 Deepak Sumbria
Deepak Sumbria Engin Berber
Engin Berber Manikannan Mathayan
Manikannan Mathayan Barry T. Rouse
Barry T. Rouse
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 February 2021
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.594963
When viruses infect cells, they almost invariably cause metabolic changes in the infected cell as well as in several host cell types that react to the infection. Such metabolic changes provide potential targets for therapeutic approaches that could reduce the impact of infection. Several examples are discussed in this review, which include effects on energy metabolism, glutaminolysis and fatty acid metabolism. The response of the immune system also involves metabolic changes and manipulating these may change the outcome of infection. This could include changing the status of herpesviruses infections from productive to latency. The consequences of viral infections which include coronavirus disease 2019 (COVID-19), may also differ in patients with metabolic problems, such as diabetes mellitus (DM), obesity, and endocrine diseases. Nutrition status may also affect the pattern of events following viral infection and examples that impact on the pattern of human and experimental animal viral diseases and the mechanisms involved are discussed. Finally, we discuss the so far few published reports that have manipulated metabolic events in-vivo to change the outcome of virus infection. The topic is expected to expand in relevance as an approach used alone or in combination with other therapies to shape the nature of virus induced diseases.
Viruses are obligate intracellular parasites and usually cause changes and often times death to cells that support their replication. In the infected host, this may manifest as disease which in some cases is the direct result of virus replication events in target cells. In other circumstances, clinical consequences of viral infection are attributed, fully or in part, to the host immune response to infected cells, or to extracellular virus (1–3). Whatever the means by which viruses act as pathogens, the infection results in changes in metabolism in the cells they infect and also in multiple cell types of the host that react to the infection. In addition, innate responses set off by viral infections, such as interferons type I and II, also act to modulate some aspects of metabolism (4). The consequences of many viral infections can differ in hosts that have abnormal metabolism such as diabetes mellitus (DM). This has become an important issue in the case of coronavirus disease 2019 (COVID-19) infection where diabetics are more likely to suffer from more severe disease (5). In addition, accumulating evidence shows that the state of nutrition can influence the outcome of a viral infection by causing changes in one or more aspect of metabolism (6). All of these observations imply that adjusting metabolic events during the course of a viral infection could represent a valuable approach to reshape the outcome of infections (7, 8). For example, when disease lesions result from immune-inflammatory reactions to infected tissues, changing the metabolic environment that limits the function of inflammatory cell subsets may change the reaction from being highly tissue damaging to one that acts to resolve lesions (8). In this brief review, we discuss evidence showing that manipulating metabolism can represent a useful approach to control the outcome of a virus infection.
Viruses themselves are metabolically inert and must rely on metabolic events in the cell to generate its component parts and to replicate new viral copies. Oftentimes, the cell at the time of infection is in a quiescent state, but the infection acts to change the cell’s metabolic activity. There are multiple mechanisms by which a virus infection can induce metabolic changes in infected cell and these are listed in Table 1. A common consequence of infection by many viruses is to induce high glucose metabolism (causing aerobic glycolysis, the so-called Warburg effect) in cells and to change the nature of lipid metabolism usually from fatty acid oxidation (FAO) to fatty acid synthesis (FAS) (16, 22). Increased FAS is particularly necessary for those viruses that are enveloped. With some viruses, infection only occurs in cells that are already metabolically active with the infection often serving to downregulate one or more metabolic events. This state of affairs occurs with HIV, which preferentially infects cells that are in an activated metabolic state (23). Accordingly, HIV infects T cells undergoing the highest levels of metabolic activity, which includes elevated aerobic glycolysis (24), TCA (tricarboxylic acid cycle) cycle activity, oxidative phosphorylation (OXPHOS), and glutaminolysis (25–28). The result is cell destruction and the onset of immunosuppression. The multiple metabolic events that can be set into play by different types of virus infection are discussed in detail in an excellent recent review by Eisenreich et al. (29).
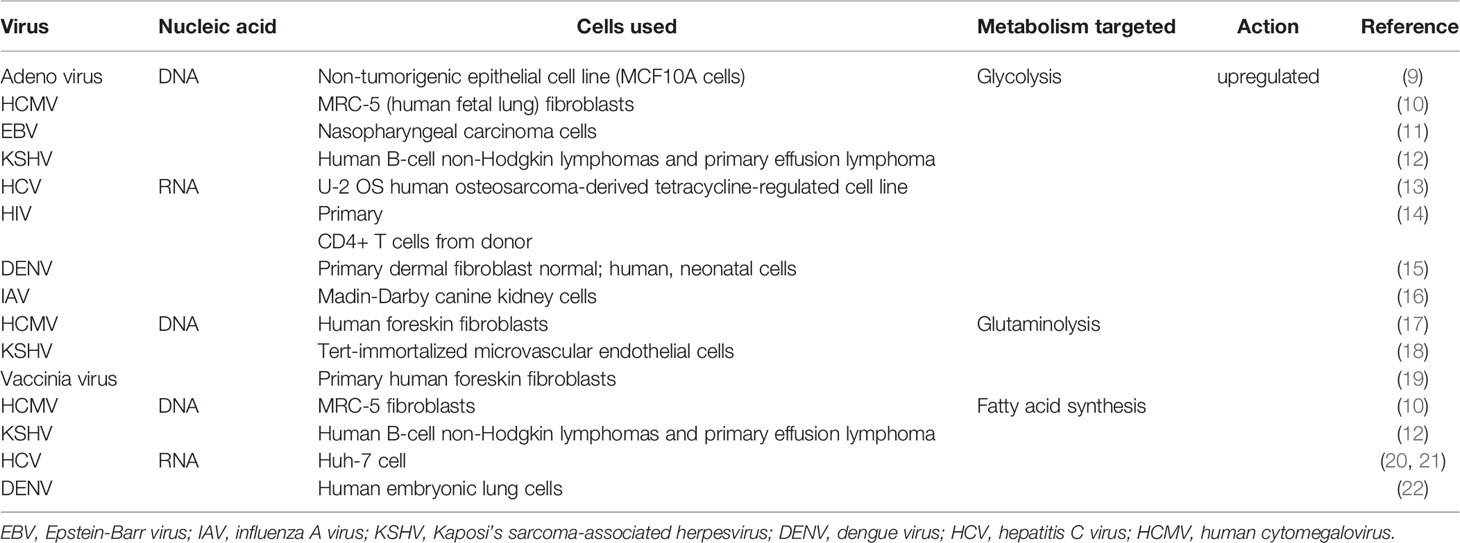
Table 1 Metabolic alteration in various virus infection.
Curiously, a wide range of mechanisms are known whereby viruses influence metabolic events in their target cells. Of particular interest is the variable events that occur following infection with different flaviviruses. With dengue virus infection, for example, the virus taps into the host cell’s lipid reserves which are held in lipid droplets. The droplets are broken down by an autophagy type mechanism releasing fatty acids. These undergo oxidation which fuels the TCA cycle providing ATP and TCA intermediates needed for viral replication (30).
The Dengue related virus Zika (ZIKV) employs an interesting metabolic manipulation, although only when it infects neuronal cells, an important event in ZIKV pathogenesis that does not occur in Dengue. Whereas in all cell types, ZIKV virus enhances the expression of proteins, such as the cell death proteins ZBP1, RIPK 1 and 3, in neuronal cells an additional event also occurs. Thus ZIKV infected neuronal cells express immune responsive gene 1 (IRG1) whose product generates itaconate from cis-aconite, a component of the TCA cycle (31). The itaconate in turn competitively inhibits succinate dehydrogenase (SDH) thus maintaining succinate levels and this keeps neuronal cells alive (31–34). In addition, the inhibition of SDH activity inhibits ZIKV replication (31). Why the IRG1 mediated effect only occurs in neuronal cells is unclear, but could be related to the increased resistance of mature neuronal cells to apoptosis. This effect is thought to occur because neurons have some protein expression differences. These includes low level of apoptotic protease activating factor 1, increased X-linked inhibitor of apoptosis protein associated factor, and reduced caspase-3 involved in programmed cell death (35).
Hepatitis C virus (HCV) is another human pathogen that causes metabolic effects but these change at different times after infection. In the first 24 h after infection, almost all pathways used to produce the macromolecules needed for the synthesis of new viruses are elevated (36). These include both FAS and FAO with the simultaneous elevation of both FAO and FAS likely needed to provide intermediates for the TCA cycle required for virus envelop formation. In later stages (48 h), several pathways are suppressed and then the infected cell’s integrity is sustained by metabolizing host cell amino acids, especially glutamine, to sustain the TCA cycle and avoid cell death (36).
Viruses may show marked differences in the metabolic changes they impose on target cells. For example, the two herpesviruses human cytomegalovirus (HCMV) and herpes simplex virus (HSV) induce quite different effects on cellular metabolism. Infection with HCMV increases glycolytic flux, lactate excretion, FAS, and profoundly increases the level of intermediary molecules involved in the TCA cycle (37). The latter are needed so that the virus can make new viral envelopes, which with HCMV are always produced de novo (10). In contrast, infection with HSV, which replicates more rapidly (24 h) than HCMV (96 h), causes minimal effects on glycolysis and FAS but nucleotide synthesis is markedly affected with the process commandeered by HSV to produce new viral genomes (37). With HSV, this occurs by the virus directing the pentose pathway as well as the TCA cycle to produce the purines and pyrimidines needed to construct viral genomes (37). Compared to HCMV, HSV relies less on FAS since its envelope is mainly made from preexisting cellular envelope material.
All herpesviruses employ two lifestyles when interacting with the host. These are productive infection, which generates new virions and oftentimes results in the infected cell being destroyed. Herpesviruses also set up a state of latency, which permits the virus to remain indefinitely in the host representing the usual source of infection to others once viral reactivation and viral excretion occurs. Metabolic events during the two lifestyles are certain to be different, since latency involves minimal or even no synthesis of new viral proteins and oftentimes almost minimal effects on cellular metabolism. Productive infections, in contrast, can provoke profound changes, as already mentioned. There is also gathering evidence that changes in metabolism can be a lifestyle changing event, causing a latently infected cell to reenter the productive cycle, frequently a step needed to permit viral transmission to other hosts. This issue is under investigation in our laboratory. Some also speculate that differences in the metabolism of immune cells at local sites may influence the clinical consequences when reactivation from latency does occur (38). Such ideas have been advocated to explain why reactivation of HSV from latency is sometimes subclinical, but on other occasions results in significant tissue damage. Could it be, for example, that the deficiency of local fuels to support glycolysis would result in changes in viral pathogenesis? This concept is currently being explored. Metabolic differences between productive and latent infections have been described for other herpesviruses, some of which could account for pathogenesis changes such as contributing to neoplasia. With the herpes virus Epstein–Barr virus (EBV), some genes are expressed during latency that include LMP1, LMP2, and EBNA1. These may act to change aerobic glycolysis via effects on glucose uptake and metabolism. This topic is comprehensively discussed in a review by Piccaluga et al (39).. Inhibition of fatty acid synthesis helps to control the excessive multiplication of EBV associated nasopharyngeal carcinoma. Moreover, inhibition of hexokinase 2 (HK2) also provides a useful treatment modality for nasopharyngeal carcinoma (11, 40).
The relevance of understanding the nature of metabolic changes imposed by different virus infections is that targeted molecular therapies may be devised that can change the outcome of infection. For example, inhibiting glucose metabolism in cells infected with HIV promotes viral elimination by accelerating the death of infected cells (41). During the late stages of HIV replication FAS is elevated, which is needed for its envelope formation. Conceivably targeting lipid metabolism could also represent a means of counteracting HIV infection and this topic has been investigated (42). It was shown that inhibition of FAS with Fasnall and C75 inhibits HIV-1 replication in-vitro, as did Fasnall knockdown with small interfering RNA (siRNA). The outcome of such procedures in-vivo are not available. Currently, there is minimal information regarding manipulating metabolic pathways to influence the outcome of COVID-19 infection.
A common outcome of many viral infection is to induce the production of one or more types of IFNs in the cells they infect. Curiously, many of the metabolic reprogramming events imposed by virus infections on cells that were discussed previously can be reversed by the IFN response. For instance, the increased energy and lipid metabolism essential for the replication of many viruses is readily reversed by IFN, with this representing one of the ways by which the IFN response acts to control virus infections (43). Characteristically, IFNs are released and bind to nearby cells that express specific receptors. This interaction signals the cell to express multiple (>400) so-called interferon stimulated genes (ISG) whose gene products express a wide range of biological activities (44, 45). One common effect is that the cell produces molecules that have antiviral activity. Other ISGs may influence cellular metabolism that in turn can affect the outcome of a virus infection. For example, a response in cells whose IFN-γ receptors are engaged is that transcription factor expression, related with steroid biosynthesis, are changed (46). Transcription levels of the sterol regulatory binding protein (SREGP), which acts to downregulate cholesterol biosynthesis, are suppressed and this limits the availability of lipids which are needed particularly to generate new enveloped viruses. The SREGP can also inhibit cell entry of some viruses such as HIV-1 and it also impairs intracellular virus budding through the endoplasmic and cell membranes (47, 48).
Another ISG induced by type I IFN, that affects cellular metabolism encodes the enzyme cholesterol-25-hydroxylase (CH25H). This enzyme converts cholesterol to soluble oxysterol 25-hydroxycholesterol (25HC) that in turn serves to decrease cholesterol accumulation within cells (Figure 1). The overall effect is increased resistance to several viruses such as ZIKV and other flaviviruses as well as with some togaviruses (49, 50). The inhibitory effect is the consequence of inhibition of viral fusion needed to enter and transport within cells as well as for the formation of new virions (51). Upon administration to animals, the molecule 25HC can also exert antiviral effects. For example, the pups of pregnant mice given 25HC are protected from the neurological consequences of ZIKV infection (49). In addition, in humanized mice treatment with 25HC increased resistance of their T cells to HIV infection (51). Finally, by in-vitro studies 25HC was shown to have antiviral effects against several viruses that include vesicular stomatitis virus (VSV) and HSV from Herpesviridae, HIV from Retroviridae, Ebola virus (EBOV) from Filoviridae, Rift Valley fever virus (RVFV) from Bunyaviridae, Russian spring-summer encephalitis virus (RSSEV) Flavirviridae, and Nipah virus within Paramyxoviridae family (51).
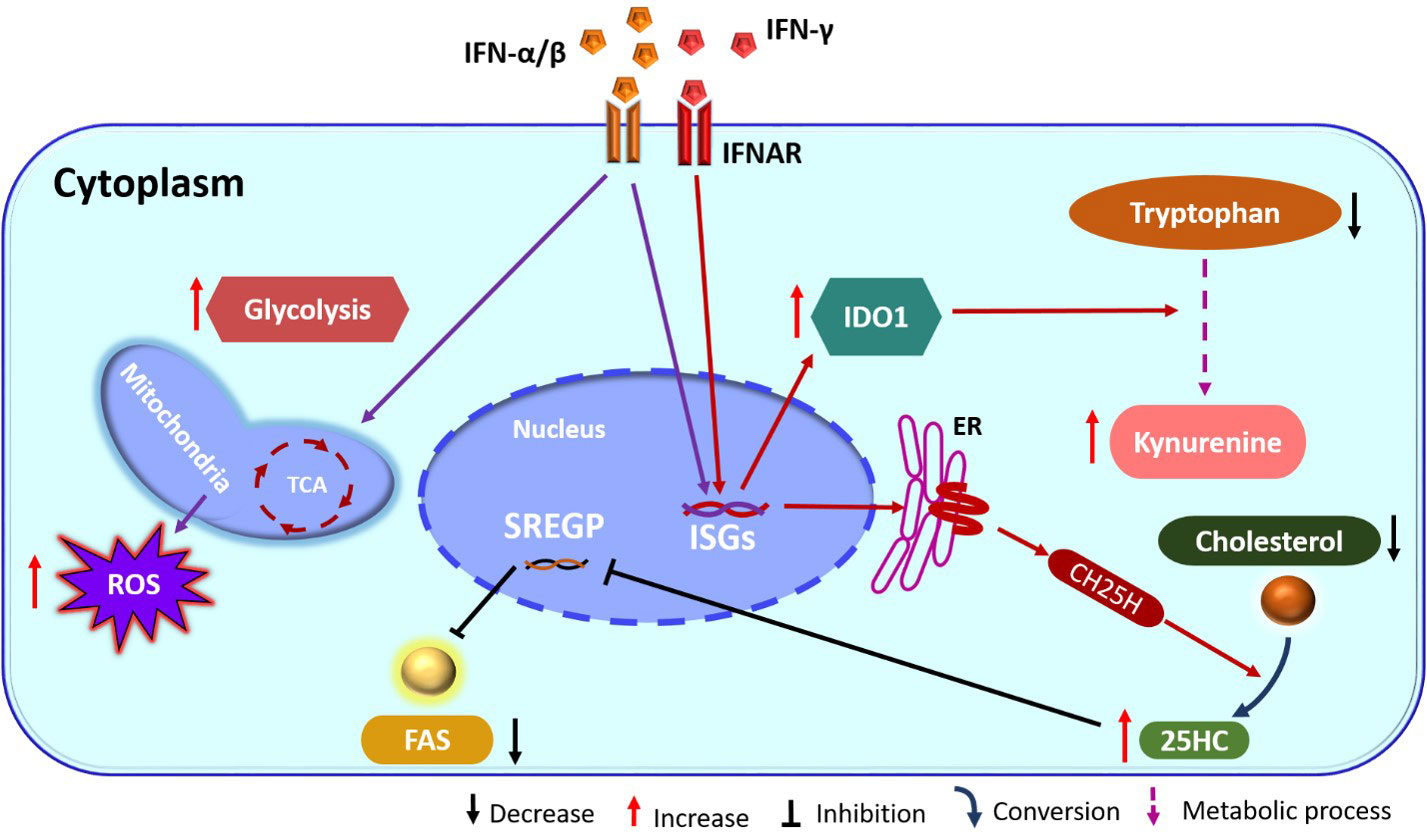
Figure 1 IFN action on cellular metabolism pathways. IFN binds its receptor (IFNAR) and induces ISGs, which includes the enzyme CH25H. This enzyme catalyzes the formation of 25HC from cholesterol. 25HC has inhibitory action on SREGP and as a consequence FAS is reduced. In addition, IFN also stimulates TCA and glycolysis. Stimulation of ISGs also increase IDO1 and enhances the kynurenine pathway from tryptophan (ER, endoplasmic reticulum; IFN, interferon; IFNAR, IFN-receptor; TCA, tricarboxylic acid cycle; CH25H, cholesterol 25-hydroxylase; ISGs, interferon stimulated genes; 25HC, 25-hydroxycholesterol; SREGP, sterol regulatory binding protein; IDO1, indoleamine 2,3 dioxygenase1; FAS, fatty acid synthesis; ROS, reactive oxygen species).
Other ISGs can also influence lipid metabolism which then acts to limit infection and/or change the inflammatory function of cells such as macrophages. For example, monocytes exposed to IFN-α change their lipid metabolism and undergo enhanced reactive oxygen species (ROS) production, which is essential for the antiviral effector functions of macrophages (52). Another critical influence on the outcome of virus infection set off by IFN-γ triggering is induction of the enzyme indoleamine 2,3 dioxygenase (IDO) (Figure 1) (53). This effect is usually mediated by IFN-γ and occurs mainly in macrophages and dendritic cells. The IDO enzyme converts tryptophan to kynurenine, which has several effects on immune function. These include inhibitory effects on some innate immune activities and suppression of T cell proliferation and function (54). Cells with upregulated IDO also increase their uptake of tryptophan that acts also to inhibit the effector activity of T cells (54). IDO may also have direct inhibitory effects on some viruses that include HSV (55), HCMV (56) and human parainfluenza virus type 3 (57) perhaps mediated by depriving infected cells of adequate tryptophan. Some have suggested tryptophan deprivation could also be one means by which herpes simplex virus type 2 latency is maintained and the lytic cycle suppressed (58).
As discussed in the previous section, when most viruses infect target cells they induce changes in metabolism that are needed for the virus to be replicated. Examples includes HCMV which upregulate glycolysis in cultured cells (10) and KSHV, which upregulate glutaminolysis in microvascular endothelial cells (18). A complete list of metabolic changes caused by various viruses is given in Table 1. Accordingly, one expects that, in situations where an aspect of host metabolism is malfunctioning, because of genetic or extrinsic reasons, the outcome of a virus infection may be affected. This topic has been mainly explored with diabetes mellitus (DM), which affects sugar metabolism. There are several reports that show that immune defense mechanisms may be less effective as a consequence of diabetes so increased susceptibility to infections is to be anticipated (59–61). Several innate immune activities are diminished during diabetes. These include reduced levels of the antibacterial molecule beta defensin (62), reduction in neutrophil traps (63), reduced capacity to undergo the respiratory burst (64), and to generate antimicrobial reactive radicals such as superoxide (65). In DM, reduced production of some enzymes such as elastase and myeloperoxidase also occur and monocyte phagocytosis may be compromised as well (66, 67). All suppressive effects observed on innate defenses during DM may be caused by hyperglycemia, but in most instances, details of molecular events involved still need to be elucidated. In the case of beta defensin production, reduced levels during diabetic hyperglycemia were attributed to the production of dicarbonyl methylglyoxal from glucose, which changes the surface charge of beta defensin and reduces its antibacterial activity (62). The effects of DM on innate immunity explains why many bacterial infections are more common during DM, but also increased susceptibility to some virus infections has been observed (68). Most reports about changed susceptibility to virus infection in DM patients involves influenza (Flu) but it has recently become evident that diabetics (both the mostly studied type II as well as type I) may experience more severe consequences of COVID-19 infection (69, 70). Rising evidence also shows that COVID-19 infection may worsen the signs of diabetes (71) and also the infection may cause the onset of DM (72).
During flu epidemics, diabetics may experience more clinical problems than non-diabetics as noted in several studies (73, 74). Thus, diabetics require more frequent hospitalization and adult diabetics suffer more severe consequences and higher mortality rates than non-diabetics (75, 76). However, it is far from clear as to the mechanisms which explain increased lesions, although decreased activity of one or more immune components is usually advocated (77–80). Another confounding issue is defining which aspect of DM accounts for susceptibility. Several studies link suppressed immunity with hyperglycemia and others to immune problems caused by obesity, which is a common outcome particularly with insulin resistant type 2 DM. The immunological consequences of obesity as regards effects on the pattern of viral infections is further discussed in a later section.
A better understanding of how DM can cause increased susceptibility to Flu comes from studies of type 1 DM in mouse models, which do show enhanced susceptibility (81). Mice with DM may develop higher levels of virus in their lungs compared to normal mice (82). In one model, the increased susceptibility was associated with excessive levels of glucose which acted to block the function of a surface protein lectin (SP-L) on endothelial cells that normally plays a protective role against Flu. SP-L may also act on neutrophils changing many of their functions and their ability to bind to virus.
In a drug induced model of type 1 DM, diabetic mice compared to non-diabetic controls had higher numbers of infected lung epithelial cells and developed more severe lung and pancreatic damage upon infection with several subtypes of influenza A virus (H1N1, H5N1 and H7N2) (83). In addition, higher mortality levels occurred upon infection with virulent strains of H1N1 as well as with the H5N1 subtype (83). However, this study provided no mechanistic explanation for the findings.
As we write this review, we are deep into the COVID-19 pandemic which raises the issue if diabetics are prone to develop more severe consequences of infection. Several reports indicate this is happening. For instance, in a study in China, out of 52 persons admitted to the intensive care unit (ICU), 32 died and 22% of these patients were diabetics (69). In another study of 173 patients, 16.2% were diabetics and these experienced severe disease consequences (84). In a study of 1,527 patients, two-fold higher numbers of diabetics required admittance to the ICU than normal patients (85). In New York, a recent study reported that, out of 5,700 COVID-19 patients, 33.8% had a history of diabetes which compares to around 10% in the uninfected population with diabetes (86). Thus, evidence accumulates to show that susceptibility to COVID-19 infection is increased in those with diabetes, but so far a mechanistic understanding of why this occurs remains to be shown. However, conceivably one explanation might be associated with the main receptor COVID-19 uses to enter cells, the angiotensin-converting enzyme 2 (ACE2). Curiously, patients with diabetes type 2 upregulate ACE2 at least in renal tissues (87, 88) and expression is further increased upon treatment with ACE inhibitors (89, 90). It is not clear in humans if ACE2 is upregulated in lung tissue where COVID-19 replicates, but in diabetic mice ACE2 levels are increased in lung tissues compared to healthy controls (91, 92). The protein component of COVID-19 that binds ACE2 is the spike protein and soluble forms of ACE2 can block virus infection (93, 94). Finally, trials are underway to determine if ACE2 inhibitor drugs such as the commonly used drug to control high blood pressure, losartan, has any beneficial effect on the outcome of COVID-19 infection in healthy patients. Very recently, it was observed that the incidence of Flu, which uses the ACE2 receptor to mediate lung damage, was reduced in persons that used ACE2 inhibitor drugs to control their blood pressure (95–97). Moreover, prolonged drug use improved the outcome of the Flu infection (95). For the expanding knowledge concerning COVID-19 and DM and management of this interaction, an excellent review was recently published (72).
Other coronaviruses associated with human disease may also be more severe in diabetics. For example, in a study in China of 135 persons that died of severe acute respiratory syndrome (SARS), 21.5% were diabetic, whereas in 385 survivors only 3.9% had diabetes (98). With Middle East respiratory syndrome (MERS) patients in the Middle East, 50.9% of those who died were diabetic, whereas in those that recovered only 22.9% were diabetics (99). In the case of MERS, experimental studies were done in diabetic and non-diabetic control mice. The diabetic animals had more extended disease, diminished innate immune responses, and also had lower T cell responses to infection (100). It is also worth mentioning that with the SARS coronavirus, which has many similarities to COVID-19, there is evidence that the infection could damage islet cells and help precipitate DM (101). Additionally, very recently COVID-19 was advocated to upregulate ACE2 receptors on pancreatic cells making them a target for COVID-19 infection (102).
In addition to coronaviruses and flu, other virus infections may have a different, usually more severe outcome in diabetic compared to non-diabetics. One example is HSV-1 infection which causes increased facial nerve damage in diabetic mice after auricular infection (103, 104). There is also the suggestion that being diabetic is a risk factor for developing herpes zoster caused by varicella-zoster virus (VZV) (105), perhaps explained by diminished T cell responses in DM patients.
It would be also interesting to know if the outcome of chronic virus infections such as HIV and hepatitis are more consequential in those with diabetes (106). There is surprisingly little information on that topic, but what has been more investigated is whether HIV infection or treatments used to control HIV can act as triggering factors to set off DM. For example, one study on HIV-seropositive patients treated with highly active antiretroviral therapy showed that four times more patients developed type 2 DM than occurred in the HIV-seronegative group (107). Treatment of HIV with protease inhibitor drugs can also result in developing DM, perhaps explained by the inhibitory effects of the drugs on proteins involved in glucose metabolism such as Glut-4 (108) and cellular retinoic acid–binding protein type-I (109). Additionally, protease inhibitors may hamper pancreatic beta cell function (110) and also reduce insulin secretion (111). However, it seems that the issue of whether diabetics are more prone to develop chronic viral infections requires more research.
Obesity can also be considered as a metabolic disease and evidence accumulates to show that the outcome of a virus infection can differ in obese compared to non-obese humans and animals (112, 113). The topic of obesity and its influence on the outcome of virus infection has been reviewed (114). Reports document that severely obese persons may respond poorly to several vaccines and generate reduced antibody and T cell responses to infection and vaccination (115). For example, in the USA during the 2009 H1N1 pandemic, morbidly obesity patients (i.e., body mass index ≥ 40) were more prone to hospitalization and death (116) than non-obese persons. In China in the same pandemic, 19% of 9,966 patients admitted to the ICU were diabetic and many died (117). We are also realizing that obesity may predispose persons to more severe consequences of COVID-19 infection. For example, in China obese persons had a 2.42 fold higher chance of developing pneumonia upon COVID-19 infection than did the non-obese (118). Similarly, in France obese persons infected with COVID-19 had higher rates of admission to the ICU and greater needs for mechanical ventilation, outcomes directly proportional with their increased body mass indices (85). In a New York study involving 5,700 COVID-19 patients, 41.7% were obese (86). Explanations why obese persons become more susceptible to COVID-19 infection needs to be established. However, one consequence of obesity is increased expression of the ACE2 receptor on adipocytes making these cells potent targets for COVID-19, which could serve to increase the viral load (119, 120). Additionally, there are several changes in immune function that occur in obese subjects. These include higher ratios of M1:M2 macrophages (121), reduced T cells but elevated B cell numbers in lymph nodes (122) decreases bone marrow hematopoiesis (123) and reduced production of IFN-γ, TNF-α, IL-6, TGF-β1 by T cells (124).
Studies in mice models of obesity have revealed mechanisms that could explain their increased susceptibility to virus infection. These include effects on N-acyltransferase metabolism, fatty acid pathways (10-fold increase in 3-oxododecanoic acid and 4-hydroxyisovaleric acid) and nucleotide metabolism pathways. In obese mice, Flu can induce more oxidative stress than in lean animals and mice develop diminished T cell memory responses to infection (125).
One could anticipate that diseases of the thyroid gland, either hypothyroidism or Graves’ disease (hyperthyroidism), might be associated with changed responses to infections since thyroid hormones (TH) have a significant effect on general metabolism. For example, TH can affect liver enzymes involved in FAO and also impact on the uptake of free fatty acids by muscle and fat cells (126). Additionally, some have shown that TH can directly influence glucose utilization (127, 128). No reports have directly linked TH dysfunctions with changed susceptibility to a virus infection. However, some observations have tried to associate thyroid disease problems with the control of latency with some herpesviruses. One topic for evaluation has been herpes zoster, the lesion that occurs at a neurodermatome when VZV reactivates from its latent infection state (129). One study reported that when TH levels were low herpes zoster lesions were observed more frequently and showed that lesion outbreaks were less frequent in TH treated patients than in untreated persons, although differences did not reach statistical significance (130). One might also anticipate, based on mouse studies which showed that the TH receptor can influence the expression in-vitro of the latency associated transcript involved in HSV latency, that breakdown of HSV latency could be affected by TH levels, but such an effect has not been reported (131).
In today’s world, many persons adjust their diet to lose or gain weight. They may also favor one form of diet over another, or use non-prescription herbal remedies. The objective is usually to improve their health and well-being, but evidence for the success of such maneuvers is less than compelling, at least when it come to the outcome of human virus infections. However, it is generally agreed that malnutrition, especially marked calorie deficiency, can increase the susceptibility of children to virus diseases such as measles and respiratory syncytial virus (132, 133). There is also evidence that vitamin A deficiency, which is prevalent in malnourished children in South-East Asia, results commonly in increased susceptibility to measles as well as to both respiratory and enteric viral infections. One explanation could be that several cells of the immune system particularly dendritic cells and T cells are activated and hence more protective when their promoter regions bind to the vitamin A derivative retinoic acid (134–136). Other ideas include an effect of the vitamin A metabolites changing the nature of inflammatory reactions to became less tissue damaging via effects on regulatory T cells (137). In several studies, supplementation with vitamin A may increase the recovery rate and reduce mortality from measles (138, 139). Treated children develop both improved T cell and antibody responses (140). Vitamin A supplements are effective, but too expensive for general use in resource poor countries. However, a solution could be the use of genetically modified cereals such as golden rice that produce vitamin A precursors. This approach is resisted in most societies for non-scientific reasons.
Our most complete understanding of how nutrition can influence the outcome of virus infections comes from diet manipulation studies in animals, most commonly Flu infections in mice. Several different approaches have been used. These include comparing the outcome of a virus infection in animals or the offspring of pregnant mothers that received a so-called Mediterranean diet (~10% fat) vs. a Western (~40% fat) diet, comparing the effect of feeding diets with different fat levels and fat composition, as well as differences in fiber content (141). The consequences of nutritional changes on viral pathogenesis are often mediated by the effects of dietary intake on the composition and activity of the enteric microflora, a topic of intense interest at present. Most studies conclude that a high fat diet (HFD) can result in more severe Flu and higher mortality than occurs in recipients of a low fat diet (LFD), or normal chow diet (NCD). For example, in a study comparing responses to the pandemic Flu strain H1N1 in mice that received either a HFD, LFD, or NCD, significant differences were observed (142). Mice fed the HFD suffered 80% mortality along with enhanced lung destruction, inflammation and excess inflammatory cytokine and chemokine responses. Recipients of the LFD and NCD had mortality rates of 40 and 0% respectively. Compared to the other groups, the HFD mice showed marked changes in some metabolites in their blood. These included metabolites related to N-acyltransferase metabolism, fatty acid pathways (10-fold increase in 3-oxododecanoic acid and 4-hydroxyisovaleric acid) and nucleotide metabolism pathways. The recipients of the HFD also showed evidence of high oxidative stress, such as elevated oxidized glutathione in the lungs and the presence of high taurine levels in the urine (142). Even LFD recipients were more susceptible to influenza infection compared with mice on the NCD diet, perhaps explained by low fiber levels in the LFD (142).
The level of fat is not the only factor that affects Flu susceptibility; the chemical nature of fat in the diet also has an influence. Studies have shown that fatty acid chain length and major composition of either omega 3 or omega 6 FA (fatty acid) can also have notable effects. With regard to the latter, a diet high in omega 3 FA favors the production of anti-inflammatory activities, such as proresolving mediator synthesis (143), whereas diets rich in omega 6 FA favors proinflammatory mediator production such as prostaglandins and leukotrienes (144). Omega 3 predominance is beneficial to control inflammatory diseases that include situations where virus lesions result from a reaction dominated by inflammatory cytokine producing T cells (143). However, with viruses that cause disease by direct effects, the omega 3 rich diet may increase susceptibility. This has been noted with Flu where an omega 3 rich diet, such as feeding fish oil, results in increased severity and mortality following intranasal infection (145). The recipients developed reduced NK cell and neutrophil responses as well as reduced inflammatory mediator production involved in viral control (145). Their CD8 T cell responses were also reduced compared to animals fed the control diet.
Many studies have shown that the chain length of FAs included in the diet can play a major role in affecting the expression of inflammatory lesions caused by viruses (146, 147). Most studies have focused on the influence of fiber composition since high fiber diets generate an abundance of short chain fatty acids (SCFA) as a consequence of microbial digestion in the gut. However, feeding a SCFA such as butyrate as a dietary supplement can also boost the resistance of mice to Flu challenge, particularly in the early stages of infection. In one study, whereas all control mice were dead by 8 days’ post infection (PI), those fed additional butyrate showed 100, 75, and 40% survival rates at 8, 12, and 18 days’ PI respectively (147). The main effect of SCFA feeding is to expand anti-inflammatory aspects of the immune response that include Treg and M2 macrophages.
As mentioned previously, the outcome of a virus infection can be changed if animals are given a high fiber diet. This has been observed with Flu, HSV, and RSV infection all of which cause less disease when animals receive a high fiber diet (146–148). The effect is now generally accepted to be explained by the expansion of bacteria such as Bifidobacteria and Bacteroides species in the gut microflora that metabolize fiber into SCFA (butyrate, acetate and propionate) (147). The resulting protective effect against flu was the consequence of an increase in the number and activation status of CD8 T cell responses and effects on the bone marrow that favored M2 macrophage differentiation, cells which produce less tissue damaging cytokines and chemokines (147). The SCFA molecules mediate such effects by binding to specific fatty acid receptors on responding cells. One consequence is an increase in mitochondrial mass and an increase in metabolites for the TCA cycle resulting enhancement of the OXPHOS energy pathway (147).
In the case of respiratory syncytial virus (RSV), protective effects were observed in mice that received a high fiber diet as well as a diet supplemented with acetate (146). In those experiments, the protective effects were attributed to the enhanced production of IFN-β from lung epithelial cells, serving to inhibit viral replication. High fiber diets may also protect against the inflammatory effects of HSV infection (148). In this instance, the protective effects were attributed to the generation of a more effective CD8 T cell response which included an expansion of memory cells to counteract future infection. Recently, our laboratory has shown beneficial effects of supplementing the diet with sodium propionate (SP) to reduce the tissue damage caused by HSV-1 infection in the eye (149).
Other studies on the effects of diet on the outcome of virus infections have evaluated the influence of supplementing the diet for certain amino acids. For example, the effect of additional glutamine and leucine has been shown to influence the outcome of vaginal infection of mice with HSV (150). Mice receiving the additional amino acids had reduced viral burdens and less mortality than those on the control diet. The supplement recipients also had elevated NK cell and IFN-γ producing CD4 Th1 T cell responses (150). Adding glutamine to the diet could also influence the stability of HSV latency. This might occur as a consequence of HSV specific CD8 T cells being expanded in the latently infected trigeminal ganglia where they are thought to function by prevented reactivation from latency (151). Another interesting observation on the value of SCFA feeding was that this could reduce the consequence of secondary bacterial infections which commonly cause Flu-associated respiratory infections to be more severe. Some have reported that a consequence of flu infection can be a change in bacterial types present in the gut, although it is not clear mechanistically how this dysbiosis occurs. The outcome is reduced presence of those bacteria species that generate SCFA especially acetate, an effect overcome by SCFA feeding (152).
Finally, with regard to nutritional influences on viral infections is the interesting observation that feeding additional glucose or SCFA can overcome the detrimental effects that high ambient temperature may exert on susceptibility to some virus infections, an issue expected to become more problematic in a warming world. Thus, when influenza infected mice were kept at 36°C compared to room temperature, they showed more susceptibility and mounted impaired immune responses that included reduced virus specific CD8 and CD4 T cells, IgG levels, and reduction in several inflammatory cytokines. The outcome of high temperature exposure was overcome either by feeding extra glucose or supplementing the diet with additional SCFA (153). Impaired immunity at high temperature has also been observed with ZIKV and Bunyavirus infections (153).
In previous sections, we have discussed how metabolic pathways can be changed upon virus infection and how host intrinsic metabolic activities and extrinsic events that affect metabolism can influence the expression of infection. These situations raise the issue as to whether manipulating one or more aspects of host metabolism represent useful and perhaps convenient approaches to control the outcome of virus infections. They might be especially useful with infections not well controlled by vaccines or antiviral drugs. Currently, the majority of reports that target metabolism for disease control have dealt with autoimmunity or cancer, but viral diseases especially chronic persistent infections, could be a fruitful field for investigation. With respect to virus infections, the great majority of studies on metabolic manipulations have been performed in-vitro with various drugs (Figure 2) and many of these are recorded in Table 2. As mentioned in-vivo studies are sparse. An early report came from the Medzhitov laboratory which was designed to compare the effects of calorie restriction on some viral and bacterial infections. The study showed that anorexia made neurogenic flu infections more severe, but protected against bacterial sepsis infections. Moreover, nutritional supplementation with glucose was detrimental to bacterial disease, but protected against lethal flu infection. In their models, the nutritional effects were explained not by effects on immune function, but by an ER stress effects on the brain with this causing more apoptosis in neuronal cells (167).
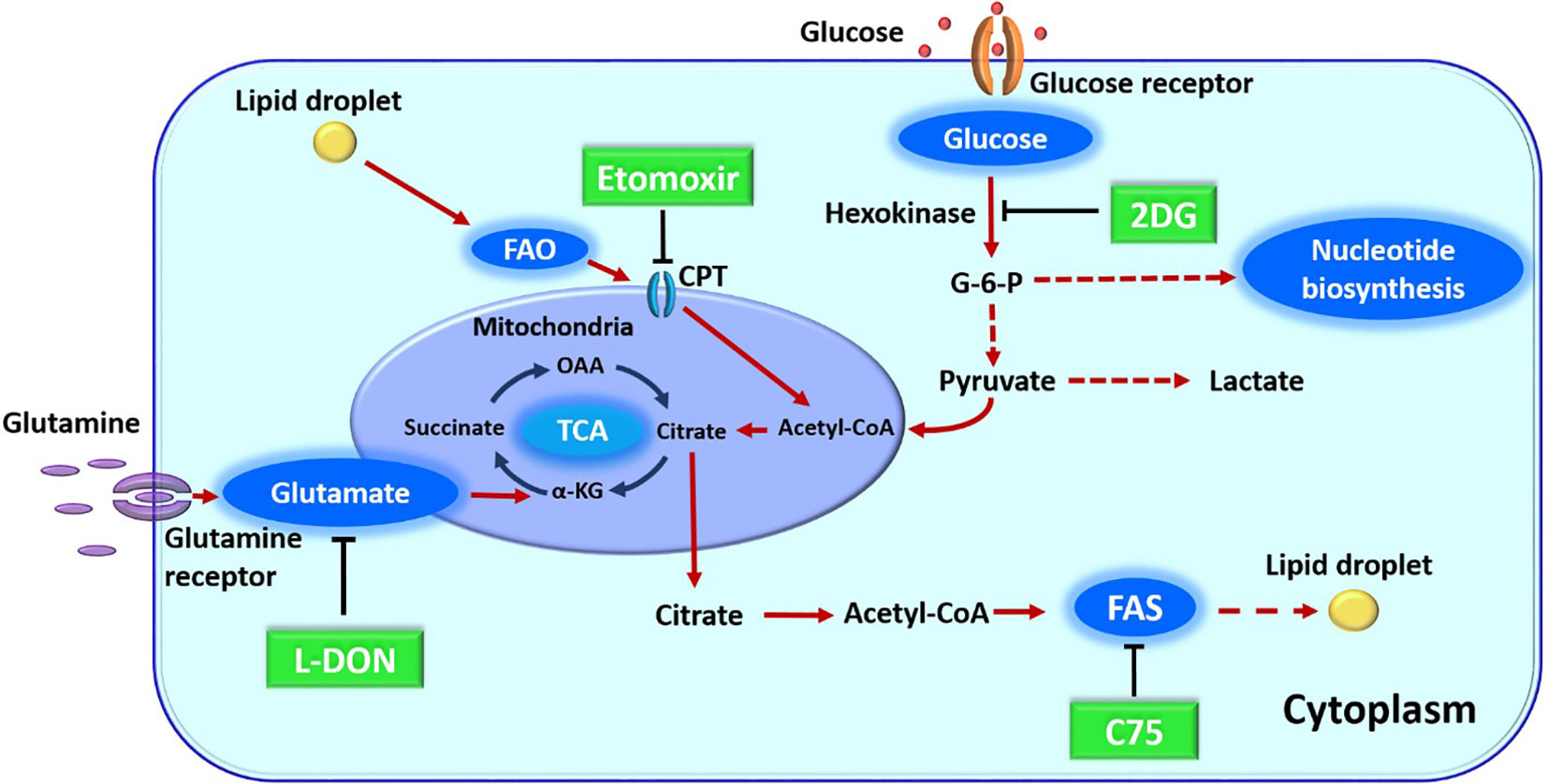
Figure 2 Major metabolic pathway along with their inhibitors. 2DG (2-deoxy-D-glucose) blocks glycolysis. L-DON (6-diazo-5-oxo-L-norleucine) blocks glutaminolysis by acting on glutamate. Etomoxir blocks FAO (fatty acid oxidation) by its inhibitor action on CPT1a (carnitine palmitoyltransferase), C75 (fatty acid synthase inhibitor) blocks fatty acid synthesis (FAS) by inhibiting a fatty acid synthase enzyme. OAA, oxaloacetic acid; alpha KG, alpha-ketoglutarate; G-6-P, glucose 6-phosphate; TCA, tricarboxylic acid cycle.
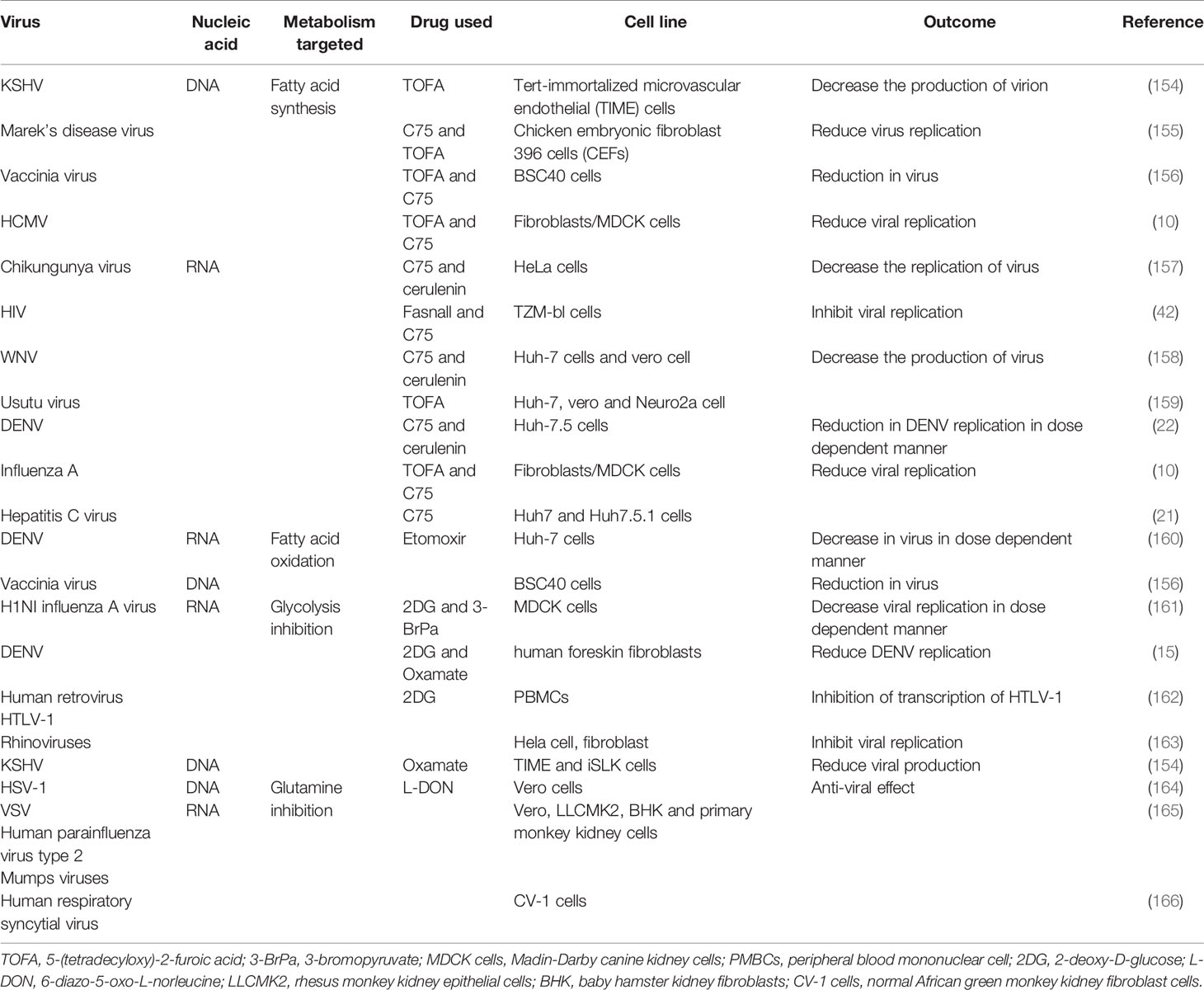
Table 2 Effect of the metabolic inhibitor drug on various virus infected cell line.
Another study looked at the in-vivo consequences of interfering with glucose utilization using 2DG (2-deoxy-D-glucose) therapy in a viral disease model (168). Inhibition of glucose utilization resulted in diminished HSV induced inflammatory lesions in the eye (168). The protective therapy was explained by a reduction in proinflammatory T cell activity that primarily use glycolysis to supply their energy. In this study, the Treg responses, which were shown previously to suppress the severity of ocular lesions (168), was not affected by 2DG therapy. Accordingly, metabolic manipulation, such as reprogramming glucose metabolism, can represent an approach to control the expression of viral inflammatory disease. Other reports on this topic have also appeared (163).
Another potential metabolic target that has promise to control the extent of a virus infection is fatty acid metabolism which is a crucial event for many viruses such as flaviviruses that depend on cellular lipids to complete their life cycle. Success at controlling several human flavivirus disease agents has been achieved in-vitro using inhibitors of acetyl coenzyme carboxylase, which is essential for de novo lipogenesis (169). So far studies on in-vivo effects have only been done using a mouse model of West Nile fever (WNF). In such studies, the infection was successfully controlled including lesions in the kidney (169). The latter is relevant since renal complications are a prominent feature of persistent WNF infection in humans (170).
Another metabolic step accessible for inhibition to suppress viral diseases is glutamine metabolism. Thus this amino acid is used to support proliferation and cytokine producing activity of inflammatory T cells and in addition is a precursor of a neurotoxic molecule, glutamate. Inhibiting glutamine metabolism was shown to be valuable to diminish CNS inflammatory lesions caused by Sindbis virus infection (171). Inhibiting glutamine metabolism could well be an approach to control other immunoinflammatory viral lesions and our group is exploring its values to limit herpetic stromal keratitis lesions.
We anticipate that additional approaches to manipulate metabolism will be useful to shape the outcome of virus infections particularly when used along with other therapies such as antiviral and anti-inflammatory drugs.
Controlling virus infections is most effectively achieved using vaccines and with a few viruses specific anti-viral drugs. However, such approaches are not available for many infections, which includes COVID-19, the cause of a current pandemic. We need new approaches to control virus infections and this review focusses on changing the metabolic events that occur during viral infections. We advocate that manipulating metabolic activities represent a useful approach to control the outcome of some viral infections that may include COVID-19 infection. We described how metabolic changes are set into play by different viral infections and point out that changing the metabolic environment might be one means of controlling if the virus host relationship is productive and tissue damaging or inapparent. We also review how the outcome of virus infections can be affected by the metabolic status of the host. Particularly relevant are DM and obesity that impact on the clinical consequences of infections such as Flu and COVID-19 infection. In addition, the nutritional status of the host may also influence the expression of some virus infections and changing the diet holds promise as one way to control the outcome of some viral infections. The major question addressed was whether it is possible to reshape the outcome of a viral infection, particularly those where the host response contributes to tissue damage. The topic has received minimal investigation, but some studies do show that controlling events such as glycolysis, glutaminolysis and fatty acid metabolism are showing promise as an approaches to limit the severity of some viral infections. We presume that manipulating metabolism to reshape the nature of some virus infections will become a valuable addition to the current approaches available to control virus infections.
BR conceptualized the manuscript. BR, DS, EB, and MM wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the NIH 2020 R21 AI (number: 5R21AI142862-02) and NIH 2020 R01 (number: EY5R01EY005093-35).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Trends Microbiol (2000) 8:410–8. doi: 10.1016/S0966-842X(00)01830-8
2. Rouse BT, Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol (2010) 10:514–26. doi: 10.1038/nri2802
3. Sumbria D, Berber E, Rouse BT. Factors affecting the tissue damaging consequences of viral infections. Front Microbiol (2019) 10:2314. doi: 10.3389/fmicb.2019.02314
4. Wu D, Sanin David E, Everts B, Chen Q, Qiu J, Buck Michael D, et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity (2016) 44:1325–36. doi: 10.1016/j.immuni.2016.06.006
5. Apicella M, Campopiano MC, Mantuano M, Mazoni L, Coppelli A, Del Prato S. COVID-19 in people with diabetes: understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol (2020) 8:782–92. doi: 10.1016/S2213-8587(20)30238-2
6. Smith M, Honce R, Schultz-Cherry S. Metabolic Syndrome and Viral Pathogenesis: Lessons from Influenza and Coronaviruses. J Virol (2020) 94:e00665–00620. doi: 10.1128/JVI.00665-20
7. Ikeda M, Kato N. Modulation of host metabolism as a target of new antivirals. Adv Drug Delivery Rev (2007) 59:1277–89. doi: 10.1016/j.addr.2007.03.021
8. Varanasi SK, Rouse BT. How host metabolism impacts on virus pathogenesis. Curr Opin Virol (2018) 28:37–42. doi: 10.1016/j.coviro.2017.11.003
9. Thai M, Graham Nicholas A, Braas D, Nehil M, Komisopoulou E, Kurdistani Siavash K, et al. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab (2014) 19:694–701. doi: 10.1016/j.cmet.2014.03.009
10. Munger J, Bennett BD, Parikh A, Feng X-J, McArdle J, Rabitz HA, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol (2008) 26:1179–86. doi: 10.1038/nbt.1500
11. Xiao L, Zy H, Dong X, Tan Z, Li W, Tang M, et al. Targeting Epstein–Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene (2014) 33:4568–78. doi: 10.1038/onc.2014.32
12. Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, et al. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc Natl Acad Sci USA (2012) 109:11818–23. doi: 10.1073/pnas.1205995109
13. Ripoli M, D’Aprile A, Quarato G, Sarasin-Filipowicz M, Gouttenoire J, Scrima R, et al. Hepatitis C Virus-Linked Mitochondrial Dysfunction Promotes Hypoxia-Inducible Factor 1α-Mediated Glycolytic Adaptation. J Virol (2010) 84:647–60. doi: 10.1128/jvi.00769-09
14. Hegedus A, Kavanagh Williamson M, Huthoff H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T cells. Retrovirology (2014) 11:98. doi: 10.1186/s12977-014-0098-4
15. Fontaine KA, Sanchez EL, Camarda R, Lagunoff M. Dengue virus induces and requires glycolysis for optimal replication. J Virol (2015) 89:2358–66. doi: 10.1128/jvi.02309-14
16. Ritter JB, Wahl AS, Freund S, Genzel Y, Reichl U. Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling. BMC Syst Biol (2010) 4:61. doi: 10.1186/1752-0509-4-61
17. Chambers JW, Maguire TG, Alwine JC. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol (2010) 84:1867–73. doi: 10.1128/jvi.02123-09
18. Sanchez EL, Carroll PA, Thalhofer AB, Lagunoff M. Latent KSHV infected endothelial cells are glutamine addicted and require glutaminolysis for survival. PLoS Pathog (2015) 11:e1005052. doi: 10.1371/journal.ppat.1005052
19. Fontaine KA, Camarda R, Lagunoff M. Vaccinia virus requires glutamine but not glucose for efficient replication. J Virol (2014) 88:4366–74. doi: 10.1128/jvi.03134-13
20. Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci USA (2005) 102:2561–6. doi: 10.1073/pnas.0409834102
21. Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, et al. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology (2008) 48:1396–403. doi: 10.1002/hep.22508
22. Heaton NS, Perera R, Berger KL, Khadka S, LaCount DJ, Kuhn RJ, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci USA (2010) 107:17345–50. doi: 10.1073/pnas.1010811107
23. Hollenbaugh JA, Munger J, Kim B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC–MS/MS analysis. Virology (2011) 415:153–9. doi: 10.1016/j.virol.2011.04.007
24. Kang S, Tang H. HIV-1 infection and glucose metabolism reprogramming of T cells: another approach toward functional cure and reservoir eradication. Front Immunol (2020) 11:572677. doi: 10.3389/fimmu.2020.572677
25. Clerc I, Abba Moussa D, Vahlas Z, Tardito S, Oburoglu L, Hope TJ, et al. Entry of glucose- and glutamine-derived carbons into the citric acid cycle supports early steps of HIV-1 infection in CD4 T cells. Nat Metab (2019) 1:717–30. doi: 10.1038/s42255-019-0084-1
26. Datta PK, Deshmane S, Khalili K, Merali S, Gordon JC, Fecchio C, et al. Glutamate metabolism in HIV-1 infected macrophages: Role of HIV-1 Vpr. Cell Cycle (2016) 15:2288–98. doi: 10.1080/15384101.2016.1190054
27. Loisel-Meyer S, Swainson L, Craveiro M, Oburoglu L, Mongellaz C, Costa C, et al. Glut1-mediated glucose transport regulates HIV infection. Proc Natl Acad Sci USA (2012) 109:2549–54. doi: 10.1073/pnas.1121427109
28. Palmer CS, Ostrowski M, Gouillou M, Tsai L, Yu D, Zhou J, et al. Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. Aids (2014) 28:297–309. doi: 10.1097/qad.0000000000000128
29. Eisenreich W, Rudel T, Heesemann J, Goebel W. How Viral and Intracellular Bacterial Pathogens Reprogram the Metabolism of Host Cells to Allow Their Intracellular Replication. Front Cell Infect Microbiol (2019) 9:42. doi: 10.3389/fcimb.2019.00042
30. Heaton NS, Randall G. Dengue Virus-induced autophagy regulates lipid metabolism. Cell Host Microbe (2010) 8:422–32. doi: 10.1016/j.chom.2010.10.006
31. Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity (2019) 50:64–76.e64. doi: 10.1016/j.immuni.2018.11.017
32. de Sousa JR, Azevedo R, Quaresma JAS, Vasconcelos P. Cell death and Zika virus: An integrated network of the mechanisms of cell injury. Infect Drug Resist (2019) 12:2917–21. doi: 10.2147/IDR.S209213
33. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature (2020) 580:391–5. doi: 10.1038/s41586-020-2129-8
34. Newton K. Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harb Perspect Biol (2020) 12:a036368. doi: 10.1101/cshperspect.a036368
35. Kole AJ, Annis RP, Deshmukh M. Mature neurons: equipped for survival. Cell Death Dis (2013) 4:e689–9. doi: 10.1038/cddis.2013.220
36. Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters K-A, Proll SC, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog (2010) 6:e1000719. doi: 10.1371/journal.ppat.1000719
37. Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PloS Pathog (2011) 7:e1002124. doi: 10.1371/journal.ppat.1002124
38. Johnston C, Corey L. Current concepts for genital Herpes Simplex Virus infection: Diagnostics and pathogenesis of genital tract shedding. Clin Microbiol Rev (2016) 29:149–61. doi: 10.1128/cmr.00043-15
39. Piccaluga PP, Weber A, Ambrosio MR, Ahmed Y, Leoncini L. Epstein-Barr virus-induced metabolic rearrangements in human B-cell lymphomas. Front Microbiol (2018) 9:1233. doi: 10.3389/fmicb.2018.01233
40. Daker M, Bhuvanendran S, Ahmad M, Takada K, Khoo AS. Deregulation of lipid metabolism pathway genes in nasopharyngeal carcinoma cells. Mol Med Rep (2013) 7:731–41. doi: 10.3892/mmr.2012.1253
41. Valle-Casuso JC, Angin M, Volant S, Passaes C, Monceaux V, Mikhailova A, et al. Cellular metabolism is a major determinant of HIV-1 reservoir seeding in CD4+ T Cells and offers an opportunity to tackle infection. Cell Metab (2019) 29:611–26.e615. doi: 10.1016/j.cmet.2018.11.015
42. Kulkarni MM, Ratcliff AN, Bhat M, Alwarawrah Y, Hughes P, Arcos J, et al. Cellular fatty acid synthase is required for late stages of HIV-1 replication. Retrovirology (2017) 14:45. doi: 10.1186/s12977-017-0368-z
43. Fritsch SD, Weichhart T. Effects of interferons and viruses on metabolism. Front Immunol (2016) 7:630. doi: 10.3389/fimmu.2016.00630
44. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12:125–35. doi: 10.1038/nri3133
45. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
46. Johnson HM, Noon-Song EN, Kemppainen K, Ahmed CM. Steroid-like signalling by interferons: making sense of specific gene activation by cytokines. Biochem J (2012) 443:329–38. doi: 10.1042/BJ20112187
47. Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses (2001) 17:1009–19. doi: 10.1089/088922201300343690
48. Nguyen DH, Hildreth JEK. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol (2000) 74:3264–72. doi: 10.1128/jvi.74.7.3264-3272.2000
49. Li C, Deng YQ, Wang S, Ma F, Aliyari R, Huang XY, et al. 25-Hydroxycholesterol protects host against zika virus infection and its associated microcephaly in a mouse model. Immunity (2017) 46:446–56. doi: 10.1016/j.immuni.2017.02.012
50. Ng CG, Coppens I, Govindarajan D, Pisciotta J, Shulaev V, Griffin DE. Effect of host cell lipid metabolism on alphavirus replication, virion morphogenesis, and infectivity. Proc Natl Acad Sci (2008) 105:16326. doi: 10.1073/pnas.0808720105
51. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity (2013) 38:92–105. doi: 10.1016/j.immuni.2012.11.005
52. Pulliam L, Calosing C, Sun B, Grunfeld C, Rempel H. Monocyte activation from interferon-α in HIV infection increases acetylated LDL uptake and ROS production. J Interferon Cytokine Res (2014) 34:822–8. doi: 10.1089/jir.2013.0152
53. Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today (1999) 20:469–73. doi: 10.1016/S0167-5699(99)01520-0
54. Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing Indoleamine 2,3-Dioxygenase inhibit T cell responses. J Immunol (2002) 168:3771–6. doi: 10.4049/jimmunol.168.8.3771
55. Adams O, Besken K, Oberdörfer C, MacKenzie CR, Takikawa O, Däubener W. Role of indoleamine-2,3-dioxygenase in alpha/beta and gamma interferon-mediated antiviral effects against herpes simplex virus infections. J Virol (2004) 78:2632–6. doi: 10.1128/jvi.78.5.2632-2636.2004
56. Bodaghi B, Goureau O, Zipeto D, Laurent L, Virelizier J-L, Michelson S. Role of IFN-γ-Induced indoleamine 2,3 dioxygenase and inducible nitric oxide synthase in the replication of Human Cytomegalovirus in retinal pigment epithelial cells. J Immunol (1999) 162:957–64.
57. Rabbani MAG, Barik S. 5-Hydroxytryptophan, a major product of tryptophan degradation, is essential for optimal replication of human parainfluenza virus. Virology (2017) 503:46–51. doi: 10.1016/j.virol.2017.01.007
58. Adams O, Besken K, Oberdörfer C, MacKenzie CR, Rüssing D,WD. Inhibition of human herpes simplex virus type 2 by interferon gamma and tumor necrosis factor alpha is mediated by indoleamine 2,3-dioxygenase. Microbes Infect (2004) 6:806–12. doi: 10.1016/j.micinf.2004.04.007
59. Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol (2011) 186:1162–72. doi: 10.4049/jimmunol.1002615
60. Sireesh D, Dhamodharan U, Ezhilarasi K, Vijay V, Ramkumar KM. Association of NF-E2 Related Factor 2 (Nrf2) and inflammatory cytokines in recent onset Type 2 Diabetes Mellitus. Sci Rep (2018) 8:5126. doi: 10.1038/s41598-018-22913-6
61. Zeng C, Shi X, Zhang B, Liu H, Zhang L, Ding W, et al. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med (2012) 90:175–86. doi: 10.1007/s00109-011-0816-5
62. Kiselar JG, Wang X, Dubyak GR, El Sanadi C, Ghosh SK, Lundberg K, et al. Modification of β-Defensin-2 by Dicarbonyls Methylglyoxal and Glyoxal inhibits antibacterial and chemotactic function in vitro. PLoS One (2015) 10:e0130533. doi: 10.1371/journal.pone.0130533
63. Joshi MB, Lad A, Bharath Prasad AS, Balakrishnan A, Ramachandra L, Satyamoorthy K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett (2013) 587:2241–6. doi: 10.1016/j.febslet.2013.05.053
64. Nelson CP, Hindson DA. Inhibition of polymorphonuclear leukocyte respiratory burst by elevated glucose concentrations in vitro. Diabetes (1989) 38:1031–5. doi: 10.2337/diab.38.8.1031
65. Perner A, Nielsen SE, Rask-Madsen J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med (2003) 29:642–5. doi: 10.1007/s00134-002-1628-4
66. Jafar N, Edriss H, Nugent K. The effect of short-Term hyperglycemia on the innate immune system. Am J Med Sci (2016) 351:201–11. doi: 10.1016/j.amjms.2015.11.011
67. Stegenga ME, van der Crabben SN, Blümer RME, Levi M, Meijers JCM, Serlie MJ, et al. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood (2008) 112:82–9. doi: 10.1182/blood-2007-11-121723
68. Kanafani ZA, Kourany WM, Fowler VG, Levine DP, Vigliani GA, Campion M, et al. Clinical characteristics and outcomes of diabetic patients with Staphylococcus aureus bacteremia and endocarditis. Eur J Clin Microbiol Infect Dis (2009) 28:1477. doi: 10.1007/s10096-009-0808-3
69. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
70. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
71. Muniangi-Muhitu H, Akalestou E, Salem V, Misra S, Oliver NS, Rutter GA. Covid-19 and diabetes: A complex bidirectional relationship. Front Endocrinol (2020) 11:582936. doi: 10.3389/fendo.2020.582936
72. Lim S, Bae JH, Kwon H-S, Nauck MA. COVID-19 and diabetes mellitus: from pathophysiology to clinical management. Nat Rev Endocrinol (2021) 17:11–30. doi: 10.1038/s41574-020-00435-4
73. Fleming DM, Crombie DL, Cross KW. Disease concurrence in diabetes mellitus: a study of concurrent morbidity over 12 months using diabetes mellitus as an example. J Epidemiol Community Health (1991) 45:73–7. doi: 10.1136/jech.45.1.73
74. Lenzi L ÂMM, Silva L, Grochocki M, Pontarolo R. Pandemic influenza A (H1N1) 2009: risk factors for hospitalization. J Bras Pneumol (2012) 38:57–65. doi: 10.1590/S1806-37132012000100009
75. Allard R, Leclerc P, Tremblay C, Tannenbaum T-N. Diabetes and the severity of pandemic influenza A (H1N1) infection. Diabetes Care (2010) 33:1491–3. doi: 10.2337/dc09-2215
76. Louie JK, Acosta M, Winter K, Jean C, Gavali S, Schechter R, et al. Factors Associated With Death or Hospitalization Due to Pandemic 2009 Influenza A(H1N1) Infection in California. JAMA (2009) 302:1896–902. doi: 10.1001/jama.2009.1583
77. Abdi A, Jalilian M, Sarbarzeh PA, Vlaisavljevic Z. Diabetes and COVID-19: A systematic review on the current evidences. Diabetes Res Clin (2020) 166:108347. doi: 10.1016/j.diabres.2020.108347
78. Azar WS, Njeim R, Fares AH, Azar NS, Azar ST, El Sayed M, et al. COVID-19 and diabetes mellitus: how one pandemic worsens the other. Rev Endocr Metab Disord (2020) 21:451–63. doi: 10.1007/s11154-020-09573-6
79. de Siqueira JVV, Almeida LG, Zica BO, Brum IB, Barceló A, de Siqueira Galil AG. Impact of obesity on hospitalizations and mortality, due to COVID-19: A systematic review. Obes Res Clin Pract (2020) 14:398–403. doi: 10.1016/j.orcp.2020.07.005
80. Popkin BM, Du S, Green WD, Beck MA, Algaith T, Herbst CH, et al. Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships. Obes Rev (2020) 21:e13128. doi: 10.1111/obr.13128
81. Wu J, Zhang F, Fang F, Chang H, Wang F, Yang Z, et al. Efficacy of inactivated vaccine against H5N1 influenza virus infection in mice with type 1 diabetes. Vaccine (2010) 28:2775–81. doi: 10.1016/j.vaccine.2010.01.037
82. Reading PC, Allison J, Crouch EC, Anders EM. Increased susceptibility of diabetic mice to Influenza virus infection: Compromise of collectin-mediated host defense of the lung by glucose? J Virol (1998) 72:6884–7. doi: 10.1128/jvi.72.8.6884-6887.1998
83. Huo C, Zhang S, Zhang S, Wang M, Qi P, Xiao J, et al. Mice with type 1 diabetes exhibit increased susceptibility to influenza A virus. Microb Pathog (2017) 113:233–41. doi: 10.1016/j.micpath.2017.10.026
84. Guan W-j, Ni Z-y, Hu Y, Liang W-h, Ou C-q, He J-x, et al. Clinical characteristics of Coronavirus disease 2019 in China. N Engl J Med (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
85. Simonnet A, Chetboun M, Poissy J, Raverdy V, Noulette J, Duhamel A, et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity (2020). 28:1195–99. doi: 10.1002/oby.22831. n/a.
86. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York city area. JAMA (2020) 323:2052–9. doi: 10.1001/jama.2020.6775
87. Konoshita T, Wakahara S, Mizuno S, Motomura M, Aoyama C, Makino Y, et al. Tissue gene expression of renin-angiotensin system in human type 2 diabetic nephropathy. Diabetes Care (2006) 29:848–52. doi: 10.2337/diacare.29.04.06.dc05-1873
88. Mizuri S, Aoki T, Hemmi H, Arita M, Sakai K, Aikawa A. Urinary angiotensin-converting enzyme 2 in patients with CKD. Nephrology (2011) 16:567–72. doi: 10.1111/j.1440-1797.2011.01467.x
89. Ferrario CM, Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Ann Tallant E, et al. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int (2005) 68:2189–96. doi: 10.1111/j.1523-1755.2005.00675.x
90. Furuhashi M, Moniwa N, Mita T, Fuseya T, Ishimura S, Ohno K, et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am J Hypertens (2015) 28:15–21. doi: 10.1093/ajh/hpu086
91. Roca-Ho H, Riera M, Palau V, Pascual J, Soler MJ. Characterization of ACE and ACE2 expression within different organs of the NOD mouse. Int J Mol Sci (2017) 18:563. doi: 10.3390/ijms18030563
92. Wysocki J, Garcia-Halpin L, Ye M, Maier C, Sowers K, Burns KD, et al. Regulation of urinary ACE2 in diabetic mice. Am J Physiol Renal Physiol (2013) 305:F600–611. doi: 10.1152/ajprenal.00600.2012
93. Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell Chem Biol (2020) 181:905–13. doi: 10.1016/j.cell.2020.04.004
94. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the Novel Coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS Coronavirus. J Virol (2020) 94:e00127–00120. doi: 10.1128/jvi.00127-20
95. Chung S-C, Providencia R, Sofat R. Association between Angiotensin Blockade and Incidence of Influenza in the United Kingdom. N Engl J Med (2020) 383:397–400. doi: 10.1056/NEJMc2005396
96. Fedson DS. Treating influenza with statins and other immunomodulatory agents. Antiviral Res (2013) 99:417–35. doi: 10.1016/j.antiviral.2013.06.018
97. Yang P, Gu H, Zhao Z, Wang W, Cao B, Lai C, et al. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci Rep (2014) 4:7027. doi: 10.1038/srep07027
98. Yang JK, Feng Y, Yuan MY, Yuan SY, Fu HJ, Wu BY, et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabetic Med (2006) 23:623–8. doi: 10.1111/j.1464-5491.2006.01861.x
99. Alqahtani FY, Aleanizy FS, Mohamed RAEH, Alanazi MS, Mohamed N, Alrasheed MM, et al. Prevalence of comorbidities in cases of Middle East respiratory syndrome coronavirus: a retrospective study. Epidemiol Infect (2018) 147:e35. doi: 10.1017/S0950268818002923
100. Kulcsar KA, Coleman CM, Beck SE, Frieman MB. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS-CoV infection. JCI Insight (2019) 4:e131774. doi: 10.1172/jci.insight.131774
101. Yang J-K, Lin S-S, Ji X-J, Guo L-M. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol (2010) 47:193–9. doi: 10.1007/s00592-009-0109-4
102. Fignani D, Licata G, Brusco N, Nigi L, Grieco GE, Marselli L, et al. SARS-CoV-2 receptor angiotensin I-converting enzyme type 2 (ACE2) is expressed in human pancreatic β-cells and in the human pancreas microvasculature. Front Endocrinol (2020) 11:596898. doi: 10.3389/fendo.2020.596898
103. Esaki S, Yamano K, Kiguchi J, Katsumi S, Keceli S, Okamoto H, et al. Diabetic mice show an aggravated course of herpes-simplex virus-induced facial nerve paralysis. Otol Neurotol (2012) 33:1452–7. doi: 10.1097/MAO.0b013e318268d54d
104. McCormick D. Herpes-simplex Virus as a cause of Bell’s Palsy. Lancet (1972) 299:937–9. doi: 10.1016/S0140-6736(72)91499-7
105. Okamoto S, Hata A, Sadaoka K, Yamanishi K, Mori Y. Comparison of varicella-zoster virus-specific immunity of patients with diabetes mellitus and healthy individuals. J Infect Dis (2009) 200:1606–10. doi: 10.1086/644646
106. Mehta SH, Moore RD, Thomas DL, Chaisson RE, Sulkowski MS. The Effect of HAART and HCV Infection on the Development of Hyperglycemia Among HIV-Infected Persons. J Acquir Immune Defic Syndr (2003) 33:577–84. doi: 10.1097/00126334-200308150-00005
107. Brown TT, Cole SR, Li X, Kingsley LA, Palella FJ, Riddler SA, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med (2005) 165:1179–84. doi: 10.1001/archinte.165.10.1179
108. Hresko RC, Hruz PW. HIV Protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PloS One (2011) 6:e25237. doi: 10.1371/journal.pone.0025237
109. Carr A. HIV Protease Inhibitor-Related Lipodystrophy Syndrome. Clin Infect Dis (2000) 30:S135–42. doi: 10.1086/313854
110. Zhang S, Carper MJ, Lei X, Cade WT, Yarasheski KE, Ramanadham S. Protease inhibitors used in the treatment of HIV+ induce β-cell apoptosis via the mitochondrial pathway and compromise insulin secretion. Am J Physiol Endocrinol Metab (2009) 296:E925–35. doi: 10.1152/ajpendo.90445.2008
111. Koster JC, Remedi MS, Qiu H, Nichols CG, Hruz PW. HIV Protease inhibitors acutely impair glucose-stimulated insulin release. Diabetes (2003) 52:1695–700. doi: 10.2337/diabetes.52.7.1695
112. Maier HE, Lopez R, Sanchez N, Ng S, Gresh L, Ojeda S, et al. Obesity increases the duration of influenza A virus shedding in adults. J Infect Dis (2018) 218:1378–82. doi: 10.1093/infdis/jiy370
113. O’Brien KB, Vogel P, Duan S, Govorkova EA, Webby RJ, McCullers JA, et al. Impaired wound healing predisposes obese mice to severe influenza virus infection. J Infect Dis (2012) 205:252–61. doi: 10.1093/infdis/jir729
114. Honce R, Schultz-Cherry S. Impact of Obesity on Influenza A Virus Pathogenesis, Immune Response, and Evolution. Front Immunol (2019) 10:1071. doi: 10.3389/fimmu.2019.01071
115. Sheridan PA, Paich HA, Handy J, Karlsson EA, Hudgens MG, Sammon AB, et al. Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes (2012) 36:1072–7. doi: 10.1038/ijo.2011.208
116. Morgan OW, Bramley A, Fowlkes A, Freedman DS, Taylor TH, Gargiullo P, et al. Morbid Obesity as a Risk Factor for Hospitalization and Death Due to 2009 Pandemic Influenza A(H1N1) Disease. PLoS One (2010) 5:e9694. doi: 10.1371/journal.pone.0009694
117. Yu H, Feng Z, Uyeki TM, Liao Q, Zhou L, Feng L, et al. Risk factors for severe illness with 2009 pandemic Influenza A (H1N1) virus infection in China. Clin Infect Dis (2011) 52:457–65. doi: 10.1093/cid/ciq144
118. Cai Q, Chen F, Wang T, Luo F, Liu X, Wu Q, et al. Obesity and COVID-19 Severity in a Designated Hospital in Shenzhen, China. Diabetes Care (2020). 43:1392–98. doi: 10.2337/dc20-0576
119. Al-Benna S. Association of high level gene expression of ACE2 in adipose tissue with mortality of COVID-19 infection in obese patients. Obes Med (2020) 19:100283–3. doi: 10.1016/j.obmed.2020.100283
120. Ryan PM, Caplice NM. Is adipose tissue a reservoir for viral spread, immune activation, and cytokine amplification in coronavirus disease 2019? Obes (Silver Spring) (2020) 28:1191–4. doi: 10.1002/oby.22843
121. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi: 10.1172/JCI29881
122. Weitman ES, Aschen SZ, Farias-Eisner G, Albano N, Cuzzone DA, Ghanta S, et al. Obesity impairs lymphatic fluid transport and dendritic cell migration to lymph nodes. PLoS One (2013) 8:e70703. doi: 10.1371/journal.pone.0070703
123. Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature (2009) 460:259–63. doi: 10.1038/nature08099
124. Yang H, Youm Y-H, Vandanmagsar B, Rood J, Kumar KG, Butler AA, et al. Obesity accelerates thymic aging. Blood (2009) 114:3803–12. doi: 10.1182/blood-2009-03-213595
125. Rebeles J, Green WD, Alwarawrah Y, Nichols AG, Eisner W, Danzaki K, et al. Obesity-induced changes in T-cell metabolism are associated with impaired memory T-cell response to influenza and are not reversed with weight loss. J Infect Dis (2018) 219:1652–61. doi: 10.1093/infdis/jiy700
126. Sinha RA, Singh BK, Yen PM. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat Rev Endocrinol (2018) 14:259–69. doi: 10.1038/nrendo.2018.10
127. Castillo M, Hall JA, Correa-Medina M, Ueta C, Kang HW, Cohen DE, et al. Disruption of thyroid hormone activation in type 2 deiodinase knockout mice causes obesity with glucose intolerance and liver steatosis only at thermoneutrality. Diabetes (2011) 60:1082–9. doi: 10.2337/db10-0758
128. Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev (2014) 94:355–82. doi: 10.1152/physrev.00030.2013
129. Ajavon A, Killian D, Odom R, Figliozzi RW, Chen F, Balish M, et al. Influence of thyroid hormone disruption on the incidence of shingles. Epidemiol Infect (2015) 143:3557–71. doi: 10.1017/s0950268815000655
130. Hsia SV, Chen LH, Tseng HF. Receipt of thyroid hormone deficiency treatment and risk of herpes zoster. Int J Infect Dis (2017) 59:90–5. doi: 10.1016/j.ijid.2017.04.016
131. Bedadala GR, Pinnoji RC, Palem JR, Hsia S-CV. Thyroid hormone controls the gene expression of HSV-1 LAT and ICP0 in neuronal cells. Cell Res (2010) 20:587–98. doi: 10.1038/cr.2010.50
132. Dossetor J, Whittle HC, Greenwood BM. Persistent measles infection in malnourished children. Br Med J (1977) 1:1633–5. doi: 10.1136/bmj.1.6077.1633
133. Tupasi TE, Lucero MG, Magdangal DM, Mangubat NV, Sunico ME, Torres CU, et al. Etiology of acute lower respiratory tract infection in children from Alabang, Metro Manila. Rev Infect Dis (1990) 12 Suppl 8:S929–39. doi: 10.1093/clinids/12.supplement_8.s929
134. Darmanin S, Chen J, Zhao S, Cui H, Shirkoohi R, Kubo N, et al. All-trans Retinoic Acid Enhances Murine Dendritic Cell Migration to Draining Lymph Nodes via the Balance of Matrix Metalloproteinases and Their Inhibitors. J Immunol (2007) 179:4616. doi: 10.4049/jimmunol.179.7.4616
135. Geissmann F, Revy P, Brousse N, Lepelletier Y, Folli C, Durandy A, et al. Retinoids regulate survival and antigen presentation by immature dendritic cells. J Exp Med (2003) 198:623–34. doi: 10.1084/jem.20030390
136. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci USA (2014) 111:E3432–40. doi: 10.1073/pnas.1408780111
137. Jaggi U, Varanasi SK, Bhela S, Rouse BT. On the role of retinoic acid in virus induced inflammatory response in cornea. Microbes Infect (2018) 20:337–45. doi: 10.1016/j.micinf.2018.04.007
138. Butler JC, Havens PL, Day SE, Chusid MJ, Sowell AL, Huff DL, et al. Measles severity and serum retinol (Vitamin A) concentration among children in the United States. Pediatrics (1993) 91:1176–81.
139. Coutsoudis A, Broughton M, Coovadia HM. Vitamin A supplementation reduces measles morbidity in young African children: a randomized, placebo-controlled, double-blind trial. Am J Clin Nutr (1991) 54:890–5. doi: 10.1093/ajcn/54.5.890
140. Coutsoudis A, Kiepiela P, Coovadia HM, Broughton M. Vitamin A supplementation enhances specific IgG antibody levels and total lymphocyte numbers while improving morbidity in measles. Pediatr Infect Dis J (1992) 11:203–9. doi: 10.1097/00006454-199203000-00006
141. Myles IA, Fontecilla NM, Janelsins BM, Vithayathil PJ, Segre JA, Datta SK. Parental dietary fat intake alters offspring microbiome and immunity. J Immunol (2013) 191:3200–9. doi: 10.4049/jimmunol.1301057
142. Milner JJ, Rebeles J, Dhungana S, Stewart DA, Sumner SCJ, Meyers MH, et al. Obesity increases mortality and modulates the lung metabolome during pandemic H1N1 influenza virus infection in mice. J Immunol (2015) 194:4846–59. doi: 10.4049/jimmunol.1402295
143. Kim JY, Lim K, Kim KH, Kim JH, Choi JS. Shim S-C. N-3 polyunsaturated fatty acids restore Th17 and Treg balance in collagen antibody-induced arthritis. PLoS One (2018) 13:e0194331. doi: 10.1371/journal.pone.0194331
144. Innes JK, Calder PC. Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids (2018) 132:41–8. doi: 10.1016/j.plefa.2018.03.004
145. Schwerbrock NMJ, Karlsson EA, Shi Q, Sheridan PA, Beck MA. Fish oil-fed mice have impaired resistance to influenza infection. J Nutr (2009) 139:1588–94. doi: 10.3945/jn.109.108027
146. Antunes KH, Fachi JL, de Paula R, da Silva EF, Pral LP, dos Santos AÁ, et al. Microbiota-derived acetate protects against respiratory syncytial virus infection through a GPR43-type 1 interferon response. Nat Commun (2019) 10:3273. doi: 10.1038/s41467-019-11152-6
147. Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, Pernot J, et al. Dietary fiber confers protection against Flu by shaping Ly6c– patrolling Monocyte hematopoiesis and CD8+ T cell metabolism. Immunity (2018) 48:992–1005.e1008. doi: 10.1016/j.immuni.2018.04.022
148. Bachem A, Makhlouf C, Binger KJ, de Souza DP, Tull D, Hochheiser K, et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T cells. Immunity (2019) 51:285–97.e285. doi: 10.1016/j.immuni.2019.06.002
149. Sumbria D, Berber E, Rouse BT. Supplementing the diet with sodium propionate suppresses the severity of viral immuno-inflammatory lesions. J Virol (2020) 95:e02056–20. doi: 10.1128/JVI.02056-20. VI.02056-02020.
150. Uyangaa E, Lee H-K, Eo SK. Glutamine and leucine provide enhanced protective immunity against mucosal infection with herpes simplex virus type 1. Immune Netw (2012) 12:196–206. doi: 10.4110/in.2012.12.5.196
151. Wang K, Hoshino Y, Dowdell K, Bosch-Marce M, Myers TG, Sarmiento M, et al. Glutamine supplementation suppresses herpes simplex virus reactivation. J Clin Investig (2017) 127:2626–30. doi: 10.1172/JCI88990
152. Sencio V, Barthelemy A, Tavares LP, Machado MG, Soulard D, Cuinat C, et al. Gut dysbiosis during influenza contributes to pulmonary pneumococcal superinfection through altered short-chain fatty acid production. Cell Rep (2020) 30:2934–47.e2936. doi: 10.1016/j.celrep.2020.02.013
153. Moriyama M, Ichinohe T. High ambient temperature dampens adaptive immune responses to influenza A virus infection. Proc Natl Acad Sci USA (2019) 116:3118–25. doi: 10.1073/pnas.1815029116
154. Sanchez EL, Pulliam TH, Dimaio TA, Thalhofer AB, Delgado T, Lagunoff M. Glycolysis, glutaminolysis, and fatty acid synthesis are required for distinct stages of kaposi’s sarcoma-associated herpesvirus lytic replication. J Virol (2017) 91:e02237–02216. doi: 10.1128/jvi.02237-16
155. Boodhoo N, Kamble N, Kaufer BB, Behboudi S. Targeted induction of de novo Fatty acid synthesis enhances MDV replication in a COX-2/PGE2α dependent mechanism through EP2 and EP4 receptors engagement. bioRxiv (2018), 323840. doi: 10.1101/323840
156. Greseth MD, Traktman P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog (2014) 10:e1004021. doi: 10.1371/journal.ppat.1004021
157. Zhang N, Zhao H, Zhang L. Fatty acid synthase promotes the palmitoylation of Chikungunya Virus nsP1. J Virol (2019) 93:e01747–01718. doi: 10.1128/jvi.01747-18
158. Martín-Acebes MA, Blázquez A-B, Jiménez de Oya N, Escribano-Romero E, Saiz J-C. West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS One (2011) 6:e24970. doi: 10.1371/journal.pone.0024970
159. Merino-Ramos T, Vázquez-Calvo Á, Casas J, Sobrino F, Saiz J-C, Martín-Acebes MA. Modification of the host cell lipid metabolism induced by hypolipidemic drugs targeting the Acetyl Coenzyme A Carboxylase impairs West Nile virus replication. Antimicrob Agents Chemother (2016) 60:307–15. doi: 10.1128/aac.01578-15
160. Fernandes-Siqueira LO, Zeidler JD, Sousa BG, Ferreira T, Da Poian AT. Anaplerotic role of glucose in the oxidation of endogenous fatty acids during dengue virus infection. mSphere (2018) 3:e00458–00417. doi: 10.1128/mSphere.00458-17
161. Kohio HP, Adamson AL. Glycolytic control of vacuolar-type ATPase activity: A mechanism to regulate influenza viral infection. Virology (2013) 444:301–9. doi: 10.1016/j.virol.2013.06.026
162. Kulkarni A, Mateus M, Thinnes CC, McCullagh JS, Schofield CJ, Taylor GP, et al. Glucose metabolism and oxygen availability govern reactivation of the latent human retrovirus HTLV-1. Cell Chem Biol (2017) 24:1377–87.e1373. doi: 10.1016/j.chembiol.2017.08.016
163. Gualdoni GA, Mayer KA, Kapsch A-M, Kreuzberg K, Puck A, Kienzl P, et al. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc Natl Acad Sci USA (2018) 115:E7158–65. doi: 10.1073/pnas.1800525115
164. Cinatl J, Vogel J-U, Cinatl J, Kabickova H, Kornhuber B, Doerr HW. Antiviral effects of 6-diazo-5-oxo-l-norleucin on replication of herpes simplex virus type 1. Antiviral Res (1997) 33:165–75. doi: 10.1016/S0166-3542(96)01012-1
165. Nishio M, Tsurudome M, Bando H, Komada H, Ito Y. Antiviral Effect of 6-diazo-5-oxo-l-Norleucine, Antagonist of γ-glutamyl Transpeptidase, on Replication of Human Parainfluenza Virus Type 2. J Gen Virol (1990) 71:61–7. doi: 10.1099/0022-1317-71-1-61
166. Huang RC, Panin M, Romito RR, Huang YT. Inhibition of replication of human respiratory syncytial virus by 6-diazo-5-oxo-l-norleucine. Antiviral Res (1994) 25:269–79. doi: 10.1016/0166-3542(94)90009-4
167. Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot J-D, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell Chem Biol (2016) 166:1512–25.e151. doi: 10.1016/j.cell.2016.07.026
168. Varanasi SK, Donohoe D, Jaggi U, Rouse BT. Manipulating glucose metabolism during different stages of viral pathogenesis can have either detrimental or beneficial effects. J Immunol (2017) 199:1748–61. doi: 10.4049/jimmunol.1700472
169. Jiménez de Oya N, Esler WP, Huard K, El-Kattan AF, Karamanlidis G, Blázquez A-B, et al. Targeting host metabolism by inhibition of acetyl-Coenzyme A carboxylase reduces flavivirus infection in mouse models. Emerg Microbes Infect (2019) 8:624–36. doi: 10.1080/22221751.2019.1604084
170. Barzon L, Pacenti M, Palù G. West Nile virus and kidney disease. Expert Rev Anti Infect Ther (2013) 11:479–87. doi: 10.1586/eri.13.34
Keywords: virus, metabolism, interferon, diabetes, obesity, short chain fatty acids, metabolic blockers
Citation: Sumbria D, Berber E, Mathayan M and Rouse BT (2021) Virus Infections and Host Metabolism—Can We Manage the Interactions? Front. Immunol. 11:594963. doi: 10.3389/fimmu.2020.594963
Received: 14 August 2020; Accepted: 16 December 2020;
Published: 03 February 2021.
Edited by:
Neal A. DeLuca, University of Pittsburgh, United StatesReviewed by:
François J. M. A. Meurens, UMR INRAE-Oniris 1300 Oniris - Nantes Atlantic National College of Veterinary Medicine, FranceCopyright © 2021 Sumbria, Berber, Mathayan and Rouse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barry T. Rouse, YnRyQHV0ay5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.