 Bella Guerrouahen1†
Bella Guerrouahen1† Muhammad Elnaggar1†
Muhammad Elnaggar1† Anjud Al-Mohannadi1†Dhanya Kizhakayil1
Anjud Al-Mohannadi1†Dhanya Kizhakayil1 Chiara Bonini2
Chiara Bonini2 Reuben Benjamin3
Reuben Benjamin3 Renier Brentjens4
Renier Brentjens4 Christian J. Buchholz5
Christian J. Buchholz5 Giulia Casorati6
Giulia Casorati6 Soldano Ferrone7Frederick L. Locke8
Soldano Ferrone7Frederick L. Locke8 Francisco Martin9
Francisco Martin9 Axel Schambach10,11
Axel Schambach10,11 Cameron Turtle12
Cameron Turtle12 Paul Veys13
Paul Veys13 Hans J. van der Vliet14,15
Hans J. van der Vliet14,15 Cristina Maccalli1* and The EICCI Faculty Group
Cristina Maccalli1* and The EICCI Faculty Group- 1Research Department, Sidra Medicine, Doha, Qatar
- 2Experimental Hematology Unit, University Vita-Salute San Raffaele and Hospital San Raffaele Scientific Institute, Milan, Italy
- 3Division of Cancer Studies, King’s College Hospital, London, United Kingdom
- 4Cellular Therapeutics, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 5Research Unit for Molecular Biotechnology and Gene Therapy, Paul-Ehrlich-Institut, Langen, Germany
- 6Experimental Immunology Unit, University Vita-Salute San Raffaele and Hospital San Raffaele Scientific Institute, Milan, Italy
- 7Department of Surgery, Massachusetts General Hospital, Harvard Medical School, Boston, MA, United States
- 8Department of Blood and Marrow Transplant and Cellular Immunotherapy, Moffitt Cancer Center, Tampa, FL, United States
- 9Pfizer/University of Granada/Andalusian Regional Government, Genomic Medicine Department, Granada, Spain
- 10Institute of Experimental Hematology, Hannover Medical School, Hannover, Germany
- 11Division of Hematology/Oncology, Boston Children’s Hospital, Harvard Medical School, Boson, MA, United States
- 12Clinical Research Division, Fred Hutchinson Cancer Center, Seattle, WA, United States
- 13Bone Marrow Transplant Unit, Great Ormond Street (GOS) Hospital, and University College London GOS Institute of Child Health, London, United Kingdom
- 14Hans van Der Vliet, Department of Medical Oncology, Amsterdam UMC, VU University and Cancer Center, Amsterdam, Netherlands
- 15Lava Therapeutics, Utrecht, Netherlands
The progress in the isolation and characterization of tumor antigen (TA)-specific T lymphocytes and in the genetic modification of immune cells allowed the clinical development of adoptive cell therapy (ACT). Several clinical studies highlighted the striking clinical activity of T cells engineered to express either Chimeric Antigen (CAR) or T Cell (TCR) Receptors to target molecularly defined antigens expressed on tumor cells. The breakthrough of immunotherapy is represented by the approval of CAR-T cells specific for advanced or refractory CD19+ B cell malignancies by both the Food and Drug Administration (FDA) and the European Medicinal Agency (EMA). Moreover, advances in the manufacturing and gene editing of engineered immune cells contributed to the selection of drug products with desired phenotype, refined specificity and decreased toxicity. An important step toward the optimization of CAR-T cell therapy is the development of “off-the shelf” T cell products that allow to reduce the complexity and the costs of the manufacturing and to render these drugs available for a broad number of cancer patients. The Engineered Immune Cells in Cancer Immunotherapy (EICCI) workshop hosted in Doha, Qatar, renowned experts, from both academia and industry, to present and discuss the progress on both pre-clinical and clinical development of genetically modified immune cells, including advances in the “off-the-shelf” manufacturing. These experts have addressed also organizational needs and hurdles for the clinical grade production and application of these biological drugs.
Introduction
Cancer immunotherapy is aimed at a driving patient’s immune system to attack tumor cells. The great advances achieved in this field during the last two decades, lead to the emerging role of immunotherapy as the “fifth pillar” of cancer treatment, together with surgery, chemotherapy, radiotherapy and targeted therapy (1).
Adoptive cell therapy (ACT) with tumor antigen (TA)-specific T lymphocytes has been clinically developed at an unparalleled pace (2–4). In particular, the approach of the genetic engineering of T or NK cells to target and destroy cancer cells, revealed as powerful and, in some cases, unprecedented in term of clinical success for the treatment of patients with aggressive malignancies, refractory to other therapeutic interventions (5–7).
T cells engineered with chimeric antigen receptors (CARs), that combine the antigen binding region of antibodies and T cell signaling domains responsible for activation (8), represented the breakthrough of cell-based immunotherapy (1, 2, 3). Different CARs have been engineered to target a variety of antigens expressed by either hematologic or solid tumors, that are listed by Sadelain and colleagues (9). The initial clinical application of CAR-T cells was quite disappointing in terms of patients’ responses, due to the inefficient expansion and persistence of CAR-T cells in vivo (10–12). Additional modifications of the structure of CARs, by including co-stimulatory domains allowed the achievement of clinical benefit through the treatment of patients with B cell malignancies overexpressing CD19 (13–18). Strikingly response rates in the range of 57%–82%, with complete response rate of 52-60%, were detected upon the infusion of CD19-CAR-T cells in patients with B cell malignancies refractory to prior treatments (7, 8, 9). These results led to the accelerated approval by both FDA and EMA of two drug products: 1. tisagenlecleucel/Kymriah for the treatment of children and young adult with acute lymphoblastic leukemia (ALL) (13, 14, 19–21), and for the treatment of adults with relapsed/refractory Diffuse Large B cell lymphoma (DLBCL) (22).
Axicabtagene Ciloleucel/Yescarta for the Treatment of Adult Patients With Relapsed/Refractory Non-Hodgkin Lymphoma (NHL), including Table 1 (14, 16, 18).
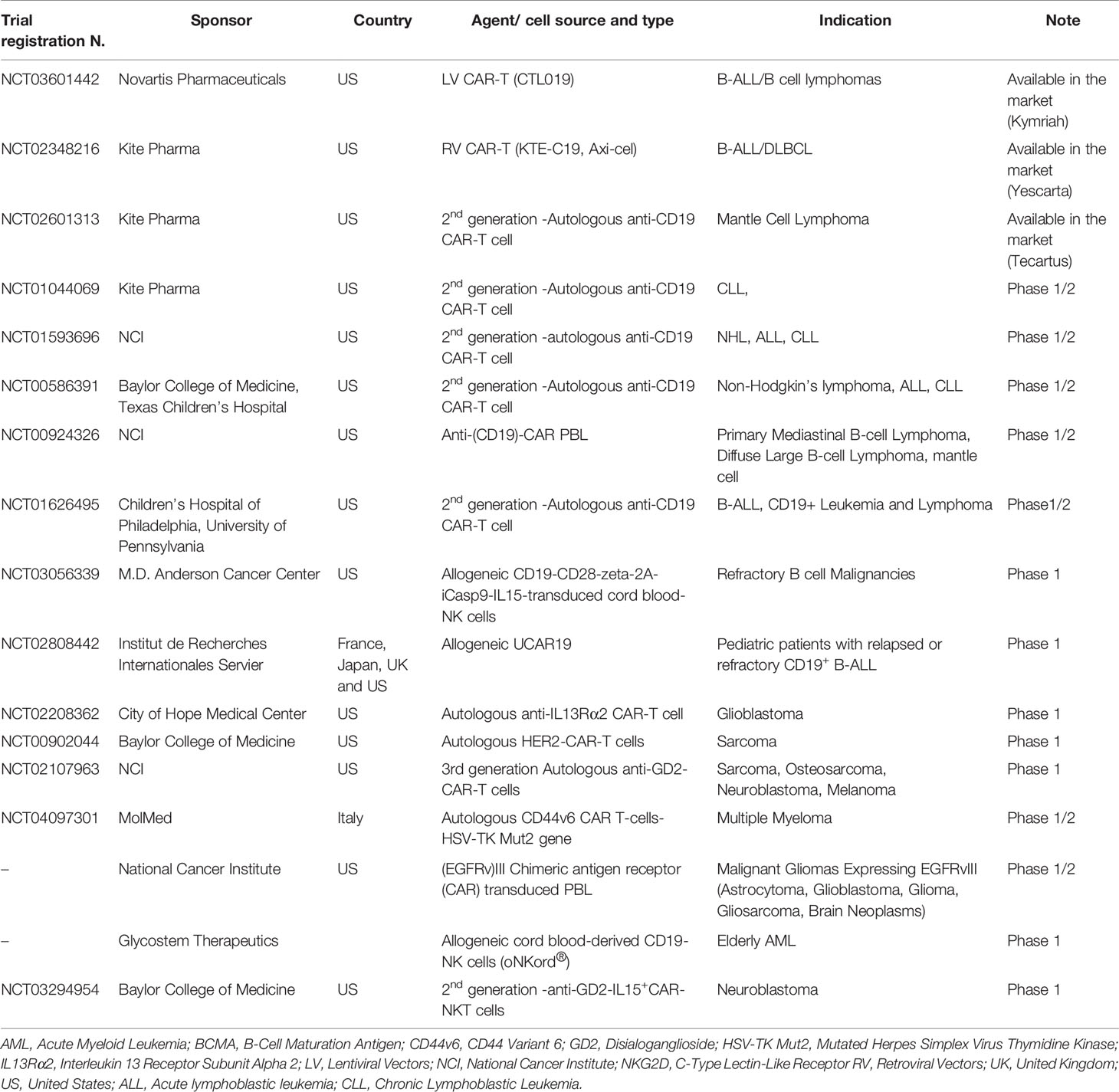
Table 1 Summary of principal clinical studies of CAR-T/NK cells.
More recently, a third product, brexucabtagene autolucel (Tecartus) has been approved for the treatment of relapsed or refractory mantle cell lymphoma. This approval was granted based on the results of the ZUMA-2 (NCT02601313) clinical trial, showing 87% of ORR, with a complete remission (CR) rate of 62% (23).
CAR-T cells represent promising therapeutic options also for solid tumors, although they are still under clinical development and do not yet have proven their clinical efficacy (Table 1) (24). The principal limitations for their clinical activity are: i. paucity of tumor specific antigens and ii. the low efficiency of T cells in penetrating the tumor microenvironment and homing to the tumor site, and iii. their limited functional activity within the tumor (24).
In addition, efforts are ongoing at different leading groups to develop allogeneic CAR-T cell therapies, in order to simplify the manufacturing process, to reduce the costs and rendering these drugs available to larger cohorts of patients (Table 1).
Promising results have been obtained also for the alternative approach of engineering T lymphocytes with TA-specific T Cell Receptor (TCR) for the treatment of solid tumors and multiple myeloma, including patients with advanced metastatic malignancies (Table 2) (2, 4, 25–31). This methodology has been applied for the generation of high avidity tumor-specific T cells. The choice of the targeted antigens is critical for the efficacy of this type of therapy and to prevent off-target reactivity (32, 33). The targeting of overexpressed antigens and neo-antigens, that are not shared with non-embryonic normal tissues, can lead to the cell-mediated killing of tumor cells without severe toxicities due to the targeting of normal tissue(s) (27–29, 34, 35).
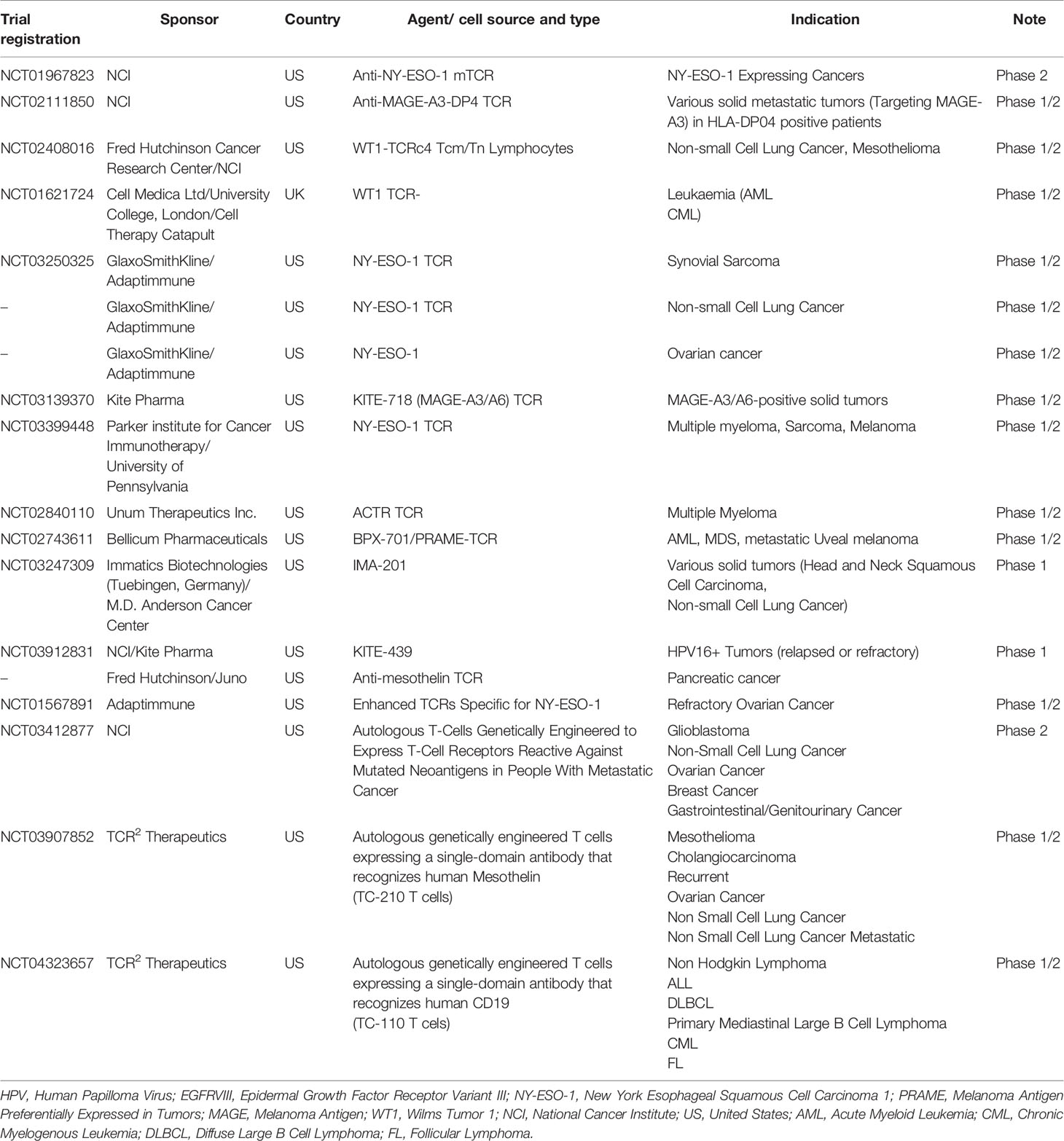
Table 2 Summary of principal clinical studies of TCR-engineered T cells.
Gene editing through the targeted knock out of genes or the insertion of suicide genes represents an innovative strategy that has been applied to tumor-specific engineered immune cells with the aims of either increasing their specificity and in vivo persistence or decreasing the induction of possible toxicities or allogeneic rejection, respectively (36–39). The application of this technique allowed also to generate universal/”off-the-shelf” CAR-T cells using the peripheral blood of healthy volunteers as source of immune cells (40–47). This strategy is currently under clinical development with few clinical trials ongoing in EU and USA (42, 44, 48).
All the topics mentioned above have been addressed in a comprehensive manner in the context of the first international workshop in Doha “Engineered immune cells in cancer immunotherapy: from discovery to off-the-shelf development” (15th–16th February 2019, Doha, Qatar). Renowned speakers from both academia and industry who pioneered the field gathered in Doha bringing high level discussions on scientific and clinical advances and bringing the participants at the forefront of this rapidly evolving topic. A satellite mini-symposium at the initial opening of the workshop, through its educational contents, has provided basic knowledge of cancer immunology, immunotherapy and cell-based therapies to health care practitioners, researchers and students. Poster sessions have also offered the opportunity for active involvement of young researchers and under-graduate students. This report summarizes key data and highlights from each session.
The Clinical Evolution of CAR-T Cell Therapy
During the past few years, CAR-T cells, either CD28/CD3ζ (4, 5) or 4-1BB/CD3ζ CARs (6), targeting CD19+ B cell malignancies have demonstrated safety and clinical activity in the context of multiple Phase I/II clinical trials (49). These products have been used for the treatment of either pediatric and adult patients with relapsed or refractory ALL, showing high CR rate (50–55). Similarly, CD19-CAR-T cells showed impressive clinical activity in relapsed/refractory pediatric-adolescent or adult Non-Hodgkin Lymphoma (NHL) with 40-63% of CR (49, 56–59). These unprecedented results lead to the rapid approval by both FDA and EMA and the commercialization of these Advanced Therapeutic Medicinal Products (ATMPs) (60, 61).
Associate Prof. Cameron Turtle (Fred Hutchinson Cancer Research Center, USA) kicked off the first session of the workshop with a keynote lecture summarizing the aforementioned results and focusing on the factors impacting the response to CD19 targeting CAR-T cell immunotherapy in adults with ALL and NHL in a clinical trial at Fred Hutchinson Cancer Research Center, Seattle, WA. Different factors were found to affect the patients’ clinical responses, including the dose of infusion of CAR–T cells, their cellular expansion and persistence in vivo, the tumor burden, and the conditioning regimen used for lymphodepletion (50, 60). Low levels of circulating CAR-T cells were identified as one of the mechanisms of resistance to CAR-T cell therapies. In some ALL patients, antigen loss was observed in tumor cells following the treatment with CD19-CAR-T cells, as a consequence of either missense mutations or alternative splicing of the CD19 encoding gene (62–64). In ALL patients low levels of lactate dehydrogenase (LDH) and normal platelet counts in the blood prior to lymphodepletion followed by the treatment with cyclophosphamide/fludarabine (Cy/Flu) compared to Cy regimen alone were identified as factors associated with better disease-free survival (DFS) (60). Furthermore, DFS was improved in ALL patients who achieved minimal residual disease (MRD)-negative CR after CAR-T cell therapy and underwent allogeneic hematopoietic stem cell transplantation compared to those who did not undergo transplantation. In NHL patients, high levels of serum LDH before lymphodepletion, and low MCP-1 and IL-7 before or shortly after CAR-T cell infusion were associated with shorter progression-free survival after treatment with CD19-CAR-T cells (65).
CD19-CAR-T cells represented salvage therapy also for patients diagnosed with NHL, including advanced DLBCL, primary mediastinal B-cell lymphoma (PMBCL) and follicular lymphoma (FL). Complete responses (CR), 49%–71%, have been observed in these patients, with a median (overall survival) OS greater than 2 years (16, 18, 22, 66, 67). FDA and EMA approval for axicabtagene ciloleucel in relapsed and refractory DLBCL was granted based on results of the multi-center Phase 2 ZUMA-1 trial, presented in a keynote session by Dr. Frederick L. Locke (Moffitt Cancer Center). Lisocabtagene maraleucel (Liso-cel; JCAR017) represents another CAR-T-cell-based therapy for aggressive NHL patients that is composed by 1:1 ratio of both CD4+ and CD8+ T cells. In the context of the multicenter TRANSCEND-NHL-001 trial, an overall response rate (ORR) of 73% [95% CI, (49, 58, 59, 67–75)] was observed. The CR rate was 53% [95% CI, (47, 48, 50–56, 66, 76–78)] with a median follow-up of 12 months (67). It appears lower frequencies of toxicities may have occurred in the TRANSCEND trial, as well as the JULIET trial that tested tisagenlecleucel in DLBCL; both of these therapeutic products contain the 4-1BBζ costimulatory molecule, in contrast to axi-cel which contains a CD28ζ costimulatory molecule (49, 56, 58, 67). It should be noted that these trials used different toxicity grading systems, so comparisons should be interpreted with caution.
The usage of mathematical and statistical modeling to evaluate the efficacy of CD19-CAR-T cell-based clinical trials, revealed that multiple factors, including the frequency of memory T cells in the circulation, the tumor burden and the inflammatory cytokine profile, can affect the extent and duration of patients’ clinical responses (59). The most frequent toxicities attributed to CAR-T cells are the cytokine release syndrome (CRS) and neurologic events. In the majority of cases they are reversible. CRS can usually be controlled by the administration of tocilizumab (anti-IL-6 mAb) with or without corticosteroids (58, 59, 68). Therapies for neurotoxicity are less well-defined, but usually involve anti-seizure prophylaxis and corticosteroids, with addition of tocilizumab (anti-IL-6 mAb) only if there is concurrent CRS. Patients presenting with severe pro-inflammatory conditions, quantified by serological increase in C-reactive protein and ferritin, before the injection of CAR-T cells appear to be at higher risk for toxicities and might benefit from treatment with immunomodulatory agents before the toxicities become severe (18, 65, 68–70). Safety cohorts of ZUMA-1 tested at early stage and the prophylactic administration of tocilizumab, showed reduced frequency of severe CRS but not neurotoxicity. In this study, the development of neurological toxicity was associated with higher level of CD14+ myeloid cells and CD4+ CAR-T cells in the cerebrospinal fluid (CSF), again implicating a pro-myeloid inflammatory state associated with CAR T toxicity (65, 68–71). These observations highlight that multiple factors could affect patients’ outcome and the development of toxicities; further studies are required to confirm the role of the association of these parameters as biomarkers predicting the risk to develop toxicities upon CAR-T cell treatment.
The Hematology/Oncology Landscape in Qatar
According to registry data from 2015, leukemia represents the 5th most common malignancy in Qatar across genders and nationalities. NHL, including DLBCL, is the 4th most common cancer among males and represents the most frequent lymphoid malignancy in Qatar with about 46% of all lymphoid malignancies. The average OS of DLBCL patients has been reported as 64% between 2013 and 2017. However, 30% to 40% of patients has a high risk of relapse from the first line of chemotherapy treatment, and 10% to develop refractory disease, with poor prognosis and a median survival lower than 1 year (72). Dr. Ruba Y. Taha (NCCCR-HMC, Qatar) summarized the available therapeutic options for these patients, including the treatment with platinum- and cytarabine-based regimens alone or prior to autologous HSCT (PARMA prospective multicenter study) (73). Patients enrolled in this study with early relapses showed similar clinical outcome as incomplete responders (74). However, these patients have superior clinical benefit from the usage of monoclonal antibodies targeting antigens expressed by blasts (CD19, CD20, CD22, and CD52) as compared to chemotherapy or targeted therapy (54). Therefore, the availability in Qatar of CAR-T cell therapy targeting the aforementioned antigens will provide further options to improve the OS of patients with relapsed/refractory DLBCL. A limited number of patients can benefit from this type of therapy only upon admittance to international clinical centers.
Qatar Cancer Society (QCS) is a charity that was founded in 1997 dedicated to implement the research in this field and orchestrate a community partnership platform to make Qatar among the leading countries in cancer prevention and burden control (Dr. Hadi Mohamad Abu Rasheed, QCS, Qatar). In this context, QCS is facilitating the access of Qatari patients to innovative therapies such as CAR-T cells. A testimonial of a patient with a diagnosis of NHL who underwent a successful CD19-CAR-T cell treatment in US, was presented during the conference, corroborating the need of implementing local efforts and research in this field.
The pediatric cancer genome project showed the differential make-up of driver mutations and related aberrant molecular pathways in these patients as compared to adult cancer patients’ profile (75). These evidences indicate the need to deeply understand the disease’s mechanisms of children with cancer in an attempt to allow early detection and intervention. The comprehensive biological characterization of pediatric leukemia ongoing at Sidra Medicine (Dr. Chiara Cugno, Sidra Medicine, Qatar) is aimed at identifying dysregulated signaling pathways to contribute to the development of precision medicine treatments. These investigations might also lead to identifying novel target molecules for CAR-T cells or the optimization of targeting multiple antigens in relationship with the evolution of the disease and possible development of resistance to this therapy (79, 80).
Targeting Tumors With TCR Engineered T Lymphocytes
ACT studies with T cells engineered with TA-specific TCR, provided evidences of their clinical success (Table 2) (2, 28–31, 34, 81, 82). Nevertheless, the improvement of the efficacy, the tumor specificity and strategies to overcome the competition in the synthesis of TCR between exogenous and endogenous alpha and beta chains are subjects of multiple novel investigations. In addition, the usage in most of the cases of epithelial-derived TAAs, that are shared with normal tissues can induce “off-target” toxic effects. The accurate choice of TAAs selectively expressed by tumor cells and/or by the associated blood vessels and microenvironment can overcome the unwanted unspecific targeting and the related toxicities.
These topics were addressed by the presentations, that are summarized in paragraphs 4 and 5, discussing advanced tools to implement the engineering of T and NKT lymphocytes and refining the selection of TAs to redirect the cell-mediated responses.
Prof. Chiara Bonini, (San Raffaele Scientific Institute, Milan, Italy) presented the application of genetic knock-out (KO) and gene substitution approach to introduce TA-specific TCRs in T lymphocytes. This aim was achieved through the transient exposure of T cells to alpha and beta chain specific Zinc Finger Nucleases (ZFNs), followed by the introduction of lentiviral vectors (LV) encoding for a “novel” TCR (83). This study was focused on targeting the leukemia-associated TA Wilms’ tumor 1 (WT1), that is a zinc finger DNA-binding protein acting as transcription factor and playing an important role in cell growth and differentiation of cells (84). The enrichment of T cell memory stem (TSCM) (85, 86) cells that are “younger” cells with persistence and survival properties and delayed ageing and exhaustion (87) occurred for TA-specific T cells. This population of T cells was found to be enriched in the circulation of patients following HSCT and the infusion with T lymphocytes genetically modified to express the thymidine kinase (TK) suicide gene (88, 89). This phenomenon was observed also following long term treatment with engineered T cells (87), indicating that both antigen exposure and the phenotype of the infused cells can affect their in vivo survival and the diversification of their immunological memory (89). ACT can benefit from HSCT platforms followed by the infusion of TCR engineered with gene editing technology in inducing the Graft-versus-tumor (GvT) effect in hematological malignancies (90).
The transfer of lipid specific TCRs into T cells represents another innovative approach for the ACT of leukemia. T cells can recognize lipid antigens presented by MHC class I-related CD1 molecules (CD1a, b, c, d). These T cells are involved in antimicrobial immunity and, in case of reactivity against CD1-presented self-lipids, in autoimmunity and cancer immunosurveillance (91). Dr. Giulia Casorati (San Raffaele Scientific Institute, Milan, Italy) demonstrated that primary acute myelocytic leukemia (AML) leukemia and B-ALL blasts express CD1c and are recognized by a group of CD1c self-reactive T cells specific for methyl-lysophosphatidic acids (mLPAs). mLPAs is a self-lipid antigen highly enriched in malignant cells that might play a role in leukemia growth. Lipid antigens are also less susceptible to mutation and immune mediated selection (92, 93). The anti-tumor activity in vitro and the control of the progression in vivo of leukemia cells by T cells engineered with mLPA-specific TCRs has been assessed, showing that these T cells could represent novel and efficient tools for ACT for leukemia patients either in the prophylactic setting at the time of HSCT or at post-transplant relapse of the disease (93).
A novel high-throughput technology, XPRESIDENT®, has been applied to identify and qualify peptides derived from TAAs (Dr. Ali Mohamed, Immatics, Tubingen, Germany and Houston, TX, USA). This platform, through the combination of ultra-sensitive mass spectrometry (LC-MS/MS) with quantitative transcriptomics, was able to identify the differential expression of HLA-bound peptides in tumor vs. normal tissues. Taking advantage of this platform, several T cell-based therapy programs have entered clinical development. These include the targeting of multiple TAAs with T cells expressing either native or exogeneous TCR. A robust clinical grade manufacturing process has been developed for autologous TCR transduced T cells that implied 7–10 days of manufacturing, (IMA201 and IMA202) or 5–6 days (IMA203), shortening the timeline required for the production in autologous setting. An interesting strategy was based on the engineering of allogeneic γδ T cells, that display inherent anti-tumor activity, the ability to infiltrate tumor tissues and MHC-independent reactivity, with exogenous TA-specific αβ TCR (ACTallo® IMA301). Therefore, these cells acquire αβT cell features in the absence of the risk to develop allogeneic reactions or GvHD due to HLA mismatching (94, 95). Additionally, the development pipeline includes T-cell Engaging Receptors (TCER®s). These bispecific TCR molecules are soluble fusion proteins with two binding domains: (i) a TCR domain that recognizes a tumor-specific peptide presented by HLA class I, and (ii) a T-cell recruiting antibody domain, which allows the recruitment and activation of T cells to attack the tumor.
Optimizing the Delivery In Vivo of CAR-T Cells and Their Redirection to Tumors
The gene transfer platforms for therapeutic application can occur either by isolating ex vivo the cells for modification and the subsequent re-infusion in the patients or in vivo through viral vectors (LV or adenovirus, AAV). The vector delivery can be controlled via particle-receptor interactions. The engineering process for specific targeting involves the mutation of the receptor binding sites and adding the desired target domains in these regions (96). Designed ankyrin repeat proteins (DARPins) can recognize target receptors with high specificities and affinities and when ligated into vectors mediate cellular binding (97). Prof. Christian Buchholz (Paul-Ehrlich-Institut, Federal Institute for Vaccines and Biomedicines, Langen, Germany) discussed the selective delivery of CARs into CD8+ T lymphocytes for the in vivo generation of CAR-T cells. LVs that can specifically transduce human CD8+ cells have been used to deliver the CD19-CAR in vivo into immunodeficient mice transplanted with human peripheral blood mononuclear cells (PBMCs). The vector administration in fully humanized mice transplanted with human CD34+ cells induced TA-specific CAR-T cells depleting the CD19+ B lymphocytes. Signs of CRS, the presence of tissue‐invading CAR-T cells and the complete elimination of the B‐lymphocyte‐rich zones in spleens were detected (98). More recently, efficient eradication of tumors in transplanted mice was demonstrated upon a single vector injection (99). Although further investigations are desirable, these promising results have demonstrated that the in vivo engineering of human CD8+ CAR-T cells could substantially simplify their manufacturing and the development of future cell-based immunotherapies.
Prof. Soldano Ferrone (Massachusetts General Hospital, Harvard Medical School, Boston MA, USA) has dedicated many efforts to elucidate the immunogenic properties of human chondroitin sulfate proteoglycan (CSPG)-4 and its expression on the membrane of different types of cancer cells, including cancer initiating cells (100–104). This molecule is also known as high molecular weight-melanoma associated antigen (HMW-MAA), or neuron-glial antigen 2 (NG2). CSPGs are key bioactive molecules which play a role in cancer cell growth, migration and neo-angiogenesis (105). This antigen can be upregulated on cancer cells incubated under hypoxic conditions and is not or poorly expressed on normal cells with the exception of pericytes in the TME. Prof. Ferrone’s team has previously isolated the mAb scFv-Fc C21 specifically reacting with CSPG4 (100, 101) and he pointed out that some of the commercially available antibodies, including those used to generate the information about CSPG4 presented in the Atlas of Proteins, have the wrong specificity. This has generated some of the misleading information presented in the Atlas of Proteins. CSPG4 represents a target TA for T cell-mediated responses (101, 105–107). In addition, the availability of specific mAbs for this antigen has allowed the generation of CAR-T cells targeting either epithelial cancers, such as triple negative breast cancer, squamous cell carcinoma of head and neck and glioblastoma (106, 108–110). Interestingly, cancer initiating cells isolated from these types of cancers could be efficiently recognized and killed in vitro by CSPG4-CAR-T cells (108, 110). The anti-tumor activity of CAR-T cells targeting CSPG4 was potentiated through the intratumoral injection of effector cells. These pieces of evidence suggest the relevance of CSPG4 as target TA of CAR-T cells.
The development of novel scFvs, through the generation and screening of monoclonal antibodies targeting TAAs, can be applied to redirect engineered T cells for the targeting of colorectal cancer stem-like cells (CRC-SC; Dr. Eleonora Ponterio, Catholic University and Fondazione Policlinico Universitario “A. Gemelli”-I.R.C.C.S. Rome, Italy; Oral poster presentation). This approach represents a novel framework to implement the efficacy of CAR-T cells for solid tumors. Through the injection into mice of primary CRC-SC, antibody-mediated immune responses selectively binding to CRC tissues and not to normal mucosa have been generated. Upon sequencing of the variable chains of these mAbs, the scFv was tagged with a constant fragment (Fc) and used to isolate, through immunoprecipitation, the recognized TAs. The scFvs have been then shuttled into LVs encoding for either 2nd or 3rd generation CARs and used to transduce the immortalized T cell line Jurkat. These cells, upon the activation of the CAR through the co-culture with CRC-SC, secreted IL-2. The isolated mAbs could represent a powerful new tool to direct T cells against CRC [unpublished data and see (111)].
The deep characterization of the mechanisms for differences in cancer phenotypes could contribute to understand patient’s clinical outcome and the responsiveness to immunotherapy. A pan-cancer analysis of the TCGA dataset has revealed that a pre-existing intratumoral T helper (Th-1) immune response can affect the OS of cancer patients with variable outcome depending on the histological origin of tumor. This indicates that cancer-specific pathways modulate the prognostic power of anti-tumor immune response and shape the TME and the type of immune infiltrating cells (Jessica Roelands, PhD Student, Sidra 805 Medicine, Qatar; Oral poster presentation) (112–115). These observations can play a role in the stratification of cancer patients. The clinical relevance of these findings was demonstrated using a dataset of melanoma patients treated with checkpoint inhibitors. A high expression of genes reflecting Th-1 anti-tumor responses in pretreatment samples was associated with improved survival only for samples with high proliferation scores, or with low TGF-β expression (115). The investigation of these parameters in tissues form large cohorts (N=366) of CRC patients represents a valuable tool to either predict the responsiveness of patients to immunotherapy or to identify novel target molecules for immune-based therapies.
Manufacturing Implementation of Engineered T Cells and Pharma/Biotech Perspectives
The manufacturing of CAR-T cells over the time underwent a rapid evolution, from the 1st until the 3rd generation of CAR-T cells, depending on the structure and number of co-stimulatory molecules included in the signaling portion of the receptor. The choice of 2nd generation CAR-T cell for therapeutic application resulted from the practical need to offset toxicity rather than to achieve greater anti-tumor efficacy. Further advances are the “armored” CAR-T cells. These represent genetically modified T cells bearing CARs that comprise in their structures immunomodulating agents such as: i. proinflammatory cytokines; ii. secreted antibodies or their part bearing antigen specificity; iii. costimulatory ligands (Dr. Renier Brentjens, Memorial Sloan Kettering Cancer Center, USA). Armored CD19-CAR-T cells bearing the murine IL-18 (mIL-18 CAR-T) showed in in vivo models enhanced expansion and persistence, anti-tumor activity and prolonged B cell aplasia (up to 150 days after treatment), dependent on autocrine IL-18R signaling (116), even without prior chemotherapy conditioning. Whereas, without preconditioning of mice, the anti-CD19 CAR-T cells failed to prolong the survival of animals in a syngeneic tumor model (117). Through targeted delivery of IL-18 to the tumor, mIL-18 CAR-T cells can modulate the tumor microenvironment, recruit, and activate endogenous anti-tumor immune effector cells, which in turn orchestrate an effective anti-tumor response beyond the CAR-specific targets (116). Programmed cell death-1 (PD-1)/ligand (PD-L1) signaling represents one of the immune checkpoints that can modulate CAR-T cell-mediated responses when they encounter either tumor cells or TME. Armored CAR-T cells endowed with the capacity to secrete anti-PD-L1 scFv showed enhanced in vivo anti-tumor activity, persistence and survival in a PD-L1+ tumor model. PD-1-blocking Ab produced by CAR-T cells can also enhance the activity of tumor-specific “bystander” T cells present in TME. Unlike the usage of systemic checkpoint blockade therapy with monoclonal antibodies, the scFv secreted by CAR-T cells penetrate and remain in the TME improving their anti-tumor activity (118). Moreover, the safety of these armored CAR-T cells is superior than checkpoint blockade systemic therapy (118). The third example of armored CAR-T cells comprise the immunostimulatory CD40 ligand (CD40L), that is a type II transmembrane protein belonging to the TNF receptor family. CD40L expressing CAR-T cells displayed enhanced proliferation and secretion of proinflammatory cytokines, e.g., IFN-γ and GM-CSF (119). These armored effector T cells demonstrated enhanced cytotoxic activity both in vitro and in vivo for both CLL and NHL models (119, 120). All together, these evidences, indicate the promising clinical efficacy of next generation armored CAR-T cells.
The clinical grade manufacturing of CAR-T cells can be challenging due to the complexity of the process. The collaboration between biotech and researchers in academia can result in the development of novel technological platforms to support researchers and physicians from discovery to the clinical development of cell therapy (64, 121). One example is represented by the automated closed system production of engineered T cells that can perform all phases of CAR-T cell generation, from magnetic T-cell enrichment, activation, viral transduction and expansion to downstream harvest for cryopreservation/infusion (CliniMACS Prodigy system). The progress in the generation of this type of system allow standardization of processes facilitating transfer between different manufacturing facilities while the flexibility of the manufacturing platform supports the rapid transfer of innovative cell therapies to the clinic (Dr. Ian Johnston, Miltenyi Biotec B.V. & Co. KG, Germany).
Genetic Engineering of Cells and Gene Editing
Genetic engineering can act on the core functioning of cells, including reprogramming, differentiation, migration, fitness, and proliferation. In addition, gene therapy is aimed at correcting genetic and degenerative diseases. Novel tools have been developed for the precise engineering of immune cells.
Retroviral vectors (RVs), one of the most effective approaches to the modification of genes, display the ability to integrate at various positions in the genome. This implies the risk of mutagenesis in the course of the integration of vectors into the genome. Studies on animal models have revealed evidence of leukemia induction in a process of insertional mutagenesis mediated by retroviral vector (122). The utilization of constructs with improved safety, such as the low-risk recombinant alpharetroviral SIN vectors (aRV) can prevent the genotoxicity caused by insertional mutagenesis (Prof. Axel Schambach, Hannover Medical School, Germany), as SIN-aRV exhibit a neutral integration pattern, lowering the risk of insertional mutagenesis as compared to gammaretroviral and LV (123, 124). Given that aRV vectors do not have the tendency to integrate their cargo into promoter/enhancer regions as well as gene bodies, insertional damages arising from the integration of aRV are lower as compared with other RVs. LVs are considered relatively safe due to their tendency to insert into genes instead to the transcriptional sites that may lead to insertional mutagenesis. The usage of transient retroviral platform, such as the retroviral episome transfer (RET), allows a controllable transgene expression, low or absence of cytotoxicity and the option for cell targeting (123, 124).
Genome editing tools like CRISPR/Cas9, transcription activator-like effector nuclease (TALENs), zinc fingers nuclease (ZFNs), Mega-tal and HEs provide a precise method for the genetic engineering of cells. These platforms introduce double strand breaks (DSBs) in the genome that can be repaired either by non-homologous end-joining (NHEJ) causing disruption of the genes or by homologous recombination which happens at lower rates allowing nucleotide modification of the genome. CRISPR/CAS9 platform requires the co-delivery of a single guide RNA (sgRNA), representing the bottleneck of this strategy. The MS2 bacteriophage RNA packaging mechanism has been applied to the production of non-integrating RVs, GV.MS2-CRISPR/Cas9 all-in-one particles, for efficient gene editing (125, 126). This strategy led to efficient KO of target genes, such as TP53 in human fibroblasts or CXCR4 or CCR7 in Jurkat or T cells, respectively (125–127). These platforms are applicable to the generation of allogeneic CAR-T cells to prevent GvHD and/or to engineer their immune functions.
The usage of ribonucleoproteins (RNPs) as the primary mode of gene editing variants of high-fidelity Cas9 (IDT HiFi SpCas9) enabled better gene-editing by reducing levels off-target INDELs as identified by bioinformatics tools (e.g., COSMID, guide-seq genome-wide profiling of target cells or circle-seq in vitro screen for genome-wide CRISPR/Cas9 nuclease off target) (Prof. Matthew Porteus, Stanford University, San Francisco, CA, USA) (128). This represents an efficient tool for CD19-CAR-T cell to lower the associated toxicities and implement the anti-tumor activity. CD19-CAR-T cells employed with the KO of TCRab to avoid GvHD were assessed in the context of infusions before and following TCRab-depleted haploidentical-HSCT (129). This was achieved through the targeting of the locus of TCR (TRAC) with CRISPR/CAS9 non-integrating AAV6 encoding forCD19-CARs. The engineered T cells showed active anti-tumor activity against CD19+ leukemia xenografts (Nalm6 cells in NSG mice). No off-targets indels was detected in these CAR-T cells through the usage of HiFi Cas9 (129). In order to improve further CAR-T cell efficacy and safety, in 2017, Dr Eyquem et al. (130) used complex genome editing tool to demonstrate that TCR-like expression of the CAR improve CAR-T therapeutic activity. However, genome editing tool to demonstrate that TCR-like expression of the CAR improves CAR-T therapeutic activity. In this direction, Prof. Francisco Martin (Pfizer-University of Granada-Junta de Andalucía Centre for Genomics and Oncological Research, GENYO, Spain) presented an interesting approach to facilitate clinic translation of TCR-like CAR-T cells. Dr. Martin´s team constructed LVs that closely followed the expression profile of the TCR upon CD3 stimulation and generated CAR-T cells using the LVs (Patent PCT/EP2019/081346, “Polynucleotide for safer and more effective immunotherapy” and unpublished data). These physiological CAR-T cells showed strong resistance to exhaustion upon repeated stimulation and showed potent in vivo anti-tumor activity. The author proposes the use of these physiological LVs (similar to the one already approved by FDA) to translate TCR-like CAR-T cells into clinic. Another important aspect to improve the potency and safety of CAR-T cells stands on the design of new tools that allow the clinicians to be able to externally control their activity. Dr. Martin presented their latest development on their transactivator-independent doxycycline-regulated LVs (Lent-On-Plus). They showed the potency of this system to induce any transgene in T cells both in vitro and in vivo with very low doses of doxycycline. As proof of concept, he also presented the ability to generate inducible CAR-T cells that selectively kill CD19+ cells only in the presence of doxycycline. However, the author envision that this approach will be more applicable for future development of inducible TRUCKs (iTRCKs), expressing any desired cytokine (factor) upon the addition of doxycycline.
Allogeneic/”Off-the-Shelf” Therapy With CAR-T Cells
The generation of autologous CAR-T cells for therapeutic treatment implies many challenges related to i. the isolation of circulating lymphocytes from heavy pre-treated patients who might have lymphopenia; ii. the manufacturing of the cells; iii. the logistics of collection of the starting material, preparation and delivery for infusion of the medicinal product into patients; iv the inter-patient variability in the yield and quality of the cell product. The major advantage of generating universal CAR-T cells is their “off-the-shelf availability, without delay in the treatment and suitability for multiple infusions of engineered T cells. Moreover, the manufacturing of universal CAR-T cells allows the implementation of the standardization of batches of cells and to reduce the costs of production. The strategy to generate anti-tumor CAR-T cells starting from peripheral blood lymphocytes isolated from healthy donors, has also the potential to overcome the impairment of effector functions of patient-derived CAR-T cells.
Universal CD19-CAR-T cells (UCART19) represent the first “off-the-shelf” CAR-T cell product targeting CD19 expressing malignancies (Dr. Reuben Benjamin, King’s College Hospital, UK). In this product, anti-CD19 scFv-41BB-CD3ζ CAR is expressed in T cells, with KO of TRAC and CD52 genes to prevent GvHD in HLA mismatched patients. The TRAC and CD52 gene KO has been performed through the usage of TALEN (41). In addition, RQR8 epitope, that is a mimotope of CD20 has been introduced into engineered T cells as a safety switch (131). The safety and tolerability of UCART19 in pediatric and adult patients with diagnosis of ALL have been tested in the context of the PALL/CALM study following lymphodepleting regimens and to evaluate the maximum tolerated dose (MTD). The study revealed that UCART19 has a safety profile, with no acute GvHD and severe toxicities, leading to 67% of CR (40, 43, 132).
Different strategies of gene editing are in place for allogeneic CAR-T cells for the knock-out of genes involved in either GvHD, such as TCRαβ - TRAC, TCRBC1, TCRBC2, rejection molecules (e.g., CD52, β2 microglobulin), or molecules associated with the exhaustion of T cells (e.g., PD-1, CTLA-4) (42, 44). CAR-T cell approaches in infant ALL patients have sometimes encountered difficulties in generating autologous CAR-T cells and, moreover, are associated with the risk of developing CD19-tumor cell escape (Prof. Paul Veys, Great Ormond Street Hospital For Children & UCL GOS Institute of Child Health, UK). UCART19 represented a bridge between lymphodepletion/CAR-T cell therapy and allogeneic HSCT. This strategy allowed the eradication of CD19- malignant cells following CAR-T cell therapy during the allogeneic HSCT, followed by reconstitution of donor immune cells and elimination of residual UCART cells. This procedure was followed by a 30-day transient GCSF-dependent recovery of neutrophils and protracted multilineage cytopenia until the second allo-SCT after 12 weeks. The TALEN trial achieved 60% clinical responses in pediatric ALL patients. The effectiveness of TALEN versus CRISPR/Cas9 was compared, indicating that the risk of GvHD might be reduced with CRISPR/Cas9, as CAR insertion and editing of TCR were coupled, leading to reduced potential contamination of the final product with TCR positive cells. The generation of CAR-T cells from umbilical cord blood (UCB) coupled with gene editing has been explored in pre-clinical models as alternative method to generate allogeneic CAR-T cells with increased early differentiation features, including increased proliferative capacity (133). The unmet need is to define the optimal lymphodepletion regimen(s) and whether allo-SCT is required post UCART infusion to extend the clinical benefit of this cell-based therapy.
The usage of UCB as starting material to generate CAR-T cells represented the object of one study ongoing at Sidra Medicine (Dr. Cristina Maccalli, Sidra Medicine, Qatar). UCB offers the unique capacity of broad HLA mismatch between donor and host. The frequency of T lymphocytes in cord blood is lower as compared to peripheral blood representing a limiting factor for the generation of CAR-T cells. The CD19-CD28ζ and CD19- 4-1BBζ CAR-T cells have been isolated and phenotypically characterized utilizing an in-house designed multiparametric IF (including 28 markers) panels. The overall phenotype at different time points (Day 0, + 7, +9, +14) of in vitro culture of UCB vs. peripheral blood-CAR-T cells varied in terms of the percentage of CD4+ and CD8+ stem/central memory T cells. In addition, immunomodulating molecules such as PD-1, TIM3, and LAG3 were differentially expressed in CAR-T cells with different origins. The integration of different platforms, such a flow cytometry, EliSpot, and FluoroSpot based cytokine release and transcriptomic profiling (134) allowed the development of a proof of principle study aimed at identifying the UCB-derived CAR-T cell population endowed with superior anti-tumor activity. Further investigations are warranted to validate these results (unpublished data).
CAR-NK cells represent a promising strategy for allogeneic and universal cell-therapy (Dr. Tomas Bos, Glycostem Therapeutics, The Netherlands). NK cells represent the 5%–10% of circulating immune cells and are members of innate immune control with low risk, due to their independence on the recognition of MHC/peptide complexes, of allogeneic rejections or GvHD upon their infusion in cancer patients. The generation of clinical grade NK cells has been optimized in the presence of a liquid cell culture containing differentiation factors and an artificial niche composed of glycosaminoglycans. The NK cell product, denominated oNKord®, showed strong cytotoxicity against CRC, regardless of the mutational status of EGFR and RAS (135), and leukemia both in vitro and in mouse models (136). Interestingly, this type of treatment in a phase I clinical trial for AML patients following conditioning with Flu/Cy, showed safety, no toxicities or sign of GvHD and clinical efficacy with improvement of survival (34 months) (137).
The repertoire of available gene editing tools for CAR-T cells has been discussed by Dr. Karim Benabdellah (GENYO, Spain; Oral Abstract Presentation), highlighting that the insertional deficient LV episomes (IDLV) combined with CRISPR/CAS-9 can implement the effciency of CD19-CAR-T cells for the KO of TCR to prevent in vivo allogeneic rejection and GvHD. These CAR-T cells can maintain both in vitro and in vivo the reactivity against tumor cells.
The Usage Of maB N Cancer Therapy As A Complementary Or Integrative Approach For Engineered Immune Cells
Several mAbs have been registered as standard of care for different types of tumors. Some of them, through the binding to the Fcγ receptor (FcγR) on effector cells, such as NK and γδ T cells, can activate the antibody-dependent cell-mediated cytotoxicity (ADCC) that can lead to the killing of malignant cells (Prof. Hans van der Vliet, Amsterdam University Medical Center, Cancer Center Amsterdam, The Netherlands). Bispecific Ab, bearing two epitope binding regions, have been designed to bind simultaneously to two target molecules and can be used to target immune effector cells to tumor cells. Trifunctional hybrid antibodies (Triomabs) are formed by combining two halves of distinct mAbs, for example of rat and mouse mAb origin, which, as a result of species preferential heavy-light chain pairing, reduces random pairing of heavy and light chains simplifying manufacturing. In addition, to facilitate the interaction of immune cells with target cells, Triomabs (e.g., catumaxomab, ertumaxomab) can also mediate ADCC through the FcγR (138–141). Notably, other bispecific antibody formats utilize a linking of single chain Fv (scFv) fragments with different antigen specificity, for which several formats can be used. An example of a scFv based bispecific antibody in a tandem orientation (TaFv) is blinatumomab, an anti-CD3 and anti-CD19 bispecific T cell engager (BiTE) used for the treatment of, e.g., relapsed or refractory B-ALL, that showed an improvement of complete remission rate and of OS as compared with chemotherapy (53, 142). Multiple alternative bispecific antibody platforms (e.g., bispecific diabodies, Fc-containing bispecific antibodies, immunocytokines, prodrug bispecifics, and MHC class I targeting bispecifics) are under development in an effort to further improve the clinical efficacy of such approaches and to extend their application towards other disease indications (143–145). The generation of novel antigen-specific scFv could also complement the function of CARs, either by enhancing the binding to target molecules or expanding the tools to redirect CAR-T cells to tumor sites. In addition, the usage of mAbs, including the bispecific, either in combination with CAR-T cells or encoded simultaneously with CARs by LV vectors, can represent an interesting approach to enhance the homing of CAR-T cells to the tumor site, their penetration into the TME and the anti-tumor activity (e.g., armored CAR-T cells discussed in paragraph 6).
Conclusions
The development of engineered immune cells, T lymphocytes and NK cells, represents a breakthrough of cancer immunotherapy for both blood and solid tumors (see Tables 1 and 2). For the first time, one of these innovative therapeutic strategies (CAR-T cells) has been approved as standard of care for some hematological malignancies. Nevertheless, the optimization of these interventions, in terms of identification of novel TAs, the improvement of the survival and the limitation of differentiation, aging and exhaustion of T cells, as well as of associated toxicities upon in vivo infusion and their capacity to penetrate TME are under investigation. Several platforms and approaches have been developed to mitigate the aforementioned limitations of anti-tumor cell-based therapies, contributing to the rapid evolution of the field. Furthermore, novel Ab-based tools designed on tumor cell phenotype and cancer immune setpoints are warranted to design more effective personalized Ab-based weapons for cancer therapy. The development of these approaches has required long path for R&D, however, the gained expertise could facilitate the implementation of the molecular and genomic tools to generate anti-tumor cell therapy with superior clinical efficacy. Nevertheless, the discussion occurring in the context of the conference highlighted the need of specialized manufacturing facilities and clinical centers, including multidisciplinary personnel, for the production, delivery and infusion into patients, as well as their clinical monitoring and follow-up. The development of inter- and intra-regional networks is of high relevance to facilitate the establishment of these cell-therapy centers and to expand the accessibilities to these therapeutic interventions to a broad number of cancer patients. The costs associated with the development of this type of centers and with the clinical grade manufacturing of these cellular medicinal products should also be taken into account. The access of patients to these therapies and their attractiveness for clinical application might increase whether a point-of-care manufacturing model would be designed and established. The role of pharma and biotech has been shown to be critical and to facilitate the clinical grade development of these approaches. The interaction and cooperation between academia and industry were emphasized during the sessions’ discussions as in need of being potentiated and initiated even at early phase of R&D. Further investigations are also required to simplify the manufacturing of engineered immune cells and potentiate the development and safety of allogeneic products. The exploration of the combination therapies (such as with immunomodulating agents and/or standard therapies) could represent a strategy to overcome the development of resistance to cell-based therapies and to target a broad variety of tumors. A consensus regarding the need of continuous exchange of information among investigators and clinicians and of the development of international networks and collaborations resulted from the participants to the workshop.
The EICCI Faculty Group
Karim Benabdellah, Pfizer/University of Granada/Andalusian Regional Government, Genomic Medicine Department, Granada, Spain; Tomas Bos, Glycostem Therapeutics, AB Oss, The Netherlands; Chiara Cugno, Research Department, Sidra Medicine, Doha, Qatar; Ian Johnston, R&D, Miltenyi Biotec, Bergisch Gladbach, Germany; Ali Mohamed, Immatics Biotechnologies, Tubingen, Germany; Eleonora Ponterio, Catholic University and Fondazione Policlinico Universitario "A. Gemelli"-I.R.C.C.S. Rome, Italy; Hadi Mohamad Abu Rasheed, Qatar Cancer Society, Doha, Qatar; Jessica Roelands, Research Department, Sidra Medicine, Doha, Qatar; Ruba Y. Taha, Hematology-BMT Department, National Cancer Center for Care and Research (NCCCR), Hamad Medical Corporation, Doha, Qatar.
Author Contributions
BG, ME, AA-M, and DK prepared the draft of the manuscript and the final editing. CM contributed to the preparation, revision, and editing of the manuscript. RBe, CBo, RBr, CBu, GC, SF, IJ, AM, FL, FM, AS, CT, PV, and HV provided critical inputs and revised the manuscript. KBL, TB, EP, JR, HMAR, and RYT contributed to the editing of the text. All authors contributed to the article and approved the submitted version.
Funding
The EICCI workshop was funded by the Qatar National Research Fund (QNRF)- Conference Workshop Sponsorship Program grant CWSP15- W-0911-18001 (Doha, Qatar). CBo received funding from AIRC (Ig 18458) and AIRC 5 per Mille, Rif. 22737, Italian Ministry of Research and University (PRIN 2017WC8499) and Italian Ministry of Health (Research project on CAR T cells for hematological malignancies and solid tumors).
Conflict of Interest
CBo received a research contract from Intellia Therapeutics and participated to the advisory boards of Molmed, Intellia Therapeutics, TxCell, Novartis, GSK, Allogene, Kiadis. CBu is the inventor of patents in the field of adoptive T cell therapy. RB is a recipient of research funding from Servier. IJ is employed at Miltenyi Biotec. FL has scientific advisory role for Kite, a Gilead Company, Novartis, Celgene/Bristol-Myers Squibb, GammaDelta Therapeutics, Wugen, Amgen, Calibr, Amgen, and Allogene; is a consultant with grant options for Cellular Biomedicine Group, Inc.; and has research support from Kite, a Gilead Company. AM is employed at Immatics. CT received research funding from Juno Therapeutics/BMS, Nektar Therapeutics, Minerva, AstraZeneca, and TCR2 Therapeutics. He is a member of Scientific Advisory Boards of Precision Biosciences, Eureka Therapeutics, Caribou Biosciences, T-CURX, Myeloid Therapeutics, ArsenalBio, and Century Therapeutics, and ad hoc advisory boards (last 12 months) of Nektar Therapeutics, Allogene, PACT Pharma, Astra Zeneca, and Amgen. He has stock/options of Precision Biosciences, Eureka Therapeutics, Caribou Biosciences, Myeloid Therapeutics, and ArsenalBio. CT is the inventor of a patent licensed to Juno Therapeutics. PV is a recipient of a grant from SERVIER to investigate Universal chimeric antigen receptor T cells for ALL (UCAR19-PALL). HV is employed as a chief scientific officer (CSO) of Lava Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the contributions of the EICCI scientific committee, Drs. Christof von Kalle, Sara Deola, Catherine Cole, (Sidra Medicine), Drs. Alexander Knuth, Gianfranco Pittari, Javid Gaziev; Said Dermime, (NCCCR, HMC); the organizing committee, Ms. Nelly EL Mistekawy, Maricris Salud, Nevin Amin, Hamda Zain Al Abdeen (Sidra Medicine); Prof. Matthew Porteus (Stanford University, San Francisco, CA, USA) as faculty member of the workshop and all the chairs of the workshop’s sessions. This workshop was organized with the partnership of Hamad Medical Corporation and Qatar Cancer Society. We would like to recognize Miltenyi Biotech (Germany), Beckman Coulter/Medtech (Germany/Qatar), Therumo BCT (Germany) and Ideal Medical Solution (Qatar) for cash or in kind supports to the conference.
Abbreviations
AAV, Adeno-Associated Virus; ACT, Adoptive Cell Therapy; ADCC, Antibody-Dependent Cell-Mediated Cytotoxicity; BMT, Bone Marrow Transplant; CAR, Chimeric Antigen Receptor; CCR7, C-C Chemokine Receptor Type 7; CD1 a, b, c, d, family of glycoproteins expressed on the surface of various human antigen-presenting cells and related to HLA class I molecules; CD19, IgSF surface glycoprotein of 95 kDa expressed on B cells; CD20, B-lymphocyte antigen CD20; CD22, B-lymphocyte cell adhesion molecule; Sialic acid-binding Ig-like lectin 2 (SIGLEC-2); CD28, Cluster of Differentiation 28, T-cell-specific surface glycoprotein; CD40/CD40L, Cluster of differentiation 40/Ligand of CD40; CD52, Campath-1 antigen; CD54, Cluster of Differentiation 54 or Intercellular Adhesion Molecule 1 (ICAM-1); CD58, Lymphocyte function-associated antigen 3 (LFA-3) CD70, Cluster of Differentiation 70; CXCR4, C-X-C Chemokine Receptor Type 4; CD80, Cluster of differentiation 80 (also B7-1); CD86, Cluster of differentiation 86 (also B7-2); Cy, Cyclophosphamide; COSMID, Database of Genomic Structural Variation; CRC, Colorectal Cancer; CRISPR/Cas9, (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas9); CTLA-4, Cytotoxic T lymphocyte Antigen 4; EGFR, Epidermal Growth Factor, Fas, Death Receptor that regulate apoptosis; FcγR, Fc Gamma Receptor; Flu, Fludarabine; GMP, Good Manufacturing Practice; GvHD, Graft versus Host Disease; HLA, Human Leukocyte Antigen; HSCT, Allogenic Hematopoietic Stem Cell Transplant; IL-6, Interleukin 6; IL-7, Interleukin 7; IL-18, Interleukin 18; KO, Knock out; LAG-3, Lymphocyte Activation Gene-3; LDH, Lactate Dehydrogenase; LV, Lentiviral vector; MCP-1, Monocyte Chemoattractant Protein-1; MEGATAL, Meganucleases that have been fused with a Transcription Activator-Like (TA) containing Repeat Variable Residues (RVD); NK, Natural Killer; OS, Overall Survival; PD-/PD-L1, Programmed Cell Death-1/Ligand; RAS, Rat Sarcoma; RV, Retroviral vectors; scFV, Single-Chain Variable Fragment; TAA, Tumor-Associated Antigen; TCGA, The Cancer Genome Atlas; Tp53, Tumor Protein 53; TSCM, T Cell Memory Stem; TCR, T Cell Receptor; TCRαβ, T Cell Receptor alpha beta; TME, Tumor Microenvironment; TRAC, T-cell receptor α constant locus; TRBC1, T Cell Receptor Beta Constant 1; TRBC2, T Cell Receptor Beta Constant 2; TIM3, T cell immunoglobulin and mucin domain- containing protein 3; UCAR, Universal Chimeric Antigen Receptor; UCB, Umbilical Cord Blood; 4-1BB, activation-induced costimulatory molecule (CD137).
References
1. Tang J, Shalabi A, Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol (2018) 29(1):84–91. doi: 10.1093/annonc/mdx755
2. Yang JC, Rosenberg SA. Adoptive T-Cell Therapy for Cancer. Adv Immunol (2016) 130:279–94. doi: 10.1016/bs.ai.2015.12.006
3. Grupp SA, June CH. Adoptive cellular therapy. Curr Top Microbiol Immunol (2011) 344:149–72. doi: 10.1007/82_2010_94
4. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi: 10.1126/science.aaa4967
5. Brenner MK. Adoptive Cell Therapy: ACT-Up or ACT-Out? Mol Ther (2019) 27(4):693–4. doi: 10.1016/j.ymthe.2019.02.017
6. Rezvani K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant (2019) 54(Suppl 2):785–8. doi: 10.1038/s41409-019-0601-6
7. Sharma P, Kranz DM. T Cell Receptors for Gene Transfer in Adoptive T Cell Therapy. Crit Rev Immunol (2019) 39(2):105–22. doi: 10.1615/CritRevImmunol.2019030788
8. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA (1993) 90(2):720–4. doi: 10.1073/pnas.90.2.720
9. Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol (2009) 21(2):215–23. doi: 10.1016/j.coi.2009.02.009
10. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
11. Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther (2007) 15(4):825–33. doi: 10.1038/sj.mt.6300104
12. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood (2008) 112(6):2261–71. doi: 10.1182/blood-2007-12-128843
13. Boyiadzis MM, Dhodapkar MV, Brentjens RJ, Kochenderfer JN, Neelapu SS, Maus MV, et al. Mackall CL et al: Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J Immunother Cancer (2018) 6(1):137. doi: 10.1186/s40425-018-0460-5
14. Hay KA, Turtle CJ. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs (2017) 77(3):237–45. doi: 10.1007/s40265-017-0690-8
15. Kohn DB, Dotti G, Brentjens R, Savoldo B, Jensen M, Cooper LJ, et al. CARs on track in the clinic. Mol Ther (2011) 19(3):432–8. doi: 10.1038/mt.2011.1
16. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
17. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7(303):303ra139. doi: 10.1126/scitranslmed.aac5415
18. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther (2017) 25(1):285–95. doi: 10.1016/j.ymthe.2016.10.020
19. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Wright JF et al: Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
20. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359(6382):1361–5. doi: 10.1126/science.aar6711
21. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med (2011) 3(95):95ra73. doi: 10.1126/scitranslmed.3002842
22. Schuster SJ, Investigators J. Tisagenlecleucel in Diffuse Large B-Cell Lymphoma. Reply. N Engl J Med (2019) 380(16):1586. doi: 10.1056/NEJMc1901464
23. Beyar-Katz O, Gill S. Advances in chimeric antigen receptor T cells. Curr Opin Hematol (2020) 27(6):368–77. doi: 10.1097/MOH.0000000000000614
24. Kosti P, Maher J, Arnold JN. Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front Immunol (2018) 9:1104. doi: 10.3389/fimmu.2018.01104
25. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23(10):1043–53. doi: 10.1089/hum.2012.041
26. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res (2015) 21(5):1019–27. doi: 10.1158/1078-0432.CCR-14-2708
27. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. doi: 10.1038/nm.3161
28. Tran E, Rosenberg SA. T-cell therapy against cancer mutations. Oncotarget (2014) 5(13):4579–80. doi: 10.18632/oncotarget.2234
29. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med (2018) 24(6):724–30. doi: 10.1038/s41591-018-0040-8
30. Anderson KG, Voillet V, Bates BM, Chiu EY, Burnett MG, Garcia NM, et al. Engineered Adoptive T-cell Therapy Prolongs Survival in a Preclinical Model of Advanced-Stage Ovarian Cancer. Cancer Immunol Res (2019) 7(9):1412–25. doi: 10.1158/2326-6066.CIR-19-0258
31. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med (2019) 25(7):1064–72. doi: 10.1038/s41591-019-0472-9
32. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol (2014) 11(9):509–24. doi: 10.1038/nrclinonc.2014.111
33. Parmiani G, Russo V, Maccalli C, Parolini D, Rizzo N, Maio M. Peptide-based vaccines for cancer therapy. Hum Vaccin Immunother (2014) 10(11):3175–8. doi: 10.4161/hv.29418
34. Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev (2014) 257(1):56–71. doi: 10.1111/imr.12132
35. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36(2):133–51. doi: 10.1097/CJI.0b013e3182829903
36. Liu X, Zhao Y. CRISPR/Cas9 genome editing: Fueling the revolution in cancer immunotherapy. Curr Res Transl Med (2018) 66(2):39–42. doi: 10.1016/j.retram.2018.04.003
37. Ren J, Zhao Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein Cell (2017) 8(9):634–43. doi: 10.1007/s13238-017-0410-x
38. Singh N, Shi J, June CH, Ruella M. Genome-Editing Technologies in Adoptive T Cell Immunotherapy for Cancer. Curr Hematol Malig Rep (2017) 12(6):522–9. doi: 10.1007/s11899-017-0417-7
39. Zhang Y, Mu W, Wang H. Gene editing in T cell therapy. J Genet Genomics (2017) 44(9):415–22. doi: 10.1016/j.jgg.2017.09.002
40. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol (2018) 11(1):132. doi: 10.1186/s13045-018-0677-2
41. Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR-T Cells: More than Ease of Access? Cells (2018) 7(10):19–23. doi: 10.3390/cells7100155
42. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery (2020) 19. doi: 10.1038/s41573-019-0051-2
43. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med (2017) 9(374):1–8. doi: 10.1126/scitranslmed.aaj2013
44. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res (2017) 23(9):2255–66. doi: 10.1158/1078-0432.CCR-16-1300
45. Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood (2012) 119(24):5697–705. doi: 10.1182/blood-2012-01-405365
46. Osborn MJ, Webber BR, Knipping F, Lonetree CL, Tennis N, DeFeo AP, et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol Ther (2016) 24(3):570–81. doi: 10.1038/mt.2015.197
47. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Pham CD et al: Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol Ther (2017) 25(4):949–61. doi: 10.1016/j.ymthe.2017.02.005
48. Qasim W. Allogeneic CAR T cell therapies for leukemia. Am J Hematol (2019) 94(S1):S50–4. doi: 10.1002/ajh.25399
49. Chavez JC, Locke FL. CAR T cell therapy for B-cell lymphomas. Best Pract Res Clin Haematol (2018) 31(2):135–46. doi: 10.1016/j.beha.2018.04.001
50. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126(6):2123–38. doi: 10.1172/JCI85309
51. Turtle CJ, Maloney DG. Clinical trials of CD19-targeted CAR-modified T cell therapy; a complex and varied landscape. Expert Rev Hematol (2016) 9(8):719–21. doi: 10.1080/17474086.2016.1203251
52. Turtle CJ, Riddell SR, Maloney DG. CD19-Targeted chimeric antigen receptor-modified T-cell immunotherapy for B-cell malignancies. Clin Pharmacol Ther (2016) 100(3):252–8. doi: 10.1002/cpt.392
53. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
54. Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med (2017) 376(9):836–47. doi: 10.1056/NEJMoa1609783
55. Kansagra AJ, Frey NV, Bar M, Laetsch TW, Carpenter PA, Savani BN, et al. Gastineau DA et al: Clinical utilization of Chimeric Antigen Receptor T-cells (CAR-T) in B-cell acute lymphoblastic leukemia (ALL)-an expert opinion from the European Society for Blood and Marrow Transplantation (EBMT) and the American Society for Blood and Marrow Transplantation (ASBMT). Bone Marrow Transplant (2019) 54(11):1868–80. doi: 10.1038/s41409-019-0451-2
56. Neelapu SS, Locke FL, Go WY. CAR T-Cell Therapy in Large B-Cell Lymphoma. N Engl J Med (2018) 378(11):1065. doi: 10.1056/NEJMc1800913
57. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med (2016) 8(355):355ra116. doi: 10.1126/scitranslmed.aaf8621
58. Locke FL, Haroun F. Advances in aggressive lymphoma from the 2017 American Society of Hematology Annual Meeting and Exposition: commentary. Clin Adv Hematol Oncol (2018) 16 Suppl 5(2):19–23.
59. Altrock PM, Kimmel G, Locke FL. Abstract 1791: Evolutionary dynamics of non-Hodgkin’s lymphoma CAR T cell therapy. Cancer Res (2018) 78(13 Supplement):1791–1. doi: 10.1158/1538-7445.AM2018-1791
60. Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood (2019) 133(15):1652–63. doi: 10.1182/blood-2018-11-883710
61. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discovery (2015) 5(12):1282–95. doi: 10.1158/2159-8290.CD-15-1020
62. Fischer J, Paret C, El Malki K, Alt F, Wingerter A, Neu MA, et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J Immunother (2017) 40(5):187–95. doi: 10.1097/CJI.0000000000000169
63. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med (2018) 24(10):1499–503. doi: 10.1038/s41591-018-0201-9
64. Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood (2019) 133(17):1876–87. doi: 10.1182/blood-2018-11-887067
65. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
66. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
67. Abramson JS, Gordon LI, Palomba ML, Lunning MA, Arnason JE, Forero-Torres A, et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J Clin Oncol (2018) 36(15_suppl):7505–5. doi: 10.1200/JCO.2018.36.15_suppl.7505
68. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
69. Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv (2020) 4(19):4898–911. doi: 10.1182/bloodadvances.2020002394
70. Oluwole OO, Jansen JP, Lin VW, Chan K, Keeping S, Navale L, et al. Comparing Efficacy, Safety, and Preinfusion Period of Axicabtagene Ciloleucel versus Tisagenlecleucel in Relapsed/Refractory Large B Cell Lymphoma. Biol Blood Marrow Transplant (2020) 26(9):1581–8. doi: 10.1016/j.bbmt.2020.06.008
71. Nastoupil LJ, Jain MD, Spiegel JY, Ghobadi A, Lin Y, Dahiya S, et al. Axicabtagene Ciloleucel (Axi-cel) CD19 Chimeric Antigen Receptor (CAR) T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma: Real World Experience. Blood (2018) 132(Supplement 1):91–1. doi: 10.1182/blood-2018-99-114152
72. Tilly H, Gomes da Silva M, Vitolo U, Jack A, Meignan M, Lopez-Guillermo A, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol (2015) 26 Suppl 5:v116–125. doi: 10.1093/annonc/mdv304
73. Philip T, Chauvin F, Bron D, Guglielmi C, Hagenbeek A, Coiffier B, et al. PARMA international protocol: pilot study on 50 patients and preliminary analysis of the ongoing randomized study (62 patients). Ann Oncol (1991) 2 Suppl 1:57–64. doi: 10.1093/annonc/2.suppl_1.57
74. Philip T, Guglielmi C, Hagenbeek A, Somers R, Van der Lelie H, Bron D, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med (1995) 333(23):1540–5. doi: 10.1056/NEJM199512073332305
75. Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, et al. The Pediatric Cancer Genome Project. Nat Genet (2012) 44(6):619–22. doi: 10.1038/ng.2287
76. Neelapu SS. An interim analysis of the ZUMA-1 study of KTE-C19 in refractory, aggressive non-Hodgkin lymphoma. Clin Adv Hematol Oncol (2017) 15(2):117–20.
77. Neelapu SS, Jacobson CA, Oluwole OO, Munoz J, Deol A, Miklos DB, et al. Outcomes of older patients in ZUMA-1, a pivotal study of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood (2020) 135(23):2106–9. doi: 10.1182/blood.2019004162
78. Abramson JS, McGree B, Noyes S, Plummer S, Wong C, Chen YB, et al. Anti-CD19 CAR T Cells in CNS Diffuse Large-B-Cell Lymphoma. N Engl J Med (2017) 377(8):783–4. doi: 10.1056/NEJMc1704610
79. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol (2019) 16(6):372–85. doi: 10.1038/s41571-019-0184-6
80. Shah NN, Maatman T, Hari P, Johnson B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front Oncol (2019) 9:146. doi: 10.3389/fonc.2019.00146
81. Rice J, Dossett ML, Ohlen C, Buchan SL, Kendall TJ, Dunn SN, et al. DNA fusion gene vaccination mobilizes effective anti-leukemic cytotoxic T lymphocytes from a tolerized repertoire. Eur J Immunol (2008) 38(8):2118–30. doi: 10.1002/eji.200838213
82. Schmitt TM, Aggen DH, Ishida-Tsubota K, Ochsenreither S, Kranz DM, Greenberg PD. Generation of higher affinity T cell receptors by antigen-driven differentiation of progenitor T cells in vitro. Nat Biotechnol (2017) 35(12):1188–95. doi: 10.1038/nbt.4004
83. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med (2012) 18(5):807–15. doi: 10.1038/nm.2700
84. Sugiyama H. WT1 (Wilms’ tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol (2010) 40(5):377–87. doi: 10.1093/jjco/hyp194
85. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi: 10.1038/nm.2446
86. Gupta B, Iancu EM, Gannon PO, Wieckowski S, Baitsch L, Speiser DE, et al. Simultaneous coexpression of memory-related and effector-related genes by individual human CD8 T cells depends on antigen specificity and differentiation. J Immunother (2012) 35(6):488–501. doi: 10.1097/CJI.0b013e31826183a7
87. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12(11):749–61. doi: 10.1038/nri3307
88. Cieri N, Oliveira G, Greco R, Forcato M, Taccioli C, Cianciotti B, et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood (2015) 125(18):2865–74. doi: 10.1182/blood-2014-11-608539
89. Oliveira G, Ruggiero E, Stanghellini MT, Cieri N, D’Agostino M, Fronza R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med (2015) 7(317):317ra198. doi: 10.1126/scitranslmed.aac8265
90. Greco R, Oliveira G, Stanghellini MT, Vago L, Bondanza A, Peccatori J, et al. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol (2015) 6:95. doi: 10.3389/fphar.2015.00095
91. Lepore M, de Lalla C, Mori L, Dellabona P, De Libero G, Casorati G. Targeting leukemia by CD1c-restricted T cells specific for a novel lipid antigen. Oncoimmunology (2015) 4(3):e970463. doi: 10.4161/21624011.2014.970463
92. Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, et al. A novel self-lipid antigen targets human T cells against CD1c(+) leukemias. J Exp Med (2014) 211(7):1363–77. doi: 10.1084/jem.20140410
93. Consonni M, de Lalla C, Bigi A, Dellabona P, Casorati G. Harnessing the CD1 restricted T cell response for leukemia adoptive immunotherapy. Cytokine Growth Factor Rev (2017) 36:117–23. doi: 10.1016/j.cytogfr.2017.06.007
94. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med (2015) 21(8):938–45. doi: 10.1038/nm.3909
95. Legut M, Cole DK, Sewell AK. The promise of gammadelta T cells and the gammadelta T cell receptor for cancer immunotherapy. Cell Mol Immunol (2015) 12(6):656–68. doi: 10.1038/cmi.2015.28
96. Buchholz CJ, Friedel T, Buning H. Surface-Engineered Viral Vectors for Selective and Cell Type-Specific Gene Delivery. Trends Biotechnol (2015) 33(12):777–90. doi: 10.1016/j.tibtech.2015.09.008
97. Hartmann J, Munch RC, Freiling RT, Schneider IC, Dreier B, Samukange W, et al. A Library-Based Screening Strategy for the Identification of DARPins as Ligands for Receptor-Targeted AAV and Lentiviral Vectors. Mol Ther Methods Clin Dev (2018) 10:128–43. doi: 10.1016/j.omtm.2018.07.001
98. Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol Med (2018) 10(11):e9158–79. doi: 10.15252/emmm.201809158
99. Agarwal S, Weidner T, Thalheimer FB, Buchholz CJ. In vivo generated human CAR T cells eradicate tumor cells. Oncoimmunology (2019) 8(12):e1671761. doi: 10.1080/2162402X.2019.1671761
100. Wang X, Katayama A, Wang Y, Yu L, Favoino E, Sakakura K, et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res (2011) 71(24):7410–22. doi: 10.1158/0008-5472.CAN-10-1134
101. Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A, et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst (2010) 102(19):1496–512. doi: 10.1093/jnci/djq343
102. Campoli M, Ferrone S, Wang X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv Cancer Res (2010) 109:73–121. doi: 10.1016/B978-0-12-380890-5.00003-X
103. Mittelman A, Chen ZJ, Yang H, Wong GY, Ferrone S. Human high molecular weight melanoma-associated antigen (HMW-MAA) mimicry by mouse anti-idiotypic monoclonal antibody MK2-23: induction of humoral anti-HMW-MAA immunity and prolongation of survival in patients with stage IV melanoma. Proc Natl Acad Sci USA (1992) 89(2):466–70. doi: 10.1073/pnas.89.2.466
104. Wang X, Ko EC, Peng L, Gillies SD, Ferrone S. Human high molecular weight melanoma-associated antigen mimicry by mouse anti-idiotypic monoclonal antibody MK2-23: enhancement of immunogenicity of anti-idiotypic monoclonal antibody MK2-23 by fusion with interleukin 2. Cancer Res (2005) 65(15):6976–83. doi: 10.1158/0008-5472.CAN-04-2328
105. Price MA, Colvin Wanshura LE, Yang J, Carlson J, Xiang B, Li G, et al. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res (2011) 24(6):1148–57. doi: 10.1111/j.1755-148X.2011.00929.x
106. Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res (2014) 20(4):962–71. doi: 10.1158/1078-0432.CCR-13-2218
107. Maciag PC, Seavey MM, Pan ZK, Ferrone S, Paterson Y. Cancer immunotherapy targeting the high molecular weight melanoma-associated antigen protein results in a broad antitumor response and reduction of pericytes in the tumor vasculature. Cancer Res (2008) 68(19):8066–75. doi: 10.1158/0008-5472.CAN-08-0287
108. Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer (2014) 2:25. doi: 10.1186/2051-1426-2-25
109. Wang Y, Geldres C, Ferrone S, Dotti G. Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen receptor-based T-cell immunotherapy of solid tumors. Expert Opin Ther Targets (2015) 19(10):1339–50. doi: 10.1517/14728222.2015.1068759
110. Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patane M, et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med (2018) 10(430):1–14. doi: 10.1126/scitranslmed.aao2731
111. Ponterio E, De Maria R, Haas TL. Identification of Targets to Redirect CAR T Cells in Glioblastoma and Colorectal Cancer: An Arduous Venture. Front Immunol (2020) 11:565631. doi: 10.3389/fimmu.2020.565631
112. Bertucci F, Finetti P, Simeone I, Hendrickx W, Wang E, Marincola FM, et al. The immunologic constant of rejection classification refines the prognostic value of conventional prognostic signatures in breast cancer. Br J Cancer (2018) 119(11):1383–91. doi: 10.1038/s41416-018-0309-1
113. Hendrickx W, Simeone I, Anjum S, Mokrab Y, Bertucci F, Finetti P, et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. Oncoimmunology (2017) 6(2):e1253654. doi: 10.1080/2162402X.2016.1253654
114. Roelands J, Kuppen PJK, Vermeulen L, Maccalli C, Decock J, Wang E, et al. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int J Mol Sci (2017) 18(10):2229–49. doi: 10.3390/ijms18102229
115. Roelands J, Hendrickx W, Zoppoli G, Mall R, Saad M, Halliwill K, et al. Oncogenic states dictate the prognostic and predictive connotations of intratumoral immune response. J Immunother Cancer (2020) 8(1):e000617–34. doi: 10.1136/jitc-2020-000617
116. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep (2018) 23(7):2130–41. doi: 10.1016/j.celrep.2018.04.051
117. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119(18):4133–41. doi: 10.1182/blood-2011-12-400044
118. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol (2018) 36(9):847–56. doi: 10.1038/nbt.4195
119. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther (2015) 23(4):769–78. doi: 10.1038/mt.2015.4
120. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell (2019) 35(3):473–88.e476. doi: 10.1016/j.ccell.2019.02.006
121. Lock D, Mockel-Tenbrinck N, Drechsel K, Barth C, Mauer D, Schaser T, et al. Automated Manufacturing of Potent CD20-Directed Chimeric Antigen Receptor T Cells for Clinical Use. Hum Gene Ther (2017) 28(10):914–25. doi: 10.1089/hum.2017.111
122. Li Z, Dullmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, et al. Murine leukemia induced by retroviral gene marking. Science (2002) 296(5567):497. doi: 10.1126/science.1068893
123. Kaufmann KB, Brendel C, Suerth JD, Mueller-Kuller U, Chen-Wichmann L, Schwable J, et al. Alpharetroviral vector-mediated gene therapy for X-CGD: functional correction and lack of aberrant splicing. Mol Ther (2013) 21(3):648–61. doi: 10.1038/mt.2012.249
124. Suerth JD, Maetzig T, Brugman MH, Heinz N, Appelt JU, Kaufmann KB, et al. Alpharetroviral self-inactivating vectors: long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol Ther (2012) 20(5):1022–32. doi: 10.1038/mt.2011.309
125. Krooss SA, Dai Z, Schmidt F, Rovai A, Fakhiri J, Dhingra A, et al. Ex Vivo/In vivo Gene Editing in Hepatocytes Using “All-in-One” CRISPR-Adeno-Associated Virus Vectors with a Self-Linearizing Repair Template. iScience (2020) 23(1):100764. doi: 10.1016/j.isci.2019.100764
126. Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-GD2 CAR Expression and Inducible Cytokines. Cancers (Basel) (2020) 12(2):375–97. doi: 10.3390/cancers12020375
127. Schott JW, Leon-Rico D, Ferreira CB, Buckland KF, Santilli G, Armant MA, et al. Enhancing Lentiviral and Alpharetroviral Transduction of Human Hematopoietic Stem Cells for Clinical Application. Mol Ther Methods Clin Dev (2019) 14:134–47. doi: 10.1016/j.omtm.2019.05.015
128. Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med (2018) 24(8):1216–24. doi: 10.1038/s41591-018-0137-0
129. Wiebking V, Lee CM, Mostrel N, Lahiri P, Bak R, Bao G, et al. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing alphabeta T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica (2020) 2. doi: 10.3324/haematol.2019.233882
130. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nat 2017 (7643) 543:113–7. doi: 10.1038/nature21405
131. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood (2014) 124(8):1277–87. doi: 10.1182/blood-2014-01-545020
132. Benjamin R, Graham C, Yallop D, Jozwik A, Ciocarlie O, Jain N, et al. Preliminary Data on Safety, Cellular Kinetics and Anti-Leukemic Activity of UCART19, an Allogeneic Anti-CD19 CAR T-Cell Product, in a Pool of Adult and Pediatric Patients with High-Risk CD19+ Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Blood (2018) 132(Supplement 1):896–6. doi: 10.1182/blood-2018-99-111356
133. Admiraal R, Chiesa R, Lindemans CA, Nierkens S, Bierings MB, Versluijs AB, et al. Leukemia-free survival in myeloid leukemia, but not in lymphoid leukemia, is predicted by early CD4+ reconstitution following unrelated cord blood transplantation in children: a multicenter retrospective cohort analysis. Bone Marrow Transplant (2016) 51(10):1376–8. doi: 10.1038/bmt.2016.116
134. Chaussabel D, Baldwin N. Democratizing systems immunology with modular transcriptional repertoire analyses. Nat Rev Immunol (2014) 14(4):271–80. doi: 10.1038/nri3642
135. Veluchamy JP, Lopez-Lastra S, Spanholtz J, Bohme F, Kok N, Heideman DA, et al. In Vivo Efficacy of Umbilical Cord Blood Stem Cell-Derived NK Cells in the Treatment of Metastatic Colorectal Cancer. Front Immunol (2017) 8:87. doi: 10.3389/fimmu.2017.00087
136. Cany J, van der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, de Vries J, et al. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow-residing human leukemia cells in NOD/SCID/IL2Rg(null) mice. PloS One (2013) 8(6):e64384. doi: 10.1371/journal.pone.0064384
137. Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, et al. Successful Transfer of Umbilical Cord Blood CD34(+) Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin Cancer Res (2017) 23(15):4107–18. doi: 10.1158/1078-0432.CCR-16-2981
138. Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer (2010) 127(9):2209–21. doi: 10.1002/ijc.25423
139. Wimberger P, Gilet H, Gonschior AK, Heiss MM, Moehler M, Oskay-Oezcelik G, et al. Deterioration in quality of life (QoL) in patients with malignant ascites: results from a phase II/III study comparing paracentesis plus catumaxomab with paracentesis alone. Ann Oncol (2012) 23(8):1979–85. doi: 10.1093/annonc/mds178
140. Bokemeyer C, Stein A, Ridwelski K, Atanackovic D, Arnold D, Wöll E, et al. A phase II study of catumaxomab administered intra- and postoperatively as part of a multimodal approach in primarily resectable gastric cancer. Gastric Cancer (2015) 18(4):833–42. doi: 10.1007/s10120-014-0423-6
141. Mau-Sorensen M, Dittrich C, Dienstmann R, Lassen U, Buchler W, Martinius H, et al. A phase I trial of intravenous catumaxomab: a bispecific monoclonal antibody targeting EpCAM and the T cell coreceptor CD3. Cancer Chemother Pharmacol (2015) 75(5):1065–73. doi: 10.1007/s00280-015-2728-5
142. Topp MS, Gokbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol (2015) 16(1):57–66. doi: 10.1016/S1470-2045(14)71170-2
143. Clynes RA, Desjarlais JR. Redirected T Cell Cytotoxicity in Cancer Therapy. Annu Rev Med (2019) 70(1):437–50. doi: 10.1146/annurev-med-062617-035821
144. van Brummelen EMJ, Huisman MC, de Wit-van der Veen LJ, Nayak TK, Stokkel MPM, Mulder ER, et al. (89)Zr-labeled CEA-targeted IL-2 variant immunocytokine in patients with solid tumors: CEA-mediated tumor accumulation and role of IL-2 receptor-binding. Oncotarget (2018) 9(37):24737–49. doi: 10.18632/oncotarget.25343
Keywords: cancer, immunotherapy, CAR-T cells, TCR engineered lymphocytes, CAR-NK cells, monoclonal antibody, clinical trial, off-the-shelf development
Citation: Guerrouahen B, Elnaggar M, Al-Mohannadi A, Kizhakayil D, Bonini C, Benjamin R, Brentjens R, Buchholz CJ, Casorati G, Ferrone S, Locke FL, Martin F, Schambach A, Turtle C, Veys P, van der Vliet HJ, Maccalli C and The EICCI Faculty Group (2021) Proceedings From the First International Workshop at Sidra Medicine: “Engineered Immune Cells in Cancer Immunotherapy (EICCI): From Discovery to Off-the-Shelf Development”, 15th–16th February 2019, Doha, Qatar. Front. Immunol. 11:589381. doi: 10.3389/fimmu.2020.589381
Received: 30 July 2020; Accepted: 30 November 2020;
Published: 14 January 2021.
Edited by:
Yoshihiko Hirohashi, Sapporo Medical University, JapanReviewed by:
Bipulendu Jena, Independent Researcher, San Diego, United StatesKawaljit Kaur, University of California, Los Angeles, United States
Copyright © 2021 Guerrouahen, Elnaggar, Al-Mohannadi, Kizhakayil, Bonini, Benjamin, Brentjens, Buchholz, Casorati, Ferrone, Locke, Martin, Schambach, Turtle, Veys, van der Vliet, Maccalli and The EICCI Faculty Group. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Maccalli, Y21hY2NhbGxpQHNpZHJhLm9yZw==
†These authors have contributed equally to this work