 Marion Anliker-Ort
Marion Anliker-Ort Jasper Dingemanse
Jasper Dingemanse John van den Anker
John van den Anker Priska Kaufmann
Priska Kaufmann
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 December 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.599417
The complement system comprises the frontline of the innate immune system. Triggered by pathogenic surface patterns in different pathways, the cascade concludes with the formation of a membrane attack complex (MAC; complement components C5b to C9) and C5a, a potent anaphylatoxin that elicits various inflammatory signals through binding to C5a receptor 1 (C5aR1). Despite its important role in pathogen elimination, priming and recruitment of myeloid cells from the immune system, as well as crosstalk with other physiological systems, inadvertent activation of the complement system can result in self-attack and overreaction in autoinflammatory diseases. Consequently, it constitutes an interesting target for specialized therapies. The paradigm of safe and efficacious terminal complement pathway inhibition has been demonstrated by the approval of eculizumab in paroxysmal nocturnal hematuria. In addition, complement contribution in rare kidney diseases, such as lupus nephritis, IgA nephropathy, atypical hemolytic uremic syndrome, C3 glomerulopathy, or antineutrophil cytoplasmic antibody-associated vasculitis has been demonstrated. This review summarizes the involvement of the terminal effector agents of the complement system in these diseases and provides an overview of inhibitors for complement components C5, C5a, C5aR1, and MAC that are currently in clinical development. Furthermore, a link between increased complement activity and lung damage in severe COVID-19 patients is discussed and the potential for use of complement inhibitors in COVID-19 is presented.
The complement system, as part of the innate immune system (1), comprises several important functions beyond fighting microbial infections, which was assumed to be the only role in the early days following its discovery (2). Nowadays, the complement system is known for being a critical part in various pathways of the immune system, such as opsonization of pathogens or damaged host cells, chemotaxis, modulating smooth muscle contraction and vascular permeability (3), pathogen lysis, removal of immune complexes, provision of proliferative signals for adaptive immune cells (4), angiogenesis, tissue regeneration, wound healing, fibrosis, or lipid metabolism (5).
Overall, the complement system (Figure 1) is a key player in host defense with a strong influence on many other physiological systems, such as the coagulation cascade (6). However, the potential for inadequate regulation or aberrant activity also exists, which can tip the equilibrium towards self-attack and, consequently, contribute to disease (7). Especially the anaphylatoxin C5a has been associated with inflammatory disorders (8, 9), in which the kidney is particularly vulnerable (10). For many years, treatment of rare inflammatory kidney diseases consisted of supportive measures, such as plasma exchange (11), or strong nonspecific immunosuppression, including corticosteroids, which is limited by underlying toxicity and adverse events (12), and many patients progressed to end-stage renal disease (ESRD) requiring dialysis a few years after diagnosis (13, 14).
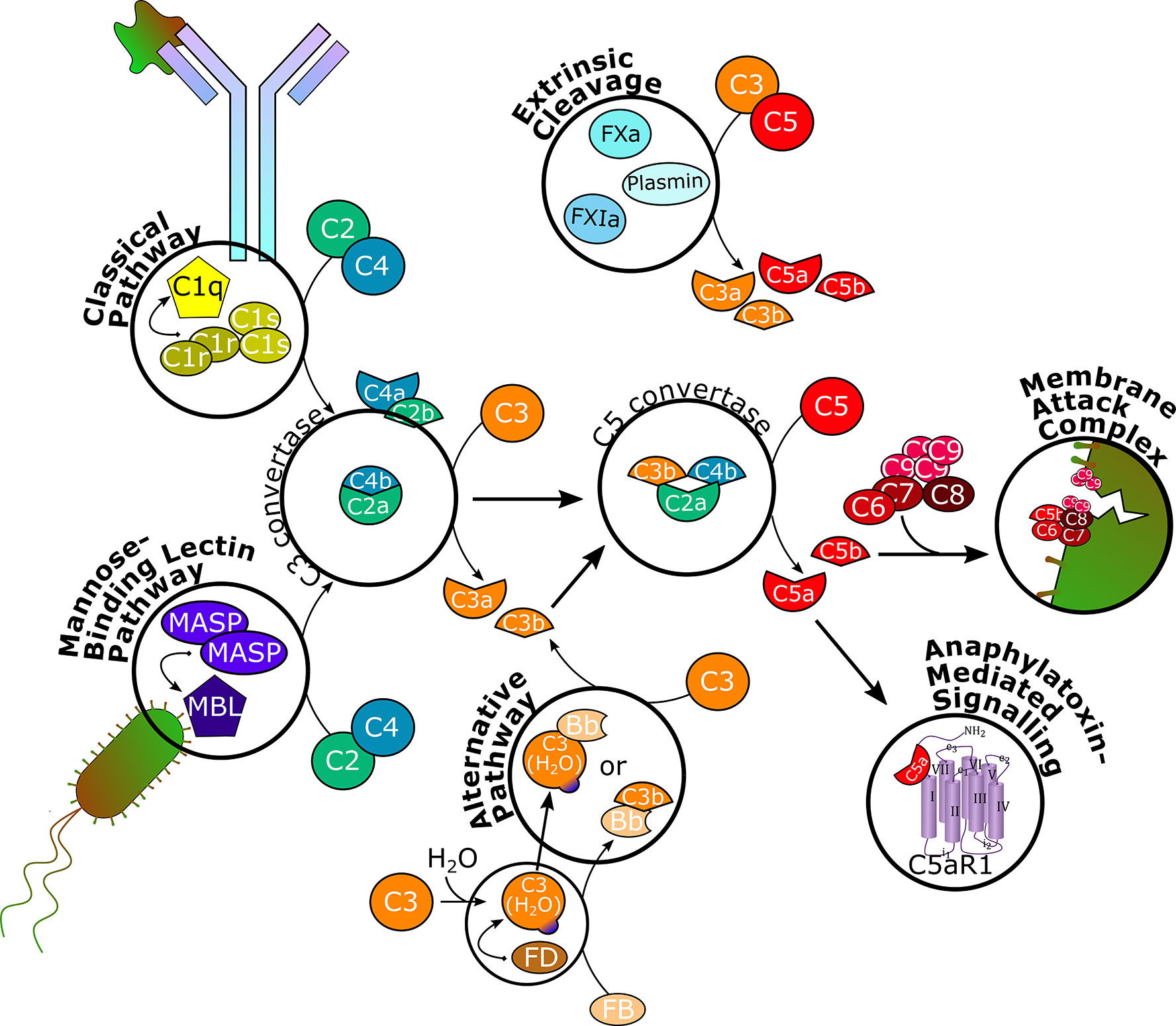
Figure 1 Schematic overview of the complement cascade. The classical pathway is initiated upon binding to antibody-antigen complexes and the mannose-binding lectin pathway is triggered by mannose residues on foreign surfaces. Through recruitment of serine proteases and splitting of C2 and C4, C3 convertase is formed, which splits C3 into C3a and C3b. The alternative pathway is constantly hydrolyzing and cleaving C3 on a low level to allow for rapid self-amplification of the signal. C3b associates with C3 convertase to form C5 convertase, which splits C5 into C5a and C5b. Serine proteases of the coagulation system can independently cleave C3 and C5. C5b recruits C6, C7, C8, and several C9 units to form the membrane attack complex, which inserts into cell membranes to induce cell lysis. C3a and C5a bind to their respective receptors C3aR and C5aR1 to execute various effector functions in different physiological systems. C5aR1, C5a receptor 1; MASP, MBL-associated serine protease; MBL, mannose-binding lectin; FB, factor B; FD, factor D; FXa, factor Xa; FXIa, factor XIa.
Of the plethora of molecules involved in the complement cascade, a growing number has been found to be druggable in recent years. This review focuses on the terminal effector function, i.e., C5 and corresponding cleavage products, and aims at providing an overview on rare inflammatory kidney diseases with complement contribution and the current status of compounds in clinical development that target the terminal pathway of the complement system (see Table 1 and Figure 2 for an overview of clinical studies listed on clinicaltrials.gov). The pioneering drug in the field is eculizumab, a monoclonal antibody C5 inhibitor that was approved by the U.S. Food and Drug Administration (FDA) for treatment of the rare hemolytic disease paroxysmal nocturnal hematuria (PNH) in 2007, which represented a major breakthrough in this previously untreatable disease (15). The successful complement-targeting therapy has subsequently been explored in other diseases and has encouraged many companies to develop their own complement modulators, on one hand to offer therapies with improved properties compared to eculizumab, but also to target other diseases with a high unmet medical need (16).
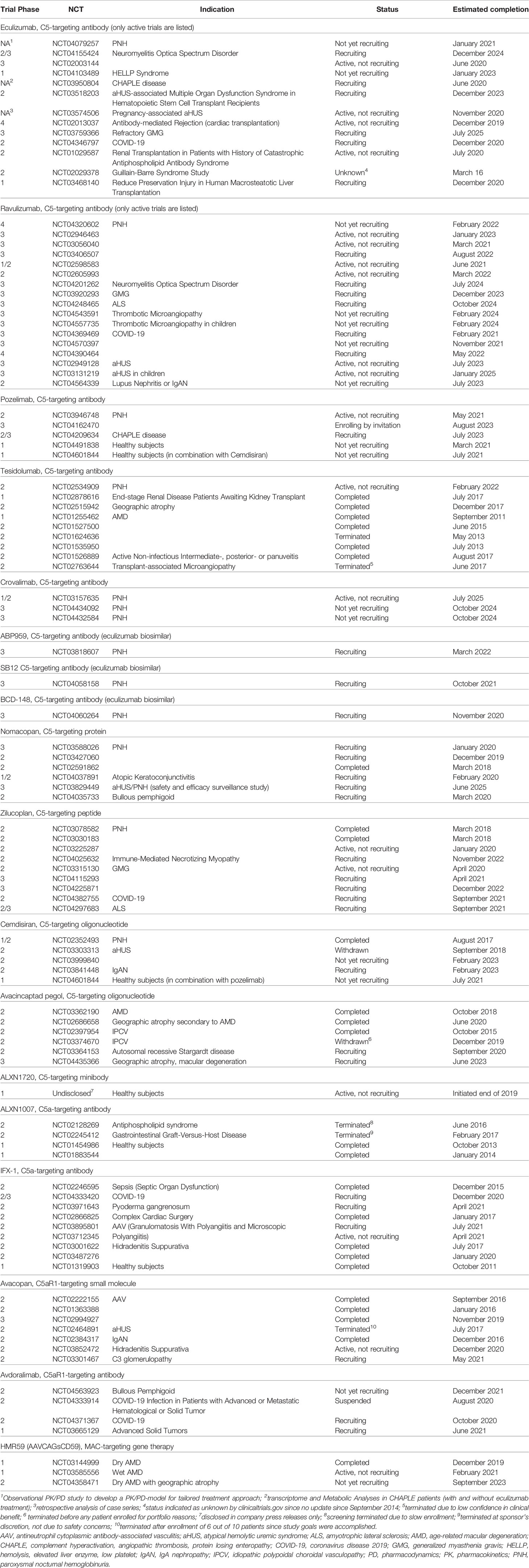
Table 1 Clinical studies of terminal complement pathway inhibitors, as disclosed on clinicaltrials.gov (for eculizumab and ravulizumab, only studies with an active (not recruiting), recruiting, or not yet recruiting status are listed).
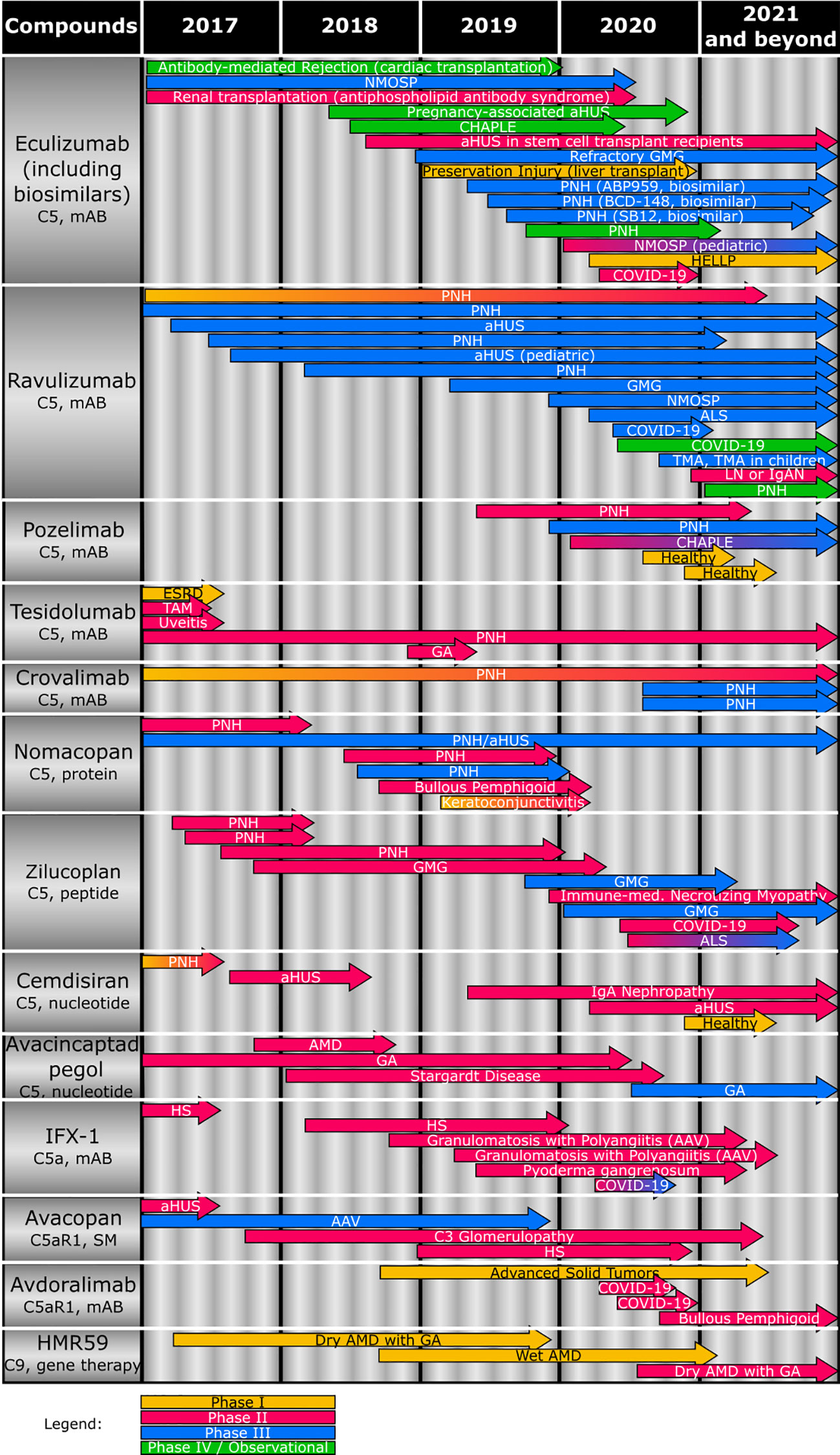
Figure 2 Overview of the clinical status of compounds (including target and compound class) targeting terminal complement effector functions. Studies completed or terminated before 2017 are not depicted. AAV, antineutrophil cytoplasmic antibody-associated vasculitis; aHUS, atypical hemolytic uremic syndrome; ALS, amyotrophic lateral sclerosis; AMD, age-related macular degeneration; CHAPLE, complement hyperactivation, angiopathic thrombosis, protein losing enteropathy; COVID-19, coronavirus disease 2019; ESRD, end-stage renal disease; GA, geographic atrophy; GMG, generalized myasthenia gravis; HELLP, hemolysis, elevated liver enzyme, low platelet; HS, hidradenitis suppurativa; IgAN, IgA nephropathy; IPCV, idiopathic polypoidal choroidal vasculopathy; mAB, monoclonal antibody; NMOSP, neuromyelitis optica spectrum disorder; PNH, paroxysmal nocturnal hemoglobinuria; SM, small molecule; TAM, Transplant-associated microangiopathy.
Besides a differentiation from state-of-the-art therapy in target, epitope, or indication, physicochemical and pharmacokinetic (PK) properties could be a potential advantage of a new compound resulting in a more convenient administration route and regimen for patients with a life-long disease. In addition, to provide an example of the versatility of complement inhibitors and in light of the outbreak of the COVID-19 pandemic, implications of complement overreaction to infectious diseases are summarized, including the potential of complement therapies in COVID-19 (17, 18).
The functions of the complement system are executed through a complex enzymatic cascade of more than 30 activating, amplifying, or regulating proteins or effectors (Figure 1) (19, 20). In brief, the complement is activated by three major pathways. The classical pathway is triggered mainly by antibody-antigen complexes, but also viral envelopes, cell envelopes of Gram-negative bacteria, C-reactive protein, or apoptotic cells. Specific moieties of these endo- or exogenous triggers are bound by the multimeric pattern-recognition molecule C1q, which recruits and activates serine proteases C1r and C1s. These, in turn, cleave complement proteins C2 and C4 into two fragments each. While C4a acts as anaphylatoxin inducing mast cell degranulation, C2b is suggested to increase vascular permeability (21). C4b and C2a form the C3 convertase, which splits C3 into C3a and C3b (22, 23).
Through the lectin pathway, the complement cascade is initiated by recognition of surface patterns on bacterial and fungal pathogens, as well as certain viruses. Thereby, mannose-binding lectins (MBLs) and ficolins are adopting the role of C1q to bind mannose residues on bacterial surfaces, while the proteolytic activity of C1r and C1s is covered by MBL-associated serine proteases (MASPs) to split C2 and C4 and generate the C3 convertase and, consequently, form C3a and C3b (23–25).
In contrast, the alternative pathway is constitutively active on a below-threshold level (tick-over), which requires active elimination of C3b deposition on endogenous cells and constant regulation (23). Therefore, native C3 is spontaneously hydrolyzed to C3(H2O), which recruits factor D, which cleaves factor B into the Bb fragment. Although C3(H2O) can associate with Bb to form a protein complex (C3(H2O)Bb) with similar function as the C3 convertase, the C3bBb complex is more efficient in cleaving C3 into C3a and C3b, indicating that the alternative pathway can also act as an amplification loop of complement effector function initiated by the classical and lectin pathways (19, 26).
Overall, the three pathways conclude in the formation of a C5 convertase, which is built through C3b binding to the existing C3 convertase complex. Therewith, C5 is cleaved into C5a and C5b, the terminal effector proteins of the complement system. Recently, a fourth way of complement activation has been introduced. Proteases otherwise not involved in the complement cascade, e.g., coagulation factors, have been found to directly cleave C3 and C5 (27, 28).
C5b recruits and binds complement proteins C6, C7, C8, and multiple units of C9 to form the membrane attack complex (MAC), a pore that inserts into lipid bilayer membranes of certain bacteria, parasites, enveloped viruses (29, 30), infected host cells, or, pathogenically, endogenous cells such as erythrocytes, to induce lysis (1, 19, 28). C5a, together with C3a, are potent anaphylatoxins modulating a plethora of cell types expressing G-protein coupled C5a or C3a receptors (C5aR1 and C3aR, respectively). In addition to increasing vasodilation and vascular permeability, inducing oxidative bursting of myeloid cells and, consequently, release of pro-inflammatory cytokines and chemokines, especially C5a has shown chemotactic properties for myeloid and certain lymphoid cells and to activate macrophages. The anaphylatoxins also interact with the coagulation system by modulating platelet response and the adaptive immune system by modulating cytokine synthesis of B-cells and initiating and regulating T-cell activation, including crosstalk with T-cell and Toll-like receptors. Furthermore, C3a and C5a effects on cell proliferation and tissue regeneration have been observed (4, 19, 28, 31–36).
Apart from C5aR1, a second C5a receptor, C5aR2 (or C5L2), was discovered, which is expressed less abundantly than C5aR1. Although few reports exist of C5aR2 modulating C5aR1 activity or exacerbating inflammation in disease models, it was initially mainly described to elicit anti-inflammatory effects, either as decoy C5a receptor or by independently downregulating cytokine responses and signaling (8, 37–40).
With involvement in numerous other physiological systems throughout the body, the complement cascade requires sensitive regulation. Besides decoy receptors, such as C5aR2, membrane-bound proteins that allow for differentiation of host and foreign cells as well as fluid-phase based proteins and cofactors prevent excessive activation and tissue damage. Most prominent, also in the context of rare inflammatory kidney diseases, is complement factor H (CFH), which disturbs C3 convertase formation of the alternative pathway, accelerates its decay, and binds to complement factor I (CFI) to lyse C3b (41). Many of them are involved in further biological processes, which again highlights the large network of the complement system throughout the body (5, 42, 43). Deficiencies in the complement pathways are known to lead to increased susceptibility to bacterial infections, but deficiencies in complement regulators either induce a depletion of certain complement components or inappropriate inflammation (44). Consequently, the potential for aberrant or inadequately regulated complement activity paves the way for disease (7, 36, 45). As one of the key effector functions, C5a-induced C5aR1 activation, is involved in numerous kidney diseases, but also cardiac remodeling in hypertension (46, 47), psoriasis (48), rheumatoid arthritis (49), complications in sepsis (50), neuropathic pain (51), or hidradenitis suppurativa (52). During the recent outbreak of SARS-CoV-2, the potential protective role of C5aR1 blockage in COVID-19-induced cytokine release and endothelial injury is being discussed (17).
The kidney is considered to be particularly vulnerable to damage caused by complement dysfunction (10, 53). Although the precise reason thereof is not completely understood, several factors might collude. While the majority of complement components is synthesized in the liver (90%), the kidney is one of the largest extrahepatic production sites (54–56). Due to the kidney’s predominant role in hemofiltration, the organ is prone to deposition of circulating immune complexes, which triggers inflammation and, subsequently, infiltration of immune cells (57). Its function in filtration and its continuous direct contact with antigens (23, 58) and a lower baseline abundance of complement regulators (59) is also thought to play a role. Overall, a significant body of evidence demonstrates that aberrant complement activation is involved in a number of kidney diseases such as lupus nephritis (60), immunoglobulin A nephropathy (IgAN) (5, 14, 61, 62), atypical hemolytic uremic syndrome (aHUS) (63, 64), C3 glomerulopathy (65), and antineutrophil cytoplasmic antibody (ANCA)-associated vasculopathy (AAV) (66).
Renal involvement in the chronic autoimmune disease systemic lupus erythematosus (SLE), manifesting as lupus nephritis, is common, with a prevalence of up to 48% of SLE patients (67), and is usually associated with poor outcome. Pathological features consist of glomerulonephritis and glomerular crescents, infiltration of inflammatory cells in the kidney’s epithelium, podocyte injury, immune complex deposition on mesangial cells, and vascular and tubulointerstitial lesions (68). The interrelationships between complement and SLE are complex and the pathogenesis of lupus nephritis is not fully elucidated. Genetic deficiency of C1q is a strong predictor for developing SLE. Moreover, anti-C1q autoantibodies and corresponding immune complexes relate to renal involvement, highlighting the protective function of the classical pathway in clearing immune complexes. However, once the disease is triggered and immune complexes are insufficiently cleared, their deposition in tissue leads to local complement activation, mainly driven by terminal complement components, indicated by decreased systemic levels of C3 and C4, suggesting high inflammatory activity and tissue damage (69–73). Furthermore, complement has been shown to contribute to the disease by increased C3aR and C5aR activity (74, 75), and elevated C5a levels (76). State-of-the-art clinical management of lupus nephritis consists of nonselective immunosuppressive treatment with corticosteroids (68). In addition, several selective immunosuppressors are currently in clinical development, e.g., B-cell activating factor inhibitors or anti-CD20, -CD40L, and -type 1 interferon monoclonal antibodies (77), which act through modes of action different from complement inhibitors and thereby could provide synergistic benefit.
Although IgAN with a yearly incidence of 0.2 to 2.8 per 100,000 as the most common cause of glomerulonephritis (78), it is an orphan disease with a prevalence of 4 in 10,000 people by the European Medicines Agency (79). The disease is caused by abnormally glycosylated IgA, recognized as autoantigens by properly functioning IgA and IgG. Subsequently formed immunocomplexes deposit in the glomerular mesangium and locally activate the complement cascade, which eventually leads to podocyte and glomerular injury, fibrosis, and ESRD in up to 40% of patients (80). Several complement proteins, most frequently C3 and alternative-pathway regulators, have been found to co-deposit with IgA deposits, contributing to renal injury. In addition, also C4, together with MBL and ficolins, are detected in mesangial deposits, whereas C1q is rarely found, suggesting that the lectin rather than the classical pathway plays a role in IgAN (81, 82). Genome-wide association studies indicated a link between rare genetic variants of CFH-related (CFHR) protein 5 and IgAN susceptibility, although their contribution to the disease is unknown (83). An IgAN induction protocol, in which mice are immunized to Sendai virus to develop an IgA-mediated immune response and corresponding immune complex deposition (84), was applied in C3aR- and C5aR-deficient mice. Anaphylatoxin deficiency resulted in decreased renal IgA and C3 deposition, less secretion of proinflammatory cytokines, and a protective effect on the kidney as indicated by reduced proteinuria (85), while urinary and renal C3a and C5a levels correlated with disease severity (14). Besides diet and lifestyle modifications, blockage of the renin-angiotensin system (RAS) has been shown to significantly reduce the decline in renal function (86). Nonselective immunosuppression, particularly corticosteroid therapy, is used as second-line management, however, improved long-term outcome could not be demonstrated in the majority of patients, while safety and tolerability are compromised with the use of corticosteroids (87).
In the majority of patients diagnosed before 16 years of age, aHUS is a rare disease that presents with microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure (64) with a prevalence of 2.2–9.4 per 1,000,000 (88). A number of genetic deficiencies of complement components lead to impaired regulation of the alternative pathway, either through loss-of-function mutations of regulating proteins, i.e., CFH or CFI, or through gain-of-function mutations of C3, leading to reduced affinity of CFH, or complement factor B, forming a hyperactive C3 convertase, thereby overwhelming regulatory capacity (89). Moreover, certain genetic variants of CFH induce generation of autoantibodies that neutralize CFH and prevent complement regulation (90). Mutated CFH and CFHR hybrid proteins are unable to protect endogenous cells from MAC-induced phenotype alteration of endothelial cells, favoring platelet aggregation (91). C3a- and, most excessively, C5a-mediated inflammation and myeloid cell immigration lead to a prothrombotic state, especially in the endothelium, since the fenestrated structure favors exposure to circulating complement components. Eventually, endothelial swelling manifests in collapsed capillaries, which is fatal in 20%–80% of cases, depending on the mutated complement regulation gene (92, 93). Treatment options consist of plasma exchange combined with nonselective immunosuppression with the goal to replace mutant CFH, however, therapy benefit was only temporary and not as successful in patients with CFI or C3 mutations. After kidney transplantation, disease recurrence rates were as high as 50% (92). More recently, eculizumab was shown efficacious in aHUS by interrupting further uncontrolled complement activation through endothelial injury. This resulted in normalized platelet counts and hematologic values, no thrombotic microangiopathic events during treatment, and improved kidney function reflected, among others, as being dialysis-free in a significantly larger proportion of patients compared to previous treatment options (94, 95).
C3 glomerulopathy consists of several subforms that are characterized by continuous local complement activity, resulting in C3 deposition in renal tissue, without co-deposition of immunoglobulin, with clinical signs of glomerulonephritis, i.e., hematuria, hypertension, and acute and chronic kidney injury. The disease is extremely rare, with a prevalence of 0.1–5 per 1,000,000, and up to 70% of patients progress to ESRD within 10 years after diagnosis (96). Two main subtypes within the term of membranoproliferative glomerulonephritis (MPGN) are distinguished: C3 glomerulonephritis (C3GN) is identified by subendothelial and mesangial depositions, while in dense-deposit disease (DDD), the deposits are found intramembranous and more electron-dense (13). In most patients, the alternative pathway is dysregulated through C3 convertase stabilization by autoantibodies such as C3 nephritic factor, but also C5 convertase binding autoantibodies are reported. Both lead to chronic and unregulated complement activation in the fluid phase. Genetic deficiencies of CFH prevent its regulatory function, either by defective protein processing or lacking secretion. In addition, a variety of deletions, duplications, and chromosomal rearrangements in 5 CFHR protein genes, which are located in the same gene cluster, are known to disturb dimerization of CFHR proteins, thereby disturbing CFHR protein equilibrium and CFH activity in the fluid phase or are associated with CFH autoantibodies. Moreover, mutated C3 forms are reported that can generate C3 convertases resistant to CFH or CFI-mediated decay (96–100). Besides these complement-mediated MPGN forms, immune complex-mediated MPGN has been differentiated, in which the classical pathway is the driver of disease and immune complexes are found to deposit in glomeruli (101). Patients are mainly treated with RAS blockers, since no therapy is specifically approved for C3 glomerulopathy. Plasma exchange and nonspecific immunosuppression have also been assessed in several patients, however, results remain controversial. C5-targeting eculizumab improved the clinical response in a subset of patients only, which might be due to the antibody not improving the C3 dysregulation (96), and C5 convertase overactivity being observed mainly in C3GN compared to DDD (13). Although C3 is the main driver of the disease, C5aR blockage is suggested to add substantial benefit for C3 glomerulopathy patients, which is supported by positive findings with eculizumab in prospective clinical trials (99, 100, 102, 103).
AAV includes three different conditions, i.e., granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis. Annual incidence rates are between 0.5–14.4 per 1,000,000 in Europe and 5-year survival is around 75% (104). The exact etiology remains unresolved, but several genetic mutations and epigenetic modifications have been associated with increased susceptibility to an increased responsiveness of the immune system and to develop ANCA. Certain infections, drugs, or airborne particles can trigger AAV by inducing an initially normal immune response, in which C5a–C5aR1 binding and pro-inflammatory cytokines from macrophages prime neutrophils to express bacteria-degrading proteins, such as myeloperoxidase (MPO) or proteinase 3 (PR3), and form neutrophil extracellular traps (NETs). In AAV patients, NET degradation is decreased and, after prolonged exposure, tolerance to self-antigens on neutrophil surfaces wanes. Consequently, ANCAs bind MPO and PR3 and further activate neutrophils. This excessive production of cytokines and reactive oxygen species induces vascular injury and necrosis in a vicious circle (105). AAV is treated with high-dose corticosteroids with subsequent tapering, in combination with cyclophosphamide or, due to cyclophosphamide toxicity, rituximab or mycophenolate mofetil (106). However, an increasing number of complement-targeting therapies have been tested in AAV in recent years (107). A first step in this direction was taken with avacopan, a small molecule C5aR1 antagonist that induces vasculitis remission while replacing corticosteroids (see Avacopan (CCX-168)) (108, 109).
As a summary, treatment options in above-mentioned diseases mainly consisted of strong, nonspecific immunosuppressive agents, including high-dose corticosteroids (68, 86). Though dampening immune system-related symptoms, corticosteroids bear considerable treatment-related morbidities. The consequences are adverse events (AEs) that increase with treatment of longer duration, including risk of infection, weight gain, new onset of diabetes, hypertension, and/or osteoporosis (12, 110). In the STOP-IgAN trial, safety and efficacy in IgAN patients on corticosteroids (and nonspecific immunosuppressive agents) compared to supportive care were assessed for 3 years. Eventually, immunosuppression by corticosteroids did not yield any substantial clinical benefits, while more infections and comorbidities were reported (87). Since complement has been shown to be involved in these diseases, compounds that selectively target complement components might provide a valuable treatment alternative.
As a first breakthrough in complement-targeting therapy, the recombinant humanized monoclonal antibody eculizumab received accelerated approval as first-in-class C5 inhibitor in PNH in 2007 (111). PNH is a rare hemolytic disease, in which mutations in the genes for hematopoietic stem cells lead to a lack of complement regulatory protein expression on the surface of erythrocytes. Consequently, these are attacked by a constantly activated alternative complement pathway, inducing vascular hemolysis and thrombosis (45). Prior to eculizumab approval only supportive measures were available to increase resynthesis of erythrocytes and prognosis was poor, whereas eculizumab successfully and over a prolonged time frame prevents erythrocyte lysis (112, 113). The selective binding of eculizumab to C5 prevents C5 cleavage and subsequent formation of proinflammatory C5a and cell lysing MAC, while preserving upstream C3a- and C3b-mediated effects (114). After several promising off-label uses of eculizumab in aHUS (115–117), the drug was approved for aHUS based on 5 single-arm studies in 2011 (94, 118, 119). Subsequently, it received approval for the neuromuscular disease generalized myasthenia gravis (120) in 2017 (121) and the eye disorder neuromyelitis optica (122) in 2019 (123). Currently, eculizumab is assessed or used off-label in a range of other indications (124), such as cold agglutinin disease (125), membranoproliferative glomerulonephritis (126), antibody-mediated rejection of transplants (127), and, as mentioned above, C3 glomerulopathy (115). In adult patients, eculizumab is administered as 35 min intravenous infusion of 900 and 1,200 mg in PNH and aHUS, respectively, in weekly intervals in the first four weeks, followed by maintenance doses every 2 weeks (118).
The success of eculizumab clinically validated the inhibition of complement and paved the way for further therapeutic agents against complement components. However, eculizumab’s route of administration and PK properties require a bimonthly infusion and, therefore, a significant burden for patients (6). Also, due to a large inter-individual variability of PK parameters among patients, several studies assessed therapeutic drug monitoring and tailored dosing regimens to maintain efficient complement suppression while avoiding excessive trough concentrations (121, 128, 129). In addition, at the time of market entry, the antibody was the most expensive therapy globally, which fueled many debates on the ethics of cost-effectiveness (130, 131). Although Alexion, the drug manufacturer, secured a patent extension for eculizumab to 2027 (Alexion press release, 15 August 2017), other companies are trying to conquer their share of the PNH market through various biosimilars that are clinically assessed, such as ABP 959 or BCD-148 (132, 133), or already received approval in certain countries, such as Elizaria® (134) and SB12 (Samsung Bioepis press release, 10 January 2020). Further, eculizumab, as a C5-neutralizing antibody, inherently blocks the MAC formation, which increases the susceptibility to bacterial infections. An increased risk of infections, mainly with Neisseria meningitidis, but also case reports of meningococcal or P. aeruginosa infections (135, 136), has been observed, requiring patients to be vaccinated (137–139). In addition, a complete suppression of complement activity has not been observed in all eculizumab-treated patients, resulting in breakthrough hemolysis either at the end of the eculizumab dosing interval or, irrespective of the antibody’s plasma concentration, during pathogenic infection provoking strong complement activity (140). Also, polymorphisms of the C5 gene have been identified that lead to C5 variants with an epitope that does not allow eculizumab binding and subsequent C5 blockade (141).
Therefore, a variety of novel therapies is currently being developed against complement-mediated diseases, filling the legacy that eculizumab provided and aiming to overcome the shortcomings of eculizumab (11, 119, 123, 142). An overview of therapeutics targeting the terminal complement pathway that are in clinical development and their intended indications is provided (Figure 2).
As a follow-up treatment to eculizumab, Alexion developed ravulizumab to target the same epitope of C5, but extended antibody recycling through enhanced affinity to the neonatal Fc receptor, which prolonged the terminal half-life by 4-fold and, consequently, the infusion intervals from 2 to 8 weeks (143). However, C5 affinity is 17-fold lower compared to eculizumab (144). Nonetheless, non-inferiority compared with eculizumab, including safe and effective switch from eculizumab to ravulizumab therapy, was established in two Phase 3 trials in PNH (145, 146). In addition, fewer events of breakthrough hemolysis were reported for the follow-up antibody, indicating a more stable C5 inhibition and improved patient safety (147). After a successful prospective open-label Phase 3 trial, ravulizumab was approved also in aHUS (148). On 14 January 2020, Alexion announced that a Phase 3 trial in amyotrophic lateral sclerosis is planned to be initiated, thereby expanding the scope of terminal complement inhibitors to neurological diseases (Alexion press release, 14 January 2020). The FDA label foresees maintenance doses of up to 3,600 mg (depending on body weight) ravulizumab administered intravenously up to 2 h every 8 weeks, starting 2 weeks after a loading dose (149). However, the risk of infections by encapsulated bacteria, inherent to the inhibition of MAC, despite patient vaccination also exists with ravulizumab (150). Nonetheless, ravulizumab is planned to be explored in further indications, since clinical trials in thrombotic microangiopathy, lupus nephritis, and IgAN have recently been set up (151–153).
Further to eculizumab and ravulizumab, Alexion is developing ALXN1720, a bi-specific anti-C5 mini-body that binds human C5 and prevents its activation, currently assessed in a Phase 1 healthy subject study (Alexion press release, 6 May 2020). However, no indications or further details have been released.
This fully humanized antibody against C5 has been shown to suppress C5 levels and hemolysis in human serum and mice models in a more potent manner compared to eculizumab and ravulizumab (154, 155). In a Phase 1 study in healthy subjects, pozelimab also showed 70% bioavailability after subcutaneous administration, allowing for more versatile administration options, such as weekly subcutaneous 400 mg administrations after a 15 mg/kg intravenous loading dose (156). At the end of 2019, positive results of pozelimab in a Phase 2 study in PNH were announced (Regeneron press release, 5 December 2019). The same treatment regimen is also being applied in a recently started Phase 2/3 study in patients with CD55-deficient protein-losing enteropathy, an intestinal inflammatory disease with complement contribution (157, 158).
This human monoclonal IgG1-antibody against C5 was originally developed against the eye disease age-related macular degeneration (AMD), one of the major causes of blindness in the world, in which C5a and C5b are deposited in retinal cells and Bruch’s membrane, causing inflammation and cell lysis (159). However, effectiveness in AMD could not be established (159). Trials with tesidolumab in transplantation-associated microangiopathy and several forms of uveitis have only shown limited clinical benefit (160, 161). So far, tesidolumab has been administered as intravitreal injection every 28 days (162). Novartis is currently assessing intravenously administered tesidolumab in a Phase 2 study in PNH, however, without indicating the treatment regimen (163). Interestingly, in ESRD patients awaiting kidney transplantation, drug-drug interactions between high-dose intravenous immunoglobulin, a common therapy to reduce antibody-mediated rejection after transplantation, and intravenous tesidolumab were reported, though the latter was more rapidly eliminated (164).
Similar to ravulizumab, crovalimab was engineered to employ pH-dependent antibody recycling for an increased half-life (165, 166). Crovalimab binds to C5 on a different epitope than eculizumab, which would enable C5-neutralization in patients carrying certain genetic polymorphisms that prevent eculizumab binding (167). In addition, crovalimab was also found to bind C5b, thereby inhibiting MAC formation, which could prevent cell lysis in cases where C5 is cleaved by proteases of the coagulation system (168). Pharmacokinetically, crovalimab can be administered subcutaneously with a bioavailability of 90%, which allows for self-administration and reduces treatment burden (169). In a combined Phase 1/2 study in PNH patients, efficacy and tolerability of subcutaneous dosing regimens of weekly up to once every 4 weeks were assessed and switching from eculizumab to crovalimab was considered safe (170).
Monoclonal antibodies excel through their target specificity and allow for targeting complex protein-protein interactions or conformational changes that remained inaccessible for small molecules for a long time, while only few safety issues owing to off-target effects are expected (171). However, high plasma concentrations are usually necessary, which require high-dose bolus injections that can trigger hypersensitivity and infusion reactions, such as dyspnea, nausea, headache, or skin reactions (172). Neutralizing immune responses such as anti-drug antibodies may develop which reduce efficiency, influence the PK properties and toxicity profile, and corresponding immune complexes can induce anaphylaxis or allergic reactions (173). In addition, mechanism-dependent toxicities cannot be avoided, in which case a longer half-life compared to oral treatments is a disadvantage (174). Administration routes and treatment regimens also present a downside. Even though larger antibodies usually present longer half-lives, which would decrease treatment frequency and facilitate compliance, their ability to penetrate into tissue is limited, which narrows down the range of accessible targets. Subcutaneous administration is also restricted by the size of the molecule (173). In diseases with incisive implications related to survival and quality of life, stationary treatment is common and, therefore, parenteral administration is not considered a limitation. Nonetheless, an orally available treatment option would present more flexibility and convenience in a chronic disease (119). Furthermore, monoclonal antibodies present a significant financial burden in long-term therapies (115), as their production based on cell lines and subsequent purification processes remains tedious and expensive (174). As for any biological macromolecule, a properly folded tertiary structure of a monoclonal antibody is crucial for its activity, however, these structures can be prone to denaturation under stress conditions, which further complicates formulation efforts (173).
In a first step to decrease the molecular weight of a potential complement inhibitor, while preserving the target specificity, proteins, peptides, and oligonucleotides are bridging the way to small molecules and expanding treatment options in complement-mediated diseases (6).
Nomacopan is a recombinant small protein derived from a tick C5 inhibitor with a binding epitope opposite of eculizumab’s (140, 175). Besides C5, it has also been shown to inhibit leukotriene B4, a contributor to skin inflammation. Nomacopan was successfully tested in a mouse model of the skin disease bullous pemphigoid, which is mediated by C5a and leukotriene B4 (176). On 1 May 2020, Akari Pharmaceuticals announced positive results from a Phase 2 study in pemphigoid diseases (Akari press release, 1 May 2020). Recently, case reports of successful inhibition of complement-mediated hemolysis by nomacopan in a PNH patient non-responsive to eculizumab (177) and a thrombotic microangiopathy patient (178) were reported. In May 2018 and July 2018, a Phase 2 study in PNH patients with resistance to eculizumab and a Phase 3 study in PNH were initiated, respectively (179–181). According to a press release from November 2018, nomacopan is also assessed in aHUS and post-transplant thrombotic microangiopathy patients (Akari press release, 20 November 2018). Besides applications in kidney and hematologic diseases, nomacopan was efficacious in preclinical models of sepsis (182), while it also showed preliminary efficacy in atopic keratoconjunctivitis patients (Akari press release, 14 October 2019). The diverse potential of nomacopan currently is only dulled by its unfavorable PK. Due to the short half-life (10 h), daily subcutaneous injections would be necessary. A modification of the protein with an N-terminal fusion tag of 600 amino acids (PASylation) could extend the half-life and allow for once weekly dosing, without compromising C5 inhibition potency (183, 184).
Cemdisiran is the first compound applying the concept of RNA interference to complement inhibition. Subcutaneous administration of small interfering RNA sequences matching C5 mRNA, conjugated with N-acetylgalactosamine for targeted delivery to hepatocytes, silenced C5 synthesis and reduced hemolytic activity in monkeys by 80% in weekly or twice weekly treatment regimens (185). Weekly dosing in PNH patients in a Phase 1/2 study decreased serum C5 levels by 98% and allowed for a subsequent reintroduction of eculizumab in a sparing dosing regimen of once monthly (Alnylam press release, 5 December 2016). However, low levels of residual hemolysis were observed due to remaining extrahepatic C5 synthesis and even after addition of eculizumab (123, 186). As a potential option to prevent residual hemolysis, cemdisiran was announced to be assessed as combination therapy with pozelimab (Regeneron press release, 8 April 2019) (187). In addition, cemdisiran is being investigated in IgAN (185) and aHUS patients (Alnylam press release, 26 September 2016) at 600 mg once every 4 weeks with option to decrease dosing frequency. In the latter, an initial Phase 2 study was terminated due to lack of enrollment (188), but efforts are continued in a new study evaluating the switch from eculizumab to the oligonucleotide in aHUS patients (189).
Even though C5 inhibition provided sweeping success in many complement-mediated diseases, the inherent inhibition of MAC formation requires vaccination of patients to Neisseria meningitidis. Additionally, increased predisposition to infections with other bacteria has been reported (137). To overcome this safety burden, direct inhibition of C5a or C5aR1 would allow for anti-inflammatory activity in diseases mediated by the anaphylatoxin without compromising MAC formation. In addition, inhibition of the C5a–C5aR1 axis has been found to alleviate Neisseria-induced sepsis in mice (190). Moreover, C5a generated through direct C5 cleavage by proteases other than C5 convertase can be neutralized with agents directly targeting the C5a-C5aR1 axis (191).
Not surprisingly, Alexion has applied its knowledge in complement-mediated disease to develop ALXN1007, a once or twice weekly intravenously administered monoclonal antibody targeting C5a that has been granted orphan drug designation for graft-versus-host disease by the FDA (Alexion press release, 19 October 2016). However, after promising results with a 77% complete response from the first cohort of weekly 10 mg administrations (192), the study was terminated early without stating the underlying reason, although not due to safety concerns. A possible explanation could be the overall disease response rate being lower at higher dose levels (193). In another Phase 2a study, ALXN1007 was assessed in antiphospholipid syndrome, which was terminated early as well, this time due to slow enrollment (194).
The chimeric monoclonal antibody IFX-1 is directed against C5a without interfering with C5 cleavage, thereby leaving MAC formation intact (191). Initially assessed in sepsis and septic shock (195) and as single intravenous dose in complex cardiac surgery (196) without reporting any results, IFX-1 showed most promising efficacy in hidradenitis suppurativa. This is a chronic inflammatory skin disease presenting with painful and stigmatizing abscesses and significant comorbidities. Many links to immune dysregulation have been found, but the exact etiology remains unknown and, consequently, therapeutic options are limited (52, 197, 198). In a prospective open-label Phase 2a study, twice weekly followed by once weekly intravenous infusions of IFX-1 were well tolerated and improved hidradenitis suppurativa clinical response scores were reached by 83% of patients (199). In the following confirmatory placebo-controlled Phase 2b study, IFX-1 failed to meet the primary endpoint of a dose-dependent effect on the hidradenitis suppurativa clinical response rate after once or twice monthly administrations for 16 weeks (InflaRx press release, 5 June 2019). However, secondary endpoint, post-hoc, and long-term extension results suggested multiple efficacy signals (InflaRx press releases, 18 July and 6 November 2019). In addition, InflaRx reported preliminary positive results of an ongoing Phase 2a study in pyoderma gangraenosum, an autoinflammatory ulcerating skin disease without approved therapy (InflaRx press release, 26 February 2020). Besides skin disorders, IFX-1 is currently tested in systemic versions of AAV on top of standard of care (200) as well as replacement for corticosteroid therapy (201). In addition, as IFX-1 administered in African green monkeys infected with H7N9 influenza virus alleviated acute lung injury and systemic inflammation (202), an open-label Phase 2/3 study in COVID-19-infected severe pneumonia patients has been initiated (203).
With avacopan, the first low molecular-weight orally available C5aR1 antagonist has reached clinical development. Avacopan potently and selectively inhibits C5a-mediated effector functions in vitro, as well as in mice glomerulonephritis (204) and monkey neutropenia models after daily oral administration (205). In a Phase 1 study in healthy subjects, single and multiple doses up to 100 mg and 50 mg twice daily, respectively, were well tolerated. The small molecule was rapidly absorbed after oral administration, reaching peak plasma concentrations after 1 to 2 h, but a similarly fast clearance from plasma in a biphasic manner was observed. At 12 h post-dose, plasma levels decreased to around 80% of peak plasma concentrations, although thereafter, the terminal elimination half-life was more than 120 h after multiple administrations above 10 mg (205). Consequently, a twice daily avacopan dosing regimen was selected for the Phase 2 studies in AAV. Since no C5aR1 antagonist had been administered to AAV patients before, the safety of twice daily 30 mg avacopan during corticosteroid therapy tapering, followed by 30 mg avacopan as corticosteroid replacement in a new cohort, both on top of background cyclophosphamide or rituximab therapy, was evaluated, before an efficacy endpoint was added in the Phase 2 CLEAR study. The avacopan arm presented a reduction in the Birmingham vasculitis score (BVAS) in a larger number of patients compared to the corticosteroid arm, while fewer AEs were reported (108). Subsequently, the Phase 2 CLASSIC study further characterized the safety profile of 10 and 30 mg avacopan in addition to standard of care (high-dose corticosteroids plus cyclophosphamide or rituximab) (109).
Recently, preliminary results of the Phase 3 study in AAV patients (206) became available, indicating that avacopan was non-inferior and superior after 26 and 52 weeks, respectively, compared to corticosteroids, in the BVAS-assessed vasculitis remission, while renal function and corticosteroid-induced toxicity were significantly improved under avacopan (207).
Besides being successfully developed in AAV and providing a valuable treatment alternative to corticosteroids, avacopan is being investigated in Phase 2 studies in several other indications. In an open-label pilot study in IgAN patients on full RAS blockade, twice daily avacopan administration led to a clinically meaningful decrease (50%) of the primary endpoint, urinary protein to creatinine ratio, after 12 weeks in 3 of 7 patients, while being well tolerated (208). Another open-label Phase 2 study of 15-day avacopan treatment in aHUS patients with ESRD was terminated after enrollment of 6 patients without disclosing any results (209). Furthermore, topline results from a placebo-controlled Phase 2 study in hidradenitis suppurativa were recently announced, indicating statistically significant improvements in severe cases compared to placebo (Chemocentryx press release, 28 October 2020). Another Phase 2 study in C3 glomerulopathy is currently ongoing (210), for which Chemocentryx was granted orphan drug designation by the EMA and the FDA (Chemocentryx press release, 23 May 2017).
The small macrocyclic peptide zilucoplan specifically and potently binds C5 at an epitope located on the C5b part, thereby also binding C5b fragments. Daily subcutaneous self-administrations of zilucoplan were efficacious in a Phase 2 study in generalized myasthenia gravis (211), after which a Phase 3 study was initiated at the end of 2019 (212). In addition, the peptide is assessed in immune-mediated necrotizing myopathy (213) and as part of a perpetual platform study to test several treatments in amyotrophic lateral sclerosis (214).
With the success in myasthenia gravis, Ra Pharmaceuticals, recently acquired by UCB Pharma (UCB press release, 2 April 2020), announced to postpone developing zilucoplan in PNH. Although preliminary positive results of the Phase 2 study are reported (215), residual hemolytic activity and breakthrough hemolysis was still observed under zilucoplan treatment (169). In addition, an extended release formulation is being developed to improve the treatment regimen (Ra Pharmaceuticals press release, 9 April 2019). Positive results in Phase 1b studies in renal indications are announced on their webpage, however, no further details are provided (216). On 11 May 2020, a prospective clinical study of zilucoplan in COVID-19 was initiated (217).
Similar to cemdisiran, avacincaptad pegol is a single-stranded oligonucleotide (also termed aptamer), but instead of silencing C5 protein synthesis, avacincaptad pegol directly binds and inhibits C5 (218). Aptamer technology is reported to have high target affinity and specificity without requiring expensive biosynthesis, which also allows for more flexibility in structural adaptations during in vitro selection compared to antibodies. Specific modifications introduced resistance to nuclease-dependent degradation, which increased the half-life of aptamers to several days (219). Avacincaptad pegol is applied as monthly intravitreal injections in the complement-mediated eye disorders Stargardt Macular Dystrophy, idiopathic polypoidal choroidal vasculopathy (IPCV), and geographic atrophy in AMD (159). In the latter, Iveric bio Inc announced statistically significant dose-dependent reduction in geographic atrophy growth with avacincaptad pegol in a Phase 2b study, meeting its primary endpoint (Iveric bio press release, 28 October 2019). Early 2020, the company launched a Phase 3 study in said eye disease (Iveric bio press release, 27 February 2020), for which fast track designation was received from the FDA (Iveric bio press release, 3 April 2020). Besides, results of a Phase 2b study in Stargardt Macular Dystrophy are expected mid 2020 (Iveric bio press release, 27 February 2020). Efforts in IPCV were terminated for portfolio reasons (220). Overall, avacincaptad pegol offers a well-tolerated treatment option in eye diseases with great unmet medical need, although the route of administration of monthly intravitreal injections still implies inconvenience, since most AEs in the Phase 2b study in AMD were due to the injection procedure (Iveric bio press release, 28 October 2019).
Avdoralimab, developed by Innate Pharma, underlines the versatility of the complement system. This fully human anti-C5aR1 antibody is being developed in advanced solid tumors, in which C5aR1 activity is suggested to suppress T cell and natural killer (NK) cell activity in tumor immune surveillance (221). In a Phase 1 study in patients with advanced solid tumors in combination with anti-PD-1 therapy, avdoralimab showed full C5aR1 blockage and early efficacy signals (222). On its webpage, the company disclosed the initiation of clinical trials of avdoralimab in chronic spontaneous urticaria and bullous pemphigoid (223). Furthermore, the effect of anti-C5aR1 activity in COVID-19 patients with severe pneumonia is currently being assessed in a Phase 2 study with avdoralimab (224).
Hemera Biosciences has taken a different approach to complement inhibition, although at the moment applied to AMD only. HMR59 is an intravitreally injected gene therapy that increases production of soluble recombinant CD59, a regulator of C9 of the MAC, in retina cells to protect them from MAC-induced cell lysis (225). The effect of a single injection of HMR59, which is expected to suffice for a life-long effect, was assessed over 18 months in an open-label Phase 1 study in AMD patients (226). Although no results are disclosed, the subsequent placebo-controlled Phase 2 study in AMD patients is estimated to start in June 2020 (227).
Besides inflammatory kidney, hematologic, neurologic, and eye diseases discussed above, there is evidence accumulating to suggest that complement is involved in SARS-CoV-2 pathology (18). Particularly, the concept of cytokine storms within the body damaging organs and tissue recently gained fresh attention with the outbreak of the COVID-19 pandemic (228, 229).
SARS-CoV-2 infects cells by endocytosis through binding to the angiotensin-converting enzyme 2 (ACE2), which is expressed on cells in the human upper airway and pneumocytes (230). In the incubation and initial (i.e., non-severe) phase, a specific adaptive immune response is required to control the virus and to preclude disease progression to the more severe phase (229), mainly consisting of cytotoxic T cells and NK cells (231). Overall, 80% of SARS-CoV-2 infected individuals present mild symptoms. In some patients, the absence of effective activation of T cells and NK cells leads to ineffective SARS-CoV-2 viral clearance and weak antibody production (230). The patient enters a severe phase and develops COVID-19 pneumonia, characterized by a destructive inflammatory response through myeloid cell (monocytes and macrophages) infiltration and, consequently, cytokine release syndrome, particularly in the lungs, as lung epithelium and endothelium are primary targets of SARS-CoV-2 (229).
In the context of influenza viral infection, the complement pathway is activated and in animal models, inhibition of C5a has been shown to be beneficial in improving disease outcome (202, 232, 233). Furthermore, for other species of the coronavirus family, complement contribution to disease starting from Day 1 post infection has been shown earlier (234). Inhibition of C5aR1 activity has been shown to alleviate disease severity and organ damage by reducing inflammation in MERS-CoV infections (235). A similar effect has been suggested in SARS-CoV (228, 234) and increased C5a levels have been found in COVID-19 patients (18). Consequently, several C5, C5a, and C5aR1 inhibitors are currently undergoing clinical studies in COVID-19, such as eculizumab, which has already showed preliminary efficacy (236), ravulizumab (237), zilucoplan (217), IFX-1 (203), and avdoralimab (224).
Current measures to contain the spread of SARS-CoV-2 include social and physical distancing to protect vulnerable populations, until a treatment or a vaccine is shown to be efficacious. In that sense, complement inhibitors could potentially reduce the severity of COVID-19-induced complications and the number of patients in intensive care or fatalities. Consequently, the impact on the health care system would be reduced, improving the overall disease burden on society, particularly since the long-term implications of severe COVID-19 cases and intubation are heavily discussed.
Through the introduction of eculizumab to the market in 2007, the paradigm of complement inhibitors as a successful therapy in rare (auto)inflammatory diseases has been validated. Many of these debilitating disorders did not have an approved treatment available and either were connected with early mortality or inconvenient stationary therapy (plasma exchange, dialysis) (238). Otherwise, physicians were relying on strong nonspecific immunosuppression by cyclophosphamide, azathioprine, methotrexate, rituximab, and mycophenolate, for which the dose has to be carefully adjusted and monitored due to inherent toxicity (12). In addition, corticosteroids were abundantly used due to good accessibility, inexpensive treatment, and the possibility of oral therapy. However, corticosteroids bear well-known and significant comorbidities for long-term administration, such as an increased infection risk, diabetes, osteoporosis, weight gain, myopathy, and psychiatric disturbances (239).
Eculizumab significantly improved quality of life and provided relief for patients in several complement-driven indications (118). Nevertheless, the monoclonal antibody presents with some unfavorable properties. Besides the increased risk of meningococcal infections due to inhibition of MAC formation, a number of patients remain unresponsive or only achieve an incomplete response, which can be unacceptable in specific indications (147). Furthermore, treatment costs are very high, especially in view of life-long therapy, which has spiked ethical and economical discussions in healthcare (131).
It is evident that many companies are aiming to take a share of the large therapeutic potential of eculizumab in PNH. Second and later generation terminal complement pathway inhibitors are trying to deal with PK challenges, for example through structural modifications, which allow for antibody recycling and a sustained exposure leading to a lower frequency of administration. An improved route of administration and treatment regimen, and eventually a more convenient therapy, would further increase the quality of life of patients (167). Moving from the intravenous to the subcutaneous route opens the possibility of self-administration, which leaves patients less dependent on hospital proximity (211). Moreover, transition from monoclonal antibodies via proteins, peptides, and oligonucleotides towards small molecules will gradually ease administration regimens and decreases production efforts (174, 240). A versatile spectrum of mechanisms of action, targets, and target epitopes increases the chance that patients with varying underlying disease causes achieve remission, while the move from general C5 to specific C5a-C5aR1-axis-targeting could be beneficial in avoiding a treatment-induced infection risk.
Besides the above elaborated complement inhibitors in clinical development, several novel agents have been described in preclinical studies that are summarized elsewhere (123). However, usually only sparse data is made available about innovations in the field before clinical development is reached (241). Consequently, the pipeline of terminal complement pathway inhibitors will continue to present novel therapeutic agents in this exciting field providing a plethora of potentially efficacious treatment options to improve patients’ lives in a growing number of rare diseases.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
MA-O, JD, and PK were employed by the company Idorsia Pharmaceuticals Ltd. The views expressed in this article are those of the authors and do not necessarily reflect those of their company.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Janeway C, Travers P, Walport M, Shlomchik M. The complement system and innate immunity. 5th ed. (2001) Immunobiology: The Immune System in Health and Disease. New York: Garland Science.
2. Fearon DT, Locksley RM. The Instructive Role of Innate Immunity in the Acquired Immune Response. Science (5258) (80- ). 1996 272:50–4. doi: 10.1126/science.272.5258.50
3. Ajona D, Ortiz-Espinosa S, Pio R. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin Cell Dev Biol (2019) 85:153–63. doi: 10.1016/j.semcdb.2017.11.023
4. Thurman JM, Nester CM. All Things Complement. Clin J Am Soc Nephrol (2016) Oct 711(10):1856–66. doi: 10.2215/CJN.01710216
5. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi: 10.1038/ni.1923
6. Ricklin D, Lambris JD. New milestones ahead in complement-targeted therapy. Semin Immunol (2016) 28(3):208–22. doi: 10.1016/j.smim.2016.06.001
7. Ricklin D, Lambris JD. Complement in Immune and Inflammatory Disorders: Pathophysiological Mechanisms. J Immunol (2013) 190(8):3831–8. doi: 10.4049/jimmunol.1203487
8. Monk PN, Scola A-M, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol (2007) 152(4):429–48. doi: 10.1038/sj.bjp.0707332
9. Wood AJT, Vassallo A, Summers C, Chilvers ER, Conway-Morris A. C5a anaphylatoxin and its role in critical illness-induced organ dysfunction. Eur J Clin Invest (2018) 48(12):e13028. doi: 10.1111/eci.13028
10. Willows J, Brown M, Sheerin NS. The role of complement in kidney disease. Clin Med J R Coll Physicians London (2020) 20(2):156–60. doi: 10.7861/clinmed.2019-0452
11. Harris CL, Pouw RB, Kavanagh D, Sun R, Ricklin D. Developments in anti-complement therapy; from disease to clinical trial. Mol Immunol (2018) 102(June):89–119. doi: 10.1016/j.molimm.2018.06.008
12. King C, Harper L. Avoidance of Harm From Treatment for ANCA-Associated Vasculitis. Curr Treat Opt Rheumatol (2017) 3(4):230–43. doi: 10.1007/s40674-017-0082-y
13. Angioi A, Fervenza FC, Sethi S, Zhang Y, Smith RJ, Murray D, et al. Diagnosis of complement alternative pathway disorders. Kidney Int (2016) 89(2):278–88. doi: 10.1016/j.kint.2015.12.003
14. Liu L, Zhang Y, Duan X, Peng Q, Liu Q, Zhou Y, et al. C3a, C5a renal expression and their receptors are correlated to severity of IgA nephropathy. J Clin Immunol (2014) 34(2):224–32. doi: 10.1007/s10875-013-9970-6
15. Thompson CA. FDA approves drug to treat rare hemoglobinuria. Am J Health Syst Pharm (2007) 64(9):906–6. doi: 10.2146/news070038
16. Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov (2019) 18(9):707–29. doi: 10.1038/s41573-019-0031-6
17. Campbell CM, Kahwash R. Will Complement Inhibition be the New Target in Treating COVID-19 Related Systemic Thrombosis? Circulation (2020). 141(22):1739–41. doi: 10.1161/CIRCULATIONAHA.120.047419
18. Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, et al. Complement activation in patients with COVID-19: A novel therapeutic target. J Allergy Clin Immunol (2020) 146(1):215–7. doi: 10.1016/j.jaci.2020.05.006
19. Mathern DR, Heeger PS. Molecules great and small: The complement system. Clin J Am Soc Nephrol (2015) 10(9):1636–50. doi: 10.2215/CJN.06230614
20. Thurman JM, Holers VM. The Central Role of the Alternative Complement Pathway in Human Disease. J Immunol (2006) 176(3):1305–10. doi: 10.4049/jimmunol.176.3.1305
21. Krishnan V, Xu Y, Macon K, Volanakis JE, Narayana SVL. The structure of C2b, a fragment of complement component C2 produced during C3 convertase formation. Acta Crystallogr D Biol Crystallogr (2009) 65(3):266–74. doi: 10.1107/S0907444909000389
22. Rus H, Cudrici C, Niculescu F. The role of the complement system in innate immunity. Immunol Res (2005) 33(2):103–12. doi: 10.1385/IR:33:2:103
23. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol (2013) 33(6):479–92. doi: 10.1016/j.semnephrol.2013.08.001
24. Wallis R, Mitchell DA, Schmid R, Schwaeble WJ, Keeble AH. Paths reunited: Initiation of the classical and lectin pathways of complement activation. Immunobiology (2010) 215(1):1–11. doi: 10.1016/j.imbio.2009.08.006
25. Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol (2007) 25(11):1265–75. doi: 10.1038/nbt1342
26. Fromell K, Adler A, Åman A, Manivel VA, Huang S, Dührkop C, et al. Assessment of the Role of C3(H2O) in the Alternative Pathway. Front Immunol (2020) 11(March):1–13. doi: 10.3389/fimmu.2020.00530
27. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol (2008) 632(Davis 2004):71–9. doi: 10.1007/978-0-387-78952-1_6
28. Nesargikar P, Spiller B, Chavez R. The complement system: History, pathways, cascade and inhibitors. Eur J Microbiol Immunol (2012) 2(2):103–11. doi: 10.1556/EuJMI.2.2012.2.2
29. Mellors J, Tipton T, Longet S, Carroll M. Viral Evasion of the Complement System and Its Importance for Vaccines and Therapeutics. Front Immunol (2020) 11(July):1–20. doi: 10.3389/fimmu.2020.01450
30. Bayly-Jones C, Bubeck D, Dunstone MA. The mystery behind membrane insertion: A review of the complement membrane attack complex. Philos Trans R Soc B Biol Sci (2017) 372(1726). doi: 10.1098/rstb.2016.0221
31. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol (2009) 46(14):2753–66. doi: 10.1016/j.molimm.2009.04.027
32. Guo R-F, Ward PA. Role of C5a in Inflammatory Responses. Annu Rev Immunol (2005) 23(1):821–52. doi: 10.1146/annurev.immunol.23.021704.115835
33. Manthey HD, Woodruff TM, Taylor SM, Monk PN. Complement component 5a (C5a). Int J Biochem Cell Biol (2009) 41(11):2114–7. doi: 10.1016/j.biocel.2009.04.005
34. Fischer WH, Hugli TE. Regulation of B cell functions by C3a and C3a(desArg): suppression of TNF-alpha, IL-6, and the polyclonal immune response. J Immunol (1997) 159(9):4279–86.
35. Reis ES, Mastellos DC, Hajishengallis G, Lambris JD. New insights into the immune functions of complement. Nat Rev Immunol (2019) 19(8):503–16. doi: 10.1038/s41577-019-0168-x
36. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol (2016) 12(7):383–401. doi: 10.1038/nrneph.2016.70
37. Ward PA. Sepsis, apoptosis and complement. Biochem Pharmacol (2008) 76(11):1383–8. doi: 10.1016/j.bcp.2008.09.017
38. Lee H, Whitfeld PL, Mackay CR. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol (2008) 86(2):153–60. doi: 10.1038/sj.icb.7100166
39. Li XX, Lee JD, Kemper C, Woodruff TM. The Complement Receptor C5aR2: A Powerful Modulator of Innate and Adaptive Immunity. J Immunol (2019) 202(12):3339–48. doi: 10.4049/jimmunol.1900371
40. Li XX, Clark RJ, Woodruff TM. C5aR2 Activation Broadly Modulates the Signaling and Function of Primary Human Macrophages. J Immunol (2020) 205(4):1102–12. doi: 10.4049/jimmunol.2000407
41. Barbour TD, Pickering MC, Terence Cook H. Recent insights into C3 glomerulopathy. Nephrol Dial Transplant (2013) 28(7):1685–93. doi: 10.1093/ndt/gfs430
42. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9(10):729–40. doi: 10.1038/nri2620
43. Pangburn MK, Ferreira VP, Cortes C. Discrimination between host and pathogens by the complement system. Vaccine (2008) 26(SUPPL. 8):1–17. doi: 10.1016/j.vaccine.2008.11.023
44. Pettigrew HD, Teuber SS, Gershwin ME. Clinical significance of complement deficiencies. Ann N Y Acad Sci (2009) 1173:108–23. doi: 10.1111/j.1749-6632.2009.04633.x
45. Wong EKS, Kavanagh D. Diseases of complement dysregulation—an overview. Semin Immunopathol (2018) 40(1):49–64. doi: 10.1007/s00281-017-0663-8
46. Zhang C, Li Y, Wang C, Wu Y, Du J. Antagonist of C5aR prevents cardiac remodeling in angiotensin II-induced hypertension. Am J Hypertens (2014) 27(6):857–64. doi: 10.1093/ajh/hpt274
47. Chen XH, Ruan CC, Ge Q, Ma Y, Xu JZ, Zhang ZB, et al. Deficiency of Complement C3a and C5a Receptors Prevents Angiotensin II-Induced Hypertension via Regulatory T Cells. Circ Res (2018) 122(7):970–83. doi: 10.1161/CIRCRESAHA.117.312153
48. Zheng QY, Liang SJ, Xu F, Li GQ, Luo N, Wu S, et al. C5a/C5aR1 pathway is critical for the pathogenesis of psoriasis. Front Immunol (2019) 10(AUG):1–12. doi: 10.3389/fimmu.2019.01866
49. Sadik CD, Miyabe Y, Sezin T, Luster AD. The critical role of C5a as an initiator of neutrophil-mediated autoimmune inflammation of the joint and skin. Semin Immunol (2018) 37(December 2017):21–9. doi: 10.1016/j.smim.2018.03.002
50. Fattahi F, Frydrych LM, Bian G, Kalbitz M, Herron TJ, Malan EA, et al. Role of complement C5a and histones in septic cardiomyopathy. Mol Immunol (2018) 102:32–41. doi: 10.1016/j.molimm.2018.06.006
51. Moriconi A, Cunha TM, Souza GR, Lopes AH, Cunha FQ, Carneiro VL, et al. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. Proc Natl Acad Sci U S A (2014) 111(47):16937–42. doi: 10.1073/pnas.1417365111
52. Kanni T, Zenker O, Habel M, Riedemann N, Giamarellos-Bourboulis EJ. Complement activation in hidradenitis suppurativa: a new pathway of pathogenesis? Br J Dermatol (2018) 179(2):413–9. doi: 10.1111/bjd.16428
53. Tan Y, Zhao MH. Complement in glomerular diseases. Nephrology (2018) 23:11–5. doi: 10.1111/nep.13461
54. Zhou W, Marsh JE, Sacks SH. Intrarenal synthesis of complement. Kidney Int (2001) 59(4):1227–35. doi: 10.1046/j.1523-1755.2001.0590041227.x
55. Brooimans RA, Stegmann APA, Van Dorp WT, Van der Ark AAJ, Van der Woude FJ, Van Es LA, et al. Interleukin 2 mediates stimulation of complement C3 biosynthesis in human proximal tubular epithelial cells. J Clin Invest (1991) 88(2):379–84. doi: 10.1172/JCI115314
56. Sheerin NS, Zhou W, Adler S, Sacks SH. TNF-α regulation of C3 gene expression and protein biosynthesis in rat glomerular endothelial cells. Kidney Int (1997) 51(3):703–10. doi: 10.1038/ki.1997.101
57. Morgan BP, Gasque P. Extrahepatic complement biosynthesis: where, when and why? Clin Exp Immunol (1997) 107(1):1–7. doi: 10.1046/j.1365-2249.1997.d01-890.x
58. Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: Basic concepts and clinical implications. Nat Rev Immunol (2013) 13(10):738–53. doi: 10.1038/nri3523
59. De Vriese AS, Sethi S, Van Praet J, Nath KA, Fervenza FC. Kidney disease caused by dysregulation of the complement alternative pathway: An etiologic approach. J Am Soc Nephrol (2015) 26(12):2917–29. doi: 10.1681/ASN.2015020184
60. Song D, Guo W-Y, Wang F-M, Li Y-Z, Song Y, Yu F, et al. Complement Alternative Pathway‘s Activation in Patients With Lupus Nephritis. Am J Med Sci (2017) 353(3):247–57. doi: 10.1016/j.amjms.2017.01.005
61. Salvadori M, Rosso G, Bertoni E. Complement involvement in kidney diseases: From physiopathology to therapeutical targeting. World J Nephrol (2015) 4(2):169–84. doi: 10.5527/wjn.v4.i2.169
62. Thurman JM. Complement in Kidney Disease: Core Curriculum 2015. Am J Kidney Dis (2015) 65(1):156–68. doi: 10.1053/j.ajkd.2014.06.035
63. Liszewski MK, Java A, Schramm EC, Atkinson JP. Complement Dysregulation and Disease: Insights from Contemporary Genetics. Annu Rev Pathol Mech Dis (2017) 12(1):25–52. doi: 10.1146/annurev-pathol-012615-044145
64. Noris M, Remuzzi G. Terminal complement effectors in atypical hemolytic uremic syndrome: C5a, C5b-9, or a bit of both? Kidney Int (2019) 96(1):13–5. doi: 10.1016/j.kint.2019.02.038
65. Zipfel PF, Wiech T, Rudnick R, Afonso S, Person F, Skerka C. Complement Inhibitors in Clinical Trials for Glomerular Diseases. Front Immunol (2019) 10:2166. doi: 10.3389/fimmu.2019.02166
66. Chen M, Jayne DRW, Zhao MH. Complement in ANCA-associated vasculitis: Mechanisms and implications for management. Nat Rev Nephrol (2017) 13(6):359–67. doi: 10.1038/nrneph.2017.37
67. Wang H, Ren Y, Chang J, Gu L, Sun LY. A systematic review and meta-analysis of prevalence of biopsy-proven lupus nephritis. Arch Rheumatol (2018) 33(1):17–25. doi: 10.5606/ArchRheumatol.2017.6127
68. Yu F, Haas M, Glassock R, Zhao MH. Redefining lupus nephritis: Clinical implications of pathophysiologic subtypes. Nat Rev Nephrol (2017) 13(8):483–95. doi: 10.1038/nrneph.2017.85
69. Bao L, Cunningham PN, Quigg RJ. Complement in Lupus Nephritis: New Perspectives. Kidney Dis (2015) 1(2):91–9. doi: 10.1159/000431278
70. Stojan G, Petri M. Anti-C1q in systemic lupus erythematosus. Lupus (2016) 25(8):873–7. doi: 10.1177/0961203316645205
71. Pinheiro SVB, Dias RF, Fabiano RCG, Araujo S de A, Silva ACSE. Pediatric lupus nephritis. J Bras Nefrol (2019) 41(2):252–65. doi: 10.1590/2175-8239-jbn-2018-0097
72. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: An update. Ann Rheum Dis (2014) 73(9):1601–6. doi: 10.1136/annrheumdis-2014-205287
73. Rother RP, Mojcik CF, McCroskery EW. Inhibition of terminal complement: A novel therapeutic approach for the treatment of systemic lupus erythematosus. Lupus (2004) 13(5):328–34. doi: 10.1191/0961203303lu1021oa
74. Bao L, Osawe I, Haas M, Quigg RJ. Signaling through Up-Regulated C3a Receptor Is Key to the Development of Experimental Lupus Nephritis. J Immunol (2005) 175(3):1947–55. doi: 10.4049/jimmunol.175.3.1947
75. Bao L, Osawe I, Puri T, Lambris JD, Haas M, Quigg RJ. C5a promotes development of experimental lupus nephritis which can be blocked with a specific receptor antagonist. Eur J Immunol (2005) 35(8):2496–506. doi: 10.1002/eji.200526327
76. Sakuma Y, Nagai T, Yoshio T, Hirohata S. Differential activation mechanisms of serum C5a in lupus nephritis and neuropsychiatric systemic lupus erythematosus. Mod Rheumatol (2017) 27(2):292–7. doi: 10.1080/14397595.2016.1193965
77. Samotij D, Reich A. Biologics in the treatment of lupus erythematosus: A critical literature review. BioMed Res Int (2019) 2019(Dc):1–17. doi: 10.1155/2019/8142368
78. Rodrigues JC, Haas M, Reich HN. IgA Nephropathy. Clin J Am Soc Nephrol (2017) 12(4):677–86. doi: 10.2215/CJN.07420716
79. European Medicines Agency. Public summary of opinion on orphan designation. Recombinant human monoclonal antibody against mannan-binding lectinassociated serine protease-2 for the treatment of primary IgA nephropathy. Available at: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/18/1984-public-summary-opinion-orphan-designation-recombinant-human-monoclonal-antibody-against-mannan_en.pdf (Accessed 2020 Oct 9).
80. Lai KN, Tang SCW, Schena FP, Novak J, Tomino Y, Fogo AB, et al. IgA nephropathy. Nat Rev Dis Primers (2016) 2:16001. doi: 10.1038/nrdp.2016.1
81. Tortajada A, Gutierrez E, Pickering MC, Praga Terente M, Medjeral-Thomas N. The role of complement in IgA nephropathy. Mol Immunol (2019) 114(June):123–32. doi: 10.1016/j.molimm.2019.07.017
82. Floege J, Daha MR. IgA nephropathy: new insights into the role of complement. Kidney Int (2018) 94(1):16–8. doi: 10.1016/j.kint.2018.03.009
83. Zhai YL, Meng SJ, Zhu L, Shi SF, Wang SX, Liu LJ, et al. Rare variants in the complement factor H-related protein 5 gene contribute to genetic susceptibility to IgA nephropathy. J Am Soc Nephrol (2016) 27(9):2894–905. doi: 10.1681/ASN.2015010012
84. Emancipator SN. Animal Models of Ig A Nephropathy. Curr Protoc Immunol (1999) 29(1):1–18. doi: 10.1002/0471142735.im1511s29
85. Zhang Y, Yan X, Zhao T, Xu Q, Peng Q, Hu R, et al. Targeting C3a/C5a receptors inhibits human mesangial cell proliferation and alleviates immunoglobulin A nephropathy in mice. Clin Exp Immunol (2017) 189(1):60–70. doi: 10.1111/cei.12961
86. Selvaskandan H, Cheung CK, Muto M, Barratt J. New strategies and perspectives on managing IgA nephropathy. Clin Exp Nephrol (2019) 23(5):577–88. doi: 10.1007/s10157-019-01700-1
87. Rauen T, Eitner F, Fitzner C, Sommerer C, Zeier M, Otte B, et al. Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med (2015) 373(23):2225–36. doi: 10.1056/NEJMoa1415463
88. Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of atypical hemolytic uremic syndrome: A systematic literature review. Clin Epidemiol (2020) 12:295–305. doi: 10.2147/CLEP.S245642
89. Avila Bernabeu AI, Cavero Escribano T, Cao Vilarino M. Atypical Hemolytic Uremic Syndrome: New Challenges in the Complement Blockage Era. Nephron (2020) 144:537–49. doi: 10.1159/000508920
90. Yoshida Y, Kato H, Ikeda Y, Nangaku M. Pathogenesis of atypical hemolytic uremic syndrome. J Atheroscler Thromb (2019) 26(2):99–110. doi: 10.5551/jat.RV17026
91. De Jorge EG, Tortajada A, García SP, Gastoldi S, Merinero HM, García-Fernández J, et al. Factor H competitor generated by gene conversion events associates with atypical hemolytic uremic syndrome. J Am Soc Nephrol (2018) 29(1):240–9. doi: 10.1681/ASN.2017050518
92. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med (2009) 361(17):1676–87. doi: 10.1056/NEJMra0902814
93. Andrighetto S, Leventhal J, Zaza G, Cravedi P. Complement and Complement Targeting Therapies in Glomerular Diseases. Int J Mol Sci (2019) 20(24):6336. doi: 10.3390/ijms20246336
94. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic–Uremic Syndrome. N Engl J Med (2013) 368(23):2169–81. doi: 10.1056/NEJMoa1208981
95. Berger BE. Atypical hemolytic uremic syndrome: A syndrome in need of clarity. Clin Kidney J (2019) 12(3):338–47. doi: 10.1093/ckj/sfy066
96. Smith RJH, Appel GB, Blom AM, Cook HT, D’Agati VD, Fakhouri F, et al. C3 glomerulopathy — understanding a rare complement-driven renal disease. Nat Rev Nephrol (2019) 15(3):129–43. doi: 10.1038/s41581-018-0107-2
97. Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, et al. The role of complement in C3 glomerulopathy. Mol Immunol (2015) 67(1):21–30. doi: 10.1016/j.molimm.2015.03.012
98. Chauvet S, Roumenina LT, Bruneau S, Marinozzi MC, Rybkine T, Schramm EC, et al. A familial C3GN secondary to defective C3 regulation by complement receptor 1 and complement factor H. J Am Soc Nephrol (2016) 27(6):1665–77. doi: 10.1681/ASN.2015040348
99. Zhao F, Afonso S, Lindner S, Hartmann A, Löschmann I, Nilsson B, et al. C3-Glomerulopathy Autoantibodies Mediate Distinct Effects on Complement C3- and C5-Convertases. Front Immunol (2019) 10(MAY):1030. doi: 10.3389/fimmu.2019.01030
100. Schena FP, Esposito P, Rossini M. A narrative review on C3 glomerulopathy: A rare renal disease. Int J Mol Sci (2020) 21(2):1–19. doi: 10.3390/ijms21020525
101. Sethi S, Fervenza FC. Membranoproliferative Glomerulonephritis - A New Look at an Old Entity. N Engl J Med (2012) 366(12):1119–31. doi: 10.1056/NEJMra1108178
102. Williams AL, Gullipalli D, Ueda Y, Sato S, Zhou L, Miwa T, et al. C5 inhibition prevents renal failure in a mouse model of lethal C3 glomerulopathy. Kidney Int (2017) 91(6):1386–97. doi: 10.1016/j.kint.2016.11.018
103. Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol (2012) 7(5):748–56. doi: 10.2215/CJN.12901211
104. Yates M, Watts R. ANCA-associated vasculitis. Clin Med (Northfield Il) (2017) 17(1):60–4. doi: 10.7861/clinmedicine.17-1-60
105. Nakazawa D, Masuda S, Tomaru U, Ishizu A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol (2019) 15(2):91–101. doi: 10.1038/s41584-018-0145-y
106. Geetha D, Jefferson JA. ANCA-Associated Vasculitis: Core Curriculum 2020. Am J Kidney Dis (2020) 75(1):124–37. doi: 10.1053/j.ajkd.2019.04.031
107. Misra DP, Naidu GSRSNK, Agarwal V, Sharma A. Vasculitis research: Current trends and future perspectives. Int J Rheum Dis (2019) 22(July):10–20. doi: 10.1111/1756-185X.13370
108. Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J Am Soc Nephrol (2017) 28(9):2756–67. doi: 10.1681/ASN.2016111179
109. Tesar V, Hruskova Z. Avacopan in the treatment of ANCA-associated vasculitis. Expert Opin Invest Drugs (2018) 27(5):491–6. doi: 10.1080/13543784.2018.1472234
110. Brilland B, Garnier AS, Chevailler A, Jeannin P, Subra JF, Augusto JF. Complement alternative pathway in ANCA-associated vasculitis: Two decades from bench to bedside. Autoimmun Rev (2019) 19(1):102424. doi: 10.1016/j.autrev.2019.102424
111. Dmytrijuk A, Robie-Suh K, Cohen MH, Rieves D, Weiss K, Pazdur R. FDA Report: Eculizumab (Soliris®) for the Treatment of Patients with Paroxysmal Nocturnal Hemoglobinuria. Oncologist (2008) 13(9):993–1000. doi: 10.1634/theoncologist.2008-0086
112. Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Gaya A, et al. Multicenter phase 3 study of eculizumab for the treatment of PNH. Blood (2008) 111(4):1840–7. doi: 10.1182/blood-2007-06-094136
113. Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med (2006) 355(12):1233–43. doi: 10.1056/NEJMoa061648
114. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol (2007) 25(11):1256–64. doi: 10.1038/nbt1344
115. Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol (2012) 8(11):643–57. doi: 10.1038/nrneph.2012.214
116. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med (2009) 360(5):544–6. doi: 10.1056/NEJMc0809959
117. Chatelet V, Frémeaux-Bacchi V, Lobbedez T, Ficheux M, De Ligny BH. Safety and long-term efficacy of eculizumab in a renal transplant patient with recurrent atypical hemolytic-uremic syndrome. Am J Transplant (2009) 9(11):2644–5. doi: 10.1111/j.1600-6143.2009.02817.x
118. FDA. SOLIRIS® (eculizumab) injection, for intravenous use, US Food and Drug Administration. Vol. p. (2019). pp. 1–42.
119. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol (2017) 14(1):26–47. doi: 10.1038/nrneph.2017.156
120. Oyama M, Okada K, Masuda M, Shimizu Y, Yokoyama K, Uzawa A, et al. Suitable indications of eculizumab for patients with refractory generalized myasthenia gravis. Ther Adv Neurol Disord (2020) 13(6):175628642090420. doi: 10.1177/1756286420904207
121. Wijnsma KL, ter Heine R, Moes DJAR, Langemeijer S, Schols SEM, Volokhina EB, et al. Pharmacology, Pharmacokinetics and Pharmacodynamics of Eculizumab, and Possibilities for an Individualized Approach to Eculizumab. Clin Pharmacokinet (2019) 58(7):859–74. doi: 10.1007/s40262-019-00742-8
122. Frampton JE. Eculizumab: A Review in Neuromyelitis Optica Spectrum Disorder. Drugs (2020) 80(7):719–27. doi: 10.1007/s40265-020-01297-w
123. Zelek WM, Xie L, Morgan BP, Harris CL. Compendium of current complement therapeutics. Mol Immunol (2019) 114(August):341–52. doi: 10.1016/j.molimm.2019.07.030
124. Castañeda-Sanabria J, Hajage D, Le Jouan M, Perozziello A, Tubach F. Off-label use of the expensive orphan drug eculizumab in France 2009–2013 and the impact of literature: focus on the transplantation field. Eur J Clin Pharmacol (2016) 72(6):737–46. doi: 10.1007/s00228-016-2027-z
125. Makishima K, Obara N, Ishitsuka K, Sukegawa S, Suma S, Kiyoki Y, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol (2019) 98(4):1031–2. doi: 10.1007/s00277-018-3521-4
126. Chanchlani R, Thorner P, Radhakrishnan S, Hebert D, Langlois V, Arora S, et al. Long-term Eculizumab Therapy in a Child With Refractory Immune Complex–Mediated Membranoproliferative Glomerulonephritis. Kidney Int Rep (2018) 3(2):482–5. doi: 10.1016/j.ekir.2017.08.019
127. Glotz D, Russ G, Rostaing L, Legendre C, Tufveson G, Chadban S, et al. Safety and efficacy of eculizumab for the prevention of antibody-mediated rejection after deceased-donor kidney transplantation in patients with preformed donor-specific antibodies. Am J Transplant (2019) 19(10):2865–75. doi: 10.1111/ajt.15397
128. Jodele S, Fukuda T, Mizuno K, Vinks AA, Laskin BL, Goebel J, et al. Variable Eculizumab Clearance Requires Pharmacodynamic Monitoring to Optimize Therapy for Thrombotic Microangiopathy after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant (2016) 22(2):307–15. doi: 10.1016/j.bbmt.2015.10.002
129. Gatault P, Brachet G, Ternant D, Degenne D, Récipon G, Barbet C, et al. Therapeutic drug monitoring of eculizumab: Rationale for an individualized dosing schedule. MAbs (2015) 7(6):1205–11. doi: 10.1080/19420862.2015.1086049
130. Coyle D, Cheung MC, Evans GA. Opportunity cost of funding drugs for rare diseases: The cost-effectiveness of eculizumab in paroxysmal nocturnal hemoglobinuria. Med Decis Making (2014) 34(8):1016–29. doi: 10.1177/0272989X14539731
131. Khedraki R, Noor Z, Rick J. The Most Expensive Drug in the World: To Continue or Discontinue, That Is the Question. Fed Pract (2016) 33(7):22–8.
132. Chow V, Pan J, Chien D, Mytych DT, Hanes V. A randomized, double-blind, single-dose, three-arm, parallel group study to determine pharmacokinetic similarity of ABP 959 and eculizumab (Soliris ® ) in healthy male subjects. Eur J Haematol (2020) 105(1):66–74. doi: 10.1111/ejh.13411
133. NCT04060264. Clinical Trial of BCD-148 and Soliris® for the Treatment of Patients With Paroxysmal Nocturnal Hemoglobinuria. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04060264 (Accessed 2020 Jun 9).
134. Kulagin A, Ptushkin V, Lukina E, Gapchenko E, Markova O, Zuev E, et al. Phase III Clinical Trial of Elizaria® and Soliris® in Adult Patients with Paroxysmal Nocturnal Hemoglobinuria: Results of Comparative Analysis of Efficacy, Safety, and Pharmacological Data. Blood (2019) 134(Supplement_1):3748–8. doi: 10.1182/blood-2019-125693
135. Kawakami T, Nakazawa H, Kurasawa Y, Sakai H, Nishina S, Senoo N, et al. Severe Infection of Pseudomonas aeruginosa during Eculizumab Therapy for Paroxysmal Nocturnal Hemoglobinuria. Intern Med (2018) 57(1):127–30. doi: 10.2169/internalmedicine.9151-17
136. Hawkins KL, Hoffman M, Okuyama S, Rowan SE. A Case of Fulminant Meningococcemia: It Is All in the Complement. Case Rep Infect Dis (2017) 2017:1–3. doi: 10.1155/2017/6093695
137. Heesterbeek DAC, Angelier ML, Harrison RA, Rooijakkers SHM. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J Innate Immun (2018) 10(5–6):455–64. doi: 10.1159/000491439
138. McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High Risk for Invasive Meningococcal Disease Among Patients Receiving Eculizumab (Soliris) Despite Receipt of Meningococcal Vaccine. Am J Transplant (2017) 17(9):2481–4. doi: 10.1111/ajt.14426
139. Konar M, Granoff DM. Eculizumab treatment and impaired opsonophagocytic killing of meningococci by whole blood from immunized adults. Blood (2017) 130(7):891–9. doi: 10.1182/blood-2017-05-781450
140. Harder MJ, Kuhn N, Schrezenmeier H, Höchsmann B, Von Zabern I, Weinstock C, et al. Incomplete inhibition by eculizumab: Mechanistic evidence for residual C5 activity during strong complement activation. Blood (2017) 129(8):970–80. doi: 10.1182/blood-2016-08-732800
141. Nishimura JI, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med (2014) 370(7):632–9. doi: 10.1056/NEJMoa1311084
142. Reddy YNV, Siedlecki AM, Francis JM. Breaking down the complement system: A review and update on novel therapies. Curr Opin Nephrol Hypertens (2017) 26(2):123–8. doi: 10.1097/MNH.0000000000000305
143. Stern RM, Connell NT. Ravulizumab: a novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Ther Adv Hematol (2019) 10(6):204062071987472. doi: 10.1177/2040620719874728
144. Sheridan D, Yu Z-X, Zhang Y, Patel R, Sun F, Lasaro MA, et al. Design and preclinical characterization of ALXN1210: A novel anti-C5 antibody with extended duration of action. Stepkowski S, editor. PloS One (2018) 13(4):e0195909. doi: 10.1371/journal.pone.0195909
145. Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez-Fernandez FA, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor–experienced adult patients with PNH: the 302 study. Blood (2019) 133(6):540–9. doi: 10.1182/blood-2018-09-876805
146. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, Pessoa V, Gualandro S, Füreder W, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood (2019) 133(6):530–9. doi: 10.1182/blood-2018-09-876136
147. Brodsky RA, Peffault de Latour R, Rottinghaus ST, Röth A, Risitano AM, Weitz IC, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica (2020), haematol.2019.236877. doi: 10.3324/haematol.2019.236877
148. Rondeau E, Scully M, Ariceta G, Barbour T, Cataland S, Heyne N, et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int (2020) 97(6):1287–96. doi: 10.1016/j.kint.2020.01.035
149. Food and Drug Administration. Ultomiris: Full Prescribing Information (Ravulizumab-cwvz). (2018), 1–18.
150. Yu Z-Y, Tsai M-J, Lin Y-J, Liu W-D, Chou S-C, Hung C-C. Disseminated gonococcal infection in a patient with paroxysmal nocturnal hemoglobinuria having received ravulizumab and meningococcal vaccine. J Microbiol Immunol Infect (2020) 53(4):660–2. doi: 10.1016/j.jmii.2020.06.013
151. NCT04557735. Study of Ravulizumab in Pediatric Participants With HSCT-TMA. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04557735 (Accessed 2020 Oct 30).
152. NCT04564339. Study of Ravulizumab in Proliferative Lupus Nephritis (LN) or Immunoglobulin A Nephropathy (IgAN). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04564339 (Accessed 2020 Oct 30).
153. NCT04543591. Ravulizumab in Thrombotic Microangiopathy After Hematopoietic Stem Cell Transplant. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04543591 (Accessed 2020 Oct 30).
154. Devalaraja-Narashimha K, Ni YG, Huang C, Wang M-D, Chaudhari U, Prasad S, et al. Pozelimab, a Human Antibody Against Complement Factor C5, Demonstrates Robust Inhibition of Alternative Complement Activity Both in Normal Human Serum and in Phase I Normal Healthy Volunteers. Blood (2019) 134(Supplement_1):2278–8. doi: 10.1182/blood-2019-129865
155. Latuszek A, Liu Y, Olsen O, Foster R, Cao M, Lovric I, et al. Inhibition of complement pathway activation with Pozelimab, a fully human antibody to complement component C5. PLoS One (2020) 15(5):1–23. doi: 10.1371/journal.pone.0231892
156. Weyne J, Ni Y, DelGizzi R, Godin S, Morton L, Prasad S, et al. A Randomized, Double-Blind, Placebo-Controlled Phase 1 Study of the Pharmacokinetics and Pharmacodynamics of REGN3918, a Human Antibody Against Complement Factor C5, in Healthy Volunteers. Blood (2018) 132(Supplement 1):1039–9. doi: 10.1182/blood-2018-99-112262
157. Ozen A, Comrie WA, Ardy RC, Domínguez Conde C, Dalgic B, Beser ÖF, et al. CD55 Deficiency, Early-Onset Protein-Losing Enteropathy, and Thrombosis. N Engl J Med (2017) 377(1):52–61. doi: 10.1056/NEJMoa1615887
158. NCT04209634. Open-Label Efficacy and Safety Study of Pozelimab in Patients With CD55-Deficient Protein-Losing Enteropathy (CHAPLE Disease). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04209634?term=pozelimab&draw=2&rank=1 (Accessed 2020 May 27).
159. Kassa E, Ciulla TA, Hussain RM, Dugel PU. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin Biol Ther (2019) 19(4):335–42. doi: 10.1080/14712598.2019.1575358
160. NCT02763644. Efficacy and Safety of LFG316 in Transplant Associated Microangiopathy (TAM) Patients. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT02763644?term=LFG316&draw=2&rank=1 (Accessed 2020 May 15).
161. NCT01526889. Safety,Tolerability and Efficacy of Intravitreal LFG316 in Patients With Active Non-infectious Intermediate-, posterior-or Panuveitis. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/results/NCT01526889?term=LFG316&draw=2&rank=9 (Accessed 2020 May 15).
162. Nebbioso M, Lambiase A, Cerini A, Limoli PG, La Cava M, Greco A. Therapeutic Approaches with Intravitreal Injections in Geographic Atrophy Secondary to Age-Related Macular Degeneration: Current Drugs and Potential Molecules. Int J Mol Sci (2019) 20(7):1693. doi: 10.3390/ijms20071693
163. NCT02534909. Proof of Concept Study to Assess the Efficacy, Safety and Pharmacokinetics of LFG316 in Patients With Paroxysmal Nocturnal Hemoglobinuria. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT02534909 (Accessed 2020 May 15).
164. Jordan SC, Kucher K, Bagger M, Hockey H, Wagner K, Ammerman N, et al. Intravenous immunoglobulin significantly reduces exposure of concomitantly administered anti-C5 monoclonal antibody tesidolumab. Am J Transplant (2020) 20(9):2581–8. doi: 10.1111/ajt.15922
165. Fukuzawa T, Nezu J. SKY59, A Novel Recycling Antibody for Complement-mediated Diseases. Curr Med Chem (2020) 27(25):4157–64. doi: 10.2174/0929867326666191016115853
166. Sampei Z, Haraya K, Tachibana T, Fukuzawa T, Shida-Kawazoe M, Gan SW, et al. Antibody engineering to generate SKY59, a long-acting anti-C5 recycling antibody. PLoS One (2018) 13(12):1–20. doi: 10.1371/journal.pone.0209509
167. Fukuzawa T, Sampei Z, Haraya K, Ruike Y, Shida-Kawazoe M, Shimizu Y, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci Rep (2017) 7(1):1–12. doi: 10.1038/s41598-017-01087-7
168. Zelek WM, Stott M, Walters D, Harris CL, Morgan BP. Characterizing a pH-switch anti-C5 antibody as a tool for human and mouse complement C5 purification and cross-species inhibition of classical and reactive lysis. Immunology (2018) 155(3):396–403. doi: 10.1111/imm.12982
169. Risitano AM, Marotta S, Ricci P, Marano L, Frieri C, Cacace F, et al. Anti-complement Treatment for Paroxysmal Nocturnal Hemoglobinuria: Time for Proximal Complement Inhibition? A Position Paper From the SAAWP of the EBMT. Front Immunol (2019) 10(JUN):1157. doi: 10.3389/fimmu.2019.01157
170. Röth A, Nishimura JI, Nagy Z, Gaàl-Weisinger J, Panse J, Yoon SS, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood (2020) 135(12):912–20. doi: 10.1182/blood.2019003399
171. Arkin MMR, Wells JA. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat Rev Drug Discov (2004) 3(4):301–17. doi: 10.1038/nrd1343
172. Guan M, Zhou Y-P, Sun J-L, Chen S-C. Adverse Events of Monoclonal Antibodies Used for Cancer Therapy. BioMed Res Int (2015) 2015:1–13. doi: 10.1155/2015/428169
173. Awwad S, Angkawinitwong U. Overview of antibody drug delivery. Pharmaceutics (2018) 10(3):1–24. doi: 10.3390/pharmaceutics10030083
174. Samaranayake H, Wirth T, Schenkwein D, Räty JK, Ylä-Herttuala S. Challenges in monoclonal antibody-based therapies. Ann Med (2009) 41(5):322–31. doi: 10.1080/07853890802698842
175. Harder MJ, Höchsmann B, Dopler A, Anliker M, Weinstock C, Skerra A, et al. Different Levels of Incomplete Terminal Pathway Inhibition by Eculizumab and the Clinical Response of PNH Patients. Front Immunol (2019) 10(JULY):1–7. doi: 10.3389/fimmu.2019.01639
176. Sezin T, Murthy S, Attah C, Seutter M, Holtsche MM, Hammers CM, et al. Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight (2019) 4(15):1–13. doi: 10.1172/jci.insight.128239
177. Schols S, Nunn MA, Mackie I, Weston-Davies W, Nishimura J II, Kanakura Y, et al. Successful treatment of a PNH patient non-responsive to eculizumab with the novel complement C5 inhibitor coversin (nomacopan). Br J Haematol (2020) 188(2):334–7. doi: 10.1111/bjh.16305
178. Goodship THJ, Pinto F, Weston-Davies WH, Silva J, Nishimura JI, Nunn MA, et al. Use of the complement inhibitor Coversin to treat HSCT-associated TMA. Blood Adv (2017) 1(16):1254–8. doi: 10.1182/bloodadvances.2016002832
179. NCT03427060. Coversin in PNH in Patients With Resistance to Eculizumab Due to Complement C5 Polymorphisms (CONSENTII). ClinicalTrials.gov. A service of the U.S. National Institutes of Health . Available at: https://clinicaltrials.gov/ct2/show/NCT03427060?term=coversin&draw=2&rank=2 (Accessed 2020 May 18).
180. NCT03588026. Treating Paroxysmal Nocturnal Haemoglobinuria Patients With rVA576 (CAPSTONE). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT03588026?term=coversin&draw=2&rank=5 (Accessed 2020 May 18).
181. NCT03829449. rVA576 (Coversin) Long Term Safety and Efficacy Surveillance Study (CONSERVE). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT03829449?term=coversin&draw=2&rank=1 (Accessed 2020 May 18).
182. Barratt-Due A, Thorgersen EB, Egge K, Pischke S, Sokolov A, Hellerud BC, et al. Combined Inhibition of Complement (C5) and CD14 Markedly Attenuates Inflammation, Thrombogenicity, and Hemodynamic Changes in Porcine Sepsis. J Immunol (2013) 191(2):819–27. doi: 10.4049/jimmunol.1201909
183. Kuhn N, Schmidt CQ, Schlapschy M, Skerra A. PASylated Coversin, a C5-Specific Complement Inhibitor with Extended Pharmacokinetics, Shows Enhanced Anti-Hemolytic Activity in Vitro. Bioconjug Chem (2016) 27(10):2359–71. doi: 10.1021/acs.bioconjchem.6b00369
184. Miles N, Skerra A. Therapeutic Development of Complement C5 Inhibitor CoversinTM with Extended Half-Life Via PASylation®. Blood (2016) 128(22):5900–5900. doi: 10.1182/blood.V128.22.5900.5900
185. Kusner LL, Yucius K, Sengupta M, Sprague AG, Desai D, Nguyen T, et al. Investigational RNAi Therapeutic Targeting C5 Is Efficacious in Pre-clinical Models of Myasthenia Gravis. Mol Ther Methods Clin Dev (2019) 13(June):484–92. doi: 10.1016/j.omtm.2019.04.009
186. NCT02352493. A Phase 1/2 Study of an Investigational Drug, ALN-CC5, in Healthy Adult Volunteers and Patients With PNH. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/results/NCT02352493?term=Cemdisiran&draw=2&rank=4 (Accessed 2020 May 19).
187. NCT04601844. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Pozelimab in Combination With Cemdisiran in Healthy Adult Volunteers. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04601844 (Accessed 2020 Oct 30).
188. NCT03303313. A Study of an Investigational Drug, Cemdisiran (ALN-CC5), in Patients With Atypical Hemolytic Uremic Syndrome. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT03303313?term=Cemdisiran&draw=1&rank=3 (Accessed 2020 May 19).
189. NCT03999840. Eculizumab to Cemdisiran Switch in aHUS (DANCE). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT03999840?term=Cemdisiran&draw=1&rank=1 (Accessed 2020 May 19).
190. Muenstermann M, Strobel L, Klos A, Wetsel RA, Woodruff TM, Köhl J, et al. Distinct roles of the anaphylatoxin receptors C3aR, C5aR1 and C5aR2 in experimental meningococcal infections. Virulence (2019) 10(1):677–94. doi: 10.1080/21505594.2019.1640035
191. Riedemann NC, Habel M, Ziereisen J, Hermann M, Schneider C, Wehling C, et al. Controlling the anaphylatoxin C5a in diseases requires a specifically targeted inhibition. Clin Immunol (2017) 180:25–32. doi: 10.1016/j.clim.2017.03.012
192. Alousi AM, Yao B, Overman B, Ratanatharathorn V, Gill S, Socié G, et al. (2016). Phase 2A Study of ALXN1007, A Novel C5A Inhibitor, in Subjects with Newly Diagnosed Acute Graft-Versus-Host Disease (GVHD) Involving the Lower Gastrointestinal Tract, in: 21st Congress of European Hematology Association, p. LB2269.
193. NCT02245412. A Phase 2A Study of ALXN1007 in Participants With Newly Diagnosed Acute Lower Gastrointestinal Graft-Versus-Host Disease (GIGVHD). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT02245412 (Accessed 2020 May 22).
194. NCT02128269. Phase IIa Trial of ALXN1007 for the Treatment of Non-criteria Manifestations of Antiphospholipid Syndrome. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT02128269?term=ALXN1007&draw=2&rank=1 (Accessed 2020 May 23).
195. NCT02246595. Studying Complement Inhibition in Early, Newly Developing Septic Organ Dysfunction (SCIENS). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT02246595?term=ifx-1&draw=1&rank=8 (Accessed 2020 May 23).
196. NCT02866825. Studying Complement Inhibition in Complex Cardiac Surgery (CARDIAC). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT02866825?term=ifx-1&draw=1&rank=7 (Accessed 2020 May 23).
197. Lim SYD, Oon HH. Systematic review of immunomodulatory therapies for hidradenitis suppurativa. Biol Targets Ther (2019) 13:53–78. doi: 10.2147/BTT.S199862
198. Theut Riis P, Thorlacius LR, Jemec GB. Investigational drugs in clinical trials for Hidradenitis Suppurativa. Expert Opin Invest Drugs (2018) 27(1):43–53. doi: 10.1080/13543784.2018.1412430
199. Giamarellos-Bourboulis EJ, Argyropoulou M, Kanni T, Spyridopoulos T, Otto I, Zenker O, et al. Clinical efficacy of complement C5a inhibition by IFX-1 in hidradenitis suppurativa: an open-label single-arm trial in patients not eligible for adalimumab. Br J Dermatol (2020) 183(1):176–8. doi: 10.1111/bjd.18877
200. NCT03712345. Safety and Efficacy Study of IFX-1 in add-on to Standard of Care in Granulomatosis With Polyangiitis (GPA) and Microscopic Polyangiitis (MPA). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT03712345?term=ifx-1&draw=1&rank=2 (Accessed 2020 May 23).
201. NCT03895801. Study of IFX-1 to Replace Steroids in Patients With Granulomatosis With Polyangiitis and Microscopic Polyangiitis. (IXchange). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT03895801?term=ifx-1&draw=2&rank=5 (Accessed 2020 May 23).
202. Sun S, Zhao G, Liu C, Fan W, Zhou X, Zeng L, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis (2015) 60(4):586–95. doi: 10.1093/cid/ciu887
203. NCT04333420. Open-label, Randomized Study of IFX-1 in Patients With Severe COVID-19 Pneumonia (PANAMO). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04333420?term=ifx-1&draw=2&rank=4 (Accessed 2020 May 23).
204. Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol (2014) 25(2):225–31. doi: 10.1681/ASN.2013020143
205. Bekker P, Dairaghi D, Seitz L, Leleti M, Wang Y, Ertl L, et al. Characterization of pharmacologic and pharmacokinetic properties of CCX168, a potent and selective orally administered complement 5a receptor inhibitor, based on preclinical evaluation and randomized Phase 1 clinical study. PLoS One (2016) 11(10):1–19. doi: 10.1371/journal.pone.0164646
206. Merkel PA, Jayne DR, Wang C, Hillson J, Bekker P. Evaluation of the Safety and Efficacy of Avacopan, a C5a Receptor Inhibitor, in Patients With Antineutrophil Cytoplasmic Antibody–Associated Vasculitis Treated Concomitantly With Rituximab or Cyclophosphamide/Azathioprine: Protocol for a Randomized, Doubl. JMIR Res Protoc (2020) 9(4):e16664. doi: 10.2196/16664
207. ChemoCentryx Inc. ChemoCentryx and VFMCRP Announce Positive Topline Data from Pivotal Phase III ADVOCATE Trial Demonstrating Avacopan’s Superiority Over Standard of Care in ANCA-Associated Vasculitis (2019). Available at: https://www.globenewswire.com/news-release/2019/11/25/1952242/0/en/ChemoCentryx-and-VFMCRP-Announce-Positive-Topline-Data-from-Pivotal-Phase-III-ADVOCATE-Trial-Demonstrating-Avacopan-s-Superiority-Over-Standard-of-Care-in-ANCA-Associated-Vasculiti.html (Accessed 2020 May 6).
208. Bruchfeld A, Nachman P, Parikh S, Lafayette R, Potarca A, Diehl J, et al. C5a Receptor Inhibitor Avacopan in IgA Nephropathy Study. Nephrol Dial Transplant (2017) 32(suppl_3):iii82–3. doi: 10.1093/ndt/gfx129.TO012
209. NCT02464891. Complement Inhibition in aHUS Dialysis Patients (ACCESS). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT02464891?term=ccx168&draw=2&rank=4.
210. NCT03301467. Controlled Trial Evaluating Avacopan in C3 Glomerulopathy (ACCOLADE). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT03301467?term=ccx168&draw=2&rank=2 (Accessed 2020 May 24).
211. Howard JF, Nowak RJ, Wolfe GI, Freimer ML, Vu TH, Hinton JL, et al. Clinical Effects of the Self-administered Subcutaneous Complement Inhibitor Zilucoplan in Patients With Moderate to Severe Generalized Myasthenia Gravis. JAMA Neurol (2020) 77(5):582. doi: 10.1001/jamaneurol.2019.5125
212. NCT04115293. Safety, Tolerability, and Efficacy of Zilucoplan in Subjects With Generalized Myasthenia Gravis (RAISE). ClinicalTrials.gov. A service of the U.S. National Institutes of Health . Available at: https://clinicaltrials.gov/ct2/show/NCT04115293?term=zilucoplan&draw=2&rank=4 (Accessed 2020 May 18).
213. NCT04025632. Safety and Efficacy Study of Zilucoplan in Subjects With Immune-Mediated Necrotizing Myopathy. ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04025632 (Accessed 2020 May 18).
215. Hill A, Schrezenmeier H, Hillmen P, Szer J, Pullon H, Spearing R, et al. Ra101495, a subcutaneously-administered peptide inhibitor of complement component C5, for the treatment of paroxysmal nocturnal hemoglobinuria: Phase 2 results. HemaSphere (2018) 2(Supplement 2):105.
216. UCB Pharma. R&D pipeline. (2020). Available at: https://www.ucb.com/our-science/pipeline (Accessed 2020 May 19).
217. Wilkinson T, Dixon R, Page C, Carroll M, Griffiths G, Ho L-P, et al. ACCORD: A Multicentre, Seamless, Phase 2 Adaptive Randomisation Platform Study to Assess the Efficacy and Safety of Multiple Candidate Agents for the Treatment of COVID-19 in Hospitalised Patients: A structured summary of a study protocol for a randomised. Trials (2020) 21(1):691. doi: 10.1186/s13063-020-04584-9
218. Drolet DW, Green LS, Gold L, Janjic N. Fit for the Eye: Aptamers in Ocular Disorders. Nucleic Acid Ther (2016) 26(3):127–46. doi: 10.1089/nat.2015.0573
219. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov (2010) 9(7):537–50. doi: 10.1038/nrd3141
220. NCT03374670. ZIMURA in Combination With Eylea in Patients With Idiopathic Polypoidal Choroidal Vasculopathy (IPCV). ClinicalTrials.gov. A service of the U.S. National Institutes of Health . Available at: https://clinicaltrials.gov/ct2/show/NCT03374670?term=zimura&draw=1&rank=2 (Accessed 2020 May 22).
221. Demaria O, Rubio L, Belaid N, Habif G, Bonnafous C, Zerbib R, et al. (2017). Characterization of anti-C5aR antibodies for specific targeting of myeloid cells and neutrophils in the TME, in: Third CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference: Translating Science into Survival, . p. B184.
222. Massard C, Cassier P, Bendell JC, Marie DB, Blery M, Morehouse C, et al. Preliminary results of STELLAR-001, a dose escalation phase I study of the anti-C5aR, IPH5401, in combination with durvalumab in advanced solid tumours. Ann Oncol (2019) 30(Supplement 5):v492. doi: 10.1093/annonc/mdz253.029
223. NCT04563923. Treatment of Bullous Pemphigoid With Avdoralimab (IPH5401), an Anti-C5aR1 Monoclonal Antibody (IPH). Available at: https://clinicaltrials.gov/ct2/show/NCT04563923?term=Avdoralimab&draw=2&rank=2 (Accessed 2020 Oct 20).
224. NCT04371367. Avdoralimab an Anti-C5aR Antibody, in Patients With COVID-19 Severe Pneumonia (FORCE). ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available at: https://clinicaltrials.gov/ct2/show/NCT04371367 (Accessed 2020 May 23).
225. Hemera Biosciences. What is HMR59? Available at: https://www.hemerabiosciences.com/hmr59/ (Accessed 2020 May 26).
226. NCT03144999. Treatment of Advanced Dry Age Related Macular Degeneration With AAVCAGsCD59. ClinicalTrials.gov. A service of the U.S. National Institutes of Health . Available at: https://clinicaltrials.gov/ct2/show/NCT03144999?term=AAVCAGsCD59&draw=2&rank=3 (Accessed 2020 May 26).
227. NCT04358471. Intravitreal AAVCAGsCD59 for Advanced Dry Age-related Macular Degeneration (AMD) With Geographic Atrophy (GA). ClinicalTrials.gov. A service of the U.S. National Institutes of Health (Accessed 2020 May 26).
228. Wang R, Xiao H, Guo R, Li Y, Shen B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg Microbes Infect (2015) 4(1):1–7. doi: 10.1038/emi.2015.28
229. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Differ (2020) 27(5):1451–4. doi: 10.1038/s41418-020-0530-3
230. Liu PP, Blet A, Smyth D, Li H. The Science Underlying COVID-19. Circulation (2020) 142(1):68–78. doi: 10.1161/CIRCULATIONAHA.120.047549
231. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol (2020) 17(5):533–5. doi: 10.1038/s41423-020-0402-2
232. Garcia CC, Weston-Davies W, Russo RC, Tavares LP, Rachid MA, Alves-Filho JC, et al. Complement C5 Activation during Influenza A Infection in Mice Contributes to Neutrophil Recruitment and Lung Injury. PloS One (2013) 8(5):1–11. doi: 10.1371/journal.pone.0064443
233. Ohta R, Torii Y, Imai M, Kimura H, Okada N, Ito Y. Serum concentrations of complement anaphylatoxins and proinflammatory mediators in patients with 2009 H1N1 influenza. Microbiol Immunol (2011) 55(3):191–8. doi: 10.1111/j.1348-0421.2011.00309.x
234. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio (2018) 9(5):1–15. doi: 10.1128/mBio.01753-18
235. Jiang Y, Li J, Teng Y, Sun H, Tian G, He L, et al. Complement Receptor C5aR1 Inhibition Reduces Pyroptosis in hDPP4-Transgenic Mice Infected with MERS-CoV. Viruses (2019) 11(1):39. doi: 10.3390/v11010039
236. Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci (2020) 24(7):4040–7. doi: 10.26355/eurrev_202004_20875
237. Kulkarni S, Fisk M, Kostapanos M, Banham-Hall E, Bond S, Hernan-Sancho E, et al. Repurposed immunomodulatory drugs for Covid-19 in pre-ICu patients - mulTi-Arm Therapeutic study in pre-ICu patients admitted with Covid-19 – Repurposed Drugs (TACTIC-R): A structured summary of a study protocol for a randomised controlled trial. Trials (2020) 21(1):626. doi: 10.1186/s13063-020-04535-4
238. Walters GD, Willis NS, Cooper TE, Craig JC. Interventions for renal vasculitis in adults. Cochrane Database Syst Rev (2020) 1:CD003232. doi: 10.1002/14651858.CD003232.pub4
239. Liu D, Ahmet A, Ward L, Krishnamoorthy P, Mandelcorn ED, Leigh R, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol (2013) 9(1):1–25. doi: 10.1186/1710-1492-9-30
240. Samanen J. Similarities and differences in the discovery and use of biopharmaceuticals and small-molecule chemotherapeutics. In: Introduction to Biological and Small Molecule Drug Research and Development. Oxford: Elsevier (2013). p. 161–203. doi: 10.1016/B978-0-12-397176-0.00005-4
Keywords: complement system, C5 antagonist, C5aR1 antagonist, IgA nephropathy, aHUS, ANCA-associated vasculitis, lupus nephritis, C3 glomerulopathy
Citation: Anliker-Ort M, Dingemanse J, van den Anker J and Kaufmann P (2020) Treatment of Rare Inflammatory Kidney Diseases: Drugs Targeting the Terminal Complement Pathway. Front. Immunol. 11:599417. doi: 10.3389/fimmu.2020.599417
Received: 27 August 2020; Accepted: 09 November 2020;
Published: 10 December 2020.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Marina Noris, Mario Negri Pharmacological Research Institute (IRCCS), ItalyCopyright © 2020 Anliker-Ort, Dingemanse, van den Anker and Kaufmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marion Anliker-Ort, bWFyaW9uLm9ydEBpZG9yc2lhLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.