
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 20 October 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.592949
This article is part of the Research Topic The Immunomodulatory Roles of Adipocytes View all 11 articles
 Peng Zhao1
Peng Zhao1 Alan R. Saltiel1,2*
Alan R. Saltiel1,2*Adipocytes and adipose tissue play critical roles in the regulation of metabolic homeostasis. In obesity and obesity-associated metabolic diseases, immune cells infiltrate into adipose tissues. Interaction between adipocytes and immune cells re-shapes both metabolic and immune properties of adipose tissue and dramatically changes metabolic set points. Both the expression and activity of the non-canonical IKK family member TBK1 are induced in adipose tissues during diet-induced obesity. TBK1 plays important roles in the regulation of both metabolism and inflammation in adipose tissue and thus affects glucose and energy metabolism. Here we review the regulation and functions of TBK1 and the molecular mechanisms by which TBK1 regulates both metabolism and inflammation in adipose tissue. Finally, we discuss the potential of a TBK1/IKKε inhibitor as a new therapy for metabolic diseases.
Obesity has reached a pandemic (1). The complications of obesity, including type 2 diabetes, cardiovascular diseases, neurodegenerative diseases, non-alcoholic fatty liver diseases, and cancer, have become leading health threats. Obesity is caused by a positive energy balance, leading to excess lipid accumulation in adipose and other tissues (2–5). In addition to being an inert site for energy storage, adipose tissues play essential roles in metabolic homeostasis (6, 7). As the major cell type within adipose tissue, adipocytes are responsible for lipid storage and mobilization in response to insulin and sympathetic activation respectively. However, these cells can also sense their nutrient status, and respond by secreting a series of hormones known as “adipokines” (6–8). Upon food intake, the resulting elevation of nutrients in the circulation stimulates insulin production. Insulin in turn lowers glucose and fatty acid levels in part by instructing fat and muscle tissue to increase glucose uptake and storage, while reducing lipolysis in fat, glycogenolysis in muscle and liver and gluconeogenesis in liver (9, 10). In adipocytes, nutrients are largely stored as triglycerides. Upon reaching a threshold of lipogenesis, adipocytes trigger the production of adipokines such as leptin, to suppress food consumption and activate the sympathetic nervous system, thus closing a loop to ensure energy homeostasis (9, 11–15). Excessive energy intake or low energy expenditure could lead to a sustained positive energy balance and consequently cause increased adiposity in obesity (3–5).
Obesity is associated with low-grade chronic inflammation in adipose tissue, featured by an increased number of macrophages and an elevated ratio of proinflammatory macrophages (16–20). Although the immediate trigger for obesity-associated inflammation in adipose tissue remains unclear, multiple factors, including hypoxia, mechanical stress, lipotoxicity, adipocyte death, and bacterial toxins may contribute to this process (9, 21–27). Inflammation has been reported to affect several properties of adipocytes. The activation of proinflammatory pathways has been shown to disrupt glucose uptake and insulin responsiveness and alter adipokine production (28–31), suggesting that inflammation plays an essential role in the pathological response to obesity.
The nuclear factor kappa B (NFκB) is a widely expressed transcription factor that mediates inflammatory responses in numerous tissues. The NFκB signaling pathway plays a key role in the development of inflammation and insulin resistance in adipose tissue (32–34). Transcription through NFκB is mainly controlled by the phosphorylation of inhibitor of NFκB (IκB) by the upstream IκB kinases (IKKs). The canonical IKKs, IKKα, and IKKβ, phosphorylate IκB, and other NFκB subunits to induce the expression of NFκB target genes (35). Besides IKKα and β, the IKK family also includes two non-canonical members, IKKϵ and TANK-binding kinase 1 (TBK1). Interestingly, despite their sequence similarity to the canonical IKK isoforms, TBK1 and IKKε do not appear to play important roles in NFκB activation in response to proinflammatory cytokines (36). However, expression of Ikke and Tbk1 mRNAs are induced by NFκB (37). Moreover, IKKϵ and TBK1 are activated by protein phosphorylation in response to proinflammatory cytokines or other substances that bind to Toll-like receptors 3 and 4 (38). It was reported that activities of IKKϵ and TBK1 are significantly increased in adipose tissue of obese mice (37). We review here the functions of the noncanonical IKKs in inflammation and metabolic regulation in adipose tissue, with a major focus on the roles of TBK1 in crosstalk between inflammation and metabolism.
NFκB plays a central role in the transcriptional response to proinflammatory stimuli. In the absence of stimuli, IκB binds to NFκB to sequester the transcription factor in the cytoplasm (39). Inflammatory stimuli increase the phosphorylation and activation of IKKs, which in turn phosphorylate IκB and NFκB to activate the expression of NFκB target genes (35, 39, 40). An IKK complex formed by IKKα, IKKβ, and the NFκB essential modifier (NEMO) directly phosphorylates IκB at Ser32 and Ser36 to induce ubiquitin-associated degradation. Consequently, NFκB is released to activate gene expression. This pathway represents the canonical NFκB signaling pathway (41, 42). Both IKKα and IKKβ possess a kinase domain (KD), a scaffold dimerization domain (SDD), and a NEMO-binding domain (NBD). A ubiquitin-like domain (ULD) is found in IKKβ but not in IKKα. In contrast to the canonical IKKs, IKKϵ and TBK1 have similar SD, ULD, and SDD, but lack the NBD. Human TBK1 shares 49% identity and 65% similarity to IKKϵ, but only 27% identity with IKKα and IKKβ (43–45). Unlike the canonical IKKs, the roles of IKKϵ and TBK1 in the NFκB signaling pathways remain uncertain. Early studies demonstrated that TBK1 phosphorylates IKKβ to increase its activity, while IKKϵ phosphorylates RelA at Ser468 to induce its nuclear translocation (44, 46, 47). However, subsequent studies found that TBK1 or IKKϵ deficiency has no effect on LPS, TNFα, interleukin-1β, or poly(I:C)-induced activation of NFκB (38, 48). Thus, it appears that IKKϵ and TBK1 are not required for the activation of NFκB in response to proinflammatory cytokines (36). Instead, studies showed that the expression of Ikke and Tbk1 are induced by NFκB under proinflammatory conditions (37). Interestingly, two separate studies demonstrated that TBK1 and IKKϵ mediate NFκB activation downstream of the cGAS-STING pathway in response to cytosolic DNA or STING ligand (49, 50).
Multiple studies demonstrated that non-canonical IKKs play important roles in metabolic regulation. The expression of Ikke was upregulated in the liver, adipocytes, and adipose tissue macrophages during diet-induced obesity (34). Knockout of Ikke reduced inflammation and improved insulin sensitivity in adipose tissue and liver. Hepatic steatosis was largely attenuated by IKKϵ deficiency as well. Ikke knockout mice gained less weight and were resistant to high fat diet-induced obesity due to the increased energy expenditure and thermogenesis (34). The expression of Uncoupling protein 1 (Ucp1), a major uncoupler utilizing the mitochondrial proton gradient to generate heat, was significantly upregulated in white adipose tissue in these mice (34).
Energy expenditure is largely controlled by sympathetic signals. Catecholamines induce Ucp1 expression and increase thermogenesis in both brown and subcutaneous white fat (51, 52). During high fat diet-induced obesity, adipose tissue becomes resistant to catecholamines, resulting in decreased energy expenditure (9, 53–55). Mowers et al. demonstrated that IKKϵ directly phosphorylates and activates phosphodiesterase 3B (PDE3B) to reduce intracellular cAMP levels and thus represses cAMP-mediated β-adrenergic signaling (55). Ikke knockout restored catecholamine sensitivity, leading to an upregulation of Ucp1 expression and an increase of thermogenesis (34, 55, 56). Therefore, during obesity, the inflammation-induced expression of Ikke represses sympathetic signal and further promotes energy storage (Figure 1). IKKϵ mediates the interaction between inflammatory and catecholamine signals, representing one example of how inflammation modulates metabolism in adipose tissue.
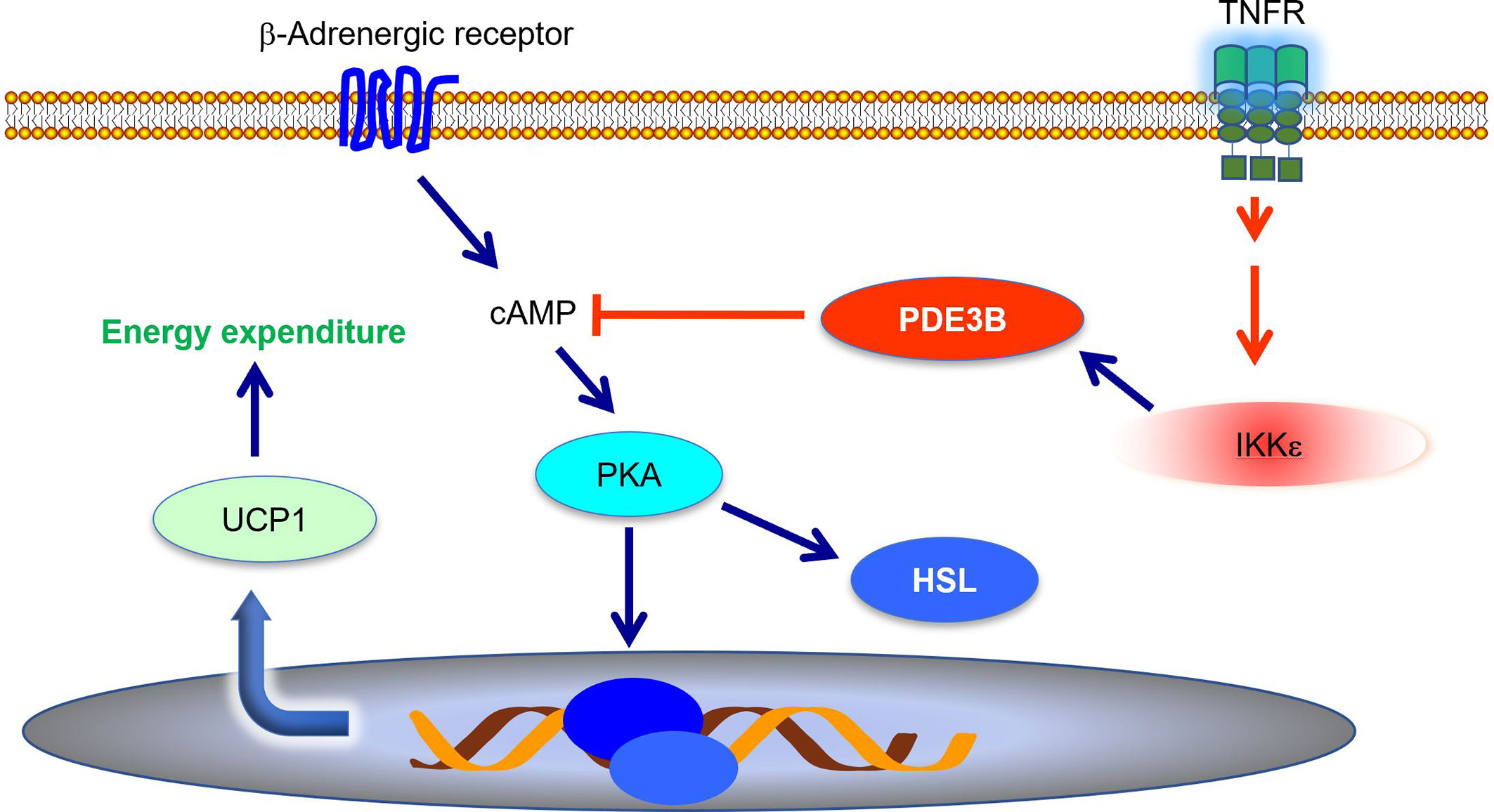
Figure 1 IKKϵ inhibits adrenergic signaling to repress thermogenesis. IKKϵ activity is induced by proinflammatory stimuli. Active IKKϵ directly phosphorylates and activates PDE3B to reduce cAMP levels. Consequently, IKKϵ inhibits cAMP-mediated adrenergic signaling pathway and represses energy expenditure in adipocytes. PDE3B, phosphodiesterase 3B; cAMP, cyclic AMP; PKA, protein kinase A; HSL, hormone sensitive lipase; UCP1, uncoupling protein 1.
Although the role of TBK1 in NFκB activation remains unclear, its function in the innate immune response has been well-recognized. In response to infection, pattern recognition receptors (PRRs) sense the pathogen-associated molecular patterns (PAMPs) on bacteria or viruses to activate TBK1-mediated signaling pathways (57, 58). Two major types of PRRs participate in this action. Toll-like receptors (TLRs), especially TLR3 and TLR4, are cell surface receptors that utilize adaptor proteins such as TIR-domain-containing adaptor-inducing interferon-β (TRIF) and Myeloid differentiation primary response 88 (MyD88). Ligands of TLRs, such as lipopolysaccharides (LPSs), bind to their receptors to induce the activation of TBK1. Retinoic acid-inducible gene I (RIG-I)-like receptors, NOD-like receptors (NLRs), and cytosolic DNA sensors are the PRRs in the cytoplasm (36, 59, 60). Cyclic-GMP-AMP (cGAMP) synthase (cGAS) is a cytosolic DNA sensor. cGAS utilizes cytosolic DNA to generate cGAMP, which in turn binds to the adaptor protein Stimulator of interferon genes (STING). Consequently, STING interacts with and activates TBK1 (61). Besides pathogen infection, proinflammatory cytokines such as tumor necrosis factor α (TNFα) also produces TBK1 activation (62, 63). Upon activation, TBK1 directly phosphorylates interferon regulatory factor 3 (IRF3) and IRF7 at multiple serine and threonine residues to induce their nuclear translocation (64–67). Consequently, these transcription factors upregulate the expression of type I interferon (Ifna, Ifnb) genes in the innate immune response. TBK1 is indispensable for the antiviral immune response (61, 68).
The activity of TBK1 is acutely controlled by phosphorylation on Ser172 within the kinase domain (63, 69, 70). However, the molecular mechanism by which this activating phosphorylation occurs is still unclear. Structural studies suggest that TBK1 undergoes multi-order oligomerization. While the kinase usually exists as a homodimer, the kinase domains face outward and are generally not capable of phosphorylation in this configuration (70). However, adapter proteins bring together these homodimers in larger heteromeric complexes, leading to Ser172 phosphorylation via transautophosphorylation (70). Moreover, recent investigations demonstrated that Unc-51 like autophagy activating kinase 1 (ULK1) can directly phosphorylate Ser172 (63). This is consistent with the observations that both ULK1 and TBK1 play essential roles in autophagy (71–75). TBK1 regulates autophagy via phosphorylating optineurin on Ser177 and SQSTM1/p62 on Ser403 to clear pathogen or damaged mitochondria (76, 77). Interestingly, the activation of NFκB also upregulates the expression of Sqstm1/p62 to induce mitophagy in response to LPS (78, 79). These studies suggest that NFκB and TBK1 may function synergistically to promote the clearance of damaged mitochondria and pathogens during infection.
Understanding the functions of TBK1 in vivo have been hampered by the lethality of global Tbk1 knockout. Whole-body knockout of Tbk1 leads to enhanced apoptotic liver degeneration and embryonic lethality at approximately E14.5 (80). In this regard, TBK1 directly phosphorylates receptor-interacting serine/threonine-protein kinase 1 (RIPK1) on Thr189 to prevent cell death. TBK1 deficiency substantially increases RIPK1-mediated cell death, resulting in embryonic lethality between embryonic day 13.5 and embryonic day 14.5 (81). In line with this finding, another study found that both TBK1 and IKKϵ phosphorylate RIPK1 on multiple sites, including Thr189, to prevent TNF-induced cell death (62, 81). To conduct in vivo studies on the roles of TBK1 in inflammation, Marchlik et al., generated (Tbk1Δ/Δ) mice expressing a TBK1 inactive mutant with the deletion of exon 2 (82). Tbk1Δ/Δ C57BL/6J mice were still embryonic lethal. However, Tbk1Δ/Δ 129S5 mice were fertile and viable, but born at a decreased Mendelian frequency. Tbk1Δ/Δ mice had increased mononuclear and granulomatous cell infiltration into multiple tissues, along with elevated circulating monocytes. This is consistent with another study reporting that Tbk1Δ/Δ mice die faster and in larger numbers in response to LPS (82).
Although it was reported that TBK1 expression and activity are induced in adipose tissues during obesity and insulin resistance (34, 37, 63), the role of TBK1 in the pathogenesis of metabolic disease was unclear. A recent study revealed that TBK1 mediates crosstalk between inflammation and metabolism in adipose tissue (Figure 2) (63). During high fat diet-induced obesity, chronic inflammation leads to an increase of proinflammatory cytokines in the adipose tissue (9, 30, 83). Consequently, these cytokines, such as TNFα, produce the activation of TBK1 (63). At the same time, the inflammatory environment also results in enhanced NFκB activity, resulting in an increase in Tbk1 expression (34, 63). Thus, high fat diet feeding substantially induces TBK1 activity in the adipose tissue through both transcriptional and posttranslational regulation (34, 37, 63). Upon activation, TBK1 attenuates adipose tissue inflammation via repressing the atypical NFκB pathway (63). In this pathway, the NFκB-inducing kinase (NIK) phosphorylates Ser176 to activate IKKα, which largely resides as a homodimer (84). IKKα in turns phosphorylates the RelB (NFκB2) precursor p100, resulting in the cleavage and maturation of RelB (85). Thus, NIK is responsible for activation of the atypical NFκB pathway, which induces the expression of target genes, such as Ccl2 (C-C motif chemokine ligand 2), to promote macrophage infiltration and inflammation (86–88). Interestingly, TBK1 directly phosphorylates NIK, leading to its degradation (62, 63). Tbk1 knockout causes hyperactivation of the atypical NFκB pathway and exacerbates macrophage infiltration and inflammation in adipose tissue of obese mice (63). Moreover, the loss of TBK1 in adipocytes attenuates HFD-induced obesity via increasing mitochondrial biogenesis and energy expenditure. TBK1 inhibits AMP-activated protein kinase (AMPK) by catalyzing phosphorylation on inhibitory sites in AMPKα subunit, Ser459 and Ser476. Tbk1 knockout thus ameliorates AMPK repression in adipose tissues of high fat diet-fed mice (63), revealing that TBK1 mediates crosstalk from inflammation to energy metabolism. The inflammation-induced TBK1 activity produced during obesity represses energy expenditure and promotes anabolism, which further enhances obesity through a feedforward loop.
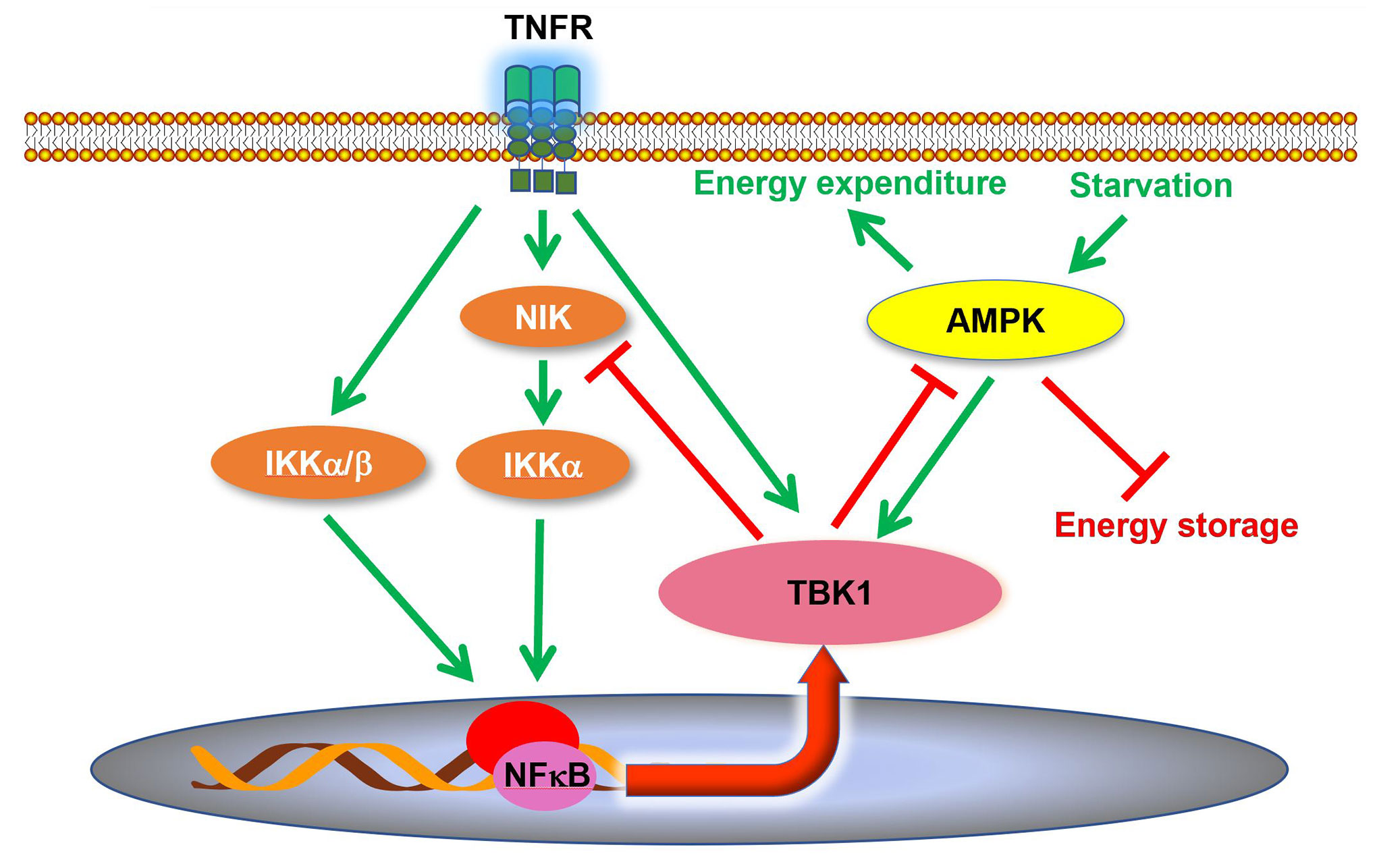
Figure 2 TBK1 regulates inflammation and energy metabolism in adipocytes. TBK1 activity is induced by proinflammatory stimuli and undernutrition. Although TBK1 is not directly involved in TNFα-induced activation of NFκB, active TBK1 phosphorylates NIK to induce its degradation and thus attenuates atypical NFκB pathway in a negative feedback loop. Moreover, TBK1 inhibits AMPK to repress energy expenditure in adipocytes. AMPK, AMP-activated protein kinase; NIK, NFκB inducing kinase.
In addition to inflammation-induced TBK1 activation, it has also been reported that TBK1 Ser172 phosphorylation is induced in adipocytes during glucose deprivation, which creates an energy shortage condition (63). Thus, TBK1 is activated not only during overnutrition, but also during undernutrition. Mechanistically, energy shortage leads to an increase of AMP/ATP ratio, which in turns activates AMPK. AMPK directly phosphorylates ULK1 at multiple residues to induce its activity (89, 90). ULK1 is able to phosphorylate Ser172 to activate TBK1 (63). Similar observations on AMPK-dependent TBK1 activation have been reported in myotubes and Hela cells as well (91). Furthermore, prolonged fasting induced Tbk1 expression in different depots of white adipose tissues (63). However, the molecular mechanism of this transcriptional regulation is still unknown. Studies on animal models and human subjects reported that fasting or undernutrition leads to a reduction of basal metabolic rate and energy expenditure (92, 93). Given the effects of TBK1 on energy metabolism, fasting likely activates a TBK1-mediated feedback loop to repress energy expenditure in response to undernutrition. The activation of TBK1 could be a protective mechanism to attenuate the loss of body weight during fasting. Moreover, reduced caloric intake has been demonstrated to attenuate adipose tissue inflammation in obesity (94–97). The anti-inflammatory function of TBK1 at least partially contributes to this effect and mediates crosstalk from undernutrition to inflammation.
In summary, TBK1 plays a central role in the regulation of both inflammation and energy metabolism in adipose tissue. It is activated during both overnutrition and undernutrition and mediates a negative feedback loop to repress inflammation and energy expenditure under certain conditions (63). More importantly, TBK1 is responsible for the bidirectional crosstalk between energy metabolism and inflammation. The deficiency of TBK1 in adipocytes leads to the attenuation of high fat diet-induced obesity, but the exaggeration of adipose tissue inflammation (63), indicating a loss of the positive correlation between adiposity and adipose tissue inflammation.
Furthermore, in response to proinflammatory stimuli, TBK1 has been shown to affect metabolic reprogramming in different cell types. Upon the activation of TLRs, active TBK1 was recruited to the myddosome and thus promotes glycolysis in macrophages (98). Another two studies also reported that TBK1 activation mediates TLR ligand-induced glycolytic reprogramming (99, 100). The rapid induction of glycolysis is critical for the production of succinate and inflammatory cytokines in the immune response (99). These findings demonstrate another TBK1-mediated pathway that regulates the crosstalk between inflammation and metabolism. However, further studies are needed to compare the cell type specific roles of TBK1.
Insights into the critical roles of the noncanonical IKKs in the pathogenesis of obesity and insulin resistance led to a screen of chemical inhibitors, identifying amlexanox as an inhibitor for both TBK1 and IKKϵ (37). Daily gavage of amlexanox in obese mice prevents genetic and high fat diet-induced obesity. The inhibition of weight gain by amlexanox is reversible after withdrawal of the drug. Amlexanox improved insulin sensitivity, reduced adipose tissue inflammation, increased energy expenditure, and attenuated hepatic steatosis in these obese animal models (37). Considering the phenotypes observed in Ikke knockout mice and adipose Tbk1 knockout mice, the beneficial effects of amlexanox is likely the combined outcomes from the inhibition of both kinases. The inhibition of IKKϵ increases cAMP and catecholamine sensitivity to upregulate thermogenesis and attenuates adipose tissue inflammation (34). On the other hand, loss of TBK1 activity de-represses AMPK to increase mitochondrial biogenesis and other catabolic functions (63). The TBK1 deficiency-induced adipose tissue inflammation is likely compensated by the anti-inflammatory effects of IKKϵ inhibition.
In a proof-of-concept randomized, double-blinded clinical study, 42 obese and diabetic patients received placebo or amlexanox treatment for 12 weeks. Amlexanox significantly reduced hemoglobin A1c levels (101), indicating an improvement of glucose metabolism. Further study found that patients with higher serum C-reactive protein (CRP) levels and higher adipose tissue inflammation were more responsive to the drug. In the responder group, amlexanox improved insulin sensitivity and hepatic steatosis. The expression of thermogenic genes, including Ucp1, Dio2 and Fgf21, was upregulated by the treatment as well in these patients. Within the responders, a transient increase of serum Interleukin 6 (IL-6) within 2–4 weeks of amlexanox treatment was reported (101). This observation is consistent with a previous mouse study showing that amlexanox upregulated Il6 expression and secretion via cAMP/Mitogen-activated protein kinase (MAPK) p38 pathway in inguinal white adipose tissue. The increase of circulating IL-6 activates Signal transducer and activator of transcription 3 (STAT3) in the liver to inhibit the expression of the gluconeogenic gene Glucose-6-phosphatase (G6pc). As a result, amlexanox represses hepatic glucose output and thus improves glucose tolerance (102).
Although the causal relationship between inflammation and obesity-associated metabolic disorders remains uncertain, there is little doubt that adipose tissue inflammation correlates well with the occurrence of insulin resistance and type 2 diabetes (16, 17, 19, 20). The crosstalk between inflammation and metabolism in adipose tissue plays a critical role in the pathogenesis of metabolic diseases. Overnutrition causes metabolic stress, which induces the initiation of inflammation to restore the metabolic homeostasis (9). The activation of proinflammatory signaling pathways attenuates insulin responsive signals to prevent further energy storage in adipocytes (103, 104). Both of these effects are the physiological/adaptive responses to overnutrition. However, along the progression of obesity, sustained inflammation causes a shift of homeostatic setpoints, leading to hyperglycemia, hyperinsulinemia, and reduced energy expenditure (9). At this stage, the inflammation causes a pathological/maladaptive response that further exaggerates obesity and obesity-associated metabolic disorders. Therefore, sustained inflammation results in a transition from an adaptive response to a maladaptive response that accelerates the progression of metabolic disorders.
NFκB signals mediate inflammatory responses and interact with metabolic pathways in adipose tissue (33, 105, 106). The activities of non-canonical IKKs, TBK1, and IKKϵ are induced during inflammation (34, 37, 63). TBK1 represses energy expenditure via inhibiting AMPK, while IKKϵ desensitizes sympathetic signals (34, 55, 63). The activation of these kinases exacerbates adiposity accumulation and promotes obesity. A recent study reported that escaped mitochondrial DNA activates TBK1 and IKKϵ to repress energy expenditure during metabolic stress (56). Amlexanox, a drug with outstanding safety record, was identified as an inhibitor of TBK1 and IKKϵ. Thus far, multiple studies on both experimental mouse models and human subjects suggest its potential as a new treatment for metabolic diseases (37, 101).
In addition to modulating metabolic pathways in adipocytes, metabolic and inflammatory signals interact at systemic level in other cell types. Metabolic stress has the potential to increase the production of adipokines, including leptin, adiponection, and others (28–31). It has been reported that leptin induces inflammation, while adiponectin attenuates inflammation (107–110). Moreover, metabolic status could affect the functions of immune cells. Caloric restriction has exhibited systemic anti-inflammatory effects, along with attenuated terminal differentiation of immune cells (111). Given the energy sensing properties of AMPK, the AMPK–ULK1–TBK1 axis may also function in immune cells to mediate anti-inflammatory effects. Nonetheless, the precise roles of adipose tissue inflammation in the progression of obesity and obesity-associated insulin resistance remains unclear. Indeed, more efforts are needed to understand the systemic interactions between immune and metabolic responses, which are essential for the maintenance of homeostasis.
PZ prepared the manuscript. AS reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
The study is supported by NIH P30DK063491, R01DK076906, R01DK117551, R01DK122804, and R01DK125820 to AS.; NIH K99HL143277 to PZ.
AS is a founder of Elgia Therapeutics.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Popkin BM. Is the obesity epidemic a national security issue around the globe? Curr Opin Endocrinol Diabetes Obes (2011) 18:328–31. doi: 10.1097/MED.0b013e3283471c74
2. Hall KD, Heymsfield SB, Kemnitz JW, Klein S, Schoeller DA, Speakman JR. Energy balance and its components: implications for body weight regulation. Am J Clin Nutr (2012) 95:989–94. doi: 10.3945/ajcn.112.036350
3. Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation (2012) 126:126–32. doi: 10.1161/CIRCULATIONAHA.111.087213
4. Ravussin E, Lillioja S, Knowler WC, Christin L, Freymond D, Abbott WG, et al. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med (1988) 318:467–72. doi: 10.1056/NEJM198802253180802
5. Roberts SB, Leibel RL. Excess energy intake and low energy expenditure as predictors of obesity. Int J Obes Relat Metab Disord (1998) 22:385–6. doi: 10.1038/sj.ijo.0800640
6. Richard AJ, Stephens JM. The role of JAK-STAT signaling in adipose tissue function. Biochim Biophys Acta (2014) 1842:431–9. doi: 10.1016/j.bbadis.2013.05.030
7. Sarjeant K, Stephens JM. Adipogenesis. Cold Spring Harb Perspect Biol (2012) 4:a008417. doi: 10.1101/cshperspect.a008417
8. Zhao P, Stephens JM. Identification of STAT target genes in adipocytes. JAKSTAT (2013) 2:e23092. doi: 10.4161/jkst.23092
9. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol (2017) 13:633–43. doi: 10.1038/nrendo.2017.90
10. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature (2001) 414:799–806. doi: 10.1038/414799a
11. Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med (2008) 14:667–75. doi: 10.1038/nm1775
12. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med (1996) 334:292–5. doi: 10.1056/NEJM199602013340503
13. Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med (1995) 1:1311–4. doi: 10.1038/nm1295-1311
14. Scarpace PJ, Matheny M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. Am J Physiol (1998) 275:E259–264. doi: 10.1152/ajpendo.1998.275.2.E259
15. Shen J, Tanida M, Niijima A, Nagai K. In vivo effects of leptin on autonomic nerve activity and lipolysis in rats. Neurosci Lett (2007) 416:193–7. doi: 10.1016/j.neulet.2007.02.003
16. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi: 10.1038/nature05485
17. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell (2015) 160:816–27. doi: 10.1016/j.cell.2015.02.010
18. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi: 10.1172/JCI29881
19. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121:2111–7. doi: 10.1172/JCI57132
20. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
21. Fischer-Posovszky P, Wang QA, Asterholm IW, Rutkowski JM, Scherer PE. Targeted deletion of adipocytes by apoptosis leads to adipose tissue recruitment of alternatively activated M2 macrophages. Endocrinology (2011) 152:3074–81. doi: 10.1210/en.2011-1031
22. Hara Y, Wakino S, Tanabe Y, Saito M, Tokuyama H, Washida N, et al. Rho and Rho-kinase activity in adipocytes contributes to a vicious cycle in obesity that may involve mechanical stretch. Sci Signal (2011) 4:ra3. doi: 10.1126/scisignal.2001227
23. Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, et al. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem (2004) 279:16971–9. doi: 10.1074/jbc.M312990200
24. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell (2014) 157:1339–52. doi: 10.1016/j.cell.2014.05.012
25. Li Q, Hata A, Kosugi C, Kataoka N, Funaki M. The density of extracellular matrix proteins regulates inflammation and insulin signaling in adipocytes. FEBS Lett (2010) 584:4145–50. doi: 10.1016/j.febslet.2010.08.033
26. Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem (2007) 282:35279–92. doi: 10.1074/jbc.M706762200
27. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes (2007) 56:2910–8. doi: 10.2337/db07-0767
28. Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol (2005) 115:911–9; quiz 920. doi: 10.1016/j.jaci.2005.02.023
29. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol (2011) 11:85–97. doi: 10.1038/nri2921
30. Unamuno X, Gomez-Ambrosi J, Rodriguez A, Becerril S, Fruhbeck G, Catalan V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur J Clin Invest (2018) 48:e12997. doi: 10.1111/eci.12997
31. Vachharajani V, Granger DN. Adipose tissue: a motor for the inflammation associated with obesity. IUBMB Life (2009) 61:424–30. doi: 10.1002/iub.169
32. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med (2005) 11:191–8. doi: 10.1038/nm1185
33. Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab (2011) 13:11–22. doi: 10.1016/j.cmet.2010.12.008
34. Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell (2009) 138:961–75. doi: 10.1016/j.cell.2009.06.046
35. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
36. Shin CH, Choi DS. Essential Roles for the Non-Canonical IkappaB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes. Cells (2019) 8:178. doi: 10.3390/cells8020178
37. Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med (2013) 19:313–21. doi: 10.1038/nm.3082
38. Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med (2004) 199:1651–8. doi: 10.1084/jem.20040528
39. Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene (1999) 18:6867–74. doi: 10.1038/sj.onc.1203219
40. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol (2009) 1:a000034. doi: 10.1101/cshperspect.a000034
41. Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol (2010) 2:a000158. doi: 10.1101/cshperspect.a000158
42. Solt LA, May MJ. The IkappaB kinase complex: master regulator of NF-kappaB signaling. Immunol Res (2008) 42:3–18. doi: 10.1007/s12026-008-8025-1
43. Durand JK, Zhang Q, Baldwin AS. Roles for the IKK-Related Kinases TBK1 and IKKepsilon in Cancer. Cells (2018) 7:139. doi: 10.3390/cells7090139
44. Pham AM, Tenoever BR. The IKK Kinases: Operators of Antiviral Signaling. Viruses (2010) 2:55–72. doi: 10.3390/v2010055
45. Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J (1999) 18:6694–704. doi: 10.1093/emboj/18.23.6694
46. Mattioli I, Geng H, Sebald A, Hodel M, Bucher C, Kracht M, et al. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J Biol Chem (2006) 281:6175–83. doi: 10.1074/jbc.M508045200
47. Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K, et al. NAK is an IkappaB kinase-activating kinase. Nature (2000) 404:778–82. doi: 10.1038/35008109
48. Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med (2004) 199:1641–50. doi: 10.1084/jem.20040520
49. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
50. Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, et al. TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep (2020) 31:107492. doi: 10.1016/j.celrep.2020.03.056
51. Collins S. beta-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front Endocrinol (Lausanne) (2011) 2:102. doi: 10.3389/fendo.2011.00102
52. Ramseyer VD, Granneman JG. Adrenergic regulation of cellular plasticity in brown, beige/brite and white adipose tissues. Adipocyte (2016) 5:119–29. doi: 10.1080/21623945.2016.1145846
53. Guo T, Marmol P, Moliner A, Bjornholm M, Zhang C, Shokat KM, et al. Adipocyte ALK7 links nutrient overload to catecholamine resistance in obesity. Elife (2014) 3:e03245. doi: 10.7554/eLife.03245
54. Komai AM, Musovic S, Peris E, Alrifaiy A, El Hachmane MF, Johansson M, et al. White Adipocyte Adiponectin Exocytosis Is Stimulated via beta3-Adrenergic Signaling and Activation of Epac1: Catecholamine Resistance in Obesity and Type 2 Diabetes. Diabetes (2016) 65:3301–13. doi: 10.2337/db15-1597
55. Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang SH, et al. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKepsilon and TBK1. Elife (2013) 2:e01119. doi: 10.7554/eLife.01119
56. Bai J, Cervantes C, He S, He J, Plasko GR, Wen J, et al. Mitochondrial stress-activated cGAS-STING pathway inhibits thermogenic program and contributes to overnutrition-induced obesity in mice. Commun Biol (2020) 3:257. doi: 10.1038/s42003-020-0986-1
57. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22:240–73. Table of Contents. doi: 10.1128/CMR.00046-08
58. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol (2012) 4:a006049. doi: 10.1101/cshperspect.a006049
59. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol (2011) 30:16–34. doi: 10.3109/08830185.2010.529976
60. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
61. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54:289–96. doi: 10.1016/j.molcel.2014.03.040
62. Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol (2018) 20:1389–99. doi: 10.1038/s41556-018-0229-6
63. Zhao P, Wong K I, Sun X, Reilly SM, Uhm M, Liao Z, et al. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell (2018) 172:731–743 e712. doi: 10.1016/j.cell.2018.01.007
64. Caillaud A, Hovanessian AG, Levy DE, Marie IJ. Regulatory serine residues mediate phosphorylation-dependent and phosphorylation-independent activation of interferon regulatory factor 7. J Biol Chem (2005) 280:17671–7. doi: 10.1074/jbc.M411389200
65. Clement JF, Bibeau-Poirier A, Gravel SP, Grandvaux N, Bonneil E, Thibault P, et al. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of dimerization and CBP association. J Virol (2008) 82:3984–96. doi: 10.1128/JVI.02526-07
66. Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem (2004) 279:9698–702. doi: 10.1074/jbc.M310616200
67. Panne D, McWhirter SM, Maniatis T, Harrison SC. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J Biol Chem (2007) 282:22816–22. doi: 10.1074/jbc.M703019200
68. Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res (2005) 15:407–22. doi: 10.1038/sj.cr.7290309
69. Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, et al. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J Biol Chem (2002) 277:13840–7. doi: 10.1074/jbc.M110474200
70. Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U.S.A. (2012) 109:9378–83. doi: 10.1073/pnas.1121552109
71. Lin MG, Hurley JH. Structure and function of the ULK1 complex in autophagy. Curr Opin Cell Biol (2016) 39:61–8. doi: 10.1016/j.ceb.2016.02.010
72. Liyana A, Vanessa SS. The emerging role of human TBK1 in virus-induced autophagy. Autophagy (2019) 15:917–8. doi: 10.1080/15548627.2019.1580513
73. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity (2012) 37:223–34. doi: 10.1016/j.immuni.2012.04.015
74. Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal (2011) 4:pe39. doi: 10.1126/scisignal.2002355
75. Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy (2013) 9:124–37. doi: 10.4161/auto.23323
76. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet (2015) 24:4429–42. doi: 10.1093/hmg/ddv179
77. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A (2016) 113:4039–44. doi: 10.1073/pnas.1523926113
78. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell (2016) 166:288–98. doi: 10.1016/j.cell.2016.05.051
79. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell (2016) 164:896–910. doi: 10.1016/j.cell.2015.12.057
80. Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J (2000) 19:4976–85. doi: 10.1093/emboj/19.18.4976
81. Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell (2018) 174:1477–91.e1419. doi: 10.1016/j.cell.2018.07.041
82. Marchlik E, Thakker P, Carlson T, Jiang Z, Ryan M, Marusic S, et al. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol (2010) 88:1171–80. doi: 10.1189/jlb.0210071
83. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front Physiol (2019) 10:1607. doi: 10.3389/fphys.2019.01607
84. Ling L, Cao Z, Goeddel DV. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc Natl Acad Sci U S A (1998) 95:3792–7. doi: 10.1073/pnas.95.7.3792
85. Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev (2012) 246:125–40. doi: 10.1111/j.1600-065X.2011.01088.x
86. Shen H, Sheng L, Chen Z, Jiang L, Su H, Yin L, et al. Mouse hepatocyte overexpression of NF-kappaB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology (2014) 60:2065–76. doi: 10.1002/hep.27348
87. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol (2017) 17:545–58. doi: 10.1038/nri.2017.52
88. Thu YM, Richmond A. NF-kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev (2010) 21:213–26. doi: 10.1016/j.cytogfr.2010.06.002
89. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (2011) 331:456–61. doi: 10.1126/science.1196371
90. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi: 10.1038/ncb2152
91. Seabright AP, Fine NHF, Barlow JP, Lord SO, Musa I, Gray A, et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J (2020) 34:6284–301. doi: 10.1096/fj.201903051R
92. Fothergill E, Guo J, Howard L, Kerns JC, Knuth ND, Brychta R, et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring) (2016) 24:1612–9. doi: 10.1002/oby.21538
93. Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med (1995) 332:621–8. doi: 10.1056/NEJM199503093321001
94. Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiol (Bethesda) (2011) 26:214–24. doi: 10.1152/physiol.00010.2011
95. Larson-Meyer DE, Heilbronn LK, Redman LM, Newcomer BR, Frisard M II, Anton S, et al. Effect of calorie restriction with or without exercise on insulin sensitivity, beta-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes Care (2006) 29:1337–44. doi: 10.2337/dc05-2565
96. Lijnen HR, Van Hul M, Hemmeryckx B. Caloric restriction improves coagulation and inflammation profile in obese mice. Thromb Res (2012) 129:74–9. doi: 10.1016/j.thromres.2011.05.023
97. Park S, Park NY, Valacchi G, Lim Y. Calorie restriction with a high-fat diet effectively attenuated inflammatory response and oxidative stress-related markers in obese tissues of the high diet fed rats. Mediators Inflammation (2012) 2012:984643. doi: 10.1155/2012/984643
98. Tan Y, Kagan JC. Innate Immune Signaling Organelles Display Natural and Programmable Signaling Flexibility. Cell (2019) 177:384–398 e311. doi: 10.1016/j.cell.2019.01.039
99. Balic JJ, Albargy H, Luu K, Kirby FJ, Jayasekara WSN, Mansell F, et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1beta expression. Nat Commun (2020) 11:3816. doi: 10.1038/s41467-020-17669-5
100. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15:323–32. doi: 10.1038/ni.2833
101. Oral EA, Reilly SM, Gomez AV, Meral R, Butz L, Ajluni N, et al. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab (2017) 26:157–70.e157. doi: 10.1016/j.cmet.2017.06.006
102. Reilly SM, Ahmadian M, Zamarron BF, Chang L, Uhm M, Poirier B, et al. A subcutaneous adipose tissue-liver signalling axis controls hepatic gluconeogenesis. Nat Commun (2015) 6:6047. doi: 10.1038/ncomms7047
103. Chen L, Chen R, Wang H, Liang F. Mechanisms Linking Inflammation to Insulin Resistance. Int J Endocrinol (2015) 2015:508409. doi: 10.1155/2015/508409
104. Zand H, Morshedzadeh N, Naghashian F. Signaling pathways linking inflammation to insulin resistance. Diabetes Metab Syndr (2017) 11 Suppl 1:S307–9. doi: 10.1016/j.dsx.2017.03.006
105. Catrysse L, van Loo G. Inflammation and the Metabolic Syndrome: The Tissue-Specific Functions of NF-kappaB. Trends Cell Biol (2017) 27:417–29. doi: 10.1016/j.tcb.2017.01.006
106. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther (2017) 2:e17023. doi: 10.1038/sigtrans.2017.23
107. Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gomez-Reino JJ, et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol (2017) 13:100–9. doi: 10.1038/nrrheum.2016.209
108. Fantuzzi G. Adiponectin in inflammatory and immune-mediated diseases. Cytokine (2013) 64:1–10. doi: 10.1016/j.cyto.2013.06.317
109. Iikuni N, Lam QL, Lu L, Matarese G, La Cava A. Leptin and Inflammation. Curr Immunol Rev (2008) 4:70–9. doi: 10.2174/157339508784325046
110. Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta (2007) 380:24–30. doi: 10.1016/j.cca.2007.01.026
Keywords: TBK1, IKK, inflammation, metabolism, obesity, adipose tissue, overnutrition, undernutrition
Citation: Zhao P and Saltiel AR (2020) Interaction of Adipocyte Metabolic and Immune Functions Through TBK1. Front. Immunol. 11:592949. doi: 10.3389/fimmu.2020.592949
Received: 08 August 2020; Accepted: 30 September 2020;
Published: 20 October 2020.
Edited by:
Willa Ann Hsueh, The Ohio State University, United StatesReviewed by:
Arvand Haschemi, Medical University of Vienna, AustriaCopyright © 2020 Zhao and Saltiel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alan R. Saltiel, YXNhbHRpZWxAaGVhbHRoLnVjc2QuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.