 Gunnar Houen
Gunnar Houen Nicole Hartwig Trier
Nicole Hartwig Trier
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 January 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.587380
This article is part of the Research Topic Pathogens, Pathobionts and Autoimmunity View all 13 articles
Epstein-Barr Virus (EBV) is an extremely successful human herpes virus, which infects essentially all human beings at some time during their life span. EBV infection and the associated immune response results in production of antibodies (seroconversion), which occurs mainly during the first years of life, but may also happen during adolescence or later in life. Infection of adolescents can result in infectious mononucleosis, an acute serious condition characterized by massive lymphocytosis. Transmission of EBV mainly occurs through saliva but can rarely be spread through semen or blood, e.g. through organ transplantations and blood transfusions. EBV transmission through oral secretions results in infection of epithelial cells of the oropharynx. From the epithelial cells EBV can infect B cells, which are the major reservoir for the virus, but other cell types may also become infected. As a result, EBV can shuttle between different cell types, mainly B cells and epithelial cells. Moreover, since the virus can switch between a latent and a lytic life cycle, EBV has the ability to cause chronic relapsing/reactivating infections. Chronic or recurrent EBV infection of epithelial cells has been linked to systemic lupus erythematosus and Sjögren’s syndrome, whereas chronic/recurrent infection of B cells has been associated with rheumatoid arthritis, multiple sclerosis and other diseases. Accordingly, since EBV can shuttle between epithelial cells and B cells, the systemic autoimmune diseases often occur as overlapping syndromes with symptoms and characteristic autoantibodies (e.g. antinuclear antibodies and rheumatoid factors) reflecting epithelial and/or B cell infection.
Epstein-Barr Virus (EBV) is a lymphotropic herpes virus and the causative agent of infectious mononucleosis (IM) (1–4). It was originally discovered in cells isolated from African Burkitt’s lymphoma and first later on, was it recognized that EBV is highly prevalent worldwide (5).
EBV is a member of the Human Herpes Viruses (HHVs) family, comprising eight viruses distributed on three subfamilies (Alpha, Beta, Gamma). EBV, which is also called HHV4, belongs to the Gammaherpesviridae, genus Lymphocryptovirus (6, 7). The circular double-stranded genome of EBV is approximately 172 kilobases, with more than hundred genes coding for approximately 85 proteins (Table 1) and approximately 50 non-coding RNAs (8–12).
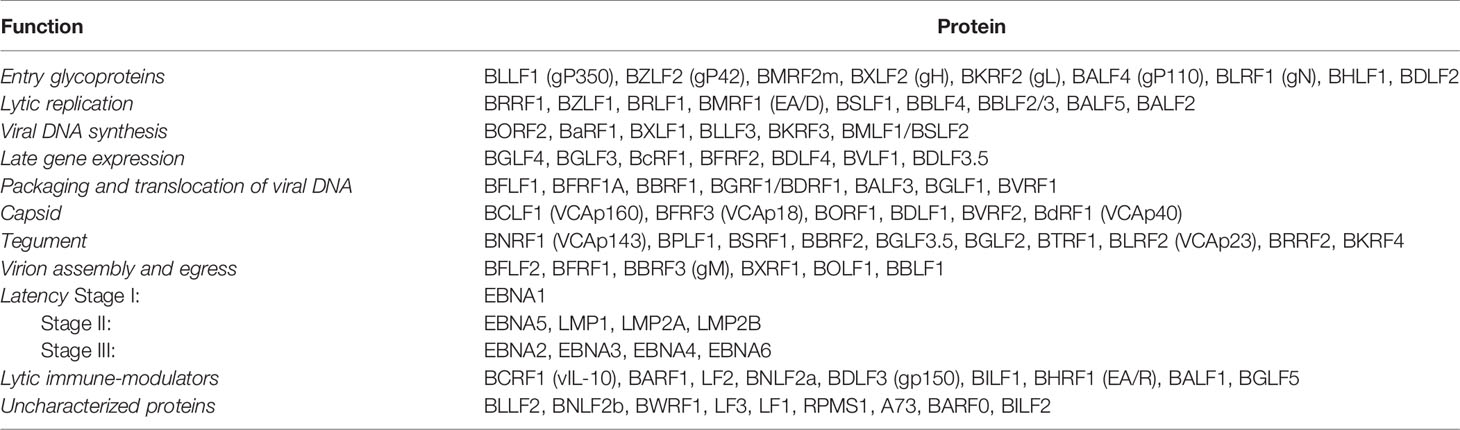
Table 1 Epstein-Barr virus (EBV) proteins and their functions.
Several strains of EBV exist. The first EBV variants identified were type 1 (type A) and type 2 (type B). While type 1 (B95-8, GD1, and Akata) is the main EBV type prevalent worldwide, type 2 (AG876 and P3HR-1) is as abundant as type 1 in sub-Saharan Africa (13). The EBV variants have different replicative properties and individuals may become superinfected with two or more strains (14–16).
The structure of EBV is typical of HHVs and related viruses (Figure 1) (17). It has an outer lipid envelope, derived from the producing host cell, wherein several viral proteins are embedded in addition to host cell-derived membrane proteins. Many of the viral envelope membrane proteins are glycoproteins (gPs). Currently, 13 gPs have been identified, 12 of which are expressed only during the productive, lytic replication cycle and one of which (BARF1, a decoy viral colony-stimulating factor 1 receptor (vCSF1R)) may be expressed during latency as well. Some of these are listed in Table 1 (18). Inside the envelope is the viral tegument, in which the capsid is embedded with its enclosed genome and associated proteins.
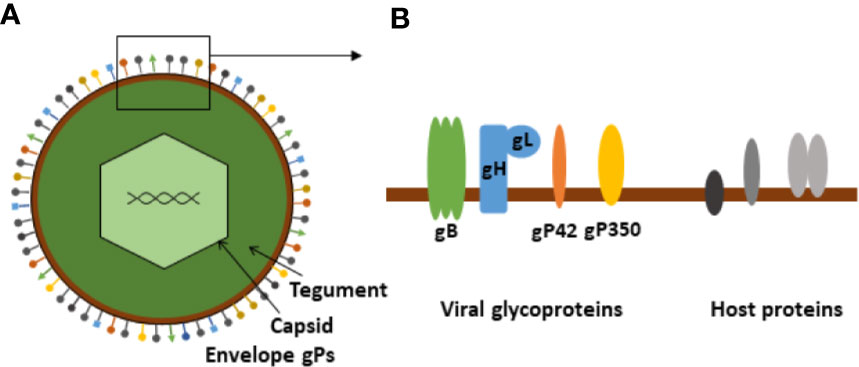
Figure 1 Schematic presentation of Epstein-Barr virus. (A) Schematic illustration of the basic EBV structure. (B) Enlargement of membrane section showing viral envelope glycoproteins (entry complex) and putative host-derived membrane proteins.
The life cycle of EBV is characteristic of a large enveloped DNA virus, being composed of primary infection, latency, and lytic reactivation phases. In addition, EBV has an ability to infect several cell types (19). The EBV genome encodes 9 different envelope entry gPs (Table 1). The functions of all of these are not completely understood, but the roles of the most important gPs are known in much detail. The tropism of newly released EBV virions is determined by the envelope gPs, which in turn vary somewhat depending on the host cell (20). The major cell types infected by EBV are epithelial cells and B cells. Epithelial cells are the first cell type to be infected, as EBV is transmitted to recipients through saliva. Next, B cells are infected when EBV gains access to the underlying tissue after release from the oropharyngeal epithelium (21–25). EBV virions released from epithelial cells have a preference for B cells and EBV virions released from B cells have a preference for epithelial cells, due to the composition of the envelope gPs (20, 26, 27).
Epithelial cell infection may occur by direct fusion of the viral envelope membrane with the plasma membrane of the target cell. Attachment of the virus to the cell surface primarily occurs via gH/gL interaction with Ephrin A2 (EphA2) and αvβ5/αvβ6/αvβ8 integrins and via BMRF1, which interacts with β1 integrins, but EBV gP350/220, which interact with complement receptor (CR)2 (CD21) and CR1 (CD35) also plays a role in epithelial cell attachment. The gH/gL interaction with integrins is mediated by a KGD motif on gH, and the interaction between gH/gL and EphA2 occurs through the receptor’s ligand binding and fibronectin type III repeats and is mediated by the gP42 binding site on gH. Upon attachment and interaction with integrins or EphA2, a conformational change in gH/gL allows interaction with the trimeric gB, which in turn changes conformation and facilitates viral entry by acting as a fusogen (20, 28–32).
Other EBV proteins may also play a role during infection of epithelial cells, e.g. BMRF2, which can bind integrin αvβ1 and BDLF2, which can bind non-muscle myosin heavy chain IIA. Moreover, gB, itself can bind neuropilin-1 and IgA directed to EBV envelope proteins may enhance infection through the polymeric IgA receptor (28, 32–36).
B cell infection is mediated by gP350/220, which binds CR2 and CR1, together with gP42, leading to the formation of a complex together with major histocompability complex (MHC)-II (37). Upon attachment, the virion is endocytosed and gH/gL can form a fusion complex with gP42-MHC-II, thus inducing a conformational change in gH/gL (similar to what happens upon gH/gL interaction with integrins and/or EphA2). As a result, trimeric gB changes conformation and promotes fusion of the viral membrane with the endosome membrane, thus releasing the virus to the cytoplasm (Figures 1–3) (20, 26, 27). The structural and mechanistic basis of B cell entry has been elucidated in much detail by solving the structures of gB, gP42, complexes of gH/gL, gP42/MHC-II (human leukocyte antigen (HLA)-DR1) and of gH/gL/gP42/MHC-II in pre- and post-fusion conformations (20, 38, 39). This has allowed modelling not only of the EBV B cell entry complex, with the involved gPs acting sequentially and in concert, but also of the epithelial cell entry complex. Thus, gH/gL/gB appears to constitute a core entry machinery and gP42 seems to be a primary determinant of EBV tropism, since it participates in and promotes B cell infection but inhibits epithelial cell infection by binding to the EphA2/integrin-binding site(s) on gH/gL (20).
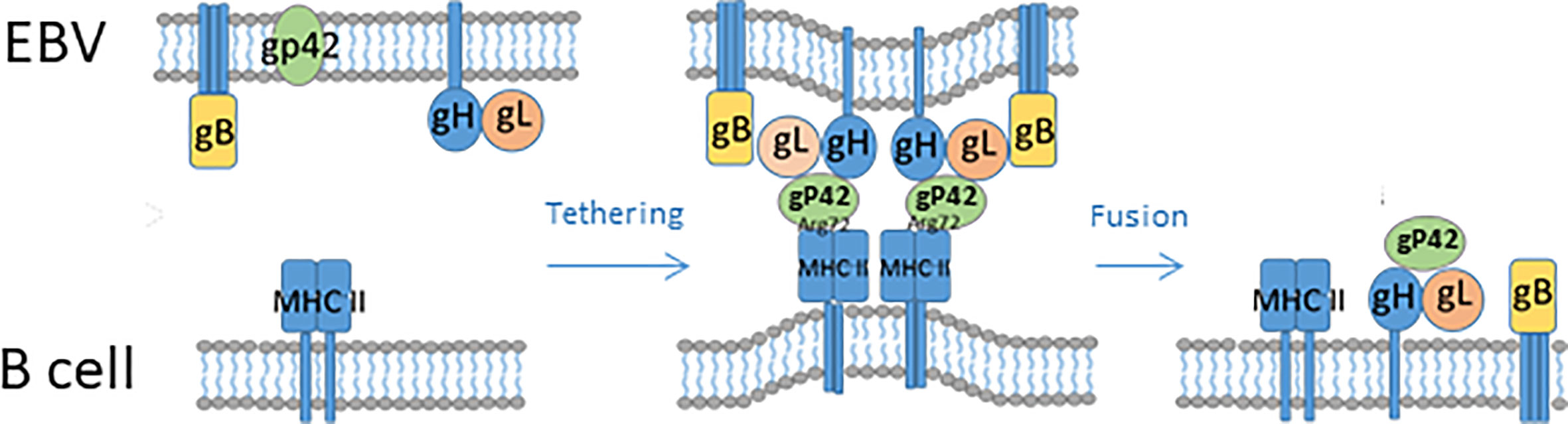
Figure 2 Schematic illustration of EBV fusion with the cellular lipid bilayer of B cells. For gP42 to become active, the protein is cleaved N-terminally. gP42 interacts with gH/gL, and the complex interacts with gB. gP42 interacts with the β1 domain of MHC-II, which ultimately results in membrane fusion.
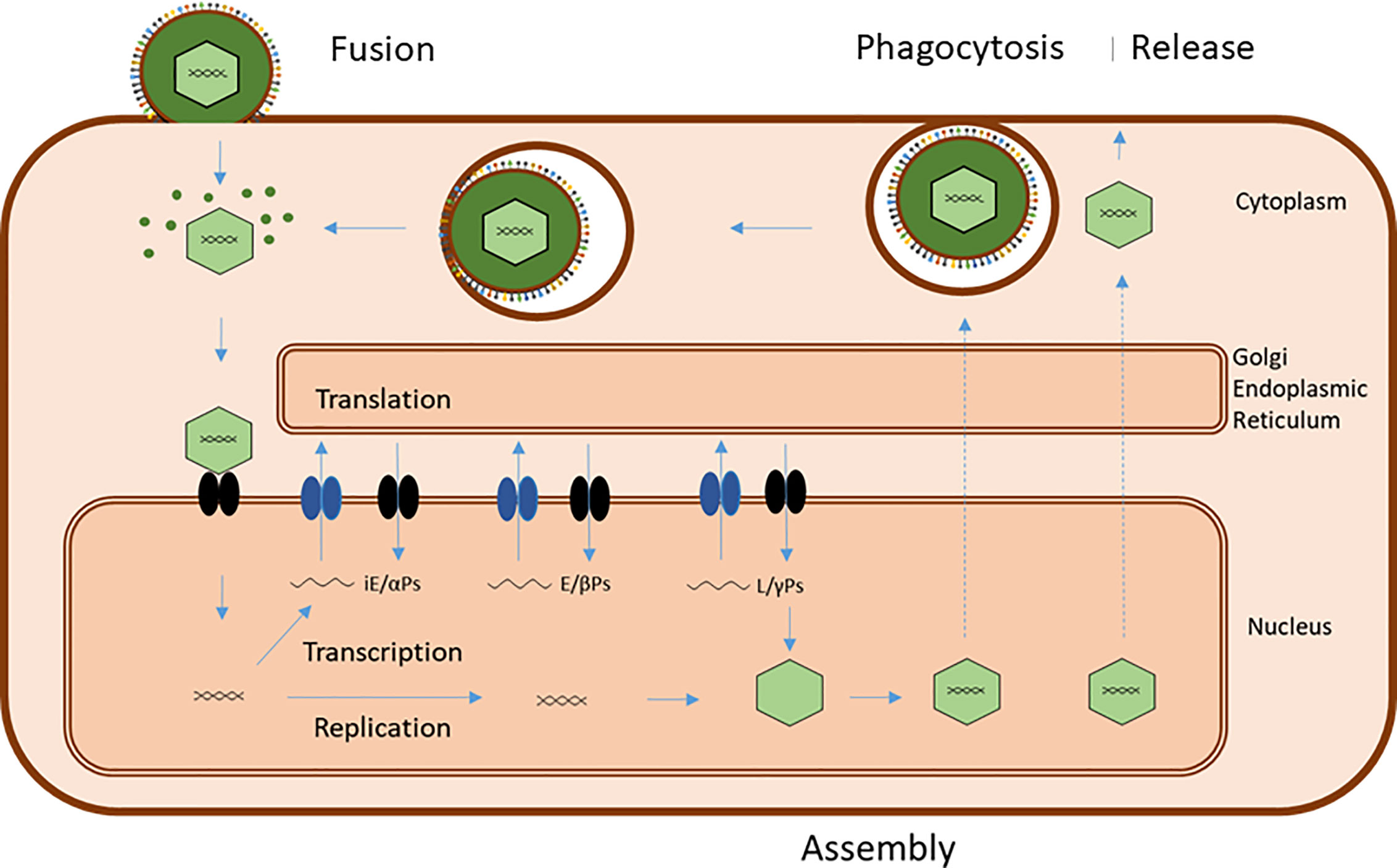
Figure 3 Common basic EBV infection scheme. Viral entry can occur by direct fusion of the viral plasma membrane-derived envelope with the target cell membrane or by endocytosis/phagocytosis of virus followed by fusion of virus envelope and endosome/phagosome membrane. Both processes release virions and viral tegument proteins into the cytoplasm. Released virions are transported to the nuclear membrane and the viral genome introduced into the nucleus together with associated proteins. This initiates transcription of viral genes in a sequence of immediate-early (iE) genes, coding for regulatory alfa-proteins, early genes, coding for catalytic beta-proteins, and late (L) genes, coding for structural gamma proteins. Translation of viral messenger RNAs takes place on ribosomes in the cytoplasm and on the endoplasmic reticulum, and the viral proteins are routed to different locations for subsequent virus assembly. Successful replication of viral genomes and transport of capsid proteins to the nucleus results in assembly of virions, which travel to the plasma membrane by a series of envelopment/fusion events involving intracellular membranes (stippled lines) ending with budding of mature virus with a plasma membrane envelope, containing viral glycoproteins and host-derived membrane proteins. Premature cell death releases a mixture of “naked” virions and diffentially enveloped viruses.
Successful entry and viral take-over of cellular control leads to an ordered sequence of transcription of viral genes, translation of viral mRNAs and finally, replication of the viral DNA and assembly of new virus (Figure 3). The virion assembly and egress from the host cell utilizes the host cell exocytosis machinery and involves several viral proteins apart from the structural, tegument and envelope proteins (Table 1) but is less understood than the entry process (40, 41). Collectively, EBV utilizes several characteristic major host cell membrane proteins for entry and release and due to the properties of gP42, it has a preference for epithelial cell infection when produced by B cells and vice versa, assuring that some virions will eventually return to salivary gland cells and be able to be transmitted to new individuals.
Infection activates the intracellular antiviral mechanisms and induces an extracellular immune response against EBV antigens, with generation of specific helper T cells, antibodies and cytotoxic T cells and activation of natural killer (NK) and NK T cells (NKT) (42–46). In response to this, EBV has evolved mechanisms for evading the extracellular innate immune system and the host cell’s innate antiviral systems together with adaptive immune system evasion mechanisms and the virus devotes a substantial part of its proteins and non-coding RNAs to this (47–51). Together, the innate and adaptive immune evasion mechanisms of EBV assure its persistence in the host. A major aspect of the immune evasion strategy is EBV’s ability to enter a latent state with minimal expression of viral genes and minimal presentation of viral peptides to the immune system (19, 52–54). This mainly occurs in (memory) B cells, but latency may also take place in epithelial cells. From the latent state, EBV can occasionally reactivate, e.g. in response to antigen stimulation of memory B cells, resulting in lytic production of virions upon expression of an ordered sequence of viral genes (55–57). This in turn mounts an increased immune response against EBV, neutralizing infected cells and forcing the virus into latency again. Reactivation may also occur upon “waning” of the cellular immunity to the virus and infected individuals through the rest of their lives experience a persistent “battle” with EBV. Depending on the host immune system and environmental factors, some individuals may eventually suffer from EBV-related diseases, either as a result of EBV immune evasion or as a result of EBV infection of other cell types (T cells, NK cells, NKT cells, monocytes/macrophages, and others), which may take place in some instances.
As a part of the common evolutionary history of humans and EBV, the virus has evolved a multitude of immune evasion mechanisms, including wrapping itself in host cell-derived membranes (envelopment) and the ability to switch between latent and lytic life stages (50, 58). Most of the immune evasion proteins of EBV are expressed during the lytic cycle and some are shown in Table 1 as “immune modulators”. More EBV proteins are presumably involved in immune evasion and many EBV proteins serve two or more functions.
In the latent state, as mentioned above, there is minimal expression of viral genes and minimal presentation of viral peptides to the immune system (19, 52–54). In the “deep” latency state, only EBV nuclear antigen (EBNA)1, which assures maintenance and replication of the EBV genome along with host cell chromosomes, is expressed. In order to avoid presentation of EBNA1-derived peptides on MHC-I, the EBNA1 protein contains a characteristic AG repeat sequence, which interferes with proteasome processing and which interacts with nucleolin to restrain its expression. Moreover, EBNA1 also contains characteristic RG repeat sequences, which may play a role in immune evasion (59–61). Upon switching to lytic cycle with production of viral proteins, EBV downregulates MHC-I and interferes with presentation of viral peptides on MHC-I via BDLF3-induced ubiquitination of MHC-I (62). Likewise, in B cells, EBV can also downregulate MHC-II by BDLF3-induced ubiquitination of MHC-II (62) and gP42 can be released in a soluble form, which inhibits interaction between MHC-II and the T cell receptor (63, 64). Other EBV proteins are involved in minimization of MHC-I expression, including BNLF2a, BILF1, BGLF5. The exonuclease BGLF5 degrades cellular mRNAs including those for MHC-I and BILF1 associates with cell surface MHC-I and enhances its degradation, while BNLF2a prevents MHC-I peptide loading by inhibiting the transporter associated with peptide loading (TAP) (65–68). As a means to avoid NK cell recognition, EBV upregulates non-classical MHC during the phase of viral protein synthesis. Lytic production of viral proteins and RNAs as well as replication of viral DNA requires that EBV can prevent cellular apoptosis and EBV has evolved an elaborate set of proteins for pacifying intracellular virus-sensing apoptosis-inducing mechanisms including downregulation and inhibition of toll-like receptors (47, 49, 50, 62, 68–71).
EBV also produces soluble mediators, which interfere with mobilization of the adaptive immune system. BCRF1 encodes a viral IL10 homologue (vIL10), which dampens inflammation (72–75) and, as mentioned above, BARF1, encodes a decoy vCSF1R, which binds CSF1 and thereby limits mobilization of hemopeoietic stem cells (76, 77).
The viral envelope derived from the host cell (Figure 2) offers substantial protection to the enclosed viral particle by mimicking a host extracellular vesicle. In principle, the viral envelope may contain all host-derived membrane proteins relevant for “disguise” and immune evasion (e.g. MHC molecules, complement regulators, Fc receptors, phagocytosis-inhibitory (“don’t-eat-me”) molecules, etc.). However, to be able to exit from the host cell in a controlled process, and to be able to infect other cells, several viral gPs have to be inserted into the envelope membrane as mentioned above. These proteins are targets for innate immune recognition and antibody (Ab) production, as described in the preceding paragraph, but extensive glycosylation with host-derived glycans affords considerable protection against pattern recognition (scavenger) receptor (including complement) and Ab recognition (“glycan shielding”). Moreover, as described, some of the immune reactions may actually be exploited for viral infection and spreading, f.ex. “hitchhiking” with complement/CRs (e.g. EBV entry in B cells) or with Abs bound to viral envelope gPs/Fc receptors (FcRs) (e.g. cytomegalovirus entry in monocytes/macrophages or EBV entry in B cells with cell surface immunoglobulins (Igs) against EBV envelope gPs) (78–80). Despite the many immune evasion mechanisms of EBV, the normal healthy human immune system is able to eradicate active virus and force it into a quiescent (“immune silent”) state (latency). Since EBV appears to be able to evade most or all innate immune system components, the final “victory” of the immune system must rely on cellular immune control of EBV involving a combination of T cells, NK cells and NKT cells, in accordance with all available evidence of EBV immunity. The molecular details of how this results in EBV latency instead of cell killing are not known, but it is firmly established that EBV has evolved mechanisms of latency as an ultimate, opportunistic and effective immune evasion strategy.
A majority of children becomes infected with EBV early in life and seroconversion, the appearance of Abs to EBV peaks around 1–2 years of life, where the majority of infectious cases is non-complicated and may even go unnoticed. A second peak in seroconversion is seen in puberty, due to increased frequency of close social contact with already infected persons. Infection in adolescence is more problematic and may result in IM in many cases, popularly denoted “kissing disease” (1–4). For the majority of infected individuals latent infection does not appear to influence the general health, however, dysregulation of latency or inability to control the lytic infection may lead to development of lymphoproliferative diseases and lymphoma (81).
The course of EBV infection is determined by the virus load and an individuals’ immune system state, which in turn is determined by the person’s gene composition, other infection history and several environmental factors, which all may influence the immune capacity of a person to various degrees.
Genetic factors influencing EBV control are in principle all genes of the immune system. In practice, T cells, NK cells and NKT cells have turned out to be of utmost importance (42–44, 46). Relatively few studies have addressed genetic factors associated with EBV infection, presumably due to the ubiquitous occurrence of EBV. Consequently, since essentially all persons eventually become infected, genetic associations will only relate to the age of infection. Epidemiological studies have indicated an association of some MHC-II and -I alleles and EBV seropositivity. Moreover, mannan-binding lectin insufficiency has been linked to EBV seropositivity as well (82). Also, some polymorphisms in the (IL) 10 gene and other immune system genes have been linked with EBV seropositivity (83). However, all these studies are hampered by a relative scarcity of seronegative persons.
Besides from genetic factors, environmental factors are known to affect a person’s EBV status. Currently identified factors are sunlight/Vitamin D (VitD), smoking and body mass index (BMI) (84, 85). These factors may be assumed to influence the general immune status of individuals and thereby affect susceptibility to EBV infection. E.g. sunlight/VitD has been proposed to protect against autoimmunity by increasing the number of CD8+ T cells available to control EBV infection (84). Moreover, obesity has been proposed to impact the cellular immune response to infections and induce a state of chronic immune-mediated inflammation (85), but more studies are required to understand these associations. Finally, prior infections may play a role in shaping an individual’s immune repertoire and resulting capacity to combat later infections, as evidenced by the more serious course of EBV infection in adolescence or later in life.
The presence of EBV nucleic acid material in infected persons can be determined by numerous methods, e.g. by direct sequencing, fluorescence in-situ hybridization (FISH) and polymerase chain reaction (PCR) analysis of blood samples for EBV-derived DNA or RNA, while (prior) infection/reactivation may also be demonstrated by PCR analysis of saliva (86–91). In relation to testing of EBV in biopsy tissues, molecular detection of EBV-encoded RNA transcripts by FISH remains the gold standard. Moreover, EBV-encoded RNA hybridization and EBV LMP1 immunostains are used routinely to detect latent EBV in tissues affected by posttransplant lymphoproliferative disorder (PTLD) or in enlarged nodes from IM patients (92). Traditionally, serology is the simplest way to test for EBV infection and even for evaluating acute versus remote infection in healthy individuals. High serological titers serve as a tumor marker for some EBV-related malignancies, but titers are not a dependable tumor marker in immunocompromised hosts. EBV viral load testing by quantitative DNA amplification of blood samples has proven useful for early diagnosis and monitoring patients with PTLD (92).
Acute infection may also be inferred from analysis of IgM to viral antigens, while prior infection may be inferred from the presence of IgG to EBV antigens, and IgA can be used as a measure of epithelial infection load (45). Using three EBV antigens, viral capsid antigen (VCA) IgG, VCA IgM and EBNA1 IgG, it is normally possible to distinguish an acute from a past infection. While the presence of VCA IgM and VCA IgG without EBNA-1 IgG indicates a current acute infection, does the presence of VCA IgG and EBNA1 IgG without VCA IgM typically indicate a past infection (93).
Among the 85 proteins encoded in the EBV genome, several have been used for detection of Abs to EBV including EBNA1, EBNA2, VCAp23, VCAp18, early antigen diffuse (EAD), gP350, BARF1 (Table 1) (15, 94–97). IM has previously been associated with the presence of so-called heterophile Abs, however, this test has a rather low specificity and it remains unclear, what the test actually measures (2, 98).
Since induction of Abs follows a pattern of viral Ag production, seropositivity will depend on a person’s ability to control EBV and the balance between latent and lytic EBV infection. Moreover, any assay has a characteristic sensitivity and specificity for EBV detection, and some individuals may be judged false negative or positive. Thus, to fully define the incidence and prevalence of EBV infection in a population, several assays should be used, preferably combining assays for detection of viral nucleic acids, Abs to different viral antigens and the frequency of virus-specific T cells. Optimally, different detection principles may also be used; e.g. for Ab detection: enzyme-linked immunosorbent assay (ELISA) and immunoblotting, for T cell detection: antigen-induced cytokine release and peptide-MHC tetramer assays, and the assays should target different parts of the viral genome or different viral antigens representing both latent and lytic states. This is evidently very labor-intensive but may be realized by using multiplex techniques.
Many diseases are known to be associated with EBV infection and prior IM increases the risk of many of these diseases (2, 99). IM itself is a prolonged state of fever, swollen lymph nodes, fatigue, malaise and various other symptoms. Few studies have focused on genetic factors associated with IM. Similar to EBV infection itself, some MHC-I and -II alleles and polymorphisms in the IL10 gene have been associated with IM development (82).
In contrast to the scarcity of information about genetic factors involved in EBV infection itself, several data has been published relating to EBV involvement in diseases and genetic factors associated with these. Several types of cancer, notably B cell lymphomas and nasopharyngeal epithelial carcinomas, affecting the two primary cell types targeted by the virus, are caused by EBV (99–102). This can be ascribed to EBV’s ability to evade cellular antiviral mechanisms and control cellular apoptotic pathways and to its capacity for immune evasion (103). However, several other diseases affecting other cell types, which may become infected by EBV are known, including T cell lymphomas, NK cell leukemias and other T cells, NKT cells and NK cell lymphoproliferative diseases (101, 104, 105). Moreover, several systemic autoimmune diseases (SADs) and multiple sclerosis (MS) have been demonstrated to be associated with chronically relapsing EBV infection and inefficient immune control of the virus.
SADs are a group of partly overlapping syndromes, also called connective tissue diseases, since they often are accompanied by inflammation of connective tissues. The SADs include the relatively common rheumatoid arthritis (RA) and the more rare conditions Sjögren’s syndrome (SS), systemic lupus erythematosus (SLE), systemic scleroderma (SSc), and others (Table 2) (106, 107).

Table 2 Systemic autoimmune diseases (SADs) and their characteristics.
The clinical characteristics of RA are swollen and painful joints, caused by synovial inflammation eventually resulting in exaggerated connective tissue deposition (pannus formation) and bone erosion, with resulting disability. Moreover, RA is frequently accompanied by systemic complications such as vascular disease, osteoporosis, and others (108–110). Most RA patients have characteristic autoantibodies (AuAbs) including rheumatoid factors (RFs) and anti-citrullinated protein antibodies (ACPA)s, but many also have anti-nuclear Abs (ANAs) (111, 112). The etiology of RA is commonly ascribed to genetically determined defective self-tolerance, but environmental factors are known to play a dominating role, including EBV infection (113–116). Alleles of many genes are known to contribute to RA, notable HLA-DRB1 alleles containing shared epitope (SE) motives, but many other genes affecting the immune system and in particular lymphocytes have an impact (108, 110, 117). Tumor necrosis factor (TNF) plays an important role in a large proportion, if not most RA patients, and therapeutic Abs targeting TNF have good therapeutic efficacy in many patients (109, 118).
EBV evidently plays an important role in the etiology of RA, although not all evidence indicates an association between RA and EBV (119). Mechanisms behind the role of EBV in RA may include either molecular mimicry in the initiation of RA, bystander activation effects or chronic recurrent infection of joint epithelial cells and synovial B cells. The characteristic ACPAs seen in a major proportion of RA patients have been found to represent Abs to a citrullinated region of EBNA2, an important transcription factor of EBV expressed in lytic phases (120). Presumably, EBNA2 and possibly also other EBV proteins become citrullinated by peptidyl arginine deiminase (PAD) enzymes during the inflammatory process in RA joints (121, 122). RFs have been found to target cryptic epitopes of IgG heavy chains, presumably being released by lysis of EBV-infected B cells (123) and MHC-II molecules with SE motives (certain HLA-DRB1 alleles) have been found to be optimal ligands for EBV gP42, thus favoring EBV infection of B cells with these forms of MHC-II (31). Thus, the major characteristics of RA can be related to chronic EBV infection, and actually, serum EBV DNA has been found to correlate with disease activity (124). Furthermore, EBV has been demonstrated to be present in the synovium of RA patients (115, 125, 126).
SS is a disease resulting in progressive destruction of exocrine salivary and lacrimal gland tissue. The major clinical characteristics are xerostomia and xeropthalmia in addition to fatigue and various other symptoms, which may also affect other organ systems (127, 128). Patients most often have ANAs and characteristic AuAbs are Ro60 and La Abs, but various other AuAbs may also be present. In addition, RFs are present in a majority of patients, whereas ACPAs are usually absent (128).
The etiology of SS has been suggested to involve several environmental and genetic factors, molecular mimicry and bystander activation (129, 130). Genetic factors include certain MHC-II (especially some HLA-DRB1) alleles, some MHC-I alleles and components of the interferon regulatory system (131). Environmental factors include vitD deficiency, smoking, silica dust exposure and virus infections (129). Especially EBV infection has been associated with SS (132, 133). The mechanisms involved in SS are presumably similar to RA and other SADs, but are much less studied. RA and SS often co-exist and SS primarily affects the epithelial tissues targeted by EBV, i.e. salivary and lacrimal glands, making the association with EBV infection particularly attractive.
SLE is a disease, which clinically presents with a heterogenous array of symptoms, often evaluated by the SLE disease activity index (SLEDAI) or similar indexes, including complementemia, DNA Abs, leukopenia, thrombocytemia, fever, fatigue, skin rash, UV sensitivity, mucosal ulcers, alopecia, pleuriris or pericarditis, proteinuria, hematuria, nephritis, myositis, arthritis, vasculitis, headache, stroke, and more rarely, neuropsychiatric symptoms (134–137). The disease may show a relapsing/remitting course, depending on the efficacy of treatments (138, 139).
SLE has been described as an immune complex disease, since it is often associated with decreased levels of complement components (140). Other characteristics are the presence of ANAs, notably DNA Abs, which are included in the SLEDAI, but in many cases AuAbs to a heterogenous panel of AuAgs are present and changes in the AuAb profile may reflect changes in disease activity (141–144).
Genetically predisposing factors are first of all certain HLA-DRB1 alleles, but multiple immune system genes, including other MHC-II alleles and some MHC-I alleles, as well as genes affecting cellular waste removal, have been found to influence disease development (145, 146).
Major environmental factors promoting development of SLE are silica dust exposure, sun burn, smoking, vitD deficiency and EBV infection (147–153). The etiology has been suggested to involve molecular mimicry between EBV EBNA1 and cellular Ro 60, and/or bystander activation (154, 155).
Decreased immune control of chronic EBV infection has been found to be a contributing factor, if not a major cause (152, 156, 157), but other infections may also play a role in SLE development or exacerbation (158, 159). The presence of DNA Abs and other ANAs would seem to be compatible with infection by a DNA virus in combination with inefficient removal of apoptotic and necrotic material.
SADs constitute a group of partly overlapping autoimmune disease syndrome and include systemic sclerosis (SSc), mixed connective tissue disease (MCTD) and polymyositis/dermatomyositis (PM/DM) in addition to RA, SS, and SLE (Table 2). These diseases share several genetic and environmental factors, in particular the predisposing effect of certain HLA-DRB1 alleles (although not exactly identical alleles), the predisposing effect of EBV infection and of factors, which can be related to EBV infection (e.g. vitD deficiency) (Table 2) (106, 107, 160–171).
The evidence for a major etiological role of EBV is particularly strong for RA, where several of the clinical characteristics can be related to EBV as described above (RFs, ACPAs, SE-allele disposition). Current treatments can also be related to EBV infection, e.g. CD20 monoclonal antibodies (MAbs), which presumably diminish the burden of EBV-infected (memory) B cells, and TNF MAbs, which possibly diminish the burden of EBV infection by an anti-inflammatory effect (172–174). The evidence for an etiological role of EBV in SLE is also strong and seems to point to EBV infection of epithelial cells in combination with decreased removal of apoptotic/necrotic cell debris (175). Thus, these two prototype SADs can be seen as the results of a chronic, poorly controlled, relapsing/remitting EBV infection targeting the two major host cells of EBV; B cells in RA and epithelial cells in SLE. In RA, relapses most likely follow re-activation of EBV in (memory) B cells upon Ag stimulation. This results in production of EBV-transformed B cell blasts, which by their very nature will attempt homing to bones and therefore will have a tendency to populate joints, where the concomitant lytic EBV production may also result in EBV infection of synovial epithelial cells. In SLE, B cells will also be involved, thus accounting for the common involvement of joints and other symptoms overlapping with RA, however, the major target cells affected are epithelial cells, thus accounting for the common skin and mucosal pathology, while the defective removal of EBV and cellular debris results in immune complex deposition in affected organs and in particular kidneys, by virtue of their filtrating actions. SS has been studies less intensively than RA and SLE but the relation to EBV is nevertheless even more obvious. In SS, pathological symptoms reminiscent of both RA and SLE are seen. This again reflects the tendency of EBV to “shuttle” between B cells and epithelial cells and in particular the ability of EBV to return to salivary (and lacrimal) gland epithelial cells as part of its natural life cycle (Figure 4). Thus, SS may in some respects be thought of as SLE effecting the exocrine glands, while SS also has many characteristics in common with RA.
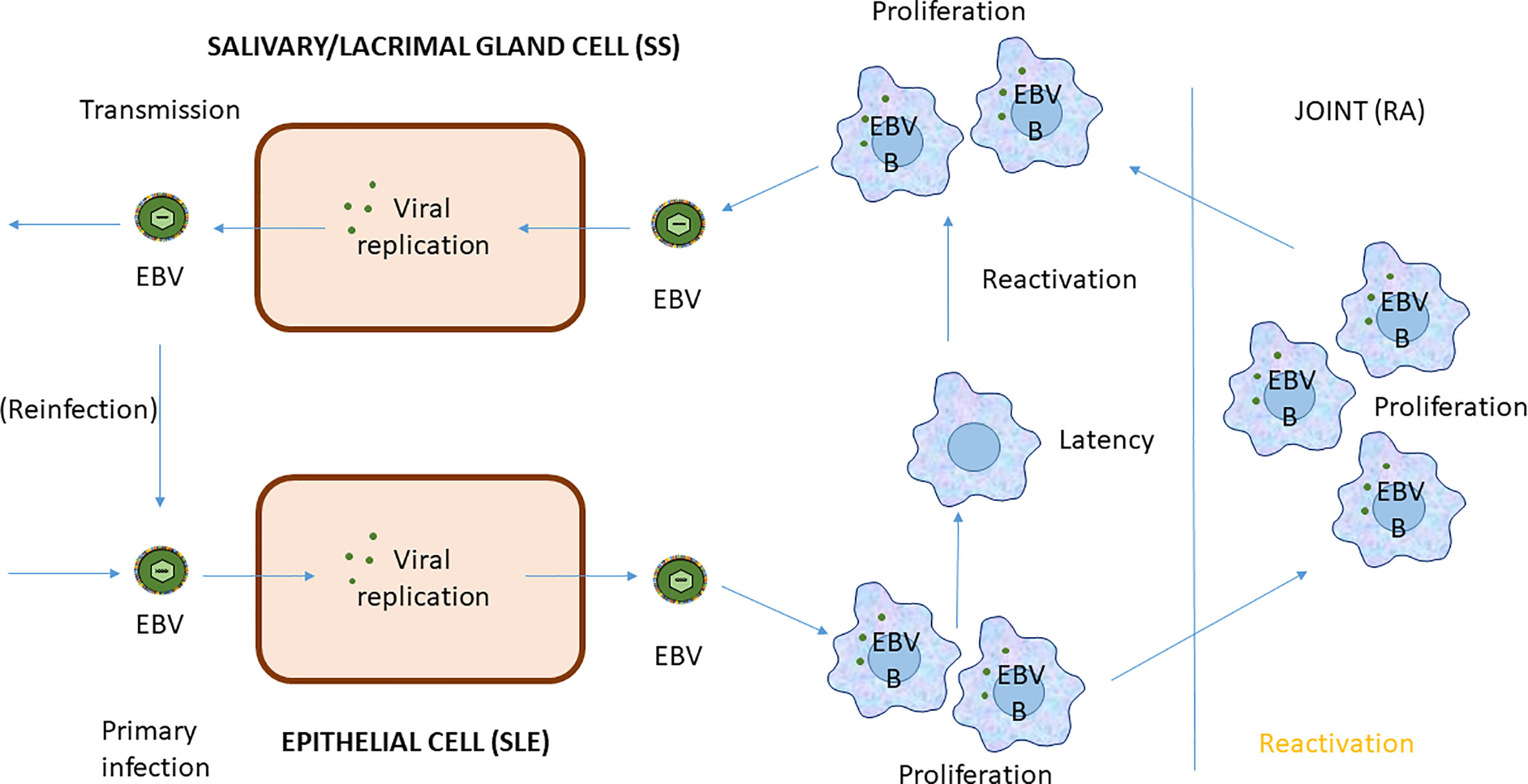
Figure 4 EBV Infection cycle in B cells of systemic autoimmune diseases.
Other autoimmune diseases, especially MS have also been found to depend on EBV infection in several aspects (176). The question therefore arises, how EBV can be involved in these apparently diverse diseases? A common feature seems to be decreased immune control of EBV. T cells are crucial for the control of EBV (and other viruses) and defective/exhausted T cell repertoires are characteristic of SADs (177). This allows for chronic infections with continuous cycles of relapses and remissions. However, while this may explain a common involvement of EBV (or other viruses) in disease etiology, it does not explain the different clinical appearances and the differences in e.g. association with different HLA alleles. A plausible explanation is that the role of EBV does not depend solely on e.g. entry, which in RA seems to be facilitated by SE-containing HLA alleles. Other HLA interactions must also be involved, e.g. presentation of EBV and/or host peptides, interactions with the peptide loading complex, interaction with other EBV or host proteins, etc. In general will the genetic composition of the host determine the fate of EBV in different cell types, including the interactions of EBV attachment and entry proteins with the target cell membrane proteins, the ability of the host cell to undergo apoptosis and the possibility to support lytic production of virus, and the efficiency of adaptive immune control of EBV. Since there are large differences in individual immune systems and in infection histories, one possibility for the different appearances of EBV-related diseases could also be individual mutations in EBV genomes during chronic infections and/or re-infections, and/or different rates of co-infection with other viruses. Patients with SADs are often prone to various infections, possibly due to inherent or acquired immune deficiencies, which predispose to coinfection with other viruses e.g. cytomegalovirus and others, which have been suggested to play a role in SAD development (178–181).
Patients with SADs also have increased tendency to develop cancer, including various forms of lymphoma. This may relate to secondary effects of treatment with immuno-suppressive drugs but may also reflect an inherent ability of EBV to cause transformation of B cells and epithelial cells (13, 99–102, 160).
EBV has been found to play a role in several, if not all SADs. It remains unclear, whether the role of EBV is primarily in initiation of disease (e.g. by molecular mimicry) or is simply due to the chronic relapsing-remitting nature of EBV infections. Many characteristics of especially RA can be ascribed to EBV infection, but this may also be the case for other SADs. Future studies should focus on interaction of EBV proteins and non-coding RNAs with host molecules and on the role of other viruses in relation to EBV infection.
All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the contributions of all students and collaborators, who have participated in our studies on SADs.
1. Rostgaard K, Balfour HH Jr, Jarrett R, Erikstrup C, Pedersen O, Ullum H, et al. Primary Epstein-Barr virus infection with and without infectious mononucleosis. PLoS One (2019) 12:e0226436. doi: 10.1371/journal.pone.0226436
2. Dunmire SK, Hogquist KA, Balfour HH. Infectious Mononucleosis. Curr Top Microbiol Immunol (2015) 390:211. doi: 10.1007/978-3-319-22822-8_9
3. Dunmire SK, Verghese PS, Balfour HH Jr. Primary Epstein-Barr virus infection. J Clin Virol (2018) 102:84. doi: 10.1016/j.jcv.2018.03.001
4. Chan KH, Tam JS, Peiris JS, Seto WH, Ng MH. Epstein-Barr virus (EBV) infection in infancy. J Clin Virol (2001) 1:57. doi: 10.1016/s1386-6532(01)00149-4
5. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet (1964) 7335:702. doi: 10.1016/s0140-6736(64)91524-7
6. Jouanguy E, Béziat V, Mogensen TH, Casanova JL, Tangye SG, Zhang SY. Human inborn errors of immunity to herpes viruses. Curr Opin Immunol (2020) 62:106. doi: 10.1016/j.coi.2020.01.004
7. Siakallis G, Spandidos DA, Sourvinos G. Herpesviridae and novel inhibitors. Antivir Ther (2009) 8:1051. doi: 10.3851/IMP1467
8. Majerciak V, Yang W, Zheng J, Zhu J, Zheng ZM. A Genome-Wide Epstein-Barr Virus Polyadenylation Map and Its Antisense RNA to EBNA. J Virol (2019) 2:e01593. doi: 10.1128/JVI.01593-18
9. Sakamoto K, Sekizuka T, Uehara T, Hishima T, Mine S, Fukumoto H, et al. Next-generation sequencing of miRNAs in clinical samples of Epstein-Barr virus-associated B-cell lymphomas. Cancer Med (2017) 3:605. doi: 10.1002/cam4.1006
10. Moss WN, Lee N, Pimienta G, Steitz JA. RNA families in Epstein-Barr virus. RNA Biol (2014) 1:10. doi: 10.4161/rna.27488
11. Tarbouriech N, Buisson M, Géoui T, Daenke S, Cusack S, Burmeister WP. Structural genomics of the Epstein-Barr virus. Acta Crystallogr D Biol Crystallogr (2006) 10:1276. doi: 10.1107/S0907444906030034
12. Longnecker R, Neipel F. Introduction to the human γ-herpesviruses. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al. editors. Human Herpesviruses. Cambridge, UK: Cambridge University Press (2007). p. 341–59.
13. Kanda T, Yajima M, Ikuta K. Epstein-Barr virus strain variation and cancer. Cancer Sci (2019) 4:1132. doi: 10.1111/cas.13954
14. Delecluse S, Poirey R, Zeier M, Schnitzler P, Behrends U, Tsai MH, et al. Identification and Cloning of a New Western Epstein-Barr Virus Strain That Efficiently Replicates in Primary B Cells. J Virol (2020) 94:e01918. doi: 10.1128/JVI.01918-19
15. Smith NA, Baresel PC, Jackson CL, Ogolla S, Toko EN, Heit S, et al. Differences in the Epstein-Barr Virus gp350 IgA Antibody Response Are Associated With Increased Risk for Coinfection With a Second Strain of Epstein-Barr Virus. J Infect Dis (2019) 6:955. doi: 10.1093/infdis/jiy601
16. Neves M, Marinho-Dias J, Ribeiro J, Sousa H. Epstein-Barr virus strains and variations: Geographic or disease-specific variants? J Med Virol (2017) 3:373. doi: 10.1002/jmv.24633
17. Liu F, Zhou ZH. Comparative virion structures of human herpesviruses. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al., editors. Human Herpesviruses, Cambridge, UK: Cambridge University Press (2007). p. 27–43.
18. Hutt-Fletcher LM. EBV glycoproteins: where are we now? Future Virol (2015) 10:1155. doi: 10.2217/fvl.15.80
19. Buschle A, Hammerschmidt W. Epigenetic lifestyle of Epstein-Barr virus. Semin Immunopathol (2000) 42:131. doi: 10.1007/s00281-020-00792-2
20. Möhl BS, Chen J, Sathiyamoorthy K, Jardetzky TS, Longnecker R. Structural and Mechanistic Insights into the Tropism of Epstein-Barr Virus. Mol Cells (2016) 4:286. doi: 10.14348/molcells.2016.0066
21. Hammerschmidt W. The Epigenetic Life Cycle of Epstein-Barr Virus. Curr Top Microbiol Immunol (2015) 390:103. doi: 10.1007/978-3-319-22822-8_6
22. Hatton OL, Harris-Arnold A, Schaffert S, Krams SM, Martinez OM. The interplay between Epstein-Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunol Res (2014) 2-3:268. doi: 10.1007/s12026-014-8496-1
23. Odumade OA, Hogqu KA. Progress and Problems in Understanding and Managing Primary Epstein-Barr Virus Infections. Clin Microbiol Rev (2011) 1:193. doi: 10.1128/CMR.00044-10
24. Crawford DH. Biology and disease associations of Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci (2001) 1408:461. doi: 10.1098/rstb.2000.0783
25. Thorley-Lawson DA, Babcock GJ. A model for persistent infection with Epstein-Barr virus: the stealth virus of human B cells. Life Sci (1999) 65:1433. doi: 10.1016/s0024-3205(99)00214-3
26. Shannon-Lowe C, Rowe M. Epstein Barr virus entry; kissing and conjugation. Curr Opin Virol (2014) 4:78. doi: 10.1016/j.coviro.2013.12.001
27. Heldwein EE. gH/gL supercomplexes at early stages of herpesvirus entry. Curr Opin Virol (2016) 18:1. doi: 10.1016/j.coviro.2016.01.010
28. Chesnokova LS, Hutt-Fletcher LM. Epstein-Barr virus infection mechanisms. Chin J Cancer (2014) 33:545. doi: 10.5732/cjc.014.10168
29. Zhang H, Li Y, Wang HB, Zhang A, Chen ML, Fang ZX, et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat Microbiol (2018) 3:1. doi: 10.1038/s41564-017-0080-8
30. Chen J, Sathiyamoorthy K, Zhang X, Schaller S, Perez White BE, Jardetzky TS, et al. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat Microbiol (2018) 3:172. doi: 10.1038/s41564-017-0081-7
31. Trier N, Izarzugaza J, Chailyan A, Marcatili P, Houen G. Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands-Relation to rheumatoid arthritis. Int J Mol Sci (2018) 1:317. doi: 10.3390/ijms19010317
32. Chen J, Longnecker R. Epithelial cell infection by Epstein-Barr virus. FEMS Microbiol Rev (2019) 43:674. doi: 10.1093/femsre/fuz023
33. Gan YJ, Chodosh J, Morgan A, Sixbey JW. Epithelial cell polarization is a determinant in the infectious outcome of immunoglobulin A-mediated entry by Epstein-Barr virus. J Virol (1997) 71:519. doi: 10.1128/JVI.71.1.519-526.1997
34. Xiao J, Palefsky JM, Herrera R, Berline J, Tugizov SM. The Epstein-Barr virus BMRF-2 protein facilitates virus attachment to oral epithelial cells. Virology (2008) 370:430. doi: 10.1016/j.virol.2007.09.012
35. Wang HB, Zhang H, Zhang JP, Li Y, Zhao B, Feng GK, et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat Commun (2015) 6:6240. doi: 10.1038/ncomms7240
36. Xiong D, Du Y, Wang HB, Zhao B, Zhang H, Li Y, et al. Nonmuscle myosin heavy chain IIA mediates Epstein-Barr virus infection of nasopharyngeal epithelial cells. Proc Natl Acad Sci U S A (2015) 112:11036. doi: 10.1073/pnas.1513359112
37. Möhl BS, Chen J, Park SJ, Jardetzky TS, Longnecker R. Epstein-Barr Virus Fusion with Epithelial Cells Triggered by gB Is Restricted by a gL Glycosylation Site. J Virol (2017) 91:e01255. doi: 10.1128/JVI.01255-17
38. Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Möhl BS, Chen J, et al. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog (2014) 10:e1004309. doi: 10.1371/journal.ppat.1004309
39. Sathiyamoorthy K, Hu YX, Möhl BS, Chen J, Longnecker R, Jardetzky TS. Structural basis for Epstein-Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat Commun (2016) 7:13557. doi: 10.1038/ncomms13557
40. Farina A, Feederle R, Raffa S, Gonnella R, Santarelli R, Frati L, et al. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J Virol (2005) 79:3703. doi: 10.1128/JVI.79.6.3703-3712.2005
41. Granato M, Feederle R, Farina A, Gonnella R, Santarelli R, Hub B, et al. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J Virol (2008) 82:4042. doi: 10.1128/JVI.02436-07
42. Long HM, Meckiff BJ, Taylor GS. The T-cell Response to Epstein-Barr Virus-New Tricks From an Old Dog. Front Immunol (2019) 10:2193. doi: 10.3389/fimmu.2019.02193
43. Latour S, Fischer A. Signaling pathways involved in the T-cell-mediated immunity against Epstein-Barr virus: Lessons from genetic diseases. Immunol Rev (2019) 1:174. doi: 10.1111/imr.12791
44. Münz C. Epstein-Barr Virus-Specific Immune Control by Innate Lymphocytes. Front Immunol (2017) 8:1658. doi: 10.3389/fimmu.2017.01658
45. Middeldorp JM. Epstein-Barr Virus-Specific Humoral Immune Responses in Health and Disease. Curr Top Microbiol Immunol (2015) 391:289. doi: 10.1007/978-3-319-22834-1_10
46. Chijioke O, Azzi T, Nadal D, Münz C. Innate immune responses against Epstein Barr virus infection. J Leukoc Biol (2013) 6:1185. doi: 10.1189/jlb.0313173
47. Iizasa H, Kim H, Kartika AV, Kanehiro Y, Yoshiyama H. Role of Viral and Host microRNAs in Immune Regulation of Epstein-Barr Virus-Associated Diseases. Front Immunol (2020) 11:367. doi: 10.3389/fimmu.2020.00367
48. Jangra S, Yuen KS, Botelho MG, Jin DY. Epstein-Barr Virus and Innate Immunity: Friends or Foes? Microorganisms (2019) 6:183. doi: 10.3390/microorganisms7060183
49. Albanese M, Tagawa T, Buschle A, Hammerschmidt W. MicroRNAs of Epstein-Barr Virus Control Innate and Adaptive Antiviral Immunity. J Virol (2017) 16:e01667. doi: 10.1128/JVI.01667-16
50. Ressing ME, van Gent M, Gram AM, Hooykaas MJ, Piersma SJ, Wiertz EJ. Immune Evasion by Epstein-Barr Virus. Curr Top Microbiol Immunol (2015) 391:355. doi: 10.1007/978-3-319-22834-1_12
51. Rowe M, Zuo J. Immune responses to Epstein-Barr virus: molecular interactions in the virus invasion of CD8+ T cell immunity. Microbes Infect (2010) 3:173. doi: 10.1016/j.micinf.2009.12.001
52. Münz C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat Rev Microbiol (2019) 17:691. doi: 10.1038/s41579-019-0249-7
53. Kempkes B, Robertson ES. Epstein-Barr virus latency: current and future perspectives. Curr Opin Virol (2015) 14:138. doi: 10.1016/j.coviro.2015.09.007
54. Liebermann PM, Hu J, Renne R. Maintenance and replication during latency. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al., editors. Human Herpesviruses. Cambridge, UK: Cambridge University Press (2007). p. 379–402.
55. Kerr JR. Epstein-Barr virus (EBV) reactivation and therapeutic inhibitors. J Clin Pathol (2019) 10:651. doi: 10.1136/jclinpath-2019-205822
56. Murata T. Regulation of Epstein-Barr virus reactivation from latency. Microbiol Immunol (2014) 6:307. doi: 10.1111/1348-0421.12155
57. Kenney SC. Reactivation and lytic replication of EBV. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al., editors. Human Herpesviruses. Cambridge, UK: Cambridge University Press (2007). p. 403–33.
58. Price AM, Luftig MA. Dynamic Epstein-Barr virus gene expression on the path to B-cell transformation. Adv Virus Res (2014) 88:279. doi: 10.1016/B978-0-12-800098-4.00006-4
59. Wilson JB, Manet E, Gruffat H, Busson P, Blondel M, Fahraeus R. EBNA1: Oncogenic Activity, Immune Evasion and Biochemical Functions Provide Targets for Novel Therapeutic Strategies against Epstein-Barr Virus- Associated Cancers. Cancers (Basel) (2018) 10:109. doi: 10.3390/cancers10040109
60. Lista MJ, Martins RP, Billant O, Contesse MA, Findakly S, Pochard P, et al. Nucleolin directly mediates Epstein-Barr virus immune evasion through binding to G-quadruplexes of EBNA1 mRNA. Nat Commun (2017) 8:16043. doi: 10.1038/ncomms16043
61. Daskalogianni C, Pyndiah S, Apcher S, Mazars A, Manoury B, Ammari N, et al. Epstein-Barr virus-encoded EBNA1 and ZEBRA: targets for therapeutic strategies against EBV-carrying cancers. J Pathol (2015) 235:334. doi: 10.1002/path.4431
62. Quinn LL, Williams LR, White C, Forrest C, Zuo J, Rowe M. The Missing Link in Epstein-Barr Virus Immune Evasion: the BDLF3 Gene Induces Ubiquitination and Downregulation of Major Histocompatibility Complex Class I (MHC-I) and MHC-II. J Virol (2015) 90:356. doi: 10.1128/JVI.02183-15
63. Ressing ME, van Leeuwen D, Verreck FA, Gomez R, Heemskerk B, Toebes M, et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc Natl Acad Sci U S A (2003) 100:11583. doi: 10.1073/pnas.2034960100
64. Ressing ME, van Leeuwen D, Verreck FA, Keating S, Gomez R, Franken KL, et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J Virol (2005) 79:841. doi: 10.1128/JVI.79.2.841-852.2005
65. Hislop AD, Ressing ME, van Leeuwen D, Pudney VA, Horst D, Koppers-Lalic D, et al. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J Exp Med (2007) 204:1863. doi: 10.1084/jem.20070256
66. Rowe M, Glaunsinger B, van Leeuwen D, Zuo J, Sweetman D, Ganem D, et al. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc Natl Acad Sci U S A (2007) 104:3366. doi: 10.1073/pnas.0611128104
67. Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME, et al. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog (2009) 5:e1000255. doi: 10.1371/journal.ppat.1000255
68. van Gent M, Gram AM, Boer IGJ, Geerdink RJ, Lindenbergh MFS, Lebbink RJ, et al. Silencing the shutoff protein of Epstein-Barr virus in productively infected B cells points to (innate) targets for immune evasion. J Gen Virol (2015) 96:858. doi: 10.1099/jgv.0.000021
69. van Gent M, Braem SG, de Jong A, Delagic N, Peeters JG, Boer IG, et al. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog (2014) 10:e1003960. doi: 10.1371/journal.ppat.1003960
70. Means RE, Lang SM, Jung JU. Human gammaherpesvirus immune evasion strategies. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al., editors. Human Herpesviruses. Cambridge, UK: Cambridge University Press (2007). p. 559–86.
71. Fares S, Spiess K, Olesen ETB, Zuo J, Jackson S, Kledal TN, et al. Distinct Roles of Extracellular Domains in the Epstein-Barr Virus-Encoded BILF1 Receptor for Signaling and Major Histocompatibility Complex Class I Downregulation. mBio (2019) 10:e01707. doi: 10.1128/mBio.01707-18
72. Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science (1990) 248:1230.
73. Salek-Ardakani S, Arrand JR, Mackett M. Epstein-Barr virus encoded interleukin-10 inhibits HLA-class I, ICAM-1, and B7 expression on human monocytes: implications for immune evasion by EBV. Virology (2002) 304:342. doi: 10.1006/viro.2002.1716
74. Yoon SI, Jones BC, Logsdon NJ, Harris BD, Kuruganti S, Walter MR. Epstein-Barr virus IL-10 engages IL-10R1 by a two-step mechanism leading to altered signaling properties. J Biol Chem (2012) 287:26586. doi: 10.1074/jbc.M112.376707
75. Jog NR, Chakravarty EF, Guthridge JM, James JA. Epstein Barr Virus Interleukin 10 Suppresses Anti-inflammatory Phenotype in Human Monocytes. Front Immunol (2018) 9:2198. doi: 10.3389/fimmu.2018.02198
76. Hoebe EK, Le Large TY, Tarbouriech N, Oosterhoff D, De Gruijl TD, Middeldorp JM, et al. Epstein-Barr virus-encoded BARF1 protein is a decoy receptor or macrophage colony stimulating factor and interferes with macrophage differentiation and activation. Viral Immunol (2012) 25:461. doi: 10.1089/vim.2012.0034
77. Elegheert J, Bracke N, Pouliot P, Gutsche I, Shkumatov AV, Tarbouriech N, et al. Allosteric competitive inactivation of hematopoietic CSF-1 signaling by the viral decoy receptor BARF1. Nat Struct Mol Biol (2012) 19:938. doi: 10.1038/nsmb.2367
78. Lubinski JM, Jiang M, Hook L, Chang Y, Sarver C, Mastellos D, et al. Herpes simplex virus type 1 evades the effects of antibody and complement in vivo. J Virol (2002) 76:9232. doi: 10.1128/jvi.76.18.9232-9241.2002
79. Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, et al. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press (2007).
80. Gram AM, Oosenbrug T, Lindenbergh MF, Büll C, Comvalius A, Dickson KJ, et al. The Epstein-Barr Virus Glycoprotein gp150 Forms an Immune-Evasive Glycan Shield at the Surface of Infected Cells. PLoS Pathog (2016) 12:e1005550. doi: 10.1371/journal.ppat.1005550
81. Dugan JP, Coleman CB, Haverkos B. Opportunities to Target the Life Cycle of Epstein-Barr Virus (EBV) in EBV-Associated Lymphoproliferative Disorders. Front Oncol (2019) 9:127. doi: 10.3389/fonc.2019.00127
82. Houldcroft CJ, Kellam P. Host genetics of Epstein-Barr virus infection, latency and disease. Rev Med Virol (2015) 20152:71. doi: 10.1002/rmv.1816
83. Helminen M, Lahdenpohja N, Hurme M. Polymorphism of the interleukin-10 gene is associated with susceptibility to Epstein-Barr virus infection. J Infect Dis (1999) 180:496. doi: 10.1086/314883
84. Pender MP. The Essential Role of Epstein-Barr Virus in the Pathogenesis of Multiple Sclerosis. Neuroscientist (2011) 17:351. doi: 10.1177/1073858410381531
85. Hedström AK, Bonfim IL, Hillert J, Olsson T, Alfredsson L. Obesity interacts with infectious mononucleosis in risk of multiple sclerosis. Eur J Neurol (2015) 22:578. doi: 10.1111/ene.12620
86. Kasifoglu N, Oz S, Dinleyici EC, Us T, Bor O, Durmaz G, et al. Comparison of Methods Used for the Diagnosis of Epstein-Barr Virus Infections in Children. Pol J Microbiol (2018) 1:81. doi: 10.5604/01.3001.0010.6287
87. Lam WKJ, Jiang P, Chan KCA, Cheng SH, Zhang H, Peng W, et al. Sequencing-based counting and size profiling of plasma Epstein-Barr virus DNA enhance population screening of nasopharyngeal carcinoma. Proc Natl Acad Sci U S A (2018) 22:E5115. doi: 10.1073/pnas.1804184115
88. Cao P, Zhang M, Wang W, Dai Y, Sai B, Sun J, et al. Fluorescence in situ hybridization is superior for monitoring Epstein Barr viral load in infectious mononucleosis patients. BMC Infect Dis (2017) 1:323. doi: 10.1186/s12879-017-2412-y
89. Fagin U, Nerbas L, Vogl B, Jabs WJ. Analysis of BZLF1 mRNA detection in saliva as a marker for active replication of Epstein-Barr virus. J Virol Methods (2017) 244:11. doi: 10.1016/j.jviromet.2017.02.016
90. Kwok H, Chan KW, Chan KH, Chiang AK. Distribution, persistence and interchange of Epstein-Barr virus strains among PBMC, plasma and saliva of primary infection subjects. PLoS One (2015) 3:e0120710. doi: 10.1371/journal.pone.0120710
91. Fafi-Kremer S, Brengel-Pesce K, Barguès G, Bourgeat MJ, Genoulaz O, Seigneurin JM, et al. Assessment of automated DNA extraction coupled with real-time PCR for measuring Epstein-Barr virus load in whole blood, peripheral mononuclear cells and plasma. J Clin Virol (2004) 2:157. doi: 10.1016/j.jcv.2003.10.002
92. Gulley ML. Molecular Diagnosis of Epstein-Barr Virus-Related Diseases. J Mol Diagn (2001) 3:1. doi: 10.1016/S1525-1578(10)60642-3
93. De Paschale M, Clerici P. Serological diagnosis of Epstein-Barr virus infection: Problems and solutions. World J Virol (2012) 1:31. doi: 10.5501/wjv.v1.i1.31
94. Sternbæk L, Draborg AH, Østerlund MT, Iversen LV, Troelsen L, Theander E, et al. Increased antibody levels to stage-specific Epstein-Barr virus antigens in systemic autoimmune diseases reveal a common pathology. Scand J Clin Lab Invest (2019) 1-2:7. doi: 10.1080/00365513.2018.1550807
95. Liu Z, Coghill AE, Pfeiffer RM, Proietti C, Hsu WL, Chien YC, et al. Patterns of Interindividual Variability in the Antibody Repertoire Targeting Proteins Across the Epstein-Barr Virus Proteome. J Infect Dis (2018) 12:1923. doi: 10.1093/infdis/jiy122
96. Färber I, Hinderer W, Rothe M, Lang D, Sonneborn HH, Wutzler W. Serological diagnosis of Epstein-Barr virus infection by novel ELISAs based on recombinant capsid antigens p23 and p18. J Med Virol (2001) 63:271. doi: 10.1002/1096-9071(200104)63:4<271::aid-jmv1001>3.0.co;2-t
97. van Grunsven WM, Spaan WJ, Middeldorp JM. Localization and diagnostic application of immunodominant domains of the BFRF3-encoded Epstein-Barr virus capsid protein. J Infect Dis (1994) 1:13. doi: 10.1093/infdis/170.1.13
98. Linde A. Diagnosis of Epstein-Barr virus-related diseases. Scand J Infect Dis Suppl (1996) 100:83.
99. Kutok JL, Wang F. Spectrum of Epstein-Barr virus-associated diseases. Annu Rev Pathol (2006) 1:375. doi: 10.1146/annurev.pathol.1.110304.100209
100. Farrell PJ. Epstein-Barr Virus and Cancer. Annu Rev Pathol (2019) 14:29. doi: 10.1146/annurev-pathmechdis-012418-013023
101. Jha HC, Pei Y, Robertson ES. Epstein-Barr Virus: Diseases Linked to Infection and Transformation. Front Microbiol (2016) 7:1602. doi: 10.3389/fmicb.2016.01602
102. Saha A, Robertson ES. Epstein-Barr virus-associated B-cell lymphomas: pathogenesis and clinical outcomes. Clin Cancer Res (2011) 10:3056. doi: 10.1158/1078-0432.CCR-10-2578
103. Wang M, Yu F, Wu W, Wang Y, Ding H, Qian L. Epstein-Barr virus-encoded microRNAs as regulators in host immune responses. Int J Biol Sci (2018) 5:565. doi: 10.7150/ijbs.24562
104. Hue SS, Oon ML, Wang S, Tan SY, Ng SB. Epstein-Barr virus-associated T- and NK-cell lymphoproliferative diseases: an update and diagnostic approach. Pathology (2020) 1:111. doi: 10.1016/j.pathol.2019.09.011
105. Iwatsuki K, Miyake T, Hirai Y, Yamamoto T. Hydroa vacciniforme: a distinctive form of Epstein-Barr virus-associated T-cell lymphoproliferative disorders. Eur J Dermatol (2019) 1:21. doi: 10.1684/ejd.2018.3490
106. Morell M, Varela N, Marañón C. Myeloid Populations in Systemic Autoimmune Diseases. Clin Rev Allergy Immunol (2017) 2:198. doi: 10.1007/s12016-017-8606-7
107. Draborg AH, Duus K, Houen G. Epstein-Barr virus in systemic autoimmune diseases. Clin Dev Immunol (2013) 2013:535738. doi: 10.1155/2013/535738
108. Scherer HU, Häupl T, Burmester GR. The etiology of rheumatoid arthritis. J Autoimmun (2020) 110:102400. doi: 10.1016/j.jaut.2019.102400
109. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet (2017) 10086:2328. doi: 10.1016/S0140-6736(17)31472-1
110. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 23:2205. doi: 10.1056/NEJMra1004965
111. van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. J Autoimmun (2020) 110:102392. doi: 10.1016/j.jaut.2019.102392
112. Volkov M, van Schie KA, van der Woude D. Autoantibodies and B Cells: The ABC of rheumatoid arthritis pathophysiology. Immunol Rev (2020) 1:148 doi: 10.1111/imr.12829
113. Klareskog L, Rönnelid J, Saevarsdottir S, Padyukov L, Alfredsson L. The importance of differences; On environment and its interactions with genes and immunity in the causation of rheumatoid arthritis. J Intern Med (2020) 5:514. doi: 10.1111/joim.13058
114. Balandraud N, Roudier J. Epstein-Barr virus and rheumatoid arthritis. Joint Bone Spine (2018) 2:165. doi: 10.1016/j.jbspin.2017.04.011
115. Masuoka S, Kusunoki N, Takamatsu R, Takahashi H, Tsuchiya K, Kawai S, et al. Epstein-Barr virus infection and variants of Epstein-Barr nuclear antigen-1 in synovial tissues of rheumatoid arthritis. PLoS One (2018) 12:e0208957. doi: 10.1371/journal.pone.0208957
116. Edwards CJ, Cooper C. Early environmental factors and rheumatoid arthritis. Clin Exp Immunol (2006) 1:1. doi: 10.1111/j.1365-2249.2005.02940.x
117. Dedmon LE. The genetics of rheumatoid arthritis. Rheumatol (Oxford) (2020) 59:2661. doi: 10.1093/rheumatology/keaa232
118. Alonso-Ruiz A, Pijoan JI, Ansuategui E, Urkaregi A, Calabozo M, Quintana A. Tumor necrosis factor alpha drugs in rheumatoid arthritis: systematic review and metaanalysis of efficacy and safety. BMC Musculoskelet Disord (2008) 9:52. doi: 10.1186/1471-2474-9-52
119. Ball RJ, Avenell A, Aucott L, Hanlon P, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and rheumatoid arthritis. Arthritis Res Ther (2015) 17:274. doi: 10.1186/s13075-015-0755-6
120. Trier NH, Holm BE, Heiden J, Slot O, Locht H, Lindegaard H, et al. Antibodies to a strain-specific citrullinated Epstein-Barr virus peptide diagnoses rheumatoid arthritis. Sci Rep (2018) 1:3684. doi: 10.1038/s41598-018-22058-6
121. Sakkas LI, Daoussis D, Liossis SN, Bogdanos DP. The Infectious Basis of ACPA-positive Rheumatoid Arthritis. Front Microbiol (2017) 8:1853. doi: 10.3389/fmicb.2017.01853
122. Roudier J, Balandraud N, Auger I. Anti PAD autoimmunity and rheumatoid arthritis. Joint Bone Spine (2018) 85:659. doi: 10.1016/j.jbspin.2018.03.017
123. Maibom-Thomsen SL, Trier NH, Holm BE, Hansen KB, Rasmussen MI, Chailyan A, et al. Immunoglobulin G structure and rheumatoid factor epitopes. PLoS One (2019) 6:e0217624. doi: 10.1371/journal.pone.0217624
124. Kuusela E, Kouri VP, Olkkonen J, Koivuniemi R, Äyräväinen L, Rajamäki K, et al. Serun Epstein-Barr virus DNA, detected by droplet digital PCR, correlates with disease activity in patients with rheumatoid arthritis. Clin Exp Rheumatol (2018) 5:778.
125. Croia C, Serafini B, Bombardieri M, Kelly S, Humby F, Severa M, et al. Epstein-Barr virus persistence and infection of autoreactive plasma cells in synovial lymphoid structures in rheumatoid arthritis. Ann Rheum Dis (2013) 9:1559. doi: 10.1136/annrheumdis-2012-202352
126. Mehraein Y, Lennerz C, Ehlhardt S, Remberger K, Ojak A, Zang KD. Latent Epstein-Barr virus (EBV) infection and cytomegalovirus (CMV) infection in synovial tissue of autoimmune chronic arthritis determined by RNA- and DNA-in situ hybridization. Mod Pathol (2004) 7:781. doi: 10.1038/modpathol.3800119
127. Wang J, Zhou L, Liu B. Update on disease pathogenesis, diagnosis, and management of primary Sjögren’s syndrome. Int J Rheum Dis (2020) 6:723. doi: 10.1111/1756-185X.13839
128. Bowman SJ. Primary Sjögren’s syndrome. Lupus (2018) 27(1_suppl):32. doi: 10.1177/0961203318801673
129. Björk A, Mofors J, Wahren-Herlenius M. Environmental factors in the pathogenesis of primary Sjögren’s syndrome. J Intern Med (2020) 5:475. doi: 10.1111/joim.13032
130. Nakamura H, Kawakami A, Eguchi K. Mechanisms of autoantibody production and the relationship between autoantibodies and the clinical manifestations in Sjögren’s syndrome. Transl Res (2006) 6:281. doi: 10.1016/j.trsl.2006.07.003
131. Teos LY, Alevizos I. Genetics of Sjögren’s syndrome. Clin Immunol (2017) 182:41. doi: 10.1016/j.clim.2017.04.018
132. Tsubota K, Fujishima H, Toda I, Katagiri S, Kawashima Y, Saito I. Increased levels of Epstein-Barr virus DNA in lacrimal glands of Sjogren’s syndrome patients. Acta Ophthalmol Scand (1995) 73:425. doi: 10.1111/j.1600-0420.1995.tb00302.x
133. Maślińska M. The role of Epstein-Barr virus infection in primary Sjögren’s syndrome. Curr Opin Rheumatol (2019) 5:475. doi: 10.1097/BOR.0000000000000622
134. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol (2019) 9:1400. doi: 10.1002/art.40930
135. Fava A, Petri M. Systemic lupus erythematosus: Diagnosis and clinical management. J Autoimmun (2019) 96:1. doi: 10.1016/j.jaut.2018.11.001
136. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol (2016) 12:716. doi: 10.1038/nrrheum.2016.186
137. Ceccarelli F, Perricone C, Massaro L, Cipriano E, Alessandri C, Spinelli FR, et al. Assessment of disease activity in Systemic Lupus Erythematosus: Lights and shadows. Autoimmun Rev (2015) 7:601. doi: 10.1016/j.autrev.2015.02.008
138. Durcan L, O’Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet (2019) 10188:2332. doi: 10.1016/S0140-6736(19)30237-5
139. Tselios K, Gladman DD, Touma Z, Su J, Anderson N, Urowitz MB. Disease course patterns in systemic lupus erythematosus. Lupus (2019) 1:114. doi: 10.1177/0961203318817132
140. Sharma M, Vignesh P, Tiewsoh K, Rawat A. Revisiting the complement system in systemic lupus erythematosus. Expert Rev Clin Immunol (2020) 4:397. doi: 10.1080/1744666X.2020.1745063
141. Pisetsky DS. Evolving story of autoantibodies in systemic lupus erythematosus. J Autoimmun (2020) 110:102356. doi: 10.1016/j.jaut.2019.102356
142. Prado MS, Dellavance A, Rodrigues SH, Marvulle V, Andrade LEC. Changes in the result of antinuclear antibody immunofluorescence assay on HEp-2 cells reflect disease activity status in systemic lupus erythematosus. Clin Chem Lab Med (2020) 8:1271. doi: 10.1515/cclm-2019-0638
143. Fu SM, Dai C, Zhao Z, Gaskin F. Anti-dsDNA Antibodies are one of the many autoantibodies in systemic lupus erythematosus. F1000Res (2015) 4:939. doi: 10.12688/f1000research.6875.1
144. Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, et al. A volcanic exPLoSion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev (2015) 1:75. doi: 10.1016/j.autrev.2014.10.003
145. Kwon YC, Chun S, Kim K, Mak A. Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond. Cells (2019) 10:1180. doi: 10.3390/cells8101180
146. Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J Autoimmun (2015) 64:125. doi: 10.1016/j.jaut.2015.08.004
147. Islam MA, Khandker SS, Alam SS, Kotyla P, Hassan R. Vitamin D status in patients with systemic lupus erythematosus (SLE): A systematic review and meta-analysis. Autoimmun Rev (2019) 11:102392. doi: 10.1016/j.autrev.2019.102392
148. Li ZX, Zeng S, Wu HX, Zhou Y. The risk of systemic lupus erythematosus associated with Epstein-Barr virus infection: a systematic review and meta-analysis. Clin Exp Med (2019) 1:23. doi: 10.1007/s10238-018-0535-0
149. Parisis D, Bernier C, Chasset F, Arnaud L. Impact of tobacco smoking upon disease risk, activity and therapeutic response in systemic lupus erythematosus: A systematic review and meta-analysis. Autoimmun Rev (2019) 11:102393. doi: 10.1016/j.autrev.2019.102393
150. Parks CG, de Souza Espindola Santos A, Barbhaiya M, Costenbader KH. Understanding the role of environmental factors in the development of systemic lupus erythematosus. Best Pract Res Clin Rheumatol (2017) 3:306. doi: 10.1016/j.berh.2017.09.005
151. Barbhaiya M, Costenbader KH. Environmental exposures and the development of systemic lupus erythematosus. Curr Opin Rheumatol (2016) 5:497. doi: 10.1097/BOR.0000000000000318
152. Draborg AH, Duus K, Houen G. Epstein-Barr virus and systemic lupus erythematosus. Clin Dev Immunol (2012) 2012:370516. doi: 10.1155/2012/370516
153. James JA, Harley JB, Scofield RH. Epstein-Barr virus and systemic lupus erythematosus. Curr Opin Rheumatol (2006) 5:462. doi: 10.1097/01.bor.0000240355.37927.94
154. Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity (2006) 1:63. doi: 10.1080/08916930500484849
155. McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med (2005) 1:85. doi: 10.1038/nm1167
156. Draborg AH, Jacobsen S, Westergaard M, Mortensen S, Larsen JL, Houen G, et al. Reduced response to Epstein-Barr virus antigens by T-cells in systemic lupus erythematosus patients. Lupus Sci Med (2014) 1:e000015. doi: 10.1136/lupus-2014-000015
157. Draborg AH, Sandhu N, Larsen N, Lisander Larsen J, Jacobsen S, Houen G. Impaired Cytokine Responses to Epstein-Barr Virus Antigens in Systemic Lupus Erythematosus Patients. J Immunol Res (2016) 2016:6473204. doi: 10.1155/2016/6473204
158. Caza T, Oaks Z, Perl A. Interplay of infections, autoimmunity, and immunosuppression in systemic lupus erythematosus. Int Rev Immunol (2014) 4:330. doi: 10.3109/08830185.2013.863305
159. Esposito S, Bosis S, Semino M, Rigante D. Infections and systemic lupus erythematosus. Eur J Clin Microbiol Infect Dis (2014) 9:1467. doi: 10.1007/s10096-014-2098-7
160. Cappelli LC, Shah AA. The relationships between cancer and autoimmune rheumatic diseases. Best Pract Res Clin Rheumatol (2020) 1:101472. doi: 10.1016/j.berh.2019.101472
161. Bank I. The Role of Gamma Delta T Cells in Autoimmune Rheumatic Diseases. Cells (2020) 9:462. doi: 10.3390/cells9020462
162. Erten Ş, Şahin A, Altunoğlu A, Gemcioğlu E, Koca C. Comparison of plasma vitamin D levels in patients with Sjögren’s syndrome and healthy subjects. Int J Rheum Dis (2015) 18:70. doi: 10.1111/1756-185X.12298
163. Servioli L, Maciel G, Nannini C, Crowson CS, Matteson EL, Cornec D, et al. Association of Smoking and Obesity on the Risk of Developing Primary Sjögren Syndrome: A Population-based Cohort Study. J Rheumatol (2019) 7:727. doi: 10.3899/jrheum.180481
164. Vlachogiannis NI, Nezos A, Tzioufas AG, Koutsilieris M, Moutsopoulos HM, Mavragani CP. Increased frequency of the PTPN22W* variant in primary Sjogren’s Syndrome: Association with low type I IFN scores. Clin Immunol (2016) 173:157. doi: 10.1016/j.clim.2016.10.015
165. Paradowska-Gorycka A, Stypińska B, Olesińska M, Felis-Giemza A, Mańczak M, Czuszynska Z, et al. Association of HLA-DRB1 alleles with susceptibility to mixed connective tissue disease in Polish patients. HLA (2016) 1:13. doi: 10.1111/tan.12698
166. Flåm ST, Gunnarsson R, Garen T, Norwegian MCTD Study Group, BA L, Molberg Ø. The HLA profiles of mixed connective tissue disease differ distinctly from the profiles of clinically related connective tissue diseases. Rheumatol (Oxford) (2015) 3:528. doi: 10.1093/rheumatology/keu310
167. Nguyen B, Mayes MD, Arnett FC, del Junco D, Reveille JD, Gonzalez EB, et al. HLA-DRB1*0407 and *1304 are risk factors for scleroderma renal crisis. Arthritis Rheum (2011) 2:530. doi: 10.1002/art.30111
168. Agarwal SK, Reveille JD. The genetics of scleroderma (systemic sclerosis). Curr Opin Rheumatol (2010) 2:133. doi: 10.1097/BOR.0b013e3283367c17
169. Zheng Q, Zhu K, Gao CN, Xu YP, Lu MP. Prevalence of Epstein-Barr virus infection and characteristics of lymphocyte subsets in newly onset juvenile dermatomyositis. World J Pediatr (2019). doi: 10.1007/s12519-019-00314-7
170. Schiffenbauer A, Faghihi-Kashani S, O’Hanlon TP, Flegel WA, Adams SD, Targoff IN, et al. The effect of cigarette smoking on the clinical and serological phenotypes of polymyositis and dermatomyositis. Semin Arthritis Rheum (2018) 3:504. doi: 10.1016/j.semarthrit.2018.02.003
171. Gao X, Han L, Yuan L, Yang Y, Gou G, Sun H, et al. HLA class II alleles may influence susceptibility to adult dermatomyositis and polymyositis in a Han Chinese population. BMC Dermatol (2014) 14:9. doi: 10.1186/1471-5945-14-9
172. Abbasi M, Mousavi MJ, Jamalzehi S, Alimohammadi R, Bezvan MH, Mohammadi H, et al. Strategies toward rheumatoid arthritis therapy; the old and the new. J Cell Physiol (2019) 7:10018. doi: 10.1002/jcp.27860
173. Faurschou M, Jayne DR. Anti-B cell antibody therapies for inflammatory rheumatic diseases. Annu Rev Med (2014) 65:263. doi: 10.1146/annurev-med-070912-133235
174. Willrich MA, Murray DL, Snyder MR. Tumor necrosis factor inhibitors: clinical utility in autoimmune diseases. Transl Res (2015) 2:270. doi: 10.1016/j.trsl.2014.09.006
175. Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev (2005) 4:189. doi: 10.1016/j.autrev.2004.10.007
176. Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung HP, Maniar T, et al. Epstein-Barr Virus in Multiple Sclerosis: Theory and Emerging Immunotherapies. Trends Mol Med (2020) 26:296. doi: 10.1016/j.molmed.2019.11.003
177. Schaier M, Gottschalk C, Kälble F, Uhlmann L, Eckstein V, Müller-Tidow C, et al. The onset of active disease in systemic lupus erythematosus patients is characterised by excessive regulatory CD4+-T-cell differentiation. Clin Exp Rheumatol (2020). Online ahead of print.
178. Khasnis AA, Schoen RT, Calabrese LH. Emerging viral infections in rheumatic diseases. Semin Arthritis Rheumatol (2011) 2:236. doi: 10.1016/j.semarthrit.2011.01.008
179. Sakkas LI, Bogdanos DP. Infections as a cause of autoimmune rheumatic diseases. Auto Immun Highlights (2016) 1:13. doi: 10.1007/s13317-016-0086-x
180. Becker J, Winthrop KL. Update on rheumatic manifestations of infectious diseases. Curr Opin Rheumatol (2010) 1:72. doi: 10.1097/BOR.0b013e328333b9f5
Keywords: antibodies, Epstein-Barr virus, connective tissue disease, systemic autoimmune diseases, human herpes virus
Citation: Houen G and Trier NH (2021) Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 11:587380. doi: 10.3389/fimmu.2020.587380
Received: 25 July 2020; Accepted: 19 November 2020;
Published: 07 January 2021.
Edited by:
Linda Ann Spatz, City University of New York, United StatesReviewed by:
Cheryl Rockwell, Michigan State University, United StatesCopyright © 2021 Houen and Trier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gunnar Houen, Z3VubmFyaEBibWIuc2R1LmRr; Nicole Hartwig Trier, bmljb2xlLmhhcnR3aWcudHJpZXJAcmVnaW9uaC5kaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.