
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 January 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.595818
This article is part of the Research TopicNew Insights into the Complexity of Tumor Immunology in B-cell Malignancies: Tumor Immunology and ImmunotherapyView all 13 articles
 Ebru Aydin1,2
Ebru Aydin1,2 Sebastian Faehling1,3
Sebastian Faehling1,3 Mariam Saleh1,4
Mariam Saleh1,4 Laura Llaó Cid1,5
Laura Llaó Cid1,5 Martina Seiffert1
Martina Seiffert1 Philipp M. Roessner1*
Philipp M. Roessner1*Phosphoinositide 3-kinases (PI3Ks) and their downstream proteins constitute a signaling pathway that is involved in both normal cell growth and malignant transformation of cells. Under physiological conditions, PI3K signaling regulates various cellular functions such as apoptosis, survival, proliferation, and growth, depending on the extracellular signals. A deterioration of these extracellular signals caused by mutational damage in oncogenes or growth factor receptors may result in hyperactivation of this signaling cascade, which is recognized as a hallmark of cancer. Although higher activation of PI3K pathway is common in many types of cancer, it has been therapeutically targeted for the first time in chronic lymphocytic leukemia (CLL), demonstrating its significance in B-cell receptor (BCR) signaling and malignant B-cell expansion. The biological activity of the PI3K pathway is not only limited to cancer cells but is also crucial for many components of the tumor microenvironment, as PI3K signaling regulates cytokine responses, and ensures the development and function of immune cells. Therefore, the success or failure of the PI3K inhibition is strongly related to microenvironmental stimuli. In this review, we outline the impacts of PI3K inhibition on the tumor microenvironment with a specific focus on CLL. Acknowledging the effects of PI3K inhibitor-based therapies on the tumor microenvironment in CLL can serve as a rationale for improved drug development, explain treatment-associated adverse events, and suggest novel combinatory treatment strategies in CLL.
The phosphoinositide 3-kinase (PI3K) family mediates nutrient sensing and metabolic control to prevent cell growth and proliferation in conditions of energy or nutrient deficiency. Several roles are ascribed to PI3K signaling both in normal cell function and cancer. The four best-characterized pathways that activate PI3K are cytokine/chemokine receptors, receptor tyrosine kinases (RTK), B-cell receptor (BCR), and G-protein coupled receptors (GPCR) (Figure 1). Stimulation of these receptors causes the autophosphorylation of tyrosine residues on immunoreceptor tyrosine-based activation motifs (ITAMs) that are important mediators of signal transduction in immune cells, leading to the activation of a PI3K p110 catalytic isoform.
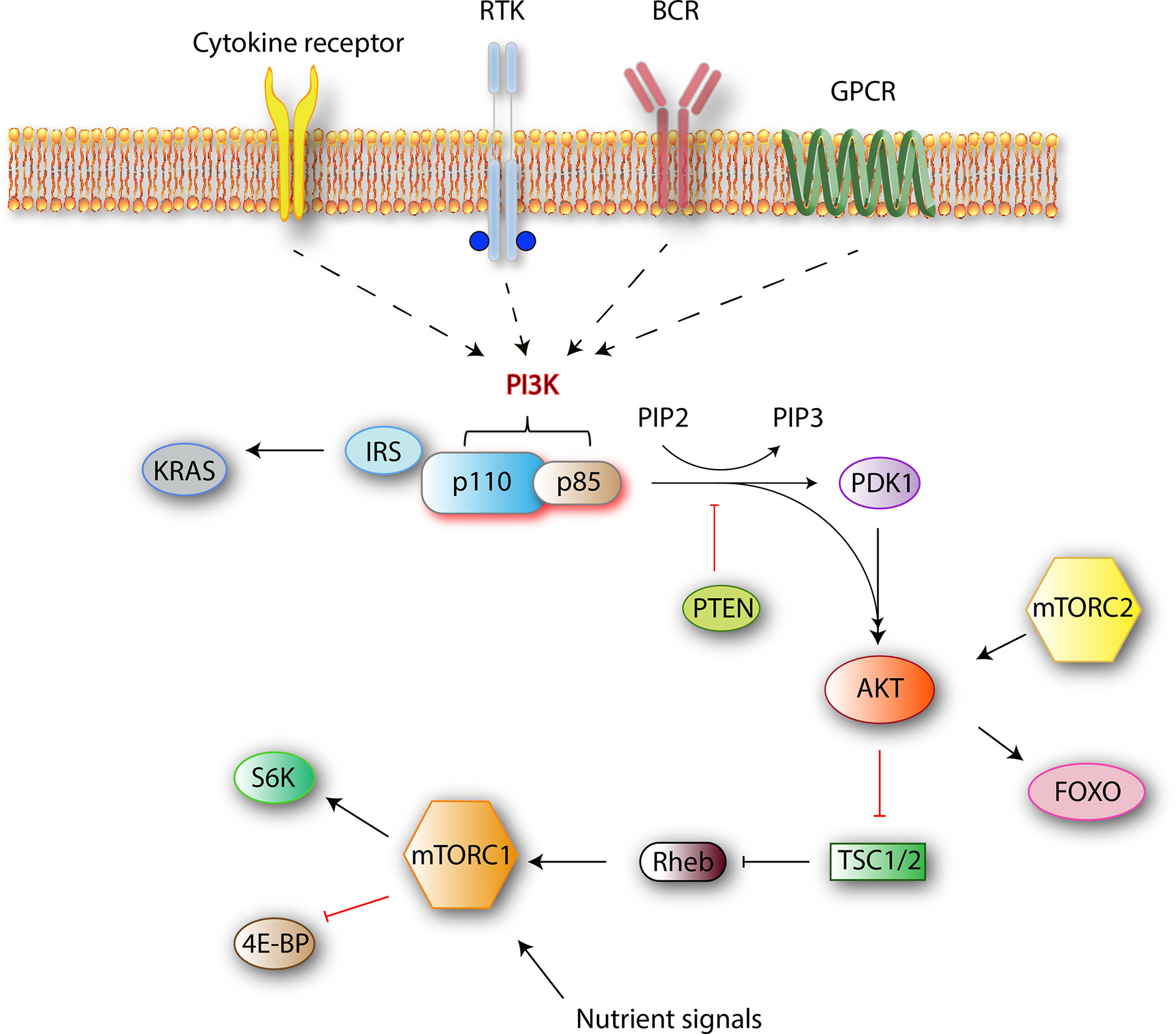
Figure 1 Class I PI3K signaling pathway simplified. Class I PI3Ks reside as dimers in the cells. After receiving an external stimulus, the regulatory p85 subunit interacts with the intracellular section of the activated receptor. The activation of a catalytic p110 isoform triggers a lipid membrane-associated cascade of phosphorylations (PIP2 to PIP3). The PI3K pathway is mediated mainly by PIP3, which is a secondary messenger that acts as a docking site. In the PI3K-AKT pathway, PIP3 can bind to both downstream effector proteins PDK1 and AKT. AKT modification activates the mammalian target of rapamycin complex 1 (mTORC1) by direct phosphorylation, which results in synthesis of growth-, proliferation-, and survival-related proteins (1).
PI3Ks phosphorylate the 3-position hydroxy group of the phosphatidylinositol ring. The resulting products are the secondary messengers phosphatidylinositol(4,5)P2 (PIP2) and phosphatidylinositol(3,4,5)P3 (PIP3), which trigger important physiological changes in cells through AKT/Protein Kinase B (PKB) phosphorylation and downstream kinases. One of those, mammalian target of rapamycin complex 1 (mTORC1), which is a central mediator of the metabolic control, combines signals from nutrients and PI3K to induce cell growth and proliferation. Major components of this pathway are outlined in Figure 1.
When a ligand binds to RTK, two RTK monomers dimerize, which results in the activation of intracellular tyrosine kinase domain and auto phosphorylation followed by indirect PI3K activation via adapter molecules such as the insulin receptor substrate (IRS). Alternatively, BCR-dependent activation of PI3Ks is mediated by Src and Syk family of receptor-associated tyrosine kinases. First, Src-family proteins phosphorylate the tyrosine residues of ITAMs that reside on the cytoplasmic part of the signal transducing subunits of the BCR-associated Ig-α and Ig-β. Phosphorylated ITAMs serve as binding sites for Src-homology 2 (SH2) domain-containing proteins such as B-cell PI3K adaptor protein (BCAP) and CD19 (Figure 2). With the help of these proteins, PI3Ks are recruited to the BCR signalosome (2, 3). Next, the regulatory subunit interacts with the intracellular section of the activated receptor via its SH2 domain and this event leads to the activation of catalytic p110 isoform which triggers a lipid membrane-associated cascade of phosphorylations (PIP2 to PIP3). The PI3K pathway is mediated mainly by PIP3, which is a secondary messenger that acts as a docking site. In the PI3K-AKT pathway, PIP3 can bind to both downstream effector proteins phosphoinositide dependent kinase (PDK1) and AKT. AKT modification activates the mTORC1 by direct phosphorylation, which results in synthesis of growth, proliferation-, and survival-related proteins (1).
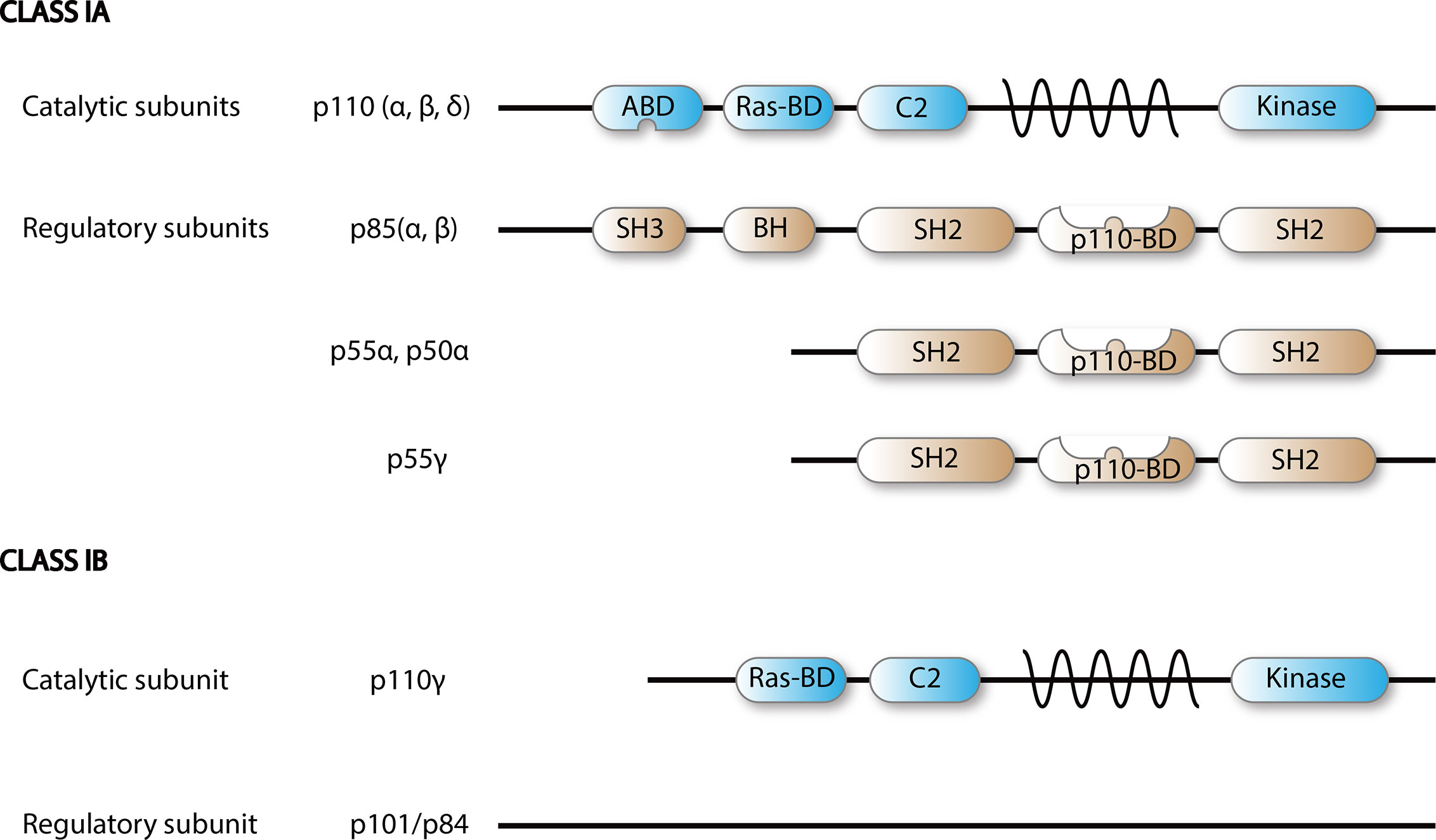
Figure 2 Scheme of PI3K class I isoforms. Functional PI3K is built upon dimerization of a catalytic p110 and a regulatory subunit.
There are eight different isoforms of PI3Ks that are grouped into three classes (class I, II, and III) based on their primary structure and regulation. Class I PI3Ks, which this review is focused on, consist of four catalytic isoforms, namely p110α, -β, -γ, and -δ. Class IA PI3Ks are dimers of a p110 catalytic protein and a p85 or p55 regulatory adapter subunit as detailed in Figure 2 (4). Each regulatory subunit can associate with any of the three catalytic class IA isoforms: p110α, p110β, and p110δ (5).
The expression of distinct catalytic isoforms is cell type specific. P110α and -β are expressed in all cell types. In mice, homozygous knockouts of p110α and p110β are embryonic lethal, emphasizing their physiological importance (6–8). In contrast, p110δ expression is mainly confined to leukocytes (4). Genetically modified mice expressing catalytically inactive PI3Kδ (PI3KδKI), manifest impaired B-cell, NK cell, and T-cell function (9, 10).
Class IB PI3Ks, which are built by a p110γ catalytic and a p101 regulatory subunit, are selectively expressed by leukocytes (4, 11) and their activation is mediated by GPCRs (4). Knockdown of p110γ in mice causes altered migration and recruitment of myeloid cell populations (12–14), which is in concordance with other reports indicating that the PI3Kγ isoform is expressed mostly in the myeloid cell lineage (15–17).
One of the best-characterized hallmarks of cancer cells is their ability to sustain chronic proliferation (18). While healthy cells can strictly regulate the production and release of growth-promoting signals, cancer cells often fail to do so and malignant transformation occurs as a result of mutations in one or more components of these signaling pathways (18). There are several pathways that influence each other through multifaceted interactions and contribute to tumor development. Some of the major signaling pathways whose components are likely to be cancer drivers include proteins like growth factor receptor tyrosine kinases (e.g., EGFR), lipid kinases (e.g., PI3K), small GTPases (e.g., RAS), oncogenes and tumor suppressors (e.g., MYC, P53), serine/threonine kinases (e.g., RAF, AKT), and cytoplasmic tyrosine kinases (e.g., SRC, ABL) (19–24). Among these, involvement of the PI3K signaling pathway in cancer has been revealed first when PI3K signaling was found to be hyperactive in phosphatase and tensin homologue (PTEN)-deficient tumors (25). Independent of stimulating receptors, PI3Ks can be activated directly via oncogenes like RAS (26) and their activity is negatively regulated by the tumor suppressor PTEN, that acts as a PI3K phosphatase (Figure 1) (27).
Altered PI3K/AKT signaling has been associated with many types of cancer (28). Chronic lymphocytic leukemia (CLL), breast, ovarian, liver, lung cancers, and glioblastomas are well-studied examples in which altered PI3K activity was observed (29–32). Genetic alterations such as amplifications, gain of copy numbers and increased gene expression have been observed for all class I PI3Ks in many different cancer entities (26). P110α (PIK3CA) is affected most frequently and a direct oncogenic potential was attributed to it (33), as the signaling of mutant p110α is active and less dependent on external stimuli (34).
Compared to the mutation rates in other cancer entities, activating mutations of PI3K are scarce in CLL (35, 36). Genomic analysis revealed an amplification of PIK3CA in 3.5% of CLL patients, which is perhaps surprising when all the significant pharmaceutical effort to inhibit the PI3K pathway in this malignancy is considered (37). However, PI3K signaling is constitutively active in CLL cells, even in the absence of genetic drivers (38, 39). There are three well-studied pathways that are capable of activating and maintaining PI3K signaling in CLL cells; BCR, RTKs and cytokine/chemokine receptors (40–42). Although the stimulation of these pathways initiates different cascades of downstream events, they converge at a final point, that is, generation of a domain that can bind to the SH2 region of the regulatory p85 subunit. If the SH2 domain remains occupied, p85 fails to bind a p110 catalytic isoform and inactivate it, which results in constitutively active PI3K (43). The importance of these three pathways varies for different cell types, however, BCR is shown to be the dominant contributor to PI3K activity in B-cell malignancies. Autonomous events or continuous BCR activation by autoantigens have been reasoned for tonic BCR stimulation in CLL (44, 45).
In conclusion, the highly active PI3K signaling pathway in CLL serves as an ideal therapeutic target to inhibit CLL cell proliferation and survival directly.
Although CLL is a genetically heterogeneous disease, it is widely accepted that CLL cells are highly dependent on supporting stimuli of bystander cells in their microenvironment for their survival, proliferation, homing to lymphoid tissues as well as chemo-resistance. Different types of stromal cells, myeloid, NK and T-cells were shown to have dual functions in the tumor microenvironment (TME) of CLL both in patient samples and more recently the Eμ-TCL1 mouse model as reviewed extensively by us and others (46, 47).
In the last 20 years, many mouse models were generated in which different components of the PI3Ks were knocked out (6, 48–51). The results of the studies using these mice need careful interpretation as the alteration of one isoform’s expression can impact on those of other isoforms. As the different catalytic and regulatory subunits have the ability to interact in various combinations (4), the outcome of the expression variations in these knockout models might be far from reflecting the true functions of these proteins. For instance, an increased expression of p85β in addition to decreased expression of the three p110 isoforms (α, β, δ) has been shown in NK cells from p85α−/− mice (5, 52). In another study where p110δ was knocked out, levels of p85α were decreased, p110β and -γ were increased but p110α remained unchanged in NK cells (53). Methodologies involving the generation of point mutations that completely inactivate the catalytic function of isoforms without altering the normal expression levels may provide more reliable conclusions compared to the knockout models that alter the expression of all isoforms, as the former may mimic the normal physiological state better and make it easier to assign functions directly.
In the TME, complex interactions between cancer associated fibroblasts (CAFs), immune cells, signaling molecules, extracellular matrix (ECM), vasculature-associated cells, and malignant cells take place (54). Cancer cells are known to orchestrate the host tissue to facilitate tumor progression and regulate the recruitment of immune cells to the TME. PI3K signaling is engaged in almost all these components, including the stromal compartment. As discussed above, CLL cells require strong support from stromal cells for their proliferation and survival (55). Similarly, Bertrand et al. have shown that stromal cells protect the B-cell acute lymphoblastic leukemia (B-ALL) cells from apoptosis via PI3K and MEK pathway dependent mechanisms and co-inhibition of these pathways can synergistically break this apoptotic resistance (56).
The ability of solid tumors to grow depends strictly on oxygen and nutrient delivery through newly developing blood vessels. Accumulating evidence indicates that angiogenesis is also important in hematologic malignancies, including CLL (57). Neovascularization is a cooperative event involving endothelial cells, myeloid cells, and the tumor cells that secrete angiogenic factors such as vascular endothelial growth factor (VEGF). Inhibitors of different PI3K isoforms were shown to generate different vascular responses as the dominant PI3K isoform diverges among these cells (58). Up to date, most of the research that investigates the effect of PI3K inhibition on tumor vascular function was performed with high doses of pan-PI3K inhibitors and these studies are pointing towards a vascular dysfunction which is associated with decreased blood flow and vessel density (59–61). As the anti-angiogenic effect of PI3K inhibition is outcompeted by anti-VEGF therapies (62, 63), PI3K inhibition has not been considered specifically for targeting tumor neoangiogenesis. However, recent preclinical studies show that low doses of PI3K inhibition contribute to vessel normalization which might enable more efficient delivery of chemotherapy drugs and immune infiltration to the tumor sites (64, 65). Overall, these studies support that targeting PI3Ks in tumor and endothelial cells selectively hold the potential to synergize with conventional therapies.
CAFs constitute one of the most abundant but poorly defined cell populations in the TME. A commonly held view is that CAFs promote tumorigenesis via remodeling of ECM components, facilitating metastasis, promoting vascularization, and secreting cytokines and growth factors (66). This view is supported also for CLL, where leukemic cells are able to induce the transformation of stromal cells to a CAF-like phenotype by an exosome-mediated fashion (67). In this study, it has been shown that the CLL-derived exosomes are actively taken up by stromal cells both in vitro and in vivo. Upon uptake, stromal cells gained pro-tumoral characteristics, evident by enhanced proliferation, migration, angiogenesis, and secretion of inflammatory cytokines via mechanisms involving activation of AKT and NF-κB. These events are known to be partly involving PI3K signaling (68–70). Thus, it is expected that PI3K inhibition might dampen the pro-tumoral activity of CAFs.
All immune cells express receptors that can initiate PI3K signaling, but the components and wiring of the pathway are different in several aspects compared to other cells (1). The major difference is the expression of the catalytic isoform. P110δ and p110γ comprise the dominantly expressed isoforms while p110α and p110β are rarely engaged in immune cells (71). In non-immune cells, a higher degree of PI3K activation usually correlates with an increased downstream function such as apoptosis, survival, proliferation, and growth. However, in immune cells, an increased activation of the PI3K pathway does not necessarily mean a stronger immune reaction. Depending on the cell type, the pathway has the potential to augment or alleviate the response to pathogens as well as tumors. The network of the PI3K pathway downstream of AKT seems to be different for lymphoid cells subsets. MTORC1 phosphorylates a wide range of downstream proteins when stimulated via PI3K/AKT and RAS/ERK pathways but it also requires signals delivered through nutrient-sensing pathways to ensure that cells do not intent to proliferate under stress conditions like nutrient deprivation (72, 73). In lymphocytes, mTORC1 was shown to be more dependent on nutrient inputs than on PI3K/AKT activity (74–77). The two best-characterized mTORC1 substrates in mammals are p70 ribosomal S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) (78). 4E-BP/Eukaryotic translation initiation factor 4E (eIF4E) arm is known to be responsible for regulation of growth and proliferation in lymphocytes (79), whereas in other cell types, S6Ks are the main mediators of cell growth (80). It has also been hypothesized that S6Ks might be involved in lymphocyte differentiation, but to our knowledge this has not been validated yet (81–83) (Figure 1). Overall, considering these immune cell-specific features might be useful while estimating the overall impact of PI3K-mTOR inhibition on the TME.
Innate immune cells such as monocytes, neutrophils, DCs, mast cells, and macrophages are the key components that mediate the initial phase of acute inflammation via cytokine/chemokine secretion, extravasation, migration, and phagocytosis. Each of these events requires class I PI3K activation (84). Upon detection of a stimulus, tissue resident macrophages immediately start producing cytokines and chemokines to attract neutrophils and macrophages to the site of infection, which requires p110δ expression (84). These chemoattractants are known to activate neutrophil p110γ through GPCR (14). After reaching the site of inflammation, macrophages and neutrophils phagocyte and lyse the pathogens with nicotinamide adenine dinucleotide phosphate oxidase (NOX)-derived ROS. It has been shown that p110β and p110δ are involved in the regulation of phagocytosis (85–87). In line with this, p110β-deficient mice fail to mediate Fc gamma receptor (FcγR)-dependent activation of neutrophils and produce ROS (88). In addition to initiating ROS production, maintenance of NOX activity for ROS production also requires the activation of p110δ (89).
All class I PI3K catalytic unit isoforms are expressed in myeloid cells. However, current literature suggests that p110δ and p110γ are the critical isoforms for mediating their immune functions. Deficiency of these two isoforms has been reported to induce neutrophilia in mice (90), while inhibition of only one of them did not have a striking effect. Although the p110γ isoform is expressed in many hematopoietic cells, highest level of expression was detected in cells of the myeloid lineage and the inhibition of p110γ resulted in dampened inflammatory responses (14, 91). Of note, neither neutrophil nor macrophage development was impaired in p110γ−/− mice. However, chemoattractant-stimulated neutrophils failed to produce PIP3 and implement respiratory burst (14). Motility and migration mechanisms have also been noted to be defective, both in neutrophils and macrophages (14). Similarly, the migration of antigen-loaded DCs from the tissue to draining lymph nodes was impaired demonstrating the non-redundant functions of p110γ (92).
PI3K signaling mediates an immunosuppressive phenotype in myeloid cells to prevent excessive innate immunity in chronic infections and inflammation (89, 93–95). This phenomenon supported the idea to pharmacologically target the PI3K p110γ isoform with specific inhibitors for the treatment of inflammatory diseases but the possibility of dampening the host defense has slowed down the development of this idea.
However, the pro-inflammatory potential of PI3K inhibitors might provide new opportunities in cancer treatment. In many cancer entities, pro-inflammatory cytokine expression is correlated with better prognosis. High expression levels of p110γ that induces immunosuppression in TAMs, were reported in several studies (15, 96, 97). Selective inhibition of PI3Kγ in mouse models of melanoma and breast cancer revealed a downregulation of M2 markers (Tgfb1, Arg1, Ido1) and an upregulation of M1 markers (Il12a and Nos2) (98), demonstrating the influence of this isoform on the phenotype and polarization of TAMs. Interestingly, reduced tumor growth as well as enhanced anti-tumoral T-cell responses have been noted upon genetic or pharmacological p110γ inhibition (15, 96, 98). Of note, cancer cells of most solid tumors do not express p110γ and anti-tumoral effects of p110γ inhibition might be attributed to TAMs and the TME.
Recently, the immune microenvironment has gained attention in cancer therapy. Although the approval of checkpoint inhibitors has been a significant breakthrough in immunotherapy, multiple resistance mechanisms exist, and the presence of tumor infiltrating myeloid cells are among these. Following encouraging pre-clinical results, a phase 1 trial combining immune checkpoint blockade and inhibition of p110γ has been initiated, but results are pending (NCT02637531) (98).
Inactivation of p110δ isoforms mostly interferes with the functions of adaptive immune cells but this isoform has also been implicated in controlling the pro-tumorigenic effect of myeloid cells in cancer (99, 100). In one study, histological analysis of patient breast tumors demonstrated a correlation between the disease progression and levels of PI3Kδ in tumor cells. In addition to the tumor cells, CD68+ macrophages also manifested strong PI3Kδ staining (101). Treatment with a PI3Kδ-specific inhibitor, IC87114, reduced the volume of human breast tumors in xenograft models when administered intratumorally or orally (101). Intratumoral treatment only slowed down tumor growth, as a small but significant proliferation of tumor cells was still detectable (101). However, oral administration of the inhibitor totally stopped tumor growth and reduced macrophage infiltration (101), suggesting that PI3Kδ inhibition in these cells might be a contributing factor to the anti-tumoral effect. To further evaluate the impact of macrophage PI3Kδ inhibition on tumor growth, the authors adoptively transferred either PI3Kδ-knockout (KO) or wildtype (WT) macrophages to tumor bearing NOD/SCID-gamma null (NSG) mice (101). Tumor growth was significantly reduced in the mice that received PI3Kδ-KO macrophages compared to the ones that received WT macrophages (101). In addition, PI3Kδ-KO macrophages were less competent in infiltrating the tumors compared to WT macrophages (101), demonstrating that inhibition of macrophage PI3Kδ might be sufficient to reduce the infiltration of macrophages to tumors and thereby impact on tumor growth. In the final set of experiments, the researchers inoculated PI3Kδ expressing tumors to macrophage-deficient NSG mice and PI3Kδ-deficient tumors to Balb/c nude mice with functional and PI3Kδ-proficient macrophages. While treatment with the PI3Kδ inhibitor IC87114 reduced tumor growth in both groups, complete abrogation was only seen in mice with PI3Kδ-expressing tumors and WT macrophages (101). Overall, these results suggest that targeting PI3Kδ both in tumor cells and TAMs is likely to be a promising strategy for treatment of breast cancer.
The exact function of PI3K isoforms in DCs is unclear. To study the impact of PI3K inhibition in DCs, Marshall et al. have employed different mouse tumor models and reported that either broad or β-, δ-, or γ-isoform-specific inhibition reduced levels of IL-10 and TGFβ but enhanced IL-12 expression (99). This caused a less immunosuppressive TME and facilitated the priming of Th1 T-cells in vivo. An important finding of this study is that selective inhibition of PI3Kβ and -δ isoforms reduced IL-10 and TGFβ expression in toll-like receptor (TLR)-activated DCs pulsed with dead tumor cells in vitro. Adoptive transfer of pulsed DCs into tumor-bearing mice in combination with pan-PI3K inhibition resulted in a significant increase in anti-tumoral T-cell response, decrease in tumor growth and prolonged overall survival, demonstrating the potential of PI3K inhibitors for the development of tumor DC-based vaccines.
Twenty years ago, Burger et al. discovered that peripheral blood monocytes differentiate in co-culture with CLL cells to adherent cells that support the survival of CLL cells in vitro (102). These cells were named as nurse-like cells (NLCs), and only later on, it was understood that they resembled tumor-associated macrophages (TAMs) in solid tumors (103, 104). NLCs exhibit an M2-like phenotype and besides delivering survival signals, they were suggested to have immunosuppressive functions (55, 105).
Research investigating the effect of PI3K inhibition on myeloid cells in CLL is scarce. Thus far, efforts have been dedicated to identifying the effect of clinically approved PI3K inhibitors in modulating the effect of type I anti-CD20 monoclonal antibody (e.g., rituximab and ofatumumab) treatment. More specifically, the role of PI3K in regulating antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent phagocytosis (ADP) responses has been investigated. Da Roit et al. described an effect of the PI3Kδ inhibitor idelalisib on inhibiting activation of polymorphonuclear neutrophils (PMN) and phagocytosis of anti-CD20 opsonized CLL cells by macrophages (106). Based on these observations, Enya Chen et al. investigated the role of PI3K-p110 in modulating antibody-mediated responses by macrophages in CLL (107). The authors demonstrated that class I PI3K p110 isoforms α, β, γ, and δ are expressed in nurse-like cells (NLCs) of both anti-CD20 antibody-sensitive and -resistant CLL patients (107). The effect of PI3K inhibition on ADCC and ADP was found to be FcγR-mediated and independent of downstream spleen tyrosine kinase (SYK) and Bruton’s tyrosine kinase (BTK) signaling. Inhibition of p110δ, both pharmacologically and via siRNA knockdown, decreased ADCC and ADP in CLL-derived NLCs (107), suggesting that this catalytic isoform is essential for CLL-derived NLC as well as macrophage response to therapeutic antibodies.
In a recent study, idelalisib has been shown to prevent AKT-dependent phosphorylation of the p47phox subunit of NOX2, resulting in suppressed rituximab-induced ROS formation in human monocytes (108). In co-cultures of monocytes and NK cells, idelalisib rescued NK cells from ROS-induced toxicity and thus improved NK cell ADCC against primary CLL cells and 221 B-lymphoblastoid cells (108).
In addition to its role in regulating ADCC, the PI3K pathway is also involved in migration and homing of CLL cells which circulate between peripheral blood and secondary lymphoid organs (109). Trafficking of CLL cells is regulated by stromal cells and NLCs in the lymph nodes that secrete chemokines including C-X-C motif chemokine ligand (CXCL)12, CXCL13, C-C motif ligand (CCL)19, and CCL21 (110). Upon the activation of chemokine receptors, PI3K signaling directs the CLL cells towards stromal cells that provide tumor supportive stimuli (111). As expected, inhibition of the PI3K pathway in vitro with PI3Kδ-specific idelalisib was shown to result in impaired homing of CLL cells to stromal cells, due to their reduced response to CXCL12 and CXCL13 (109). These findings partly explain the rapid lymph node shrinkage in CLL patients treated with PI3Kδ inhibitor idelalisib (112).
Overall, current research highlights the importance of PI3K activity in immune responses to therapeutic antibodies and suggests that both enhancing and inhibiting this pathway might improve these responses (Figure 3). Nevertheless, further in vitro and in vivo studies are needed to unravel the diverse and complex effects of PI3K inhibition in the myeloid compartment of CLL.
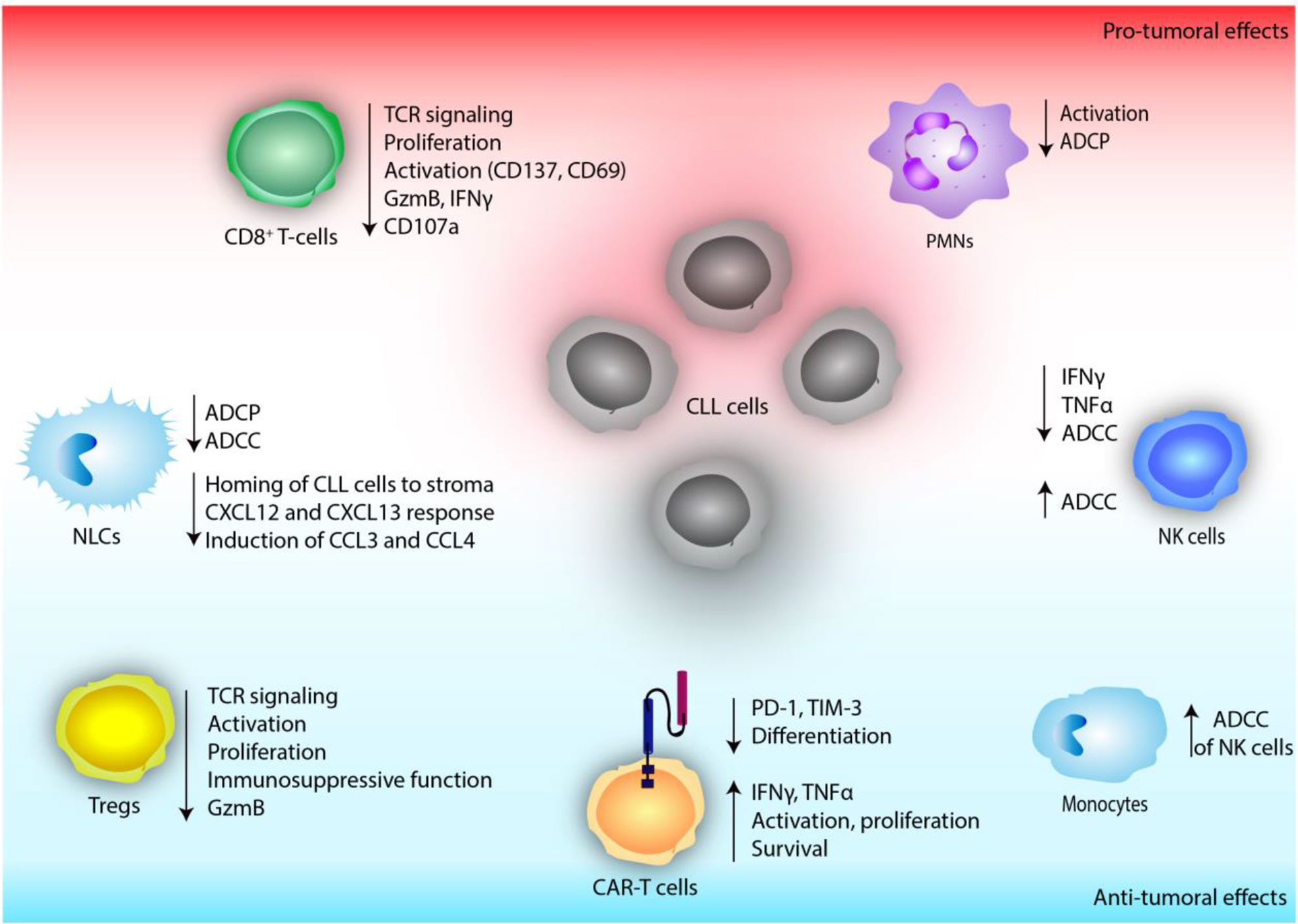
Figure 3 The effect of defective PI3K signaling on immune cells of the tumor microenvironment in chronic lymphocytic leukemia (CLL).
Information about how NK cells are affected by PI3K inhibition is relatively sparse and derived mainly from knockouts of PI3K isoforms in mice. NK cells are critical mediators of innate immunity that do not require prior sensitization or antigenic stimulation. Upon activation by tumor cells, they can exert their cytolytic functions via granzyme (Gzm)- or receptor-mediated fashion (113, 114). They are also producers of an array of cytokines including interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα), which act on other immune cells to enhance the immune response. There are several studies providing evidence that PI3K isoforms control signaling checkpoints in cytokine expression and secretion in mouse NK cells (115). PI3Ks are involved in the signaling downstream of different NK cell activation receptors such as NKG2D, CD16, and CD28. In addition, PI3K–mTOR pathway is indispensable for efficient NK cell activity, specifically for cytokine responses in IL-15 primed NK cells (116). PI3K inhibitors were also shown to have an impact on NK cell granule polarization. Upon tumor contact, NK cells start to mobilize perforins and GzmB towards the tumor cells (117). Zhong et al. have shown that pre-treatment of NK cells with pan-PI3K inhibitors (wortmannin or LY294002) effectively stopped this intracellular polarization of cytotoxic granules (118). A complementary study has also shown that mice lacking the regulatory p85α subunit and its alternatively spliced variants (p55α/p50α) had impaired lineage commitment leading to reduced NK cell numbers in the bone marrow and liver. In addition to genetic inhibition, broad pharmacological PI3K inhibition with wortmannin also reduced the cytotoxicity against EL-4 lymphoma cells and cytokine/chemokine generation was significantly compromised (119). Other studies that support these findings exist, in which treatment with different PI3K inhibitors (wortmannin, LY294002, IPI-145) quenched mouse and human NK cell cytotoxicity against tumor cells (120–122) while inhibition of a single p110 isoform did not have a profound effect. This phenomenon brings up the question whether different p110 isoforms can compensate for/replace each other in NK cells. Kim et al. have investigated the relative contribution of p110δ and -γ isoforms on NK cell anti-tumor cytotoxicity and provided an answer to this question to some extent (123). They reported a reduction in NK cell numbers and production of the cytokines IFNγ, TNFα, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in p110δ-deficient, but not p110γ-deficient NK cells. When both p110δ and p110γ isoforms were inactivated by a combination of genetic and biochemical approaches, NK cell cytotoxicity was attenuated, which was not observed upon inhibition of a single isoform (123). Therefore, it is likely that cooperation of isoforms can compensate for the maintenance of cytotoxic function in NK cells. However, the overall results suggest that p110δ is the dominant PI3K isoform for cytokine secretion by NK cells. In line with this study, Saudemont et al., have also proposed that P110δ is the dominantly expressed and preferentially activated isoform in NK cells (124).
Using a murine model of catalytically inactive p110δ (PI3KδKI), Guo et al. have shown that p110δ plays a crucial role in NK cell terminal maturation and cytokine/chemokine generation (10). Moreover, the PI3KδKI mice had reduced NK cell numbers and maturation. NK-mediated cytotoxicity was slightly impaired against EL-4, YAC-1, and RMA-S tumor cells in vitro and in vivo. However, cytokine and chemokine generation by NK cells was severely affected accompanied by a reduced c-Jun N-terminal kinase (JNK) 1/2 phosphorylation in response to NKG2D-mediated activation. The study provides evidence for the role of PI3Kδ in downstream JNK activation that regulates NK cell cytokine generation (10).
Another study using the same mouse model showed that the NK cells derived from PI3KδKI mice showed comparable cytotoxicity to WT NK cells, suggesting that p110δ is not indispensable to kill RMA-S lymphoma cells in vitro (115). On the contrary, mice lacking a functional p110δ failed to eliminate i.p. administered syngeneic tumor cells. The authors hypothesized that the failure might be specific to the intraperitoneal model they have used, or it may possibly indicate a defect of NK cell migration to the tumor site (115). Two years later, the same group has tested whether the p110δ isoform was essential for NK cell trafficking. Using genetically modified mice, they observed that p110δ was indispensable for chemotaxis to sphingosine-1-phosphate (S1P) and CXCL10, NK cell distribution throughout lymphoid and non-lymphoid tissues and for extravasation to tumors (124).
NK cells are not able to kill CLL cells directly, however they contribute substantially to ADCC (125, 126). Rituximab, a chimeric monoclonal antibody that targets CD20, was approved in 2010 and served as standard therapy in combination with chemotherapy for the treatment of CLL. Fc region of the antibody communicates with the NK cell surface Fc-γ-receptor CD16 (FcγRIIIA) (127). Fc receptor ligation leads to PI3K activation in NK cells, triggering release of cytotoxic granules and ADCC (123, 128). Earlier in vitro studies using idelalisib have not reported any significant cytotoxicity towards NK cells (38). Interestingly, pre-treatment of NK cells with idelalisib inhibited their production of inflammatory cytokines, such as TNFα and IFNγ but did not alter their ADCC against CLL cells (38) (Figure 3).
Considering that the ADCC of NK cells is mediated through PI3K activation, it might be expected that inhibition of PI3Kδ would limit the clinical efficacy of rituximab. For clinical use, idelalisib is only approved in the combination with αCD20 antibodies and this treatment is efficacious (129). To investigate whether idelalisib dampens NK cell mediated ADCC, a direct comparison of idelalisib versus idelalisib + rituximab would be necessary.
PI3K signaling is involved in several T-cell specific functions spanning from their activation to apoptosis. The activity of PI3K is regulated by the co-stimulatory receptors CD28 (4, 9) and inducible T-cell co-stimulator (ICOS) (4). Activation of these receptors results in subsequent activation of PI3Kδ, the dominantly expressed PI3K isoform, which leads to proliferation and differentiation into regulatory, effector, and memory T-cell phenotypes (4, 9). In addition, PI3Ks regulate survival and apoptosis by increase of B-cell lymphoma-extra large (BCL-XL) expression or IL-2 secretion in response to T-cell receptor (TCR) activation (4). These functions of T-cells involve calcium flux, in which PI3Ks are also involved (4). Resting T-cells sustain a low level of Ca2+. Upon TCR activation, the cells experience an influx of Ca2+ (130). It is reported that PI3Ks are involved in the regulation of calcium signaling via IL-2-inducible T-cell kinase (ITK), indicating the significance of the crosstalk between these pathways (4).
PI3Kδ is crucial for the function of T-cells, as reduced activity of PI3Kδ has been shown to diminish TCR signaling of murine (9, 131–134) as well as human T-cells (135–137) in vitro, including all major CD4+ and CD8+ T-cell subsets. Subsequently, reduced proliferation of murine (9, 132, 133, 135, 137, 138) as well as human T-cells (134–137) has been observed upon abrogated PI3Kδ signaling, without induction of apoptosis (133). Of note, some combinatory approaches targeting the TCR as well as the co-stimulatory receptor CD28 or supplying IL-2 have been shown to overcome the negative effects of PI3Kδ inhibition on T-cell activation (9, 133, 135).
T-cells differentiate after the activation of their TCR into antigen-experienced effector and memory cells (139, 140). During this process, surface marker expression as well as transcriptional programs change, which for example enables effector and memory T-cells to home to distinct organs. Memory T-cells are equipped to provide fast cytotoxic functions and proliferation upon secondary recall infections (140, 141). In line with reduced TCR-activation, impaired PI3Kδ signaling has been shown to result in a higher expression of CD62L (138, 142, 143), CD127 (143) as well as CCR7 (9, 143), and therefore, less terminally differentiated T-cells. Long-term culture of T-cells using repeated TCR stimulations in the presence of PI3Kδ inhibitors resulted in higher proliferation rates and IL-2 secretion (138), likely caused by initial blockade of T-cell activation and differentiation. The typical memory T-cell markers such as CD62L or CCR7 are important mediators of migration and homing. In contrast to the higher expression of these markers in PI3Kδ-defective T-cells, transfer of WT, or PI3KδKI T-cells into syngeneic hosts did result in comparable homing to lymph nodes (133), suggesting that the expression of naïve/memory markers does not affect the migration of these cells.
Following the clonal expansion of T-cells, particularly their production of effector molecules is essential to control pathogens as well as cancer. After in vitro or in vivo stimulation of PI3Kδ-defective T-cells, a lower cytokine production of IFNγ, IL-2, IL-17, and TNFα (135) has been observed compared to WT cells. This was confirmed using an antigen-specific stimulation of T-cells of WT or PI3KδKI mice, in which a reduced expansion as well as IFNγ production of mutant T-cells was noted (133).
In conclusion, PI3Kδ is essential for TCR signaling and subsequent activation, proliferation, and cytokine production of T-cells. However, it is likely that the negative effects of PI3Kδ inhibition in T-cells can be partially overcome by applying strong or combinatory stimuli.
CD4+ helper T-cells (Th cells) are involved in multiple immune functions, such as control of immune reactions by Tregs as well as providing help in the clearance of pathogens by Th cells (46).
IFNγ, IL-4 and IL-17 are the key cytokines of Th1-, Th2-, and Th17-mediated responses, respectively (46). Following in vitro stimulation in the presence of PI3Kδ inhibitors, a reduced production of IFNγ (133, 135), IL-4 (133), and IL-17 have been noted (135). Of interest, memory CD4+ T-cells were more susceptible to abrogated PI3Kδ signaling than their naïve counterparts (135). These data highlight that PI3Kδ inhibition affects the function of Th cell subsets and suggest that antigen-experienced T-cell subsets might be more dependent on PI3Kδ signaling than naïve T-cells.
Tregs are crucial in maintaining orchestrated immune reactions by paracrine secretion of IL-10 or direct, contact-mediated inhibition of effector responses (46). In line with the reduced proliferation of this cell type upon PI3Kδ inhibition in vitro (136, 137), lower frequencies of Tregs in lymphoid organs and blood of p110δKI mice have been observed (100, 144), and shown to be most likely due to an increased rate of apoptosis (137). In line with reduced TCR activation upon PI3Kδ inhibition, lower expression levels of FOXP3, CD25, ICOS, and PD-1 have been noted in human Tregs in vitro (136), indicating a reduced activation state and immunosuppressive potential of this cell type. Accordingly, PI3Kδ-inhibited murine (144) as well as human Tregs (136) were reported to be less efficient in suppressing effector T-cell proliferation in vitro, and lower concentrations of the Treg-specific cytokine IL-10 but higher concentrations of the effector cytokines IL-2 and TNFα have been observed in the culture supernatants upon PI3Kδ inhibition (136). Interestingly, the negative effect of PI3Kδ inhibition could be overcome upon co-culture with antigen-presenting cells (APCs) (144), which could provide strong co-stimulatory signals to compensate for the inhibitory effects of abrogated PI3Kδ signaling. Comparing the sensitivity of Tregs and effector T-cell populations to PI3Kδ inhibition, in vitro assays showed that Tregs seem to be more sensitive (134, 136, 137). In sum, the functions of helper T-cells as well as Tregs are dependent on PI3Kδ signaling. Moreover, it is likely that Tregs are more sensitive to PI3Kδ inhibition than effector T-cells, which could unleash immunosuppressive bonds from effector cells and enhance their effector function.
CD8+ T-cells are important for the control of pathogens as well as anti-tumoral immunity (46). In addition to the previously mentioned reduced in vitro proliferation of T-cells, the numbers of pathogen-specific CD8+ T-cells was also reduced in PI3Kδ-deficient mouse models of infections (131, 132). Therefore, a thorough analysis of CD8+ T-cell function upon PI3Kδ inhibition is essential, as enhanced effector functions could compensate for the reduced expansion of these cells and result in similar immunological properties.
The expression of early activation markers is often used to analyze TCR signaling activity and the activation status of T-cells. Upon TCR stimulation reduced expression of the activation markers CD25 (132), CD69 (132, 134, 143), killer cell lectin-like receptor subfamily G member 1 (KLRG1) (143) as well as PD-1 (143) have been noted in murine, PI3Kδ-inhibited CD8+ T-cells compared to untreated cells. Similarly, abrogated PI3Kδ signaling resulted in a reduced expression of CD69 (134, 135) as well as T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) (143), a marker of activation-induced dysfunction, on human CD8+ T-cells after TCR engagement in comparison to PI3Kδ-proficient T-cells, suggesting that not only numbers, but also activation of CD8+ T-cells could be altered by PI3Kδ inhibitors.
Once activated, CD8+ T-cells secrete granzymes and cytokines such as IFNγ or TNFα, which is essential for their immune control of pathogens and cancer (46). In vitro analysis of CD8+ T-cell function after antigen-specific or antibody-mediated TCR stimulation revealed lower expression of the effector molecules IFNγ (131, 135), TNFα (132), GzmB (100, 131), GzmA (100, 142), perforin (100), and IL-2 (135) upon PI3Kδ inhibition.
In summary, PI3Kδ is essential for the activation and cytotoxic function of CD8+ T-cells, which could in turn result in a reduced immune surveillance and anti-tumoral immunity in individuals that are treated with PI3Kδ inhibitors.
The function of PI3Kδ in T-cells has extensively been investigated in many murine cancer models. According to previously mentioned results, global PI3Kδ abrogation in mice caused a reduced frequency and absolute number of Tregs at tumor sites (137, 142, 145) and in draining lymph nodes (100). Of interest, reduced frequencies of Tregs have already been found in spleens of mice as early as 3 days after a single dose of idelalisib (137), suggesting a very fast reaction to PI3Kδ inhibition. Concomitantly with reduced Treg numbers, improved tumor control has been noted in various tumor models using either pharmacological or genetic inhibition of PI3Kδ (100, 142, 145). Utilizing FoxP3Cre x δflox/flox mice, in which PI3Kδ-deficiency is specific to Tregs, a reduced tumor growth and improved survival was noted in comparison to systemic PI3Kδ-deficient or PI3Kδ-proficient mice (100). The improved tumor control in the presence of PI3Kδ-deficient Tregs was further confirmed by transfer of WT or PI3KδKI Tregs into PI3KδKI-mice (100). This data highlight that inhibition of Treg-PI3Kδ reduces the accumulation and immunosuppressive function of Tregs which results in an enhanced anti-tumoral immunity in mouse models of cancer.
Supported by reduced Treg functions upon PI3Kδ inhibition, enhanced tumor-infiltration, and cytotoxic function of CD8+ T-cells were expected. Accordingly, systemic PI3Kδ inhibition resulted in an increased ratio of CD8+ T-cells to Tregs and increased numbers of CD8+ tumor-infiltrating lymphocytes (TIL) in mouse models of carcinoma (100, 145). Further, transfer of WT but not PI3KδKI Tregs into PI3KδKI mice resulted in reduced infiltration of CD8+ TILs (100). These data strongly support the hypothesis that reduced Treg function, caused by defective PI3Kδ signaling, increases TIL infiltration and enhances anti-tumoral immunity.
Seminal work by Lim et al. investigated several tumor models expressing the ovalbumin (OVA) antigen, namely EL-4 lymphoma, MC-38 colon, and LLC lung carcinoma (142). For EL-4 and MC-38 cancer models, tumor growth was dependent on Treg-PI3Kδ, as depletion of Tregs using FoxP3DTR mice, as well as Treg-specific, genetic PI3Kδ deficiency resulted in a reduced tumor burden (142). In line, transfer of WT or PI3KδKI, tumor-specific CD8+ T-cells into EL-4 tumor-bearing mice did not impact on the disease progression (100). This data suggest that CD8+ T-cells are not sufficient to mediate disease control in the EL4 model while targeting PI3Kδ in Tregs has a high potential.
In contrast, growth of LLC- and MC-38 tumors was neither altered by depletion of Tregs, nor using a systemic PI3KδKI model (142), suggesting that tumor control in these models relies at least partially on functional PI3Kδ in other immune cells, likely CD8+ T-cells. In line, LLC tumor cells were most susceptible to CD8+ T-cell mediated killing in vitro, followed by MC-38 and EL-4 tumors (142). This anti-tumoral immunity was abolished if CD8+ T-cells harbored a PI3KδKI mutation (142). Similarly, ex vivo analysis of CD8+ TILs of LLC-OVA or MC-38 tumors revealed a reduced expression of the cytotoxic molecules GzmA and GzmB if PI3Kδ-signaling was abrogated (142).
In conclusion, the effects of PI3Kδ inhibition on T-cells and subsequently on tumor control are highly contextual. In tumors that are controlled by an immunosuppressive, Treg-dependent microenvironment, PI3Kδ inhibition can succumb the pro-tumoral TME and result in slowed tumor progression. On the contrary, in tumors that are controlled by CD8+ T-cells, PI3Kδ inhibition reduces the tumor infiltration, activity as well as tumor control of this cell type. Of note, even though PI3Kδ deficiency does not have a significant impact on tumors that are resistant to CD8+ T-cell mediated killing, it is likely that abolished CD8+ T-cell function could cause a reduced response to pathogens and therefore, limit the success of combining immunotherapeutic treatment approaches and PI3Kδ inhibitors.
T-cells, their subsets and function are profoundly altered in CLL and known to be of pathological relevance, as recently reviewed by us (46), emphasizing the need to evaluate the effect of PI3K inhibitors on these cells.
Similar as in the above-mentioned tumor models, PI3Kδ inhibition by idelalisib treatment of CLL patient-derived T-cells in vitro also resulted in reduced proliferation, expression of activation markers (CD69, CD25) (146), and cytokine production, including IL-2, TNFα, and IFNγ (146). Using Boyden-chamber migration assays, a reduced migration of idelalisib pre-treated T-cells obtained from CLL patients has been observed (146). This data highlight that idelalisib pre-treatment reduces the function of CLL patient-derived T-cells, emphasizing the need for more detailed investigations.
Tregs have repeatedly been reported to be more abundant in CLL patients than healthy individuals, as recently reviewed by us (46). As expected, PI3Kδ inhibition decreased TCR signaling of Tregs, which were isolated from TCL1-leukemia bearing mice, in a dose-dependent manner (134). Accordingly, reduced proliferation and numbers of Tregs have been noted in vivo using either pharmacological PI3Kδ inhibition in the TCL1 AT model of CLL, in which leukemic splenocytes of Eµ-TCL1 mice were transplanted into syngeneic WT mice (134) or PI3KδKI x Eµ-TCL1 mice (147). Functionally, PI3Kδ inhibition reduced the expression of activation markers and their immunosuppressive function, as a reduced secretion of GzmB was noted (134). These findings indicate that preclinical PI3Kδ inhibition in CLL results not only in defects of Treg proliferation but also induces their reduced activation and immunosuppressive phenotype.
Until now, reports analyzing Tregs in samples of CLL patients undergoing idelalisib treatment are scarce. Comparison of blood samples before idelalisib treatment initiation and during the treatment revealed a reduced frequency of circulating Tregs (148) as well as a reduced fraction of KI-67+, proliferating Tregs, in comparison to pre-treatment samples (136), implicating that idelalisib treatment of CLL patients likely reduces accumulation and proliferation of Tregs (Figure 3). Although effects of reduced CLL burden cannot be excluded to contribute to these observations, these results are concordant with the findings of other pre-clinical models of different cancer entities.
In CLL, CD8+ T-cells have been proposed to contribute to the control of disease progression (46, 149), thus, their function has to be maintained during treatment. As suggested by other models, PI3Kδ inhibition ex vivo decreased TCR signaling of CD8+ T-cells, which were isolated from TCL1-leukemia bearing mice, in a dose-dependent manner (134). And a reduced proliferation of CD8+ T-cells has been observed after PI3Kδ inhibitor treatment in vivo in the preclinical TCL1 AT model of CLL (134) as well as in another model, in which OVA-expressing TCL1 leukemic cells were transplanted into PI3KδKI mice (147). Concomitantly with their reduced TCR signaling activity and proliferation upon defective PI3Kδ signaling, a reduced expression of the activation markers CD137 and CD69 and an enrichment of naïve CD8+ T-cells have been noted in the TCL1 AT model (134). Functionally, PI3Kδ inhibition caused a reduced effector molecule production of CD8+ T-cells, as less IFNγ, GzmB, and CD107a were detected, even if focusing the analysis on the antigen-experienced effector population (134). In conclusion, preclinical PI3Kδ inhibition in the TCL1 AT mouse model of CLL reduces the proliferation, expression of activation markers, differentiation, as well as effector function of CD8+ T-cells, which likely contributes to a diminished CLL-control by this cell type.
In contrast to these results, Dong et al. performed a transplantation study of TCL1 leukemic cells into PI3KδKI mice, which resulted in reduced TCL1 leukemia growth in comparison to WT recipient mice (147). Of importance, tumor cell transplantation in mice with genetic differences between the donor and recipient has been shown to cause tumor rejection in the TCL1 mouse model of CLL (150) as well as a model of multiple myeloma (151), which seems a likely explanation for the lower TCL1 tumor burden in PI3KδKI versus WT mice. Re-challenge of these PI3KδKI mice, which previously rejected TCL1-leukemia, with tumor cells resulted again in tumor rejection and expansion of CD44+ effector/memory CD4+ as well as CD8+ T-cells (147). Rather than providing evidence on the role of PI3Kδ for CD8+ T-cell function in CLL, these results suggest that PI3KδKI mice are still proficient in a recall, memory T-cell response. Moreover, graft-rejection of TCL1-leukemia is based on a strong T-cell stimulus which potentially overcomes inhibitory effects of abrogated PI3Kδ signaling.
Most evidence about the impact of PI3Kδ inhibition on CD8+ T-cells in CLL patients is derived from preclinical studies. In the clinical study by Chellappa et al. analysis of blood samples of idelalisib-treated CLL patient showed a reduced frequency of KI-67 expressing, proliferating CD8+ effector T-cells during idelalisib treatment for two out of three patients (136), suggesting that PI3Kδ inhibition might not be beneficial for CD8+ T-cell mediated tumor control as well as their clearance of pathogens. But based on the very small patient cohort in this study and recent findings showing that CD8+ T-cells derived from lymphoid organs of CLL patients are more active than their blood-derived counterparts (149, 152), the provided analysis of blood T-cells during idelalisib-treatment allows only for very limited conclusions.
In summary, although there is evidence suggesting that pre-clinical as well as clinical PI3Kδ inhibition in CLL diminishes CD8+ T-cell function (Figure 3), further studies investigating patient samples are needed to fully elucidate the effects of inhibited PI3Kδ signaling on CD8+ T-cells in CLL.
Adoptive cell transfer-based therapies such as chimeric antigen receptor (CAR) T-cell therapy, are new approaches to specifically target tumor cells that are currently also investigated in CLL, as recently reviewed by us (46). So far, the effect of PI3Kδ inhibition on T-cells during the generation of the infusion T-cell product on anti-tumor activity has been investigated in different cancer entities, including CLL. Transplantation of PI3Kδ inhibitor pre-treated CD8+ T-cells into B16 melanoma-bearing WT mice, resulted in a better tumor control (138, 143), which could be further enhanced by vaccination with the tumor antigen (138). Similarly to beforehand mentioned, in vitro PI3Kδ inhibition results in a less differentiated phenotype of T-cells (143) and in increased proliferation rates of T-cells after long-term cultivation (138). Accordingly, the frequency of PI3Kδ inhibitor pre-treated T-cells within the tumor site trended to be higher in comparison to vehicle-treated controls (143). Of interest, transfer of naïve T-cells similarly reduced the tumor size as seen for PI3Kδ inhibitor pre-treated CD8+ T-cells (143). This data suggest that PI3Kδ inhibition during the generation of the T-cell product results in their reduced differentiation, activation, and subsequently higher proliferation in vivo as well as a better tumor control.
In comparison to other B-cell malignancies, the remission rates of CD19 CAR T-cell therapies in CLL have been disappointing (46). Therefore, different efforts were taken to improve treatment efficacy. Among them, pre-treatment of CAR T-cells with ibrutinib (153) or idelalisib (154) have been explored. PI3Kδ inhibition during CAR T-cell production of patient derived T-cells resulted in lower proliferation rates of CD4+ T-cells, while proliferation of CD8+ T-cells was enhanced (154). Of note, in healthy donor-derived T-cells, no difference in the proliferation between PI3Kδ inhibited and control cells was observed (154). Phenotypically, an expansion of naïve and effector T-cells, accompanied by a reduction of memory cell populations (143, 154), and lower levels of PD-1 and TIM-3 expression of idelalisib-treated compared to untreated CAR T-cells of CLL patients were seen (154). This lower state of pre-activation resulted in an increased TNFα and IFNγ production ex vivo (154). And chromium release assays showed that cytotoxicity of idelalisib-treated CAR T-cells was not altered in vitro compared to untreated CAR T-cells (154). This data suggest that priming of CAR T-cells by idelalisib causes a less differentiated and activated phenotype of the T-cell product, which could result in an enhanced anti-tumoral activity in vivo. Investigating their function in vivo, two independent groups showed an enhanced tumor control and prolonged survival of PI3Kδ inhibitor pre-treated CAR T-cells in xenograft mouse models of CLL (143, 154) (Figure 3).
In conclusion, pre-treatment of cells for adoptive transfer therapies, such as CAR T-cells, with PI3Kδ inhibitors results in a less differentiated and activated cell product accompanied by an enhanced in vivo efficiency in xenograft models. Further studies are needed to assess the long-term persistence and tumor control of PI3Kδ inhibitor-primed CAR T-cells in an immunocompetent TME. Ultimately, a confirmation of these results in patients is still pending.
PI3Kδ is the most studied isoform in T-cells. To investigate the role of other PI3K subtypes in CD4+ as well as CD8+ T-cells, initially pan-class IA PI3K inhibitors were utilized. These studies showed a similar effect as p110δ isoform specific inhibition, such as reduced TCR signaling, proliferation, and higher expression of naïve/memory T-cell markers (137, 138). Intriguingly, neither the specific inhibition of PI3Kα nor -β did result in an altered TCR signaling, proliferation (137, 138), or affected the phenotype or cytokine production of CD8+ T-cells in vitro (138). This data suggest that neither PI3Kα nor -β alone are indispensable for T-cell function. To elucidate whether PI3Kα or -β inhibition have additive effects to PI3Kδ inhibition on T-cells, combinations of Class IA inhibitors and genetically inactive isoforms have been investigated. Treatment of PI3Kδ-deficient cells with PI3Kα or -β inhibitors further reduced TCR signaling as well as proliferation of CD4+ T-cells in vitro (137, 145) and in vivo (145). Concomitantly, higher frequencies of CD8+ T-cells with lower PD-1 expression at the tumor site have been noted upon PI3Kα and -δ inhibition (145). Of importance, all results were comparable to single δ-specific inhibition (145), and differences in T-cell phenotype as well as function have not been observed upon combination of PI3Kα and -β inhibition (137).
These results highlight that PI3Kδ is indispensable for T-cell activation and proliferation and its loss of function cannot be compensated by class IA PI3K isoforms. Dual inhibition of PI3Kα and -δ likely does not have additive effects, although a direct head to head comparison is missing.
PI3Kγ inhibition is mostly targeting myeloid cells, as detailed above. Interestingly, an increased frequency of T-cells in tumor-bearing animals with either pharmacological or genetical PI3Kγ inhibition has been noted (96). To exclude bystander effects via the myeloid cell compartment, which is affected by dysfunctional PI3Kγ, myeloid cells of LLC-tumor bearing mice were depleted using clodronate liposomes and mice were treated with PI3Kγ inhibitors. In comparison to vehicle-treated animals, PI3Kγ inhibition did not result in altered T-cell numbers suggesting that it does not affect T-cell function markedly (96). In line, genetical or antibody-mediated depletion of CD8+ T-cells in PI3Kγ−/− mice caused a reduced tumor control but no alterations in proliferation or production of IFNγ or GzmB of PI3Kγ-deficient T-cells isolated from either naïve or tumor bearing mice have been observed ex vivo (96). In conclusion, PI3Kγ inhibition is likely not affecting T-cell function directly. Nevertheless, the effects of PI3Kγ inhibition on other components of the TME, such as myeloid cells, can enhance the recruitment and anti-tumoral of T-cells.
Although both isoform-specific and pan-PI3K inhibitors show promising clinical efficacy in certain human cancers, there are several intrinsic and acquired resistance mechanisms that challenge their use. The PI3K/AKT pathway involves numerous feedback loops and converges with other signaling pathways at several points. This crosstalk mechanism enables the tumors to adapt by directing the signaling to an alternative path and thereby influencing the therapeutic outcomes (155, 156). RAS oncogene that activates both RAF-MAPK and PI3K signaling is one of the striking examples. The two pathways converge at early points after the growth factor receptor stimulation and interestingly, they evolved as antagonists (156). It has been shown that inhibition of the PI3K pathway can cause over-activation of the RAF-MAPK pathway, which induces tumor growth and quenches the efficacy of PI3K inhibition (156). Dual inhibition strategies have strong potential to overcome this dilemma but likely to result in high toxicities and a narrow therapeutic window.
Presence of a genetic alteration that leads to over-activation of the targeted kinase usually predicts the success of the inhibition therapy, as proven in different malignancies (28). However, cancer cells are genetically complex and mutations downstream of the targeted node might also influence the cellular responses to inhibition resulting in reduced sensitivity. In line, activating mutations of the NOTCH pathway induced resistance to PI3K inhibition in breast cancer (157).
Besides pathway-related mutations, so-called “gatekeeper” mutations that deteriorate the sensitivity of tumor cells to the inhibitor can be acquired during the therapy. A common feature of these “gatekeeper” mutations is their presence in the kinase domain of the targeted protein kinase which hinders its binding to the inhibitor. Similar mutations in PI3K are likely to exist. However, studies analyzing the resistance mutations are scarce. Zunder et al. have detected a potential hotspot mutation in p110α that confers resistance by inhibiting the potency of several PI3K inhibitors 5- to 30-fold (158). To our knowledge, other PI3K isoforms have not been analyzed but these results might serve as a start point for the development of new generation, isoform selective PI3K inhibitors since the hotspot is conserved in the entire PI3K family (158). The nature of PI3K inhibition itself is likely to provide a supportive environment for new mutations to occur, as PI3K inhibition in vitro is not cytotoxic but mostly cytostatic for cancer cells (58, 159, 160). Upon inhibition, it has been shown that cells enter a dormant state and can survive with subtle amount of PI3K activity for a significant time (161) during which the accumulation of new mutations might be facilitated.
In CLL, treatment failure of idelalisib caused by de-novo mutations seems to be a rather rare event but disease progression is observed (162). Overall response rates (ORR) are in general relatively high in phase III trials of relapsed/refractory CLL patients with 83.6% for idelalisib/rituximab (median progression-free survival (PFS) 19.4 months) (129), and 70.0% for idelalisib/bendamustine + rituximab (median PFS 20.8 months) (163), but results of long-term follow-ups are still pending. Nevertheless, activating mutations of the BRAF-MAPK pathway have been observed in 10 non-responders to idelalisib (N=7) or voxtalisib (N=3), which targets the PI3K/mTOR pathway. Recurrent mutations were identified in BRAF (N=5), MAP2K1 (N=2), and KRAS (N=2), but no information is provided whether these mutations resulted in resistance to idelalisib or voxtalisib (164). To investigate acquired resistance to idelalisib, samples of 13 CLL patients that initially responded to idelalisib treatment and developed progressive disease were analyzed by whole exome sequencing across three phase III trials (165). Intriguingly, neither recurrent, nor “gatekeeper” mutations or genomic alterations of any other related signaling pathways were identified that could confer resistance to idelalisib treatment. Similarly, in a preclinical mouse model, long-term PI3Kδ inhibition and serial tumor transplantations caused resistance to the treatment, but no recurrent mutations were identified by whole exome sequencing (166). Intriguingly, expression of insulin-like growth factor 1 receptor (IGF1R) was upregulated due to enhanced activity of forkhead box protein O1 (FOXO1) and glycogen synthase kinase 3 beta (GSK3β) which resulted in pronounced MAPK signaling pathway activity (166).
Despite all the challenges and resistance mechanisms, pharmacological targeting of the PI3K pathway is an efficient strategy. It is highly likely that long-term follow up data of CLL patients will provide more information about resistance mechanisms and combination strategies to achieve a durable effect with minimum toxicity.
As PI3Ks are involved in many different cellular functions and crucial signaling pathways, off-target effects need to be considered when treating patients, which has been extensively reviewed (167–169). In brief, inhibition of the α-isoform with alpelisib in breast cancer frequently caused hyperglycemia, rash, diarrhea or stomatitis (167). In CLL, PI3Kδ inhibition using idelalisib caused immune-cell mediated adverse events like pneumonitis, neutropenia, diarrhea, colitis or transaminitis, and an infiltration of T-cells to inflamed tissues has been observed. These adverse events were more frequent when combining idelalisib with rituximab-bendamustine or ofatumumab (129, 148, 168, 169). Recently, Kienle and Stilgenbauer raised optimism that i) the development of more specific PI3Kδ inhibitors like umbralisib, ii) the adaptation of the scheduling of PI3K inhibitors (e.g., intermittent dosing), and iii) combinatory treatment approaches could result in a more favorable toxicity profile and deep treatment responses with PI3K inhibitors (169).
Initial success of idelalisib, the first approved PI3K-inhibitor for CLL, paved the development of a plethora of PI3K inhibitors, as recently reviewed by Kienle and Stilgenbauer (169). However already in 2016, concerns arose that clinical PI3Kδ inhibition can be associated with an increased frequency of immune-related adverse events (AE), such as colitis, pneumonitis, neutropenia as well as elevated hepatic transaminases (129, 168). This highlights the need for a detailed review of the expression as well as function of PI3Ks in components of the TME of CLL, which could serve as potential explanation for the observed AE during idelalisib treatment.
PI3Ks are involved in crucial signaling cascades of both, the non-immune and immune compartment of the TME in CLL. Some PI3K isoforms are expressed by all cell types whereas others are specific for distinct leukocyte subsets. The use of selective inhibitors gives the opportunity to enhance anti-tumoral immune responses of some cell types. An example is the effect of PI3Kγ inhibition on myeloid cells. Therefore, the challenge of subtype specific PI3K inhibition is not to dampen the function of other immune cells that are essential for tumor or pathogen control.
PI3Kδ-selective inhibition and its effect on the immune environment has so far been studied most in CLL. Apart its great success in controlling the CLL progression (129, 163, 170), adverse events, such as immune cell-mediated liver toxicities are a drawback. Reduced frequencies of circulating Tregs accompanied by an enhanced CD8+ T-cell infiltration to liver tissues have been reasoned to be involved in those cases (148). For Tregs, accumulating pre-clinical as well as clinical evidence supports that PI3Kδ inhibition reduces this population in numbers and dampens their immunosuppressive function. In contrast, the effect of PI3kδ inhibition on CD8+ T-cells is still under debate. On the one hand, diminished Treg-mediated immunosuppression was suggested to unleash CD8+ T-cell function, but on the other hand direct, negative effects of PI3Kδ inhibition on the function of this cell type have been observed. The latter could be causative for compromised clearance of pathogens, especially viruses, as well as a reduced anti-tumor control.
Neutrophils are important for the clearance of extracellular pathogens such as bacteria and fungi. Therefore, neutropenia, an AE observed in idelalisib-treated CLL patients, could be reasoned for higher rates of infections such as pneumonia. In vitro treatment of neutrophils with PI3Kδ inhibitors hindered their activation and their ADCC, supporting that inhibition of PI3Kδ in neutrophils could be involved in reduced immune function of idelalisib-treated CLL patients.
Today, little is known about the effect of other PI3K isoforms on the function of the TME in CLL. In contrast, many clinical trials investigating novel PI3Kδ inhibitors, dual inhibitors of PI3Kδ and -γ, as well as pan-Class IA inhibitors have been initiated (169). Therefore, thorough investigations of the TME in these trials are essential to elucidate the role of PI3K class I isoforms on the function of distinct cell types and to reassure that PI3K inhibitors can serve as a highly active, safe, and tolerable treatment option in CLL.
EA reviewed the literature, prepared the figures, wrote and revised the manuscript. SF, MSa, and LC reviewed the literature and prepared a draft of the manuscript. MSe reviewed the literature and revised the final version of the manuscript. PMR reviewed the literature, wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell (2017) 170(4):605–35. doi: 10.1016/j.cell.2017.07.029
2. Werner M, Hobeika E, Jumaa H. Role of PI3K in the generation and survival of B cells. Immunol Rev (2010) 237(1):55–71. doi: 10.1111/j.1600-065X.2010.00934.x
3. Lauenstein JU, Udgata A, Bartram A, De Sutter D, Fisher DI, Halabi S, et al. Phosphorylation of the multifunctional signal transducer B-cell adaptor protein (BCAP) promotes recruitment of multiple SH2/SH3 proteins including GRB2. J Biol Chem (2019) 294(52):19852–61. doi: 10.1074/jbc.RA119.009931
4. Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol (2003) 3(4):317–30. doi: 10.1038/nri1056
5. Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci (2005) 30(4):194–204. doi: 10.1016/j.tibs.2005.02.008
6. Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem (1999) 274(16):10963–8. doi: 10.1074/jbc.274.16.10963
7. Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, et al. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal (2008) 1(36):ra3. doi: 10.1126/scisignal.1161577
8. Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome (2002) 13(3):169–72. doi: 10.1007/BF02684023
9. Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science (2002) 297(5583):1031–4. doi: 10.1126/science.1073560
10. Guo H, Samarakoon A, Vanhaesebroeck B, Malarkannan S. The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J Exp Medicine (2008) 205(10):2419–35. doi: 10.1084/jem.20072327
11. Cantrell DA. Phosphoinositide 3-kinase signalling pathways. J Cell Science (2001) 114(Pt 8):1439–45.
12. Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science (2000) 287(5455):1040–6. doi: 10.1126/science.287.5455.1040
13. Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science (2000) 287(5455):1046–9. doi: 10.1126/science.287.5455.1046
14. Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science (2000) 287(5455):1049–53. doi: 10.1126/science.287.5455.1049
15. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discovery (2016) 6(8):870–85. doi: 10.1158/2159-8290.CD-15-1346
16. Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carre C, et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Reports (2019) 26(6):1627–40.e7. doi: 10.1016/j.celrep.2019.01.041
17. Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell (2018) 175(6):1701–15.e16. doi: 10.1016/j.cell.2018.10.022
18. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
19. Capra M, Nuciforo PG, Confalonieri S, Quarto M, Bianchi M, Nebuloni M, et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res (2006) 66(16):8147–54. doi: 10.1158/0008-5472.CAN-05-3489
20. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer (2003) 3(1):11–22. doi: 10.1038/nrc969
21. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev (2004) 30(2):193–204. doi: 10.1016/j.ctrv.2003.07.007
22. Hantschel O. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer (2012) 3(5-6):436–46. doi: 10.1177/1947601912458584
23. Paul MK, Mukhopadhyay AK. Tyrosine kinase - Role and significance in Cancer. Int J Med Sci (2004) 1(2):101–15. doi: 10.7150/ijms.1.101
24. Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets (2012) 16(1):15–31. doi: 10.1517/14728222.2011.648617
25. Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A (1999) 96(8):4240–5. doi: 10.1073/pnas.96.8.4240
26. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer (2015) 15(1):7–24. doi: 10.1038/nrc3860
27. Bunney TD, Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer (2010) 10(5):342–52. doi: 10.1038/nrc2842
28. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discover (2009) 8(8):627–44. doi: 10.1038/nrd2926
29. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science (2004) 304(5670):554. doi: 10.1126/science.1096502
30. Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res an Off J Am Assoc Cancer Res (2005) 11(8):2875–8. doi: 10.1158/1078-0432.CCR-04-2142
31. Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene (2005) 24(8):1477–80. doi: 10.1038/sj.onc.1208304
32. Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, et al. PIK3CA as an oncogene in cervical cancer. Oncogene (2000) 19(23):2739–44. doi: 10.1038/sj.onc.1203597
33. Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A (2005) 102(3):802–7. doi: 10.1073/pnas.0408864102
34. Samuels Y, Diaz LA Jr., Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell (2005) 7(6):561–73. doi: 10.1016/j.ccr.2005.05.014
35. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med (2011) 365(26):2497–506. doi: 10.1056/NEJMoa1109016
36. Seiffert M, Dietrich S, Jethwa A, Glimm H, Lichter P, Zenz T. Exploiting biological diversity and genomic aberrations in chronic lymphocytic leukemia. Leuk Lymphoma (2012) 53(6):1023–31. doi: 10.3109/10428194.2011.631638
37. Brown JR, Hanna M, Tesar B, Werner L, Pochet N, Asara JM, et al. Integrative genomic analysis implicates gain of PIK3CA at 3q26 and MYC at 8q24 in chronic lymphocytic leukemia. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18(14):3791–802. doi: 10.1158/1078-0432.CCR-11-2342
38. Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood (2010) 116(12):2078–88. doi: 10.1182/blood-2010-02-271171
39. Ringshausen I, Schneller F, Bogner C, Hipp S, Duyster J, Peschel C, et al. Constitutively activated phosphatidylinositol-3 kinase (PI-3K) is involved in the defect of apoptosis in B-CLL: association with protein kinase Cdelta. Blood (2002) 100(10):3741–8. doi: 10.1182/blood-2002-02-0539
40. Ramadani F, Bolland DJ, Garcon F, Emery JL, Vanhaesebroeck B, Corcoran AE, et al. The PI3K isoforms p110alpha and p110delta are essential for pre-B cell receptor signaling and B cell development. Sci Signal (2010) 3(134):ra60. doi: 10.1126/scisignal.2001104
41. Koyasu S. The role of PI3K in immune cells. Nat Immunol (2003) 4(4):313–9. doi: 10.1038/ni0403-313
42. Davids MS, Brown JR. Phosphoinositide 3’-kinase inhibition in chronic lymphocytic leukemia. Hematol Oncol Clin North Am (2013) 27(2):329–39. doi: 10.1016/j.hoc.2012.12.002
43. Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem (1997) 272(31):19236–41. doi: 10.1074/jbc.272.31.19236
44. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol (2013) 34(12):592–601. doi: 10.1016/j.it.2013.07.002
45. Okkenhaug K, Burger JA. PI3K Signaling in Normal B Cells and Chronic Lymphocytic Leukemia (CLL). B Cell Receptor Signaling. Cham: Springer International Publishing (2016). p. 123–42.
46. Roessner PM, Seiffert M. T-cells in chronic lymphocytic leukemia: Guardians or drivers of disease? Leukemia (2020) 34(8):2012–24. doi: 10.1038/s41375-020-0873-2
47. Hanna BS, Ozturk S, Seiffert M. Beyond bystanders: Myeloid cells in chronic lymphocytic leukemia. Mol Immunol (2019) 110:77–87. doi: 10.1016/j.molimm.2017.11.014
48. Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science (1999) 283(5400):393–7. doi: 10.1126/science.283.5400.393
49. Suzuki H, Terauchi Y, Fujiwara M, Aizawa S, Yazaki Y, Kadowaki T, et al. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science (1999) 283(5400):390–2. doi: 10.1126/science.283.5400.390
50. Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Medicine (2002) 196(6):753–63. doi: 10.1084/jem.20020805
51. Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Carpino N, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol (2002) 22(24):8580–91. doi: 10.1128/mcb.22.24.8580-8591.2002
52. Vanhaesebroeck B, Rohn JL, Waterfield MD. Gene targeting: attention to detail. Cell (2004) 118(3):274–6. doi: 10.1016/j.cell.2004.07.018
53. Tassi I, Cella M, Gilfillan S, Turnbull I, Diacovo TG, Penninger JM, et al. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity (2007) 27(2):214–27. doi: 10.1016/j.immuni.2007.07.014
54. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi: 10.1038/ni.2703
55. Boissard F, Laurent C, Ramsay AG, Quillet-Mary A, Fournie JJ, Poupot M, et al. Nurse-like cells impact on disease progression in chronic lymphocytic leukemia. Blood Cancer J (2016) 6(1):e381. doi: 10.1038/bcj.2015.108
56. Bertrand FE, Spengemen JD, Shelton JG, McCubrey JA. Inhibition of PI3K, mTOR and MEK signaling pathways promotes rapid apoptosis in B-lineage ALL in the presence of stromal cell support. Leukemia (2005) 19(1):98–102. doi: 10.1038/sj.leu.2403560
57. Han Y, Wang X, Wang B, Jiang G. The progress of angiogenic factors in the development of leukemias. Intractable Rare Dis Res (2016) 5(1):6–16. doi: 10.5582/irdr.2015.01048
58. Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discovery (2016) 6(10):1090–105. doi: 10.1158/2159-8290.CD-16-0716
59. Kong D, Okamura M, Yoshimi H, Yamori T. Antiangiogenic effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor. Eur J Cancer (2009) 45(5):857–65. doi: 10.1016/j.ejca.2008.12.007
60. Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, et al. RAS interaction with PI3K p110alpha is required for tumor-induced angiogenesis. J Clin Invest (2014) 124(8):3601–11. doi: 10.1172/JCI74134
61. Soler A, Serra H, Pearce W, Angulo A, Guillermet-Guibert J, Friedman LS, et al. Inhibition of the p110alpha isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J Exp Medicine (2013) 210(10):1937–45. doi: 10.1084/jem.20121571
62. Soler A, Figueiredo AM, Castel P, Martin L, Monelli E, Angulo-Urarte A, et al. Therapeutic Benefit of Selective Inhibition of p110alpha PI3-Kinase in Pancreatic Neuroendocrine Tumors. Clin Cancer Res an Off J Am Assoc Cancer Res (2016) 22(23):5805–17. doi: 10.1158/1078-0432.CCR-15-3051
63. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15(3):220–31. doi: 10.1016/j.ccr.2009.01.027
64. Qayum N, Im J, Stratford MR, Bernhard EJ, McKenna WG, Muschel RJ. Modulation of the tumor microvasculature by phosphoinositide-3 kinase inhibition increases doxorubicin delivery in vivo. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18(1):161–9. doi: 10.1158/1078-0432.CCR-11-1413
65. Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res (2012) 72(1):239–48. doi: 10.1158/0008-5472.CAN-11-2263
66. Hirsch E, Ciraolo E, Franco I, Ghigo A, Martini M. PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene (2014) 33(24):3083–90. doi: 10.1038/onc.2013.265
67. Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood (2015) 126(9):1106–17. doi: 10.1182/blood-2014-12-618025
68. Ding X, Xi W, Ji J, Cai Q, Jiang J, Shi M, et al. HGF derived from cancerassociated fibroblasts promotes vascularization in gastric cancer via PI3K/AKT and ERK1/2 signaling. Oncol Rep (2018) 40(2):1185–95. doi: 10.3892/or.2018.6500
69. Li H, Zhang Q, Wu Q, Cui Y, Zhu H, Fang M, et al. Interleukin-22 secreted by cancer-associated fibroblasts regulates the proliferation and metastasis of lung cancer cells via the PI3K-Akt-mTOR signaling pathway. Am J Transl Res (2019) 11(7):4077–88.
70. Tian B, Chen X, Zhang H, Li X, Wang J, Han W, et al. Urokinase plasminogen activator secreted by cancer-associated fibroblasts induces tumor progression via PI3K/AKT and ERK signaling in esophageal squamous cell carcinoma. Oncotarget (2017) 8(26):42300–13. doi: 10.18632/oncotarget.15857
71. Okkenhaug K. Two birds with one stone: dual p110delta and p110gamma inhibition. Chem Biol (2013) 20(11):1309–10. doi: 10.1016/j.chembiol.2013.11.002
72. Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol (2015) 25(9):545–55. doi: 10.1016/j.tcb.2015.06.002
73. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
74. Donahue AC, Fruman DA. Distinct signaling mechanisms activate the target of rapamycin in response to different B-cell stimuli. Eur J Immunol (2007) 37(10):2923–36. doi: 10.1002/eji.200737281
75. Wlodarski P, Kasprzycka M, Liu X, Marzec M, Robertson ES, Slupianek A, et al. Activation of mammalian target of rapamycin in transformed B lymphocytes is nutrient dependent but independent of Akt, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase, insulin growth factor-I, and serum. Cancer Res (2005) 65(17):7800–8. doi: 10.1158/0008-5472.CAN-04-4180
76. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
77. Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity (2011) 34(2):224–36. doi: 10.1016/j.immuni.2011.01.012
78. Tatebe H, Shiozaki K. Evolutionary Conservation of the Components in the TOR Signaling Pathways. Biomolecules (2017) 7(4). doi: 10.3390/biom7040077
79. Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science (2010) 328(5982):1172–6. doi: 10.1126/science.1187532
80. So L, Lee J, Palafox M, Mallya S, Woxland CG, Arguello M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci Signal (2016) 9(430):ra57. doi: 10.1126/scisignal.aad8463
81. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Reports (2012) 1(4):360–73. doi: 10.1016/j.celrep.2012.02.007
82. Pai C, Walsh CM, Fruman DA. Context-Specific Function of S6K2 in Th Cell Differentiation. J Immunol (Baltimore Md 1950) (2016) 197(8):3049–58. doi: 10.4049/jimmunol.1600167
83. Sasaki CY, Chen G, Munk R, Eitan E, Martindale J, Longo DL, et al. p((7)(0)S(6)K(1)) in the TORC1 pathway is essential for the differentiation of Th17 Cells, but not Th1, Th2, or Treg cells in mice. Eur J Immunol (2016) 46(1):212–22. doi: 10.1002/eji.201445422
84. Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta (2015) 1851(6):882–97. doi: 10.1016/j.bbalip.2014.12.006
85. Boyle KB, Gyori D, Sindrilaru A, Scharffetter-Kochanek K, Taylor PR, Mocsai A, et al. Class IA phosphoinositide 3-kinase beta and delta regulate neutrophil oxidase activation in response to Aspergillus fumigatus hyphae. J Immunol (Baltimore Md 1950) (2011) 186(5):2978–89. doi: 10.4049/jimmunol.1002268
86. Leverrier Y, Okkenhaug K, Sawyer C, Bilancio A, Vanhaesebroeck B, Ridley AJ. Class I phosphoinositide 3-kinase p110beta is required for apoptotic cell and Fcgamma receptor-mediated phagocytosis by macrophages. J Biol Chem (2003) 278(40):38437–42. doi: 10.1074/jbc.M306649200
87. Shimizu-Hirota R, Xiong W, Baxter BT, Kunkel SL, Maillard I, Chen XW, et al. MT1-MMP regulates the PI3Kdelta.Mi-2/NuRD-dependent control of macrophage immune function. Genes Dev (2012) 26(4):395–413. doi: 10.1101/gad.178749.111
88. Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, et al. PI3Kbeta plays a critical role in neutrophil activation by immune complexes. Sci Signal (2011) 4(168):ra23. doi: 10.1126/scisignal.2001617
89. Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood (2005) 106(4):1432–40. doi: 10.1182/blood-2005-03-0944
90. Bucher K, Schmitt F, Mothes B, Blumendeller C, Schall D, Piekorz R, et al. Deficiency of PI3-Kinase catalytic isoforms p110gamma and p110delta in mice enhances the IL-17/G-CSF axis and induces neutrophilia. Cell Commun Signal (2017) 15(1):28. doi: 10.1186/s12964-017-0185-y
91. Costa C, Martin-Conte EL, Hirsch E. Phosphoinositide 3-kinase p110gamma in immunity. IUBMB Life (2011) 63(9):707–13. doi: 10.1002/iub.516
92. Del Prete A, Vermi W, Dander E, Otero K, Barberis L, Luini W, et al. Defective dendritic cell migration and activation of adaptive immunity in PI3Kgamma-deficient mice. EMBO J (2004) 23(17):3505–15. doi: 10.1038/sj.emboj.7600361
93. Steinbach EC, Kobayashi T, Russo SM, Sheikh SZ, Gipson GR, Kennedy ST, et al. Innate PI3K p110delta regulates Th1/Th17 development and microbiota-dependent colitis. J Immunol (Baltimore Md 1950) (2014) 192(8):3958–68. doi: 10.4049/jimmunol.1301533
94. Aksoy E, Taboubi S, Torres D, Delbauve S, Hachani A, Whitehead MA, et al. The p110delta isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat Immunol (2012) 13(11):1045–54. doi: 10.1038/ni.2426
95. Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol (2003) 24(7):358–63. doi: 10.1016/s1471-4906(03)00139-x
96. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature (2016) 539(7629):437–42. doi: 10.1038/nature19834
97. Usman MW, Gao J, Zheng T, Rui C, Li T, Bian X, et al. Macrophages confer resistance to PI3K inhibitor GDC-0941 in breast cancer through the activation of NF-kappaB signaling. Cell Death Disease (2018) 9(8):809. doi: 10.1038/s41419-018-0849-6
98. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature (2016) 539(7629):443–7. doi: 10.1038/nature20554
99. Marshall NA, Galvin KC, Corcoran AM, Boon L, Higgs R, Mills KH. Immunotherapy with PI3K inhibitor and Toll-like receptor agonist induces IFN-gamma+IL-17+ polyfunctional T cells that mediate rejection of murine tumors. Cancer Res (2012) 72(3):581–91. doi: 10.1158/0008-5472.CAN-11-0307
100. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature (2014) 510(7505):407–11. doi: 10.1038/nature13444
101. Goulielmaki E, Bermudez-Brito M, Andreou M, Tzenaki N, Tzardi M, de Bree E, et al. Pharmacological inactivation of the PI3K p110delta prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Disease (2018) 9(6):678. doi: 10.1038/s41419-018-0717-4
102. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell, Aquila M, et al. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell–derived factor-1. Blood (2000) 96(8):2655–63. doi: 10.1182/blood.V96.8.2655.h8002655_2655_2663
103. Tsukada N, Burger JA, Zvaifler NJ, Kipps TJ. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood (2002) 99(3):1030–7. doi: 10.1182/blood.v99.3.1030
104. Boissard F, Fournie JJ, Laurent C, Poupot M, Ysebaert L. Nurse like cells: chronic lymphocytic leukemia associated macrophages. Leuk Lymphoma (2015) 56(5):1570–2. doi: 10.3109/10428194.2014.991731
105. Cutucache CE. Tumor-induced host immunosuppression: special focus on CLL. Int Immunopharmacol (2013) 17(1):35–41. doi: 10.1016/j.intimp.2013.05.021
106. Da Roit F, Engelberts PJ, Taylor RP, Breij EC, Gritti G, Rambaldi A, et al. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica (2015) 100(1):77–86. doi: 10.3324/haematol.2014.107011
107. Enya Chen YC, Burgess M, Mapp S, Mollee P, Gill D, Blumenthal A, et al. PI3K-p110delta contributes to antibody responses by macrophages in chronic lymphocytic leukemia. Leukemia (2020) 34(2):451–61. doi: 10.1038/s41375-019-0556-z
108. Akhiani AA, Hallner A, Kiffin R, Aydin E, Werlenius O, Aurelius J, et al. Idelalisib Rescues Natural Killer Cells from Monocyte-Induced Immunosuppression by Inhibiting NOX2-Derived Reactive Oxygen Species. Cancer Immunol Res (2020) 8(12):1232–41. doi: 10.1158/2326-6066.CIR-20-0055
109. Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood (2011) 118(13):3603–12. doi: 10.1182/blood-2011-05-352492
110. Burger JA, Okkenhaug K. Haematological cancer: idelalisib-targeting PI3Kdelta in patients with B-cell malignancies. Nat Rev Clin Oncol (2014) 11(4):184–6. doi: 10.1038/nrclinonc.2014.42
111. Davids MS, Burger JA. Cell Trafficking in Chronic Lymphocytic Leukemia. Open J Hematol (2012) 3(S1). doi: 10.13055/ojhmt_3_s1_03.120221
112. Fruman DA, Cantley LC. Idelalisib–a PI3Kdelta inhibitor for B-cell cancers. N Engl J Med (2014) 370(11):1061–2. doi: 10.1056/NEJMe1400055
113. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331(6013):44–9. doi: 10.1126/science.1198687
114. Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol (2008) 8(9):713–25. doi: 10.1038/nri2381
115. Saudemont A, Okkenhaug K, Colucci F. p110delta is required for innate immunity to transplantable lymphomas. Biochem Soc Trans (2007) 35(Pt 2):183–5. doi: 10.1042/BST0350183
116. Nandagopal N, Ali AK, Komal AK, Lee SH. The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front Immunol (2014) 5:187. doi: 10.3389/fimmu.2014.00187
117. Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol (2006) 6(12):940–52. doi: 10.1038/nri1983
118. Zhong B, Liu JH, Gilvary DL, Jiang K, Kasuga M, Ritchey CA, et al. Functional role of phosphatidylinositol 3-kinase in direct tumor lysis by human natural killer cells. Immunobiology (2002) 205(1):74–94. doi: 10.1078/0171-2985-00112
119. Awasthi A, Samarakoon A, Dai X, Wen R, Wang D, Malarkannan S. Deletion of PI3K-p85alpha gene impairs lineage commitment, terminal maturation, cytokine generation and cytotoxicity of NK cells. Genes Immun (2008) 9(6):522–35. doi: 10.1038/gene.2008.45
120. Dong S, Guinn D, Dubovsky JA, Zhong Y, Lehman A, Kutok J, et al. IPI-145 antagonizes intrinsic and extrinsic survival signals in chronic lymphocytic leukemia cells. Blood (2014) 124(24):3583–6. doi: 10.1182/blood-2014-07-587279
121. Colucci F, Schweighoffer E, Tomasello E, Turner M, Ortaldo JR, Vivier E, et al. Natural cytotoxicity uncoupled from the Syk and ZAP-70 intracellular kinases. Nat Immunol (2002) 3(3):288–94. doi: 10.1038/ni764
122. Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, et al. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol (2000) 1(5):419–25. doi: 10.1038/80859
123. Kim N, Saudemont A, Webb L, Camps M, Ruckle T, Hirsch E, et al. The p110delta catalytic isoform of PI3K is a key player in NK-cell development and cytokine secretion. Blood (2007) 110(9):3202–8. doi: 10.1182/blood-2007-02-075366
124. Saudemont A, Colucci F. PI3K signaling in lymphocyte migration. Cell Cycle (2009) 8(20):3307–10. doi: 10.4161/cc.8.20.9692
125. Werlenius O, Aurelius J, Hallner A, Akhiani AA, Simpanen M, Martner A, et al. Reactive oxygen species induced by therapeutic CD20 antibodies inhibit natural killer cell-mediated antibody-dependent cellular cytotoxicity against primary CLL cells. Oncotarget (2016) 7(22):32046–53. doi: 10.18632/oncotarget.8769
126. Hofland T, Endstra S, Gomes CKP, de Boer R, de Weerdt I, Bobkov V, et al. Natural Killer Cell Hypo-responsiveness in Chronic Lymphocytic Leukemia can be Circumvented In Vitro by Adequate Activating Signaling. Hemasphere (2019) 3(6):e308. doi: 10.1097/HS9.0000000000000308
127. Weiner GJ. Rituximab: mechanism of action. Semin Hematol (2010) 47(2):115–23. doi: 10.1053/j.seminhematol.2010.01.011
128. Bonnema JD, Karnitz LM, Schoon RA, Abraham RT, Leibson PJ. Fc receptor stimulation of phosphatidylinositol 3-kinase in natural killer cells is associated with protein kinase C-independent granule release and cell-mediated cytotoxicity. J Exp Medicine (1994) 180(4):1427–35. doi: 10.1084/jem.180.4.1427
129. Sharman JP, Coutre SE, Furman RR, Cheson BD, Pagel JM, Hillmen P, et al. Final Results of a Randomized, Phase III Study of Rituximab With or Without Idelalisib Followed by Open-Label Idelalisib in Patients With Relapsed Chronic Lymphocytic Leukemia. J Clin Oncol Off J Am Soc Clin Oncol (2019) 37(16):1391–402. doi: 10.1200/JCO.18.01460
130. Friedmann KS, Bozem M, Hoth M. Calcium signal dynamics in T lymphocytes: Comparing in vivo and in vitro measurements. Semin Cell Dev Biol (2019) 94:84–93. doi: 10.1016/j.semcdb.2019.01.004
131. Pearce VQ, Bouabe H, MacQueen AR, Carbonaro V, Okkenhaug K. PI3Kdelta Regulates the Magnitude of CD8+ T Cell Responses after Challenge with Listeria monocytogenes. J Immunol (Baltimore Md 1950) (2015) 195(7):3206–17. doi: 10.4049/jimmunol.1501227
132. Gracias DT, Boesteanu AC, Fraietta JA, Hope JL, Carey AJ, Mueller YM, et al. Phosphatidylinositol 3-Kinase p110delta Isoform Regulates CD8+ T Cell Responses during Acute Viral and Intracellular Bacterial Infections. J Immunol (Baltimore Md 1950) (2016) 196(3):1186–98. doi: 10.4049/jimmunol.1501890
133. Okkenhaug K, Patton DT, Bilancio A, Garcon F, Rowan WC, Vanhaesebroeck B. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol (Baltimore Md 1950) (2006) 177(8):5122–8. doi: 10.4049/jimmunol.177.8.5122
134. Hanna BS, Roessner PM, Scheffold A, Jebaraj BMC, Demerdash Y, Ozturk S, et al. PI3Kdelta inhibition modulates regulatory and effector T-cell differentiation and function in chronic lymphocytic leukemia. Leukemia (2019) 33(6):1427–38. doi: 10.1038/s41375-018-0318-3
135. Soond DR, Bjorgo E, Moltu K, Dale VQ, Patton DT, Torgersen KM, et al. PI3K p110delta regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood (2010) 115(11):2203–13. doi: 10.1182/blood-2009-07-232330
136. Chellappa S, Kushekhar K, Munthe LA, Tjonnfjord GE, Aandahl EM, Okkenhaug K, et al. The PI3K p110delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J Immunol (Baltimore Md 1950) (2019) 202(5):1397–405. doi: 10.4049/jimmunol.1701703
137. Ahmad S, Abu-Eid R, Shrimali R, Webb M, Verma V, Doroodchi A, et al. Differential PI3Kdelta Signaling in CD4(+) T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res (2017) 77(8):1892–904. doi: 10.1158/0008-5472.CAN-16-1839
138. Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced Therapeutic Efficacy and Memory of Tumor-Specific CD8 T Cells by Ex Vivo PI3K-delta Inhibition. Cancer Res (2017) 77(15):4135–45. doi: 10.1158/0008-5472.CAN-16-1925
139. Snook JP, Kim C, Williams MA. TCR signal strength controls the differentiation of CD4(+) effector and memory T cells. Sci Immunol (2018) 3(25). doi: 10.1126/sciimmunol.aas9103
140. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature (2017) 552(7685):362–7. doi: 10.1038/nature24633
141. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature (2017) 552(7685):404–9. doi: 10.1038/nature25144
142. Lim EL, Cugliandolo FM, Rosner DR, Gyori D, Roychoudhuri R, Okkenhaug K. Phosphoinositide 3-kinase delta inhibition promotes antitumor responses but antagonizes checkpoint inhibitors. JCI Insight (2018) 3(11). doi: 10.1172/jci.insight.120626
143. Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, et al. PI3Kdelta Inhibition Enhances the Antitumor Fitness of Adoptively Transferred CD8(+) T Cells. Front Immunol (2017) 8:1221. doi: 10.3389/fimmu.2017.01221
144. Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, et al. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol (Baltimore Md 1950) (2006) 177(10):6598–602. doi: 10.4049/jimmunol.177.10.6598
145. Carnevalli LS, Sinclair C, Taylor MA, Gutierrez PM, Langdon S, Coenen-Stass AML, et al. PI3Kalpha/delta inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J Immunother Cancer (2018) 6(1):158. doi: 10.1186/s40425-018-0457-0
146. Martinelli S, Maffei R, Fiorcari S, Quadrelli C, Zucchini P, Benatti S, et al. Idelalisib impairs T-cell-mediated immunity in chronic lymphocytic leukemia. Haematologica (2018) 103(12):e598–601. doi: 10.3324/haematol.2017.187070
147. Dong S, Harrington BK, Hu EY, Greene JT, Lehman AM, Tran M, et al. PI3K p110delta inactivation antagonizes chronic lymphocytic leukemia and reverses T cell immune suppression. J Clin Invest (2019) 129(1):122–36. doi: 10.1172/JCI99386
148. Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood (2016) 128(2):195–203. doi: 10.1182/blood-2016-03-707133
149. Hanna BS, Roessner PM, Yazdanparast H, Colomer D, Campo E, Kugler S, et al. Control of chronic lymphocytic leukemia development by clonally-expanded CD8(+) T-cells that undergo functional exhaustion in secondary lymphoid tissues. Leukemia (2019) 33(3):625–37. doi: 10.1038/s41375-018-0250-6
150. Ozturk S, Roessner PM, Schulze-Edinghausen L, Yazdanparast H, Kalter V, Lichter P, et al. Rejection of adoptively transferred Emicro-TCL1 chronic lymphocytic leukemia cells in C57BL/6 substrains or knockout mouse lines. Leukemia (2019) 33(6):1514–39. doi: 10.1038/s41375-018-0332-5
151. Friend NL, Hewett DR, Panagopoulos V, Noll JE, Vandyke K, Mrozik KM, et al. Characterization of the role of Samsn1 loss in multiple myeloma development. FASEB Bioadv (2020) 2(9):554–72. doi: 10.1096/fba.2020-00027
152. de Weerdt I, Hofland T, de Boer R, Dobber JA, Dubois J, van Nieuwenhuize D, et al. Distinct immune composition in lymph node and peripheral blood of CLL patients is reshaped during venetoclax treatment. Blood Advances (2019) 3(17):2642–52. doi: 10.1182/bloodadvances.2019000360
153. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood (2016) 127(9):1117–27. doi: 10.1182/blood-2015-11-679134
154. Stock S, Ubelhart R, Schubert ML, Fan F, He B, Hoffmann JM, et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int J Cancer (2019) 145(5):1312–24. doi: 10.1002/ijc.32201
155. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res (2006) 66(3):1500–8. doi: 10.1158/0008-5472.CAN-05-2925
156. Jun T, Gjoerup O, Roberts TM. Tangled webs: evidence of cross-talk between c-Raf-1 and Akt. Sci STKE (1999) 1999(13):PE1. doi: 10.1126/stke.1999.13.pe1
157. Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol (2011) 7(11):787–93. doi: 10.1038/nchembio.695
158. Zunder ER, Knight ZA, Houseman BT, Apsel B, Shokat KM. Discovery of drug-resistant and drug-sensitizing mutations in the oncogenic PI3K isoform p110 alpha. Cancer Cell (2008) 14(2):180–92. doi: 10.1016/j.ccr.2008.06.014
159. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discover (2014) 13(2):140–56. doi: 10.1038/nrd4204
160. Yap TA, Bjerke L, Clarke PA, Workman P. Drugging PI3K in cancer: refining targets and therapeutic strategies. Curr Opin Pharmacol (2015) 23:98–107. doi: 10.1016/j.coph.2015.05.016
161. Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc Natl Acad Sci U S A (2010) 107(25):11381–6. doi: 10.1073/pnas.0906461107
162. Sedlarikova L, Petrackova A, Papajik T, Turcsanyi P, Kriegova E. Resistance-Associated Mutations in Chronic Lymphocytic Leukemia Patients Treated With Novel Agents. Front Oncol (2020) 10:894. doi: 10.3389/fonc.2020.00894
163. Zelenetz AD, Barrientos JC, Brown JR, Coiffier B, Delgado J, Egyed M, et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol (2017) 18(3):297–311. doi: 10.1016/S1470-2045(16)30671-4
164. Murali I, Kasar S, McWilliams EM, Itchaki G, Tyekucheva S, Livitz D, et al. Activating MAPK Pathway Mutations Mediate Primary Resistance to PI3K Inhibitors in Chronic Lymphocytic Leukemia (CLL). Blood (2018) 132(Supplement 1):587–. doi: 10.1182/blood-2018-99-115304
165. Ghia P, Ljungström V, Tausch E, Agathangelidis A, Scheffold A, Scarfo L, et al. Whole-Exome Sequencing Revealed No Recurrent Mutations within the PI3K Pathway in Relapsed Chronic Lymphocytic Leukemia Patients Progressing Under Idelalisib Treatment. Blood (2016) 128(22):2770–. doi: 10.1182/blood.V128.22.2770.2770
166. Scheffold A, Jebaraj BMC, Tausch E, Bloehdorn J, Ghia P, Yahiaoui A, et al. IGF1R as druggable target mediating PI3K-delta inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood (2019) 134(6):534–47. doi: 10.1182/blood.2018881029
167. Nunnery SE, Mayer IA. Management of toxicity to isoform α-specific PI3K inhibitors. Ann Oncol (2019) 30:x21–x6. doi: 10.1093/annonc/mdz440
168. Coutre SE, Barrientos JC, Brown JR, de Vos S, Furman RR, Keating MJ, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma (2015) 56(10):2779–86. doi: 10.3109/10428194.2015.1022770
169. Kienle DL, Stilgenbauer S. Approved and emerging PI3K inhibitors for the treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma. Expert Opin Pharmacother (2020) 21(8):917–29. doi: 10.1080/14656566.2020.1737010
170. Jones JA, Robak T, Brown JR, Awan FT, Badoux X, Coutre S, et al. Efficacy and safety of idelalisib in combination with ofatumumab for previously treated chronic lymphocytic leukaemia: an open-label, randomised phase 3 trial. Lancet Haematol (2017) 4(3):e114–e26. doi: 10.1016/S2352-3026(17)30019-4
Keywords: phosphoinositide 3-kinase (PI3K), chronic lymphocytic leukemia, tumor microenvironment, idelalisib, phosphoinositide 3-kinase (PI3K) inhibition
Citation: Aydin E, Faehling S, Saleh M, Llaó Cid L, Seiffert M and Roessner PM (2021) Phosphoinositide 3-Kinase Signaling in the Tumor Microenvironment: What Do We Need to Consider When Treating Chronic Lymphocytic Leukemia With PI3K Inhibitors? Front. Immunol. 11:595818. doi: 10.3389/fimmu.2020.595818
Received: 17 August 2020; Accepted: 07 December 2020;
Published: 20 January 2021.
Edited by:
Dominik Wolf, Innsbruck Medical University, AustriaReviewed by:
Dimitar G. Efremov, International Centre for Genetic Engineering and Biotechnology, ItalyCopyright © 2021 Aydin, Faehling, Saleh, Llaó Cid, Seiffert and Roessner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philipp M. Roessner, cC5yb2Vzc25lckBka2Z6LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.