 Biswas Neupane
Biswas Neupane Dhiraj Acharya
Dhiraj Acharya Alex Sutton Flynt
Alex Sutton Flynt Fengwei Bai
Fengwei Bai
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 16 November 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.588382
This article is part of the Research Topic Immune Evasion Mechanisms by RNA Viruses View all 32 articles
Interferons (IFNs) are the key components of innate immunity and are crucial for host defense against viral infections. Here, we report a novel role of interleukin-17A (IL-17A) in inhibiting IFN-α2 expression thus promoting chikungunya virus (CHIKV) infection. CHIKV infected IL-17A deficient (Il17a−/−) mice expressed a higher level of IFN-α2 and developed diminished viremia and milder footpad swelling in comparison to wild-type (WT) control mice, which was also recapitulated in IL-17A receptor-deficient (Il17ra−/−) mice. Interestingly, IL-17A selectively blocked IFN-α2 production during CHIKV, but not West Nile virus (WNV) or Zika virus (ZIKV), infections. Recombinant IL-17A treatment inhibited CHIKV-induced IFN-α2 expression and enhanced CHIKV replication in both human and mouse cells. We further found that IL-17A inhibited IFN-α2 production by modulating the expression of Interferon Regulatory Factor-5 (IRF-5), IRF-7, IFN-stimulated gene 49 (ISG-49), and Mx1 expression during CHIKV infection. Neutralization of IL-17A in vitro leads to the increase of the expression of these antiviral molecules and decrease of CHIKV replication. Collectively, these results suggest a novel function of IL-17A in inhibiting IFN-α2–mediated antiviral responses during CHIKV infection, which may have broad implications in viral infections and other inflammatory diseases.
Chikungunya virus (CHIKV) is a reemerging mosquito-transmitted alphavirus of the Togaviridae family that can cause crippling musculoskeletal inflammatory disease in humans that is characterized by fever, polyarthralgia, myalgia, rash, and headache. CHIKV, first isolated in 1953 in Tanzania, was endemic in the tropic regions of Africa and the Indian Ocean (1). Due to the increasing human travel and the rapid spread of the mosquito vectors to cooler climates, CHIKV has dramatically expanded its territory and is currently circulating in over 60 countries, including Africa, Asia, Europe, and the Americas (2). Since its introduction to the western hemisphere in 2013, CHIKV has caused an explosive epidemic in the Americas with over two million febrile cases with debilitating polyarthralgia in about 50 countries (3). However, there are no specific treatments or vaccines currently available, and the pathogenesis of CHIKV is largely unknown.
CHIKV is primarily transmitted to humans by the bites of infected female Aedes aegypti and Aedes albopictus mosquitos. After mosquito inoculation, CHIKV replicates in human epithelial cells, endothelial cells, and primary fibroblasts then enters the lymph nodes and finally disseminate to other tissues via the circulation (4, 5). Upon infection, the virus is sensed by diverse innate immune pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG I), melanoma differentiation-associated gene 5 (MDA5), and nod like receptors (NLRs). CHIKV infection induces the production of proinflammatory cytokines, chemokines, and type I interferons (IFN-α/β) (6). IFNα/β have been shown to play a key role in limiting CHIKV replication in both humans and mouse models (7). Fibroblasts are the primary target cells of CHIKV infection and also are the major producers of CHIKV-induced IFNα/β (8). Previous studies have revealed that the CHIKV infection induces antiviral responses by activating interferon regulatory factors (IRFs) (8, 9). IRF-5, along with IRF-3 and IRF-7 induces the Interferon stimulated genes (ISGs) during CHIKV infection (10). Further, IRF-7–mediated type I IFN responses provide critical antiviral protection as neonatal animals lacking either of these factors succumbed to CHIKV infection (8). Interestingly, CHIKV was one of the virus models used to discover and characterize the type I IFN (11–13). However, the regulation of type I IFN production and responses during CHIKV infection is still not clearly understood.
IL-17A, a best-characterized member of the IL-17 family, exerts diverse immune functions, including host defense from infection, tissue remodeling and repair, regulation of immune cell homing and inflammation, and cancer progression (14). For example, a high concentration of IL-17A in the joints contributes to inflammatory arthritis and other allergic and autoimmune diseases (15–20). IL-17A can facilitate WNV clearance from mouse brain by promoting CD8+ T-cell cytotoxicity (21) and also inhibit the replication of HSV by enhancing IFN-γ+ Th1 cell response (22). IL-17A signaling also has a protective role to the host during infection of adenovirus by inducing high levels of IL-7R and RORγt expression in mouse liver cells (23, 24). Similarly, IL-17A has also been suggested to facilitate the infection of coxsackievirus B3 by decreasing splenic CD8+ T cell numbers and cardiac IFN-γ production (25) and Theiler’s murine encephalomyelitis virus by up-regulating anti-apoptotic molecules (26, 27). During CHIKV infection, the serum IL-17A levels in patients with arthritis were higher than non-arthritic patients (28). In addition, strong associations between IL-17A levels and swollen joints have been identified (28, 29), suggesting IL-17A might play an important role in the pathogenesis of CHIKV; however, its detailed role has not been characterized. In this study, we identified a novel function of IL-17A signaling in promoting CHIKV infection by negatively regulating IFN-α2–mediated antiviral responses.
All the experimental procedures involving animals in this study were reviewed and approved by the Institutional Animal Care and Use Committees at The University of Southern Mississippi (USM). Experiments and animal studies involving live CHIKV were performed by certified personnel in Biosafety Level 3 (BSL-3) laboratories following the biosafety protocols approved by the USM Institutional Biosafety Committee.
CHIKV (LR OPY1 2006 strain) was provided by University of Texas Medical Branch. A single passage of parental viruses was propagated in Vero cells (ATCC CCL-81) and used as viral stock for this study. The viral stocks were titrated in Vero cells by plaque assay as previously described (30, 31).
Vero cells, NIH3T3 cells (ATCC CRL-1658), and Raw 264.7 cells (ATCC TIB-71) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) containing 1% L-glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum (FBS). Saos2 cells (ATCC HTB-85) were cultured in McCoy’s 5A medium (ATCC 30-2007) supplemented with 15% FBS. All cells were kept in an incubator at 37°C with 5% CO2, and relative humidity of about 95%. Mouse BMDMs and BMDCs were prepared according to previous publications (32–34).
WT C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Breeding pairs of Il17a−/− and Il17ra−/− mice (both in a C57BL/6J background) were provided by Richard A. Flavell (Yale University School of Medicine) and Dr. Sarah Gaffen (University of Pittsburgh), respectively. All the breeders and their pups were kept in a pathogen-free room. Viral infection studies were carried out in the BSL-3 animal facility at USM.
Seven to eight weeks old, sex-matched WT, Il17a−/− and Il17ra−/− mice were subcutaneously injected on the ventral side of the left hind footpad toward the ankle with 1 × 105 PFU of CHIKV in 50-µl phosphate buffer saline (PBS), as mentioned in the previous publications (33, 35). Starting from the day of infection (day 0) to 12 days p.i., the thickness and width of the peri-metatarsal area of the infected feet were measured daily using a digital caliper (Electron Microscopy Science). The relative increase in footpad swelling was calculated and expressed in comparison to pre-infection as previously described (33–36). Blood samples were collected in 0.5M EDTA by retro-orbital bleeding to analyze viral RNA and cytokines. Mice were sacrificed at the selected time-points for collection of the infected footpad.
Total RNA was converted into the first-strand complementary DNA (cDNA) using iSCRIPT™ cDNA synthesis kit (Bio-Rad). QPCR assays were performed in a CFX Connect Real-Time System (Bio-Rad) using SYBR Green supermix (Bio-Rad) for the detection of CHIKV-E1, immune response genes, and β-actin. Viral RNA copy numbers were expressed as the ratio of CHIKV-E1 to cellular β-actin. For QPCR assay of immune response genes, data were expressed as relative fold change (RFC) expressed by ΔΔCT method after normalizing to cellular β-actin. CHIKV-E1 and cytokine transcripts were also quantified by RT-QPCR in total RNA extracted from footpad cells. The primer sequences were designed using NCBI’s primer-designing tool and synthesized by Integrated DNA Technologies or Invitrogen (Supplementary Table 1).
Vero cells were plated in 6-welled plates at a density of 6 × 105 cells per well and incubated overnight. Cell culture supernatant or mouse serum samples were applied to the wells and incubated for 1 h at 37°C with 5% CO2. After virus adsorption, the inoculum was removed, and the cells were covered with an overlay medium containing 1% SeaPlaque agarose (Lonza). The plates were incubated for 2 to 3 days for plaque development. Plaques were visualized by staining with Neutral Red and counted.
For flow cytometric analysis of footpad immune cells, the infected footpad tissues were collected to prepare single-cell suspension as previously described protocol (37). Briefly, the infected footpads were chopped into small pieces and incubated for 1 h at 37°C in digestion medium containing hyaluronidase and collagenase type VIII. The footpad cells were collected after filtering the mixture with 70-µm strainer. The footpad cells were fixed in 2% paraformaldehyde (Electron Microscopy Science) for 15 min at room temperature (RT). The cells were then washed and blocked with Fc block for 30 min at RT. After washing, the cells were probed with mouse monoclonal anti-CD45, CD4, CD8, CD11b, and Ly6G antibodies (BD BioSciences or eBioscience) and incubated for 1 h at RT. The cells were then washed twice and resuspended in staining buffer and analyzed in a BD LSRFortessa (BD Biosciences) using FlowJo (Version 10.4).
The infected and control mouse footpads were collected on day 6 p.i. After fixation overnight in 4% PFA, the footpads were decalcified in 10% EDTA for 10 days. Tissues were then dehydrated; paraffin embedded, and sectioned (10 µm) using a microtome (American Optical Spencer 820). The sectioned tissue slides were stained with hematoxylin and eosin (H&E), and images were acquired using a bright-field microscope (Olympus BH2).
IL-17A and IFN-α2 in the cell culture supernatants of CHIKV-infected NIH3T3 cells was measured by using a commercial ELISA kit (Abcam) following the manufacturer’s instruction. NIH3T3 cells were infected with CHIKV at 1 MOI in the presence of recombinant IL-17A. After 24 h, the cells were lysed in Laemmli sample buffer (Bio-Rad), and the proteins were separated by 10% SDS-polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane (Bio-Rad). The membrane was blocked in 5% milk in Tris-buffered saline with Tween 20 (TBS-T) for 1h at RT and probed with mouse specific Rabbit primary antibody (Phospho-IRF-7, Cell Signaling; Phospho-IRF-5, Invitrogen; GAPDH, Abcam) in the ratio 1:1,000 at 4°C overnight in a rocker. After washing with TBS-T, horseradish peroxidase conjugated secondary antibody (Goat pAb to Rabbit IgG, Abcam; 1:5,000) was added for 1 h at RT. The membranes were washed and developed using SuperSignal West Pico Chemiluminiscence Substrate (Thermo Scientific) and images were acquired using a ChemiDoc MP system (Bio-Rad). Quantification of blot was performed by ImageLab.
Data analysis was performed by using either a Student’s t-test or one-way analysis of variance (ANOVA) wherever applicable in GraphPad Prism software (version 6.0). p < 0.05 was considered statistically significant.
IL-17A is a proinflammatory cytokine that plays essential roles in infections and inflammatory diseases. To test our hypothesis if IL-17A plays a role in CHIKV pathogenesis, we measured its expression in CHIKV-infected mouse and human cells. Mouse fibroblasts (NIH3T3 cells) and human bone epithelial cells (Saos2 cells) were infected with CHIKV (MOI 0.1, 0.5, and 1) for 24 h, and transcripts of Il-17a and cellular β-actin (a housekeeping gene) were measured by a reverse-transcription quantitative PCR (QPCR). The results showed an upregulation of Il-17a transcripts by CHIKV in both NIH3T3 cells (Figure 1A) and Saos2 cells (Figure 1B) in a dose-dependent manner. To relate these results in a mouse model, we infected C57BL/6J wild-type (WT) mice with CHIKV via footpad inoculation, and the blood and footpad samples were collected on days (D) 1 and 2 post-infection (p.i.). Similar to the in vitro results, Il-17a transcript levels were significantly upregulated in the blood and footpads of CHIKV-infected mice at D1 p.i. (Figures 1C, D). In addition, ELISA results showed significant increases of IL-17A at protein levels in the NIH3T3 cell culture (Figure 1E) and the plasma samples on D1 p.i. (Figure 1F). These results demonstrate that CHIKV infection induces the expression of IL-17A in physiologically relevant human and mouse cells and mice.
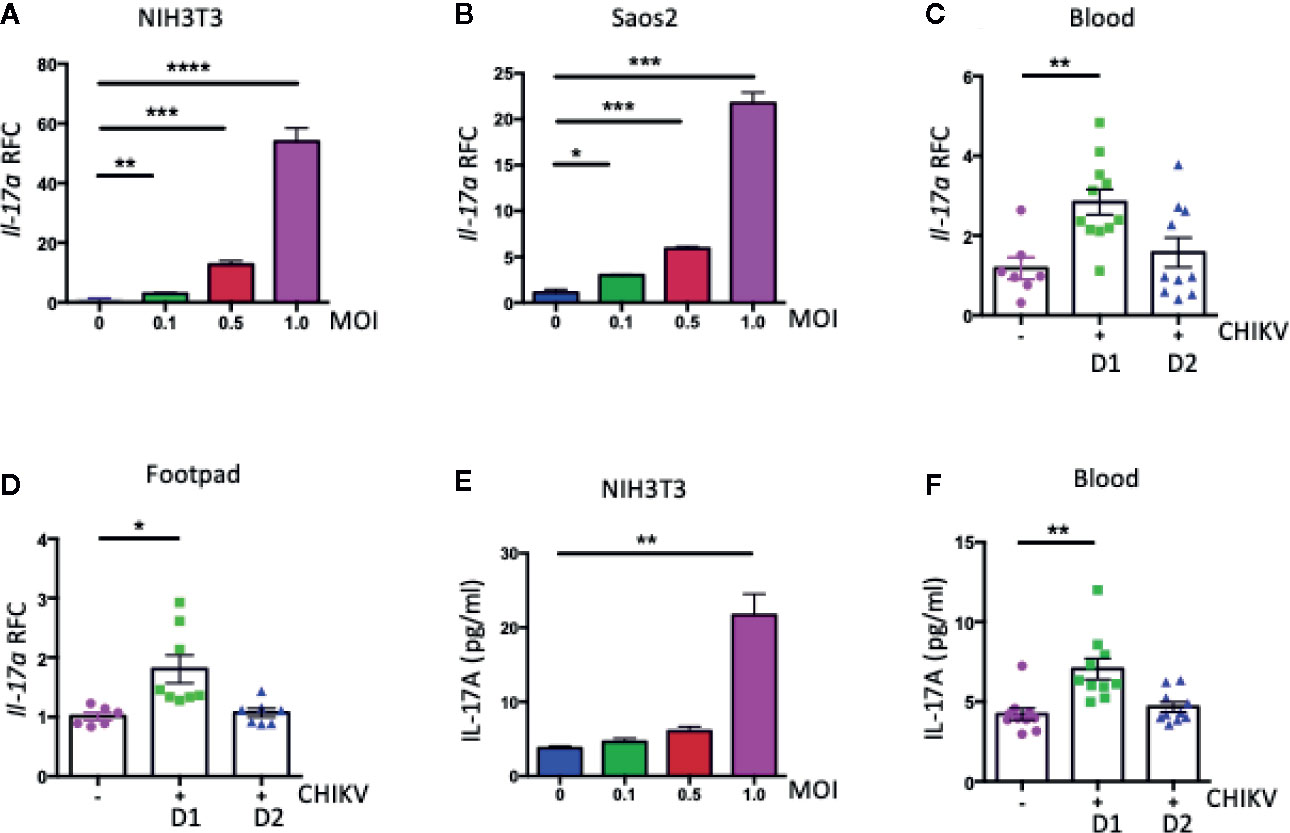
Figure 1 CHIKV infection induces the expression of Il17a in mouse and human. (A, B) The expression of Il17a transcripts was measured by QPCR in NIH3T3 (A) and Saos2 (B) cells that were infected with indicated MOIs of CHIKV for 24 h and expressed as Relative Fold Changes (RFC) after normalization to cellular β-actin transcripts. WT (C57BL/6J) mice (7 to 8 weeks old) were inoculated with CHIKV (1 × 105 PFU) or vehicle control (PBS) via footpad, and blood was collected on days (D) 1 and 2 post-infection (p.i.). (C, D) RFC in the expression of Il17a transcripts in blood (C) and footpad (D) of CHIKV-infected mice was compared with that of mock-infected control mice after normalization to β-actin. (E, F) The protein levels of IL-17A was measured by ELISA in NIH3T3 cells (E) and mice blood (F). The results were analyzed either by using one-way ANOVA followed by Tukey’s test (A, B, E) or a two-tailed student’s t-test (C, D, F). The results are representative of at least two independent experiments and are expressed as mean ± the standard errors of the mean (SEM) [n = 3 biological replicates for (A, B, E); n = 7 to 11 mice/group for (C, D, F). *, **, ***, and **** denote p < 0.05, p < 0.01, p < 0.001, and p < 0.0001, respectively.
To investigate the role of IL-17A in CHIKV-induced disease, we infected 7 to 8 weeks old, sex-matched WT, IL-17A–deficient (Il17a−/−), and IL-17A receptor-deficient (Il17ra−/−) mice with 1 × 105 PFU of CHIKV via footpad, as described in the previous reports (32, 37). Following infection, the CHIKV inoculated footpads usually swell between D1 and D12 p.i. in the adult WT C57BL/6J mice (32, 38). We, therefore, monitored footpad swelling daily for up to 12 days and calculated the relative increase in footpad swelling of CHIKV-infected WT, Il17a−/− and Il17ra−/− mice. The swelling data indicated that Il17a−/− and Il17ra−/− mice had milder inflammation in their inoculated footpads compared to WT control mice (Figure 2A). To corroborate these observations, we collected blood on D1 and 2 p.i. and the viremia was measured by both QPCR and plaque assay. The QPCR results showed a three-fold mean reduction of CHIKVE1 transcripts on D1 p.i. in the Il17a−/− and Il17ra−/− mice compared to WT mice (Figure 2B), whereas the CHIKV was mostly cleared from the blood on D2 p.i. Consistent with QPCR, the plaque assay also showed less infectious viruses in both Il17a−/− and Il17ra−/− mice (Figure 2C). After inoculation, the CHIKV initially replicates locally in the footpad tissues. Therefore, to assess whether the systemic viral replication is similar to viral replication in the infected footpad, we sacrificed selected mice and collected the tissues of the infected footpad on D1 and 2 p.i. and performed the QPCR analysis. Consistent with the viremia, there was an approximately two-fold reduction in CHIKVE1 RNA in the foot tissues of Il17a−/− and Il17ra−/− mice compared to the WT controls on D1 (Figure 2D). Despite the differences are not statistically significant, a similar trend remains on D2 p.i. (Figure 2D).
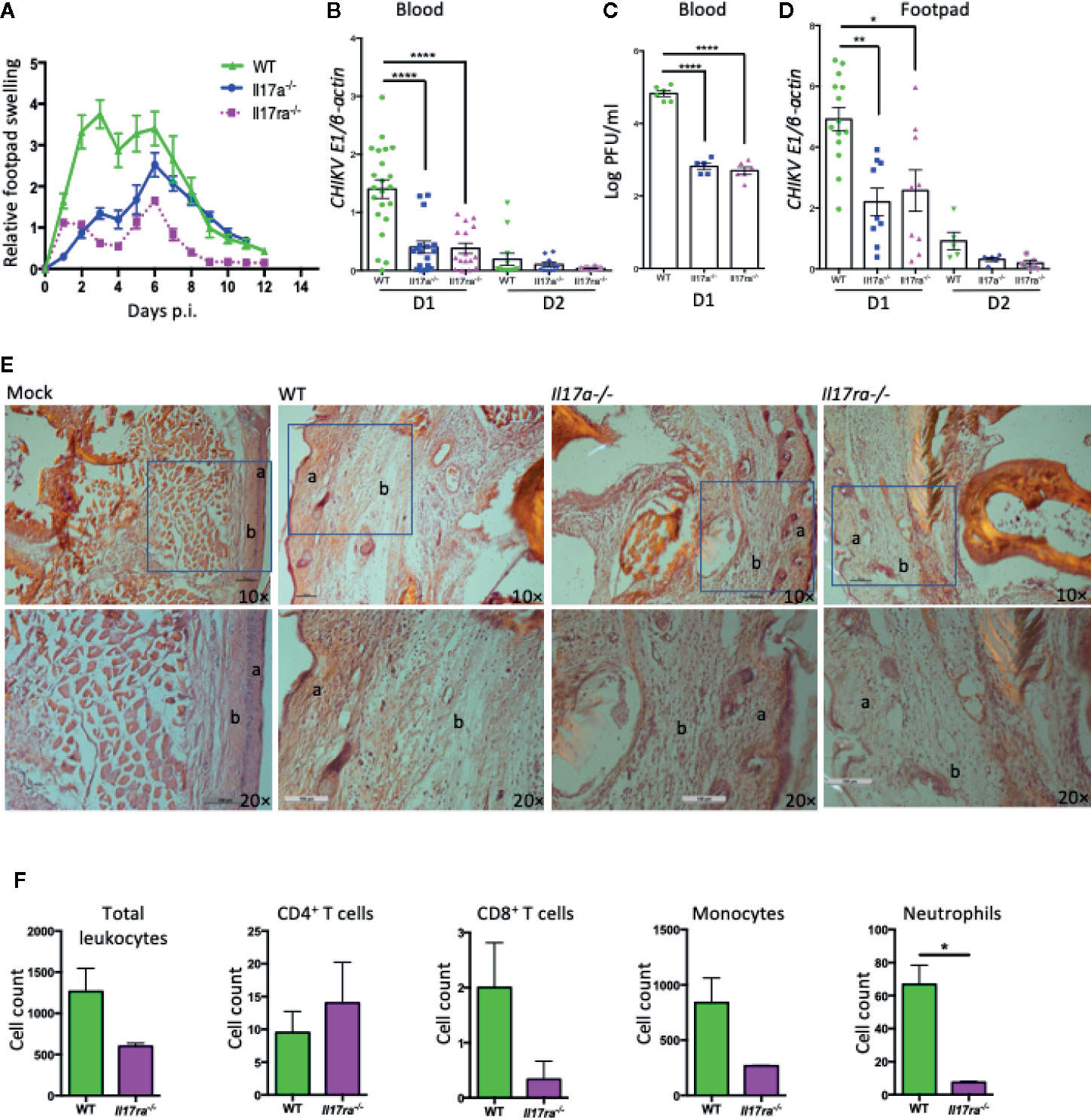
Figure 2 Il17a−/− and Il17ra−/− mice are more resistant to CHIKV infection. Seven to eight weeks old WT, Il17a−/− and Il17ra−/− mice were infected with 1 × 105 PFU of CHIKV via footpad. (A) Swelling of injected footpads (peri-metatarsal area) of mice is presented as a relative increase in swelling that was calculated by measuring the thickness and width of an inoculated footpad. (B) Blood was collected on days 1 (D1) and D2 p.i., and QPCR was performed to measure the copies of CHIKV E1 and cellular β-actin transcripts. Viral burden was expressed as a ratio of copy number of CHIKV E1 to cellular β-actin transcripts and compared between WT (n = 22) and Il17a−/− (n = 17) or Il17ra−/− (n = 16) mice. (C) Plaque assay was performed to measure the viral load in the blood samples of D1 from (B). (D) Inoculated footpads of WT (n = 14) and Il17a−/− (n = 9) or Il17ra−/− (n = 9) were collected on D1 and D2 p.i., and the viral burden was quantified by QPCR after normalization to cellular β-actin. (E) Representative H&E stained histological images (10×, upper row and 20×, lower row) of peri-metatarsal area of the inoculated foot tissue (n = 5 mice/group) on D6 p.i. The layer of epidermis (a) and dermis (b) are labeled. (F) Seven to 8 weeks old WT (n = 4) and Il17ra−/− mice (n = 3) were infected with 1 × 105 PFU of CHIKV via footpad. Footpad cells were isolated at D6 p.i., and quantified by flow cytometry after probing with antibodies against CD45, CD4, CD8, CD11b, and Ly6G. The cell counts for positive cells within the gated population are shown for WT and Il17ra−/− mice. The results are expressed as mean ± SEM (A–D, F) or a representative image (E) that represents at least three independent experiments. The data were analyzed by a two-tailed Student t-test (A–D, F). *, **, and **** denote p < 0.05, p < 0.01, and p < 0.0001 respectively, when compared to WT controls).
To further dissect this observation, we performed histological and flow cytometric analysis with the inoculated footpad tissues at peak swelling (D6 p.i.). The histological analysis showed that the peri-metatarsal area of the inoculated footpads of Il17a−/− and Il17ra−/− mice had less severe swelling and tissue damage compared to that of WT mice (Figure 2E). In addition, the flow cytometric analysis demonstrated a significantly reduced infiltration of neutrophils (CD45+CD11b+Ly6G+) in the footpad of Il17ra−/− mice compared to WT mice (p < 0.05), while the differences are not statistically significant (p > 0.05) for other immune cells including CD4 T cells (CD45+CD3+CD4+), CD8 T cells (CD45+CD3+CD8+), and monocytes (CD45+CD11b+) (Figure 2F). A similar trend, but with no statistical difference, of these infiltrated cells was also observed in Il17a−/− mice compared to WT mice (data not shown). Together, these results suggest that IL-17A signaling facilitates CHIKV replication and exacerbates CHIKV-induced footpad inflammation in mice.
As we observed that the deficiency in IL-17A signaling in mice led to a significantly reduced CHIKV replication in the blood and footpad tissues, we hypothesized that IL-17A signaling may inhibit the antiviral innate immune responses. To test this, we profiled the expression of a set of innate antiviral genes in CHIKV-infected WT and Il17ra−/− mice by QPCR analysis. Interestingly, we found that among 11 different genes tested (Ifn-α, Ifn-β, Ifn-γ, Rig-I, Mda-5, Myd88, Tlr3, Tlr9, Il-12, Cxcl2, and Cxcr3), the expression of Ifn-α was significantly increased in the blood of Il17ra−/− mice (Figure 3A). There are multiple Ifn-α subtypes in mice, and we further tested the effect of IL-17A signaling in the expression of these subtypes upon CHIKV infection. Interestingly, of the 11 Ifn-α subtypes tested, specifically the expression of Ifn-α2 transcripts was significantly inhibited in CHIKV-infected NIH3T3 cells (Supplementary Figure 1). To further validate these results, we first confirmed that the expression of Ifn-α2 was induced by CHIKV in NIH3T3, Saos2 cells, and mice (Figures 3B–D). We next tested if the expression of CHIKV-induced Ifn-α2 in NIH3T3 cells could be inhibited by recombinant mouse IL-17A (rIL-17A). Both QPCR and ELISA results indicated that rIL-17A treatment suppressed the expression of CHIKV-induced Ifn-α2 (Figures 3E, F) in NIH3T3 cells. Consistently, this phenotype was also confirmed in other cell types, i.e., Raw 264.7 cells, mouse bone marrow–derived macrophages (BMDM), and bone marrow–derived dendritic cells (BMDCs) (Figures 3G–L), suggesting a cell type-independent effect. We next measured the expression levels of Ifn-α2 in Il17a−/− and Il17ra−/− mice that lack IL-17A signaling to further determine if IL-17A signaling inhibits the expression of Ifn-α2 in vivo following CHIKV infection. Indeed, the mRNA level of Ifn-α2 in blood and footpads was increased in both Il17a−/− and Il17ra−/− mice than in WT mice, confirming the inhibitory effect of IL-17A on the CHIKV-induced expression of Ifn-α2 (Figures 3M–P).
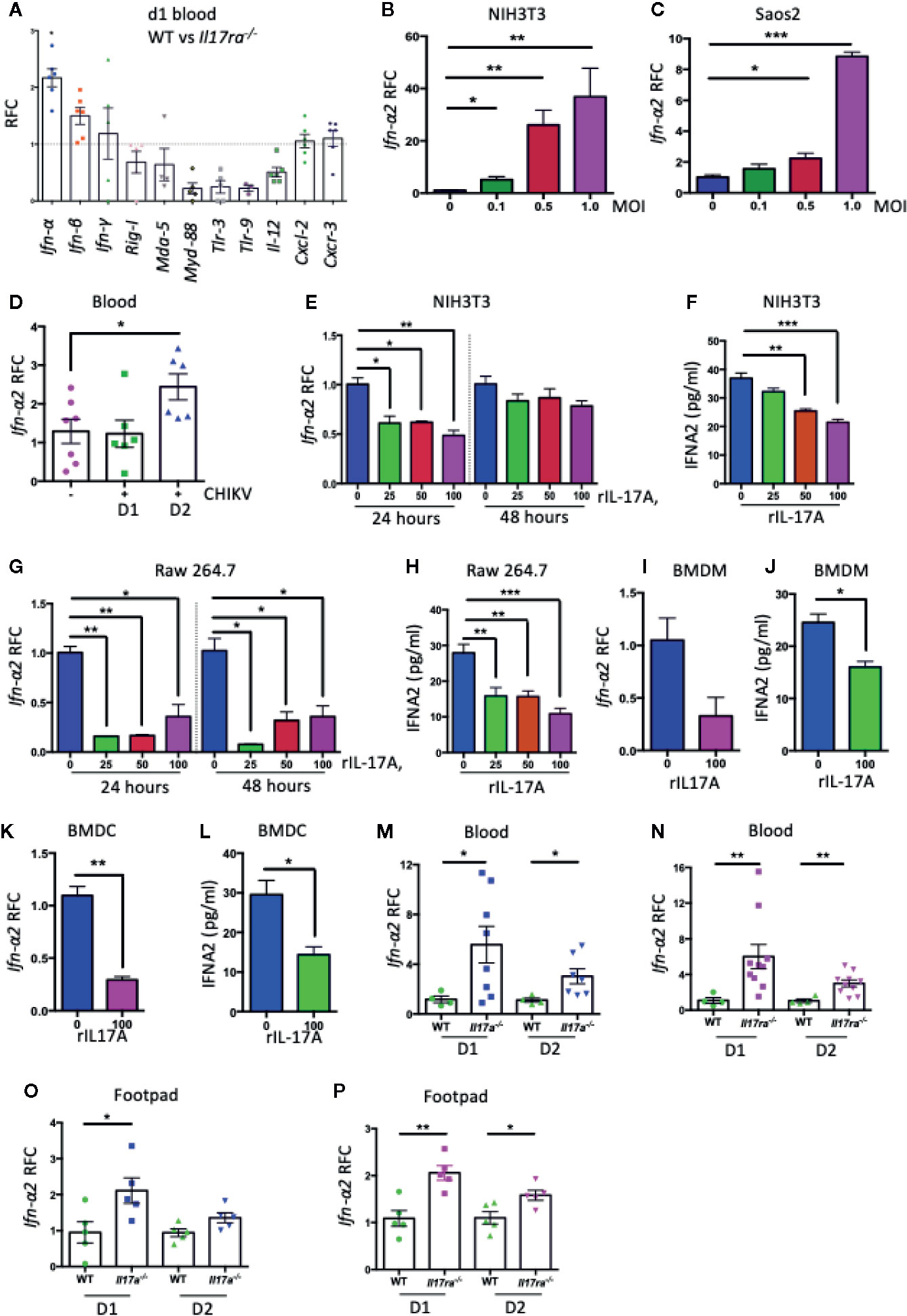
Figure 3 IL-17A down-regulates the expression of Ifn-α2. (A) Seven to eight weeks old WT (n = 5) and Il17ra−/− mice (n = 6) were infected with 1 × 105 PFU of CHIKV via footpad and the transcripts of indicated genes in blood were measured at day 1 post-infection by QPCR and normalized to β-actin mRNA. (B) NIH3T3 and (C) Saos2 cells were infected with CHIKV for 24 h, and the transcript level of Ifn-α2 was measured by QPCR and normalized to β-actin mRNA. (D) Seven to eight weeks old WT mice were mock infected (n = 7) or infected with 1 × 105 PFU of CHIKV (n = 6) via footpad and the expression of Ifn-α2 was measured by QPCR in the blood at indicated time points and normalized to β-actin mRNA. (E) NIH3T3 cells were infected with CHIKV (MOI = 1) and treated with indicated concentrations of rIL-17A for 24 and 48 h. The expression of Ifn-α2 was measured by QPCR and normalized to cellular β-actin mRNA. (F) The protein level of IFN-α2 was measured by ELISA in NIH3T3 cells infected with CHIKV (MOI = 1) and treated with different concentrations of rIL-17A. (G, H) Raw 264.7 cells were infected with CHIKV (MOI = 1) in the presence of indicated concentrations of rIL-17A and incubated for 24 and 48 h followed by the measurement of mRNA level (G) and protein level (H) of IFN-α2. (I–L) Mouse bone marrow derived-macrophages (I, J) and dendritic cells (K, L) were infected with CHIKV (MOI = 1) for 48 h in the presence of rIL-17A (100 ng/ml). The level of Ifn-α2 was measured either by QPCR normalized to cellular β-actin mRNA or by ELISA. (M, N) WT (n = 4), Il17a−/− (n = 8) and Il17ra−/− mice (n = 10) were infected with 1 × 105 PFU of CHIKV via footpad. Blood was collected on days 1 and 2 post-infection, and the mRNA level of Ifn-α2 was measured by QPCR and normalized to β-actin mRNA between WT and Il17a−/− mice (M), and WT and Il17ra−/− mice (N). Ifn-α2 mRNA levels in the footpads of Il17a−/− mice (O) and Il17ra−/− mice (P) were measured by QPCR and normalized to β-actin mRNA. All the data represent at least two independent experiments performed in triplicates and analyzed by one-way ANOVA followed by Turkey’s test, (*, **, and *** denote p < 0.05, p < 0.01, and p < 0.001 respectively, when compared to untreated control).
To test if IL-17A also inhibits the expression of Ifn-α2 during other virus infections, we infected NIH3T3 cells with West Nile virus (WNV) or Zika virus (ZIKV) in the presence of rIL-17A, and measured Ifn-α2 mRNA by QPCR or ELISA. Interestingly, the results suggested that IL-17A has no such effect on IFN-α2 production during WNV and ZIKV infections (Supplementary Figures 2A–C). IL-17F, a closely related member of IL-17 family, has related functions to IL-17A and also shares the same set of receptors IL-17RA and IL-17RC. To test if IL-17F function similarly with IL-17A in the regulation of IFN-α2, we measured the level of CHIKV-induced Ifn-α2 expression in NIH3T3 cells upon recombinant IL-17F (rIL-17F) treatment (Supplementary Figures 2D, E). Our results demonstrated that only IL-17A, but not IL-17F, inhibited the CHIKV-induced IFN-α2 expression. As a consequence, the treatment of rIL-17A after CHIKV infection significantly increases the virus replication in NIH3T3 cells (Figures 4A, B), which may be due to the reduced antiviral IFN-α2 production. In another experiment, we also tested if the cells pretreated with rIL-17A could enhance susceptibility to CHIKV infection. The results showed that the pretreatment with rIL-17A, but not rIL-17F, enhanced CHIKV replication (Figures 4C, D), implying that IL-17A constrains the production of IFN-α2 thus facilitating CHIKV replication. The antiviral activities of IFN-α2 were confirmed by the experiment showing that the presence of rIFN-α2 constrains CHIKV replication in NIH3T3 cells (Figures 4E, F), while the addition of rIL-17A neutralizes the antiviral effects of rIFN-α2 (Figure 4G). However, it does not rule out that the observed effect of IL-17A happens independently of CHIKV infection. Next, we tested if the presence of IL-17A neutralizing antibody could inhibit the effects of CHIKV-induced IL-17A. We infected NIH3T3 cells with CHIKV in the presence of IL-17A neutralizing antibody or anti-flavivirus 4G2 antibody as an isotype control. The results show that CHIKV replication is inhibited and the expression level of Ifn-α2 increases in the presence of IL-17A neutralizing antibody, but not of the control antibody (Figures 4H, I). Collectively, these results demonstrate that IL-17A signaling inhibits the production of IFN-α2, thus facilitating the CHIKV replication in vitro and mice.
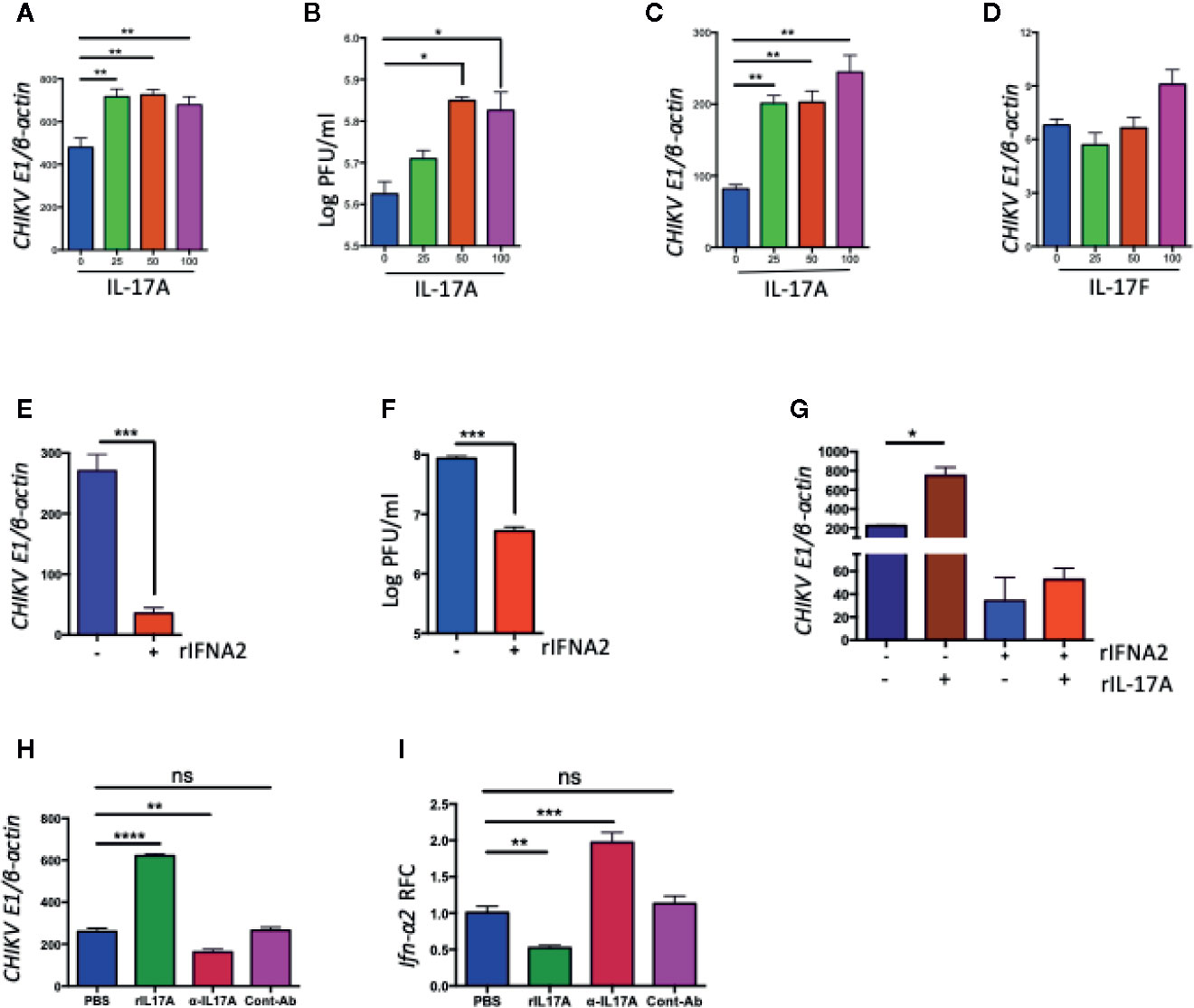
Figure 4 IL-17A, but not IL-17F, promotes CHIKV replication in NIH3T3 cells. (A, B) NIH3T3 cells were infected with CHIKV (MOI =1) in the presence of indicated concentrations of rIL-17A for 48 h. The ratio of CHIKVE1 to cellular β-actin transcripts was determined by QPCR (A) and the number of infectious virus in the culture supernatants were measured by a plaque assay (B). (C) NIH3T3 cells were preincubated for 24 h with indicated concentrations of rIL-17A, then infected with CHIKV (MOI = 1) for an additional 48 h. The ratio of CHIKVE1 RNA to cellular β-actin transcripts was determined by QPCR. (D) NIH3T3 cells were infected with CHIKV (MOI = 1) in the presence of IL-17F for 48 h, and the ratio of CHIKVE1 RNA to cellular β-actin transcripts was determined by QPCR. (E, F) NIH3T3 cells were treated with recombinant IFNA2 for 8 h and then infected with CHIKV at 1 MOI for 24 h. The ratio of CHIKVE1 to cellular β-actin transcripts was determined by QPCR (E) and the infectious virus in the cell medium were measured by a plaque assay (F). (G) NIH3T3 cells were treated with rIL-17A and IFNA2, then infected with CHIKV at 1 MOI for 24 h. The ratio of CHIKVE1 to cellular β-actin transcripts was measured by QPCR. (H, I) NIH3T3 cells were infected with CHIKV (1 MOI) in the presence of rIL-17A, IL-17A neutralizing antibody or anti-flavivirus 4G2 antibody as an isotype control for 24 h. The ratio of CHIKVE1 to cellular β-actin mRNA was determined by QPCR (H) and the expression of Ifn-α2 was measured by QPCR and normalized to cellular β-actin mRNA (I). All the data represent at least two independent experiments performed in triplicates and analyzed by one-way ANOVA followed by Turkey’s test, (*, **, and *** denote p < 0.05, p < 0.01, and p < 0.001, respectively, when compared to untreated control). ns denotes not significant.
We next hypothesized that IL-17A signaling interrupts the signaling pathway(s) that differentially regulate the expression of Ifn-α subtypes and thus inhibiting the expression of Ifn-α2. Relative amounts of IRF genes have been shown to modulate the differential expression of various IFN-A subtypes during paramyxovirus infection (39). IRF genes have been shown to play protective roles against CHIKV infection (9). Therefore, we assessed the role of IL-17A signaling in the expression of IRF genes during CHIKV infection. We infected NIH3T3 cells with CHIKV in the presence of rIL-17A for QPCR analysis, and the results showed that the expression of Irf-3, Irf-5, and Irf-7 was inhibited at the transcriptional levels (Figures 5A–C). In addition, the immunoblotting analysis suggests that IL-17A inhibits the phosphorylation IRF-5 and IRF-7 during CHIKV infection (Figures 5D, E), while the phosphorylation of IRF-3 was not detected (data not shown). Considering IL-17A inhibits the expression and phosphorylation of IRF-5 and IRF-7 in the cell culture during CHIKV infection, we measured the levels of Irf-5 and Irf-7 mRNA in mice blood and footpads following CHIKV infection. Consistent with the results, the levels of Irf-5 are significantly upregulated in the blood and footpads of Il17a−/− and Il17ra−/− mice compared to WT control mice (Figures 5F–H). A similar trend is also shown for Irf-7, although the difference is not statistically significant (Figures 5I–K). These results indicate that IL-17A may inhibit the expression of Ifn-α2 via IRF-5 and 7 pathways. We next tested if IL-17A signaling inhibits IFN-mediated antiviral responses. We tested the expression of a set of ISGs (Isg-15, Isg-49, Isg-54, Isg-56, Oas1a, Mx1, Ifitm3, and bst2) by QPCR in the blood and footpads of WT, Il17a−/− and Il17ra−/− mice following CHIKV infection. Interestingly, we found that among those test ISGs, Isg-49, and Mx1 were significantly upregulated in Il17a−/− and Il17ra−/− mice footpads compared to the WT controls (Figures 5L–M). Similar patterns were observed in the blood, although the difference was not significant (data not are shown). We further confirmed that the expression of Irf-5, Irf-7, Isg-49, and Mx1 were upregulated in the presence of IL-17A neutralizing antibody in CHIKV-infected NIH3T3 cells (Figures 5N–Q). Collectively, these results demonstrate that IL-17A signaling inhibits IFN-α2–mediated antiviral responses during CHIKV infection.
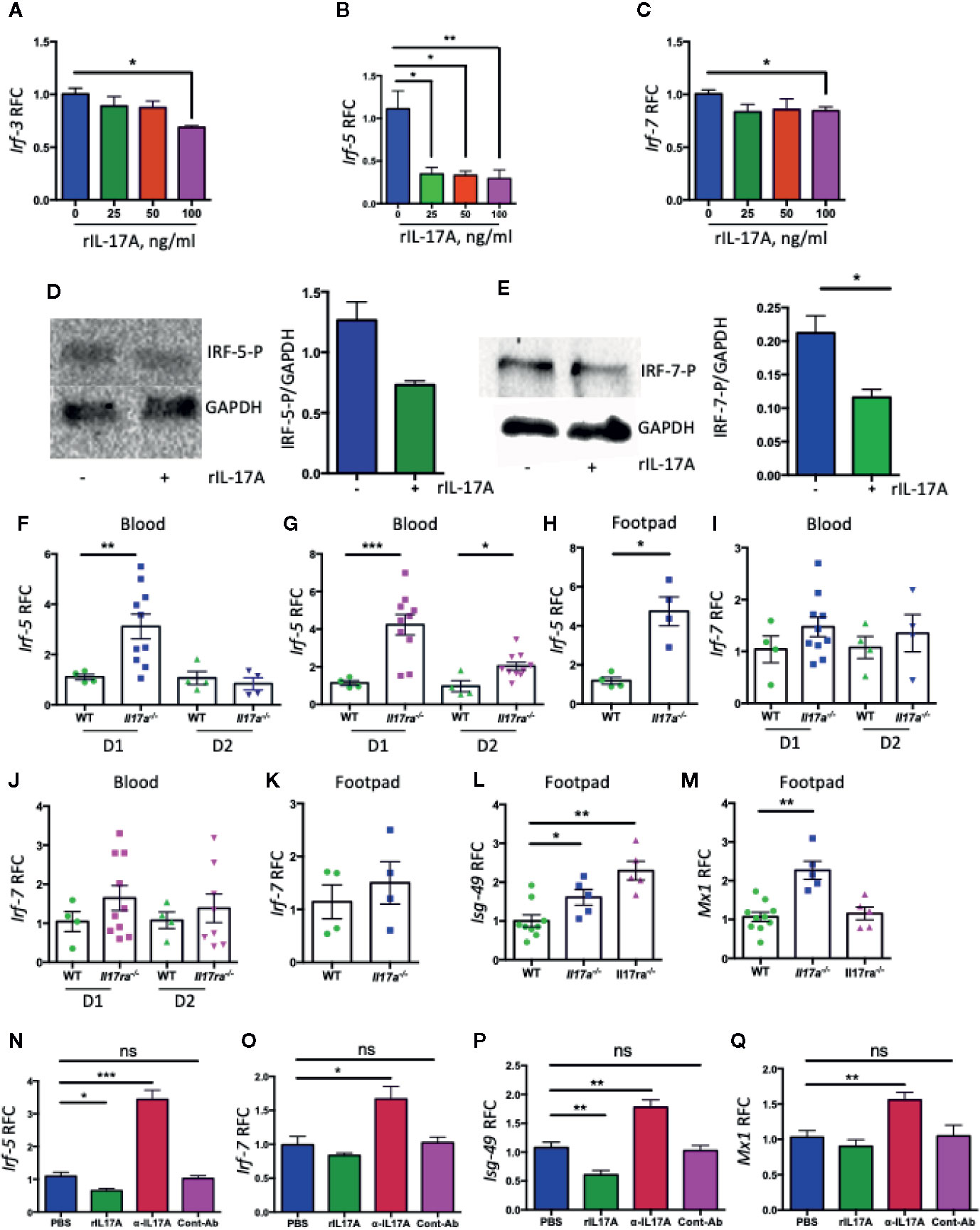
Figure 5 IL-17A downregulates the expression of Irf5 and Irf7 during CHIKV infection. NIH3T3 cells were infected with CHIKV (MOI = 1) and simultaneously treated with indicated concentrations of rIL-17A for 48 h. The transcripts of Irf-3 (A), Irf-5 (B), and Irf-7 (C) were measured by QPCR and expressed as RFC after normalization to cellular β-actin mRNA. (D) NIH3T3 cells were infected with CHIKV (MOI = 1) for 24 h in the presence of IL-17A (100 ng/ml) and indicated proteins in the cell lysates were analyzed by immunoblotting (left) and the ratio of phosphorylated IRF-5 to GAPDH was calculated by densitometric measurement by using ImageLab (right). (E) The immunoblotting analysis was done similarly for IRF-7 and GAPDH (left), and the ratio of phosphorylated IRF-7 to GAPDH was calculated by densitometric measurement by using ImageLab (right). (F, G) WT (n = 4), Il17a−/− (n = 10), and Il17ra−/− (n = 10) mice were infected with 1 × 105 PFU of CHIKV via footpad. Blood was collected on days 1 and 2 post-infection, and the mRNA levels of Irf-5 were measured and normalized to β-actin mRNA between WT and Il17a−/− mice (F), and WT and Il17ra−/− mice (G). Footpads were collected from selected WT (n = 4) and Il17a−/− (n = 4) mice on D1 p.i., and the mRNA level of Irf-5 was measured and normalized to β-actin mRNA between WT and Il17a−/− mice (H). The mRNA levels of Irf-7 were measured in the blood of Il17a−/− mice (I), the blood of Il17ra−/− mice (J), and the footpads of Il17a−/− mice (K). (L, M) Isg-49 and Mx1 mRNAs in the footpads (n = 5 to 10/group) on D1 p.i. (N–Q) NIH3T3 cells were infected with CHIKV (1 MOI) in the presence of IL-17A, anti-Il17A antibody or 4G2 control antibody for 24 h. The level of Irf-5 (N), Irf-7 (O), Isg-49 (P), and Mx1 (Q) were measured and normalized to cellular β-actin mRNA. The data (A–C and N–Q) represent the results of two independent experiments carried out in triplicates, and analyzed by one-way ANOVA followed by Turkey’s test, (*, **, and *** denote p < 0.05, p < 0.01, and p < 0.001 respectively, when compared to untreated control). The results (F–M) represent three independent experiments analyzed by a two-tailed Student t-test, (*, **, and *** denote p < 0.05, p < 0.01, and p < 0.001 respectively, when compared to WT controls). ns denotes not significant.
IL-17A signaling has been associated with several inflammatory diseases in humans, such as rheumatoid arthritis (40, 41), systemic lupus erythematosus (42), and Crohn’s disease (43, 44). Among its diverse functions, IL-17A can regulate the activities of various other inflammatory cytokines, which include TNF-α, IL-1β, and IFN-γ (45–48). For example, IL-17A signaling has also been shown to upregulate joint destructive factors by stimulating transcriptional NF-κB activity and expression of IL-1, granulocyte/macrophage colony-stimulating factor (GM-CSF), prostaglandin E2, IL-6, IL-8, and TNF-α in fibroblasts, endothelial and epithelial cells, and inducing T cell proliferation (46, 49–56). In this study, we report a novel role of IL-17A in inhibiting IFN-α2 expression during CHIKV infection.
CHIKV infection in humans can induce severe inflammatory responses and chronic arthritis, which is correlated with the increased production of IL-17A in patients (6, 57). As such, it is plausible to hypothesize that IL-17A may play an important role in the pathogenesis of CHIKV. We detected a significant up-regulation of Il-17a in both human and mouse cells, and mouse blood and footpads following CHIKV infection (Figure 1). Interestingly, the CHIKV burden was negatively regulated by IL-17A signaling as both Il17a−/− and Il17ra−/− mice exhibited lower CHIKV loads in their blood and mild swelling in inoculated footpads compared to those of WT mice (Figure 2). These results suggest that IL-17A signaling facilitates CHIKV replication and disease in mice. In mouse models, after subcutaneous inoculation of CHIKV via footpad, biphasic swelling responses usually occur in the inoculated footpad. Two peaks of swelling can be observed on around days 1–3 and 5–7 p.i., respectively. Mice also develop severe arthritis, tendonitis, and fasciitis in the inoculated foot, but these inflammatory effects are generally milder in the contralateral foot without swelling (32, 34, 38). Consistent with these results, in our study, the major peak of CHIKV-induced footpad swelling occurred on day 6 p.i. when the viruses had been cleared from the animal circulation, which suggests that the second peak of the swelling is largely mediated by pathogenic immune responses.
Type I IFN signaling has been shown to play an essential role in controlling infections of alphaviruses including CHIKV (58–60). Inhibition of type I IFN signaling in mice causes severe CHIKV-associated disease due to higher viral loads and virus dissemination to the CNS (58, 61). As the major target cells, fibroblasts can be infected by CHIKV in vitro and in vivo (4, 7) and are the major source of type I IFNs responding to CHIKV infection (7). Our results on NIH3T3 cells indicate that IL-17A inhibits IFN-α2 expression during CHIKV infection (Supplementary Figure 1 and Figure 3). We also showed the same effect of IL-17A on the other cell types, such as BMDMs and dendritic cells, as well as mouse macrophage cells (Raw 264.7), suggesting that IL-17A inhibits IFN-α2 expression during CHIKV infection in a cell-type independent manner. The blockade of IFN-α in mice results in the increases of CHIKV viral loads in the infected foot and serum demonstrating its importance in restricting viral replication and spread (62). Our in vitro results show that CHIKV replication is supported by IL-17A, which may be due to the decrease in the production of IFN-α2. We further showed that Ifn-α2 was upregulated in Il17a−/− and Il17ra−/− mice compared to WT mice after CHIKV infection, which may be the reason for lowering the level of CHIKV after infection. Our results further suggest that IL-17A, but not IL-17F, specifically inhibits the expression of IFN-α2 during CHIKV, but not WNV or ZIKV infection (Figure 4 and Supplementary Figure 2). Both IL-17A and IL-17F belong to the same cytokine family and bind to the same receptors, but their specificity and binding affinity to the receptors differ. This might account for the differences observed in the inhibition of IFN-α2 and the replication of CHIKV in the presence of IL-17A and IL-17F. Alphaviruses are known for their antiviral counter defense strategies during infection, the C-terminal domain of CHIKV nsP2 specifically inhibits IFN response by promoting the nuclear export of STAT1 (63). Further, alphaviruses have evolved mechanisms to obstruct antiviral responses by inhibiting specific signaling pathways and modulating the host cell shutoff by the inhibition of general transcription and/or translation (64, 65). These properties of CHIKV might be involved in the inhibition of IFN-α2 expression during CHIKV infection, but not during WNV and ZIKV infection.
In mice, the family of type I IFN consists of 14 IFN-α subtypes and single forms of IFN-β, IFN-ϵ, IFN-κ, and IFN-ω (66). In response to viral infection, host cell PRRs recognize the viral ligands and leads to the production of IFN-β and IFN-α4 through activation of IRF-3 (67–71). Through autocrine and paracrine signaling, these IFNs modulate the expression of various transcriptional factors and ultimately induce expression of diverse IFN subtypes and IFN stimulated genes (ISGs). One of the transcriptional regulators induced by these IFNs is IRF-7, which induces all other IFN-α subtypes in a positive feedback loop, thus amplifying the response (72–74). Thus, IRF-3, IRF-5, and IRF-7 are considered to be the master regulators of type I IFN induction and ISG expression. The combined effect of these three transcription factors has been shown to coordinately regulate IFN response during WNV infection (75). In lethal CHIKV infection, the survival time of Irf3−/− Irf7−/− double knockout mice are longer than Ifnar1−/− mice (7), thus speculating the role of additional transcription factors, such as IRF-5, contributing to the IFN response after CHIKV infection. Here, we show that IRF-3, IRF-5, and IRF-7 are inhibited by IL-17A at the mRNA levels during CHIKV infection; however, the phosphorylation of IRF-3 was not detectable by immunoblotting, suggesting IRF-3 may not play an essential role mediating IL-17A–IFN-α2 pathway. Instead, the expression and phosphorylation of Irf-5 and Irf-7 are inhibited by IL-17A, indicating that IL-17A signaling inhibits IFN-α2 mainly by affecting IRF-5 and IRF-7 mediated pathways (Figure 5). Type I IFNs bind to their receptors and initiate the JAK-STAT signaling pathways and induce the production of the ISGs that are critical to control viral replication. Our results show the transcript levels of Isg-49 and Mx1 decrease in the presence of rIL-17A while increase in the presence of IL-17A neutralizing antibody during CHIKV infection, thus suggesting IL-17A directly or indirectly inhibits the ISG-49 and Mx1-mediated antiviral responses against CHIKV replication.
Different IFN types may exert different functions during CHIKV infection, IFN-α limits early viral replication and dissemination; while IFN-β modulates neutrophil-mediated inflammation (62). Among IFN-α subtypes, IFN-α2 can be induced by Herpes Simplex Virus, Respiratory Syncytial Virus, and Newcastle Disease Virus (76). It has antiviral effects against influenza A virus and human metapneumovirus (76–78). We found that IL-17A inhibits the expression of IFN-α2 during CHIKV infection in a variety of cell types. The level of IFN-α2 was decreased and the viral replication was increased during CHIKV infection in the presence of rIL-17A. Our results also indicate only IFN-α2, but not the other 10 tested IFN-α subtypes is inhibited by IL-17A. Therefore, these results suggest that the increase of the production of IFN-α2 in the absence of IL-17A signaling may contribute to the reduced viral burden in the blood and footpad during CHIKV infection. Supporting this hypothesis, we showed that the level of CHIKV is less in the infected cells in the presence of recombinant IFNA2. Consistent with the previous research that has demonstrated that type I IFN signaling is essential to mitigate CHIKV-induced pathogenicity in mice (9, 62). Furthermore, we found that the infiltration of neutrophils into the footpads of Il17ra−/− mice was significantly reduced compared to WT mice on day 6 p.i. when the second peak of footpad swelling occurred. Neutrophils have been shown to be a major player contributing to CHIKV-induced inflammation and tissue damage in joints (79), which may be due to the increased level of inflammatory mediators, such as CXCL1, CXCL2, granulocyte colony-stimulating factor (G-CSF), IL-1β, and decreasing the level of anti-inflammatory macrophages. The lower viral burdens in the early phase of CHIKV infection seen in Il17a−/− and Il17ra−/− mice, may cause reduced levels of these inflammatory mediators, thus leading to a less severe footpad swelling on day 6 p.i.
In conclusion, our study discovers a novel role of IL-17A in inhibiting IFN-α2 production during CHIKV infection. Further studies are warranted to dissect the mechanism by which IL-17A regulates CHIKV-induced IFN-α2 expression, which may have broad implications in the understanding of IL-17A regulated immunity and development of novel IL-17A–based therapeutic strategy.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by The Institutional Animal Care and Use Committees at The University of Southern Mississippi (USM).
FB conceived the experiments. BN, DA, FN, and GG-F conducted the experiments and analyzed the results. FB and BN wrote the manuscript. AF provided experimental materials. All authors contributed to the article and approved the submitted version.
This work was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health R15AI35893 (FB). The authors also thank the Mississippi INBRE (funded by the National Institute of General Medical Sciences P20 GM103476) for use of the research facility. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.588382/full#supplementary-material
1. Ross RW. The Newala epidemic. III. The virus: isolation, pathogenic properties and relationship to the epidemic. J Hyg (Lond) (1956) 54(2):177–91. doi: 10.1017/S0022172400044442
2. Wahid B, Ali A, Rafique S, Idrees M. Global expansion of chikungunya virus: mapping the 64-year history. Int J Infect Dis (2017) 58:69–76. doi: 10.1016/j.ijid.2017.03.006
3. Silva LA, Dermody TS. Chikungunya virus: epidemiology, replication, disease mechanisms and prospective intervention strategies. J Clin Invest (2017) 127(3):737–49. doi: 10.1172/JCI84417
4. Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, et al. Characterization of reemerging chikungunya virus. PloS Pathog (2007) 3(6):e89. doi: 10.1371/journal.ppat.0030089
5. Kam YW, Ong EK, Renia L, Tong JC, Ng LF. Immuno-biology of chikungunya and implications for disease intervention. Microbes Infect (2009) 11(14-15):1186–96. doi: 10.1016/j.micinf.2009.09.003
6. Chow A, Her Z, Ong EKS, Chen JM, Dimatatac F, Kwek DJC, et al. Persistent arthralgia induced by chikungunya virus infection is associated with Interleukin-6 and Granulocyte macrophage colony-stimulating factor. J Infect Dis (2011) 203(2):149–57. doi: 10.1093/infdis/jiq042
7. Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med (2010) 207(2):429–42. doi: 10.1084/jem.20090851
8. Schilte C, Buckwalter MR, Laird ME, Diamond MS, Schwartz O, Albert ML. Cutting edge: independent roles of IRF-3 and IRF-7 in hematopoietic nonhematopoietic cells during host response to chikungunya infection. J Immunol (2012) 188(7):2967–71. doi: 10.4049/jimmunol.1103185
9. Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, et al. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J Virol (2012) 86(18):9888–98. doi: 10.1128/JVI.00956-12
10. McCarthy MK, Reynoso GV, Winkler ES, Diamond MS, Hickman HD, Morrison TE. MyD88-dependent influx of monocytes and neutrophils impairs lymph node B cell responses to chikungunya virus infection via Irf5, Nos2 ans Nox2. PloS Pathog (2020) 16(1):e1008292. doi: 10.1371/journal.ppat.1008292
11. Heller E. Enhancement of chikungunya virus replication and inhibition of interferon production by actinomycin D. Virology (1963) 21(4):652–6. doi: 10.1016/0042-6822(63)90239-3
12. Friedman RM. Role of Interferon in viral interference. Nature (1964) 201:848–9. doi: 10.1038/201848a0
13. Wagner RR. Inhibition of Interferon biosynthesis by actinomycin D. Nature (1964) 204:49–51. doi: 10.1038/204049a0
14. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 receptor-based signaling and implications for disease. Nat Immunol (2019) 20:1594–602. doi: 10.1038/s41590-019-0514-y
15. McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol (2007) 8:913–9. doi: 10.1038/ni1507
16. Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating diseases. Trends Immunol (2011) 32:232–9. doi: 10.1016/j.it.2011.02.007
17. van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol (2009) 5:549–53. doi: 10.1038/nrrheum.2009.179
18. Raychaudhuri SP. Role of IL-17 in psoriasis and psoriatic arthritis. Clin Rev Allergy Immunol (2013) 44:183–93. doi: 10.1007/s12016-012-8307-1
19. Newcomb DC, Peebles RS Jr. Th17-mediated inflammation in asthma. Curr Opin Immunol (2013) 25:755–60. doi: 10.1016/j.coi.2013.08.002
20. Siakavellas SI, Bamias G. Role of the IL-23/IL-17 axis in Crohn’s disease. Discovery Med (2012) 14:253–62.
21. Acharya D, Wang P, Paul AM, Dai J, Gate D, Lowery JE, et al. Interleukin-17A promotes CD8+ T cell cytotoxicity to facilitate West Nile virus clearance. J Virol (2017) 91:e01529–16. doi: 10.1128/JVI.01529-16
22. Bagri P, Anipindi VC, Nguyen PV, Vitali D, Stampfli MR, Kaushic C. Novel role for interleukin-17 in enhancing type 1 helper T cell immunity in the female genital tract following mucosal Herpes Simplex Virus 2 vaccination. J Virol (2017) 91(23):e01234–17. doi: 10.1128/JVI.01234-17
23. Hou L, Jie Z, Desai M, Liang Y, Soong L, Wang T, et al. Early IL-17 production by intrahepatic T cells is important for adaptive immune responses in viral hepatitis. J Immunol (2013) 190(2):621–9. doi: 10.4049/jimmunol.1201970
24. Jie Z, Liang Y, Hou L, Dong C, Iwakura Y, Soong L, et al. Intrahepatic innate lymphoid cells secrete IL-17A and IL-17F that are crucial for T cell priming in viral infection. J Immunol (2014) 192(7):3289–300. doi: 10.4049/jimmunol.1303281
25. Yuan J, Yu M, Lin QW, Cao AL, Yu X, Dong JH, et al. Th17 cells contribute to viral replication in Coxsackievirus B3-induced acute viral myocarditis. J Immunol (2010) 185(7):4004–10. doi: 10.4049/jimmunol.1001718
26. Hou W, Kang HS, Sim BS. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J Exp Med (2009) 206(2):313–28. doi: 10.1084/jem.20082030
27. Hou W, Jin YH, Kang HS, Kim BS. Interleukin-6 (IL-6) and IL-17 synergistically promote viral persistence by inhibiting cellular apoptosis and cytotoxic T cell function. J Virol (2014) 88(15):8479–89. doi: 10.1128/JVI.00724-14
28. Cavalcanti NG, MeloVilar K, Duarte ALBP, de Melo Rego MJB, Pereira MC, da Rocha Pitta I, et al. IL-27 in patients with Chikungunya fever: a possible chronicity biomarker? Acta Trop (2019) 196:48–51. doi: 10.1016/j.actatropica.2019.05.005
29. Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol (2015) 11:415–29. doi: 10.1038/nrrheum.2015.53
30. Bai F, Town T, Qian F, Wang P, Kamanaka M, Connolly TM, et al. IL-10 signaling blockade controls murine West Nile Virus infection. PloS Pathog (2009) 5(10):e1000610. doi: 10.1371/journal.ppat.1000610
31. Bai F, Wang T, Pal U, Bao F, Gould LH, Fikrig E. Use of RNA interference to prevent lethal murine West Nile Virus infection. J Infect Dis (2005) 191:1148–54. doi: 10.1086/428507
32. Acharya D, Paul AM, Anderson JF, Huang F, Bai F. Loss of glycosaminoglycan receptor binding after mosquito cell passage reduces chikungunya virus infectivity. PloS Negl Trop Dis (2015) 9(10):e0004139. doi: 10.1371/journal.pntd.0004139
33. Paul AM, Acharya D, Duty L, Thompson EA, Linda Le, Stokic DS, et al. Osteopontin facilitates West Nile Virus neuroinvasion via neutrophil “Trojan horse” transport. Sci Rep (2017) 7:4722. doi: 10.1038/s41598-017-04839-7
34. Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, et al. Chikungunya virus arthritis in adult wild-type mice. J Virol (2010) 84:8021–32. doi: 10.1128/JVI.02603-09
35. Teng TS, Foo SS, Simamarta D, Lum FM, Teo TH, Lulla A, et al. Viperin restricts chikungunya virus replication and pathology. J Clin Invest (2012) 122(12):4447–60. doi: 10.1172/JCI63120
36. Akitsu A, Iwakura Y. Isolation of Joint-Infiltrating Cells. Bio-protocol (2016) 6(17):e1911. doi: 10.21769/BioProtoc.1911
37. Teo TH, Lum FM, Claser C, Lulla V, Lulla A, Mertis A, et al. A pathogenic role for CD4+ T cells during chikungunya virus infection in mice. J Immunol (2013) 190(1):259–69. doi: 10.4049/jimmunol.1202177
38. Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, et al. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am J Pathol (2011) 178:32–40. doi: 10.1016/j.ajpath.2010.11.018
39. Genin P, Lin R, Hiscott J, Civas A. Differential regulation of human interferon A gene expression by interferon regulatory factors 3 and 7. Mol Cell Biol (2009) 29(12):3435–50. doi: 10.1128/MCB.01805-08
40. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest (1999) 103:1345–52. doi: 10.1172/JCI5703
41. van Bezooijen RL, Farih-Sips HCM, Papapoulos SE, Lowik CWGM. Interleukin-17: A new bone acting cytokine in vitro. J Bone Min Res (1999) 14:1513–23. doi: 10.1359/jbmr.1999.14.9.1513
42. Wong CK, Ho CY, Li EK, Lam CW. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus (2000) 9:583–93. doi: 10.1191/096120300678828703
43. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut (2003) 52:65–70. doi: 10.1136/gut.52.1.65
44. Yen D, Cheung J, Scheeres H, Poulet F, McClanahan T, Mckenzie B, et al. IL-23 is essential for T-cell mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest (2006) 116:1310–6. doi: 10.1172/JCI21404
45. Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: Synergistic or antagonistic effects with IFN-g and TNF-a. J Immunol (1999) 162:492–502. doi: 10.1016/S0923-1811(98)84061-9
46. Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis, synoviocytes and its regulation by Th2 cytokines. J Immunol (1998) 161:409–14.
47. Miossec P. Interleukin-17 in rheumatoid arthritis: If T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum (2003) 48:594–601. doi: 10.1002/art.10816
48. Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: Microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J Leukoc Biol (2005) 77:388–99. doi: 10.1189/jlb.0904490
49. Lubberts E, Koenders MI, van den Berg WB. The role of T cell interleukin-17 in conducting destructive arthritis: Lessons from animal models. Arthritis Res Ther (2005) 7:29–37. doi: 10.1186/ar1478
50. Dong C, Nurieva RI. Regulation of immune and autoimmune responses by ICOS. J Autoimmun (2003) 21:255–60. doi: 10.1016/S0896-8411(03)00119-7
51. Gaffen SL. Interleukin-17: A unique inflammatory cytokine with roles in bone biology and arthritis. Arth Res Ther (2004) 6:240–7. doi: 10.1186/ar1444
52. Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity (2004) 21:467–76. doi: 10.1016/j.immuni.2004.08.018
53. Koenders MI, Kolls JK, Oppers-Walgreen B, van den Bersselaar L, Joosten LA, Schurr JR, et al. Interleukin-17 receptor deficiency results in impaired synovial expression of interleukin-1 and matrix metalloproteinases 3, 9, and 13 and prevents cartilage destruction during chronic reactivated streptococcal cell wall-induced arthritis. Arthritis Rheum (2005) 52:3239–47. doi: 10.1002/art.21342
54. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med (1996) 183:2593–603. doi: 10.1084/jem.183.6.2593
55. Yao Z, Fanslow WC, Seldin MF, Rousseau A-M, Painter SL, Comeau MR, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity (1995) 3:811–21. doi: 10.1016/1074-7613(95)90070-5
56. Katz Y, Nadiv O, Beer Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukin 1, 6, and 8 in skin and synovial fibroblasts: a possible role as a ‘fine-tuning cytokine’ in inflammation processes. Arthritis Rheum (2001) 44:2176–84. doi: 10.1002/1529-0131(200109)44:9<2176::AID-ART371>3.0.CO;2-4
57. Teng TS, Kam YW, Lee B, Hapuarachchi HC, Wimal A, Ng LC, et al. A systematic meta-analysis of immune signatures in patients with acute chikungunya virus infection. J Infect Dis (2015) 211(12):1925–35. doi: 10.1093/infdis/jiv049
58. Couderc T, Chretien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PloS Pathog (2008) 4(2):e29. doi: 10.1371/journal.ppat.0040029
59. Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol (2000) 74:3366–78. doi: 10.1128/JVI.74.7.3366-3378.2000
60. Trgovcich J, Aronson JF, Johnston RE. Fatal Sindbis virus infection of neonatal mice in the absence of encephalitis. Virology (1996) 224:73–83. doi: 10.1006/viro.1996.0508
61. Laurent P, Le Roux K, Grivard P, Betil G, Naze F, Picard M, et al. Development of a sensitive real-time reverse transcriptase PCR assay with an internal control to detect and quantify Chikungunya virus. Clin Chem (2007) 53:1408–14. doi: 10.1373/clinchem.2007.086595
62. Cook LE, Locke MC, Young AR, Monte K, Hedberg ML, Shimak RM, et al. Distinct roles of interferon alpha and beta in controlling chikungunya virus replication and modulating neutrophil-mediated inflammation. J Virol (2019) 94(1):e00841-19. doi: 10.1128/JVI.00841-19
63. Goertz GP, McNally KL, Robertson SJ, Best SM, Pijlman GP, Fros JJ. The methyltransferase-like domain of chikungunya virus nsP2 inhibits the Interferon response by promoting the nuclear export of STAT1. J Virol (2018) 92(17):e01008–18. doi: 10.1128/JVI.01008-18
64. Strauss JH, Strauss EG. The alphaviruses – Gene-expression, replication and evolution. Microbiol Rev (1994) 58:491–562. doi: 10.1128/MMBR.58.3.491-562.1994
65. Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J Virol (2004) 78:8455–67. doi: 10.1128/JVI.78.16.8455-8467.2004
66. Pesch VV, Lanaya H, Renauld JC, Michiels T. Characterization of the murine alpha interferon gene family. J Virol (2004) 78(15):8219–28. doi: 10.1128/JVI.78.15.8219-8228.2004
67. Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of IRF-3 Transcription Factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol (1998) 18:2986–96. doi: 10.1128/MCB.18.5.2986
68. Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-β gene. FEBS Lett (1998) 425:112–6. doi: 10.1016/S0014-5793(98)00210-5
69. Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM. Regulation of type I Interferon gene expression by interferon regulatory factor-3. J Biol Chem (1998) 273:2714–20. doi: 10.1074/jbc.273.5.2714
70. Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol Cell (1998) 1:507–18. doi: 10.1016/S1097-2765(00)80051-9
71. Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J (1998) 17:1087–95. doi: 10.1093/emboj/17.4.1087
72. Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J (1998) 17:6660–9. doi: 10.1093/emboj/17.22.6660
73. Honda K, Takaoka A, Taniguchi T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
74. Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tamaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett (1998) 441:106110. doi: 10.1016/S0014-5793(98)01514-2
75. Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PloS Pathog (2013) 9(1):e1003118. doi: 10.1371/journal.ppat.1003118
76. Loseke S, Grage-Griebenow E, Wagner A, Gehlhar K, Bufe A. Differential expression of IFN-alpha subtypes in human PBMC: evaluation of novel real-time PCR assays. J Immunol Methods (2003) 276:207–22. doi: 10.1016/S0022-1759(03)00072-3
77. Moll HP, Maier T, Zommer A, Lavoie T, Brostjan C. The differential activity of interferon-alpha subtypes is consistent among distinct target genes and cell types. Cytokine (2011) 53:52–9. doi: 10.1016/j.cyto.2010.09.006
78. Scagnolari C, Trombetti S, Selvaggi C, Carbone T, Monteleone K, Spano L, et al. In vitro sensitivity of human metapneumovirus to type I interferons. Viral Immunol (2011) 24:159–64. doi: 10.1089/vim.2010.0073
Keywords: IL-17A, IFN-α2, chikungunya virus, IRF5/7, inflammation, mice
Citation: Neupane B, Acharya D, Nazneen F, Gonzalez-Fernandez G, Flynt AS and Bai F (2020) Interleukin-17A Facilitates Chikungunya Virus Infection by Inhibiting IFN-α2 Expression. Front. Immunol. 11:588382. doi: 10.3389/fimmu.2020.588382
Received: 28 July 2020; Accepted: 19 October 2020;
Published: 16 November 2020.
Edited by:
Bin Su, Capital Medical University, ChinaReviewed by:
Penghua Wang, University of Connecticut Health Center, United StatesCopyright © 2020 Neupane, Acharya, Nazneen, Gonzalez-Fernandez, Flynt and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengwei Bai, ZmVuZ3dlaS5iYWlAdXNtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.