
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 19 February 2021
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.587229
This article is part of the Research TopicNLRP3 Inflammasome: Regulatory Mechanisms, Role in Health and Disease and Therapeutic PotentialView all 25 articles
 Yu-Wei Chang1,2
Yu-Wei Chang1,2 Ling-Chien Hung3,4
Ling-Chien Hung3,4 Yu-Cheng Chen3,4
Yu-Cheng Chen3,4 Wen-Hung Wang3,4
Wen-Hung Wang3,4 Chun-Yu Lin1,3,4Hsin-Han Tzeng1
Chun-Yu Lin1,3,4Hsin-Han Tzeng1 Jau-Ling Suen1,5,6*†
Jau-Ling Suen1,5,6*† Yen-Hsu Chen3,4,7,8*†
Yen-Hsu Chen3,4,7,8*†The NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome is the platform for IL-1β maturation, aimed at mediating a rapid immune response against danger signals which must be tightly regulated. Insulin is well known as the critical hormone in the maintenance of glucose in physiologic response. Previous studies have proved insulin has the anti-inflammatory effect but the molecular mechanism of immunomodulation provided by insulin is not clear so far. Here we investigated whether insulin reduces inflammation by regulating the NLRP3 inflammasome. In the present study, we used LPS and ATP to induce the intracellular formation of the NLRP3 inflammasome. Insulin inhibited the secretion of IL-1β by preventing the assembly of the ASC in THP-1 cells and human CD14+ monocyte-derived macrophages. The phosphorylation status of Syk, p38 mitogen−activated protein kinase (MAPK) and ASC were altered by insulin. These effects were attenuated in THP-1 cells transfected with small interfering RNA targeting insulin receptors. In vivo, administration of glucose–insulin–potassium reduced serum IL-1β level, intestinal ASC speck formation, local macrophage infiltration and alleviated intestinal injury in mice exposed to LPS. Insulin may play an immunomodulatory role in anti-inflammation by regulating the NLRP3 inflammasome.
Sepsis is a life-threatening organ dysfunction caused by a deregulated immune response to infection. It is a leading cause of mortality in intensive care units and a major health problem worldwide (1). The host immune responses to sepsis are characterized by pro-inflammatory and anti-inflammatory responses. In general, pro-inflammatory responses are activated to eliminate pathogens but excessive inflammation mediates tissue damage in sepsis. On the other hand, anti-inflammatory reactions are responsible for limiting tissue injury but immune suppression results in the enhanced susceptibility to secondary infection (2). Upon early inflammatory response, monocytes and macrophages play a critical role in connecting the innate and adaptive immunity during sepsis. They also participate in releasing pro-inflammatory cytokines; however, dysregulated function of these important cell subsets contributes to a “cytokine storm” and causes organ dysfunction (3). Until recently, several randomized clinical trials investigating agents for the treatment of sepsis, such as anti-TNF and anti-LPS, have failed (4). Therefore, the development of new potential therapeutic strategies against sepsis is of crucial importance.
Inflammasomes, receptors and sensors of the innate immune system, play an important role in mediating a rapid immune response against infections caused by pathogens and tissue damage (5). The assembly of the complex is initiated by the activation of pattern recognition receptors (PRRs), including the AIM2-like receptors and the nucleotide binding oligomerization domain (NOD)-like receptors (NLRs). Among the numerous PRRs identified, NLRP3 is the most extensively characterized (5, 6). The NLRP3 inflammasome is the platform for caspase-1 activation and IL-1β maturation. It consists of the NLRP3 scaffold, the ASC adaptor, and pro-caspase-1 (7). The multiprotein complex is activated upon exposure to the board spectrum of stimuli, including fungi (8), bacteria (9, 10), virus (11, 12), crystal particles (13), and environmental particles (14). Although activation of NLRP3 is critical for the clearance of extracellular pathogens and induction of the innate immune response, aberrant NLRP3 activity participates in the development of inflammation-related diseases, such as type II diabetes, gout, Alzheimer’s disease, Crohn’s disease, atherosclerosis, and sepsis (15). Therefore, accumulating studies have clarified the mechanism through which the NLRP3 inflammasome is regulated (16, 17); however, the role of NLRP3 inflammasome in sepsis awaits further investigations.
Insulin is a critical hormone that maintains blood glucose levels by facilitating cellular glucose uptake, regulating carbohydrate, lipid and protein metabolism and promoting cell division and growth through its mitogenic effects (18). Moreover, numerous experiments have established that insulin exerts glucose homeostatic and anti-inflammatory effects. Pretreatment with insulin inhibits the activation of NF-κB and expression of pro-inflammatory cytokines to alleviate inflammatory response in vitro and in vivo (19–21). However, the molecular mechanism involved in the improvement of sepsis outcome by insulin remains unclear. In the present study, we explored the immuno-modulatory role of insulin in sepsis and demonstrated that insulin inhibited the inflammatory response by reducing the oligomerization of the ASC, leading to attenuation of inflammasome activation during sepsis.
The THP-1 human leukemia monocytic cell line was purchased from Biosource Collection and Research Center (Taiwan) and maintained in RPMI 1640 medium (Mediatech, Manassas, VA, USA) supplemented with 10% FBS and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 g/ml amphotericin B).
Human CD14+ monocytes were isolated from donor peripheral blood mononuclear cells (PBMCs) as previously described (22). In brief, 20 ml of each vein blood sample from volunteers was collected into sterile tubes containing EDTA. First, PBMCs were obtained by gradient centrifugation with Ficoll–Hypaque (GE Healthcare, Wauwatosa, WI, USA). Human CD14+ cells were subsequently isolated through the column with anti-CD14 microbeads (Miltenyi BioTec GmbH, Bergisch Gladbach, Germany). Cells were subsequently washed twice with cold PBS and resuspended in RPMI 1640 medium (Mediatech) supplemented with 10% FBS and antibiotics. For the purity measurement of CD14+ cells, the hematology analyzer (XS-800i™; Sysmex, Kobe, Japan) was used to evaluate the cell population of the isolated cells. CD14+ cells showed >90% of purity compared with control PBMCs. We further differentiated the purified human CD14+ monocytes into macrophages by adding recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF) (10 ng/ml) (PeproTech, Rocky Hill, NJ, USA) for 7 days at 37°C in 5% CO2. The human blood samples were collected anonymously following approval of the project by the Institutional Review Board of Kaohsiung Medical University Hospital (KMUH-IRB-20170045).
Male C57BL/6 mice (age: 6–8 weeks) were purchased from the National Laboratory Animal Center (Taipei, Taiwan), and maintained in a pathogen-free facility. The animal experimental protocol was approved by the Institutional Animal Care and Use Committee of the Kaohsiung Medical University (IACUC permit number: 106186) and was performed based on the guidelines and regulations of the institution. The mice were randomly allocated into one of four groups (n=7 per group) as follows: PBS control group, LPS exposure group, glucose–potassium (GK) solution plus LPS group (GK/LPS), and glucose–insulin–potassium (GIK) solution plus LPS group (GIK/LPS). To evaluate the insulin effect but avoid hypoglycemia and hypokalemia in vivo, we used the GIK solution, which is consisted of glucose (400 mg/kg), insulin (50 μg/kg), and potassium chloride (64 μEq/kg) (23), to observe the effect of insulin in vivo. The GK solution, the same components as those of GIK except insulin, was used as vehicle control. Briefly, naive mice (PBS control group) were primed through i.p. injection with sterile PBS (total volume: 100 μl). The GK/LPS and GIK/LPS groups were i.p. injected with GK and GIK solution for 30 min, respectively. Finally, after LPS (10 mg/kg) i.p. injection for 6 h, the mice were sacrificed.
Subsequently, a GlucoSure Blood Glucose Monitoring System (APEXBIO, Hsinchu, Taiwan) was used to monitor the blood glucose levels. The serum samples obtained from the hearts of mice were used to determine the levels of cytokines. In addition, the sera biochemistry profiles were determined using a cobas c311 analyzer (Diagnostics Roche, Rotkreuz, Switzerland). The small and large intestine of mice were obtained and fixed in 4% formaldehyde. Organ embedding in paraffin, tissue sectioning, hematoxylin and eosin (H&E) staining, and immunohistochemistry (IHC) staining were performed by Litzung Biocompany Inc. (Kaohsiung, Taiwan). The severity of intestinal injury after the i.p. administration of LPS was quantified in histological sections of the terminal ileum by a pathologist (blinded to the study groups) who assigned a score according to the International Harmonization of Nomenclature and Diagnostic Criteria scale as previously reported (24).
After pretreatment with insulin or vehicle (HEPES) for 30 min, THP-1 cells were exposed to LPS (1 μg/ml) for 24 h. Prior to cell lysate harvest, the cells were additionally stimulated with ATP (5 mM) for 30 min. Subsequently, the cells were washed in PBS and resuspended in cell lysis buffer supplemented with a protease inhibitor cocktail. After centrifugation at 15,000 rpm for 30 min, the protein lysate was harvested and the total protein concentration was determined. An equal amount (30 μg) of protein was resolved through 10%–12% SDS-PAGE electrophoresis. Subsequently, the proteins were transferred onto a PVDF membrane. After blocking in tris-buffered saline with Tween 20 buffer containing 2% bovine serum albumin, the membrane was probed with specific antibodies as listed in Table 1.
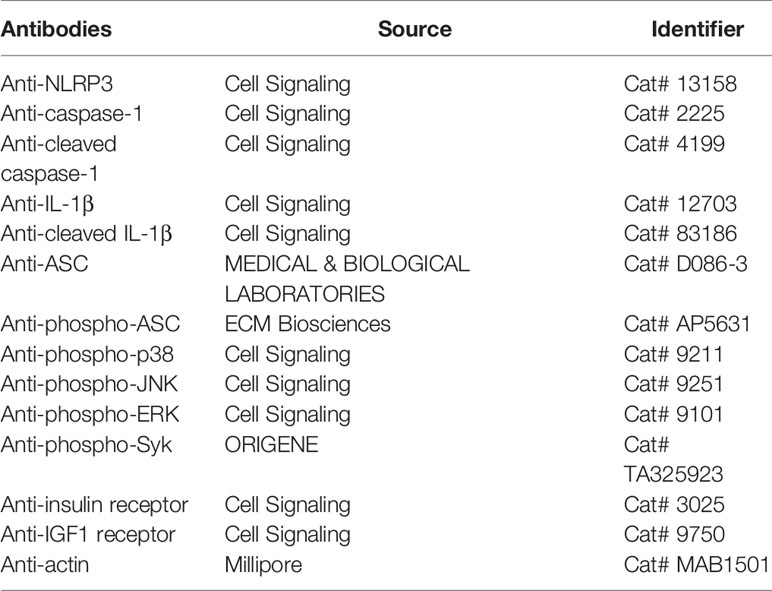
Table 1 The sources of the antibodies used in the study.
The formation of ASC specks in the cell lysates was determined as previously described (25, 26). For the harvest of lysates, THP-1 cells were lysed in Buffer A (20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 320 mM sucrose) supplemented with a protease inhibitor cocktail. After cell lysis, a 21-gage needle was used to disrupt the cells in the total lysates (30 repetitions). The lysates were subsequently centrifuged at 300×g for 8 min. The supernatants were transferred to sterile empty Eppendorf tubes without disturbing the nuclear pellets and diluted with 1 volume of CHAPS buffer (20 mM HEPES-KOH, pH 7.5, 5 mM MgCl2, 0.5 mM EGTA, 0.1% CHAPS) supplemented with a protease inhibitor cocktail. After centrifugation at 2,400×g for 8 min at 4°C, the supernatants were removed. The pellets were washed twice with 0.5 ml ice-cold PBS and resuspended in 30 μl CHAPS buffer. DSS crosslinker material was added at a final concentration of 2 mM and incubated at 37°C for 30 min. Subsequently, the protein lysates were resolved through 12% SDS-PAGE electrophoresis, and the levels of ASC were determined via western blotting.
Cells were washed, fixed, and incubated with anti-ASC antibodies overnight at 4°C, and finally incubated with an Alexa Fluor 488-conjugated secondary antibody. Cell nuclei were specifically stained with DAPI. Finally, a Zeiss LSM510 META laser scanning microscope (Carl Zeiss, Germany) was used to visualize the results under the oil immersion objective.
We used the LDH quantitative assessment assay to evaluate the level of LDH release and determine the cell death (27). Based on the instructions provided by the manufacturer, the cell supernatants were centrifuged at 500×g for 5 min at 4°C and the dead cells were subsequently removed. The LDH cytotoxicity assay kit (Pierce Biotechnology, USA) was used and the absorbance was read at 490 nm. The results were calculated and presented as the percentage of LDH release and cell death.
THP-1 cells and isolated human CD14+ monocytes were plated at a density of 4×105 cells per well in 12-well plates. After pretreatment with different concentrations of insulin for 30 min, the cells were treated with LPS (1 μg/ml) for 24 h. The cells were additionally stimulated with ATP (5 mM for 30 min) or nigericin (10 μM for 1 h), followed by harvesting of the supernatants. The levels of IL-1β (Cat# 88726188), IL-6 (Cat# 88706688), and TNF-α (Cat# 88734688) were measured using ELISA kits (Thermo Fisher).
The RNA was extracted from THP-1 cells using the RNAspin Mini RNA Isolation Kit and reverse-transcribed to cDNA using the RT reagent kit. Quantitative real-time PCR analysis was performed using a LightCycler (Roche Diagnostics, Mannheim, Germany) to evaluate the levels of target mRNA. We used the ΔΔCt method to calculate the expression of target genes. All data of the quantitative PCR were normalized to the levels of GAPDH, the housekeeping gene for THP-1 cells (28). The primer sequences used in the quantitative PCR analysis were listed in Table 2.
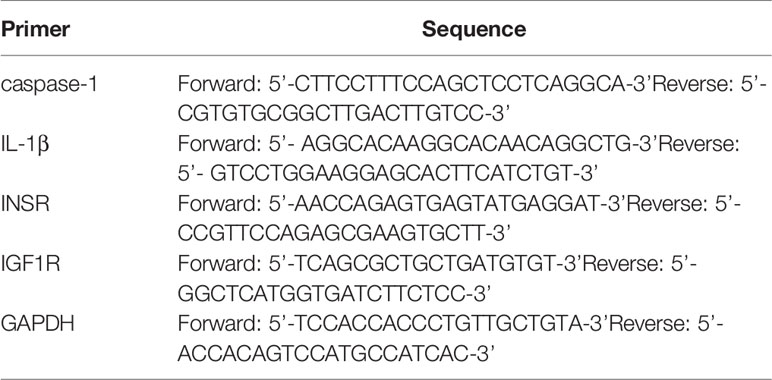
Table 2 Primer sequences for RT-qPCR.
The target siRNAs were purchased from Thermo Fisher Scientific and transfected into THP-1 cells using the Lipofectamine RNAiMAX reagent as previously described (29). For efficient knockdown, the cells were incubated for 48 h before treatment.
All quantification data from the present study are represented as mean ± standard deviation from experiments performed at least 3 independent times under identical conditions. Statistical analysis was performed by GraphPad Prism 7.0 software. Two tailed t-tests was used for two groups and ANOVA with Dunnett’s multiple comparisons test was used for more than two groups. A p-value <0.05 indicated a statistically significant difference.
NLRP3 activation requires LPS priming and ATP stimulation for the final assembly of the inflammasome complex, which cleaves pro-caspase 1 and releases the mature IL-1β (30). We used LPS plus ATP-stimulated THP-1 cells to establish an in vitro condition of inflammasome activation. Initially, we examined whether insulin pretreatment inhibits the release of pro-inflammatory cytokines via reducing inflammasome activation. Treatment with insulin significantly inhibited the expression of IL-1β in the THP-1 cells stimulated with LPS and ATP (Figure 1A). The consistent result was also observed that insulin reduced the IL-1β production of the THP-1 cell treated by LPS and nigericin, another NLRP3 activator (Supplemental Figure 1A). As a control, the production of TNF-α and IL-6 remained unchanged in insulin treatment conditions, indicating that the anti-inflammatory effect exerted by insulin is specifically associated with inflammasome activation. (Figures 1B, C). Subsequently, we evaluated the caspase-1-dependent programmed cell death (pyroptosis) through the LDH release assay. Following stimulation with LPS plus ATP, THP-1 cells treated with insulin exhibited a lower rate of pyroptosis than control cells (Figure 1D). We further used primary human macrophage to elucidate the insulin effect. CD14+ monocytes from human PBMCs were isolated and differentiated into macrophages through incubation with recombinant human GM-CSF. As shown in Figure 1E, compared with the vehicle group, a sharp reduction in IL-1β secretion was observed in the human macrophages treated with a lower concentration of insulin than those treated in THP-1 cells. This finding indicated that insulin provides more obviously effects to reduce the pro-inflammatory response in primary human cells. Consistently, insulin did not alter the expression of TNF-α and IL-6 under the same conditions (Figures 1F, G).These results suggest that insulin may effectively inhibit the production of pro-inflammatory cytokines and prevent the programmed cell death and this insulin effect may be associated with NLRP3 inflammasome activation.
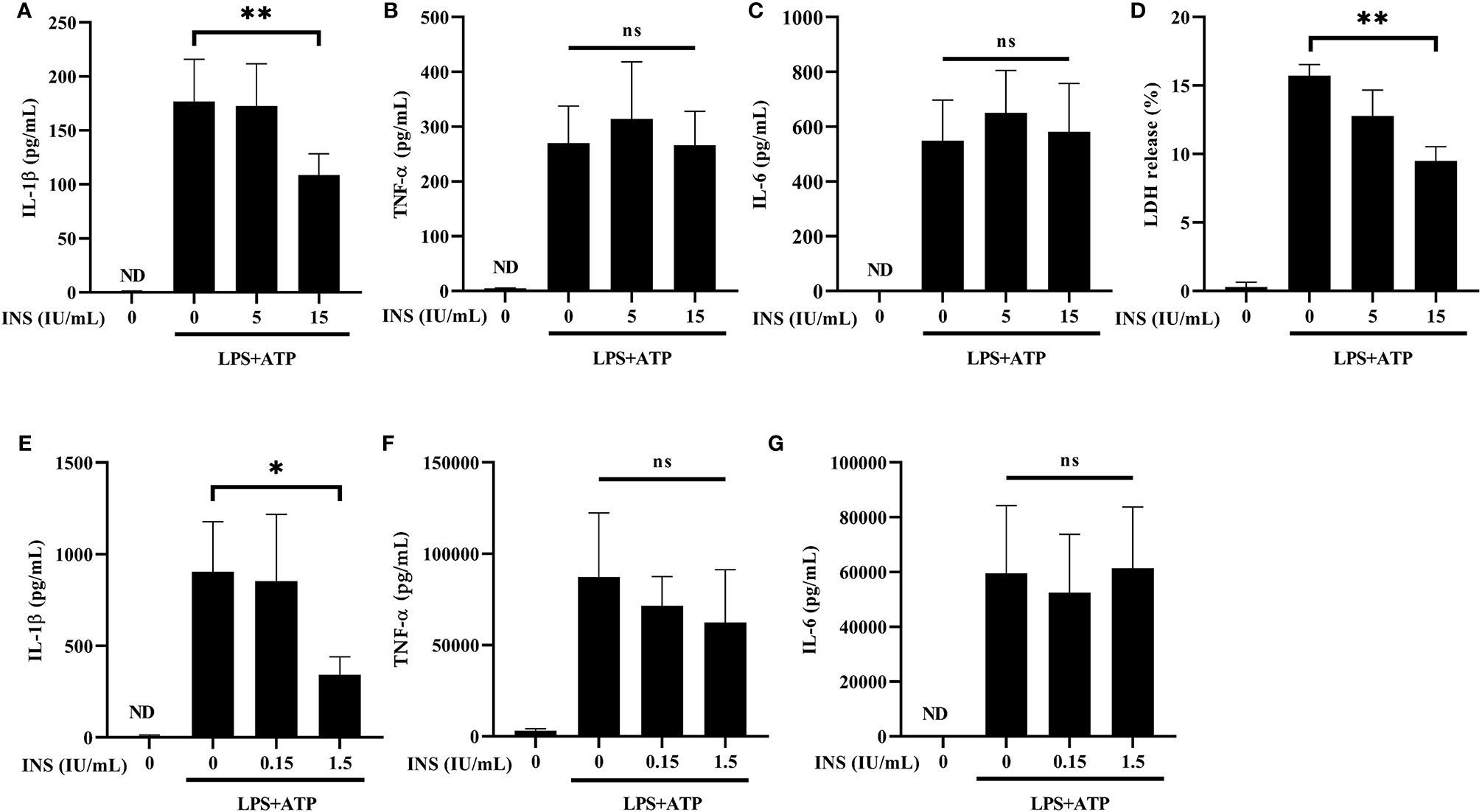
Figure 1 Insulin inhibits the secretion of NLRP3-mediated IL-1β, but not that of TLR-mediated TNF-α and IL-6. (A–C) Determination of IL-1β (A), TNF-α (B) and IL-6 (C) levels in the supernatants obtained from LPS (1 μg/ml for 24 h) plus ATP (5 mM for 30 min)-stimulated THP-1 cells pretreated with insulin in a dose-dependent manner (0, 5, and 15 IU/ml) for 30 min. (D) Pyroptosis of THP-1 cells stimulated as in (A) was evaluated by LDH release assay. The present result of LDH release (%) depicted the calculated cytotoxicity. (E-G) ELISA assay was used to determine the production of IL-1β (E), TNF-α (F), and IL-6 (G) in the CD14+ isolated primary macrophages. CD14+ cells in the treatment group were pretreated with insulin in a dose-dependent manner (0, 0.15, and 1.5 IU/ml) for 30 min and subsequently stimulated by LPS (1 μg/ml) and ATP (5 mM). The quantification data are presented as mean values (± SDs) derived from experiments performed 3 independent times under identical conditions; *p < 0.05, **p < 0.01 (one way ANOVA with Dunnett’s multiple comparisons test). INS, insulin; LPS, lipopolysaccharide; ND, non-detectable; ns, non-significance.
NLRP3 recruits pro-caspase-1 through ASC specks, leading to activation of caspase-1 and cleavage of pro-IL-1β to mature IL-1β (7). We subsequently analyzed the expression of the main components of the NLRP3 inflammasome through western blotting analysis to determine the mechanism of insulin-dependent immunomodulation during exposure to DAMPs. As expected, insulin pretreatment reduced the expression of the cleaved caspase-1 and mature IL-1β in the supernatant obtained from THP-1 cells stimulated by LPS plus ATP. In cell lysates, there were no changes in the endogenous level of the caspase-1 and IL-1β precursors (Figures 2A, B). The consistent data can be seen in the Supplementary Figure 2, mRNA levels of caspase-1 and IL-1β were not altered by insulin. The formation of ASC specks serves as a signal amplification mechanism for the inflammasome-mediated production of cytokines (31). Hence, we examined the ability of immuno-speck formation in inflammasome-associated proteins using immunofluorescence staining and the ASC oligomerization assay. Figures 2C, D showed the immunofluorescence images and quantitative evaluations. We found a sharp ASC speck signal in the cytoplasm following stimulation with LPS and ATP (condensed green points in Figure 2C). Interestingly, ASC aggregation was significantly inhibited after pretreatment with insulin (15 IU/ml). Also, ASC speck formation derived from LPS plus nigericin was inhibited by insulin (Supplemental Figures 1B, C). In support of the finding, the ASC oligomerization assay showed that insulin obviously inhibited the formation of ASC specks in a dose-dependent manner (Figure 2E). Our results suggest that insulin may prevent the formation of ASC specks without affecting the expression of the NLRP3 in inflammasome activation condition.
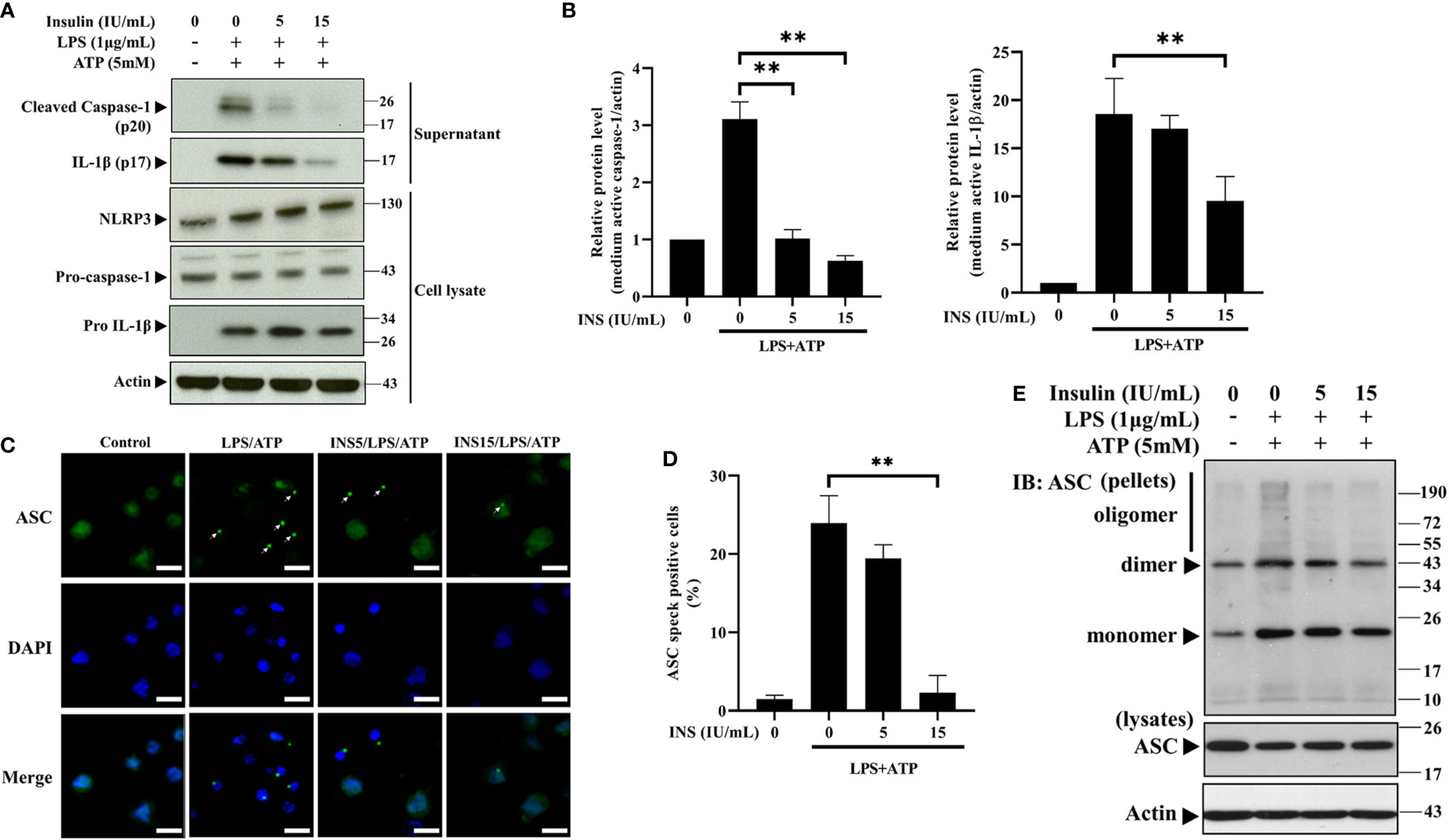
Figure 2 Insulin significantly reduces the NLRP3-mediated formation of the ASC pyroptosome. (A) Immunoblotting for caspase-1 (p20 and caspase-1 precursor), IL-1β (p17 and IL-1β precursor), and NLRP3 of the supernatant (upper panels) and cell lysates (lower panels) from THP-1 monocytes pretreated with insulin (0, 1, 5, and 15 IU/ml), followed by stimulation with LPS (1 μg/ml) for 6 h and ATP (5 mM) for 30 min. (B) Quantification of immunoblotting was determined by densitometric analysis, and each result was normalized to the levels of the vehicle control group. (C) Immunofluorescence microscopy of LPS (1 μg/ml for 24 h) and ATP (5 mM for 30 min)-stimulated THP-1 monocytes pretreated with insulin (0, 5, and 15 IU/ml) for 30 min, and subsequently immunostained for ASC (green) and DNA (DAPI, blue). Scale bars, 10 μm. White arrows indicate ASC specks. (D) The quantification represents the percentages of cells with an ASC speck, with ≥100 cells counted from 10 random fields in each experiment. (E) ASC oligomerization and redistribution assay in THP-1 cells treated as in (A). Immunoblotting analysis of ASC in cross-linked pellets (upper panels) and in cell lysates (lower panels). The quantification data are presented as mean values (± SDs) derived from experiments performed three independent times under identical conditions; Immunoblotting results as shown in (A, E) are one representative of three independent experiments; **p < 0.01 (one way ANOVA with Dunnett’s multiple comparisons test).
Syk kinase signaling in macrophages may play a critical role in immunomodulation through activation of the inflammasome (32, 33). Furthermore, both the Syk and MAPK pathways are involved in the phosphorylation and oligomerization of ASC (34, 35). Thus, we investigated the role of Syk and MAPK in the anti-inflammatory response induced by insulin. As can been seen in Figures 3A, B, the phosphoryl activities of Syk were significantly reduced after pretreatment with insulin in LPS plus ATP-stimulated THP-1 cells. While observing the MAPK activity, we found that p38 MAPK were obviously blocked following the treatment of insulin; however, there were no effects on the phosphorylation of JNK and ERK. We also confirmed the insulin significantly abrogated the phosphorylation of ASC (Figures 3C, D). Our results showed that insulin may inhibit ASC phosphorylation, by reducing the phosphorylation of Syk and p38 MAPK.
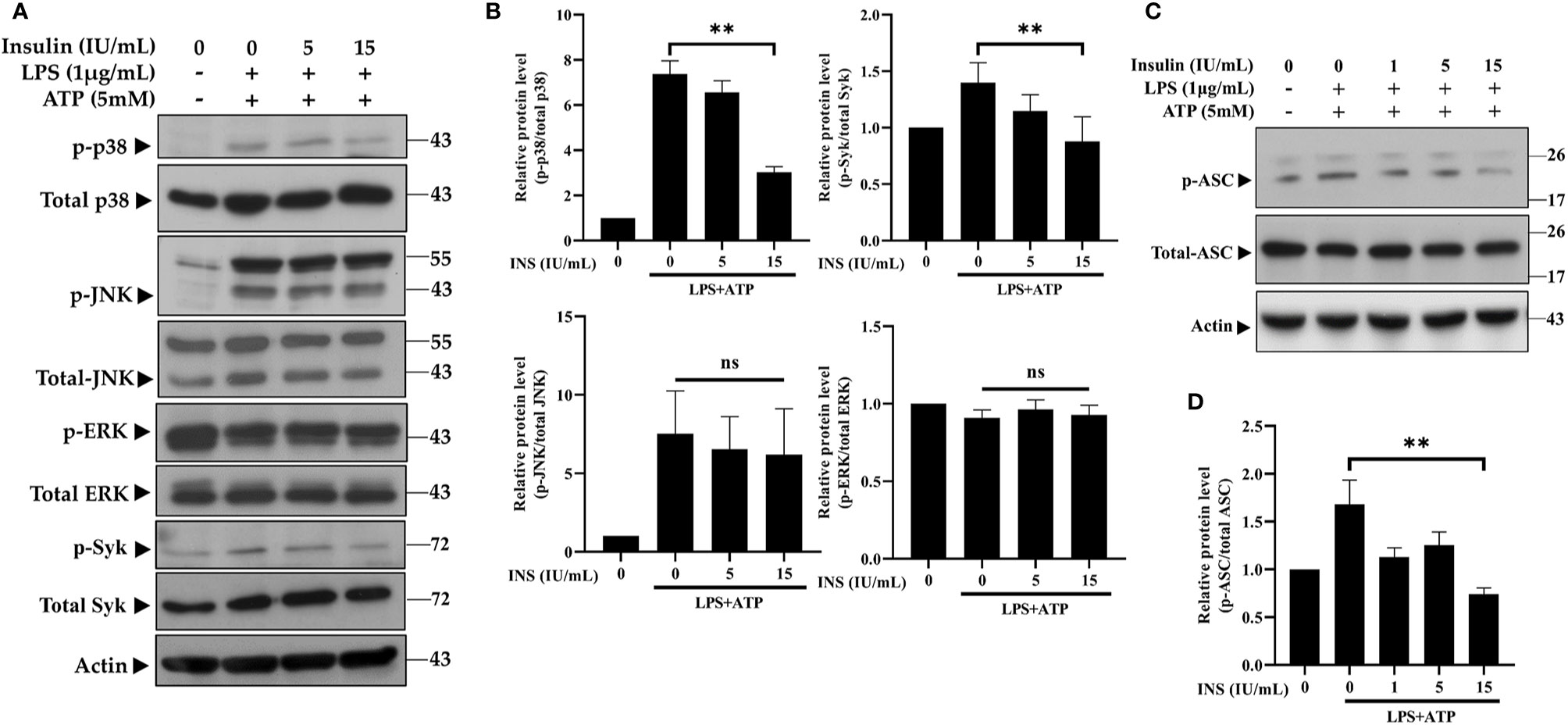
Figure 3 Insulin treatment regulates the molecular signaling in LPS plus ATP-stimulated THP-1 cells. (A) Immunoblotting analysis of MAPK and Syk kinase in the protein lysates from THP-1 cells pretreated with insulin (0, 5, and 15 IU/ml), followed by stimulation with LPS (1 μg/ml) for 2 h and ATP (5 mM) for 30 min. (B) Quantification of MAPK and Syk kinase was determined by densitometric analysis, and each result was normalized to the levels of the corresponding vehicle control group. (C) Immunoblotting analysis of phosphoryl ASC (p-ASC) in the protein lysates from THP-1 cells pretreated as in (A). (D) Quantification of p-ASC was determined by densitometric analysis, and normalized the levels of the vehicle control. The quantification data are presented as mean values (± SDs) derived from experiments performed three independent times under identical conditions; Immunoblotting results as shown in (A, C) are one representative of three independent experiments; **p < 0.01 (one way ANOVA with Dunnett’s multiple comparisons test). ns, non-significance.
To further confirm the immunomodulation of NLRP3 inflammasome derived from insulin, we evaluated IL-1β secretion in response to NLRP3 activation in THP-1 cells, in which INSR and IGF1R (major receptors of insulin) had been knocked down using small interfering RNA (siRNA). Firstly, siRNA-mediated gene silencing specifically and effeIctively inhibited the expression of the target mRNA and protein (Supplemental Figures S3A–E). Individual depletion of INSR or IGF1R significantly reversed the NLRP3 inflammasome-mediated secretion of IL-1β, respectively; however, it did not affect the expression of TNF-α and IL-6 (Figure 4A). Furthermore, the suppressive effects contributed by insulin could be reversed by silencing of INSR or IGF1R, such as the phosphorylation of p38, Syk, and ASC (Figures 4B, C), and ASC oligomerization (Figure 4D). Thus, these results suggest that insulin may play an important role in modulating the activation of the ASC-IL-1β axis in NLRP3 inflammasomes.
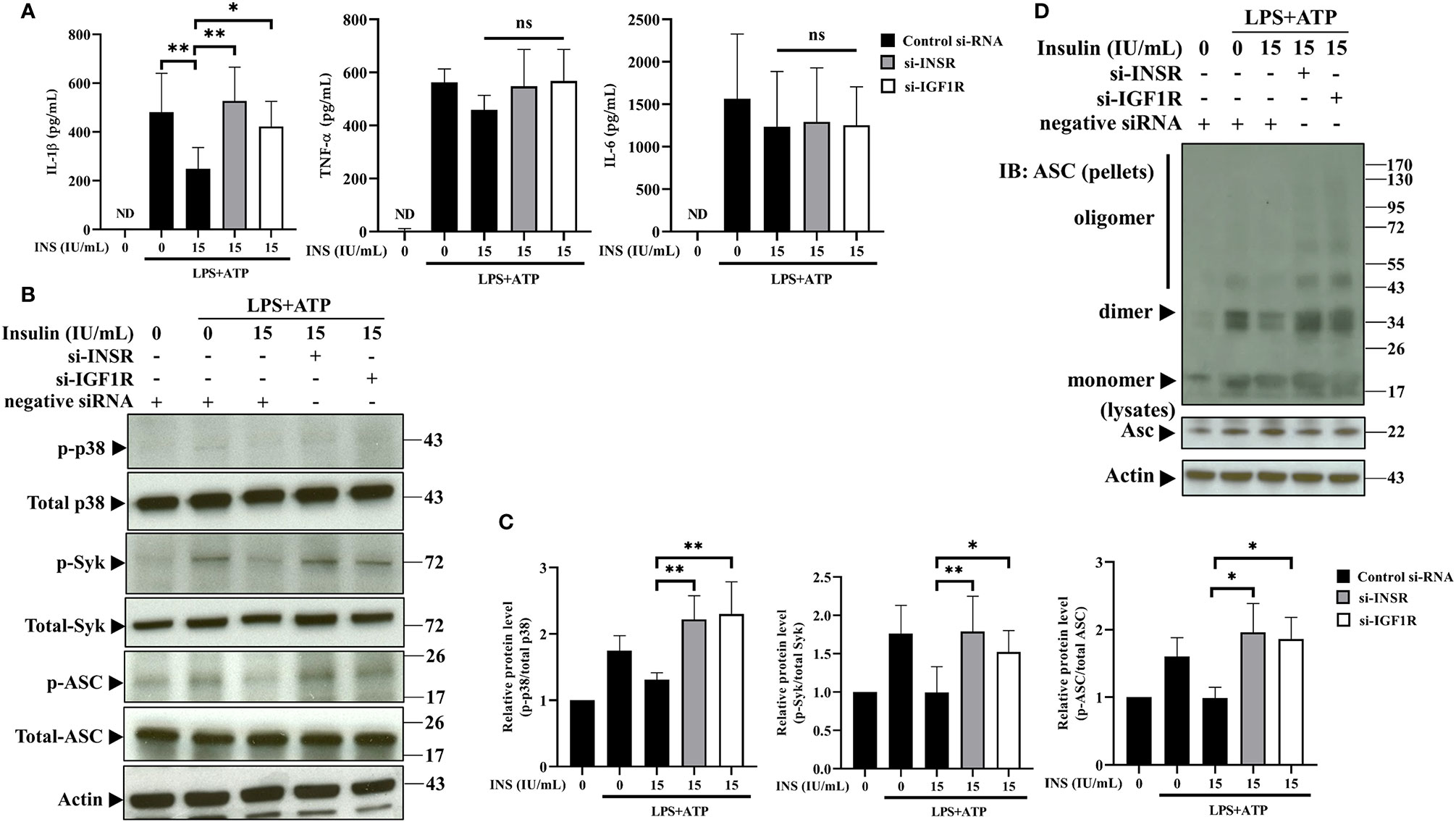
Figure 4 Insulin modulates the expression of ASC-IL-1β axis via the insulin receptor and insulin-like growth factor-1 receptor. (A) Cytokine ELISA assay for the levels of IL-1β, TNF-α, and IL-6 in the supernatants of THP-1 cells transfected with INSR-, IGF1R-, or negative control siRNA, followed by pretreatment with insulin (15 IU/ml for 30 min) and stimulation with LPS (1 μg/ml for 4 h) plus ATP (5 mM for 30 min). (B) Immunoblotting analysis of p38, Syk, and ASC in the protein lysates from THP-1 cells pretreated as in (A). (C) Quantification of p38, Syk, and ASC were determined by densitometric analysis, and each result was normalized to the levels of the corresponding vehicle control groups. (D) ASC oligomerization and redistribution assay in THP-1 cells treated as in (A). Immunoblotting analysis of ASC in cross-linked pellets (upper panels) and cell lysates (lower panels). The quantification data are presented as mean values (± SDs) derived from experiments performed three independent times under identical conditions (n=3); Immunoblotting result as shown in (B, D) are one representative of three independent experiments; *p < 0.05, **p < 0.01 (one way ANOVA with Dunnett’s multiple comparisons test). ns, non-significance.
We elucidated the in vivo immune modulatory effects of insulin using a GIK solution administered to a LPS induced murine endotoxemia model (36, 37). Serum analysis showed that pro-inflammatory cytokines (including IL-1β, TNF-α, and IL-6) were effectively induced 6 h after intraperitoneal (i.p.) injection of LPS in C57BL/6 mice (Figure 5A). Compared with the LPS group or GK/LPS group, mice treated with GIK50 (contains 50 μg insulin/kg body weight) followed by LPS injection were significantly reduced levels of the pro-inflammatory cytokines, including IL-β, TNF-α, and IL-6. The level of blood glucose was significantly reduced following the administration of LPS, whereas GK/LPS or GIK/LPS treatment did not affect the glucose level compared to LPS-treated mice. Regarding serum biochemistry, only LDH level, as an additional pyroptosis marker (38), was significantly inhibited by treatment with GIK50, but not blood urea nitrogen (BUN) and aspartate aminotransferase (AST) (Figure 5B). Furthermore, the architecture of the terminal ileum was preserved in the control group (PBS control without LPS exposure) (Figure 5C). However, mice i.p. injected with LPS showed an obvious disruption of the small intestinal mucosa, caused by the intensive infiltration of inflammatory cells into the lamina propria, edema of the submucosa, and epithelial cell loss. Interestingly, treatment with GIK50/LPS decreased the severity of LPS-mediated intestinal injury, compared to the GK/LPS group (Figures 5C, D). After that, we performed immunohistochemical assessment to further observe the mucosal cytokine expression involved in the small intestine. In line with the pathologic results, GIK50/LPS treatment sharply reduced the IL-1β expression compared to the LPS or GK/LPS groups (Figures 5E, F). It is well known that IL-1β promotes the macrophage recruitment in the context of inflammation (39). Thus, we next tested whether insulin treatment suppressed macrophage infiltration in tissue. Further evidence can be seen in the immunofluorescence images (Figure 5G). ASC specks were observed around the small intestinal mucosa from LPS administrated mice and co-localized with the infiltrated macrophages, whereas GIK/LPS treatment obviously blocked the ASC speck formation and reduced the macrophages infiltration. Collectively, insulin potentially exerts anti-inflammatory effects and in turn alleviates pyroptosis in mice with LPS-induced sepsis.
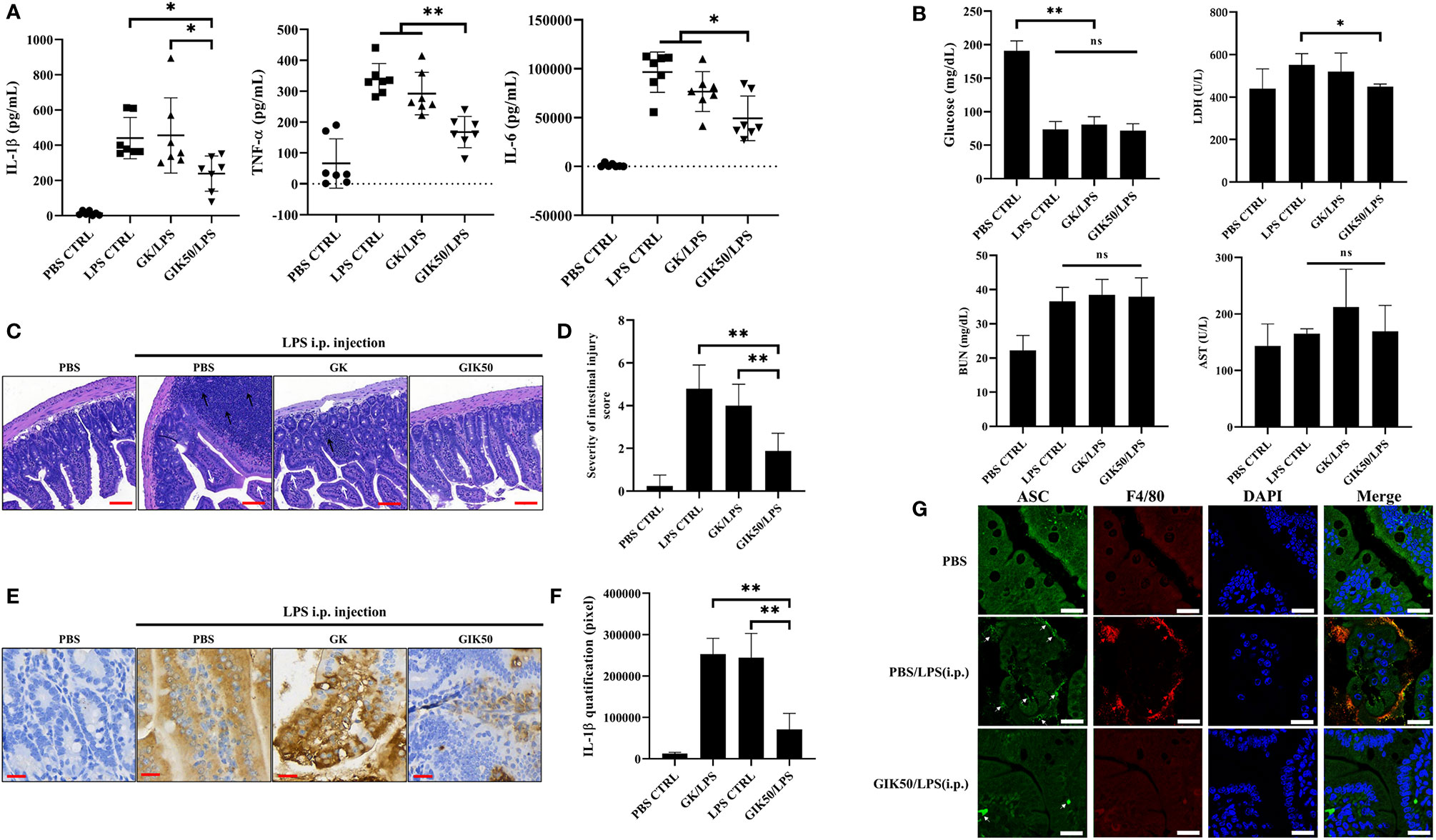
Figure 5 Immunomodulatory effects of insulin on systemic inflammation and intestinal injury in the LPS-treated mice. Serum samples were harvested 6 h after intraperitoneal injection with LPS, GK, or GIK50 in the first set of mice. (A) The serum levels of IL-1β, TNF-α, and IL-6 (n=7). (B) The levels of blood glucose (n=7), LDH (n=3), urea nitrogen (BUN) (n=3), and aspartate aminotransferase (AST) (n=3). (C) The representative images of H&E stained intestine sections depicts the infiltration of inflammatory cells (black arrows; scale bar, 100 μm) and disruption of the small intestinal mucosa (white arrows). (D) Quantification of the intestinal injury score was performed according to INHAND grades, as described in the section of Materials and Methods (n=3). (E) The images of immunohistochemical staining of intestine sections using the IL-1β mouse monoclonal antibody. (scale bar, 30 μm) (F) Quantification of IHC staining was determined in pixels (n=3). (G) Immunofluorescence images show ASC speck (green, white arrow) and F4/80 (red, red arrow) distribution in the small intestinal mucosa. Nuclei (blue) were revealed by DAPI. The merged images revealed the co-localization of ASC with F4/80, the cell surface markers of mice macrophages (Scale bars, 20 μm, n=3). The quantification data are represented as mean ± SDs, with “n” indicating the number of mice per group; **p < 0.01 (one way ANOVA with Dunnett’s multiple comparisons test). GK, glucose-potassium solution; GIK, glucose-insulin-potassium solution; i.p., intraperitoneal. *p < 0.05, **p < 0.01; ns, non-significance.
It has been shown that insulin has anti-inflammatory effects; however, the molecular mechanism remains largely unknown. The present study demonstrates that insulin exerts anti-inflammatory effect by alleviating the formation of ASC specks, to finally reduce the activation of the inflammasome and secretion of the pro-inflammatory cytokines in an INSR and IGF1R-dependent manner (Figure 6). To our best knowledge, this is the first study to reveal that insulin is a key regulator of NLRP3 inflammasome activation. Furthermore, the administration of insulin in mice with LPS-induced sepsis revealed that insulin effectively exerts an anti-inflammatory effect and prevented pyroptosis cell death. These findings suggest insulin may not only be the homeostasis endocrine but serve as an important role in immune-modulation in chronic metabolic or severe infectious diseases.
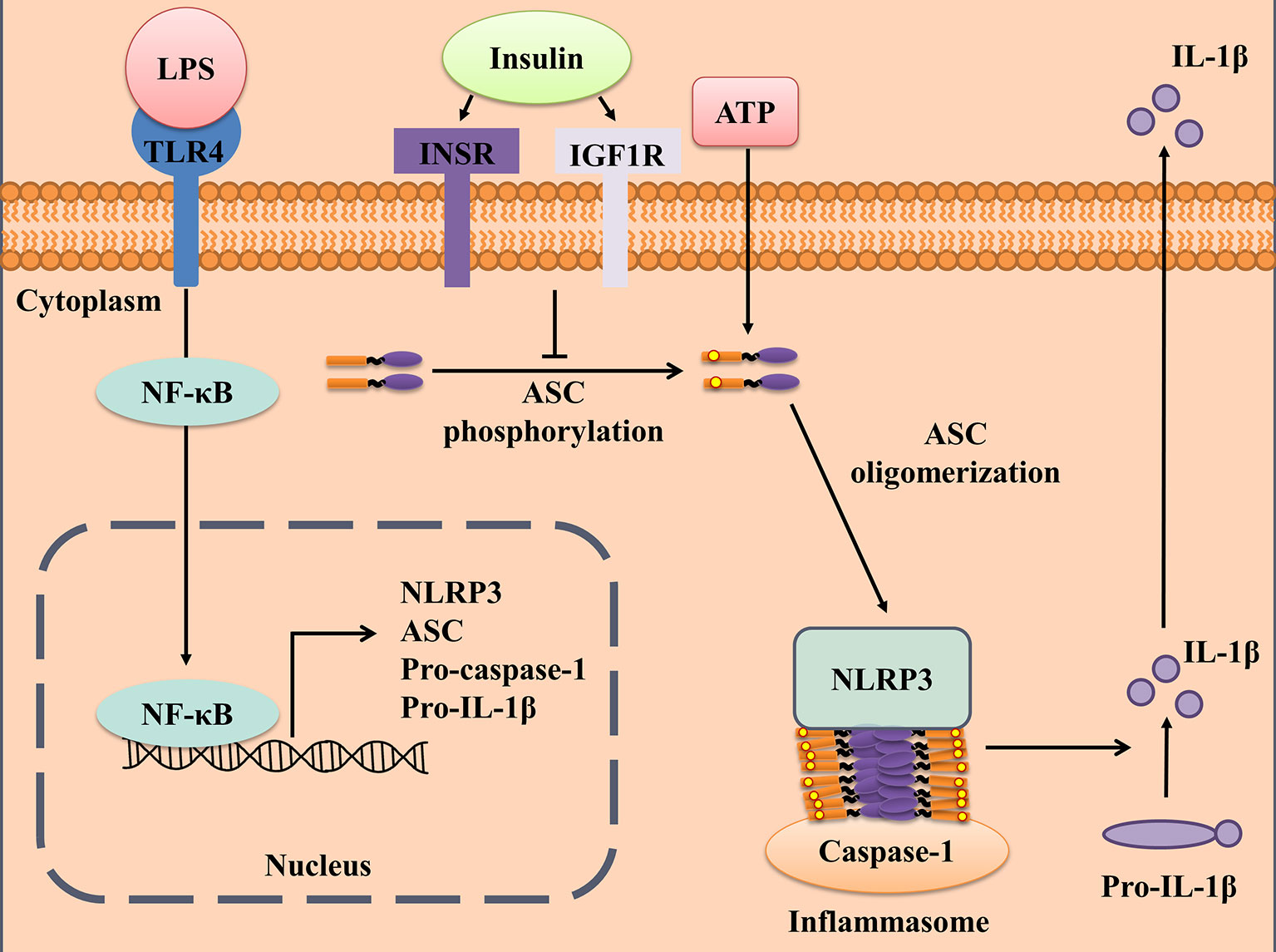
Figure 6 Schematic illustration of a possible mechanism for insulin reduced NLRP3 inflammasome activation. According to our results, we proposed that insulin inhibited ASC phosphorylation cascade and followed by reducing ASC oligomerization in the presence of NLRP3 stimulus. This immuno-modulatory effect derived from insulin reduced the IL-1β production and prevented pyroptosis cell death, which exerted the protective role during the danger signals exposure.
Insulin has been documented to exert glucose homeostatic and anti-inflammatory effects. It was capable of reducing the activation of intranuclear NF-κB activity and expression of pro-inflammatory cytokines, to alleviate inflammatory response in vitro and in vivo (19, 21, 40, 41). Furthermore, the previous studies revealed that intensive therapy with insulin improved the mortality rate in intensive care units, further indicating that treatment with insulin reduced mortality in patients with sepsis (42, 43). The evidence suggests that insulin may exert an anti-inflammatory effect by modulating the inflammatory response, and finally improves the symptoms of infection. However, studies concerning the anti-inflammatory effects of insulin have been limited in terms of the sample population or were not focused on the molecular mechanism. In the present study, we provide evidence showing the immunomodulation by insulin in the NLRP3 inflammasome pathway, which is rapidly stimulated in response to a variety of stress and infectious signals and elicits a robust inflammatory response.
Our results detail the molecular mechanism of inflammasome activation, which is regulated by treatment with insulin. The data suggest the protective role played by insulin in the inflammatory response. These findings extend the conclusion of the previous studies, suggesting that treatment with insulin therapy improves the outcome of patients with sepsis in the intensive care unit (42). In addition, the improvements noted were the results of immunomodulation contributed by insulin, which has negative feedback of inflammatory stress caused by microbes, immune disorders, and chronic diseases. Furthermore, it has been demonstrated that the effects of anti-inflammatory and organ protection gained from insulin in scalded experimental rats without controlling hyperglycemia (44). In line with the conclusion, our findings have greatly enhanced the understanding of the molecular mechanisms by which the immunomodulation derived from insulin are involved.
Binding of insulin to the α subunit of INSR induces a conformational change, followed by facilitation of the cell signal transduction, such as the binding of ATP, phosphorylation of β subunits, recruitment of intracellular substrates, and their subsequent phosphorylation (45, 46). Moreover, insulin binds to and activates the INSR, IGF1R, or INSR/IGF1R hybrid receptors with different affinities. Here, we propose that insulin modulates the activation of the NLRP3 inflammasome via INSR and IGF1R. Consistently, depletion of INSR or IGF1R by siRNA can reverse the inhibitory effect of insulin on the activation of the NLRP3 inflammasome (Figure 4). We demonstrated that INSR and IGF1R play a linked role in the dysregulation of NLRP3 inflammasome caused by insulin. Further investigations are warranted to clarify the docking conformation between insulin and the associated receptor, which can influence the development of inflammatory diseases caused by dysregulation of the NLRP3 inflammasome.
In contrast to in vitro effect, our results showed that in vivo effect of insulin on proinflammatory cytokine production is not restricted to IL-1β. The levels of TNF-α and IL-6 were also inhibited by insulin treatment in a LPS induced murine endotoxemia model. It is highly possible that the decrease of TNF-α and IL-6 was due to the significant IL-1β decrease in vivo as IL-1β release is a critical step in cascade induction of other pro-inflammatory cytokines (47–49). Another possible reason is that insulin may exert anti-inflammatory effect through another IL-1β-independent mechanism in vivo, including suppressing NF-κB pathway (21) or changing T cell polarization to Th2 (50). Thus, the levels of all three pro-inflammatory cytokines were significantly decreased in the insulin-treated model in the present study. In addition to the cascade inducing activity of IL-1β for other two proinflammatory cytokines, IL-1β can recruit macrophages in the context of inflammation (39). Therefore, the obviously less macrophage infiltration in insulin treated group is highly possible because of the decrease of IL-1β release in vivo.
However, this study had limitations. The results obtained in other kinds of cytosolic PRRs (e.g., NLRP1, AIM2, or NLRC4) were not representative of the entire pathophysiology encountered in sepsis. Future studies should include different DAMPs or PAMPs stimulations. This approach will comprehensively evaluate whether treatment with insulin plays a protective role in host cells receiving the inflammatory stimulators, and further establish the potential of insulin as a new therapeutic agent against sepsis.
In conclusion, insulin serves as a negative regulator of NLRP3 inflammasome activation. Our results provide compelling evidence for translational medicine to improve the clinical predicament in sepsis. Moreover, they suggest that treatment with insulin is effective against inflammatory stress. We expect the present evidence provides a new strategy for the therapeutic application of insulin and introduces an entirely new view for the comprehensive study of this field.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Kaohsiung Medical University Hospital (KMUH-IRB-20170045). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of the Kaohsiung Medical University (IACUC permit number: 106186). Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Y-WC, L-CH, and J-LS conceived and designed the experiments. Y-WC, L-CH, Y-CC, and H-HT performed the experimental works. Y-WC, W-HW, L-CH, C-YL, and J-LS analyzed and interpreted the results. Y-WC and J-LS drafted the manuscript. Y-HC is the guarantor of integrity of the entire study and responsible of editing and finally reviewing of the paper. All authors contributed to the article and approved the submitted version.
This study is supported partially by Kaohsiung Medical University Research Center Grant (KMU-TC108B03), Ministry of Science and Technology, Taiwan (MOST 106-2314-B-037 -087 and MOST 107-2314-B-037 -079 to Y-HC), and Ministry of Health and Welfare, Taiwan (Project No. 10645 to Y-WC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.587229/full#supplementary-material
1. Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315(8):762–74. doi: 10.1001/jama.2016.0288
2. Angus DC, van der Poll T. Severe Sepsis and Septic Shock. New Engl J Med (2013) 369(9):840–51. doi: 10.1056/NEJMra1208623
3. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev MMBR (2012) 76(1):16–32. doi: 10.1128/MMBR.05015-11
4. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med (2014) 20(4):195–203. doi: 10.1016/j.molmed.2014.01.007
5. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
6. Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell (2017) 68(1):185–197.e6. doi: 10.1016/j.molcel.2017.08.017
7. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
8. Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature (2009) 459(7245):433–6. doi: 10.1038/nature07965
9. Li L-H, Lin J-S, Chiu H-W, Lin W-Y, Ju T-C, Chen F-H, et al. Mechanistic Insight Into the Activation of the NLRP3 Inflammasome by Neisseria gonorrhoeae in Macrophages. Front Immunol (2019) 10:1815–32. doi: 10.3389/fimmu.2019.01815
10. Li L-H, Chen T-L, Chiu H-W, Hsu C-H, Wang C-C, Tai T-T, et al. Critical Role for the NLRP3 Inflammasome in Mediating IL-1β Production in Shigella sonnei-Infected Macrophages. Front Immunol (2020) 11:1115–28. doi: 10.3389/fimmu.2020.01115
11. Halfmann P, Hill-Batorski L, Kawaoka Y. The Induction of IL-1β Secretion Through the NLRP3 Inflammasome During Ebola Virus Infection. J Infect Dis (2018) 218(suppl_5):S504–s507. doi: 10.1093/infdis/jiy433
12. Shrivastava G, Visoso-Carvajal G, Garcia-Cordero J, Leon-Juarez M, Chavez-Munguia B, Lopez T, et al. Dengue Virus Serotype 2 and Its Non-Structural Proteins 2A and 2B Activate NLRP3 Inflammasome. Front Immunol (2020) 11:352–66. doi: 10.3389/fimmu.2020.00352
13. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440(7081):237–41. doi: 10.1038/nature04516
14. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci (2008) 105(26):9035–40. doi: 10.1073/pnas.0803933105
15. Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunological Rev (2011) 243(1):152–62. doi: 10.1111/j.1600-065X.2011.01043.x
16. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49(2):331–8. doi: 10.1016/j.molcel.2012.11.009
17. Martine P, Chevriaux A, Derangère V, Apetoh L, Garrido C, Ghiringhelli F, et al. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis (2019) 10(4):256. doi: 10.1038/s41419-019-1491-7
19. Chen Q, Yu W, Shi J, Shen J, Gao T, Zhang J, et al. Insulin alleviates the inflammatory response and oxidative stress injury in cerebral tissues in septic rats. J Inflammation (Lond) (2014) 11:18. doi: 10.1186/1476-9255-11-18
20. Sun Q, Li J, Gao F. New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J Diabetes (2014) 5(2):89–96. doi: 10.4239/wjd.v5.i2.89
21. Huang CT, Lue JH, Cheng TH, Tsai YJ. Glycemic control with insulin attenuates sepsis-associated encephalopathy by inhibiting glial activation via the suppression of the nuclear factor kappa B and mitogen-activated protein kinase signaling pathways in septic rats. Brain Res (2020) 1738:146822. doi: 10.1016/j.brainres.2020.146822
22. Chang Y-W, Huang W-C, Lin C-Y, Wang W-H, Hung L-C, Chen Y-H, et al. A Type of Plant Polyphenol Extracted from Trapa bispinosa Inhibits Antibiotic Resistance of Drug-Resistant Staphylococcus aureus. Int J Mol Sci (2019) 20(22):5790. doi: 10.3390/ijms20225790
23. Zhang JL, Chen YT, Chen GD, Wang T, Zhang JX, Zeng QY. Glucose-Insulin-Potassium Alleviates Intestinal Mucosal Barrier Injuries Involving Decreased Expression of Uncoupling Protein 2 and NLR Family-Pyrin Domain-Containing 3 Inflammasome in Polymicrobial Sepsis. BioMed Res Int (2017) 2017:4702067. doi: 10.1155/2017/4702067
24. Shackelford C, Long G, Wolf J, Okerberg C, Herbert R. Qualitative and quantitative analysis of nonneoplastic lesions in toxicology studies. Toxicol Pathol (2002) 30(1):93–6. doi: 10.1080/01926230252824761
25. Palazón-Riquelme P, Worboys JD, Green J, Valera A, Martín-Sánchez F, Pellegrini C, et al. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep (2018) 19(10):e44766. doi: 10.15252/embr.201744766
26. Chiu H-W, Li L-H, Hsieh C-Y, Rao YK, Chen F-H, Chen A, et al. Glucosamine inhibits IL-1β expression by preserving mitochondrial integrity and disrupting assembly of the NLRP3 inflammasome. Sci Rep (2019) 9(1):5603. doi: 10.1038/s41598-019-42130-z
27. Rayamajhi M, Zhang Y, Miao EA. Detection of pyroptosis by measuring released lactate dehydrogenase activity. Methods Mol Biol (Clifton NJ) (2013) 1040:85–90. doi: 10.1007/978-1-62703-523-1_7
28. Maess MB, Sendelbach S, Lorkowski S. Selection of reliable reference genes during THP-1 monocyte differentiation into macrophages. BMC Mol Biol (2010) 11:90–0. doi: 10.1186/1471-2199-11-90
29. Lee D-K, Kim J-H, Kim J, Choi S, Park M, Park W, et al. REDD-1 aggravates endotoxin-induced inflammation via atypical NF-κB activation. FASEB J (2018) 32(8):4585–99. doi: 10.1096/fj.201701436R
30. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol (2014) 15(8):727–37. doi: 10.1038/ni.2913
31. Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun (2016) 7:11929. doi: 10.1038/ncomms11929
32. Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol (2013) 14(12):1247–55. doi: 10.1038/ni.2749
33. Chung IC, OuYang C-N, Yuan S-N, Li H-P, Chen J-T, Shieh H-R, et al. Pyk2 activates the NLRP3 inflammasome by directly phosphorylating ASC and contributes to inflammasome-dependent peritonitis. Sci Rep (2016) 6:36214. doi: 10.1038/srep36214
34. Neumann K, Ruland J. Kinases conquer the inflammasomes. Nat Immunol (2013) 14(12):1207–8. doi: 10.1038/ni.2763
35. Li D, Ren W, Jiang Z, Zhu L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol Med Rep (2018) 18(5):4399–409. doi: 10.3892/mmr.2018.9427
36. Hagar JA, Edin ML, Lih FB, Thurlow LR, Koller BH, Cairns BA, et al. Lipopolysaccharide Potentiates Insulin-Driven Hypoglycemic Shock. J Immunol (2017) 199(10):3634–43. doi: 10.4049/jimmunol.1700820
37. Lewis AJ, Seymour CW, Rosengart MR. Current Murine Models of Sepsis. Surg Infect (2016) 17(4):385–93. doi: 10.1089/sur.2016.021
38. Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis (2019) 10(10):778. doi: 10.1038/s41419-019-2021-3
39. Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1α and IL-1β Recruit Different Myeloid Cells and Promote Different Stages of Sterile Inflammation. J Immunol (2011) 187: (9):4835–43. doi: 10.4049/jimmunol.1102048%JTheJournalofImmunology
40. Aljada A, Ghanim H, Saadeh R, Dandona P. Insulin inhibits NFkappaB and MCP-1 expression in human aortic endothelial cells. J Clin Endocrinol Metab (2001) 86(1):450–3. doi: 10.1210/jcem.86.1.7278
41. Dandona P, Chaudhuri A, Ghanim H, Mohanty P. Insulin as an Anti-Inflammatory and Antiatherogenic Modulator. J Am Coll Cardiol (2009) 53(5, Supplement):S14–20. doi: 10.1016/j.jacc.2008.10.038
42. Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, et al. Intensive Insulin Therapy in the Medical ICU. New Engl J Med (2006) 354(5):449–61. doi: 10.1056/NEJMoa052521
43. Langouche L, Vanhorebeek I, Vlasselaers D, Vander Perre S, Wouters PJ, Skogstrand K, et al. Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest (2005) 115(8):2277–86. doi: 10.1172/jci25385
44. Zhu Z, Hu T, Wang Z, Wang J, Liu R, Yang Q, et al. Anti-inflammatory and organ protective effect of insulin in scalded MODS rats without controlling hyperglycemia. Am J Emerg Med (2018) 36(2):202–7. doi: 10.1016/j.ajem.2017.07.070
45. Kohanski RA. Insulin receptor autophosphorylation. I. Autophosphorylation kinetics of the native receptor and its cytoplasmic kinase domain. Biochemistry (1993) 32(22):5766–72. doi: 10.1021/bi00073a007
46. Escribano O, Beneit N, Rubio-Longás C, López-Pastor AR, Gómez-Hernández A. The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J Diabetes Res (2017) 2017:12. doi: 10.1155/2017/1403206
47. Yang J, Hooper WC, Phillips DJ, Talkington DF. Interleukin-1beta responses to Mycoplasma pneumoniae infection are cell-type specific. Microb Pathog (2003) 34(1):17–25. doi: 10.1016/s0882-4010(02)00190-0
48. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood (1996) 87(6):2095–147. doi: 10.1182/blood.V87.6.2095.bloodjournal8762095
49. Eskan MA, Benakanakere MR, Rose BG, Zhang P, Zhao J, Stathopoulou P, et al. Interleukin-1beta modulates proinflammatory cytokine production in human epithelial cells. Infect Immun (2008) 76(5):2080–9. doi: 10.1128/IAI.01428-07
Keywords: insulin, nucleotide binding oligomerization domain (NOD)-, LRR-, and pyrin domain-containing protein 3 (NLRP3), inflammasome, ASC, immunomodulation
Citation: Chang Y-W, Hung L-C, Chen Y-C, Wang W-H, Lin C-Y, Tzeng H-H, Suen J-L and Chen Y-H (2021) Insulin Reduces Inflammation by Regulating the Activation of the NLRP3 Inflammasome. Front. Immunol. 11:587229. doi: 10.3389/fimmu.2020.587229
Received: 25 July 2020; Accepted: 24 December 2020;
Published: 19 February 2021.
Edited by:
Kuo-Feng Hua, National Ilan University, TaiwanReviewed by:
Yi-Guang Chen, Medical College of Wisconsin, United StatesCopyright © 2021 Chang, Hung, Chen, Wang, Lin, Tzeng, Suen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yen-Hsu Chen, aW5mY2hlbkBnbWFpbC5jb20=; Jau-Ling Suen, amxzdWVuQGttdS5lZHUudHc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.