. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe (2017) 22(5):627–38.e627. doi: 10.1016/j.chom.2017.10.003
24. Gupta S, Yla-Anttila P, Callegari S, Tsai MH, Delecluse HJ, Masucci MG. Herpesvirus deconjugases inhibit the IFN response by promoting TRIM25 autoubiquitination and functional inactivation of the RIG-I signalosome. PLoS Pathog (2018) 14(1):e1006852. doi: 10.1371/journal.ppat.1006852
25. Liu H, Li M, Song Y, Xu W. TRIM21 Restricts Coxsackievirus B3 Replication, Cardiac and Pancreatic Injury via Interacting With MAVS and Positively Regulating IRF3-Mediated Type-I Interferon Production. Front Immunol (2018) 9:2479. doi: 10.3389/fimmu.2018.02479
26. Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol (2006) Chapter 15:Unit 15.8. doi: 10.1002/0471142727.mb1508s73
27. Li L, Wei Z, Zhou Y, Gao F, Jiang Y, Yu L, et al. Host miR-26a suppresses replication of porcine reproductive and respiratory syndrome virus by upregulating type I interferons. Virus Res (2015) 195:86–94. doi: 10.1016/j.virusres.2014.08.012
28. Wang WX, Wilfred BR, Hu Y, Stromberg AJ, Nelson PT. Anti-Argonaute RIP-Chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA (2010) 16(2):394–404. doi: 10.1261/rna.1905910
29. Corsten MF, Heggermont W, Papageorgiou AP, Deckx S, Tijsma A, Verhesen W, et al. The microRNA-221/-222 cluster balances the antiviral and inflammatory response in viral myocarditis. Eur Heart J (2015) 36(42):2909–19. doi: 10.1093/eurheartj/ehv321
30. Corsten MF, Papageorgiou A, Verhesen W, Carai P, Lindow M, Obad S, et al. MicroRNA profiling identifies microRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myocarditis. Circ Res (2012) 111(4):415–25. doi: 10.1161/CIRCRESAHA.112.267443
31. Jiang D, Li M, Yu Y, Shi H, Chen R. microRNA-34a aggravates coxsackievirus B3-induced apoptosis of cardiomyocytes through the SIRT1-p53 pathway. J Med Virol (2019) 91(9):1643–51. doi: 10.1002/jmv.25482
32. Gou W, Zhang Z, Yang C, Li Y. MiR-223/Pknox1 axis protects mice from CVB3-induced viral myocarditis by modulating macrophage polarization. Exp Cell Res (2018) 366(1):41–8. doi: 10.1016/j.yexcr.2018.03.004
33. He F, Xiao Z, Yao H, Li S, Feng M, Wang W, et al. The protective role of microRNA-21 against coxsackievirus B3 infection through targeting the MAP2K3/P38 MAPK signaling pathway. J Transl Med (2019) 17(1):335. doi: 10.1186/s12967-019-2077-y
34. Liu F, Wang H, Du L, Wei Z, Zhang Q, Feng WH. MicroRNA-30c targets the interferon-alpha/beta receptor beta chain to promote type 2 PRRSV infection. J Gen Virol (2018) 99(12):1671–80. doi: 10.1099/jgv.0.001166
35. Zhang Q, Huang C, Yang Q, Gao L, Liu HC, Tang J, et al. MicroRNA-30c Modulates Type I IFN Responses To Facilitate Porcine Reproductive and Respiratory Syndrome Virus Infection by Targeting JAK1. J Immunol (2016) 196(5):2272–82. doi: 10.4049/jimmunol.1502006
36. Lin X, Yu S, Ren P, Sun X, Jin M. Human microRNA-30 inhibits influenza virus infection by suppressing the expression of SOCS1, SOCS3, and NEDD4. Cell Microbiol (2020) 22(5):e13150. doi: 10.1111/cmi.13150
Keywords: miR-30a, CVB3, viral replication, TRIM25, type I IFN responses
Citation: Li J, Xie Y, Li L, Li X, Shen L, Gong J and Zhang R (2021) MicroRNA-30a Modulates Type I Interferon Responses to Facilitate Coxsackievirus B3 Replication Via Targeting Tripartite Motif Protein 25. Front. Immunol. 11:603437. doi: 10.3389/fimmu.2020.603437
Received: 06 September 2020; Accepted: 23 November 2020;
Published: 14 January 2021.
Edited by:
Bin Su, Capital Medical University, ChinaReviewed by:
Sally A. Huber, University of Vermont, United StatesCopyright © 2021 Li, Xie, Li, Li, Shen, Gong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rufang Zhang, emhhbmdydWZhbmcwMTAzQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.