
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 26 January 2021
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.594603
This article is part of the Research Topic Epigenetic Regulation of Innate Immunity View all 8 articles
 Annette E. Neele1*
Annette E. Neele1* Hung-Jen Chen1
Hung-Jen Chen1 Marion J. J. Gijbels1,2Saskia van der Velden1
Marion J. J. Gijbels1,2Saskia van der Velden1 Marten A. Hoeksema1,3Marieke C. S. Boshuizen1
Marten A. Hoeksema1,3Marieke C. S. Boshuizen1 Jan Van den Bossche1,4Anton T. Tool5
Jan Van den Bossche1,4Anton T. Tool5 Hanke L. Matlung5
Hanke L. Matlung5 Timo K. van den Berg5Esther Lutgens1,6
Timo K. van den Berg5Esther Lutgens1,6 Menno P. J. de Winther1*
Menno P. J. de Winther1*Macrophages define a key component of immune cells present in atherosclerotic lesions and are central regulators of the disease. Since epigenetic processes are important in controlling macrophage function, interfering with epigenetic pathways in macrophages might be a novel approach to combat atherosclerosis. Histone H3K27 trimethylation is a repressive histone mark catalyzed by polycomb repressive complex with EZH2 as the catalytic subunit. EZH2 is described to increase macrophage inflammatory responses by supressing the suppressor of cytokine signaling, Socs3. We previously showed that myeloid deletion of Kdm6b, an enzymes that in contrast to EZH2 removes repressive histone H3K27me3 marks, results in advanced atherosclerosis. Because of its opposing function and importance of EZH2 in macrophage inflammatory responses, we here studied the role of myeloid EZH2 in atherosclerosis. A myeloid-specific Ezh2 deficient mouse strain (Ezh2del) was generated (LysM-cre+ x Ezh2fl/fl) and bone marrow from Ezh2del or Ezh2wt mice was transplanted to Ldlr-/- mice which were fed a high fat diet for 9 weeks to study atherosclerosis. Atherosclerotic lesion size was significantly decreased in Ezh2del transplanted mice compared to control. The percentage of macrophages in the atherosclerotic lesion was similar, however neutrophil numbers were lower in Ezh2del transplanted mice. Correspondingly, the migratory capacity of neutrophils was decreased in Ezh2del mice. Moreover, peritoneal Ezh2del foam cells showed a reduction in the inflammatory response with reduced production of nitric oxide, IL-6 and IL-12. In Conclusion, myeloid Ezh2 deficiency impairs neutrophil migration and reduces macrophage foam cell inflammatory responses, both contributing to reduced atherosclerosis.
Atherosclerosis is a chronic lipid-driven inflammatory disorder of the arteries. Macrophages define a key component of immune cells present in atherosclerotic lesions and are important regulators of disease development and outcome (1). Unraveling pathways that are involved in the control of macrophage inflammatory responses in atherosclerosis will lead to a better understanding of disease. Since epigenetic processes are important modulators of macrophage function, we postulate that interference with epigenetic enzymes in macrophages might be a novel approach to modify macrophage function and influence atherosclerosis development (2, 3).
Histone H3K27 trimethylation (H3K27me3) is a repressive histone mark generated by the polycomb repressive complex 2 (PRC2), which core consists of Embryonic ectoderm development (EED), SUZ12, RpAp46/48 and Enhancer of the zeste homolog 1 (EZH1) or EZH2 (4). EZH1/2 contain a SET domain, which is necessary for the methyltransferase activity of the PRC2 complex (5, 6). Both EZH1 and EZH2 homologs can form similar PRC2 complexes but their differential function remains unclear. PRC2-EZH2 catalyzes mainly H3K27me2/3 methylation and Ezh2 knockdown affects global H3K27me3 levels, while EZH1 in the PRC2 complex performs this function weakly (7). EZH2 has been studied mainly in relation to developmental biology and cancer research. In addition, EZH2 controls stem cell proliferation and differentiation and it was demonstrated that EZH2 regulates T cell differentiation and plasticity (8).
EZH2 is a strong regulator macrophage inflammatory responses (9, 10). Macrophage Ezh2 deficiency was shown to inhibit pro-inflammatory gene expression via upregulation of suppressor of cytokine signaling 3 (SOCS3). Consequently, mice lacking Ezh2 in their macrophages showed improved outcome in models for autoimmune inflammation, experimental autoimmune encephalomyelitis (EAE) and colitis. The H3K27 demethylase KDM6b (also known as Jmjd3) removes repressive histone marks and is also an important regulator of macrophage activation (11–17). We previously showed that myeloid Kdm6b deficiency in mice results in advanced atherosclerosis (18). Since the H3K27 methyltransferases have opposing effects on this histone mark and the fact that EZH2 controls macrophage inflammatory responses, we hypothesized that inhibition of the H3K27 methyltransferase EZH2 in myeloid cells improves atherosclerosis outcome. To test this, we generated myeloid specific Ezh2 knockout mice and studied atherosclerosis progression. We found that atherosclerosis was significantly reduced in Ezh2-deficient mice, correlating with impaired neutrophil migration and reduced foam cell inflammatory responses.
Low-density lipoprotein receptor knock out mice (Ldlr-/-) (C57BL/6 background, Jackson laboratories) were used to study atherosclerosis, since these mice are prone to develop atherosclerosis in the presence of a high fat diet (HFD). A bone marrow transplantation (BMT) was performed with either LysM-cre+ Ezh2fl/fl mice (Ezh2del) or LysM-cre- Ezh2fl/fl littermates (Ezh2wt). Ezh2fl/fl mice were described before and crossbreeding with LysM-cre was performed in our mice facility (19). Briefly, 40 (20 per group), 10-week-old female Ldlr-/- mice were randomly divided over filter-top cages and provided with antibiotics water [autoclaved tap water with neomycin (100 mg/l, Sigma) and polymyxin B sulfate (60,000 U/l, Invitrogen)] from 1 week pre-BMT till 5 weeks post-BMT. The animals received 2 × 6 Gy total body irradiation on two consecutive days. Bone marrow was isolated from 4 Ezh2del and 4 Ezh2wt littermates, resuspended in RPMI1640 (Gibco) with 5 U/ml heparin and 2% iFCS (Gibco), and 5×106 bone marrow cells were injected intravenously per irradiated mouse. Five weeks after the BMT, the mice were put on a HFD (0.15% cholesterol, 16% fat, Arie Blok Diets) for 9 weeks. After sacrifice, hearts were isolated and frozen in Tissue-Tek (Sakura) for histology. Blood samples were taken before the start of the diet and four days before sacrifice (week 8) for lipid profiling and immune cell flow cytometry. Blood was withdrawn from mice which were fasted for 3 h. Bone marrow transplantation efficiency was determined with quantitative PCR for the Ldlr on DNA isolated from blood (GE Healthcare). Four mice were excluded from the analysis due to inefficient bone marrow transplantation (≤80%). Three mice were euthanized before the end of experiment since they reached their humane endpoints as a result of excessive weight loss (>15% weight loss since starting weight). Two additional mice were excluded from the analysis due to insufficient tissue quality and one mouse was excluded after outlier detection. A final of 15 mice per group was used for the statistical analysis. All animal experiments were conducted at the University of Amsterdam and approved (permit: DBC10AH) by the Committee for Animal Welfare of the Amsterdam UMC, location Academic Medical Center, University of Amsterdam.
Atherosclerotic lesions from the heart were cut in sections of 7 μm on a Leica 3050 cryostat at −25°C. Cross sections of every 42 μm were stained with Toluidin Blue (0.2% in PBS, Sigma-Aldrich) to determine lesion size. Lesion size was measured by use of adobe photoshop CS4 and the sum of the three valves is presented. Sirius red staining was performed for 30 min to detect collagen (0.05% direct Red in saturated picric acid, Sigma). Collagen content was quantified as the percentage collagen of total lesion size. For immunohistochemistry, slides were fixed in acetone and blocked with Avidin/Biotin Blocking Kit (Vector Laboratories). Hereafter, cells were incubated with MOMA-2 (1:4,000, AbD Serotec) to stain macrophages. Biotin-labeled rabbit anti–rat antibody (1:300, Dako) was used as a secondary antibody. The signal was amplified using ABC kit (Vector Laboratories) and visualized with the AEC kit (Vector Laboratories). Macrophages were quantified as the percentage of total lesion size. Neutrophils were stained with Ly6G antibody (BD biosciences) and incubated overnight at 4°C. Fluorescent rabbit anti-rat antibody was used as a secondary antibody (1:500, Thermo Fisher Scientific) and incubated for 1 h at room temperature (RT). Nuclei were stained with DAPI (1:5,000, Thermo Fisher Scientific) and incubated for 15 min at RT. Slides were mounted and fluorescence was measured using a Leica DM300 microscope. Neutrophil numbers were counted manually and corrected for lesion size. Necrosis area was measured based on Toluidine Blue staining by our pathologist and corrected for total lesion area.
150 μl of blood was withdrawn from mice via tail vein incision before the start of the diet and four days prior to sacrifice and added to 20 μl of 0.5 M EDTA (Sigma-Aldrich). Blood was withdrawn from mice which were fasted for 3 h. The blood was centrifuged (10 min, 4°C, 2,000 rpm) to separate the plasma from blood cells and plasma cholesterol and triglyceride levels were enzymatically measured according to the manufacturer’s protocol (Roche). Blood was further used for flow cytometry to assess relative leukocyte counts. Red blood cells were lysed by adding 5 ml of erythrocyte lysis buffer (8.4 g of NH4Cl, 0.84 g of NaHCO3, and 0.37 g of EDTA in 1 L MilliQ) for 15 min at RT. PBS was added to stop the reaction and cells were centrifuged. This was repeated until the pellet was not red anymore. White blood cells were used for flow cytometry. First Fc receptors were blocked with CD16/CD32 blocking antibody (1:100, eBioscience) in FACS buffer (0.5% BSA, 2mM EDTA in PBS). Hereafter cells were incubated with the appropriate antibodies for 30 min at RT (Supplemental Table 1). Cells were washed once and resuspended in FACS buffer. Fluorescence was measured with a BD Canto II and analyzed with FlowJo software. Immune cells were gated based on CD45+ expression and the following cell types were distinguished: Monocytes (CD11b+ and Ly6G-), Neutrophils (CD11b+ and Ly6G+), B cells (CD19+), and T cells (CD3+).
Bone marrow was isolated from femurs and tibia of Ezh2wt or Ezh2del mice by flushing. The cells were cultured in RPMI-1640 with 25 mM HEPES and 2 mM L-glutamine (Life Technologies) which was supplemented with 10% FCS, penicillin (100 U/ml), streptomycin (100 µg/ml) (all Gibco) and 15% L929-conditioned medium. Cells were cultured for 8 days to generate bone marrow-derived macrophages (BMDMs) on bacteriologic plastic plates. On day 8, macrophages were resuspended at a density of 106 cells/ml and plated in suspension culture plates allowing adherence for 6 h (Greiner). Next, cells were stimulated with LPS (10 ng/ml) for 6 h or left unstimulated where after the supernatant was collected for neutrophil migration experiments. Cultured BMDMs were also used for qPCR analysis, western blots and the H3K27 histone methyltransferase activity assay.
RNA from BMDMs or PEMs was isolated with the High Pure RNA isolation kits (Roche) from 500,000 cells. 400 ng of RNA was used for cDNA synthesis with iScript (BioRad). qPCR was performed with 4 ng of cDNA using Sybr Green Fast on a ViiA7 PCR machine (Applied Biosystems). Ezh1, Ezh2, Cxcl1, and Cxcl2 gene expression was normalized to the mean of the two housekeeping genes Ppia and Rplp0. Primer sequences are available on request.
NP40 lysis buffer (Invitrogen) was used for whole cell lysates, supplemented with protease inhibitor cocktail (1:25; Roche). Cells were lysed for 30 min on ice. Hereafter, cells were spun down for 10 min, 4°C at maximum speed and the supernatant was collected and used for western blot analysis for EZH2 protein expression. Histone extractions were used for H3K27Me3 Western blotting. For the histone extractions, cells were resuspended in Triton Extraction Buffer [PBS containing 0.5% Triton X 100 (v/v) and 0.02% (w/v) NaN3 supplemented with protease inhibitor cocktail (1:25; Roche)] and lysed on ice for 10 min. Cells were spun down for 10 min at 4°C at 2000 rpm. The supernatant was discarded and the pellet was resuspended in 0.2N HCl and histones were extracted overnight at 4°C on a rotator. Samples were centrifuged for 10 min, 4°C at 2000 rpm and the supernatant was used. Whole cell lysates were diluted with 6x reducing sample buffer (374 mM Tris, 6% SDS, 0.05% Bromophenol Blue, 20% Glycerol, 10% β-mercaptoethanol) and histone extracts with 2x reducing sample buffer (4% SDS, 20% glycerol, 0.125 M Tris-HCl, 10% β-mercaptoethanol, and 0.004% Serva Blue, pH 6.8) and boiled for 10 min at 95°C. Hereafter samples were loaded on a NuPAGE® Novex 4-12% Bis-Tris protein gel and ran for 1.5 h in MOPS buffer for EZH2 western blotting or MES buffer for H3K27Me3 western blotting (NuPAGE® MOPS or MES SDS Running Buffer 20x (Invitrogen) in demi water) at 100 V. The gel was transferred to a nitrocellulose membrane (Bio-Rad) and blotted for 2 h at 30 V in transfer buffer (20x transferbuffer NuPAGE® (Invitrogen), 20% Methanol in demi water). The membrane was blocked in 5% milk (Milk powder (Elk) in TBS-T) for 1 h and hereafter the blot for EZH2 was cut and incubated with the primary antibodies overnight in 1% milk at 4°C. The primary antibodies we used were anti-EZH2 (1:1,000; Bioke) and anti-α-tubulin (1:5,000; Sigma) and for histone extracts anti-H3K27Me3 (1:1,000; bioconnect) and anti-H3 (1:5,000; Cell Signaling Technology)The next day, blots were washed and incubated for 1 h at RT with the appropriate HRP-conjugated secondary antibody in 1% milk (1:5000 Dako). Blots were visualized using ECL substrate kit (Thermo Scientific).
The H3K27 histone methyltransferase activity assay was performed with the EpiQuikTM assay kit on nuclear lysates following manufactures instructions (Epigentek). Absorbace was read on a microplate reader at 450 nm. HMT activity was calculated by the following formula: Activity (OD/h/mg)=(OD(sample-blank)/protein amount (µg) x incubation time substrate (h) × 1,000.
Neutrophils were isolated from female mouse bone marrow of Ezh2wt and Ezh2del mice using a Ly6G-specific antibody (anti-mouse GR-1 APC, clone 1A8, 1:200, eBioscience). Cells were incubated with anti-APC beads and separated by MACS cell separation columns following manufacturer’s instruction (Miltenyi Biotec). Neutrophils were labeled with calcein-AM (life technologies) and chemotaxis was measured in response to supernatants of unstimulated or 6 h LPS stimulated BMDMs (5× diluted) in a transwell chemotactic assay over 3-µm pore size fluoroblok filters, as described previously (20).
Five mice per group from the atherosclerosis experiment were injected with thioglycollate medium (3%, Fischer). Four days after injection, mice were sacrificed and the peritoneum was flushed once with 10 ml ice-cold PBS to collect peritoneal macrophages (PEMs). Since these mice are on a HFD, these elicited macrophages are fully loaded with lipids and can thus be considered foam cells (21). Flushed thioglycollate-elicited foam cells were pooled per group and cultured at a density of 5 × 105 cells/well in 24-well tissue culture plates in triplicate (Greiner Bio-One) and cultured in RPMI-1640 with 25 mM HEPES, 2 mM L-glutamine, 10% FCS, penicillin (100 U/ml), and streptomycin (100 μg/ml) (all Gibco) and allowed adherence for 3 h. Hereafter the cells were washed and stimulated for 24 h with LPS (10 ng/ml), LPS plus IFNγ (100 U/ml), IL-4 (20 ng/ml), or left unstimulated.
Antibody pair kits were used to measure TNF, IL-6 and IL-12p40 (Life technologies) with ELISA following manufactures instructions. The following sample dilutions were used, TNF 1:10; IL-6 and IL-12p40 1:20. Streptavidin HRP was used to detect bound antibodies and TMB (Thermo scientific) was used as a substrate for the color reaction. This reaction was stopped with 1.8 M H2SO4. Absorbance was measured on a microplate reader (Victor) at a wavelength of 450 nm. For the nitric oxide (NO) assay, 50 µl of undiluted samples was used on a flat bottom 96-wells plate. A standard curve was made with NaNO2 in culture medium. Next 50 µl of Griess reagent (2.5% H3PO4, 1% Sulfanylamide, 0.1% Napthyleene diamine in MilliQ) was added to the samples and absorbance was immediately measured on the microplate reader at a wavelength of 550 nm.
Data is presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism 5.0 software using an unpaired t-test or two-way ANOVA with bonferroni post-hoc analysis when comparing multiple groups. P-values < 0.05 were considered statistically significant.
A myeloid-specific Ezh2 deficient mouse strain (Ezh2del) was generated and cultured bone marrow-derived macrophages (BMDMs) showed a reduction of Ezh2 at both the mRNA as well as protein level, without changes in Ezh1 mRNA expression (Figures 1A, B). EZH1 is most likely not compensating for the loss of EZH2 in macrophages as global H3K27Me3 levels and H3K27 methyl transferase activity was strongly reduced in Ezh2-deficient BMDMs (Figures 1B, C). Bone marrow of Ezh2wt or Ezh2del mice was transplanted to Ldlr-/- mice, which were subsequently fed a high fat diet (HFD) for 9 weeks to induce atherosclerosis. Bone marrow of both Ezh2wt and Ezh2del mice was effectively transplanted to Ldlr-/- mice as chimerism was around 95% and not different between the groups (Figure 1D). Weight, cholesterol and triglyceride levels were similar after 8 weeks of HFD between Ezh2wt and Ezh2del transplanted mice (Figures 1E–G). Additionally, leukocyte levels in both blood and spleen were unaltered in Ezh2del transplanted mice compared to wildtype (Figures 1H, I). Overall, no differences in the lipid levels or composition of immune cell populations were seen after transplantation of Ezh2del bone marrow compared to wildtype, giving similar baseline characteristics.
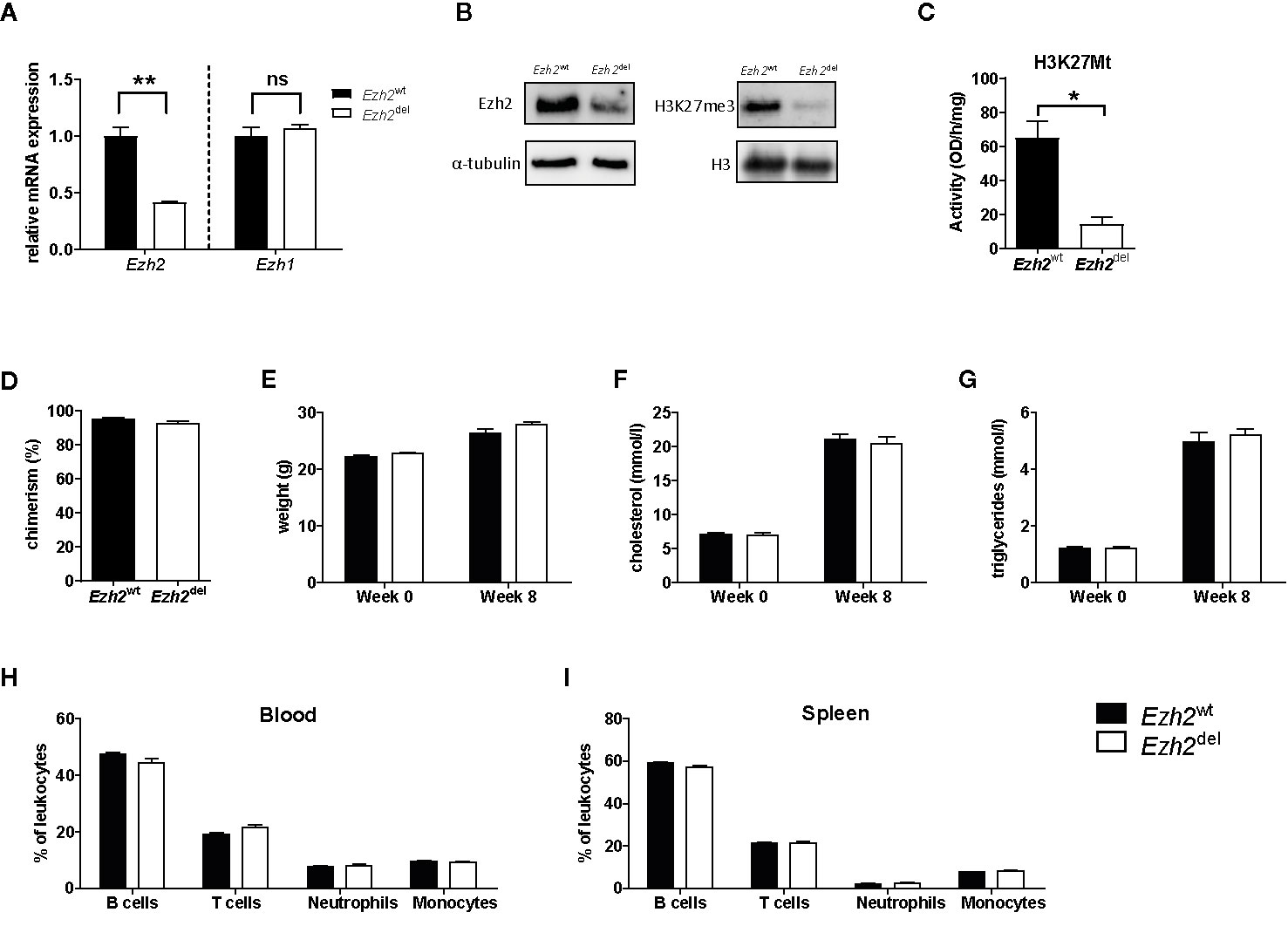
Figure 1 Bone marrow of Ezh2wt and Ezh2del mice was effectively transplanted to Ldlr-/- mice. (A) Relative normalized Ezh2 and Ezh1 mRNA expression in Ezh2wt and Ezh2del BMDMs (B) EZH2 protein expression on whole cell lysates (left) and H3K27Me3 levels on histone extracts (right) of BMDMs. α-tubulin and H3 were used as loading control (C) H3K27 methyltransferase activity in nuclear lysates of BMDMs. (D) Chimerism determination by qPCR for the Ldlr in the DNA of blood of Ezh2wt (black bars) and Ezh2del (white bars) transplanted mice. (E) Mouse weight in grams at the start of the diet (week 0) and after 8 weeks of HFD (week 8). (F) Cholesterol and (G) triglyceride levels in the plasma at the start and after 8 weeks of HFD. Percentage of (H) blood and (I) spleen leukocyte subsets assessed by flow cytometry. B cells (CD45+, CD19+), T cells (CD45+, CD3+), Neutrophils (CD45+, CD11b+, Ly6G+) and monocytes (CD45+, CD11b+, Ly6G-). N=15 each group. Data represent mean ± SEM.
Interestingly, we observed that atherosclerotic lesion size was significantly reduced in Ezh2del transplanted mice compared to Ezh2wt mice after 9 weeks of HFD (Figures 2A, B). A small but non-significant decrease in collagen content was seen in Ezh2del transplanted mice (P = 0.0528) (Figures 2C, D) and the percentage of macrophage and necrosis area were not different between Ezh2wt and Ezh2del transplanted mice (Figures 2E–H). Interestingly, the number of neutrophils was significantly lower in lesions of Ezh2del transplanted mice, even when corrected for lesion area (Figures 2I, J). These data indicate that myeloid Ezh2 deficiency leads to atherosclerotic lesions that are not only smaller but also less inflammatory.
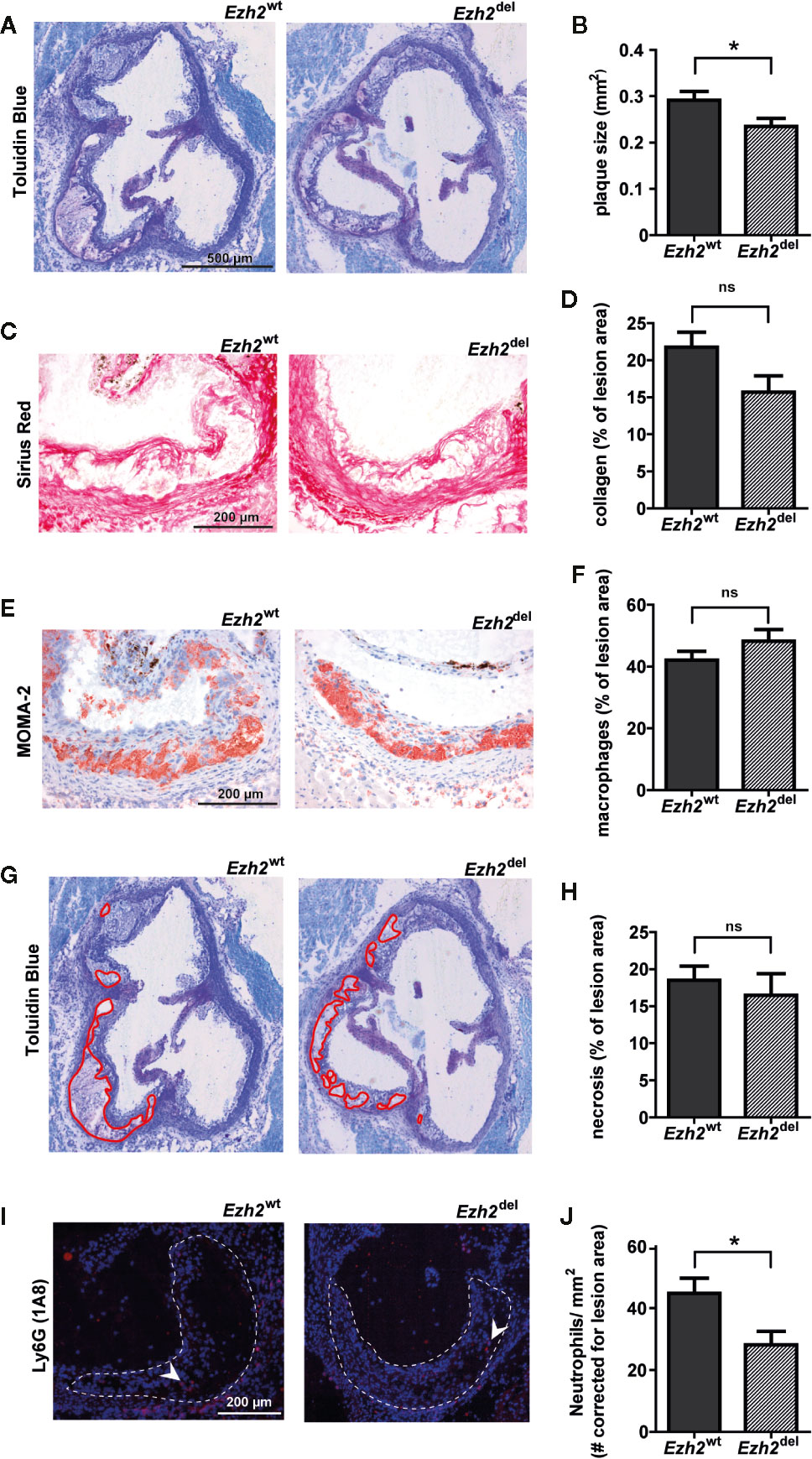
Figure 2 Atherosclerotic lesions size is reduced in Ezh2del transplanted mice. (A) Representative Toluidin Blue staining of the aortic root of Ezh2wt and Ezh2del transplanted mice. (B) Aortic lesion area presented as the sum of the three valves per mice. (C) Representative Sirius Red staining to measure collagen content. (D) Collagen content as percentage of total lesion area. (E) Representative MOMA-2 staining for macrophages. (F) Macrophage area as percentage of total lesion area. (G) Necrotic core area indicated in red on the toluidine blue staining. (H) Necrotic core as percentage of total lesion area. (I) Ly6G (1A8) neutrophil staining in red and nuclei in blue (DAPI). (J) Neutrophil counts, corrected for total lesion area. Data represent mean ± SEM of 15 mice. *P < 0.05.
Since blood neutrophil numbers were unaltered (Figure 1E) but were significantly lower in Ezh2del lesions, our data suggest that the migration or recruitment of neutrophils to atherosclerotic lesions is reduced. We next investigated if this was due to differences in the chemotactic factors secreted by Ezh2del macrophages or due to intrinsic migration defects of neutrophils. We isolated neutrophils from bone marrow of Ezh2wt and Ezh2del mice and performed chemotaxis assays. We also assessed wild type neutrophil migration toward supernatants of stimulated Ezh2wt and Ezh2del BMDMs (Figure 3A). We observed no differences in the migration of wild type neutrophils toward supernatants of unstimulated and LPS activated Ezh2wt and Ezh2del BMDMs (Figure 3B, left). Furthermore, the expression of Cxcl1 and Cxcl2 expression as the main chemoattractants for neutrophils, was not different in aortic arches of Ezh2wt and Ezh2del transplanted mice, suggesting that these are similar in the lesion environment and not causing the difference in neutrophil accumulation (Figure 3C). Analysis of Cxcl1 and Cxcl2 mRNA expression in peritoneal foam cells and BMDMs was inconclusive, as expression was partly increased, which does not support the reduced neutrophil numbers in vivo (Supplemental Figure S1). Conversely, we did observe that the migration of Ezh2del neutrophils itself was reduced compared to Ezh2wt neutrophils in vitro (Figure 3B, right). These data suggest that neutrophils of Ezh2del mice are less migratory and therefore accumulate less in the atherosclerotic lesions.
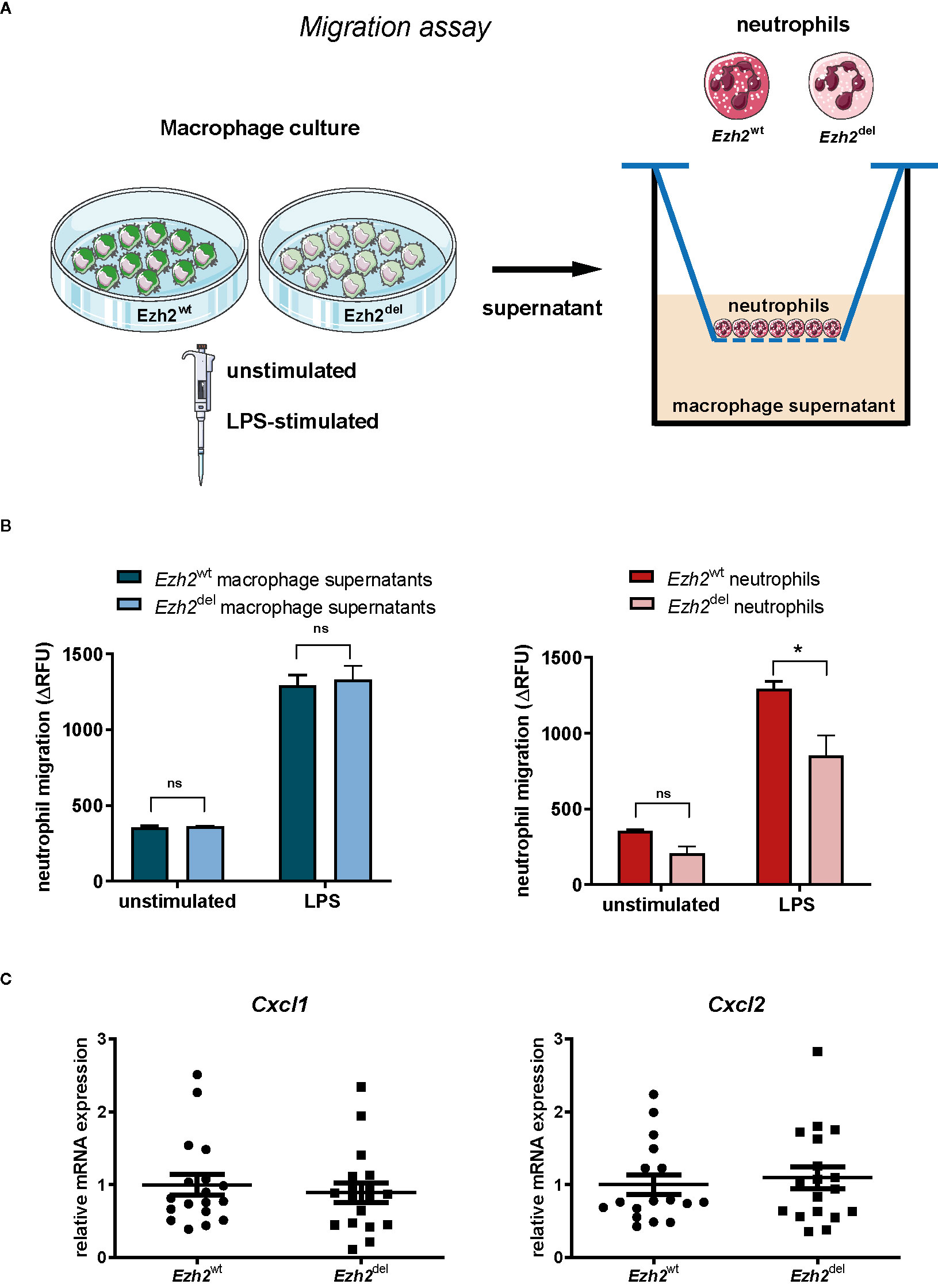
Figure 3 EZH2 regulates neutrophil migration. (A) Neutrophils were isolated from bone marrow of Ezh2wt or Ezh2del mice. Neutrophils were fluorescently labeled and chemotaxis was measured in response to supernatants of unstimulated or LPS activated Ezh2wt or Ezh2del BMDMs. (B) Neutrophil migration as the increase of RFU in time in relative fluorescence units (delta RFU). Presented is one out of three experiment of 2 pooled Ezh2wt and 2 pooled Ezh2del mice plated in duplo. Left is Ezh2wt neutrophil migration toward supernatant of Ezh2wt or Ezh2del BMDMs and right is Ezh2wt or Ezh2del neutrophil migration toward wildtype supernantants. (C) Cxcl1 and Cxcl2 mRNA expression in the aortic arch of Ezh2wt or Ezh2del transplanted mice. Statistics are performed on duplicates with a two-way anova with bonferroni correction. Data represent mean ± SEM. *P < 0.05. Panel (A) was made with use of smart servier medical art, lincesed under a Creative Common Attribution 3.0 Unported License. https://smart.servier.com/.
Since Zhang et al. showed that Ezh2 deficiency diminishes the pro-inflammatory response of microglia and macrophages, we here studied whether this is also true for macrophages under HFD conditions, i.e. foam cells. Thioglycollate elicited peritoneal macrophages were isolated from transplanted HFD-fed mice and studied ex vivo. Since these mice are on a HFD, these macrophages are fully loaded with lipids and can thus be considered foam cells (21). Peritoneal foam cells from Ezh2del mice produced less NO, IL-6, and IL-12p40 after 24 h of LPS+IFNγ stimulation compared to Ezh2wt foam cells, indicating that the inflammatory response is reduced in Ezh2del foam cells (Figure 4A). However, TNF secretion was enhanced in activated Ezh2del foam cells (Figure 4A). We also activated these foam cells with IL-4 as an inducer of alternative macrophage activation but did not observe any changes in IL-4 responsive gene expression (Figure 4B). The inflammatory response is thus partly reduced in Ezh2del foam cells without changes in IL-4 responsive genes.
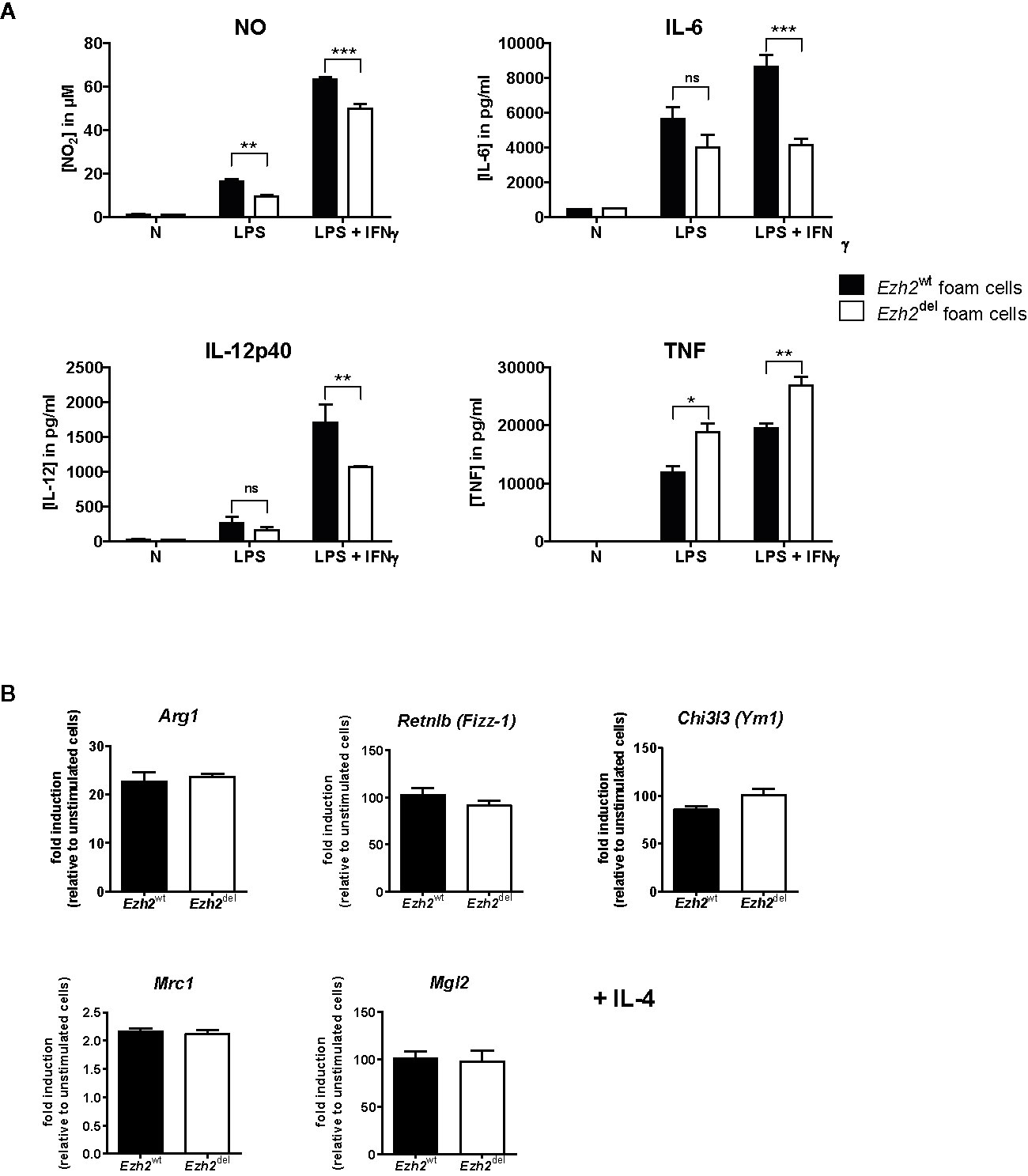
Figure 4 EZH2 affects inflammatory responses in peritoneal foam cells. (A) Cytokine production by Ezh2wt (black bars) and Ezh2del peritoneal foam cells after stimulation for 24 h with LPS or LPS+IFNγ. (B) mRNA expression in Ezh2wt and Ezh2del peritoneal foam cells after 24 h of IL-4 stimulation. Foam cells of five individual mice per group were pooled and plated in triplicate. Data represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
We here studied the role of the H3K27 methyltransferase EZH2 in atherosclerosis development by use of a myeloid-specific Ezh2 knockout mice. Ezh2-deficient mice showed reduced levels of H3K27me3 and lower H3K27 methyltransferase activity, indicating that EZH1 is not compensation for the loss of EZH2. Ezh2 mRNA levels were partly reduced (± 60%) as deletion is never complete in the used floxed LysM-cre system and recombination efficiency dependent on the genomic location and distance between the LoxP sites (22–24). As our LoxP sites flank the catalytic SET domain of Ezh2, it is sufficient to strongly reduce H3K27Me3 levels and H3K27 methyltransferase activity.
We show that myeloid Ezh2 deficiency improves atherosclerosis outcome as lesion size was significantly reduced in Ezh2del transplanted mice compared to wildtype. Overexpression of Ezh2 in all cell types was previously reported to promote macrophage foam cells formation and worsen atherosclerosis outcome (25). In line with their findings, we show that silencing of Ezh2 specifically in myeloid cells reduces atherosclerosis. Moreover, we show that atherosclerotic lesions of myeloid Ezh2-deficient mice are not only smaller but also contain less neutrophils. Neutrophils play an important role in all stages of atherosclerosis (26) and neutrophil depletion studies in atherosclerotic mice (ApoE-/-) showed a reduction in early atherosclerosis (27). Since neutrophil numbers were similar in blood, this implies that the recruitment of neutrophils toward the lesion is impaired.
We studied neutrophil migration in vitro and indeed found that Ezh2-deficient neutrophils are less migratory. This phenomenon is consistent with studies showing that neutrophils derived from Ezh2 knockout stem cells have impaired migration together with increased cell death, decreased phagocytosis and overproduction of reactive oxygen species (ROS) (28). Accordingly, Gunawan et al. observed reduced neutrophil migration and in addition found reduced dendritic cell migration in cells lacking Ezh2 (29). They propose a mechanism by which EZH2 regulates migration, independent of its H3K27 methyltransferase activity (29). They show that EZH2 regulates integrin mediated migration via methylation of Talin1. EZH2 interacts with VAV1, causing methylation of Talin1. In turn, binding to filamentous actin (F-actin) was altered affecting adhesion and migration.
Likewise, we show that the inflammatory response of peritoneal foam cells is reduced in activated Ezh2del cells compared to Ezh2wt cells. IL-6, IL-12 and NO production, which are key cytokines induced upon macrophage activation, were reduced in Ezh2del foam cells. Interestingly, the inflammatory cytokine TNF was increased in cells lacking Ezh2, indicating that not the full activation spectrum is impaired. Additionally, IL-4 responsive genes were not affected. The idea that inhibition of EZH2, a mediator of repression, results in enhancement of the inflammatory gene program in macrophages appears to be too simplistic. Zhang and colleagues nicely demonstrated one of the mechanisms by which the repressive EZH2 causes activation of inflammation in macrophages (10). By chromatin immunoprecipitation (ChIP) and RNA sequencing analysis they identified the suppressor of cytokine signaling 3 (Socs3) as being strongly upregulated in the absence of EZH2 in combination with EZH2 binding sites and reduced H3K27me3 levels in macrophages. They show that Ezh2 deficiency lowers Socs3 levels thereby enhancing Traf6 ubiquitination and degradation. The Myd88-NF-κB signaling pathway is thereby suppressed, resulting in the reduced inflammatory responses. They illustrate how a repressor like EZH2 can cause activation of inflammation by repression of another repressor, in this case SOCS3 (9, 10). Additionally, Ezh2 deficiency improved the outcome on EAE and colitis, two inflammatory mouse models. We here show that myeloid Ezh2 deficiency also improves the outcome of atherosclerosis, another chronic inflammatory disease.
In conclusion, we show that myeloid Ezh2 deficiency limits atherosclerosis due to impaired neutrophil migration and modulation of foam cell inflammatory responses. Our findings support the thought that EZH2 is not only a target for various types of cancer but may also serve as novel target for auto-immune diseases. Several EZH2 inhibitors are available and the use of these inhibitors in mouse models of atherosclerosis would be the next step to test the therapeutic potential of EZH2 inhibition to control atherosclerosis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Committee for Animal Welfare of the Amsterdam UMC, location Academic Medical Center, University of Amsterdam (permit: DBC10AH).
AN and MW conceived and designed the experiments. AN, H-JC, MG, SV, MH, MB, JB, AT, and HM performed the experiments. AN, MG, and SV analyzed the data. AN, MG, EL, and MW were involved in the interpretation of the data. AN and MW wrote the manuscript. MG, JB, TB, and EL critically read and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by The Netherlands Heart Foundation [CVON 2011/B019 and CVON 2017-20: Generating the best evidence-based pharmaceutical targets for atherosclerosis (GENIUS I&II)]. AN received a Junior Postdoc grant from the Netherlands Heart Foundation (2020T029) and a postdoc grant from Amsterdam Cardiovascular Sciences. JB received a senior postdoctoral fellowship (2017T048) from the Netherlands Heart Foundation. MW is an established investigator of the Netherlands Heart Foundation (2007T067), is supported by a Netherlands Heart Foundation grant (2010B022), The Netherlands Heart Foundation and Spark-Holding BV (2015B002, 2019B016), the European Union (ITN-grant EPIMAC), Leducq Transatlantic Network Grant, ZonMW (Open competition 09120011910025) and holds an AMC-fellowship. MW and EL are both supported by REPROGRAM (EU Horizon 2020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Ezh2-floxed mice were a gift from Dr. van Lohuizen (Netherlands Cancer Institute) and originally generated by the laboratory of Dr. Orkin, S.H. (19).
The content of this manuscript has been published in part in Chapter 7 of the thesis of Annette E. Neele (30).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.594603/full#supplementary-material
1. Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol (2010) 7:77–86. doi: 10.1038/nrcardio.2009.228
2. Neele AE, Van den Bossche J, Hoeksema MA, de Winther MP. Epigenetic pathways in macrophages emerge as novel targets in atherosclerosis. Eur J Pharmacol (2015) 763:79–89. doi: 10.1016/j.ejphar.2015.03.101
3. Van den Bossche J, Neele AE, Hoeksema MA, de Winther MP. Macrophage polarization: the epigenetic point of view. Curr Opin Lipidol (2014) 25:367–73. doi: 10.1097/MOL.0000000000000109
4. Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol (2013) 20:1147–55. doi: 10.1038/nsmb.2669
5. Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev (2002) 16:2893–905. doi: 10.1101/gad.1035902
6. Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell (2002) 111:197–208. doi: 10.1016/S0092-8674(02)00976-5
7. Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell (2008) 32:503–18. doi: 10.1016/j.molcel.2008.11.004
8. Karantanos T, Chistofides A, Barhdan K, Li L, Boussiotis VA. Regulation of T Cell Differentiation and Function by EZH2. Front Immunol (2016) 7:172. doi: 10.3389/fimmu.2016.00172
9. Neele AE, de Winther MPJ. Repressing the repressor: Ezh2 mediates macrophage activation. J Exp Med (2018) 215:1269–71. doi: 10.1084/jem.20180479
10. Zhang X, Wang Y, Yuan J, Li N, Pei S, Xu J, et al. Macrophage/microglial Ezh2 facilitates autoimmune inflammation through inhibition of Socs3. J Exp Med (2018) 215:1365–82. doi: 10.1084/jem.20171417
11. Chen S, Ma J, Wu F, Xiong LJ, Ma H, Xu W, et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev (2012) 26:1364–75. doi: 10.1101/gad.186056.111
12. De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J (2009) 28:3341–52. doi: 10.1038/emboj.2009.271
13. De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell (2007) 130:1083–94. doi: 10.1016/j.cell.2007.08.019
14. Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood (2009) 114:3244–54. doi: 10.1182/blood-2009-04-217620
15. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature (2012) 488:404–8. doi: 10.1038/nature11262
16. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol (2010) 11:936–44. doi: 10.1038/ni.1920
17. an Q, Sun L, Zhu Z, Wang L, Li S, Ye RD. Jmjd3-mediated epigenetic regulation of inflammatory cytokine gene expression in serum amyloid A-stimulated macrophages. Cell Signal (2014) 26:1783–91. doi: 10.1016/j.cellsig.2014.03.025
18. Neele AE, Gijbels MJJ, van der Velden S, Hoeksema MA, Boshuizen MCS, Prange KHM, et al. Myeloid Kdm6b deficiency results in advanced atherosclerosis. Atherosclerosis (2018) 275:156–65. doi: 10.1016/j.atherosclerosis.2018.05.052
19. Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell (2008) 32:491–502. doi: 10.1016/j.molcel.2008.10.016
20. Kuijpers TW, Maianski NA, Tool AT, Becker K, Plecko B, Valianpour F, et al. Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood (2004) 103:3915–23. doi: 10.1182/blood-2003-11-3940
21. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell (2012) 151:138–52. doi: 10.1016/j.cell.2012.06.054
22. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res (1999) 8:265–77. doi: 10.1023/A:1008942828960
23. Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods (2014) 408:89–100. doi: 10.1016/j.jim.2014.05.009
24. Vooijs M, Jonkers J, Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep (2001) 2:292–7. doi: 10.1093/embo-reports/kve064
25. Lv YC, Tang YY, Zhang P, Wan W, Yao F, He PP, et al. Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis. PloS One (2016) 11:e0157265. doi: 10.1371/journal.pone.0157265
26. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol (2020) 17:327–40. doi: 10.1038/s41569-019-0326-7
27. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation (2010) 122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714
28. Perez-Ladaga A, Caughey B, Xie H, Orkin SH, Sykes DB, Elbert BL, et al. Functional Defects In Neutrophils Derived From Ezh2 Null Mice. Blood (2013) 122:1556. doi: 10.1182/blood.V122.21.1556.1556
29. Gunawan M, Venkatesan N, Loh JT, Wong JF, Berger H, Neo WH, et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat Immunol (2015) 16:505–16. doi: 10.1038/ni.3125
Keywords: atherosclerosis, epigenetic, histone modification, H3K27, macrophage, polycomb, PRC2
Citation: Neele AE, Chen H-J, Gijbels MJJ, van der Velden S, Hoeksema MA, Boshuizen MCS, Van den Bossche J, Tool AT, Matlung HL, van den Berg TK, Lutgens E and de Winther MPJ (2021) Myeloid Ezh2 Deficiency Limits Atherosclerosis Development. Front. Immunol. 11:594603. doi: 10.3389/fimmu.2020.594603
Received: 13 August 2020; Accepted: 26 November 2020;
Published: 26 January 2021.
Edited by:
Martin Rottenberg, Karolinska Institutet (KI), SwedenReviewed by:
Ivan Lilyanov Dzhagalov, National Yang-Ming University, TaiwanCopyright © 2021 Neele, Chen, Gijbels, van der Velden, Hoeksema, Boshuizen, Van den Bossche, Tool, Matlung, van den Berg, Lutgens and de Winther. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annette E. Neele, YS5lLm5lZWxlQGFtc3RlcmRhbXVtYy5ubA==; Menno P. J. de Winther, bS5kZXdpbnRoZXJAYW1zdGVyZGFtdW1jLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.