
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 December 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.600886
This article is part of the Research Topic Deciphering the Microbiome-Immunity-Cancer Axis View all 9 articles
 Marine Fidelle1,2,3,4,5
Marine Fidelle1,2,3,4,5 Satoru Yonekura1,2,3,5
Satoru Yonekura1,2,3,5 Marion Picard1,2,3,6
Marion Picard1,2,3,6 Alexandria Cogdill7,8Antoine Hollebecque1,9
Alexandria Cogdill7,8Antoine Hollebecque1,9 Maria Paula Roberti1,2,3*Laurence Zitvogel1,2,3,4,5*
Maria Paula Roberti1,2,3*Laurence Zitvogel1,2,3,4,5*While colorectal cancers (CRC) are paradigmatic tumors invaded by effector memory lymphocytes, the mechanisms accounting for the relative resistance of MSI negative CRC to immunogenic cell death mediated by oxaliplatin and immune checkpoint inhibitors has remained an open conundrum. Here, we propose the viewpoint where its microenvironmental contexture could be explained -at least in part- by macroenvironmental cues constituted by the complex interplay between the epithelial barrier, its microbial ecosystem, and the local immune system. Taken together this dynamic ménage-à-trois offers novel coordinated actors of the humoral and cellular immune responses actionable to restore sensitivity to immune checkpoint inhibition. Solving this paradox involves breaking tolerance to crypt stem cells by inducing the immunogenic apoptosis of ileal cells in the context of an ileal microbiome shifted towards immunogenic bacteria using cytotoxicants. This manoeuver results in the elicitation of a productive Tfh and B cell dialogue in mesenteric lymph nodes culminating in tumor-specific memory CD8+ T cell responses sparing the normal epithelium.
Colorectal cancer (CRC) is the third leading malignancy worldwide and the second most common cause of cancer mortality, regardless of gender. CRC is expected to increase in incidence by 60% by 2030, posing an increasing burden on health care systems worldwide (1). The typical development of CRC takes place in aberrant crypts, evolving over time into a neoplastic precursor lesion called a “polyp.” The carcinogenic evolution is thought to occur over 10–15 years, following a traditional adenoma-carcinoma tumor progression. CRCs are often divided into two groups delineated by location: left-sided and right-sided (proximal) colon cancers. These anatomical sites help to capture the heterogeneous features of CRC in relation to key physiological landmarks (2).
CRC results from a progressive accumulation of epigenetic and genetic alterations resulting in the transformation of normal colonic mucosa to adenocarcinoma. 60%–65% of CRCs are classified as sporadic, occurring in people without a family history or genetic predisposition (3). Sporadic CRC development is often associated with numerous risk factors related to health determinants such as lifestyle, diet, smoking, and alcohol consumption (4). Key determinants such as smoking status is linked to proximal colorectal cancer (pCC) and rectal cancer development (5). While dietary habits such as overt consumption of animal fat, red and processed meat, low intake of dietary fibers, unrefined grains and vegetables promote key inflammatory pathways associated with CRC (6, 7). The current staging or classification of CRC malignancies, such as TNM (8, 9) only takes into account primary tumor size, regional lymph node involvement, and metastatic spread but fails in the ability to further delineate how best to approach therapeutic management of malignancies. Indeed, key aspects of classification, such as genetic specificities, immunological contexture, and gut dysbiosis, play a vital role in the physiopathology of CRC and should be considered in context (Figure 1) (10).
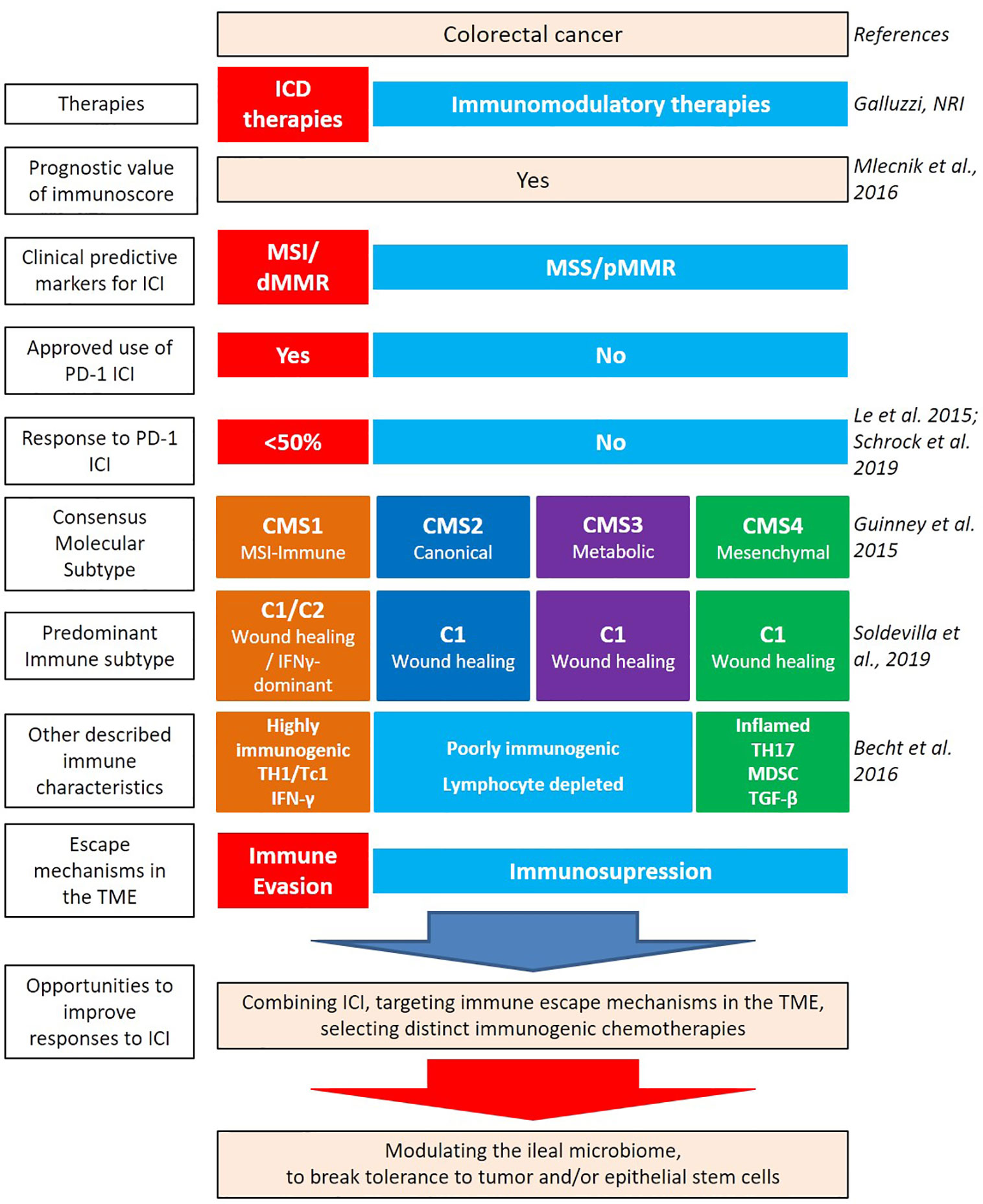
Figure 1 Heterogeneity of colorectal cancer (CRC) leading to new classifications. Several factors contribute to the intrinsic immunogenicity of CRC. There are several classifications based on anatomical, genetic, and immunological parameters which allow for the dissection of the intertwined relationships between these components and to predict clinical outcome. Therapies can contribute to gears of intrinsic immunogenicity, by providing antigens and adjuvants. While oxaliplatin (OXA)-based chemotherapeutic regimen and anti-EGFR Abs can be considered “immunogenic” therapeutics, paving the way to a better efficacy of immune checkpoint inhibitors (ICI), anti-VEGF Abs could rather modulate the vascularization and distinct immunosuppressive cues of the tumor microenvironment (TME) [such as myeloid-derived suppressor cell (MDSC)]. Despite this knowledge, the combination of immunogenic cell death-mediating compounds with ICI failed to ameliorate CRC patients’ prognosis, at least in MSS CRC. Integrating the ileal microbiome in this equation has the potential to break tolerance to self-antigens of the crypts, by priming Tfh and B cell responses, instrumental to control tumor progression.
CRC arises from mutational activation of oncogenes associated with the mutational inactivation of tumor suppressor genes (11). Three non-exclusive major types of genomic instability have been described in CRC. The first type, occurring in 85% of CRC, concerns gene mutations in APC or other tumor suppressor genes resulting in activation of the Wnt pathway characterized by a chromosomal instability (CIN) phenotype. The second type found in 20%–30% of CRC, accounts for global genome hypermethylation coinciding with the inactivation of tumor suppressor genes, known as CpG island methylator phenotype (CIMP) (12). The last type is found in ~15% of patients who encounter the loss of DNA mismatch repair (MMR), leading to a high level of microsatellite instability (MSI-High), a hypermutable phenotype (13). The MSI-H phenotype results from either a somatic inactivation of MLH1 MMR gene (sporadic cases, 12%) or from a germline mutation in MMR genes (MLH1, MSH2, MSH6, PMS2) leading to a deficient DNA mismatch repair (dMMR); such as in the case of the Lynch syndrome (3%) (14). The CIMP phenotype can lead to the MSI phenotype when hypermethylation of the MLH1 gene promoter occurs. This particular MSI phenotype generates neoantigens accounting for their intrinsic immunogenicity (15).
CRC outcomes are not only dictated by genetic features but also by the immune contexture (Figure 2). Several cell types associated with innate and adaptive immune responses cooperate and dictate the prognosis of patients diagnosed with CRC. γδT cells expressing a heterodimeric T-cell receptor (TCR) are often enriched in epithelial barriers of various mucosae to sense cellular stress at portal of entry (16). However, preclinical murine models of colitis and clinical CRC data have shown that the γδT17 cell subset, producing the IL-17A or IL-17F cytokines, promotes tumor progression through the accumulation of myeloid-derived suppressive cells (MDSC) (17, 18). MDSCs accumulate in the tumor microenvironment (TME), as compared to the adjacent healthy tissue, in patients with CRC and their circulation correlates with cancer stage and metastasis (19, 20). Moreover, Th17 cells through the secretion of IL-17A and the transduction of the STAT3 pathway lead to the downregulation of CXCR3 expression on CD8+ T cells. Consequently, these Th17 cells dampen the CXCL10-dependent recruitment of cytotoxic CD8+ T cells (CTLs) in advanced stages of CRC (21). In addition, the IL-17R signaling in tumor cells blunts CXCL10 release thereby limiting CTLs influx in tumor bed (22). Furthermore, Th17 cells secrete IL-22 which promotes colitis associated with CRC (23). Contrasting with γδT17 and Th17 cells, IFN-γ producing conventional CD4+ T cells, namely, Th1 lymphocytes, are associated with a favorable prognosis in CRC (24).
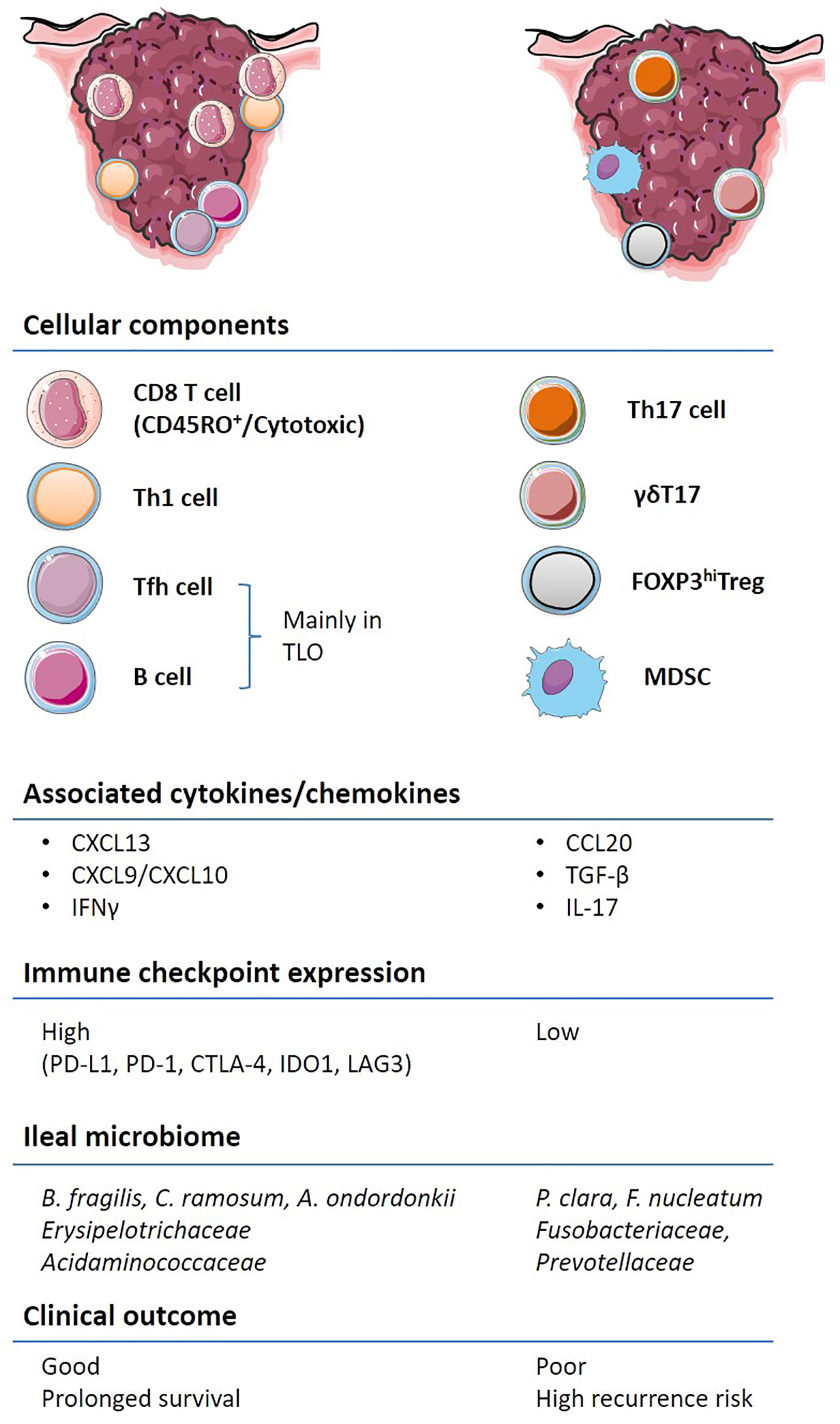
Figure 2 Immune contexture of primary and metastatic colorectal cancer (CRC). A non-exhaustive list of the main immune features contributing to the stability or acceleration CRC progression is aligned on the left and right colon respectively. Pre-existing tumor immunity, termed “immune contexture”, monitored by “immunoscoring” as well as transcriptome deconvolution represent strong and independent predictors of long-term progression free and overall survival in CRC. Tumors enriched with cytotoxic CD8+, CD4+, in particular Th1 and Tfh, and B cells, are associated with an IFN-γ response, the upregulation of immuno-inhibitory molecules and better clinical outcome (left). Other types of inflammation, characterized by IL-17 expressing T cells, FOXP3hi Tregs and immunosuppressive myeloid populations are associated with worse clinical outcome. The composition of the ileal microbiome contributes to shift the balance between Tfh and Th17 cells.
Another subset of auxiliary T cells, the T follicular helper (Tfh) CD4+ lymphocytes, defined by CXCR5 chemokine receptor expression and the Bcl6 transcription factor, are found within and around CRC tumor nests and tumor draining lymph nodes (tdLN). Their density is negatively correlated with CRC tumor progression (25, 26). A positive Tfh/B cell signature associated with increased CD8+ T cell infiltrates has been reported in CRC cases with favorable outcomes (25, 27). The Tfh/B cell dialogue is pivotal to orchestrate CD8+ T cell effector functions, which are believed to keep in check CRC at early stages of development (28, 29) and may pave the way to the efficacy of immune checkpoint inhibitors (30). Indeed, B cells, FcγR and Ig as well as Th2 and IL-4 have been associated with chronic inflammatory processes, leading to tumor development in T cell-dependent (31–33) or -independent tumor models (34). Likewise, bladder tumors, amenable to therapeutic immune checkpoint blockade, have a dismal prognosis when their TILs contain CD8+ T cells producing IL-10 and expressing inhibitory receptors (TIGIT, Lag3, Tim3, and CTLA-4), associated with GATA3+ CD4+ Th2 cells and Treg (35, 36). Moreover, PD-1 expressing B cells are memory B cells exerting suppressive activity that can be alleviated by anti-PD-1 Abs (37). Another report described PD-1+ B cells that possessed regulatory capacity toward T cell responses, and although rare in peripheral blood, they were found enriched in thyroid tumors (38).
However, recent accumulating evidence shows that B cells can orchestrate favorable TCF7+ naive/memory CD8+ T cell-based immune contexture, specifically when tertiary lymphoid organs can be formed to prime naive CD8+ T cells in human tumors, paving the way to clinical benefit to anti-PD-1 Abs (39–41). In breast tumor models genetically modified to express neoantigens, CD8+ T cell effector memory cells could be elicited with anti-PD-1 Abs when Treg were depleted (using a mouse anti-CTLA-4 Ab with ADCC properties), therefore triggering a cascade whereby B cells became activated, capable of triggering Tfh activation and IL-21 production. B cell depletion or inhibition prevented class switching, plasma cell generation, and the release of anti-tumor IgG indispensable for tumor rejection following therapy with ICBs (30). In these models of combined immune checkpoint inhibitors (ICIs), effector Tc1 CD8+ T cells cannot be generated in the absence of Tfh, IL-2, or B cells. Hence, the optimal B cell activity and therapeutic benefits derived from anti-PD-1/anti-CTLA-4 therapy correlated with concurrent Treg inhibition and Tfh activation (30) or, as described in our study, by lamina propria IL-12 and IL-1β producing DC after stimulation with immunogenic ileal commensals (26).
Tumor-associated Tfh secrete CXCL13, attracting CXCR3+ CTLs (42) and via IL-21 secretion promotes their effector functions (43). Tfh are involved in the orchestration of the humoral responses through interactions with B cells. Nevertheless, the Tfh/B cells/CTLs collaboration is limited by an increased expression of PD-1 on Tfh during tumor development, dominated by a PD-L1-associated immunosuppression (43, 44). It has been shown that germinal center B cells and/or plasma cells play a protective role, while the T cell suppressive function of regulatory B cell subsets was associated with the dissemination of metastases (45–49). The CD8+ Tfh counterpart has also been described in the tdLN of CRC. CXCR5+ CD8+ T follicular cytotoxic cells (Tfc) express high levels of effector molecules and convey a better prognosis in CRC (50). In line with these contentions, Roberti et al. have recently described the beneficial role of antibody producing B cells in cancer immunosurveillance in murine transplantable colon tumors (26). Hence, as described for other tumor types, terminally differentiated memory B cells and/or plasma cells and Tfh could predict increased patients survival in CRC (49, 51, 52).
Pagès and Galon (Figure 2) were the first to report the major role of cytotoxic and memory T lymphocytes within primary tumors in predicting survival of CRC patients (28, 53, 54). These studies included early-stage cancer patients (55) and described distinct cellular and molecular cues modulating tumor-infiltrating lymphocyte (TIL) densities (56). A scoring system ‘‘Immunoscore’’ based on the quantification of CD3 and CD8 in the core (CT) and at the invasive margin (IM) of primary tumors has a prognostic value superior to the AJCC/UICC TNM-classification (28, 54, 57, 58). Immunoscore predicts survival of non-metastatic patients, as does the immune infiltrate evaluation in the metastasis for metastatic patients (59–61). These authors have also demonstrated the superiority of Immunoscore over microsatellite instability and PD-L1 expression in predicting survival (62, 63). Focusing on metastasis profiling in CRC, Galon’s group brought forward evidence that the cytotoxic T lymphocytes-based adaptive immune response plays a key role in preventing tumor recurrence and metastatic dissemination; despite evidence for clonal expansion of distinct T lymphocyte subsets culminating in immunoediting (60, 64). Disease-free survival (DFS) and OS in stage IV patients are largely governed by the state of the local adaptive immune response within the metastases being the most likely site of tumor immune escape (63). However, further studies are ongoing to better understand how dynamics of genomic and immune patterns shape the CRC metastatic landscape.
Given the potential significance of the intertwined relationships between genetics and immunometrics, the Cancer Genome Atlas Consortium (TCGA) proposed, in 2012, a different stratification of CRC (65). Four consensus molecular subtypes (CMS1-4) have been discussed to classify CRC based on transcriptomic of the tumor microenvironment (TME) (Figure 1) (66, 67).
The CMS1 group (14% of early-stage CRC) showing hypermutation, hypermethylation, and high antigenicity (neoepitopes/neoantigens) is heavily immune infiltrated and predominately constituted by MSI-H tumors (76%). BRAF mutations are often found in the CMS1 subtype. The CMS1 group is characterized by a higher expression of immune checkpoints conveying a dismal prognosis (67–70) yet potentially favoring the relative efficacy of immune checkpoint inhibitors (ICIs) (69). CMS2 “canonical” and CMS3 “metabolic” groups (37% and 13% of early-stage tumors, respectively) correspond to “immune-neglected” CRC, in that CMS2 represents an “immune desert” and CMS3, an “immune excluded” tumor microenvironment. The CMS2 group shows WNT and MYC activation. The CMS2 “immune-desert” subtype is characterized by a poor intratumoral T cell infiltrate. After relapse, the CMS2 group has a superior survival rate than the CMS1 (67). Compared to CMS2, the CMS3 “immune-excluded” subtype often harbors KRAS-activating mutations associated with memory Th and naïve B cell-based immune components (71). The CMS4 “mesenchymal” subtype (23% of early-stage tumors) exhibits the prototypic fingerprint of the epithelial-mesenchymal transition (EMT) characterized by a stroma-related gene transcription centered by the transforming growth factor-β (TGF-β) metagene. Stromal cells secret immunosuppressive chemokines that interact with cancer cells and inhibit cytotoxic immune cells, thus contributing to the proliferation of MDSCs (66). CMS4 has the worst relapse-free and overall survival (67). A small fraction of CRC tumors have indeterminate features (13%), and possibly represent a transitional state, or result of a mixed phenotype, due to intratumoral heterogeneity (67).
Recently, based on TCGA transcriptomic analysis of more than 10.000 solid primary tumors, Thorsson et al. have identified six immune subtypes (I.S.) of the tumor microenvironment (C1-C6), namely: wound healing (C1), IFN-γ dominant (C2), inflammatory (C3), lymphocyte depleted (C4), immunologically quiet (C5), and TGF-β dominant (C6). Across all cancer types, the C3 “inflammatory” I.S. was associated with the best overall survival. C1 and C2 I.S. were characterized by a high proliferation rate and exhibited less favorable prognosis even if a higher lymphocyte signature ameliorated the clinical outcome (72). The “wound healing” (C1) and “IFN-γ dominant” (C2) subtypes are the two main I.S. represented in CRC (72, 73). The C1 “wound healing” I.S. showed a prominent expression of angiogenesis-related genes and a low Th1/Th2 ratio. The C2 “IFN-γ dominant” I.S. has the highest intratumoral heterogeneity; containing higher levels of TILs and Tfh cells (73). Nevertheless, the C1 “wound healing” I.S. harbors a better prognosis than the C2 “IFN-γ dominant” I.S. (5-year overall survival (OS) 65% and 49%, respectively), perhaps in line with the upregulation of PD-L1, PD-1, CTLA-4, IDO1, and LAG3 molecules related to immune exhaustion in C2.
Depending on the TNM score, the main treatment for colon cancers remains the surgical resection of the tumor; especially for tumors with a low risk of recurrence (74). For tumors with stage III or high-risk stage II, a (neo) adjuvant therapy based on chemotherapy is performed prior to surgery to reduce tumor burden and recurrence rates (74). For stage III or high-risk stage II CRC, adjuvant chemotherapies with FOLFOX (5-FU/leucovorin/oxaliplatin) is the standard of care (75–77). As for metastatic diseases, a form of immunotherapy called immune checkpoint inhibitors (ICIs) has transformed the standard-of-care of cancer patients (78–81).
Six months of adjuvant chemotherapy frequently benefit immunoscore-high-patients with stage III primary CRC (82). Moreover, patients treated with chemotherapy and anti-EGFR (Epidermal Growth Factor Receptor) had greater infiltration of T lymphocytes at the core of the metastatic lesion and a higher Immunoscore than patients who received chemotherapy and anti-VEGF (Vascular Endothelial Growth Factor) (63). Hence, Immunoscore® is improved by chemotherapy and predicts the beneficial effects. Oxaliplatin (OXA), a platinum salt used as a cornerstone chemotherapy for CRC (77) is one of the best cytotoxicants capable of inducing immunogenic cell death (ICD) (83). ICD triggers an endoplasmic reticulum (ER) stress response and the activation of the autophagic machinery culminating in tumor cell surface exposure and/or secretion of the mandatory damage-associated molecular patterns (DAMPs); stimulating anticancer immune responses (84). Moreover, OXA increased Tfh density in tumor nests and eventually TILs accumulation as a function of its capacity to induce ileal crypt apoptosis (26).
In CRC, MSI status constitutes a predictive biomarker for response to ICIs. Metastatic CRC with MSI-H phenotype show a high disease control rate and a favorable progression-free survival (PFS) after anti-PD-1 antibody while the 85% patients presenting with microsatellite stable (MSS) or proficient MMR (pMMR) CRC have failed to benefit from various immunotherapy approaches (85, 86). POLE proofreading domain mutations are also associated with a favorable response to ICIs (87). Hence, anti-PD-1 therapies, such as nivolumab and pembrolizumab, have been approved by the U.S. Food and Drug Administration (FDA) in 2017 for patients with MSI-H/dMMR metastatic CRC that have progressed following treatment with a fluoropyrimidine, oxaliplatin, or irinotecan-based chemotherapy (88, 89). Though, the response rate after anti-PD-1 antibodies often remains less than 50% (85, 90), in relation to tumor heterogeneity within MSI-H CRC (91), or immune evasion based on alterations in the antigen processing and presentation machinery (92). However, the immunoscore turned out to exhibit a superior prognostic value for overall survival regardless of the MSI/MMR status. MSI-H patients with a low immunoscore do not have any survival benefit compared with MSS patients. Additionally, in MSS CRC cases, the immunoscore correlates with higher disease-free survival and overall survival (54). Actually, there is no prospective study confirming the clinical significance of the immunoscore for ICI response yet. An ancillary study coinciding with a clinical trial in metastatic MSS CRC (the POCHI trial: NCT04262687) just started to analyze the predictive value of the immunoscore for the clinical benefit to expect from chemotherapy and immunotherapy (93).
The degree of microsatellite instability (MSI) and resultant tumor mutational burden (TMB) have also been shown to underlie the variable response to PD-1 blockade in dMMR, with indel mutations strongly associated with objective response (85, 94). In one of the largest number of patients treated with ICI analyzed for TILs and TMB, Loupakis et al. found that TILs correlate to TMB and the higher the number of TILs, the better the outcome (95). Thus, pembrolizumab is FDA approved for TMB-high tumors (96). In other cases, such as in dMMR/MSI-H metastatic CRC, the combination of ICIs (such as PD-1 and CTLA-4 co-blockade) are likely to be more effective than monotherapy (97). The vast majority of CRC cases, MSS or pMMR CRC, are naturally resistant to immunotherapy; at least in part due to the low antigenicity (TMB) of the malignancy. However, based on TCGA dataset analysis, a small subset of patients among MSI-Low/MSS-CRC cases exhibiting a high CD8+ T cell infiltrate and an upregulation of IFN-γ could benefit from immunotherapies (98).
To overcome the lack of efficacy of ICIs in MSS and pMMR CRC, investigators have combined ICIs with chemotherapy and targeted therapeutics (99). Despite these efforts, the combination of oxaliplatin and pembrolizumab has failed to prove superior over OXA-based chemotherapy alone in advanced CRC (100).
Taken together, CRC is the first neoplasia found to be under immunological control, and the first to be treated with OXA, a chemotherapy endowed with immunogenic cell death properties. However, we can see that most attempts to overcome this malignancy have failed, with the exception of a minority of lesions characterized by MSI. This paradox may be solved by a more complete understanding of the role played by the macroenvironment in which this cancer develops (Figure 1).
The gastrointestinal (GI) tract is the major mucosal surface of the human body and the most densely colonized organ. The overall bacterial load of the GI tract has been described to contain between 1013-1014; nearly equaling the number of the mammalian cells in the body (101, 102).
In mammals, and more specifically in humans, colonization by the intestinal microbiome occurs rapidly during and after birth (103, 104). Successful colonization is determined by microbial selection and competition that begins in the first hours of life (105). Throughout our lifetimes, the microbial population in the human GI tract is affected by a multitude of environmental factors such as age (103, 106), geography (106), dietary habits (107, 108), use of antibiotics (109, 110), host genetics (111, 112), and the pressure of the immune system (113).
The intestinal microbial ecosystem has a significant role in host physiology in multiple ways. The processing of food and xenobiotics enables a host to obtain essential nutrients (101). The intestinal microbiota promotes post-natal maturation of physiological gut functions such as the integrity of the epithelial barrier, the expression of essential enzymes (114), the development of the intestinal vascularization (114, 115), and of the enteric nervous system; all essential for motility (116). Most importantly, the gut microbiota participates in the maturation of the local and systemic immune system to ensure tolerance vis-à-vis of food antigens and the elimination of pathogens, maintaining a mutual symbiosis between commensals and self-tissues for the homeostasis of the meta-organism (117, 118).
Various bacterial communities are disseminated along the GI tract with diversity affected by anatomical and environmental differences from the esophagus to the rectum. There is a limited diversity of the microbiome in the esophagus where Streptococci remain the dominant species (119). Similarly, the stomach has limited microbial diversity due to low pH of the gastric lumen (120). While the small intestine and colon harbor a wider commensalism compared to that of the upper GI tract. Different physiologies and environments such as chemical, oxygen and nutrient gradients, and the compartmentalized immune system along the GI tract result in distinct distribution of the intestinal microbiota (121). The small intestine contains a smaller load of bacteria and with less diversity, than the colon. This is due to a number of factors such as harsh environment for bacterial communities, a shorter transit time, an increased influx of digestive enzymes of antimicrobial peptides and bile acids, and an intermittent food substrate delivery (121, 122). Although the taxonomic classification has been inconsistent across studies, several reports show the predominance of two major phyla, i.e., Firmicutes and Proteobacteria (121, 123, 124), followed by others (Bacteroidetes, Fusobacteria, Verrucomicrobia, Actinobacteria) residing in the human small intestine (124). At the genus level, several genera are commonly found in the small intestine, such as Lactobacillus, Clostridium, Staphylococcus, Streptococcus, and Bacteroides (124–127). Sample collection from the distal ileum has shown that Streptococcus, Granulicatella, Actinomyces, Solobacterium, Rothia, Gemella, and TM7(G-1) are the most frequently detected bacterial genera by 16S gene sequencing (127) with Streptococci, Actinomyces, Rothia and Lactobacillus species most frequently identified by culturomics and mass spectrometry (128). In contrast, the colon contains roughly 70% of all the bacteria of the human body (129). The microbial population in the colon is a result of environmental factors such as lower concentrations of antimicrobials, slower transit time, and fermentation of polysaccharides (121, 129). Interestingly, the two major phyla in the colon are Bacteroidetes and Firmicutes (121, 129). At the genus level, Bacteroides, Prevotella, and Ruminococcus are predominant (129) (Figure 3).
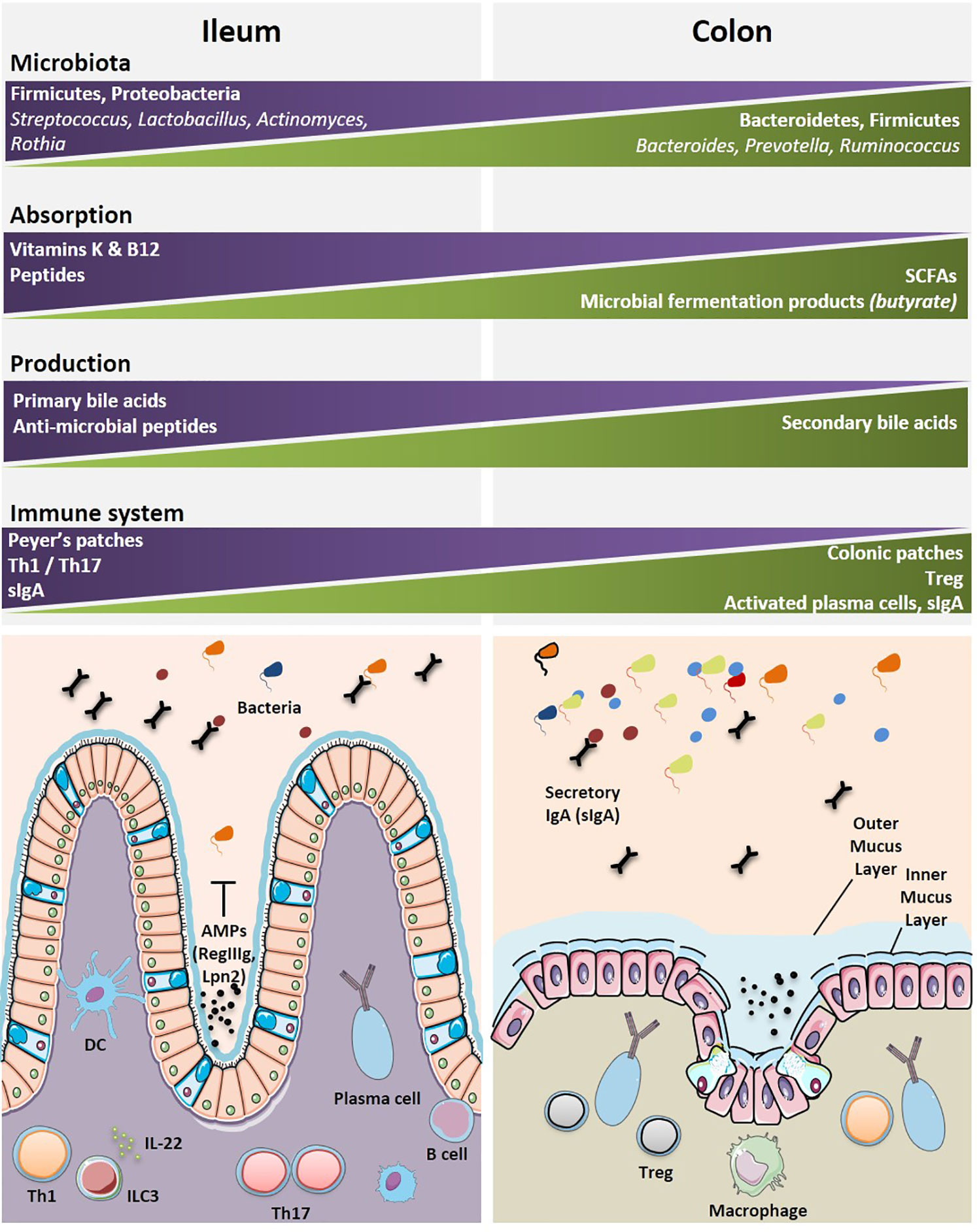
Figure 3 Geographical distributions of immune and microbial components highlight specificities between the ileal and colonic segments. The most dominant microbial and immune cells accounting for the diversity and specificity of each organ are depicted. The main specialized functions (metabolic, immune, motility, etc.) for each segment are indicated. A graphical abstract is lining below the boxes where these cells and functional specificities are listed. Immune cells are found either clustered within organized lymphoid structures or scattered within intestinal epithelium (IE) and lamina propria (LP). AMP, anti-microbial peptides; B cell, B cell lymphocyte; DC, dendritic cell; ILC3, innate lymphoid cell type 3; IL-22, interleukin-22; Th1, T helper 1 cell; Th17, T helper 17 cell; Treg, regulatory T cell.
Fecal profiling of the microbial populations is investigated most frequently because the sampling of feces is non-invasive and convenient for patients compared to performing an invasive biopsy or collecting luminal content of ilea or colons (130). However, there is increasing evidence that the observed microbial composition in feces is different from the content of mucosal samples of the GI tract (123, 131–135). Bacteroidetes and Firmicutes are the main phyla in the healthy human stool samples followed by other phyla such as Actinobacteria, Proteobacteria, Synergistetes, and Verrucomicrobia (123, 134, 136–142). At the genus level, Bacteroides is the most abundant genus (136, 140, 141) followed by Faecalibacterium, Bifidobacterium, Lachnospira, Roseburia, Subdoligranulum, Collinsella, Ruminococcus, Prevotella, Alistipes, and Akkermansia (123, 132, 136–142). Of note, mucosal microbiome of the colon differs from that of fecal composition. James et al. reported that Enterococcus was found to be more prevalent in the proximal colon while Coprobacillus and Escherichia/Shigella were more abundant in the distal colonic mucosae (143).
The symbiotic relationship between the gut microbiome and the associated local immune system is central for the maintenance of the systemic immune tonus. The lamina propria and gut-associated lymphoid tissues (GALT) contain the largest pool of cells mediating innate and cognate immune responses (Figure 3). There is marked regional variation in immune cells along the GI tract, with Th17 decreasing in number from the duodenum to the colon, and regulatory CD4+ T cell (Treg) being most abundant in the colon (144). Pioneering mouse studies demonstrated that distinct bacterial species can fine-tune intestinal immune responses, such as Th17 (145, 146), Treg (118, 147), or Th1 (148, 149), Tfh (148), and B cell activation (150). James et al. catalogued the mucosal microbiome in different regions of the human colon and reported the annotated colon immune single-cell dataset at the Gut Cell Atlas (https://www.gutcellatlas.org/) (143). There are 25 cell types in the lamina propria (LP) and mesenteric lymph nodes (mLNs), draining the healthy colons. Among these, are follicular and memory B cells, IgA+, and IgG+ plasma cells, effector and memory CD4+ T cells, Treg cells, CD8+ T cells, γδ T cells, innate lymphoid cells, natural killer cells (NK), mast cells, and myeloid cells (cDC1, cDC2, pDC, LYVE1+, or CD16+ macrophages, monocytes). B cells, dendritic cells and γδ T cells are Ki67+ cycling populations compared to other colonic immune cell populations. The cecum and the sigmoid were reciprocally enriched in CCL20+ Th17 and Th1 respectively. While mLN contained follicular B cells and memory CD4+ T cells, colonic mucosae, and most specifically the sigmoid LP is rich in effector CD4+ T cells and plasma cells. Region-specific transcriptional differences linked activation and tissue migration of Th1 and Th17 cells of the proximal and sigmoid colon as well as the identification of clonal sharing between these colonic regions plead for cell-extrinsic rather than cell-intrinsic factors regulating T cell functions. However, the relative proportion of Treg cells do not change significantly from proximal to distal colon. There is heterogeneity in Treg cell states in the mLNs and colon with a transient loss of the FoxP3-expressing cell population. Data infer a continuous activation trajectory of these Treg cell states between draining lymph nodes and colon, with genes regulating Treg cell migration and adoption of Th-like profiles in tissues (143). Treg transiently losing FoxP3 expression could re-express it transcription under activation to achieve their bona fide immunosuppressive functions (151). There is a highly activated state of plasma cells found in the distal colon compared with proximal colon plasma cells, which are characterized by greater accumulation, somatic hypermutation, clonal expansion, and stronger homing to the colonic mucosa (143). Compared with the cecum, IgA+ plasma cells of the sigmoid colon respond to a rich and unevenly represented community of bacterial species; most likely accounting for the increased activation, migratory and greater clonality status of plasma cells (143). Indeed, by means of a novel method for identifying and isolating lymphoid follicles along the length of the human intestine and IgA sequencing analysis, Agace’s group showed that IgA adaptive immune responses are initiated in an anatomically restricted manner in the human Peyer’s patches and submucosal isolated lymphoid follicules contributing to the ileal and colonic plasma cell repertoires, respectively (152).
Western style diets (high fat, high sugar) and meat consumption may in fact be major risk factors in the development of CRC (153). Diet displays a dominating role in shaping the structure of gut microbiota, occasionally leading to a detrimental alteration of microbiota ecology and functionality. Indeed, increasing dietary fiber can lead to the establishment of a favorable microbiota regulating the production of the anti-inflammatory short chain fatty acids (SCFA) (154). An unbalanced microbiota, known as dysbiosis, has been associated with many maladaptive states, including CRC (155–159). (Figure 4A) However, the contribution of the microbiota in CRC initiation and development remains a subject of debate. Pre-clinical and human clinical studies have linked the intestinal microbiota to CRC. Pre-clinical studies performed with sporadic CRC rodent models showed that germ-free mice and rats display less intestinal tumorigenesis than animals conventionally reared, highlighting the role of the microbiota in CRC emergence (160). Confirming this functionality, the first mouse model of spontaneous invasive CRC has been described, in which resident microbiota play a key role in disease outcome (161). Indeed, intestinal epithelial cell (IEC)-specific transgenic expression of the epithelial-mesenchymal transition regulator Zeb2 in mice (Zeb2IEC-Tg/+ mice) lead to increased intestinal permeability and spontaneous invasive colon carcinoma development in a microbiota-dependent manner (161). However, this work did not yet identify CRC associated- microbial entities. Several independent studies comparing tumor versus paired adjacent healthy tissues showed marked differences in the gut microbiota composition (162–165), with either depletion or enrichment of selected bacterial species. Meta-analysis of five publicly available datasets and two new cohorts with validation using two additional cohorts, considering n=969 and n=768 fecal metagenomes revealed an enrichment of bacterial species within CRC and feces of patients, not found in homeostatic controls. They identified 29 species, mostly from the oral cavity, associated with CRC development (166, 167). At the functional level, the choline trimethylamine-lyase gene (166) as well as protein and mucin catabolism genes were overabundant while carbohydrate degradation genes were depleted (167) in CRC.

Figure 4 Instrumental links between intestinal dysbiosis and colon carcinogenesis. Risk factors contributing to coloractal cancer (CRC) initiation and development encompass the life style, diet, the exposure to environment (xenobiotics), host genetics and the gut microbiome (A). Several hypotheses have been formulated to account for the toxicity of the microbiome for the epithelium. A driver bacterium could recruit a consortium of disease-facilitating microbes (B). The “keystone hypothesis” suggests that specific bacteria lead to dysbiosis and pro-carcinogenic microenvironment (C). Mucus digestion by some bacteria could expose the epithelium to toxins produced by virulent bacteria organized in biofilms (D).
Of note, this particular microbial shift distinguishes benign from malignant colon tumors (166). Alterations of microbiota composition are distinct across major stages of colorectal carcinogenesis (168, 169), suggesting a role of specific bacterial communities in this process. Moreover, besides the important shifts observed in the colonic microbiome of CRC, significant variations have been reported in the ileal microbiome. Using healthy ileal mucosa-associated microbiome, lining upstream from Bauhin’s valve and collected during hemicolectomy in patients suffering from different stages of proximal CC, Roberti et al. (26) identified distinct species and family members conveying immunogenicity to CRC by regulating the accumulation of Tfh instead of Th17 cells in the tumor microenvironment.
Furthermore microbiome diversity and richness changes during carcinogenesis, bacteria can be found with a specific organization in normal colon mucosa, mirroring the bacterial composition within the tumor. Bacterial organization in biofilms protects commensals against external agents and induces pro-oncogenic properties by inducing major deregulation of epithelial cell biology culminating in sustained inflammation as detailed below (Figure 4). Biofilms are associated with increased risk of developing sporadic CRC (170, 171). Formation of biofilms is mostly a characteristic of right-sided colon cancer (170).
Altogether, these studies showing significant variations of taxonomic footprints suggest a role of the gut microbiota in the emergence of CRC. However, whether one or more species are necessary for CRC development remains unclear. Notable steps forward have been made in the identification of single bacterial species or bacterial communities associated with CRC. Several studies have nailed down mechanistic cues that link distinct bacteria strains and species to CRC carcinogenesis. Bacteria can exert their pro-oncogenic effects through multiple mechanisms. The inflammatory contexture can modulate microbial gene functions and increase the cancer-promoting activity of some bacteria strains, as exemplified for E.coli NC101 (172). Bacteria can produce toxins and metabolites from the fermentation of by-products or induce the formation of superoxide radicals that lead to subsequent genetic mutations in the colonic epithelium (173, 174). Several theories have also been proposed to delineate the involvement of specific microbiota in CRC initiation or progression (Figure 4). The “driver-passenger” model supports the idea that a microbial leader recruits a consortium of disease-facilitating microbes to initiate the biological events causing CRC (175–177). This model suggests a chronological recruitment of specific bacteria concurring to CRC. First, “driver” bacteria create a pro-oncogenic environment through DNA-damage and malignant transformation of epithelial stem cells. After initiation of tumorigenesis, there is an emergence of “passenger” bacteria, more adapted to the tumor environment such as Fusobacterium nucleatum and Streptococcus bovis/gallolyticus (177) (Figure 4B). Secondly, the ”keystone hypothesis,” or “alpha-bug hypothesis,” suggests that certain low-abundance bacteria possessing unique virulence traits can reshuffle a benign environment into a carcinogenic one. For instance, enrichment of Fusobacterium species is associated with a depletion of the Bacteroidetes and Firmicutes, in malignant colon relative to normal colon tissue (175, 178–180). Thus far, several “alpha-bugs” that promote intestinal carcinogenesis in animal models have been described such as Fusobacterium nucleatum (181, 182), colibactin-producing Escherichia coli (183–185) and enterotoxigenic Bacteroides fragilis (ETBF) (186) (Figure 4C).
Fusobacterium nucleatum is frequently associated with advanced proximal CC tumors, metastasis, chemoresistance, and poor prognosis (26, 187–189). These pathogenic properties occur through the expression of two different virulence factors, FadA, and Fap2 on the intestinal CRC and non-CRC epithelium, which induce the expression of a large spectrum of transcription factors, oncogenes, and genes coding for inflammatory functions or tumor progression (182, 190). F. nucleatum shapes the immune environment to promote tumor growth through the elicitation of a pro-inflammatory cytokine cascade (IL-8, CXCL1) and the direct inhibition of T and NK cell functions (191, 192). Furthermore, F. nucleatum has been shown to modulate autophagy in intestinal epithelial cells (IECs) by activating regulatory microRNAs that consequently alter colorectal cancer chemotherapeutic responses (189) (Figure 4).
Regulatory T cells (Treg) can be anti-tumorigenic or pro-tumorigenic in colorectal cancer (CRC) depending on the presence of different Treg subsets with various immunosuppressive molecules. FoxP3 expression intensity dictates the prognosis of CRC; FoxP3(hi) Treg being associated with poor clinical outcome in contrast to producing-FoxP3(lo) Treg producing inflammatory cytokines (193). Earlier work described non-effector T cells comprising regulatory and anergic T lymphocytes that specifically accumulate in tumor tissues and eventually recirculate (194). Some reports suggested synergistic associations between PD-1/CTLA-4 and PD-1/CD39 within Helios+ or Heliosneg FoxP3+ CD4+ T cells to dampen T-cell activation and functions in CRC (195, 196).
Escherichia coli, although commonly found in homeostatic gut microbiota, is another bacterium frequently correlated with tumor staging and prognosis (184). The association between E. coli and CRC specifically implies E. coli strains that produce the genotoxin colibactin. This toxin accelerates tumor progression (185, 197) by damaging host DNA (172, 183, 198) and by inducing cellular senescence (Figure 4). This process may involve the production of several growth factors in human CRC (197).
ETBF is an enterotoxin-producing bacterium that is thought to play a role in the occurrence and progression of CRC (186). The toxicity of this bacterial compound is linked to its capacity to damage DNA strands via the generation of reactive oxygen species (ROS) and through the induction of Th17-dependent inflammatory responses (186). The ability of Bacteroides fragilis to promote tumor growth presumably relies on the enterotoxin production, as nontoxigenic B. fragilis (NTBF) does not induce Th17 responses and fails to facilitate cancer outgrowth (186), offering prophylactic effects against colitogenic B. fragilis (199). While IL-17 initiates a NFκB-mediated recruitment of protumoral CXCR2+ myeloid cells (200), other mechanistic events mediated by toxigenic B. fragilis contribute to colon tumor development. In particular, digestion of the mucus layer by ETBF enabled enterotoxigenic species, namely pks+ E. coli, to adhere to colonic IEC, thus facilitating access to the toxin (201).
Finally, mucus-invasive bacterial biofilms were identified on the colon mucosa of 50% of CRC patients and approximately 13% of healthy individuals. Remarkably, biofilm-positive communities from healthy colonoscopy induced colon inflammation and tumors similarly to biofilm-positive tumor tissues in three independent mouse models of CRC (202). Bacterial genera shown to be amplified in CRC patients such as Clostridium XI, Clostridium XVIII, Erysipelotrichaceae incertae sedis, Escherichia/Shigella, Eubacterium, and Parabacteroides were increased in biofilm-positive–associated mice. These bacteria may interact with one another. Collaboration between multiple types of bacteria is likely to be a contributing factor, as suggested by the identification of ETBF and pks+ E. coli within familial adenomatous polyposis mucosal biofilms. In contrast, Bifidobacterium was depleted in mice inoculated with biofilm-positive tissues, confirming the metagenomics data of stool composition in CRC patients (162, 203). Mechanistically, biofilm-positive colon cancers have been associated with decreased E-cadherin expression, an increased release of IL-6, and the polyamine metabolites N(1),N(12)-diacetylspermine, activation of STAT3, all culminating in IEC proliferation and cell transformation (170) (Figure 4D). This work converged into the demonstration that biofilm formation plays a key carcinogenic role (202).
The composition of the ileal microbiome influenced the accumulation of T follicular helper (Tfh) cells and CD8+ T cells in proximal colon cancers (26). Cremonesi et al. showed that distinct bacteria species can directly stimulate colon cancer cells producing a chemokine array closely correlating with T cell subsets expressing the appropriate chemokine receptors, which in turn, may modulate tumor immunosurveillance and dictate the prognosis of colon malignancies. Hence, Fusobacteria and Prevotella can trigger CCL20 release through tumor cells in vitro, while Th17 infiltration in vivo was associated with “cold” (poor in tumor infiltrating lymphocytes) CRC (204, 205).
Aside from Th1 and Th17 responses, the tumor contexture can also reveal the presence of beneficial Th9 that could be associated with specific local microbes. In recent reports, IL-9 producing-Th9 cells resulted from the conversion of Th2 cells into Th9 cells endowed with pro-apoptotic tumoricidal activity and immunizing capacities (206). Moreover, positive and negative correlations between intratumoral IL-9 and abundance of Prevotella or Bacteroides spp. were reported in CRC respectively (207).
Wong et al. also demonstrated a causal relationship between the composition of CRC patients’ stools and carcinogenesis. Wong et al. fed fecal samples from patients diagnosed with CRC (versus healthy volunteers) to germ-free mice and conventional mice treated with the procarcinogenic azoxymethane. CRC-derived feces promoted polyp formation, intestinal dysplasia, epithelial proliferation and stemcellness, as well as inflammation (IL-17A, IL-22, IL-23A) associated with the colonic recruitment of Th1 and Th17 cells (208).
Most pharmacological compounds in the oncological armamentarium have the capacity to perturb the delicate triangle of fitness and integrity of the intestinal barrier, the microbiota ecosystem, and the gut immune system. Such “off target” side effects of targeted therapeutics drugs may eventually affect their anticancer efficacy and/or the safety profile. Multiple chemotherapies and immune checkpoint inhibitors have been studied in preclinical models of colon carcinoma (26, 209, 210) and pancreatic cancer (211) expanding our current understanding of mechanisms underlying response.
Oxaliplatin (OXA) is a clinically effective tumoricidal platinum salt commonly used against colon, breast, ovarian and lung carcinomas (212, 213). The initial mode of action described for OXA was that it creates DNA damage, inducing formation of intra- and interstrand DNA adducts, generated by crosslinking between activated platinum species and specific base sequences (214). However, in the absence of a functional immune system, in particular a TLR4 signaling pathway and T lymphocytes, OXA fails to mediate full blown anticancer activity in tumor bearing mice and in patients diagnosed with CRC (215, 216). Indeed, following OXA-based chemotherapy, antitumor adaptive immune responses are activated (217). This activation occurs through the induction of immunogenic cell death (ICD) of colon cancer cells due to the initiation of executioner caspases (216, 218–220) and through the subsequent release of several damage associated molecular patterns (DAMPs) including surface exposed-calreticulin (220), high mobility group box 1 protein (HMGB1) (221), CXCL10 (223), and Annexin A1 (224). Moreover, the antitumor efficacy of OXA largely depends on the presence of an intact microbiota. Disruption of murine microbiota with broad-spectrum antibiotics was shown to reduce the cytotoxicity and production of reactive oxygen species (ROS) via the NADPH oxidase NOX2 by tumor-infiltrating myeloid cells after OXA treatment (209). ROS are needed for the OXA antitumor effect. Although the genotoxic effect of platinum compounds was known to require ROS and particularly H2O2 production by tumor cells in vitro, ROS is produced by tumor-associated myeloid cells in vivo. Thus, the microbiota affects OXA early tumor genotoxicity by systemically priming tumor-associated myeloid cells for ROS production (209) (Figure 5A).
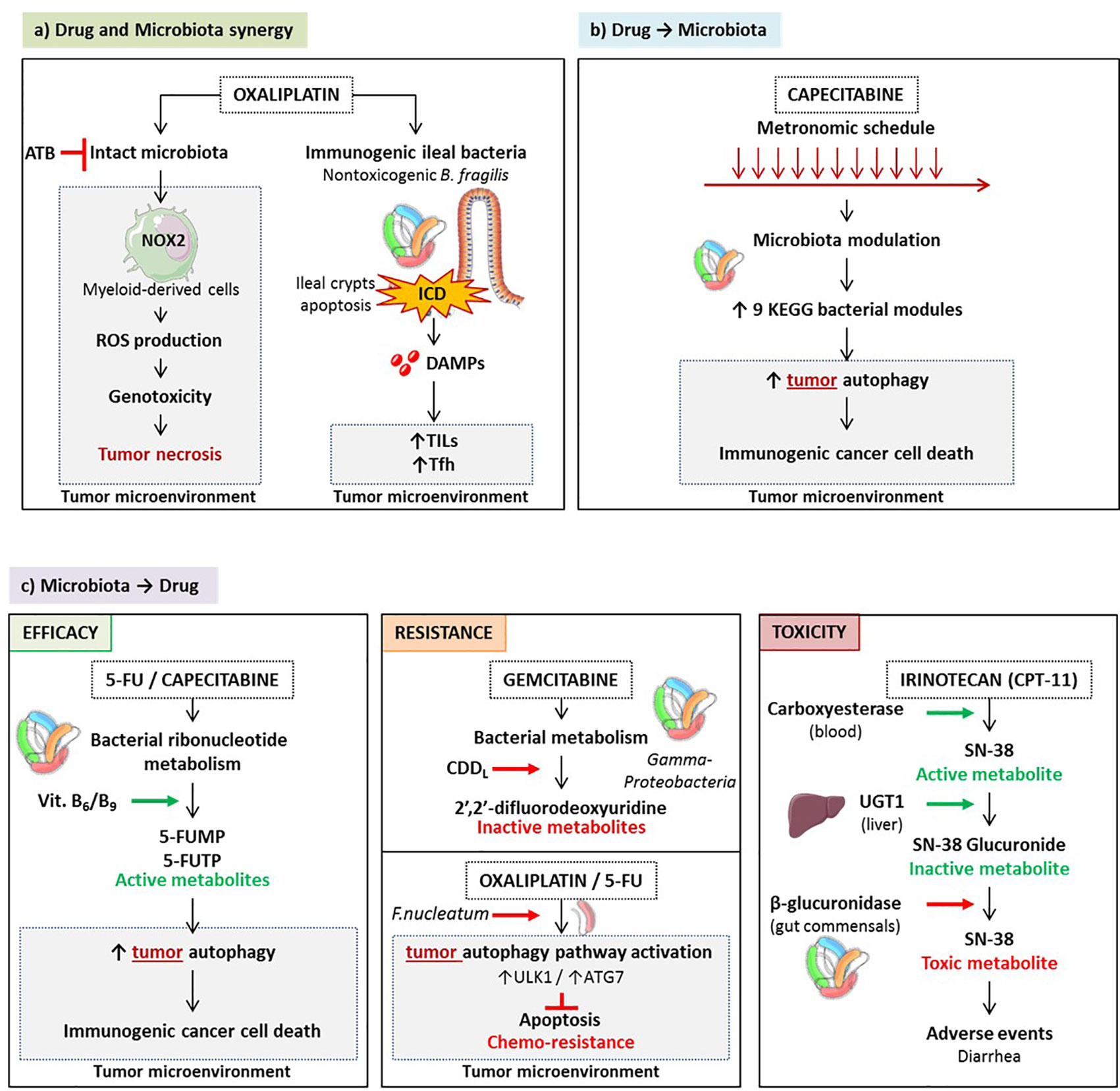
Figure 5 Pharmacological-microbiota interactions. Drugs and microbes can mutually influence each other. A synergy - exemplified by oxaliplatinum - can occur culminating in triggering the local immune system eventually controlling the tumor microenvironment. This synergy is severely compromised by antibiotics. (A) Drugs can shape gut microbiota and activate bacterial functions beneficial for the anticancer therapy. For instance, CTLA-4 blockade could enrich the ileal microbiome with distinct Bacteroides spp. that in turn fosters Th1 responses beneficial against cancer. (B) On the other hand, commensals can metabolize anticancer prodrugs, thus activating or inactivating their bioactive metabolite, sustaining or compromising the therapeutic effect. Conversely, increased recycling of bioactive compounds may be accelerated by enzymatic machineries associated with distinct microbes (such as camptothecin-11), generating severe side effects. (C).
Finally, OXA does not just induce apoptosis of tumor cells. Chemotherapy can promote apoptotic cell death in the intestinal crypts of healthy tissues, known as chemotherapy-induced mucositis (224). As many other anticancer agents, OXA compromises the integrity of the intestinal barrier which may create a dysbiosis and potentiate systemic inflammation and immunity (225–228). It has been shown that OXA is associated with cell apoptosis in ileal, but not colonic, crypts sparing the villi and the lamina propria, in tumor bearing mice and in patients diagnosed with proximal colon cancer (pCC) (26). Ileal crypt apoptosis was correlated with progression-free survival and the accumulation of TILs and Tfh in the tumor bed (26). (Figure 5A) Ileal cell apoptosis requires caspase 3 and caspase 7 executioners as well as ileal commensals. First, tumor bearing mice in which caspase 3/7 expression was ablated specifically in intestinal epithelial cells (IEC) (Casp3/7ΔIEC) failed to respond to OXA while RIPK3-deficient animals disabled for necroptosis of the IEC controlled tumor outgrowth during OXA therapy (26). Secondly, when broad spectrum antibiotics were administered along with OXA, the apoptosis of the crypts was blunted and the anticancer effects were abrogated. To consolidate the impact of patient microbiota on the efficacy of OXA, Roberti et al. utilized an avatar model (229), in which the intestines of germ free mice were colonized with human colonic content collected from pCC patients. Two weeks later, these mice were inoculated with subcutaneous MC38 and 7 days later, treated with OXA. While 66% of patient microbiota resulted in antitumor efficacy of OXA comparable to that observed in mice reared in specific pathogen-free conditions, 33% patients’ microbiota induced complete resistance to this immunogenic chemotherapy. Ileal crypt apoptosis was highly correlated with major changes in the composition of the ileal microbiome, with a dominance of Erysipelotrichaceae at the expense of Fusobacteriaceae.
Capecitabine, an oral prodrug of fluorouracil, inhibits the enzyme activity of thymidylate synthase during DNA replication (230). It has been proven effective in conjunction with OXA in metastatic colon cancers and other cytotoxicants in breast cancer and is widely used in adjuvant setting. A pioneering study explored the role of microbes in modulating the effect of 5-FU and other fluoropyrimidines on Caenorhabditis elegans and found that bacteria are key determinants of fluoropyrimidine efficacy on host metabolism (231). Microbes can boost or suppress the effects of fluoropyrimidines through metabolic drug interconversion involving bacterial vitamin B6, B9, and ribonucleotide metabolism. (Figure 5C) Also, modulations in bacterial deoxynucleotide storage amplify 5-FU-induced autophagy and cell death in host cells. A Chinese prospective study of the fecal composition of 31 females treated for HER2 negative breast cancer used 16S rRNA sequencing to analyze the variations of the gut microbiota during a maintenance chemotherapy comparing metronomic versus conventional dose of capecitabine (232). While alpha diversity was not different between the two treatment modalities, beta diversity varied significantly between the two groups, with a relative depletion of Cyanobacteria, Chloroplast, Blautia, and Streptophyta in stools of the metronomic capecitabine -treated females. Multivariate analyses of progression-free survival of all 31 patients regardless of capecitabine dosing revealed that Blautia obeum was significantly associated with clinical benefit (HR 3.4) in contrast with Slakia (HR 0.2). For the study of relative bacteria functions, the Kyoto Encyclopedia of Genes and Genomes (KEGG) modules were quantified, indicating that metronomic capecitabine tended to enrich for bacteria involved in nitrification, and putrescine, lysine/arginine/ornithine transport system. These metabolic traits are reminiscent of polyamine synthesis closely linked with the activation of the autophagic machinery mandatory for the immunogenicity of cancer cell death (233) (Figure 5B). Although concerning for breast cancer females, and underpowered, this pioneering study suggests that distinct chemotherapeutic regimen influence the composition of the gut microbiota that in turn, could impact clinical outcome (232).
Bacteria can metabolize chemotherapeutic drugs, to increase or decrease their pharmacological effect. The presence of bacteria inside human tumors may paradoxically result in drug concentrations that are lower in the tumor than in other organs. Gemcitabine is a nucleoside analog (2′,2′-difluorodeoxycytidine) used to treat patients with pancreatic, lung, breast, or bladder cancers. It has been occasionally combined with FOLFOX against colon cancer (234). Bacteria can metabolize the chemotherapeutic drug gemcitabine (2′,2′-difluorodeoxycytidine) into its inactive form, 2′,2′-difluorodeoxyuridine (211). Metabolism is dependent on the expression of a long isoform of the bacterial enzyme cytidine deaminase (CDDL), seen primarily in Gammaproteobacteria found in pancreatic ductal adenocarcinoma or colon cancers (211). In a colon cancer mouse model, the authors demonstrated that gemcitabine resistance was induced by intratumor Gammaproteobacteria, dependent on bacterial CDDL expression, and abrogated by the ciprofloxacin fluoroquinolone. Antibiotic-treated mice displayed a better antitumor response to gemcitabine than control mice (211) (Figure 5C).
Bacteria could confer drug resistance through the induction of autophagy in colorectal cancer cells, therefore interfering in the tumoricidal activity of chemotherapy. F. nucleatum is gradually increased from normal tissues to adenoma tissues and to adenocarcinoma tissues in colorectal carcinogenesis (178, 179). Moreover, the amount of F. nucleatum in CRC tissues was associated with shorter survival (187). Another independent group found that the five-year recurrence survival was substantially shorter in patients with CRC rich in F. nucleatum than in those poor in F. nucleatum (189). Multivariate regression analyses demonstrated that the amount of intratumor F. nucleatum was an independent predictor of CRC aggressiveness and recurrence post-chemotherapy with significant hazard ratios for predicting clinical outcome (189). ULK1/ATG7 -dependent autophagy contributed to F. nucleatum-mediated CRC resistance to OXA and 5-FU regimens (189). F.nucleatum induced- genomic loss of miR-18a* and miR-4802 depended upon the TLR4/MYD88 signaling pathway (189) (Figure 5C).
Camptothecin, a potent antineoplastic compound, poisons the catalytic cycle of human topoisomerase I. Camptothecin exhibited marked toxicity and poor bioavailability. Although its derivatives topotecan and CPT-11 (also called irinotecan) are now in clinical use (234), they still elicit pronounced side effects that limit efficacy. CPT-11 is one of the three commonly used chemotherapeutic agents for CRC (236). It is a prodrug that gives rise to the active metabolite SN-38 in vivo (237). Intravenously administered CPT-11 is activated by carboxylesterases to SN-38, the antineoplastic topoisomerase I poison. Liver SN-38 is inactivated via glucuronidation to SN-38G by UDP-glucuronosyltransferases and spread to the GI tract. β-Glucuronidases in the gut commensals remove the glucuronide as a carbon source, and active SN-38 in the intestinal lumen generates dose-limiting diarrhea. High throughput screening of inhibitors selectively targeting the enzyme in living bacteria, and sparing non relevant bacteria and mammalian cells led to the identification of promising compounds that could reach a better therapeutic index of camptothecin-11 (238) (Figure 5C).
DNA mismatch repair-deficient or microsatellite-instable tumors particularly benefit from the immune checkpoint blockade (85). Accumulating evidence points to the critical role of the gut microbiota in the efficacy of ICIs. First, antibiotics taken within the month preceding start of anti-CTLA-4 or anti-PD-1 antibodies, markedly attenuated the clinical benefit, reducing both progression-free and overall survival across many metastatic and stage III malignancies amenable to therapies based on immune checkpoint inhibition (239, 240). Second, fecal microbial transplantation of stools from patients prone to respond to ICI or doomed to fail first or second line ICI-based therapy confer sensitivity or resistance to tumor bearing mice treated with anti-PD-1 antibodies respectively (241). Third, shot gun metagenomics sequencing of patients’ stools at diagnosis may help predicting primary resistance to PD-1 blockade in metastatic melanoma (242), kidney (241) and lung cancers (243). Fourth, oral compensation of antibiotics-treated tumor bearers or dysbiotic hosts with monoclonal anticancer probiotics (such as Akkermansia muciniphila (243), or Bifidobacterium pseudolongum (244) restored full blown immunostimulatory activity of anti-PD-1 antibodies. (Figure 6). Conversely, ICIs also have an impact on the gut composition. Vetizou et al. reported that anti-CTLA-4 antibody modulated the ileal bacteria composition, increasing the relative abundance in Bacteroides spp. (namely B. fragilis) and Burkolderiaceae family members, involved in the IL-12-dependent priming of Th1 cells as well as the immunostimulatory and anti-cancer effects of CTLA-4 blockade (210). In line with these preclinical data, six months of therapy with nivolumab ameliorated the alpha diversity of metastatic kidney cancer bearing patients in those individuals benefiting from the antibodies (241). Such studies are awaited in MSIhigh CRC amenable to PD-1 blockade. Finally, memory Th1 and Tc1 immune responses directed against immunogenic bacteria E. hirae, A. muciniphila, and B. fragilis dictated progression free survival in cancer patients treated with anti-PD-1 or anti-CTLA-4 Abs (210, 243).
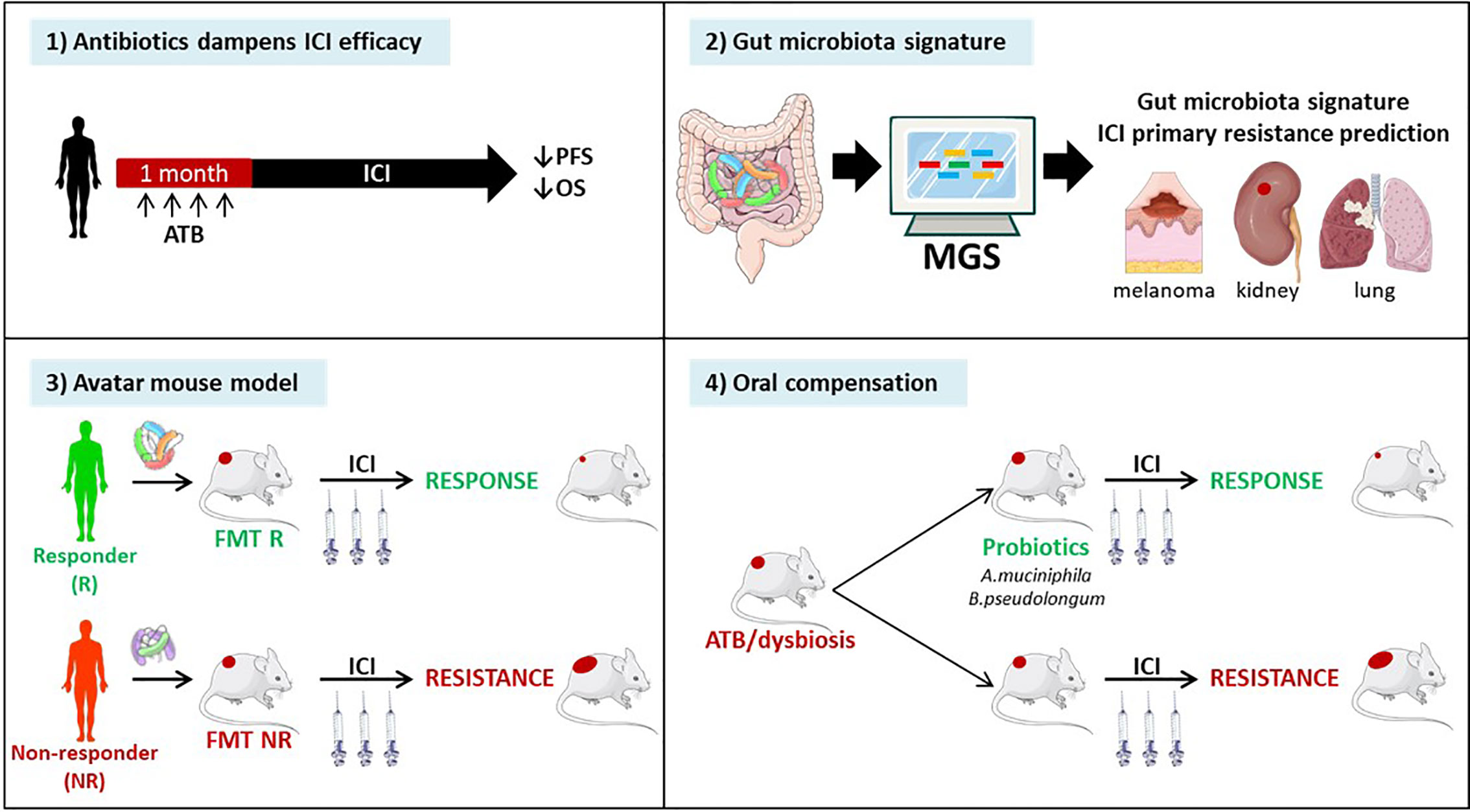
Figure 6 Evidence pointing to a key role of the gut microbiota in immune checkpoint inhibitors (ICI)-mediated anticancer effects. Recently, pre- and clinical studies have shown the clinical significance of the composition of the gut microbiota in ICI efficacy. 1) Retrospective and prospective studies have highlighted the detrimental effect of antibiotics administration within the month preceding the start of immunotherapy in multiple stage III and IV cancers. 2) Metagenomic analysis (MGS) of patients’ stool composition predicts primary resistance to ICI in 1L and 2L melanoma, kidney and lung cancers. 3) Avatar mouse models where the intestines of tumor bearing rodents are colonized by human stools from cancer patients at diagnosis allowed to predict the response to immunotherapy in patients. 4) The identification of beneficial bacteria, such as Akkermansia muciniphila or Bifidobacterium pseudolongum for immunotherapy efficacy opens up prospects to restore cancer-associated dysbiosis in patients.
As stated above, the reasons why DNA mismatch repair-proficient or microsatellite-stable CRC, endowed with tumor infiltrating lymphocytes at the primary or metastatic stage and treated with a cytotoxic compound mediating ICD, fail to benefit from immune checkpoint blockade are enigmatic (85, 100). Indeed, FOLFOX could induce T-bet-dependent PD-1 expressing CD8+ T cell infiltration and IFN-γ-mediated PD-L1 upregulation in mouse models of colon cancers, and in CRC patients (245, 246).
The molecular and cellular cues underlying this delicate equilibrium between tolerance (vis-à-vis of self and food antigens) and immunity (against pathogens and tumors) in the intestinal barrier remain an immunological challenge.
Recent work has highlighted how ileal bacteria may contribute to break tolerance to self-antigens shared between ileal crypts and cancer stem cells. Roberti et al. reported that the immunogenicity of ileal epithelial cell death mediated by OXA to treat a proximal CRC relied on two biological features: (1) antigenicity provided by the caspase 3/7-dependent apoptosis of crypt-derived IEC and (2) the adjuvanticity of selected ileal bacterial families or species (Erysipelotrichaceae, B. fragilis). These features cooperated to elicit Tfh immune responses and antibody producing cells protective against tumor progression. The serum IgG levels were increased by immunogenic bacteria but not by tolerogenic bacteria and could be associated with IgG responses directed toward bacteria or tumor cells (Figure 7A). A vaccine composed of OXA-exposed dying ileal IEC was more effective to immunize naive hosts against a lethal dose of colon cancer cells (CT26 and MC38) when harvested from SPF wild type mice than TLR2/4 or MYD88 KO mice or germ-free animals. These data suggest that self-antigens derived from ileal crypts could elicit an immune response in the presence of microbial adjuvants. Using 16S rRNA gene sequencing of patients’ ilea and culturomics, the authors concluded that distinct bacteria residing in ilea may convey the immunogenicity of apoptotic cell death of the crypt, shifting the antitumor immune response from Th17 (observed in progressive tumors) to Tfh cells accumulating in tumor draining lymph nodes (tdLN) of mice responding to OXA. Indeed, Bacteroides fragilis, or Erysipelatoclostridium ramosum could potentially restore responsiveness to chemotherapy in germ free mice or ATB-treated SPF animals or could confer immunogenicity to sterile ileal apoptosis. In contrast, Fusobacterium nucleatum or Prevotella clara failed to do so. In fact, OXA could mobilize migratory conventional type 1 DCs (cDC1) (CD103+CD11b- DC) from the ileal lamina propria to the mLN, and contribute to IL-1β and IL-12-dependent priming of CXCR5+Bcl6+ PD-1+ CD4+ Tfh cells and systemic IgG responses. In the absence of Tfh or B cells, the vaccine composed of OXA-exposed crypt ileal enterocytes failed to induce long term memory autoreactive CD4+ and CD8+ T cell responses protective against colon carcinoma. In Batf3 gene deficient animals defective in cDC1, Tfh were not primed during OXA administration. Moreover, cDC1 exposed to immunogenic ileal-residing bacteria (B. fragilis or C. ramosum) could produce IL-1β and IL-12p70 while they could not produce IL-12p70 when stimulated with F. nucleatum or P. clara. The authors postulated that PD-1 expressing Tfh induced by OXA-mediated ileal apoptosis could be bolstered by anti-PD-1 Abs and carried out to modulate the efficacy of a combination of OXA with this immune checkpoint inhibitor with a concomitant oral gavage with immunogenic (B. fragilis, C. ramosum) versus tolerogenic (F. nucleatum, P. clara) bacteria in MSS and MSI mouse colon cancers. They could bring evidence that the ileal residence and/or colonization of bacteria fostered (as for B. fragilis, C. ramosum) or blunted (as for F. nucleatum, P. clara) the immunostimulatory anticancer effects of the combinatorial regimen. These additive effects of both therapeutic modalities were accompanied by a rise in IgG2b serum levels.
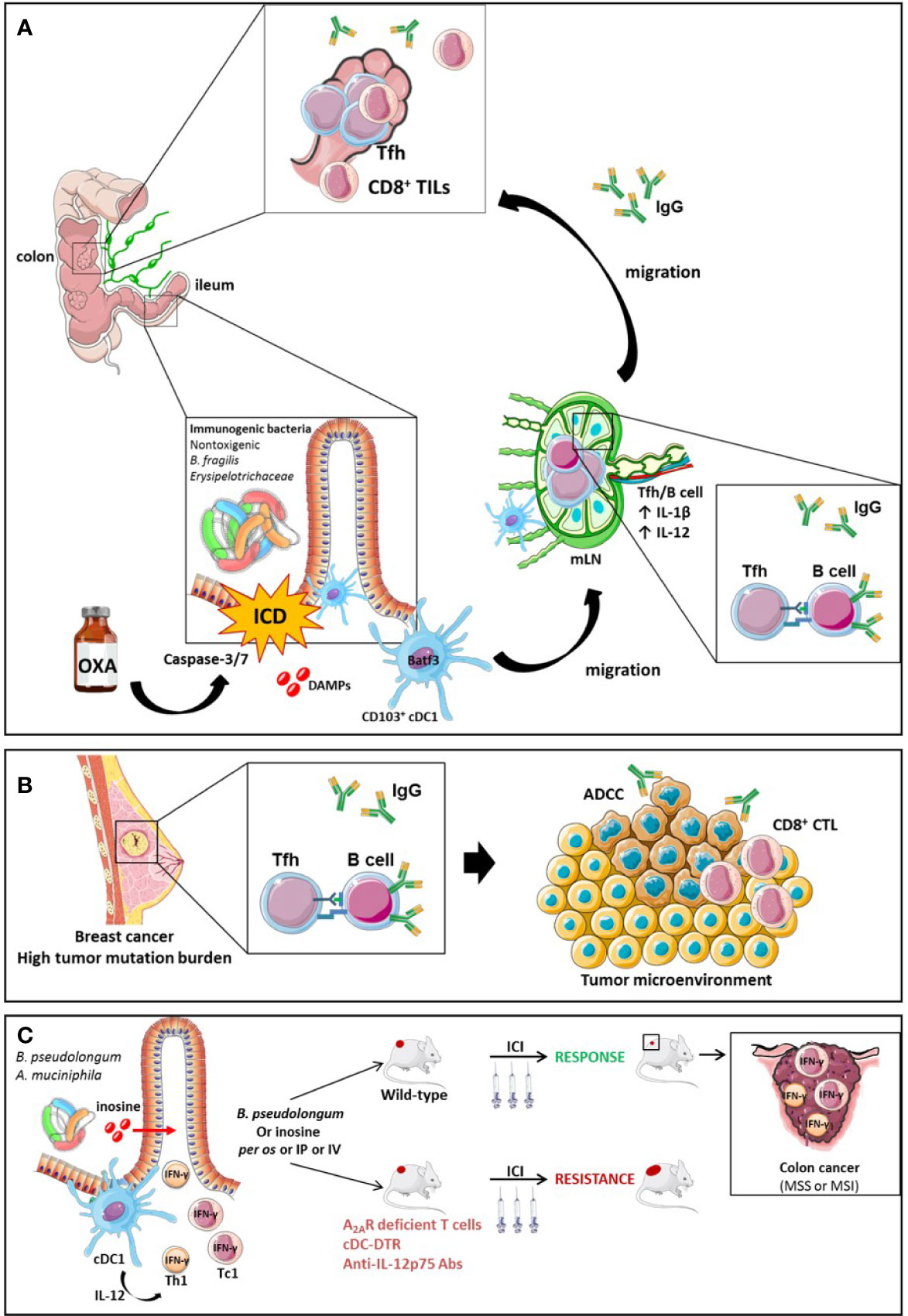
Figure 7 The role of Tfh and B cell orchestration for the efficacy of ICI in TMB high breast tumors or MSS CRC endowed with an immunogenic ileal microbiome. The efficacy of OXA in mouse model of CRC is driven by ileal features. On one hand, OXA induces apoptosis and the release of DAMPs following caspase-3/7-dependent ileal epithelial cell death of the crypts. The bacterial composition is critical to turn this cell death into immunogenic cell demise instead of a tolerogenic cell death. The ICD triggers the migration of CD103+ conventional dendritic cells (BATF3+ cDC1) to the mesenteric lymph node (mLN), which prime Tfh cells in an IL-1β and IL-12 dependent manner. Next, a crosstalk between Tfh and B cells occurs, leading to a systemic IgG2b response and an accumulation of Tfh and TILs into MSS or MSI colon tumors. The IgG2b responses are suspected to be directed either against bacteria or tumor cells. (A) Activation mechanisms between B cells and Tfh result in the generation of antibodies that elicit antibody-dependent cellular cytotoxicity (ADCC) towards tumor-associated antigens in mouse model of breast cancers endowed with a high tumor mutational burden (TMB) (B). Small intestine immunogenic species (B.pseudolongum or A.muciniphila) secrete the inosine, a metabolite activating Th1 and Tc1 cells through the A2A. Conjointly with IL-12 co-stimulatory signal produced by cDCs in presence of antigens, inosine-stimulated Th1 and Tc1 secrete IFN-γ and increase ICIs efficacy in MSS or MSI mouse models of colon cancer (C).
Initially, ileal bacteria participate in ileal crypt apoptosis but not in the release of DAMPs. Indeed, in the presence of antibiotics, OXA-induced ileal cell demise was significantly reduced. Interestingly, OXA alone could promote the release of ATP and HMGB1, two hallmarks of ICD, that were not mandatory for the immunizing capacities of dying IEC, in contrast to live bacteria or pathogen associated molecular patterns (26). This is in contrast with a report utilizing gram negative bacteria ghosts injected intraperitoneally with OXA that could turn on ICD in the peritoneal cavity and elicit potent NKT and CD8+ T cell responses associated with regression of peritoneal carcinogenesis (247). These observations emphasize that the tumor macroenvironment and organ may critically influence the requirements for efficient priming of a T cell response.
Secondly, it is intriguing that ileum may be more appropriate than the colon mucosae to elicit Tfh immunity in the mLN. As outlined above (248, 249), the immune cell types and microbiota composition are highly compartmentalized (250, 251), lymphatics draining the small and large intestines differ and ileal crypts can undergo apoptotic cell death after chemotherapy while colonic crypts failed to do so (26).
Third, breaking tolerance to self-tissues in colon cancer has been previously achieved (252). The authors showed the capacity of Stat3-deficient IEC to promote adaptive immunity in spontaneous models of carcinogenesis (252). Mitophagy, the specific degradation process of damaged or aged mitochondria, triggered lysosomal membrane permeabilization which enhanced MHC-I expression in IEC that in turn allowed cross-dressing of DC with IEC derived-MHC-I/peptide complexes. This pathway could also come into action upon OXA treatment, knowing that STAT3 deficiency in epithelial cancer cells amplified an OXA-elicited type 1 IFN response and triggered an immunogenic cell death pattern, ameliorating antitumor T cell responses (253).
Fourth, our findings raise the theoretical possibility of a self-reactive T cell-dependent immunity that spares healthy tissues to specifically combat cancer (26). Uncoupling antitumor effects from ileitis or colitis remains an open conundrum in cancer chemotherapy and immunotherapy. We postulate that Lgr5 negative crypt-derived cells devoid of stemcellness share common antigens with pCC that elicit therapeutically relevant immune responses, at least in the context of chemotherapy with OXA. As a possibility, the fragments of apoptotic ileal IEC (that contain colon cancer-crossreactive antigens) and immunogenic commensals (with their TLR ligands) could end up in the same phagosomes of antigen-presenting cells in the GALT, thus providing an opportunity for both self-and non-self-peptides to be concomitantly loaded into MHC class II molecules (254). Actually, interactions between cytokine-producing Th cells and MHC class II+ Lgr5+ intestinal stem cells were shown to be critical for the self-renewal and/or differentiation of this latter cell type (255).
Finally, although Tfh and Tfh-like populations have been associated with poor prognosis in lymphoma (256, 257) and ICI-treated melanoma (258), numerous pre-clinical and clinical studies revealed their positive impact on the outcome of many solid tumors (259), including breast cancer (260, 261), non-small-cell lung cancer (NSCLC) (262), ovarian cancer (263), and colorectal cancer (25, 26). Mechanisms underlying the protective role of Tfh cells are not fully understood, although some evidence point to cooperation with CD8+ T cells and IL-21 (25, 43). CXCL13-producing Tfh act as “organizers” of germinal centers in secondary and tertiary lymphoid organs, sustaining local humoral immunity by attracting antibody-secreting B cells (264), which control tumor progression (260, 265–268). ICIs are used to reinvigorate cellular immunity and accumulating evidence show a direct impact of ICIs on humoral response. Indeed, anti-PD-1 antibodies increase germinal center formation, CD4+ T infiltrates and terminal B cell differentiation (269). The cooperation between Tfh and B cells appears to be associated with a favorable clinical outcome in colorectal cancer (25) and in breast cancer, in both human (260, 270) and murine models of tumors harboring high tumor mutational burden (30). The humoral response in CRC may be directed towards several tumor-associated antigens, such as Carcinoembryonic antigen (CEA) (271, 272), Epidermal Growth Factor Receptor (EGFR) (273), Human Epidermal Growth Factor Receptor 2 (HER2 or ErbB2) (274), MUC5AC (275), and ribosomal P proteins (276) and facilitate antibody-dependent cellular cytotoxicity (30) (Figure 7B).
Intratumoral terminally differentiated memory B cells and/or plasma cells can predict prolonged survival (49, 51, 52). Tumor-infiltrating B cells have been associated with positive clinical outcome following therapy with ICIs in melanoma (40, 277), with (41) or without metastasis (278), in sarcoma (39) and in renal cell carcinoma (40). Of note, B cells can exert direct tumoricidal activity against cancer cells, in an antigen-specific and Fas ligand-dependent manner (279).
Intratumoral bacterial colonization and residency, whether localized within myeloid cells or in tumor cells have recently been described to modulate local metabolism, change tumor cell biology or interfere in the drug catabolism (211, 280). IgG responses directed against intratumoral bacteria may contribute to preventing tumor colonization and/or tumor killing, in both mouse models and patients (30, 281, 282).
Collectively, these studies highlight the potential clinical significance of B cells, antibody secreting cells, and antibodies as well as Tfh in dictating the efficacy of the combination of FOLFOX+ICIs.
Moreover, Mager et al. have recently shown that metabolites of small intestine-derived immunogenic species, such as Bifidobacterium pseudolongum, can act as co-stimulatory molecules on the TCR signaling of Th1 and Tc1 cells. Indeed, B. pseudolongum produces the inosine, a metabolite that acts through the adenosine A2A receptor expressed on TILs and potentiates the effect of PD-1 or CTLA-4 antibodies in mice bearing transplantable MC38 colon cancer in conjunction with co-stimulatory signal. This effect was lost in mice harboring A2A receptor-deficient T cells. The anti-tumor effect of the inosine to reduce tumor growth is still dependent on the presence of a microbiota since in germ-free mice the administration of inosine alone did not ameliorated ICIs efficacy while it did in the presence of a complex microbiota. Additionally, B. pseudolongum modulated the intrinsic local immunity as it increased the T-bet expression among CD4+ T cells in the GALT and the mLN but not in the periphery. On the other hand, B. pseudolongum induced systemic immune modulation when associated with ICIs leading to the activation of effector cells and the secretion of IFN-γ by CD4+ and CD8+ T cells. The systemic effect can be explained by the fact that anti-CTLA-4 alters the gut barrier integrity increasing the systemic translocation of metabolites from the gut. The activation of T cells requires the presence of an IL-12 co-stimulatory signal provided by cDCs. Thus, in the absence of cDCs (cDC-DRT mouse model), B. pseudolongum associated with anti-CTLA-4 failed to induce IFN-γ-T cell producers and anti-tumoral effect. Furthermore, anti-IL-12p75 neutralization dramatically dampened the effect of anti-CTLA-4 combined with immunogenic species in a genetically modified mouse model presenting dMMR intestinal tumors mismatch repair (Msh2LoxP/LoxPVillin-Cre tumors) (Figure 7C) (244).
Altogether, these studies point to the central role of the ileal, but not the colonic, epithelial cells and its natural microbial ecosystem, in mobilizing an efficient immune response against self-antigens expressed by colon cancers, presumably tumor stem cells. The solution to the paradox of colon cancers-which has the propensity to elicit and attract CD3+/CD8+ T cells (so called “high immunoscore”) but remains resistant to immunotherapy when devoid of mutated neoantigen-relies on two pillars, i) ileal apoptosis through activation of the executioner caspases, and ii) a proper ileal microbiome composition balancing immunogenic and tolerogenic commensals. These two conditions can be achieved by cytotoxic regimen inducing immunogenic cell death of tumor and/or crypt stem cells and by doing so, favor the dominance of immunogenic bacteria. Such suitable therapeutic regimens include FOLFOX (5-FU/leucovorin/OXA), FOLFIRI, or any type of antibody-dependent cytotoxicity through various payloads releasing compounds endowed with ICD properties (283). The ensuing immune response is composed of humoral (Ab secreting cell and B cell-based) and cellular (Tfh and memory CD8+ T cell-based) based-immune responses towards crypt -derived- self antigens shared between the small intestine and tumor cells. These premises allowed the demonstration of synergistic effects between ICD-mediating chemotherapy and PD-1 blockade in pMMR or MSS mouse colon tumors.
Despite tremendous advances in the classification of CRC based on genetics, transcriptomics and immunometrics, few patients benefit from combinatorial regimens that combine ICD-mediating cytotoxicants and ICIs or targeted therapies. Increasing knowledge on the macroenvironment of colon carcinoma, mainly on the connections between the local microbial ecosystem and the systemic immune tonus, has shed new light on the role of intratumoral and ileal bacteria in modulating the immune contexture as well as tumor signaling pathways. This review gives novel insights into the development of potential biomarkers of response (ileobiome, ileal immune patterns) or novel therapies based on live bio-therapeutics consisting of appropriate mixtures of immunogenic commensals or phages (284) to complement current combinatorial regimen. Moreover, attention should be paid to new actors playing a role in colon cancer immunosurveillance, including antibodies to self-antigens, B cells, and Tfh. Moreover, technological advanced such as single cell sorting and sequencing will also enable a comprehensive characterization of these actors’ functions and specificities.
The figures were conceived using https://smart.servier.com/.
MF, SY, MP, AH, AC, MR, and LZ contributed to the writing of this manuscript. MF and MR conceived the figures using https://smart.servier.com/. All authors contributed to the article and approved the submitted version.
LZ was supported by the Ligue contre le Cancer (équipe labelisée); Association pour la recherche sur le cancer (ARC); Agence Nationale de la Recherche (ANR) francogermanique ANR-19-CE15-0029, Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); Institut National du Cancer (INCa); Inserm (HTE); the LabEx Immuno-Oncology; the RHU Torino Lumière (ANR-16-RHUS-0008); H2020 ONCOBIOME, the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); FHU CARE, Dassault, and Badinter Philantropia, and the Paris Alliance of Cancer Research Institutes (PACRI). MF was supported by AP-HP. MP was supported by H2020 ONCOBIOME. SY was supported by Fondation Philanthropia, Gustave Roussy. AC is supported by the France Fulbright Comission and the Cancer Research and Prevention Insitute of Texas (CPRIT), Research Training Program (RP170067).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
5-FU, 5-fluorouracil; Ab, antibody; ADCC, antibody-dependent cellular cytotoxicity; ATB, antibiotics; Casp, caspase; cDC, conventional dendritic cell; CDDL, enzyme cytidine deaminase; CIMP, CpG island methylator phenotype; CIN, chromosomal instability; CMS, consensus molecular subtype; CPT-11, irinotecan; CRC, colorectal cancer; CTL, cytotoxic T lymphocyte; DAMP, danger associated molecular pattern; d/pMMR, deficient/proficient DNA mismatch repair; EMT, epithelial-mesenchymal transition; ETBF, enterotoxigenic Bacteroides fragilis; FMT, fecal microbial transplantation; GALT, gut-associated lymphoid tissues; GF, germ free; GI, gastrointestinal; HMGB1, high mobility group box 1 protein; I.S., immune subtypes; IS, immunoscore; ICD, immunogenic cell death; ICI, immune checkpoint inhibitors, IEC, intestinal epithelial cells; KEGG, Kyoto Encyclopedia of Genes and Genomes; KO, knock-out; LP, Lamina Propria; MDSC, myeloid-derived suppressor cell; MGS, metagenomic shotgun; mLN, mesenteric lymph node; MSI/MSS, microsatellite instability/stability; NK, natural killer; NSCLC, non-small lung cancer; NTBF, non-toxigenic Bacteroides fragilis; OS, overall survival; OXA, oxaliplatin; pCC, proximal colon adenocarcinoma; pDC, plasmacytoid dendritic cell; PFS, progression-free survival; ROS, reactive oxygen species; SCFA, short-chain fatty acid; SPF, specific pathogen free; Tc1, T cytotoxic type 1; TCGA, The Cancer Genome Atlas; tdLN, tumor draining lymph node; TFC, T follicular cell; Tfh, T follicular helper cell; Th, T helper; TILs, tumor-infiltrating lymphocytes; TLR, Toll-like receptor; TMB, tumor mutational burden; TME, tumor microenvironment; TNM, T-primary tumor, N-regional lymph nodes, M-distant metastases; Treg, T regulatory cell; WT, wild type.
1. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut (2017) 66:683–91. doi: 10.1136/gutjnl-2015-310912
2. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet (2019) 394:1467–80. doi: 10.1016/S0140-6736(19)32319-0
3. Rustgi AK. The genetics of hereditary colon cancer. Genes Dev (2007) 21:2525–38. doi: 10.1101/gad.1593107
4. Dashti SG, Buchanan DD, Jayasekara H, Ouakrim DA, Clendenning M, Rosty C, et al. Alcohol Consumption and the Risk of Colorectal Cancer for Mismatch Repair Gene Mutation Carriers. Cancer Epidemiol Prev Biomark (2017) 26:366–75. doi: 10.1158/1055-9965.EPI-16-0496
5. Murphy N, Ward HA, Jenab M, Rothwell JA, Boutron-Ruault M-C, Carbonnel F, et al. Heterogeneity of Colorectal Cancer Risk Factors by Anatomical Subsite in 10 European Countries: A Multinational Cohort Study. Clin Gastroenterol Hepatol (2019) 17:1323–31.e6. doi: 10.1016/j.cgh.2018.07.030
6. Chan AT, Giovannucci EL. Primary Prevention of Colorectal Cancer. Gastroenterology (2010) 138:2029–2043.e10. doi: 10.1053/j.gastro.2010.01.057
7. Dahm CC, Keogh RH, Spencer EA, Greenwood DC, Key TJ, Fentiman IS, et al. Dietary fiber and colorectal cancer risk: a nested case-control study using food diaries. J Natl Cancer Inst (2010) 102:614–26. doi: 10.1093/jnci/djq092
8. Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin (2017) 67:93–9. doi: 10.3322/caac.21388
9. Weiser MR. AJCC 8th Edition: Colorectal Cancer. Ann Surg Oncol (2018) 25:1454–5. doi: 10.1245/s10434-018-6462-1
10. Kroemer G, Galluzzi L, Zitvogel L, Fridman WH. Colorectal cancer: the first neoplasia found to be under immunosurveillance and the last one to respond to immunotherapy? OncoImmunology (2015) 4:e1058597. doi: 10.1080/2162402X.2015.1058597
11. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell (1990) 61:759–67. doi: 10.1016/0092-8674(90)90186-i
12. Advani SM, Advani PS, Brown DW, DeSantis SM, Korphaisarn K, VonVille HM, et al. Global differences in the prevalence of the CpG island methylator phenotype of colorectal cancer. BMC Cancer (2019) 19:964. doi: 10.1186/s12885-019-6144-9
13. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology (2010) 138:2073–2087.e3. doi: 10.1053/j.gastro.2009.12.064
14. Gatalica Z, Vranic S, Xiu J, Swensen J, Reddy S. High microsatellite instability (MSI-H) colorectal carcinoma: a brief review of predictive biomarkers in the era of personalized medicine. Fam Cancer (2016) 15:405–12. doi: 10.1007/s10689-016-9884-6
15. Kloor M, Doeberitz M von K. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer (2016) 2:121–33. doi: 10.1016/j.trecan.2016.02.004
16. Nielsen MM, Witherden DA, Havran WL. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol (2017) 17:733–45. doi: 10.1038/nri.2017.101
17. Sun X, Cai Y, Fleming C, Tong Z, Wang Z, Ding C, et al. Innate γδT17 cells play a protective role in DSS-induced colitis via recruitment of Gr-1+CD11b+ myeloid suppressor cells. Oncoimmunology (2017) 6:e1313369. doi: 10.1080/2162402X.2017.1313369
18. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity (2014) 40:785–800. doi: 10.1016/j.immuni.2014.03.013
19. Toor SM, Syed Khaja AS, El Salhat H, Bekdache O, Kanbar J, Jaloudi M, et al. Increased levels of circulating and tumor-infiltrating granulocytic myeloid cells in colorectal cancer patients. Front Immunol (2016) 7:560. doi: 10.3389/fimmu.2016.00560
20. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and Tumor-Infiltrating Myeloid-Derived Suppressor Cells in Patients with Colorectal Carcinoma. PloS One (2013) 8:e57114. doi: 10.1371/journal.pone.0057114
21. Wang D, Yu W, Lian J, Wu Q, Liu S, Yang L, et al. Th17 cells inhibit CD8+ T cell migration by systematically downregulating CXCR3 expression via IL-17A/STAT3 in advanced-stage colorectal cancer patients. J Hematol Oncol (2020) 13:68. doi: 10.1186/s13045-020-00897-z
22. Chen J, Ye X, Pitmon E, Lu M, Wan J, Jellison ER, et al. IL-17 inhibits CXCL9/10-mediated recruitment of CD8+ cytotoxic T cells and regulatory T cells to colorectal tumors. J Immunother Cancer (2019) 7:324. doi: 10.1186/s40425-019-0757-z
23. Perez LG, Kempski J, McGee HM, Pelzcar P, Agalioti T, Giannou A, et al. TGF-β signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat Commun (2020) 11:2608. doi: 10.1038/s41467-020-16363-w
24. Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res (2011) 71:1263–71. doi: 10.1158/0008-5472.CAN-10-2907
25. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity (2013) 39:782–95. doi: 10.1016/j.immuni.2013.10.003
26. Roberti MP, Yonekura S, Duong CPM, Picard M, Ferrere G, Tidjani Alou M, et al. Chemotherapy-induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nat Med (2020) 26:919–31. doi: 10.1038/s41591-020-0882-8
27. Edin S, Kaprio T, Hagström J, Larsson P, Mustonen H, Böckelman C, et al. The Prognostic Importance of CD20+ B lymphocytes in Colorectal Cancer and the Relation to Other Immune Cell subsets. Sci Rep (2019) 9:19997. doi: 10.1038/s41598-019-56441-8
28. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science (2006) 313:1960–4. doi: 10.1126/science.1129139
29. Galon J, Fridman W-H, Pagès F. The adaptive immunologic microenvironment in colorectal cancer: a novel perspective. Cancer Res (2007) 67:1883–6. doi: 10.1158/0008-5472.CAN-06-4806
30. Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, et al. B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell (2019) 179:1191–1206.e21. doi: 10.1016/j.cell.2019.10.028
31. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell (2009) 16:91–102. doi: 10.1016/j.ccr.2009.06.018
32. Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell (2010) 17:121–34. doi: 10.1016/j.ccr.2009.12.019
33. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer (2017) 17:457–74. doi: 10.1038/nrc.2017.51
34. Kammertoens T, Qin Z, Briesemeister D, Bendelac A, Blankenstein T. B-cells and IL-4 promote methylcholanthrene-induced carcinogenesis but there is no evidence for a role of T/NKT-cells and their effector molecules (Fas-ligand, TNF-α, perforin). Int J Cancer (2012) 131:1499–508. doi: 10.1002/ijc.27411
35. Liu Z, Zhou Q, Wang Z, Zhang H, Zeng H, Huang Q, et al. Intratumoral TIGIT+ CD8+ T-cell infiltration determines poor prognosis and immune evasion in patients with muscle-invasive bladder cancer. J Immunother Cancer (2020) 8:e000978. doi: 10.1136/jitc-2020-000978
36. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
37. Thibult M-L, Mamessier E, Gertner-Dardenne J, Pastor S, Just-Landi S, Xerri L, et al. PD-1 is a novel regulator of human B-cell activation. Int Immunol (2013) 25:129–37. doi: 10.1093/intimm/dxs098
38. Wang X, Wang G, Wang Z, Liu B, Han N, Li J, et al. PD-1-expressing B cells suppress CD4+ and CD8+ T cells via PD-1/PD-L1-dependent pathway. Mol Immunol (2019) 109:20–6. doi: 10.1016/j.molimm.2019.02.009
39. Petitprez F, Reyniès A, Keung E, Chen W-W, Sun C-M, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature (2020) 577:1–5. doi: 10.1038/s41586-019-1906-8
40. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
41. Cabrita R, Lauss M, Sanna A, Donia M, Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature (2020) 577:1–5. doi: 10.1038/s41586-019-1914-8
42. Jenh C-H, Cox MA, Hipkin W, Lu T, Pugliese-Sivo C, Gonsiorek W, et al. HUMAN B CELL-ATTRACTING CHEMOKINE 1 (BCA-1; CXCL13) IS AN AGONIST FOR THE HUMAN CXCR3 RECEPTOR. Cytokine (2001) 15:113–21. doi: 10.1006/cyto.2001.0923
43. Shi W, Dong L, Sun Q, Ding H, Meng J, Dai G. Follicular helper T cells promote the effector functions of CD8+ T cells via the provision of IL-21, which is downregulated due to PD-1/PD-L1-mediated suppression in colorectal cancer. Exp Cell Res (2018) 372:35–42. doi: 10.1016/j.yexcr.2018.09.006
44. Gao W, Zhou J, Ji B. Evidence of Interleukin 21 Reduction in Osteosarcoma Patients Due to PD-1/PD-L1-Mediated Suppression of Follicular Helper T Cell Functionality. DNA Cell Biol (2017) 36:794–800. doi: 10.1089/dna.2017.3669
45. Bodogai M, Lee Chang C, Wejksza K, Lai J, Merino M, Wersto RP, et al. Anti-CD20 antibody promotes cancer escape via enrichment of tumor-evoked regulatory B cells expressing low levels of CD20 and CD137L. Cancer Res (2013) 73:2127–38. doi: 10.1158/0008-5472.CAN-12-4184
46. Mao H, Pan F, Wu Z, Wang Z, Zhou Y, Zhang P, et al. Colorectal tumors are enriched with regulatory plasmablasts with capacity in suppressing T cell inflammation. Int Immunopharmacol (2017) 49:95–101. doi: 10.1016/j.intimp.2017.05.018
47. Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature (2015) 521:94–8. doi: 10.1038/nature14395
48. Shalapour S, Lin X-J, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature (2017) 551:340–5. doi: 10.1038/nature24302
49. Shimabukuro-Vornhagen A, Schlößer HA, Gryschok L, Malcher J, Wennhold K, Garcia-Marquez M, et al. Characterization of tumor-associated B-cell subsets in patients with colorectal cancer. Oncotarget (2014) 5:4651–64. doi: 10.18632/oncotarget.1701
50. Jifu E, Yan F, Kang Z, Zhu L, Xing J, Yu E. CD8+CXCR5+ T cells in tumor-draining lymph nodes are highly activated and predict better prognosis in colorectal cancer. Hum Immunol (2018) 79:446–52. doi: 10.1016/j.humimm.2018.03.003
51. Lohr M, Edlund K, Botling J, Hammad S, Hellwig B, Othman A, et al. The prognostic relevance of tumour-infiltrating plasma cells and immunoglobulin kappa C indicates an important role of the humoral immune response in non-small cell lung cancer. Cancer Lett (2013) 333:222–8. doi: 10.1016/j.canlet.2013.01.036
52. Wouters MCA, Nelson BH. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin Cancer Res (2018) 24:6125–35. doi: 10.1158/1078-0432.CCR-18-1481
53. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol (2011) 29:610–8. doi: 10.1200/JCO.2010.30.5425
54. Pagès F, Mlecnik B, Marliot F, Bindea G, Ou F-S, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet Lond Engl (2018) 391:2128–39. doi: 10.1016/S0140-6736(18)30789-X
55. Pagès F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol (2009) 27:5944–51. doi: 10.1200/JCO.2008.19.6147
56. Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci Transl Med (2014) 6:228ra37. doi: 10.1126/scitranslmed.3007240
57. Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med (2012) 10:205. doi: 10.1186/1479-5876-10-205
58. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the “Immunoscore” in the classification of malignant tumours. J Pathol (2014) 232:199–209. doi: 10.1002/path.4287
59. Berghoff AS, Fuchs E, Ricken G, Mlecnik B, Bindea G, Spanberger T, et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology (2016) 5:e1057388. doi: 10.1080/2162402X.2015.1057388
60. Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med (2016) 8:327ra26. doi: 10.1126/scitranslmed.aad6352
61. Mlecnik B, Van den Eynde M, Bindea G, Church SE, Vasaturo A, Fredriksen T, et al. Comprehensive Intrametastatic Immune Quantification and Major Impact of Immunoscore on Survival. J Natl Cancer Inst (2018) 110:97–108. doi: 10.1093/jnci/djx123
62. Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity (2016) 44:698–711. doi: 10.1016/j.immuni.2016.02.025
63. Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The Link between the Multiverse of Immune Microenvironments in Metastases and the Survival of Colorectal Cancer Patients. Cancer Cell (2018) 34:1012–1026.e3. doi: 10.1016/j.ccell.2018.11.003
64. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160:48–61. doi: 10.1016/j.cell.2014.12.033
65. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
66. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer (2017) 17:79–92. doi: 10.1038/nrc.2016.126
67. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21:1350–6. doi: 10.1038/nm.3967
68. Kuai W, Xu X, Yan J, Zhao W, Li Y, Wang B, et al. Prognostic Impact of PD-1 and Tim-3 Expression in Tumor Tissue in Stage I-III Colorectal Cancer. BioMed Res Int (2020) 2020:5294043. doi: 10.1155/2020/5294043
69. Narayanan S, Kawaguchi T, Yan L, Peng X, Qi Q, Takabe K. Cytolytic Activity Score to Assess Anticancer Immunity in Colorectal Cancer. Ann Surg Oncol (2018) 25:2323–31. doi: 10.1245/s10434-018-6506-6
70. Yang L, Xue R, Pan C. Prognostic and clinicopathological value of PD-L1 in colorectal cancer: a systematic review and meta-analysis. Onco Targets Ther (2019) 12:3671–82. doi: 10.2147/OTT.S190168
71. Karpinski P, Rossowska J, Sasiadek MM. Immunological landscape of consensus clusters in colorectal cancer. Oncotarget (2017) 8:105299–311. doi: 10.18632/oncotarget.22169
72. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, et al. The Immune Landscape of Cancer. Immunity (2018) 48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023
73. Soldevilla B, Carretero-Puche C, Gomez-Lopez G, Al-Shahrour F, Riesco MC, Gil-Calderon B, et al. The correlation between immune subtypes and consensus molecular subtypes in colorectal cancer identifies novel tumour microenvironment profiles, with prognostic and therapeutic implications. Eur J Cancer Oxf Engl 1990 (2019) 123:118–29. doi: 10.1016/j.ejca.2019.09.008
74. Horvat N, Tavares C, Rocha C, Clemente Oliveira B, Petkovska I, Gollub MJ. MRI of Rectal Cancer: Tumor Staging, Imaging Techniques, and Management. RadioGraphics (2019) 39:367–87. doi: 10.1148/rg.2019180114
75. deBraud F, Munzone E, Nolè F, De Pas T, Biffi R, Brienza S, et al. Synergistic activity of oxaliplatin and 5-fluorouracil in patients with metastatic colorectal cancer with progressive disease while on or after 5-fluorouracil. Am J Clin Oncol (1998) 21:279–83. doi: 10.1097/00000421-199806000-00015
76. Giacchetti S, Perpoint B, Zidani R, Le Bail N, Faggiuolo R, Focan C, et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J Clin Oncol (2000) 18:136–47. doi: 10.1200/JCO.2000.18.1.136
77. André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, et al. Oxaliplatin, Fluorouracil, and Leucovorin as Adjuvant Treatment for Colon Cancer. N Engl J Med (2004) 350:2343–51. doi: 10.1056/NEJMoa032709
78. Ferris RL, Blumenschein GJ, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375:1856–1867. doi: 10.1056/NEJMoa1602252
79. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N Engl J Med (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
80. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun (2020) 11:3801. doi: 10.1038/s41467-020-17670-y
81. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
82. Pagès F, André T, Taieb J, Vernerey D, Henriques J, Borg C, et al. Prognostic and predictive value of the Immunoscore in stage III colon cancer patients treated with oxaliplatin in the prospective IDEA France PRODIGE-GERCOR cohort study. Ann Oncol (2020) 31:921–9. doi: 10.1016/j.annonc.2020.03.310
83. Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, et al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev (2011) 30:61–9. doi: 10.1007/s10555-011-9273-4
84. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic Cell Death in Cancer Therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
85. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
86. Andre T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt CJA, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. J Clin Oncol (2020) 38:LBA4–4. doi: 10.1200/JCO.2020.38.18_suppl.LBA4
87. Silberman RF, Steiner D, Lo AA, Gomez A, Zehnder JL, Chu G, et al. Complete and Prolonged Response to Immune Checkpoint Blockade in POLE-Mutated Colorectal Cancer. JCO Precis Oncol (2019) 3:1–5. doi: 10.1200/PO.18.00214
88. Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability–High/Mismatch Repair–Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol (2019) 38:11–9. doi: 10.1200/JCO.19.02107
89. Morse MA, Hochster H, Benson A. Perspectives on Treatment of Metastatic Colorectal Cancer with Immune Checkpoint Inhibitor Therapy. Oncologist (2020) 25:33–45. doi: 10.1634/theoncologist.2019-0176
90. Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol (2019) 30:1096–103. doi: 10.1093/annonc/mdz134
91. Hu W, Yang Y, Qi L, Chen J, Ge W, Zheng S. Subtyping of microsatellite instability-high colorectal cancer. Cell Commun Signal (2019) 17:79. doi: 10.1186/s12964-019-0397-4
92. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov (2018) 8:730–49. doi: 10.1158/2159-8290.CD-17-1327
93. Federation Francophone de Cancerologie Digestive. Pembrolizumab in Combination With Xelox Bevacizumab in Patients With Microsatellite Stable Mestatic Colorectal Cancer and a High Immune Infiltrate : a Proof of Concept Study. clinicaltrials.gov (2020). Available at: https://clinicaltrials.gov/ct2/show/NCT04262687 (Accessed October 5, 2020).
94. Mandal R, Samstein RM, Lee K-W, Havel JJ, Wang H, Krishna C, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti–PD-1 immunotherapy response. Science (2019) 364:485–91. doi: 10.1126/science.aau0447
95. Loupakis F, Depetris I, Biason P, Intini R, Prete AA, Leone F, et al. Prediction of Benefit from Checkpoint Inhibitors in Mismatch Repair Deficient Metastatic Colorectal Cancer: Role of Tumor Infiltrating Lymphocytes. Oncologist (2020) 25:481–7. doi: 10.1634/theoncologist.2019-0611
96. Marabelle A, Fakih MG, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with select advanced solid tumours treated with pembrolizumab in KEYNOTE-158. Ann Oncol (2019) 30:v477–8. doi: 10.1093/annonc/mdz253.018
97. Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol (2018) 36:773–9. doi: 10.1200/JCO.2017.76.9901
98. Kikuchi T, Mimura K, Okayama H, Nakayama Y, Saito K, Yamada L, et al. A subset of patients with MSS/MSI-low-colorectal cancer showed increased CD8(+) TILs together with up-regulated IFN-γ. Oncol Lett (2019) 18:5977–85. doi: 10.3892/ol.2019.10953
99. Antoniotti C, Borelli B, Rossini D, Pietrantonio F, Morano F, Salvatore L, et al. AtezoTRIBE: a randomised phase II study of FOLFOXIRI plus bevacizumab alone or in combination with atezolizumab as initial therapy for patients with unresectable metastatic colorectal cancer. BMC Cancer (2020) 20:683. doi: 10.1186/s12885-020-07169-6
100. Shahda S, Noonan AM, Bekaii-Saab TS, O’Neil BH, Sehdev A, Shaib WL, et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J Clin Oncol (2017) 35:3541–1. doi: 10.1200/JCO.2017.35.15_suppl.3541
101. Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic Analysis of the Human Distal Gut Microbiome. Science (2006) 312:1355–9. doi: 10.1126/science.1124234
102. Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PloS Biol (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
103. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci (2011) 108:4578–85. doi: 10.1073/pnas.1000081107
104. Rodríguez JM, Murphy K, Stanton C, Ross RP, Kober OI, Juge N, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis (2015) 26:26050. doi: 10.3402/mehd.v26.26050
105. Browne HP, Neville BA, Forster SC, Lawley TD. Transmission of the gut microbiota: spreading of health. Nat Rev Microbiol (2017) 15:531–43. doi: 10.1038/nrmicro.2017.50
106. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature (2012) 486:222–7. doi: 10.1038/nature11053
107. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature (2014) 505:559–63. doi: 10.1038/nature12820
108. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science (2011) 334:105–8. doi: 10.1126/science.1208344
109. Koido S, Ohkusa T, Kajiura T, Shinozaki J, Suzuki M, Saito K, et al. Long-term alteration of intestinal microbiota in patients with ulcerative colitis by antibiotic combination therapy. PloS One (2014) 9:e86702. doi: 10.1371/journal.pone.0086702
110. Reese AT, Cho EH, Klitzman B, Nichols SP, Wisniewski NA, Villa MM, et al. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. eLife (2018) 7:e35987. doi: 10.7554/eLife.35987
111. Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell (2014) 159:789–99. doi: 10.1016/j.cell.2014.09.053
112. Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet (2016) 48:1413–7. doi: 10.1038/ng.3693
113. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118:229–41. doi: 10.1016/j.cell.2004.07.002
114. Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science (2001) 291:881–4. doi: 10.1126/science.291.5505.881
115. Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A (2002) 99:15451–5. doi: 10.1073/pnas.202604299
116. Barbara G, Stanghellini V, Brandi G, Cremon C, Di Nardo G, De Giorgio R, et al. Interactions between commensal bacteria and gut sensorimotor function in health and disease. Am J Gastroenterol (2005) 100:2560–8. doi: 10.1111/j.1572-0241.2005.00230.x
117. Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature (2008) 456:507–10. doi: 10.1038/nature07450
118. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
119. Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci (2004) 101:4250–5. doi: 10.1073/pnas.0306398101
120. Hollister EB, Gao C, Versalovic J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology (2014) 146:1449–58. doi: 10.1053/j.gastro.2014.01.052
121. Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol (2016) 14:20–32. doi: 10.1038/nrmicro3552
122. Kastl AJ, Terry NA, Wu GD, Albenberg LG. The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. Cell Mol Gastroenterol Hepatol (2020) 9:33–45. doi: 10.1016/j.jcmgh.2019.07.006
123. Leite GGS, Weitsman S, Parodi G, Celly S, Sedighi R, Sanchez M, et al. Mapping the Segmental Microbiomes in the Human Small Bowel in Comparison with Stool: A REIMAGINE Study. Dig Dis Sci (2020) 65:2595–2604. doi: 10.1007/s10620-020-06173-x
124. Wang M, Ahrné S, Jeppsson B, Molin G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol (2005) 54:219–31. doi: 10.1016/j.femsec.2005.03.012
125. Ahmed S, Macfarlane GT, Fite A, McBain AJ, Gilbert P, Macfarlane S. Mucosa-associated bacterial diversity in relation to human terminal ileum and colonic biopsy samples. Appl Environ Microbiol (2007) 73:7435–42. doi: 10.1128/AEM.01143-07
126. Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol (2005) 54:1093–101. doi: 10.1099/jmm.0.45935-0
127. Villmones HC, Haug ES, Ulvestad E, Grude N, Stenstad T, Halland A, et al. Species Level Description of the Human Ileal Bacterial Microbiota. Sci Rep (2018) 8:4736. doi: 10.1038/s41598-018-23198-5
128. Villmones HC, Halland A, Stenstad T, Ulvestad E, Weedon-Fekjær H, Kommedal Ø. The cultivable microbiota of the human distal ileum. Clin Microbiol Infect (2020) 0. doi: 10.1016/j.cmi.2020.08.021
129. Hillman ET, Lu H, Yao T, Nakatsu CH. Microbial Ecology along the Gastrointestinal Tract. Microbes Environ (2017) 32:300–13. doi: 10.1264/jsme2.ME17017
130. Tang Q, Jin G, Wang G, Liu T, Liu X, Wang B, et al. Current Sampling Methods for Gut Microbiota: A Call for More Precise Devices. Front Cell Infect Microbiol (2020) 10:151. doi: 10.3389/fcimb.2020.00151
131. Carroll IM, Chang Y-H, Park J, Sartor RB, Ringel Y. Luminal and mucosal-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Gut Pathog (2010) 2:19. doi: 10.1186/1757-4749-2-19
132. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the Human Intestinal Microbial Flora. Science (2005) 308:1635–8. doi: 10.1126/science.1110591
133. Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe (2014) 15:382–92. doi: 10.1016/j.chom.2014.02.005
134. Lavelle A, Lennon G, O’Sullivan O, Docherty N, Balfe A, Maguire A, et al. Spatial variation of the colonic microbiota in patients with ulcerative colitis and control volunteers. Gut (2015) 64:1553–61. doi: 10.1136/gutjnl-2014-307873
135. Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans ADL, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol (2002) 68:3401–7. doi: 10.1128/aem.68.7.3401-3407.2002
136. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature (2011) 473:174–80. doi: 10.1038/nature09944
137. Hayashi H, Sakamoto M, Benno Y. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol Immunol (2002) 46:535–48. doi: 10.1111/j.1348-0421.2002.tb02731.x
138. Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol (2002) 39:33–9. doi: 10.1111/j.1574-6941.2002.tb00904.x
139. Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring Microbial Diversity and Taxonomy Using SSU rRNA Hypervariable Tag Sequencing. PloS Genet (2008) 4(11):e1000255. doi: 10.1371/journal.pgen.1000255
140. Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res (2007) 14:169–81. doi: 10.1093/dnares/dsm018
141. Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet J-P, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol (2009) 11:2574–84. doi: 10.1111/j.1462-2920.2009.01982.x
142. Wang X, Heazlewood SP, Krause DO, Florin THJ. Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J Appl Microbiol (2003) 95:508–20. doi: 10.1046/j.1365-2672.2003.02005.x
143. James KR, Gomes T, Elmentaite R, Kumar N, Gulliver EL, King HW, et al. Distinct microbial and immune niches of the human colon. Nat Immunol (2020) 21:343–53. doi: 10.1038/s41590-020-0602-z
144. Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol Baltim Md 1950 (2011) 187:733–47. doi: 10.4049/jimmunol.1002701
145. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell (2015) 163:367–80. doi: 10.1016/j.cell.2015.08.058
146. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
147. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science (2011) 331:337–41. doi: 10.1126/science.1198469
148. Ansaldo E, Slayden LC, Ching KL, Koch MA, Wolf NK, Plichta DR, et al. Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science (2019) 364:1179–84. doi: 10.1126/science.aaw7479
149. Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science (2017) 358:359–65. doi: 10.1126/science.aan4526
150. Fanning S, Hall LJ, Cronin M, Zomer A, MacSharry J, Goulding D, et al. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc Natl Acad Sci U S A (2012) 109:2108–13. doi: 10.1073/pnas.1115621109
151. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity (2012) 36:262–75. doi: 10.1016/j.immuni.2011.12.012
152. Fenton TM, Jørgensen PB, Niss K, Rubin SJS, Mörbe UM, Riis LB, et al. Immune Profiling of Human Gut-Associated Lymphoid Tissue Identifies a Role for Isolated Lymphoid Follicles in Priming of Region-Specific Immunity. Immunity (2020) 52:557–70. doi: 10.1016/j.immuni.2020.02.001
153. Mann S, Sidhu M, Gowin K. Understanding the Mechanisms of Diet and Outcomes in Colon, Prostate, and Breast Cancer; Malignant Gliomas; and Cancer Patients on Immunotherapy. Nutrients (2020) 12:2226. doi: 10.3390/nu12082226
154. Segain JP, Raingeard de la Blétière D, Bourreille A, Leray V, Gervois N, Rosales C, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut (2000) 47:397–403. doi: 10.1136/gut.47.3.397
155. Dulal S, Keku TO. Gut microbiome and colorectal adenomas. Cancer J Sudbury Mass (2014) 20:225–31. doi: 10.1097/PPO.0000000000000050
156. Filippo CD, Cavalieri D, Paola MD, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci (2010) 107:14691–6. doi: 10.1073/pnas.1005963107
157. Shanahan F, van Sinderen D, O’Toole PW, Stanton C. Feeding the microbiota: transducer of nutrient signals for the host. Gut (2017) 66:1709–17. doi: 10.1136/gutjnl-2017-313872
158. Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J (2011) 5:220–30. doi: 10.1038/ismej.2010.118
159. Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J (2010) 4:232–41. doi: 10.1038/ismej.2009.112
160. Vannucci L, Stepankova R, Kozakova H, Fiserova A, Rossmann P, Tlaskalova-Hogenova H. Colorectal carcinogenesis in germ-free and conventionally reared rats: Different intestinal environments affect the systemic immunity. Int J Oncol (2008) 32:609–17. doi: 10.3892/ijo.32.3.609
161. Slowicka K, Petta I, Blancke G, Hoste E, Dumas E, Sze M, et al. Zeb2 drives invasive and microbiota-dependent colon carcinoma. Nat Cancer (2020) 1:620–34. doi: 10.1038/s43018-020-0070-2
162. Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat Commun (2015) 6:6528. doi: 10.1038/ncomms7528
163. Liang C, Tseng H-C, Chen H-M, Wang W-C, Chiu C-M, Chang J-Y, et al. Diversity and enterotype in gut bacterial community of adults in Taiwan. BMC Genomics (2017) 18:932. doi: 10.1186/s12864-016-3261-6
164. Marchesi JR, Dutilh BE, Hall N, Peters WHM, Roelofs R, Boleij A, et al. Towards the human colorectal cancer microbiome. PloS One (2011) 6:e20447. doi: 10.1371/journal.pone.0020447
165. Yazici C, Wolf PG, Kim H, Cross T-WL, Vermillion K, Carroll T, et al. Race-dependent association of sulfidogenic bacteria with colorectal cancer. Gut (2017) 66:1983–94. doi: 10.1136/gutjnl-2016-313321
166. Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med (2019) 25:667–78. doi: 10.1038/s41591-019-0405-7
167. Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat Med (2019) 25:679–89. doi: 10.1038/s41591-019-0406-6
168. Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WKK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun (2015) 6:8727. doi: 10.1038/ncomms9727
169. Geng J, Fan H, Tang X, Zhai H, Zhang Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog (2013) 5:2. doi: 10.1186/1757-4749-5-2
170. Dejea CM, Wick EC, Hechenbleikner EM, White JR, Welch JLM, Rossetti BJ, et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci (2014) 111:18321–6. doi: 10.1073/pnas.1406199111
171. Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab (2015) 21:891–7. doi: 10.1016/j.cmet.2015.04.011
172. Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, et al. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun (2014) 5:4724. doi: 10.1038/ncomms5724
173. Huycke MM, Gaskins HR. Commensal bacteria, redox stress, and colorectal cancer: mechanisms and models. Exp Biol Med Maywood NJ (2004) 229:586–97. doi: 10.1177/153537020422900702
174. Schwabe L, Wolf OT. Stress and multiple memory systems: from “thinking” to “doing.” Trends Cognit Sci (2013) 17:60–8. doi: 10.1016/j.tics.2012.12.001
175. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol (2012) 10:717–25. doi: 10.1038/nrmicro2873
176. Sears CL, Garrett WS. Microbes, Microbiota and Colon Cancer. Cell Host Microbe (2014) 15:317–28. doi: 10.1016/j.chom.2014.02.007
177. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol (2012) 10:575–82. doi: 10.1038/nrmicro2819
178. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res (2012) 22:299–306. doi: 10.1101/gr.126516.111
179. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res (2012) 22:292–8. doi: 10.1101/gr.126573.111
180. Sears CL, Pardoll DM. Perspective: Alpha-Bugs, Their Microbial Partners, and the Link to Colon Cancer. J Infect Dis (2011) 203:306–11. doi: 10.1093/jinfdis/jiq061
181. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology (2014) 146:1489–99. doi: 10.1053/j.gastro.2014.02.009
182. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe (2013) 14:195–206. doi: 10.1016/j.chom.2013.07.012
183. Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science (2012) 338:120–3. doi: 10.1126/science.1224820
184. Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res (2014) 20:859–67. doi: 10.1158/1078-0432.CCR-13-1343
185. Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut (2014) 63:1932–42. doi: 10.1136/gutjnl-2013-305257
186. Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, Yen H-R, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med (2009) 15:1016–22. doi: 10.1038/nm.2015
187. Mima K, Cao Y, Chan AT, Qian ZR, Nowak JA, Masugi Y, et al. Fusobacterium nucleatum in Colorectal Carcinoma Tissue According to Tumor Location. Clin Transl Gastroenterol (2016) 7:e200. doi: 10.1038/ctg.2016.53
188. Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res (2014) 74:1311–8. doi: 10.1158/0008-5472.CAN-13-1865
189. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell (2017) 170:548–563.e16. doi: 10.1016/j.cell.2017.07.008
190. Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, Grenov A, et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe (2016) 20:215–25. doi: 10.1016/j.chom.2016.07.006
191. Casasanta MA, Yoo CC, Udayasuryan B, Sanders BE, Umaña A, Zhang Y, et al. Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci Signal (2020) 13(641):eaba9157. doi: 10.1126/scisignal.aba9157
192. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity (2015) 42:344–55. doi: 10.1016/j.immuni.2015.01.010
193. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med (2016) 22:679–84. doi: 10.1038/nm.4086
194. Niccolai E, Ricci F, Russo E, Nannini G, Emmi G, Taddei A, et al. The Different Functional Distribution of “Not Effector” T Cells (Treg/Tnull) in Colorectal Cancer. Front Immunol (2017) 8:1900. doi: 10.3389/fimmu.2017.01900
195. Syed Khaja AS, Toor SM, El Salhat H, Ali BR, Elkord E. Intratumoral FoxP3+Helios+ Regulatory T Cells Upregulating Immunosuppressive Molecules Are Expanded in Human Colorectal Cancer. Front Immunol (2017) 8:619. doi: 10.3389/fimmu.2017.00619
196. Toor SM, Murshed K, Al-Dhaheri M, Khawar M, Abu Nada M, Elkord E. Immune Checkpoints in Circulating and Tumor-Infiltrating CD4+ T Cell Subsets in Colorectal Cancer Patients. Front Immunol (2019) 10:2936:2936. doi: 10.3389/fimmu.2019.02936
197. Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes (2014) 5:675–80. doi: 10.4161/19490976.2014.969989
198. Denizot J, Desrichard A, Agus A, Uhrhammer N, Dreux N, Vouret-Craviari V, et al. Diet-induced hypoxia responsive element demethylation increases CEACAM6 expression, favouring Crohn’s disease-associated Escherichia coli colonisation. Gut (2015) 64:428–37. doi: 10.1136/gutjnl-2014-306944
199. Chan JL, Wu S, Geis AL, Chan GV, Gomes TAM, Beck SE, et al. Non-toxigenic Bacteroides fragilis (NTBF) administration reduces bacteria-driven chronic colitis and tumor development independent of polysaccharide A. Mucosal Immunol (2019) 12:164–77. doi: 10.1038/s41385-018-0085-5
200. Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe (2018) 23:203–214.e5. doi: 10.1016/j.chom.2018.01.007
201. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science (2018) 359:592–7. doi: 10.1126/science.aah3648
202. Tomkovich S, Dejea CM, Winglee K, Drewes JL, Chung L, Housseau F, et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J Clin Invest (2019) 129:1699–712. doi: 10.1172/JCI124196
203. Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PloS One (2012) 7:e39743. doi: 10.1371/journal.pone.0039743
204. Cremonesi E, Governa V, Garzon JFG, Mele V, Amicarella F, Muraro MG, et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut (2018) 67:1984–94. doi: 10.1136/gutjnl-2016-313498
205. Amicarella F, Muraro MG, Hirt C, Cremonesi E, Padovan E, Mele V, et al. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut (2017) 66:692–704. doi: 10.1136/gutjnl-2015-310016
206. Liu J-Q, Li X-Y, Yu H-Q, Yang G, Liu Z-Q, Geng X-R, et al. Tumor-specific Th2 responses inhibit growth of CT26 colon-cancer cells in mice via converting intratumor regulatory T cells to Th9 cells. Sci Rep (2015) 5:10665. doi: 10.1038/srep10665
207. Niccolai E, Russo E, Baldi S, Ricci F, Nannini G, Pedone M, et al. Significant and conflicting correlation of IL-9 with Prevotella and Bacteroides in human colorectal cancer. bioRxiv (2020), 2020.04.28.066001. doi: 10.1101/2020.04.28.066001
208. Wong SH, Zhao L, Zhang X, Nakatsu G, Han J, Xu W, et al. Gavage of Fecal Samples From Patients With Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology (2017) 153:1621–1633.e6. doi: 10.1053/j.gastro.2017.08.022
209. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (2013) 342:967–70. doi: 10.1126/science.1240527
210. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350:1079–84. doi: 10.1126/science.aad1329
211. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (2017) 357:1156–60. doi: 10.1126/science.aah5043
212. Pelley RJ. Oxaliplatin: a new agent for colorectal cancer. Curr Oncol Rep (2001) 3:147–55. doi: 10.1007/s11912-001-0015-6
213. Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol (2016) 27:1386–422. doi: 10.1093/annonc/mdw235
214. Woynarowski JM, Chapman WG, Napier C, Herzig MC, Juniewicz P. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol Pharmacol (1998) 54:770–7. doi: 10.1124/mol.54.5.770
215. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
216. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene (2010) 29:482–91. doi: 10.1038/onc.2009.356
217. Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science (2012) 337:1678–84. doi: 10.1126/science.1224922
218. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202:1691–701. doi: 10.1084/jem.20050915
219. Galluzzi L, Kepp O, Kroemer G. Mitochondrial regulation of cell death: a phylogenetically conserved control. Microb Cell (2016) 3:101–8. doi: 10.15698/mic2016.03.483
220. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund A-C, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J (2009) 28:578–90. doi: 10.1038/emboj.2009.1
221. Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev (2007) 220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x
222. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med (2014) 20:1301–9. doi: 10.1038/nm.3708
223. Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science (2015) 350:972–8. doi: 10.1126/science.aad0779
224. Xian CJ. Roles of growth factors in chemotherapy-induced intestinal mucosal damage repair. Curr Pharm Biotechnol (2003) 4:260–9. doi: 10.2174/1389201033489793
225. Goldszmid RS, Dzutsev A, Viaud S, Zitvogel L, Restifo NP, Trinchieri G. Microbiota modulation of myeloid cells in cancer therapy. Cancer Immunol Res (2015) 3:103–9. doi: 10.1158/2326-6066.CIR-14-0225
226. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science (2013) 342:971–6. doi: 10.1126/science.1240537
227. Zitvogel L, Galluzzi L, Viaud S, Vétizou M, Daillère R, Merad M, et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med (2015) 7:271ps1. doi: 10.1126/scitranslmed.3010473
228. Round JL, Mazmanian SK. The gut microbiome shapes intestinal immune responses during health and disease. Nat Rev Immunol (2009) 9:313–23. doi: 10.1038/nri2515
229. Routy B, Gopalakrishnan V, Daillère R, Zitvogel L, Wargo JA, Kroemer G. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol (2018) 15:382–96. doi: 10.1038/s41571-018-0006-2
230. Walko CM, Lindley C. Capecitabine: a review. Clin Ther (2005) 27:23–44. doi: 10.1016/j.clinthera.2005.01.005
231. Scott TA, Quintaneiro LM, Norvaisas P, Lui PP, Wilson MP, Leung K-Y, et al. Host-Microbe Co-metabolism Dictates Cancer Drug Efficacy in C. elegans. Cell (2017) 169:442–456.e18. doi: 10.1016/j.cell.2017.03.040
232. Guan X, Ma F, Sun X, Li C, Li L, Liang F, et al. Gut Microbiota Profiling in Patients With HER2-Negative Metastatic Breast Cancer Receiving Metronomic Chemotherapy of Capecitabine Compared to Those Under Conventional Dosage. Front Oncol (2020) 10:902:902. doi: 10.3389/fonc.2020.00902
233. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell (2016) 30:147–60. doi: 10.1016/j.ccell.2016.05.016
234. Caraglia M, Correale P, Giannicola R, Staropoli N, Botta C, Pastina P, et al. GOLFIG Chemo-Immunotherapy in Metastatic Colorectal Cancer Patients. A Critical Review on a Long-Lasting Follow-Up. Front Oncol (2019) 9:1102. doi: 10.3389/fonc.2019.01102
235. Pizzolato JF, Saltz LB. The camptothecins. Lancet Lond Engl (2003) 361:2235–42. doi: 10.1016/S0140-6736(03)13780-4
236. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer (2006) 6:789–802. doi: 10.1038/nrc1977
237. Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicol In Vitro (2006) 20:163–75. doi: 10.1016/j.tiv.2005.06.045
238. Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science (2010) 330:831–5. doi: 10.1126/science.1191175
239. Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol (2018) 29:1437–44. doi: 10.1093/annonc/mdy103
240. Elkrief A, El Raichani L, Richard C, Messaoudene M, Belkaid W, Malo J, et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology (2019) 8:e1568812. doi: 10.1080/2162402X.2019.1568812
241. Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. Eur Urol (2020) 78:195–206. doi: 10.1016/j.eururo.2020.04.044
242. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2018) 359:97–103. doi: 10.1126/science.aan4236
243. Routy B, Chatelier EL, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science (2018) 359:91–7. doi: 10.1126/science.aan3706
244. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (2020) 369:1481–9. doi: 10.1126/science.abc3421
245. Dosset M, Vargas TR, Lagrange A, Boidot R, Végran F, Roussey A, et al. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology (2018) 7:e1433981. doi: 10.1080/2162402X.2018.1433981
246. Guan Y, Kraus SG, Quaney MJ, Daniels MA, Mitchem JB, Teixeiro E. FOLFOX Chemotherapy Ameliorates CD8 T Lymphocyte Exhaustion and Enhances Checkpoint Blockade Efficacy in Colorectal Cancer. Front Oncol (2020) 10:586. doi: 10.3389/fonc.2020.00586
247. Groza D, Gehrig S, Kudela P, Holcmann M, Pirker C, Dinhof C, et al. Bacterial ghosts as adjuvant to oxaliplatin chemotherapy in colorectal carcinomatosis. Oncoimmunology (2018) 7:e1424676. doi: 10.1080/2162402X.2018.1424676
248. Agace WW, McCoy KD. Regionalized Development and Maintenance of the Intestinal Adaptive Immune Landscape. Immunity (2017) 46:532–48. doi: 10.1016/j.immuni.2017.04.004
249. Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203:2271–9. doi: 10.1084/jem.20061308
250. Jang MH, Sougawa N, Tanaka T, Hirata T, Hiroi T, Tohya K, et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol Baltim Md 1950 (2006) 176:803–10. doi: 10.4049/jimmunol.176.2.803
251. Zhao H, Yang J, Qian Q, Wu M, Li M, Xu W. Mesenteric CD103+DCs Initiate Switched Coxsackievirus B3 VP1-Specific IgA Response to Intranasal Chitosan-DNA Vaccine Through Secreting BAFF/IL-6 and Promoting Th17/Tfh Differentiation. Front Immunol (2018) 9:2986. doi: 10.3389/fimmu.2018.02986
252. Ziegler PK, Bollrath J, Pallangyo CK, Matsutani T, Canli Ö, De Oliveira T, et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell (2018) 174:88–101.e16. doi: 10.1016/j.cell.2018.05.028
253. Yang H, Yamazaki T, Pietrocola F, Zhou H, Zitvogel L, Ma Y, et al. STAT3 Inhibition Enhances the Therapeutic Efficacy of Immunogenic Chemotherapy by Stimulating Type 1 Interferon Production by Cancer Cells. Cancer Res (2015) 75:3812–22. doi: 10.1158/0008-5472.CAN-15-1122
254. Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature (2006) 440:808–12. doi: 10.1038/nature04596
255. Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell (2018) 175:1307–20. doi: 10.1016/j.cell.2018.10.008
256. Mintz MA, Cyster JG. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol Rev (2020) 296:48–61. doi: 10.1111/imr.12860
257. Ochando J, Braza MS. T follicular helper cells: a potential therapeutic target in follicular lymphoma. Oncotarget (2017) 8:112116–31. doi: 10.18632/oncotarget.22788
258. Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, et al. Non-conventional Inhibitory CD4+Foxp3–PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell (2018) 33:1017–32.e7. doi: 10.1016/j.ccell.2018.05.009
259. Singh D, Ganesan AP, Panwar B, Eschweiler S, Hanley CJ, Madrigal A, et al. CD4+ follicular helper-like T cells are key players in anti-tumor immunity. bioRxiv (2020) 2020.01.08.898346. doi: 10.1101/2020.01.08.898346
260. Garaud S, Buisseret L, Solinas C, Gu-Trantien C, de Wind A, Van den Eynden G, et al. Tumor-infiltrating B cells signal functional humoral immune responses in breast cancer. JCI Insight (2019) 4:e129641. doi: 10.1172/jci.insight.129641
261. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest (2013) 123:2873–92. doi: 10.1172/JCI67428
262. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
263. Montfort A, Pearce O, Maniati E, Vincent BG, Bixby L, Böhm S, et al. A Strong B-cell Response Is Part of the Immune Landscape in Human High-Grade Serous Ovarian Metastases. Clin Cancer Res (2017) 23:250–62. doi: 10.1158/1078-0432.CCR-16-0081
264. Vinuesa CG, Linterman MA, Yu D, MacLennan ICM. Follicular Helper T Cells. Annu Rev Immunol (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
265. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
266. Gu-Trantien C, Migliori E, Buisseret L, de Wind A, Brohée S, Garaud S, et al. CXCL13-producing TFH cells link immune suppression and adaptive memory in human breast cancer. JCI Insight (2017) 2:e91487. doi: 10.1172/jci.insight.91487
267. Largeot A, Pagano G, Gonder S, Moussay E, Paggetti J. The B-Side of Cancer Immunity: The Underrated Tune. Cells (2019) 8:449. doi: 10.3390/cells8050449
268. Tomihara K, Shin T, Hurez VJ, Yagita H, Pardoll DM, Zhang B, et al. Aging-associated B7-DC+ B cells enhance anti-tumor immunity via Th1 and Th17 induction. Aging Cell (2012) 11:128–38. doi: 10.1111/j.1474-9726.2011.00764.x
269. Zhang M, Xia L, Yang Y, Liu S, Ji P, Wang S, et al. PD-1 blockade augments humoral immunity through ICOS-mediated CD4+ T cell instruction. Int Immunopharmacol (2019) 66:127–38. doi: 10.1016/j.intimp.2018.10.045
270. Fontsa ML, Noel G, Garaud S, de Wind A, den Eynden GV, Boisson A, et al. Immune functions of follicular helper CD4+CXCR5+ T cells in human breast cancer. Ann Oncol (2019) 30:iii45. doi: 10.1093/annonc/mdz099.001
271. Turriziani M, Fantini M, Benvenuto M, Izzi V, Masuelli L, Sacchetti P, et al. Carcinoembryonic antigen (CEA)-based cancer vaccines: recent patents and antitumor effects from experimental models to clinical trials. Recent Pat Anticancer Drug Discov (2012) 7:265–96. doi: 10.2174/157489212801820020
272. Zanetti BF, Ferreira CP, de Vasconcelos JRC, Han SW. scFv6.C4 DNA vaccine with fragment C of Tetanus toxin increases protective immunity against CEA-expressing tumor. Gene Ther (2019) 26:441–54. doi: 10.1038/s41434-019-0062-y
273. Foy KC, Wygle RM, Miller MJ, Overholser JP, Bekaii-Saab T, Kaumaya PTP. Peptide vaccines and peptidomimetics of EGFR (HER-1) ligand binding domain inhibit cancer cell growth in vitro and in vivo. J Immunol Baltim Md 1950 (2013) 191:217–27. doi: 10.4049/jimmunol.1300231
274. Wang J, Hollingshead J, El-Masry N, Horncastle D, Talbot I, Tomlinson I, et al. Expression of EGFR, HER2, phosphorylated ERK and phosphorylated MEK in colonic neoplasms of familial adenomatous polyposis patients. J Gastrointest Cancer (2012) 43:444–55. doi: 10.1007/s12029-011-9330-9
275. Kocer B, McKolanis J, Soran A. Humoral immune response to MUC5AC in patients with colorectal polyps and colorectal carcinoma. BMC Gastroenterol (2006) 6:4. doi: 10.1186/1471-230X-6-4
276. Benvenuto M, Sileri P, Rossi P, Masuelli L, Fantini M, Nanni M, et al. Natural humoral immune response to ribosomal P0 protein in colorectal cancer patients. J Transl Med (2015) 13:101. doi: 10.1186/s12967-015-0455-7
277. Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun (2019) 10:4186. doi: 10.1038/s41467-019-12160-2
278. Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med (2018) 24:1649–54. doi: 10.1038/s41591-018-0197-1
279. Tao H, Lu L, Xia Y, Dai F, Wang Y, Bao Y, et al. Antitumor effector B cells directly kill tumor cells via the Fas/FasL pathway and are regulated by IL-10. Eur J Immunol (2015) 45:999–1009. doi: 10.1002/eji.201444625
280. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell (2019) 178:795–806.e12. doi: 10.1016/j.cell.2019.07.008
281. Luna Coronell JA, Sergelen K, Hofer P, Gyurján I, Brezina S, Hettegger P, et al. The Immunome of Colon Cancer: Functional In Silico Analysis of Antigenic Proteins Deduced from IgG Microarray Profiling. Genomics Proteomics Bioinf (2018) 16:73–84. doi: 10.1016/j.gpb.2017.10.002
282. Wang H-F, Li L-F, Guo S-H, Zeng Q-Y, Ning F, Liu W-L, et al. Evaluation of antibody level against Fusobacterium nucleatum in the serological diagnosis of colorectal cancer. Sci Rep (2016) 6:33440. doi: 10.1038/srep33440
283. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17:97–111. doi: 10.1038/nri.2016.107
Keywords: colon cancer, immunity, Bacteroides fragilis, Fusobacterium nucleatum, ileum, microbiome, immune checkpoint
Citation: Fidelle M, Yonekura S, Picard M, Cogdill A, Hollebecque A, Roberti MP and Zitvogel L (2020) Resolving the Paradox of Colon Cancer Through the Integration of Genetics, Immunology, and the Microbiota. Front. Immunol. 11:600886. doi: 10.3389/fimmu.2020.600886
Received: 31 August 2020; Accepted: 09 November 2020;
Published: 14 December 2020.
Edited by:
Xin Chen, BeiGene, ChinaReviewed by:
Alexandre Corthay, Oslo University Hospital, NorwayCopyright © 2020 Fidelle, Yonekura, Picard, Cogdill, Hollebecque, Roberti and Zitvogel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Paula Roberti, TXBhdWxhLnJvYmVydGlAZ21haWwuY29t; Laurence Zitvogel, bGF1cmVuY2Uueml0dm9nZWxAZ3VzdGF2ZXJvdXNzeS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.