
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 November 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.587946
This article is part of the Research Topic Autoimmune Vasculitis: Advances in Pathogenesis and Therapies View all 29 articles
 Arun Rawat1
Arun Rawat1 Darawan Rinchai1
Darawan Rinchai1 Mohammed Toufiq1
Mohammed Toufiq1 Alexandra K. Marr1
Alexandra K. Marr1 Tomoshige Kino1
Tomoshige Kino1 Mathieu Garand1
Mathieu Garand1 Zohreh Tatari-Calderone1
Zohreh Tatari-Calderone1 Basirudeen Syed Ahamed Kabeer1
Basirudeen Syed Ahamed Kabeer1 Navaneethakrishnan Krishnamoorthy1
Navaneethakrishnan Krishnamoorthy1 Davide Bedognetti1,2
Davide Bedognetti1,2 Mohammed Yousuf Karim3
Mohammed Yousuf Karim3 Konduru S. Sastry1
Konduru S. Sastry1 Damien Chaussabel1*
Damien Chaussabel1*Transcriptome profiling approaches have been widely used to investigate the mechanisms underlying psoriasis pathogenesis. Most researchers have measured changes in transcript abundance in skin biopsies; relatively few have examined transcriptome changes in the blood. Although less relevant to the study of psoriasis pathogenesis, blood transcriptome profiles can be readily compared across various diseases. Here, we used a pre-established set of 382 transcriptional modules as a common framework to compare changes in blood transcript abundance in two independent public psoriasis datasets. We then compared the resulting “transcriptional fingerprints” to those obtained for a reference set of 16 pathological or physiological states. The perturbations in blood transcript abundance in psoriasis were relatively subtle compared to the changes we observed in other autoimmune and auto-inflammatory diseases. However, we did observe a consistent pattern of changes for a set of modules associated with neutrophil activation and inflammation; interestingly, this pattern resembled that observed in patients with Kawasaki disease. This similarity between the blood-transcriptome signatures in psoriasis and Kawasaki disease suggests that the immune mechanisms driving their pathogenesis might be partially shared.
Inflammation has an important role to play as part of the host defense against infection. However, prolonged or excessive inflammation can cause notable pathology (1–3). One example of such a pathology is psoriasis, which affects ~100 million individuals worldwide (4). This common, immune-mediated disease results in a unique skin barrier abnormality caused by excessive epidermal proliferation and inflammation (5, 6). Psoriasis pathogenesis is likely driven by many factors, including environmental triggers, genetic susceptibility, and even microbiome composition (1, 6, 7). At the cellular level, an interaction between innate and adaptive immune responses, and the activation of Th17 and Th1 cells are key to the immunopathogenesis (8). Plasmacytoid dendritic cells (DCs) found in psoriatic skin are activated by antigens and subsequently release IFN-α. Meanwhile, myeloid DCs secrete IL-23 and IL-12, which favors T-cell differentiation into Th17 and Th1 pathways, respectively (9, 10). In turn, Th17-derived cytokines including IL-17A and IL-22 play a dominant role in driving keratinocyte activation and proliferation. Finally, TNF-α secreted by DCs, Th17, and Th1 cells, and keratinocytes in psoriatic skin amplifies and perpetuates inflammation. Current treatment modalities include topical glucocorticoids, vitamin D analogues, phototherapy, conventional immunosuppressives (e.g., ciclosporin, methotrexate), and various biologics that target TNF-α (e.g., infliximab), the IL-17 pathway (e.g., secukinumab), and IL12/IL-23 (e.g., ustekinumab) (9–12). But despite such progress in understanding the molecular mechanisms driving psoriasis, we are still far from having a complete understanding of the immunopathogenesis and developing highly effective therapeutics.
To gain a better understanding of psoriasis pathophysiology, many researchers have compared the transcriptome profiles of diseased vs. healthy skin tissues isolated from affected patients (13–17). By contrast, only a few research groups have profiled genome-wide transcript abundance in blood samples from patients (18, 19). Measuring transcript abundance in the blood might seem less applicable to studies of skin diseases such as psoriasis; however, the blood presents the advantage of being highly accessible and amenable to serial sampling. Thus, blood transcriptional profiling could be harnessed to monitor dynamic treatment responses. Another advantage is the availability of numerous public blood transcriptome datasets, which allows us to make comparative analyses across various inflammatory diseases.
Here, we compared the blood transcriptome fingerprints of two publicly available psoriasis datasets (18, 19) with those derived from a collection of 16 reference patient cohort datasets (20). Our functional interpretations relied on extensive annotations and expression patterns observed in purified leukocyte populations.
The public datasets used in this re-analysis and interpretation were available in the NCBI GEO repository (21) (Supplementary Table 1). They include psoriasis blood transcriptome datasets as well as a collection of reference transcriptome datasets used for contextual interpretation. They are described in brief here:
Two psoriasis blood transcriptome datasets were identified for which data analysis was performed as detailed below. Both comprised control groups of subjects. Findings were reported in the literature by their original contributors:
● The GSE55201 dataset contributed by Wang et al. was generated using an Affymetrix U133 Plus (microarray) and consists of profiles for 81 samples. The study examined the role of IL-17 in ameliorating systemic inflammation and its impact on psoriasis complications, such as atherosclerosis and ischemic cardiovascular disease (19).
● The GSE123786 dataset contributed by Catapano et al. was generated using an Illumina HiSeq 2000 platform (RNA-Seq) and consists of profiles for 16 samples. This study examined the involvement of IL-36 in extracutaneous manifestations of psoriasis (18).
Other blood transcriptome datasets were used as reference:
● The GSE100150 dataset (20) contributed by our group was generated using an Illumina HumanWG-6 v3.0 BeadChip and consists of 16 reference patient cohorts encompassing the profiles of 985 subjects/samples.
Two other reference datasets were used to functionally interpret gene signatures:
● The GSE24759 dataset contributed by Novershtern et al. (22) was generated using Affymetrix U133A GeneChip and consists of 211 samples. The samples were collected from 4 to 7 donors and a wide range of hematopoietic cell populations from both adult and cord blood were profiled.
● The GSE60424 dataset (23) contributed by our group was generated using an Illumina RNA-Seq platform and consists of 134 subject/samples profiles. In this study, leukocyte populations were isolated from the blood of healthy individuals and patients with diabetes mellitus type 1, amyotrophic lateral sclerosis, multiple sclerosis (MS; pre- and post-interferon treatment) or sepsis.
The analysis workflow determines for each module the proportion of its constitutive transcripts that significantly differ in comparison with a given baseline (e.g., healthy controls). Thus, at the module level, changes are expressed as the proportion of transcripts constituting a given module being significantly increased (0 to 100%) or decreased (0 to −100%) compared with healthy controls. By design, changes in abundance among transcripts within a given module tend to be coordinated. However, when both significant increases and decreases are observed for the same module, the dominant trend is retained.
Data pre-processing steps for the two public psoriasis blood transcriptome datasets were performed as follows: The Wang et al. dataset (GSE55201) was generated using Affymetrix GeneChip and normalized with GCRMA (24). The Catapano et al. dataset (GSE123786) was generated via RNA-Seq data and the data are presented as RPKM values (reads per kilobase of transcript, per million mapped reads) after mapping with the HG38 genome build; the read counts were calculated using htseq-count (25).
Modular repertoire analyses were performed at the group level on both GSE55201 and GSE123786 psoriasis datasets using the BloodGen3Module R package: https://github.com/Drinchai/BloodGen3Module (26). The pre-determined repertoire of 382 co-expressed blood transcriptional modules that served as a framework for this analysis was described by Altman et al. (20). Briefly, it was constituted based on co-expression observed across a collection of 16 reference datasets encompassing 985 unique blood transcriptome profiles and 14,168 transcripts. A wide range of immune states are represented in this collection of reference datasets, including several infectious diseases, autoimmune diseases, inflammatory disorders as well as cancer, pregnancy, and solid organ transplantation. Because this module repertoire is “fixed” and destined for reuse as a generic framework for blood transcriptome analyses, considerable efforts were dedicated to its annotation and functional characterization. This work included functional enrichment analyses (ontologies, pathways, literature terms), and the generation of heatmaps representing transcript abundance patterns for reference datasets. The latter included, for instance, profiling data from isolated leukocyte populations. Interactive presentations were established to provide access to the large compendium of analysis reports and heatmaps that were generated as part of these annotation efforts. The presentation for a subset of 21 modules associated with inflammation that will be discussed in more detail as part of this work can be accessed via this link: https://prezi.com/view/GkH4wHb0jhIbDGt7Ibwi/. A demonstration video can be accessed via this link: https://youtu.be/fTfqGhcCNdE. However, it should be noted that module annotation has an element of subjectivity and is still a work-in-progress. Additionally, some of the functional “labels” that have been assigned are still tentative and subject to change as data analyses and interpretations progress across several projects.
The percent of increased or decreased transcripts computed per module were represented on a fingerprint grid plot. In brief, modules occupy a fixed position on the grid and changes for that module are indicated by a red spot (increased abundance compared to controls) or a blue spot (decreased abundance compared to controls). All fingerprints plots show changes in transcript abundance in cases compared to respective healthy controls (run concomitantly and matched for demographics). Modules arranged on the same row belong to one of 38 “module aggregates.” These aggregates are formed based on similarities in the patterns of transcript abundance changes across the 16 reference datasets. Thus, a vertical reading of the grid across the rows gives an indication of the patterns of change in a given set of patients at the least granular level (aggregates). A second horizontal reading within each row and across the columns gives an indication of the changes occurring at a more granular level (modules). Functional interpretations are indicated by a color code that is overlaid on the grid plot (Supplementary Figure 1). Because the positions of the modules on the grid are fixed, different fingerprints generated for independent groups of patients can be compared. Fingerprint grid plots for all of the 16 reference cohorts can be generated dynamically using a previously developed app: https://drinchai.shinyapps.io/dc_gen3_module_analysis/#.
Transcripts among the A35 modules were screened for the presence of drug targets using the “open targets platform” that is available via the open targets consortium at https://www.targetvalidation.org/ (27). The batch query functionality was used. The transcripts encoding targets of existing drugs (referred to in the results as “targets with clinical precedence”) were retrieved (Table 1).
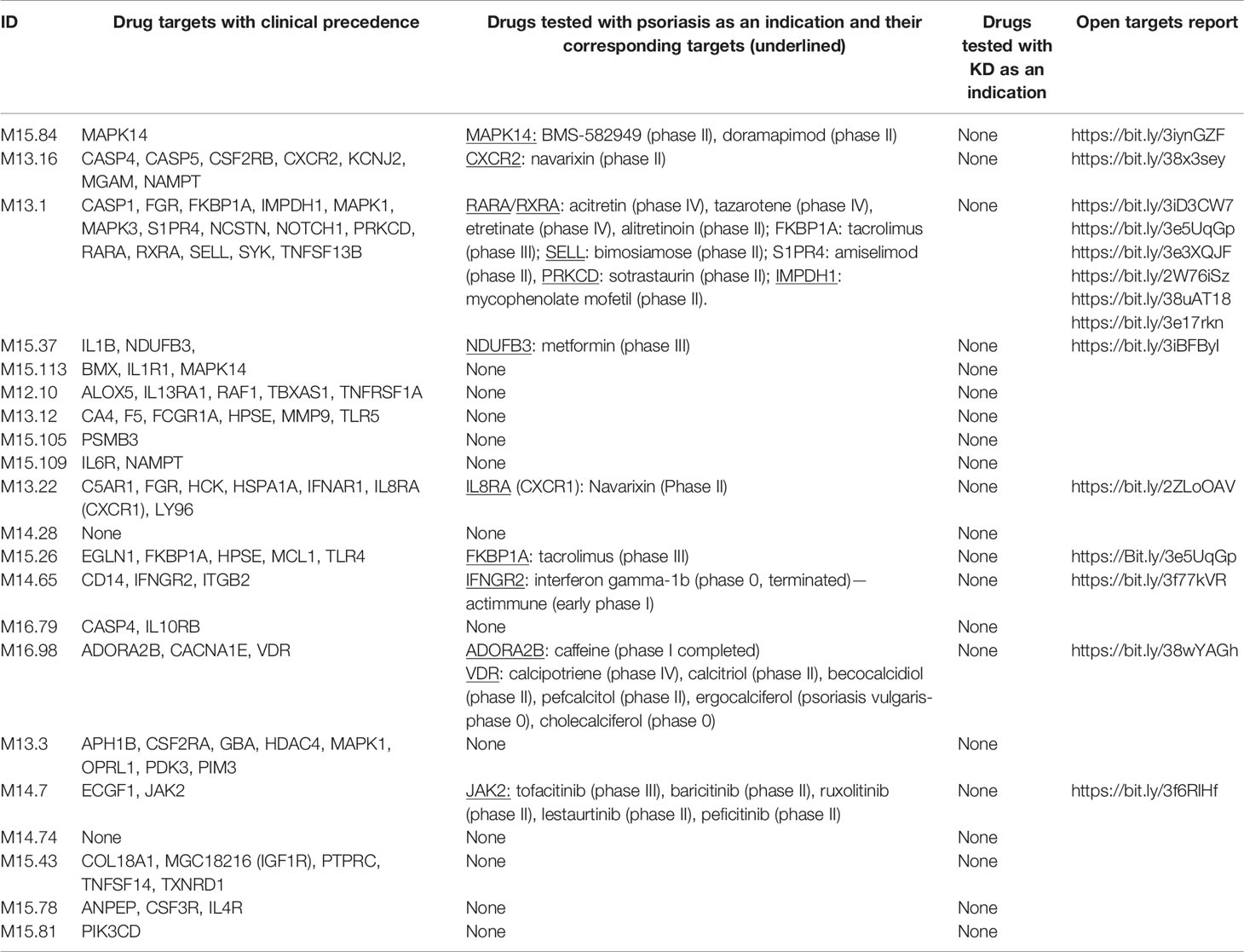
Table 1 Transcripts comprised in aggregate A35 for which the gene products are targetable by existing drugs, and the drugs tested in psoriasis or Kawasaki disease (see Methods).
We first aimed to determine whether we could measure robust changes in transcript abundance in the blood of psoriasis patients in comparison to heathy controls. We presumed that such signatures, if present, could then be “benchmarked” against that of other inflammatory or autoimmune diseases.
For this first step, we harnessed data from two psoriasis blood transcriptome datasets of a relatively modest size that have been published and made available via the NCBI GEO repository (18, 19). The technology platforms used to generate each dataset were quite dissimilar: Wang et al. used microarrays while Catapano et al. performed RNA sequencing. We previously showed that differences in transcript abundance summarized at the level of coordinately expressed gene sets (modules) are more amenable to cross-platform comparisons than when differences are expressed at the individual gene level (28). We therefore used a pre-determined repertoire of blood transcriptome modules that was recently developed and characterized by our group (20) (see Methods). Briefly, we formed this repertoire on the basis of co-expression measured across 16 reference patient cohorts, encompassing 985 unique blood transcriptome profiles. Two-dimensional reduction levels are built into the repertoire. The least reduced level has 382 variables, which are the modules that are constituted by sets of genes. The most reduced level has 38 variables, which are module aggregates that are constituted by sets of modules that altogether encompass the 382-module repertoire. Changes between cases and controls are expressed as a proportion of the transcripts constituting a given module found to be significantly increased (max +100%, all transcripts are increased) or decreased (−100%). We thus determined differences in transcript abundance for each of the 382 modules for the Wang et al. and Catapano et al. datasets. We represented these differences on a fingerprint grid plot, where the assignment of modules to a given position on the grid was fixed (Figure 1).
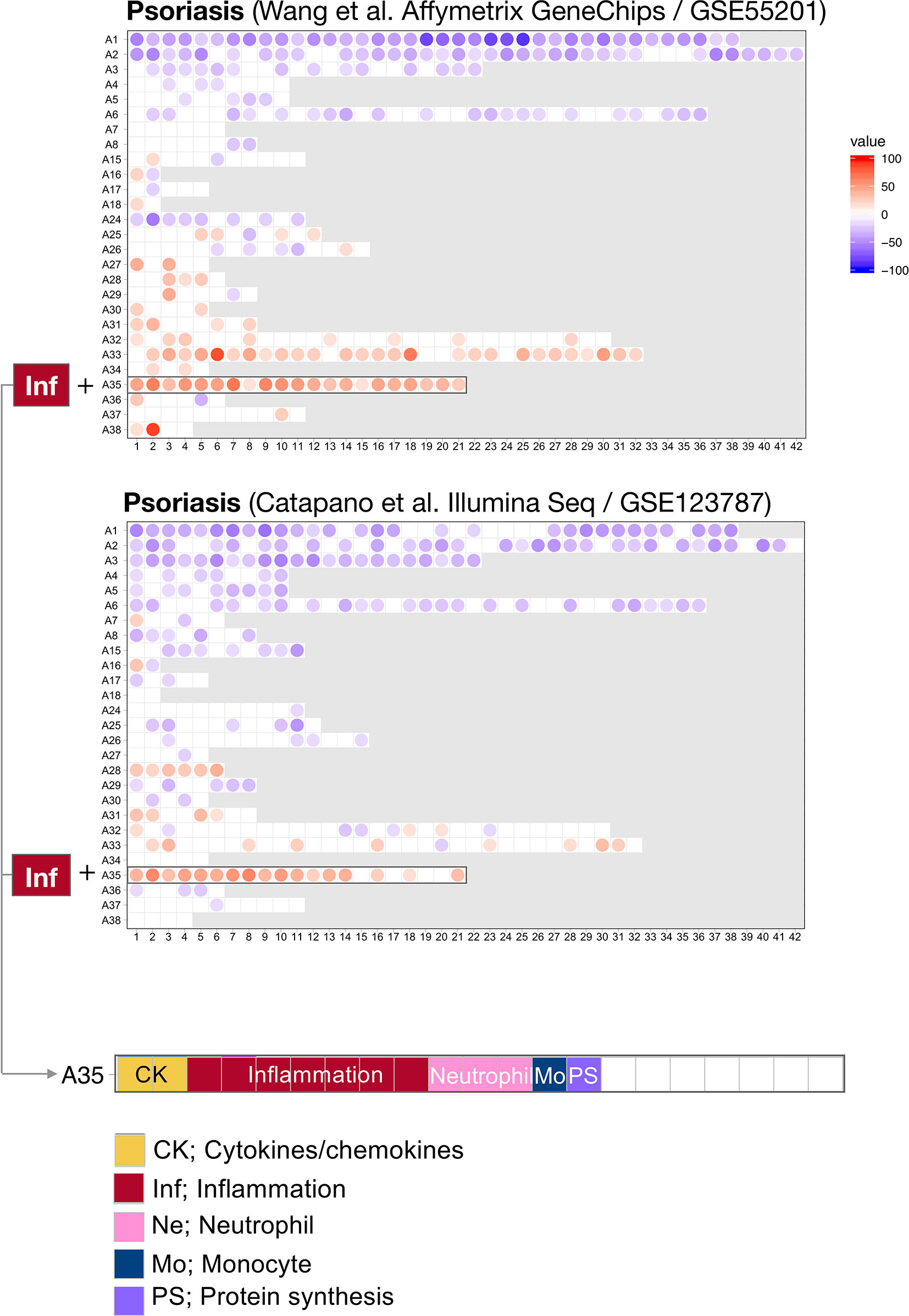
Figure 1 Blood transcriptome fingerprints of psoriasis. Differences in the levels of blood transcript abundance in patients with psoriasis and controls were mapped on a grid for the two public datasets from Wang et al. (GSE55201: 30 controls and 51 subjects) and Catapano et al. (GSE123786: 7 controls and 9 subjects). Each position on the grid is occupied by a given module (pre-determined set of co-expressed genes). A blue spot indicates a module for which constitutive transcripts are predominantly present at lower levels in patients vs. controls. Conversely, a red spot indicates a module for which constitutive transcripts are predominantly present at higher levels in patients vs. controls. No spots on a white background indicate that there are no changes for the module in question. A gray background means that there are no changes in the module at this position. The modules are arranged by rows in “module aggregates” and ordered by their similarity in expression patterns across a set of 16 disease or physiological states (reference dataset collection). A consistent increase was observed for modules constituting aggregate A35. This aggregate is highlighted on the grid and functional annotations are provided (bottom panel). Functional annotations for the entire gird are provided in Supplementary Figure 1.
The fact that positions of modules on the grid are fixed ensures that the generated fingerprints are directly comparable. Here, we found a good level of concordance between the two datasets, with both predominantly showing changes for the 21 modules forming row A35 on the grid. At a high level, the module aggregate A35 is functionally associated inflammation (detailed below). In addition, the Catapano et al. dataset showed increases for modules forming row A28. The module aggregate A28 is functionally associated with interferon responses. Notably, an interferon signature was also reported by Catapano and colleagues (18), and seems to be associated with generalized pustular psoriasis, which is a severe form of the disease (1).
The fact that an increase in abundance of A35 modules was observed in both datasets suggests that this modular signature constitutes the main component of the blood transcriptome fingerprint associated with psoriasis overall. At a high level, seven of the 21 modules forming aggregate A35 were associated with inflammation, three with neutrophils, two with cytokines/chemokines, one with macrophages, and one with protein synthesis (Figure 1). The remaining seven modules were not associated with any given functional annotations due to lack of convergence between the functional profiling results obtained via different methodologies. The reports from gene ontology (GO), pathway and literature keyword enrichment analyses upon which these determinations were made (Figure 2), are available via an interactive presentation (https://prezi.com/view/7Q20FyW6Hrs5NjMaTUyW/) and all functional annotations of the A35 module are readily available (Table 2 and Supplementary File 1).
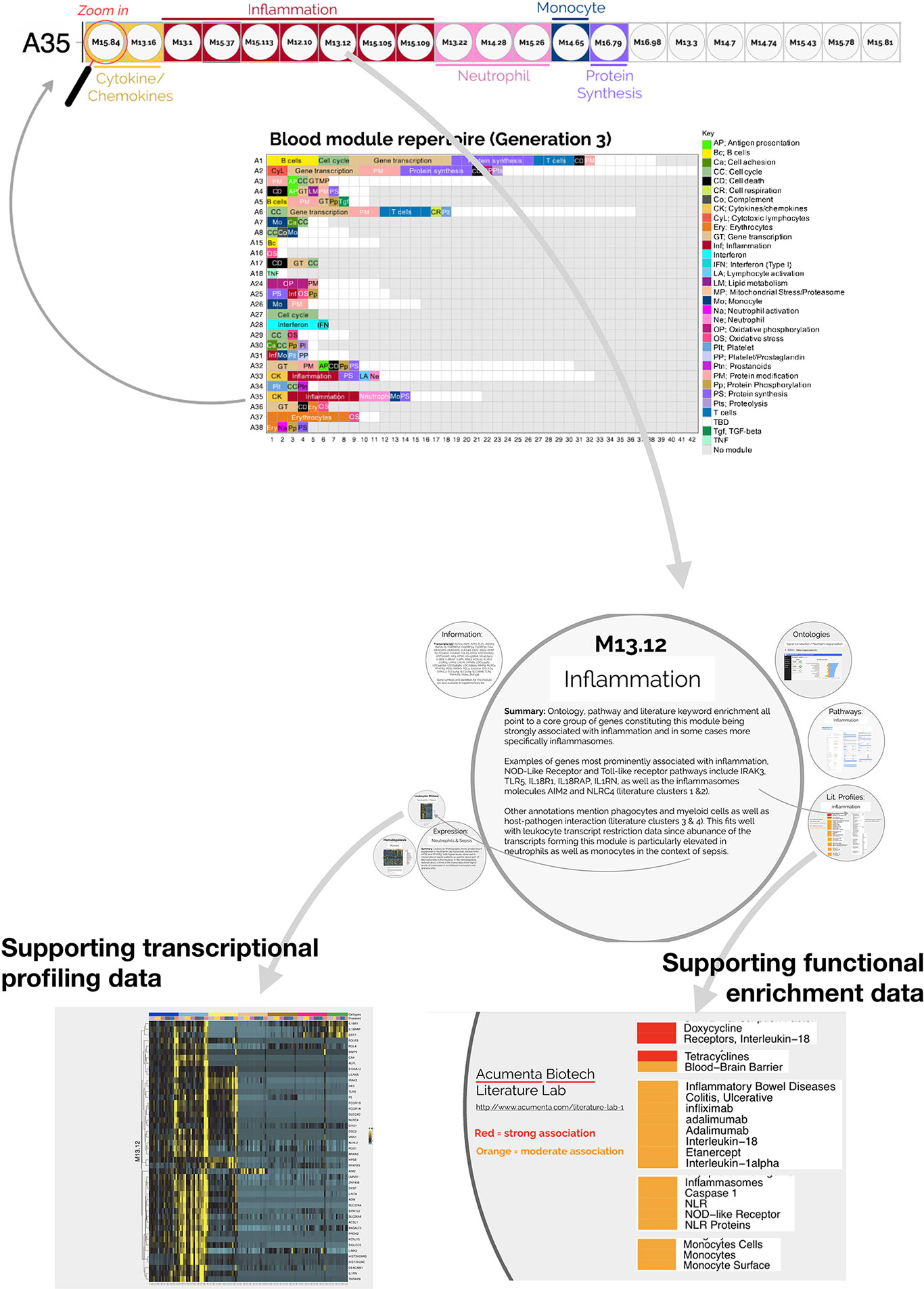
Figure 2 Interactive presentation providing transcriptional profiling and functional enrichment data for modules constituting aggregate A35. An interactive presentation has been developed that allows for exploration of the modules constituting aggregate A35. A gene list is provided for each module, along with gene ontology, pathway or literature term enrichment results and transcriptional profiling data for the reference transcriptome datasets (circulating leukocyte populations, hematopoiesis). A summary of the findings is also given. The interactive presentation is available via: https://prezi.com/view/7Q20FyW6Hrs5NjMaTUyW/. The presentation provides zoom in/out functionalities for close-up examination of the text and figures embedded in the presentation.
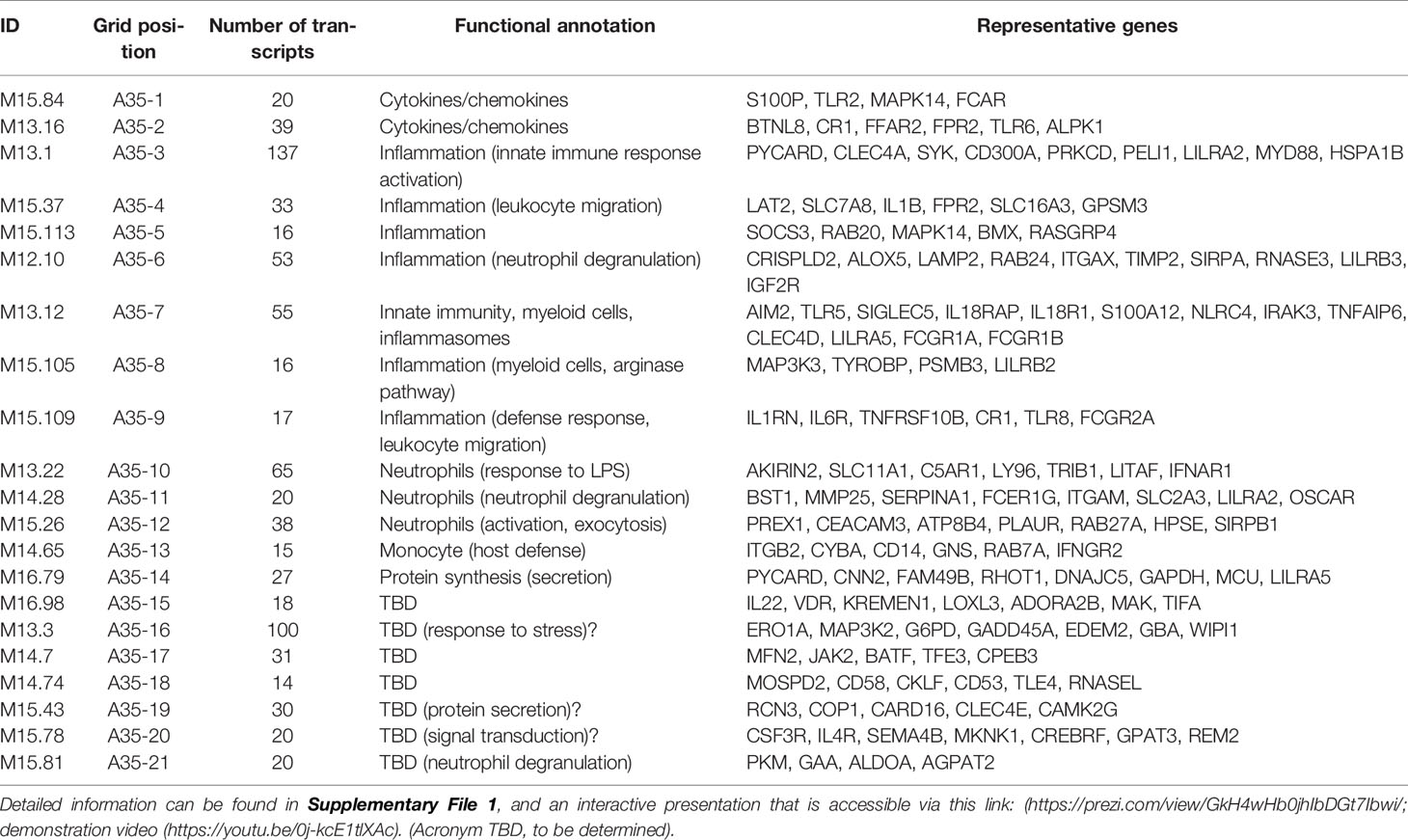
Table 2 The 21 modules constituting aggregate A35.
In summary, this step identified that modules forming aggregate A35 are conserved between two independent psoriasis blood transcriptome datasets. Notably, this convergence was evident even though distinct technology platforms were used to generate the respective datasets. Altogether, these findings indicate that a blood transcriptional signature can consistently be observed in the blood of psoriasis patients.
We next aimed to determine the relevance of the increase in A35 transcripts in the context of psoriasis pathogenesis. To do so, we proceeded with the functional interpretations of this signature.
The two converging themes that emerged through the extensive annotation work mentioned above were “neutrophil” and “inflammation.” For instance, enriched literature terms included “neutrophil degranulation,” “inflammation,” and “inflammasome.” Consistently, some of the genes in these modules are most recognizable as being involved in inflammatory processes, including those coding for inflammasome components. For example, NLR protein families were found across different modules within this aggregate, including NLRX1, NLRC4, NLRP12. Furthermore, “neutrophil activation involved in immune responses” (GO:0002283) was one of the most over-represented GO terms, with 121/784 transcripts forming modules belonging to aggregate A35. Thus, both gene composition and functional enrichment analyses suggest that this set of 21 modules constituting aggregate A35 is involved in inflammatory processes.
To complement our functional profiling analyses, we examined the expression patterns of the genes belonging to module A35 in reference transcriptome datasets (see Methods). In particular, a dataset that we previously generated and deposited in the GEO showed that among circulating leukocyte populations, the expression of A35 transcripts was predominant in neutrophils [Linsley et al. (GSE60424) (23), http://sepsis.gxbsidra.org/dm3/miniURL/view/Q0)] (Figure 3). In another dataset, also contributed by our group, we found that the expression of A35 transcripts was upregulated in neutrophils exposed to plasma from septic patients in vitro (29); http://sepsis.gxbsidra.org/dm3/miniURL/view/Q2.
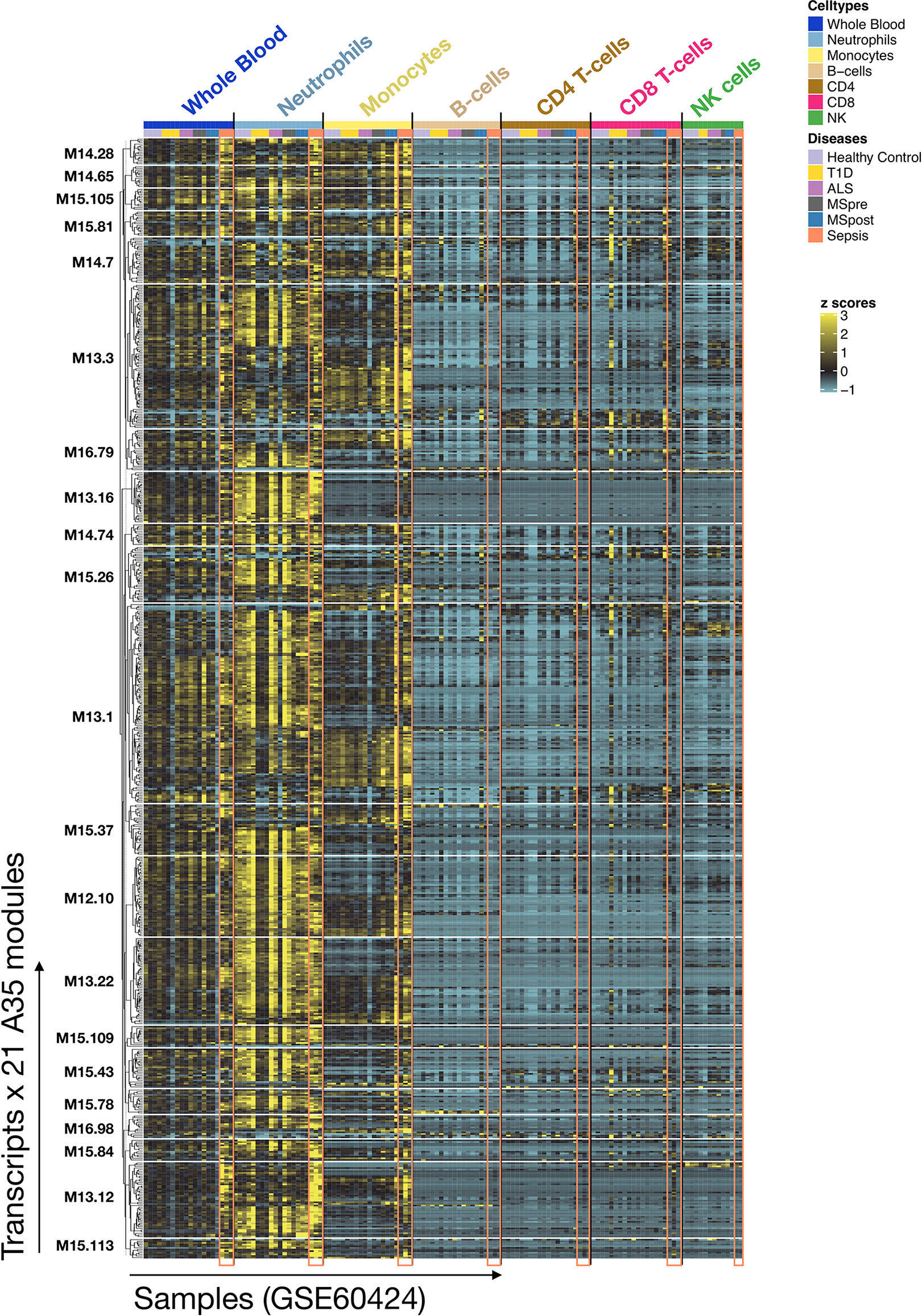
Figure 3 Expression patterns of genes constituting A35 modules across whole blood and purified leukocyte populations. The expression levels for genes constituting A35 modules are shown on a heatmap for a reference dataset comprising the profiles of isolated leukocyte populations (GSE60424). The rows represent genes, with each cluster of rows representing a module. The columns represent samples. This study compared the whole transcriptome signatures of six immune-cell subsets and whole blood from patients with one of an array of immune-associated diseases. Fresh blood samples were collected from healthy subjects and those diagnosed with type 1 diabetes, amyotrophic lateral sclerosis, sepsis, or multiple sclerosis (before and 24 h after the first dose of IFN-beta). RNA was extracted from each of the indicated cell subsets and whole blood samples, and then processed for RNA sequencing (Illumina TruSeq; 20M reads).
Altogether, functional and gene expression profiles observed in this reference dataset suggest that the A35 signature is associated with neutrophil-driven inflammation.
As mentioned, a benefit of examining transcriptome signatures in blood rather than skin samples from patients with psoriasis is that it lends itself to making comparisons across a wide range of diseases. Carrying out such comparisons allows us to draw parallels or identify differences with diseases for which the pathogenesis might be better understood and managed clinically.
To achieve this, we compared the module repertoire fingerprints of psoriasis with those of the 16 other diseases comprising the reference collection of datasets used to construct our module repertoire (20). As was the case for psoriasis, we observed an increase in abundance of A35 modules in the blood repertoire fingerprints of systemic lupus erythematous (SLE), systemic onset juvenile idiopathic arthritis (SoJIA), and Kawasaki disease (Figure 4). In the case of SLE and SoJIA, the increase in abundance of A35 transcripts was one of many “perturbations” of the blood transcriptome repertoire, which is consistent with the systemic inflammation that characterizes these two diseases [for example: modules in aggregates/rows A27-A29 (SLE) or A30-A38 (SoJIA)]. The fingerprints of patients with acute infections (e.g., bacterial sepsis, tuberculosis, or influenza infection) also showed pronounced changes (such fingerprints can be generated dynamically via our web application accessible at: https://drinchai.shinyapps.io/dc_gen3_module_analysis/#). Notably, the fingerprints of patients with sepsis closely resembled those of patients with SoJIA. In both pathologies, other modules functionally associated with inflammation, such as A33, also showed a robust increase in abundance; such increases were not observed in the context of psoriasis or Kawasaki disease. Indeed, patterns of abundance of A33 and A35 modules across the 16 reference patient cohorts and two psoriasis datasets indicated that A35 modules tend to be more ubiquitously increased in comparison to A33 modules (Figure 5). The relative difference in intensity of A33 and A35 signatures between the two psoriasis datasets also suggests that those signatures might be non-synonymous and represent distinct inflammation pathways. Conversely, the Kawasaki disease blood transcriptome repertoire fingerprint was more subtle and, like that of psoriasis, was mostly restricted to an increase in abundance of A35 module transcripts (Figure 4). This finding might reflect the fact that inflammation is typically localized at the onset of these diseases: to the skin for psoriasis and the vasculature endothelium for Kawasaki disease.

Figure 4 Blood transcriptional fingerprints of other autoimmune or autoinflammatory diseases. The differences in the levels of transcript abundance in the blood of patients with Kawasaki disease (top), systemic lupus erythematous (middle), or systemic onset juvenile idiopathic arthritis (bottom) are mapped on a grid, as described in Figure 1. The modules belonging to aggregate A35 are highlighted on this grid.

Figure 5 Patterns of abundance for modules forming aggregates A33 (A) and A35 (B). Heatmaps displaying the changes in transcript abundance for modules (columns) belonging to two aggregates associated with inflammation (A33 and A35), across 16 reference datasets and two psoriasis datasets (rows). The two psoriasis datasets are designated by the names of the researchers who contributed them and are indicated by green arrowheads. An increase or decrease in the abundance of transcripts constituting these modules is shown by a red or blue spot, respectively. The rows (datasets from each disease cohort) and columns (modules) were arranged by hierarchical clustering based on similarities in patterns of transcript abundance.
Taken together, these results suggest that the extent of changes in blood transcript abundance tends to correlate with the disease manifestation, from local (a low number of modules perturbed) to generalized inflammation (a high number of modules perturbed). Furthermore, the A35 transcriptional modules constituted the least common denominator across these inflammatory pathologies as it was the only set for which increases in transcript abundance were observed in all these immune-mediated diseases.
The identification of a robust transcriptional signature in the blood of psoriasis patients has several implications. For instance, it opens up the possibility of including blood transcript profiling assays in patient monitoring studies. For instance, such studies might be designed to predict the risk of flares or to monitor responses to therapy. We thus went on to examine the presence of transcripts among A35 modules that encode molecules that are targets for existing drugs and could be included in immune monitoring panels.
Among the 784 transcripts constituting the A35 modules, 81 are encoding targets for existing drugs (see Methods) (Table 1). Among these, we sought to identify targets for which drugs have been tested in clinical trials for psoriasis (14 targets) or Kawasaki disease (none). Notable examples for targets among A35 transcripts for which drugs have been considered for treatment of psoriasis include JAK2. This member of the Janus kinase family participates in signaling events downstream of a broad range of cytokine and hormone receptors. Drugs targeting this molecule that have been tested in the context of psoriasis include tofacitinib, which seems to be safe and to confer a clinical benefit (30, 31). Other immunosuppressive drugs have also been evaluated, such as the selective, pan-protein kinase C inhibitor sotrastaurin that inhibits the kinase PRKCD (32, 33).
Overall, we found that a sizeable number of transcripts comprised in A35 modules encode targets for existing drugs: a minority of these have already been tested in patients with psoriasis. We posit that other suitable candidates might be included in this list that have not yet been evaluated in the context of psoriasis. Furthermore, given the parallels in the blood transcriptome signatures of psoriasis and Kawasaki disease, there is good cause to consider investigating repurposing drugs showing clinical benefit in patients with psoriasis for the treatment of patients with Kawasaki disease.
Involved skin tissue is the ideal sample source to investigate psoriasis pathogenesis. However, despite being less relevant to this disease, blood presents the advantage of being amenable to repetitive sampling with minimal risk or discomfort. The blood can also harbor information regarding the immune status of affected patients. Such information can be obtained via blood transcriptome profiling, whereby all RNA species that are present in a given sample are measured simultaneously. Maybe more importantly, vast amounts of blood transcriptome profiling data are available in public repositories that can be used for contextual interpretation and “benchmarking” of blood transcriptional signatures.
Here, we compared the blood transcriptome fingerprints derived from several inflammatory diseases with those derived from patients with psoriasis. We found that blood transcriptome profiling may indeed serve to assess the extent of systemic involvement in these pathologies. Interestingly, we saw that the repertoire of changes characterizing the psoriasis blood transcriptome signature is much narrower than what is observed in other systemic inflammatory diseases. We also established that modules assigned to the aggregate A35 and associated with neutrophil-driven inflammation are a hallmark of the psoriasis blood transcriptome signature.
The role played by neutrophils in psoriasis pathogenesis has received particular attention over recent years (34, 35). Consistently, the data from our study suggest that blood transcriptome profiling studies might be of value for further patient-based investigations. While such an approach has been relatively under-utilized in this context, our findings suggest the possibility of employing blood transcriptional profiling as a means to assess the extent of systemic inflammation in psoriasis patients. Whether these measurements add value to those obtained using more traditional inflammatory markers (e.g., measurement of serum protein markers or neutrophil: lymphocyte ratios) remains to be investigated. It may be particularly relevant to assess utility of such blood transcriptional markers for the evaluation of cardiovascular diseases (CVD) risk in patients suffering from inflammatory disorders. Indeed, systemic inflammation associated with psoriasis was recently linked with development of CVD in this patient population (36, 37), while the risk of cardiovascular symptoms in Kawasaki disease is well established (38). And the question of the relative benefits of the available psoriasis treatment options with regards to addressing this risk remains to be fully addressed (36, 39).
Blood transcriptome profiling may also help stratify psoriasis patients according to molecular/immunological types. Such classification may be achieved through the delineation of distinct, biologically relevant modular A35 “sub-signatures.” For instance, our previous work identified distinct modular interferon signatures that formed the basis of a stratification system for SLE patients (20). Psoriasis classification might also be informed by measuring the changes in the abundance of other aggregates/modules. For instance, we observed changes in the abundance of transcripts comprising the A33 and A28 modules, (also associated with inflammation and interferon responses, respectively) in either one of the two psoriasis datasets. This was the case for A28 (interferon) in the GSE123787 fingerprints (Figure 1), which is in line with the interpretation contributed earlier by Catapano et al. (18).
Some of our findings may also be relevant from a drug discovery/repurposing standpoint. For one, a number of the gene products comprising the A35 signature are targeted by drug candidates for psoriasis treatment (Table 2). This finding suggests that—according to the principle of “guilt by association”—other valuable targets might be identified among the genes constituting these modules. Secondly, similarities observed between the psoriasis and Kawasaki disease fingerprints suggest that the pathogenesis and/or pathophysiology of these diseases might be driven, at least in part, by similar immune mechanisms. Kawasaki disease, also known as mucocutaneous lymph node syndrome, is a rare childhood disease that mostly affects children <5 years old (38, 40). This disease presents as an acute, self-limiting vasculitis that sometimes targets coronary arteries and causes ischemic heart disease (41–43). The parallels we drew between psoriasis and Kawasaki disease blood transcriptional signatures are consistent with the growing body of evidence showing that patients with Kawasaki disease can develop psoriasiform eruptions (44–48). Among the treatments approved for psoriasis, drugs inhibiting IL17 might be considered good candidates for repurposing in Kawasaki disease (9, 49). Independent reports have also associated Th17 responses (defined by IL17 production) with Kawasaki disease (50–53), and IL17 is a known driver of neutrophil development, recruitment, and activation (54, 55). We therefore posit that IL17 might constitute one of the factors underpinning the A35 signature that we identified here in patients with psoriasis and Kawasaki disease.
Several aspects of the benchmarking exercise that we have conducted here across independent studies are inherently limiting and need to be addressed in follow-on investigations. First, while the use of respective healthy control groups as common denominators permits comparisons across independent studies, further investigations should comprise cohorts of patients with psoriasis, Kawasaki disease, and healthy controls. Samples should be collected and processed using harmonized protocols and the generated data should be analyzed concomitantly using the same platform (RNA-seq). This approach would permit direct comparisons of psoriasis and Kawasaki disease profiles while minimizing technical sources of variation. Inclusion of healthy controls would help with the interpretation of the data and would also permit future data re-use and meta-analyses across independent studies.
Second, the cohort size should be sufficient to allow for investigations into inter-individual variability. Such investigations would permit, for instance, the identification of “endotypes” or distinct molecular phenotypes within each patient population. The relatively low cost of recently introduced RNA-seq protocols might help realize such sample sizes (e.g., QuantSeq 3’ mRNA-Seq by Lexogen: <$100/sample). Consolidating the results from multiple studies would remain feasible but any level of coordination or consultation between groups/centers could prove helpful.
Third, future studies are needed to clarify whether the A33/A35 signature observed in patients with psoriasis or Kawasaki disease is due to neutrophil priming secondary to inflammation or is a causal component of psoriasis pathophysiology. It can be difficult to ascertain in patient-based studies whether signatures are merely associated with or drive pathogenesis. Monitoring changes in transcript abundance at a high temporal frequency, either prior to a worsening of the clinical course of the disease or in response to therapy might provide useful indications in that sense. From a practical perspective, such studies could be implemented using protocols for at-home self-collection of low blood volumes and RNA stabilization (56, 57).
Finally, investigations into immune changes in the periphery and in bulk whole blood samples have inherent limitations. While systemic inflammation and interferon responses can be measured in whole blood, a dissection of the immune response at a more granular level (e.g., cellular subsets) might not be possible. Indeed, it is possible that we did not identify some responses associated with psoriasis pathogenesis (e.g., T-cell responses) for this reason. In addition, at least some immune responses may only be observed in affected skin tissues. Studies harnessing single-cell RNA-seq in a subset of patients are now warranted to further interrogate and interpret the psoriasis immune signatures measured in the peripheral blood.
Overall, our study provides a proof-of-principle for the use of fixed transcriptional module repertoires for blood transcriptome signature “benchmarking” and cross-study comparisons. It highlights the pertinence of using transcriptomic approaches for monitoring systemic inflammation. And it may also provide the necessary justification for further blood transcriptome studies in the context of psoriasis and Kawasaki disease.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/ (the NCBI Gene Expression Omnibus).
AR, and DC: conceptualization. AR, KS and DC: data curation and validation. AR and DR: visualization. AR, AM, TK, MK, and ZT-C: analysis and interpretation. AR, DR, MT, MG, BK, DB, and NK: methodology development. AR and DC: writing of the first draft. AR, DR, MT, AM, TK, MG, ZT-C, NK, DB, MK, KS, and DC: writing—review and editing. The contributor’s roles listed above follow the Contributor Roles Taxonomy (CRediT) managed by The Consortia Advancing Standards in Research Administration Information (CASRAI) (https://casrai.org/credit/). All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the fellow researchers who made public the transcriptome datasets used in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.587946/full#supplementary-material
Supplementary Figure 1 | Module annotation grid. Complete annotation for the grid used to map the differences in transcript abundance between the cases and controls shown in Figure 1. Each position on the grid corresponds to a different module. Each of the 382 modules are constituted by a set of transcripts found to be co-expressed across a range of disease and physiological states. In turn, the modules are arranged in rows based on similarities in gene expression. Gene ontology, pathway or literature keyword enrichment analyses provide the basis for attribution of biological functions to the modules; these are indicated by the color-coded abbreviations list below the grid. An interactive presentation is available that permits the exploration of functional enrichment results and expression patterns for the A35 modules: https://prezi.com/view/7Q20FyW6Hrs5NjMaTUyW/.
1. Catapano M, Vergnano M, Romano M, Mahil SK, Choon S-E, Burden DA, et al. Interleukin-36 promotes systemic Type-I IFN responses in severe psoriasis. bioRxiv (2018). p. 496851. doi: 10.1101/496851
2. Mousa A, Misso M, Teede H, Scragg R, de Courten B. Effect of vitamin D supplementation on inflammation: protocol for a systematic review. BMJ Open (2016) 6(4):e010804. doi: 10.1136/bmjopen-2015-010804
3. Paparella D, Yau TM, Young E. Cardiopulmonary bypass induced inflammation: pathophysiology and treatment. An update. Eur J Cardiothorac Surg (2002) 21(2):232–44. doi: 10.1016/S1010-7940(01)01099-5
4. Michalek I, Loring B, John S. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol (2017) 31(2):205–12. doi: 10.1111/jdv.13854
5. Wang J, Suarez-Farinas M, Estrada Y, Parker ML, Greenlees L, Stephens G, et al. Identification of unique proteomic signatures in allergic and non-allergic skin disease. Clin Exp Allergy (2017) 47(11):1456–67. doi: 10.1111/cea.12979
6. Fry L, Baker BS, Powles AV, Engstrand L. Psoriasis is not an autoimmune disease? Exp Dermatol (2015) 24(4):241–4. doi: 10.1111/exd.12572
7. Meyer O. Interferons and autoimmune disorders. Joint Bone Spine (2009) 76(5):464–73. doi: 10.1016/j.jbspin.2009.03.012
8. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol (2017) 140(3):645–53. doi: 10.1016/j.jaci.2017.07.004
9. Silfvast-Kaiser A, Paek SY, Menter A. Anti-IL17 therapies for psoriasis. Expert Opin Biol Ther (2019) 19(1):45–54. doi: 10.1080/14712598.2019.1555235
10. van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol (2009) 182(9):5836–45. doi: 10.4049/jimmunol.0802999
11. Garber K. Anti-IL-17 mAbs herald new options in psoriasis. Nat Biotechnol (2012) 30(6):475–7. doi: 10.1038/nbt0612-475
12. Conrad C, Gilliet M. Psoriasis: from Pathogenesis to Targeted Therapies. Clin Rev Allergy Immunol (2018) 54(1):102–13. doi: 10.1007/s12016-018-8668-1
13. Fyhrquist N, Muirhead G, Prast-Nielsen S, Jeanmougin M, Olah P, Skoog T, et al. Microbe-host interplay in atopic dermatitis and psoriasis. Nat Commun (2019) 10(1):4703. doi: 10.1038/s41467-019-12253-y
14. Tomalin LE, Russell CB, Garcet S, Ewald DA, Klekotka P, Nirula A, et al. Short-term transcriptional response to IL-17 receptor-A antagonism in the treatment of psoriasis. J Allergy Clin Immunol (2020) 145(3):922–32. doi: 10.1016/j.jaci.2019.10.041
15. Brodmerkel C, Li K, Garcet S, Hayden K, Chiricozzi A, Novitskaya I, et al. Modulation of inflammatory gene transcripts in psoriasis vulgaris: Differences between ustekinumab and etanercept. J Allergy Clin Immunol (2019) 143(5):1965–9. doi: 10.1016/j.jaci.2019.01.017
16. Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, et al. Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. J Invest Dermatol (2014) 134(7):1828–38. doi: 10.1038/jid.2014.28
17. Correa da Rosa J, Kim J, Tian S, Tomalin LE, Krueger JG, Suarez-Farinas M. Shrinking the Psoriasis Assessment Gap: Early Gene-Expression Profiling Accurately Predicts Response to Long-Term Treatment. J Invest Dermatol (2017) 137(2):305–12. doi: 10.1016/j.jid.2016.09.015
18. Catapano M, Vergnano M, Romano M, Mahil SK, Choon SE, Burden AD, et al. IL-36 Promotes Systemic IFN-I Responses in Severe Forms of Psoriasis. J Invest Dermatol (2020) 140(4):816–26.e3. doi: 10.1016/j.jid.2019.08.444
19. Wang CQF, Suarez-Farinas M, Nograles KE, Mimoso CA, Shrom D, Dow ER, et al. IL-17 induces inflammation-associated gene products in blood monocytes, and treatment with ixekizumab reduces their expression in psoriasis patient blood. J Invest Dermatol (2014) 134(12):2990–3. doi: 10.1038/jid.2014.268
20. Altman MC, Rinchai D, Baldwin N, Toufiq M, Whalen E, Garand M, et al. Development and Characterization of a Fixed Repertoire of Blood Transcriptome Modules Based on Co-expression Patterns Across Immunological States. bioRxiv (2020). p. 525709. doi: 10.1101/525709
21. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res (2013) 41(Database issue):D991–5. doi: 10.1093/nar/gks1193
22. Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell (2011) 144(2):296–309. doi: 10.1016/j.cell.2011.01.004
23. Linsley PS, Speake C, Whalen E, Chaussabel D. Copy number loss of the interferon gene cluster in melanomas is linked to reduced T cell infiltrate and poor patient prognosis. PloS One (2014) 9(10):e109760. doi: 10.1371/journal.pone.0109760
24. Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc (2004) 99(468):909–17. doi: 10.1198/016214504000000683
25. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics (2015) 31(2):166–9. doi: 10.1093/bioinformatics/btu638
26. Rinchai D, Roelands J, Hendrickx W, Altman MC, Bedognetti D, Chaussabel D. Blood transcriptional module repertoire analysis and visualization using R. bioRxiv (2020). doi: 10.1101/2020.07.16.205963
27. Carvalho-Silva D, Pierleoni A, Pignatelli M, Ong C, Fumis L, Karamanis N, et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res (2019) 47(D1):D1056–65. doi: 10.1093/nar/gky1133
28. Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity (2008) 29(1):150–64. doi: 10.1016/j.immuni.2008.05.012
29. Khaenam P, Rinchai D, Altman MC, Chiche L, Buddhisa S, Kewcharoenwong C, et al. A transcriptomic reporter assay employing neutrophils to measure immunogenic activity of septic patients’ plasma. J Transl Med (2014) 12:65. doi: 10.1186/1479-5876-12-65
30. Strober BE, Gottlieb AB, van de Kerkhof PCM, Puig L, Bachelez H, Chouela E, et al. Benefit-risk profile of tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: pooled analysis across six clinical trials. Br J Dermatol (2019) 180(1):67–75. doi: 10.1111/bjd.17149
31. Menter MA, Papp KA, Cather J, Leonardi C, Pariser DM, Krueger JG, et al. Efficacy of Tofacitinib for the Treatment of Moderate-to-Severe Chronic Plaque Psoriasis in Patient Subgroups from Two Randomised Phase 3 Trials. J Drugs Dermatol (2016) 15(5):568–80.
32. He X, Koenen H, Smeets RL, Keijsers R, van Rijssen E, Koerber A, et al. Targeting PKC in human T cells using sotrastaurin (AEB071) preserves regulatory T cells and prevents IL-17 production. J Invest Dermatol (2014) 134(4):975–83. doi: 10.1038/jid.2013.459
33. Wagner J, von Matt P, Faller B, Cooke NG, Albert R, Sedrani R, et al. Structure-activity relationship and pharmacokinetic studies of sotrastaurin (AEB071), a promising novel medicine for prevention of graft rejection and treatment of psoriasis. J Med Chem (2011) 54(17):6028–39. doi: 10.1021/jm200469u
34. Chiang CC, Cheng WJ, Korinek M, Lin CY, Hwang TL. Neutrophils in Psoriasis. Front Immunol (2019) 10:2376. doi: 10.3389/fimmu.2019.02376
35. Schon MP, Broekaert SMC, Erpenbeck L. Sexy again: the renaissance of neutrophils in psoriasis. Exp Dermatol (2017) 26(4):305–11. doi: 10.1111/exd.13067
36. Masson W, Lobo M, Molinero G. Psoriasis and Cardiovascular Risk: A Comprehensive Review. Adv Ther (2020) 37(5):2017–33. doi: 10.1007/s12325-020-01346-6
37. Hu SC, Lan CE. Psoriasis and Cardiovascular Comorbidities: Focusing on Severe Vascular Events, Cardiovascular Risk Factors and Implications for Treatment. Int J Mol Sci (2017) 18(10):2211. doi: 10.3390/ijms18102211
38. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation (2017) 135(17):e927–99. doi: 10.1161/CIR.0000000000000484
39. Kim J, Tomalin L, Lee J, Fitz LJ, Berstein G, Correa-da Rosa J, et al. Reduction of Inflammatory and Cardiovascular Proteins in the Blood of Patients with Psoriasis: Differential Responses between Tofacitinib and Etanercept after 4 Weeks of Treatment. J Invest Dermatol (2018) 138(2):273–81. doi: 10.1016/j.jid.2017.08.040
40. Lin MT, Wu MH. The global epidemiology of Kawasaki disease: Review and future perspectives. Glob Cardiol Sci Pract (2017) 2017(3):e201720. doi: 10.21542/gcsp.2017.20
41. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics (2004) 114(6):1708–33. doi: 10.1542/peds.2004-2182
42. Burns JC, Glode MP. Kawasaki syndrome. Lancet (2004) 364(9433):533–44. doi: 10.1016/S0140-6736(04)16814-1
43. de La Harpe M, di Bernardo S, Hofer M, Sekarski N. Thirty Years of Kawasaki Disease: A Single-Center Study at the University Hospital of Lausanne. Front Pediatr (2019) 7:11. doi: 10.3389/fped.2019.00011
44. Sillen H, Maes M, Boiy T, Wojciechowski M. Plaque psoriasis following Kawasaki disease and varicella. BMJ Case Rep (2018) 2018:bcr-2018-224539. doi: 10.1136/bcr-2018-224539
45. Ergin S, Karaduman A, Demirkaya E, Bakkaloglu A, Ozkaya O. Plaque psoriasis induced after Kawasaki disease. Turk J Pediatr (2009) 51(4):375–7.
46. Yoon SY, Oh ST, Lee JY, Cho BK. A plaque type psoriasiform eruption following Kawasaki disease. Pediatr Dermatol (2007) 24(1):96–8. doi: 10.1111/j.1525-1470.2007.00349.x
47. Mizuno Y, Suga Y, Muramatsu S, Hasegawa T, Ogawa H. Psoriasiform and palmoplanter pustular lesions induced after Kawasaki disease. Int J Dermatol (2006) 45(9):1080–2. doi: 10.1111/j.1365-4632.2005.02524.x
48. Menni S, Gualandri L, Boccardi D, Agostoni C, Sala M, Riva E. Association of psoriasis-like eruption and Kawasaki disease. J Dermatol (2006) 33(8):571–3. doi: 10.1111/j.1346-8138.2006.00134.x
49. Toussi A, Maverakis N, Le ST, Sarkar S, Raychaudhuri SK, Raychaudhuri SP. Updated therapies for the management of psoriatic arthritis. Clin Immunol (2020) 108536. doi: 10.1016/j.clim.2020.108536
50. Jia S, Li C, Wang G, Yang J, Zu Y. The T helper type 17/regulatory T cell imbalance in patients with acute Kawasaki disease. Clin Exp Immunol (2010) 162(1):131–7. doi: 10.1111/j.1365-2249.2010.04236.x
51. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17- and Treg-related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy (2015) 70(3):310–8. doi: 10.1111/all.12558
52. Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. Pathophysiology of psoriasis: recent advances on IL-23 and Th17 cytokines. Curr Rheumatol Rep (2007) 9(6):461–7. doi: 10.1007/s11926-007-0075-1
53. Marinoni B, Ceribelli A, Massarotti MS, Selmi C. The Th17 axis in psoriatic disease: pathogenetic and therapeutic implications. Auto Immun Highlights (2014) 5(1):9–19. doi: 10.1007/s13317-013-0057-4
54. Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol (2012) 188(12):6287–99. doi: 10.4049/jimmunol.1200385
55. Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA, et al. IL-17A-mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol (2017) 10(3):673–84. doi: 10.1038/mi.2016.80
56. Speake C, Whalen E, Gersuk VH, Chaussabel D, Odegard JM, Greenbaum CJ. Longitudinal monitoring of gene expression in ultra-low-volume blood samples self-collected at home. Clin Exp Immunol (2017) 188(2):226–33. doi: 10.1111/cei.12916
Keywords: psoriasis, transcriptomics, blood, Kawasaki disease, systems biology
Citation: Rawat A, Rinchai D, Toufiq M, Marr AK, Kino T, Garand M, Tatari-Calderone Z, Kabeer BSA, Krishnamoorthy N, Bedognetti D, Karim MY, Sastry KS and Chaussabel D (2020) A Neutrophil-Driven Inflammatory Signature Characterizes the Blood Transcriptome Fingerprint of Psoriasis. Front. Immunol. 11:587946. doi: 10.3389/fimmu.2020.587946
Received: 27 July 2020; Accepted: 26 October 2020;
Published: 24 November 2020.
Edited by:
Joshua Daniel Ooi, Monash University, AustraliaReviewed by:
Aditya A. Joshi, National Institutes of Health (NIH), United StatesCopyright © 2020 Rawat, Rinchai, Toufiq, Marr, Kino, Garand, Tatari-Calderone, Kabeer, Krishnamoorthy, Bedognetti, Karim, Sastry and Chaussabel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Damien Chaussabel, ZGNoYXVzc2FiZWxAc2lkcmEub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.