 Miren Zuazo1
Miren Zuazo1 Hugo Arasanz1
Hugo Arasanz1 Ana Bocanegra1
Ana Bocanegra1 Gonzalo Fernandez2
Gonzalo Fernandez2 Luisa Chocarro1Ruth Vera2*Grazyna Kochan1*
Luisa Chocarro1Ruth Vera2*Grazyna Kochan1* David Escors1*
David Escors1*- 1Oncoimmunology Group, Navarrabiomed, Fundación Miguel Servet-Complejo Hospitalario de Navarra-UPNA-IdISNA, Pamplona, Spain
- 2Department of Oncology, Complejo Hospitalario de Navarra-IdISNA, Pamplona, Spain
PD-L1/PD-1 blockade immunotherapy has significantly improved treatment outcome for several cancer types compared to conventional cytotoxic therapies. However, the specific molecular and cellular mechanisms behind its efficacy are currently unclear. There is increasing evidence in murine models and in patients that unveil the key importance of systemic immunity to achieve clinical responses under several types of immunotherapy. Indeed, PD-L1/PD-1 blockade induces the expansion of systemic CD8+ PD-1+ T cell subpopulations which might be responsible for direct anti-tumor responses. However, the role of CD4+ T cells in PD-L1/PD-1 blockade-induced anti-tumor responses has been less documented. In this review we focus on the experimental data supporting the “often suspected” indispensable helper function of CD4 T cells towards CD8 effector anti-tumor responses in cancer; and particularly, we highlight the recently published studies uncovering the key contribution of systemic CD4 T cells to clinical efficacy in PD-L1/PD-1 blockade therapies. We conclude and propose that the presence of specific CD4 T cell memory subsets in peripheral blood before the initiation of treatments is a strong predictor of responses in non-small cell lung cancer patients. Therefore, development of new approaches to improve CD4 responses before PD-L1/PD-1 blockade therapy could be the solution to increase response rates and survival of patients.
Introduction
Immunotherapies based on PD-L1/PD-1 blockade have revolutionized the treatment paradigm for several cancer types. The PD-L1/PD-1 immune checkpoint inhibitory interaction regulates the activation of immune responses and specifically of T cell responses in physiological conditions. However, cancer cells utilize this strategy to evade from anti-tumor immune responses, but also contributing to a general state of immunosuppression. Many tumor cells upregulate PD-L1, that subsequently binds to PD-1 on the surface of tumor infiltrating T cells (TILs). This interaction inhibits T cell responses by several mechanisms (1, 2). Currently, most PD-L1/PD-1 inhibitors consist of recombinant antibodies that interfere with this T cell inhibitory signal. Therefore, these inhibitors reinvigorate anti-tumor T cell responses and induce the effective elimination of tumor cells. In 2014, pembrolizumab was approved as the first PD-1 inhibitor for the treatment of metastatic melanoma. Since then, the Food and Drug Administration (FDA) has approved nivolumab and pembrolizumab as PD-1 inhibitors, and atezolizumab, durvalumab and avelumab as PD-L1 inhibitors for the treatment of several metastatic cancer types (Table 1). These treatments have achieved impressive clinical results characterized by durable responses and prolonged survival of patients compared to conventional therapies. Yet, only a small group of patients obtain clinical benefit while a significant number of patients are still refractory. In addition, there is accumulating evidence supporting that PD-L1/PD-1 blockade in a certain group of patients does accelerate tumor progression and death, an adverse event termed hyperprogressive disease (3). Therefore, there is a critical need for the identification of accurate predictive biomarkers to discriminate patients who will potentially benefit from PD-L1/PD-1 blockade immunotherapies from those that will not respond or can develop hyperprogression. Moreover, the precise molecular mechanisms by which T functions are stimulated by PD-L1/PD-1 inhibitors remain to be fully understood. Such understanding is highly relevant not only for the discovery of predictive biomarkers, but also to designing complementary approaches that may increase clinical efficacy of PD-L1/PD-1 blockade therapy. Despite the general assumption that PD-L1/PD-1 inhibitors reinvigorate pre-existing TIL immunity, recent experimental research supports systemic T cell immunity as a key contributor to PD-L1/PD-1 blockade efficacy (4). These treatments induce broad changes in CD8 immunity which correlate with clinical responses, which could be required for efficacy. However, the longstanding fact that proficient CD8 anti-tumor responses largely depend on CD4 help is often underestimated, or at least overlooked. Here, we discuss the evidence supporting the implication of systemic CD4 immunity in achieving clinical responses under PD-L1/PD-1 blockade therapies, with the proposal that indeed the pre-treatment status of CD4 immunity strongly influences therapy outcomes. We also discuss the promising emerging biomarkers based on quantification of peripheral T cell populations, highlighting CD4 T cell memory subpopulations possibly as a key reliable biomarker for patient selection. Research should go in depth into the molecular mechanisms regulating the interplay between CD4 and CD8 T cell responses under PD-L1/PD-1 blockade immunotherapies. These results will provide new insights on potential molecular targets and alternative strategies to boost CD4 immunity in patients to increase PD-L1/PD-1 blockade efficacy.
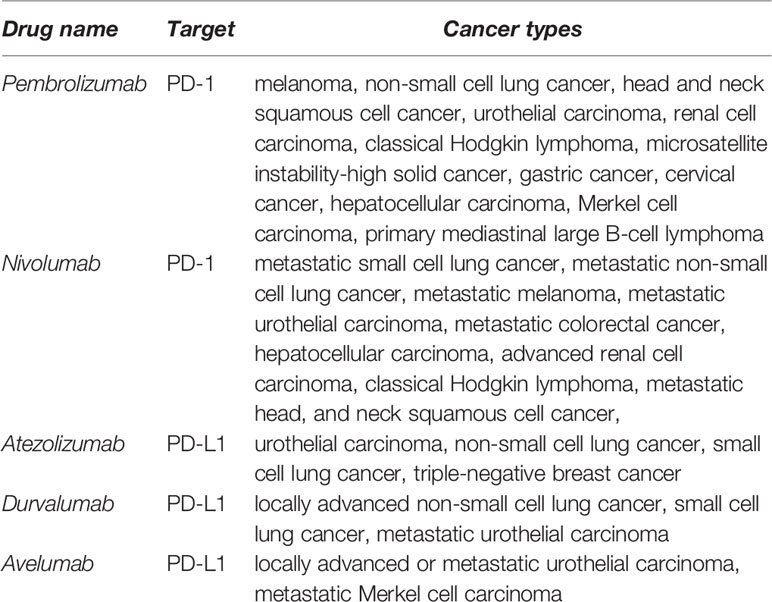
Table 1 FDA-approved PD-L1/PD-1 inhibitors for cancer treatment.
CD4 T Cells as Central Players in Anti-Tumor Immunity
The cancer immunity cycle summarizes how the development of T cell-derived specific anti-tumor responses are essential for controlling tumor growth. Dying cancer cells release tumor-specific antigens (TAA) which are captured by dendritic cells (DC) to prime tumor-specific effector T cells in secondary lymphoid organs. So far, CD8 effector responses have been classically considered as the major players in anti-tumor immunity due to their potent cytotoxicity which enables direct tumor-cell killing (5). Upon antigen recognition, CD8 T cells expand and differentiate into cytotoxic T lymphocytes (CTL) which migrate through peripheral blood and infiltrate tumors. Cancer cells can escape by several mechanisms through a process of immunological editing (6), which includes down-regulation of MHC-I to prevent CD8 T cell recognition (7). In contrast, the importance of CD4 immunity for anti-tumor responses is less recognized as a result of limited studies. Nevertheless, the existing experimental evidence supporting the important role of CD4 T cells is compelling, by promoting and providing help to innate and adaptive anti-tumor immune responses (8). This is reflected by a stronger selective pressure of mutations in MHC-II-restricted neoantigens compared to MHC-I-restricted neoantigens during tumorigenesis, which reinforces the key contribution of CD4 T cells in cancer immunosurveillance (9). The contribution of the different types of effector CD4 T cell subsets is very diverse. Depending on the cytokine milieu during TCR activation, naïve T cells can differentiate into different effector subsets, characterized by different expression of key transcription factors and cytokine profiles that distinctly influence anti-tumor immunity (10).
The CD4 helper (Th) 1 subset is the most prominent for anti-tumor immunity (Figure 1). Th1 cells promote the priming and differentiation of naïve CD8 T cells into CTLs during antigen presentation by producing cytokines such as IFN-γ and IL-2 (11–13). They also contribute to the maturation and activation of DCs in a process called “DC licensing” through the engagement of CD40L with CD40 on DCs (14–17). Such interaction promotes IL-12 and IL-15 production by DC and up-regulates co-stimulatory ligands CD80 and CD86, providing the required signals for efficient CD8 CTL priming (18, 19). DC activation also favors naïve CD8 T cell recruitment to lymph nodes for priming by yet largely unknown mechanisms (20). Importantly, a recent study has identified the specific gene expression programs by which CD4 Th1 cells provide help for the acquisition of CD8 CTL effector functions, with the CD70-CD27 co-stimulatory pathway playing a prominent role. CD70 up-regulation via CD40-CD40L signaling results in co-stimulation of CD8 T cells through binding with CD27, which contributes to CD8 CTL differentiation and clonal expansion (21, 22). In addition, Th1-mediated signaling promotes the establishment of long-lasting CD8 memory (23, 24). Indeed, memory CD8 CTLs primed in absence of CD4 help fail to expand after a second antigen reencounter, and present dysfunctional phenotypes with expression of multiple inhibitory receptors (21, 25, 26). Furthermore, CD4 Th1 cells also activate innate anti-tumor responses by NK and type-1 anti-inflammatory macrophages, promoting tumor cell killing and providing a source of TAAs for T cell priming (27, 28).
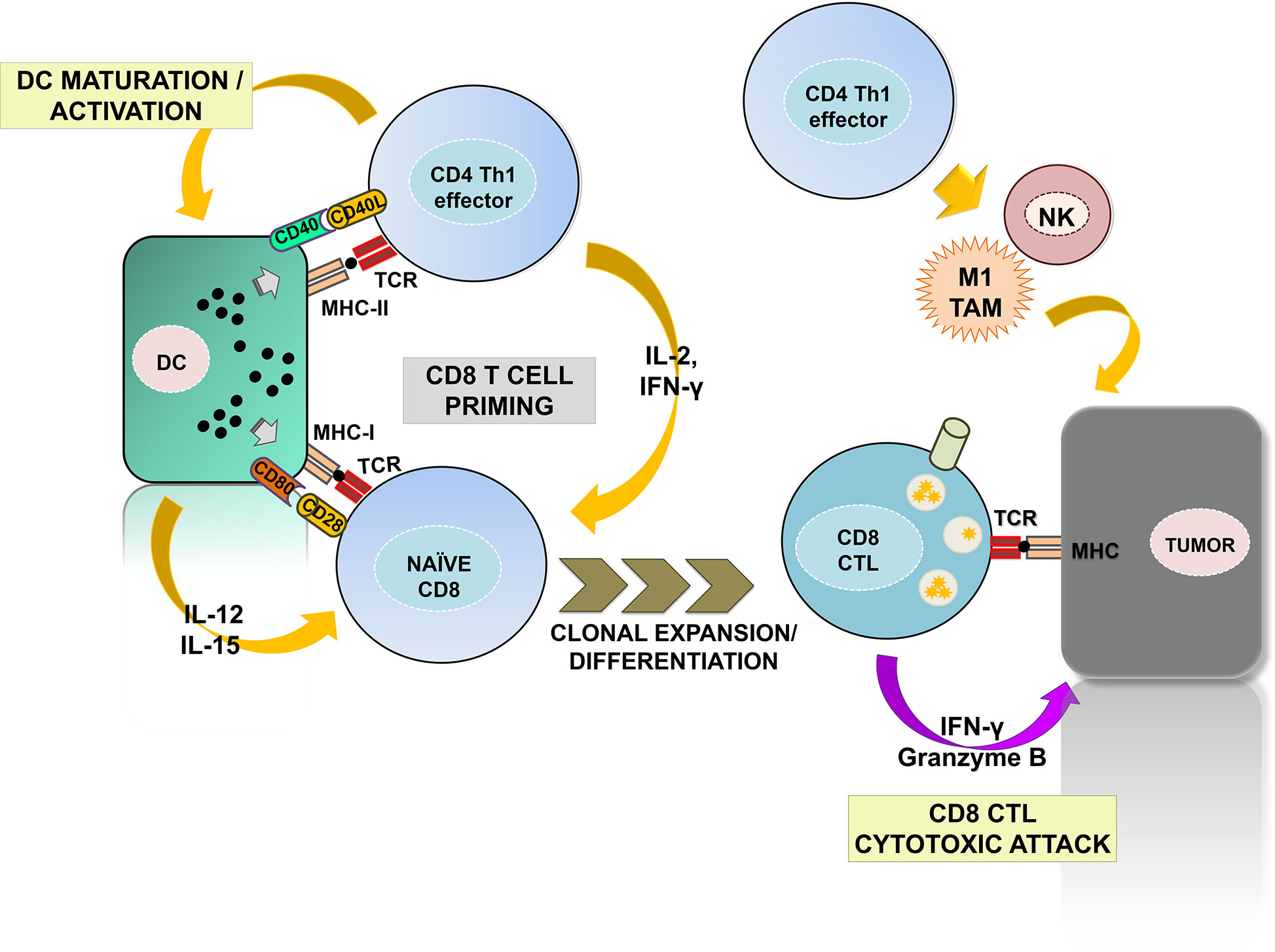
Figure 1 The contribution of CD4 Th1 subsets to anti-tumor immunity. The figure summarizes the well-established roles of CD4 Th1 subsets in anti-tumor responses. Right, CD4 Th1 cells allow the correct priming and differentiation of naïve CD8 T into CTLs by secretion of cytokines and co-stimulatory interactions with DCs within the secondary lymphoid organs. This process termed “DC licensing” leads to DC maturation by CD40L-CD40 binding. CD40-CD40L signaling on DCs induces production of IL-12 and IL-15 and up-regulates co-stimulatory ligands CD80, CD86, and CD70, providing the required signals for CD8 CTL priming. CD80, CD86, and CD70 co-stimulatory ligands on activated DC bind to their receptors CD28 and CD27 on naïve CD8 T cells leading to CTL differentiation and survival. CD8 CTLs infiltrate tumors and exert cytotoxic responses against tumor cells after TAA recognition. Within the tumors, Th1 cells activate NK and M1-macrophages enhancing their innate anti-tumor responses. Th1, T helper 1; CTL, cytotoxic T lymphocyte; DC, dendritic cell; NK, natural killer; M1 TAM, type-1 tumor associated macrophages.
Other CD4 T helper subpopulations including Th2 and Th17 have been generally associated with tumor progression. However, several recent studies also show the contrary. CD4 Th2 effector cells could be required for establishing long-term anti-tumor memory responses (29). Likewise, Th17 responses have been reported to induce potent anti-tumor responses in an IFN-γ-dependent manner, and to allow the recruitment of effector cells into the tumor microenvironment (30–34). This “duality of responses” is likely to be context-dependent. Regulatory T cells (Tregs) are key contributors of tolerance by suppressing the other immune cell populations by several means (35–38), such as cell-to-cell contact and production of anti-inflammatory cytokines including IL-10 and TGF-β (39–41). Finally, CD4 T cells can also mediate direct cytotoxic responses through IFN-γ and TNF secretion, production of cytolytic granules or expressing ligand of tumor necrosis factor (TNF) superfamily molecules including FasL or TRAIL leading to cancer cell apoptosis when engaged with their receptors (42–44).
Differentiation of Memory CD4 T Cells
Upon TAA recognition, CD4 T cells proliferate and differentiate into helper effector T cells. These T cells are short-lived, but a small proportion differentiate into long-lived memory subsets following antigen clearance. Memory T cells undergo fast activation and strong effector responses upon antigen re-encounter (45–47). In humans, the discrimination between the functionally different subsets is based on different expression profiles of cell surface receptors including CD62L and CD45RA. Naïve T cells co-express both CD62L and CD45RA. These T cells exit the thymus and migrate to secondary lymphoid organs driven by CD62L (48). Memory T cells have been divided in two subpopulations based on their location and pattern of migration, either in secondary lymphoid organs (central memory) or in inflamed tissues (effector memory). Central memory T cells express CD62L but not CD45RA, which enable them to circulate between secondary lymphoid organs. In contrast, effector memory T cells are tissue-resident and do not need CD62L nor CD45RA. Effector memory T cells express high levels of chemokine and cytokine receptors to reach inflamed tissues. Finally, the effector population which re-expresses CD45RA (EMRA) is considered a terminally differentiated phenotype which accumulates during lifetime (49). Distinct expression patterns of transcription factors regulate the acquisition of memory phenotypes.
Human CD4 T cells can also be classified in distinct differentiation stages based on CD27/CD28 expression profiles. Following the initial antigen recognition, T cell differentiation advances through the progressive loss of CD27 and CD28 co-stimulatory receptors (50, 51). Hence, human CD4 T cells can be classified into poorly differentiated (CD27+ CD28+), intermediately-differentiated (CD27negative CD28+) and highly-differentiated (CD27negative CD28low/negative, THD) subsets (51, 52). In humans, THD cells are largely composed of memory, effector, and senescent T cells.
CD4 Immunity Has an Important Contribution to PD-L1/PD-1 Blockade Efficacy
The specific molecular mechanisms behind the efficacy of PD-L1/PD-1 blockade therapy have not been fully elucidated yet. These treatments interfere with PD-L1-PD-1 inhibitory interactions by the administration of monoclonal antibodies against both molecules. PD-L1 is overexpressed by several tumor types and confers resistance to pro-apoptotic signals (53, 54); while PD-1 is expressed on T cells upon antigen recognition and interferes with T cell activation when engaged with PD-L1. TILs express high levels of PD-1 and are often functionally imparted due to several dysfunctional states (55). When efficacious, PD-L1/PD-1 blockade therapies seem to counteract tumor-induced T cell dysfunctionality by interfering with PD-1 and PD-L1 signals, and by unleashing activating pathways (1, 56). As a consequence, inhibited tumor reactive T cells reinvigorate and mount an effective anti-tumor response. Indeed, tumor resident CD8+ CD103+ CD69+ T cells which express high levels of PD-1 have been shown to proliferate after PD-L1/PD-1 blockade therapy in melanoma patients (57).
In the last years, increasing evidences have challenged the conventional view of PD-L1/PD-1 inhibitors reinvigorating pre-existing intra-tumoral immunity, supporting that systemic immunity might have a relevant role in therapy efficacy. Various groups have identified that both systemic and intra-tumor CD8 PD-1+ T cell subpopulations experience a proliferative burst after PD-L1/PD-1 blockade treatment in several cancer types (57–61). This proliferative burst may be driven by inhibition of the up-regulation of CBL-b ubiquitin ligase by interference with PD-1 signaling in T cells (62, 63). Extensive phenotypic characterization of intra-tumor PD-1+ CD8 T cells shows that this subpopulation is highly heterogeneous, with distinct exhaustion degrees and susceptibility to be reinvigorated by PD-L1/PD-1 blockade (61, 63–66). Interestingly, K.E. Yost and colleagues recently demonstrated that the ability of PD-1 blockade to rescue pre-existing TILs from exhaustion might be limited, demonstrating that expanded TIL clones post-therapy arose from novel clonotypes recruited from the periphery (4). Indeed, two different studies with NSCLC patients have shown that PD-1+ CD8 T cells expand systemically following PD-1 blockade therapy and correlated with clinical responses (58, 59). These cells exhibited an effector-like phenotype and expressed co-stimulatory molecules including CD28, CD27, and ICOS. Although it has not been directly demonstrated that this expanded subpopulation was tumor-specific, both studies showed that PD-1 blockade did not alter the proliferation of virus-specific PD-1+ CD8 systemic T cells. The same result was observed in melanoma patients treated with pembrolizumab, with a systemic PD-1+ CD8 subpopulation that expanded after treatment with similar clonotypes to those found in the tumor (60). Additionally, a recent elegant study with melanoma patients confirmed that rearrangements of the peripheral memory cytotoxic CD8 T cell repertoire correlated with response to immune checkpoint blockade (67). As CD8 T cells are possibly the main direct effectors of anti-tumor responses through cytotoxicity over cancer cells, these systemic changes are thought to be the drivers of efficacious clinical responses.
Given the importance of CD4 help function in anti-tumor CD8 responses, it is likely that CD4 responses might be required systemically to achieve efficacious CD8 responses under PD-L1/PD-1 blockade therapy. Pre-clinical studies using murine models and in patients have demonstrated the importance of CD4 immunity for immunotherapy efficacy (68–70). The systemic expansion of a CD4+ CD62Llow CD27- FOXP3- CD44+ CXCR3+ ICOS+ T-bet+ T cell subset in mice treated with anti-cancer cell immunoglobulins was correlated with tumor rejection (68). This observation was confirmed in melanoma patients treated with immune checkpoint blockade where a subset of CD4+ PD-1- CD127low T cells was increased in responders compared to non-responders (68). Alspach and colleagues confirmed previously published data on the contribution of CD4 T cells recognizing tumor neoepitopes for the efficacy of immunotherapy (71–73). Moreover, they demonstrated using murine models that both MHC-class I and II-restricted neoantigens are required to generate efficient anti-tumor responses (74). Emerging studies are evaluating the specific contribution of CD4 immunity to PD-L1/PD-1 blockade therapy efficacy which is still unknown. Two recently published studies have independently demonstrated that pre-treatment status of systemic CD4 immunity is a critical factor for determining the clinical outcome of PD-L1/PD-1 blockade therapy in NSCLC patients. Particularly, our study showed that only patients with pre-treatment high numbers of CD4 central and effector memory T cells with a highly differentiated phenotype (CD27-CD28low/-) responded to the treatment (75). Responder patients presented high proportion of CD4 memory T cells before treatment initiation. These CD4 T cells exhibited significant proliferative capacities and low co-expression of PD-1/LAG-3 at baseline, and were responsive to PD-1 blockade ex vivo and in vivo (75). In contrast, patients with low numbers of memory CD4 T cells exhibited a strong CD4 T cell dysfunctionality. Such dysfunctionality was reflected as strongly impaired proliferation capacities and high co‐expression of LAG‐3/PD‐1 associated to resistance to PD-L1/PD‐1 blockade ex vivo and in vivo. Although all patients showed a baseline dysfunctional CD8 proliferative response, this was only recovered by PD-L1/PD-1 blockade in patients with high numbers of CD4 memory T cells harboring functional proliferative CD4 responses before treatment initiation. These results strongly suggest the implication of PD-1/LAG-3 signaling on systemic T cells dysfunctionality which is under investigation in order to provide new molecular targets to restore CD4 pre-treatment immunity before PD-L1/PD-1 blockade treatment application. Kobayashi and colleagues simultaneously obtained similar conclusions by identifying an equivalent CD4+ CD62Llow T effector memory population which was present at higher numbers in responders before PD-1 blockade treatment initiation (76). Patients who maintained high numbers of CD62Llow CD4 T cells were long survivors while decreased levels after therapy resulted in acquired resistance. Gene expression analysis of the specific subset revealed that they represented TCR-engaged proliferating Th1-like cells. Indeed, these cells expressed genes related to CD4 help functions involved in the promotion of CD8 CTL responses (76). Hence, the maintenance of systemic CD4 responses over time is required for therapy efficacy. Overall, these two studies provide strong evidence that proficient systemic CD4 immunity is a key factor to achieve efficacious clinical responses under PD-L1/PD-1 blockade therapies.
Although CD4 memory help functions seem to directly influence PD-L1/PD-1 inhibitor effects on CD8 anti-tumor responses, the molecular mechanisms by which both populations interplay during PD-L1/PD-1 blockade therapy remain to be fully understood. In addition, it is not clear yet whether CD4 memory T cells could also play a role within the tumor microenvironment under PD-L1/PD-1 blockade therapy. A recent study in Classic Hodgkin lymphoma (CHL) murine models demonstrated that PD-1 blockade therapy has strong anti-tumor effects on MHC-II expressing tumors mediated by cytotoxic CD4+ T cells (77). Moreover, they observed that CHL patients responding to PD-1 blockade therapy exhibited high CD4+ T cell infiltration compared to non-responders (77). Another study using murine models demonstrated that the expansion of tumor infiltrating follicular CD4+ PD1+ T cells after PD-1 blockade therapy correlated with enhanced CD8 CTL anti-tumor responses and tumor growth control (78). They proposed that this specific population might be an important target of PD-1 blockade within the tumor microenvironment. In contrast, a clinical study with NSCLC patients revealed that the accumulation of CD4+ FOXP3- PD-1 high T cells within the tumor and peripheral blood correlated with higher tumor burden. Moreover, the decrease of such population during therapy onset was significantly associated with improved OS (79) suggesting that those cells might be exhausted. Although the data published so far suggests that exhausted T cell populations within the tumor microenvironment might not be the predominant target of PD-L1/PD-1 blockade therapy, further research is needed to reveal the specific contribution of the different intra-tumoral CD4 T cell subsets to PD-L1/PD-1 blockade efficacy in different cancer types.
CD4 Immunity as a Reliable Biomarker of Response to PD-L1/PD-1 Blockade Therapy
Most of the current research has been focused on the tumor cell and the immunological status of the tumor microenvironment to find biomarkers of response to PD-L1/PD-1 blockade. These biomarkers include tumor PD-L1 expression, mutational and neoantigen burden, TAA-specific repertoire, presence, and characterization of TILs and infiltration with immunosuppressive cells. PD-L1 tumor expression is the only predictive biomarker accepted so far, but its reliability is still under debate. Recently, tumor mutational burden (TMB) quantification and TIL gene expression have demonstrated potential predictive value in patients treated with PD-L1/PD-1 inhibitors (80–83). Nevertheless, the identification of biomarkers using tumor sampling is challenging for most cancer types, particularly in advanced stages due to limited accessibility. Moreover, single tumor samples do not often represent the tumor heterogeneity. It is important to remark that PD-L1/PD-1 inhibitors are administered systemically and will have a direct impact on systemic immunity, which in turn could correlate with clinical responses (58, 59). Hence, characterization of peripheral immune cell populations of patients undergoing PD-L1/PD-1 blockade therapy is a promising non-invasive source of biomarkers of response, more homogeneous and less costly than tumor sampling. Nevertheless, no peripheral biomarkers approved by the FDA, EMA, and PMDA have been validated for clinical application. Here, we highlight the most promising findings on peripheral biomarkers in predicting clinical outcome to PD-L1/PD-1 inhibitors (Table 2).
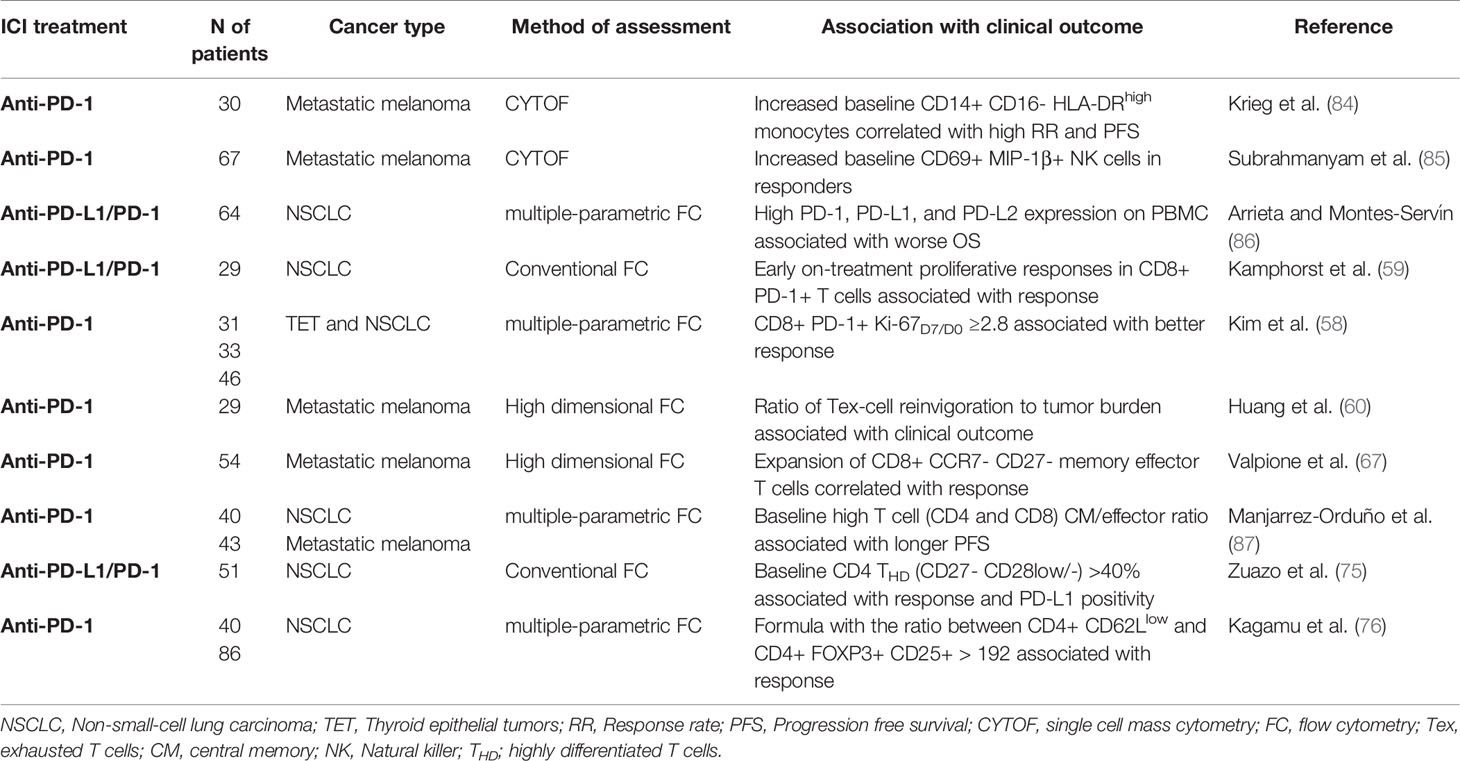
Table 2 Proposed biomarkers of clinical response to PD-L1/PD-1 blockade immunotherapy based on peripheral immune cell populations.
A study evaluating peripheral blood samples of NSCLC patients treated with anti-PD-1 by conventional flow cytometry demonstrated that early expansion of peripheral PD-1+ CD8 T cells was associated with clinical efficacy (59). Another study in patients with thymic epithelial tumors confirmed the previous observation (58). The authors correlated peripheral expansion of PD-1+ CD8 T cells with durable clinical benefit to anti-PD-1 treatment. In this case, T cell expansion was measured as the fold-change in the percentage of Ki67+ cells (Ki-67D7/D0) ≥2.8 after the first week of treatment. The predictive value of Ki-67D7/D0 ≥2.8 was validated in two independent cohorts of NSCLC patients treated with PD-1 inhibitors (58). Early clonal expansion of peripheral T populations evaluated by genome-wide sequencing was also positively associated with clinical responses in NSCLC patients under PD-1 blockade therapy (88). Studies with metastatic melanoma patients treated with PD-1 inhibitors have confirmed this observation. A high ratio of baseline Ki67+PD1+CD8 T subpopulation to tumor burden was associated with longer PFS (60). A recent study from Richard Marais and colleagues using high dimensional flow cytometry also identified the correlation between the expansion of a systemic subset of CCR7- CD27- CD8 cytotoxic memory effector T cells with response to PD-1 blockade in melanoma patients (67). All these studies support the quantification of proliferating peripheral CD8 PD-1+ CD8 T cell subpopulations as a surrogate biomarker to predict responses to PD-L1/PD-1 blockade therapy. These approaches can help decision-making in the early onset of the treatment, but do not identify deleterious adverse events such as hyperprogressive disease. Hence, the power of CD8 T cell subsets to discriminate clinical responses before PD-L1/PD-1 blockade treatment initiation are so far limited.
Detailed characterization of pre-treatment peripheral immune cell population profiles has been performed to identify potential biomarkers for patient selection before treatment initiation. For example, mass cytometry (CYTOF) allows the identification of multiple markers simultaneously. CYTOF-based analyses of peripheral blood from metastatic melanoma patients before anti-PD-1 treatment initiation have identified several potential biomarkers. Increased baseline peripheral CD14+ CD16- HLA-DRhigh monocytes correlated with a high response rate and PFS (84). High numbers of CD69+ MIP-1β+ NK cells are found in responders compared to non-responders (85). Analyses of PBMC in NSCLC patients by multi-parametric flow cytometry have identified high PD-1, PD-L1 and PD-L2 expression associated with worse OS (86). Although associations with clinical benefit and survival have been observed, none so far have been validated as predictive biomarkers in prospective studies. Moreover, the high cost of these technologies makes it difficult to standardize and implement them in clinical practice.
Work from our group and from other independent groups is demonstrating the value of baseline CD4 memory T cell quantification to predict the efficacy of PD-L1/PD-1 blockade therapy. A preliminary study analyzing 4 metastatic melanoma patients showed that patients with longer survival had an increase in central memory CD4 T cells (89). Another small-scale study with NSCLC patients treated with nivolumab also uncovered higher ratios between systemic central memory and effector subsets in the total populations of CD4 and CD8 T cells was associated with benefit (87). In the last year, two prospective studies which include our own have independently monitored the dynamics of systemic CD4 T cell populations in NSCLC patients undergoing PD-L1/PD-1 blockade therapy as second line treatments (75, 76). Apart from demonstrating that CD4 T cells play a crucial role in PD-L1/PD-1 blockade efficacy, both studies proposed similar relative percentages of peripheral CD4 memory T cells before the start of immunotherapies with strong predictive capacities for clinical benefit. We identified that relative percentages of systemic CD27- CD28low/- central and effector memory CD4 T cells discriminate NSCLC patients with differential clinical outcome (75). A cut-off value of >40% of this subset at baseline identified a subgroup of patients containing the objective responders with an ORR 50%; while patients with <40% at baseline showed an ORR of 0% and were significantly associated with a higher risk for hyperprogressive disease (75). Independently, K. Kobayashi and colleagues identified a CD4+ CD62Llow effector memory subset by multiparametric flow cytometry which also discriminates patients with distinct clinical outcome, and with similar threshold values found by Zuazo and collaborators (76). Responders to PD-1 blockade therapy presented a significantly higher proportion of baseline CD62Llow CD4 T cells. In contrast, percentages of Tregs were significantly higher in non-responders. Hence, the authors proposed an algorithm accounting for the ratio between CD62Llow and CD25+Foxp3+CD4+ T cell population percentages to discriminate responders with a cut-off value of ≥192. Long-term responders maintained a high number of CD62Llow CD4 T cell subsets during therapy. In contrast, decreased CD62Llow CD4 T cell percentages were an indicator of acquired resistance. Importantly, the results were validated in an independent NSCLC patient cohort.
Finally, multiple studies have evaluated blood parameters derived from routine clinical analyses for correlations with responses to PD-L1/PD-1 blockade immunotherapy. Some of these include absolute neutrophil counts (ANC), absolute lymphocyte counts (ALC), neutrophil-to-lymphocyte ratio (NLR), absolute eosinophil counts (AEC), absolute monocyte counts (AMC), and serum lactate dehydrogenase (LDH). In a cohort of advanced NSCLC patients treated with nivolumab, a baseline ANC ≥7,500/ul correlated with worse PFS and OS (90). Pre-treatment NLR ratios greater than five were associated with decreased PFS and OS in several studies across different cancer types treated with PD-L1/PD-1 inhibitors (91–93). The derived NLR (dNRL) seems to be more consistent as a biomarker, as this index includes monocytes and other granulocyte populations. High dNRL (> 3) was associated with worse OS in NSCLC patients treated with PD-1 blockers. dNRL greater than 3 together with lactate dehydrogenase (LDH) values greater than the upper limit of normal (ULN) have been integrated into a parameter termed “lung immune prognostic index”. This parameter efficiently identifies 3 groups of patients with good (0 factor), intermediate (1 factor), and poor survival (2 factors) under PD-L1/PD-1 blockade therapy (94). Nevertheless, parameters such as ANC, NLR, dNLR, and AMC do not accurately discriminate the wide ranges of myeloid-derived populations at different activation stages in peripheral blood which may play distinct roles in therapy.
Discussion and Future Perspectives
Most of the research is centered on the immunological status of the tumor microenvironment as a major driving force for the efficacy of PD-L1/PD-1 blockade. However, it is often ignored the significant impact that these treatments exert over systemic immunity and the diverse susceptibility of TIL populations to be reactivated by them. Indeed, several studies have demonstrated that these therapies cause systemic changes in immune cell populations which can be correlated with clinical efficacy (4, 58, 59). Here we wanted to summarize the most relevant studies in which peripheral blood immune cell populations are analyzed in patients undergoing PD-L1/PD-1 blockade therapy. Many of these studies are difficult to validate and implement in clinical routine due to the complexity and high cost of the employed analytical technology. Moreover, biomarkers based on blood cell parameters such as “lung immune prognostic index” have demonstrated to possess rather a prognostic value, while others cannot be cross-tested due to the lack to discrimination between different immune cell populations. This is especially important in studies with myeloid-derived populations (95).
Several of studies reviewed here have followed the dynamics of CD8 T cells and have demonstrated that changes on systemic PD-1+ CD8 subpopulation post-PD-L1/PD-1 blockade therapy might reflect significant anti-tumor activities in patients (59, 60). Moreover, emerging studies suggest that these changes may depend on systemic CD4 immunity (75, 76). Interestingly, they have also revealed that high systemic numbers of CD4 memory T cells assessed by peripheral blood samples before PD-L1/PD-1 blockade treatment initiation can serve as a reliable predictive biomarker in NSCLC patients. Although more extensive independent validation cohorts for distinct cancer types are required to assure its reliability as a predictive biomarker, the statistically powerful ROC curves in both studies supports its clinical application. Moreover, although the proposed CD4 memory populations are very likely equivalent, each study applied different phenotypic markers for their identification. Unification of data provided by both studies will help to better select the specific CD4 memory subpopulation with the strongest predictive value. In addition, dynamic changes of CD4 T-cell memory populations could be successfully used for “real-time” monitoring of responses from blood samples during immunotherapy, to identify early progressors and patients with a high risk of developing hyperprogression (75, 96). These studies together with emerging evidences supporting the limited susceptibility of TILs to be reinvigorated by PD-L1/PD-1 blockade, are indicating that PD-L1/PD-1 inhibitors might be predominantly targeting systemic immunity, which is then recruited into the tumor. The enhancement of systemic CD8 anti-tumor responses by PD-L1/PD-1 inhibitors could possibly be orchestrated by CD4 T cells in the periphery. Hence, these evidences provide the rationale to design new approaches to boost CD4 functionality in cancer patients before their enrolment in PD-L1/PD-1 blockade immunotherapy. Indeed, we showed that increased co-expression of immune-checkpoint molecules PD-1/LAG-3 in systemic T cells confers resistance to PD-1/PD-L1 inhibitors (75). PD-1/LAG-3 co-signaling induce dysfunctional proliferative capacities on systemic CD4 T cells, hampering the help functions over peripheral CD8 responses. The combination of PD-1 and LAG-3 blockade has been shown to reverse CD4 dysfunctionality ex vivo (75). In addition, given the contribution of CD40 and CD70 signaling pathways to CD4 helper functions, these molecules represent good molecular targets to reinvigorate CD4 helper functions. Based on data from murine models, the combination of PD-1 inhibitors with CD27 agonistic antibodies seems to be a promising approach to enhance the correct T cell priming and expansion of PD1+ CD8 circulating T cells (97). Cytokine-based therapy with IL-12 has also demonstrated to reinvigorate CD8 expansion during PD-1 blockade ex vivo (unpublished observation). Hence, deciphering the molecular mechanisms regulating CD4 helper functions under PD-L1/PD-1 blockade therapy will provide insights into new potential targets, and better approaches to increase PD-L1/PD-1 blockade therapy efficacy.
Author Contributions
The first version was drafted by MZ and HA. All authors contributed to the article and approved the submitted version.
Funding
The authors are supported and funded by the Spanish Association against Cancer (AECC, PROYE16001ESCO), Instituto de Salud Carlos III (ISCIII-FEDER, FIS. PI17/02119), and a Biomedicine Project grant from the Department of Health of the Government of Navarre (BMED 050-2019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wished to acknowledge the cancer patients and their families that have selflessly contributed to their studies.
References
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
2. Zhang Y, Huang S, Gong D, Qin Y, Shen Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell Mol Immunol (2010) 7(5):389–95. doi: 10.1038/cmi.2010.28
3. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria J-C, et al. Hyperprogressive disease: recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol (2018) 15(12):748–62. doi: 10.1038/s41571-018-0111-2
4. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
5. Zhang N, Bevan MJ. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity (2011) 35(2):161–8. doi: 10.1016/j.immuni.2011.07.010
6. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
7. Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int J Cancer (2010) 127(2):249–56. doi: 10.1002/ijc.25270
8. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol (2018) 18(10):635–47. doi: 10.1038/s41577-018-0044-0
9. Marty R, Thompson WK, Salem RM, Zanetti M, Carter H. Evolutionary Pressure against MHC Class II Binding Cancer Mutations. Cell (2018) 175(7):1991. doi: 10.1016/j.cell.2018.08.048
10. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4 +T cells: Differentiation and functions. Clin Dev Immunol (2012) 2012:925135. doi: 10.1155/2012/925135
11. Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res (2010) 70(21):8368–77. doi: 10.1158/0008-5472.CAN-10-1322
12. Wong SBJ, Bos R, Sherman LA. Tumor-Specific CD4 + T Cells Render the Tumor Environment Permissive for Infiltration by Low-Avidity CD8 + T Cells. J Immunol (2008) 180(5):3122–31. doi: 10.4049/jimmunol.180.5.3122
13. Bennett SRM, Carbone FR, Karamalis F, Miller JFAP, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med (1997) 186(1):65–70. doi: 10.1084/jem.186.1.65
14. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4 + T-helper and a T-killer cell. Nature (1998) 393(6684):474–8. doi: 10.1038/30989
15. Schoenberger SP, Toes REM, Van Dervoort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD4OL interactions. Nature (1998) 393(6684):480–3. doi: 10.1038/31002
16. Nesbeth YC, Martinez DG, Toraya S, Scarlett UK, Cubillos-Ruiz JR, Rutkowski MR, et al. CD4 + T Cells Elicit Host Immune Responses to MHC Class II – Ovarian Cancer through CCL5 Secretion and CD40-Mediated Licensing of Dendritic Cells. J Immunol (2010) 184(10):5654–62. doi: 10.4049/jimmunol.0903247
17. Baxevanis CN, Voutsas IF, Tsitsilonis OE, Gritzapis AD, Sotiriadou R, Papamichail M. Tumor-Specific CD4+ T Lymphocytes from Cancer Patients Are Required for Optimal Induction of Cytotoxic T Cells Against the Autologous Tumor. J Immunol (2014) 164(7):3902–12. doi: 10.4049/jimmunol.164.7.3902
18. Greyer M, Whitney PG, Stock AT, Davey GM, Tebartz C, Bachem A, et al. T Cell Help Amplifies Innate Signals in CD8+ DCs for Optimal CD8+ T Cell Priming. Cell Rep (2016) 14(3):586–97. doi: 10.1016/j.celrep.2015.12.058
19. Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev (2006) 211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x
20. Kumamoto Y, Mattei LM, Sellers S, Payne GW, Iwasaki A. CD4+ T cells support cytotoxic T lymphocyte priming by controlling lymph node input. Proc Natl Acad Sci USA (2011) 108(21):8749–54. doi: 10.1073/pnas.1100567108
21. Ahrends T, Busselaar J, Severson TM, Bąbała N, de Vries E, Bovens A, et al. CD4+ T cell help creates memory CD8+ T cells with innate and help-independent recall capacities. Nat Commun (2019) 10(1):5531. doi: 10.1038/s41467-019-13438-1
22. Van De Ven K, Borst J. Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: Rationale and potential. Immunotherapy (2015) 7(6):655–67. doi: 10.2217/imt.15.32
23. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat Rev Immunol (2016) 16(2):102–11. doi: 10.1038/nri.2015.10
24. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Sci (80- ) (2003) 300(5617):337–9. doi: 10.1126/science.1082305
25. Janssen EM, Lemmens EE, Wolfe T, Christen U, Von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature (2003) 421(6925):852–6. doi: 10.1038/nature01441
26. Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Sci (80- ) (2003) 300(5617):339–42. doi: 10.1126/science.1083317
27. Eisel D, Das K, Dickes E, König R, Osen W, Eichmüller SB. Cognate Interaction With CD4 + T Cells Instructs Tumor-Associated Macrophages to Acquire M1-Like Phenotype. Front Immunol (2019) 10:219. doi: 10.3389/fimmu.2019.00219
28. Shklovskaya E, Terry AM, Guy TV, Buckley A, Bolton HA, Zhu E, et al. Tumour-specific CD4 T cells eradicate melanoma via indirect recognition of tumour-derived antigen. Immunol Cell Biol (2016) 94(6):593–603. doi: 10.1038/icb.2016.14
29. Lorvik KB, Hammarström C, Fauskanger M, Haabeth OAW, Zangani M, Haraldsen G, et al. Adoptive transfer of tumor-specific Th2 cells eradicates tumors by triggering an in situ inflammatory immune response. Cancer Res (2016) 76(23):6864–76. doi: 10.1158/0008-5472.CAN-16-1219
30. Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J, et al. Cutting Edge: Th17 and Regulatory T Cell Dynamics and the Regulation by IL-2 in the Tumor Microenvironment. J Immunol (2014) 178(11):6730–3. doi: 10.4049/jimmunol.178.11.6730
31. Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood (2009) 114(6):1141–9. doi: 10.1182/blood-2009-03-208249
32. Wilke CM, Bishop K, Fox D, Zou W. Deciphering the role of Th17 cells in human disease. Trends Immunol (2011) 32(12):603–11. doi: 10.1016/j.it.2011.08.003
33. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity (2009) 31(5):787–98. doi: 10.1016/j.immuni.2009.09.014
34. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood (2008) 112(2):362–73. doi: 10.1182/blood-2007-11-120998
35. Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: Possible involvement of regulatory T cells in disease progression. Cancer (2003) 98(5):1089–99. doi: 10.1002/cncr.11618
36. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi: 10.1038/nm1093
37. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA (2005) 102(51):18538–43. doi: 10.1073/pnas.0509182102
38. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol (2006) 24(34):5373–80. doi: 10.1200/JCO.2006.05.9584
39. Larmonier N, Marron M, Zeng Y, Cantrell J, Romanoski A, Sepassi M, et al. Tumor-derived CD4+CD25+ regulatory T cell suppression of dendritic cell function involves TGF-β and IL-10. Cancer Immunol Immunother (2007) 56(1):48–59. doi: 10.1007/s00262-006-0160-8
40. Jarnicki AG, Lysaght J, Todryk S, Mills KHG. Suppression of Antitumor Immunity by IL-10 and TGF-β-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of CD4 + and CD8 + Regulatory T Cells. J Immunol (2006) 177(2):896–904. doi: 10.4049/jimmunol.177.2.896
41. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor Evasion of the Immune System by Converting CD4 + CD25 – T Cells into CD4 + CD25 + T Regulatory Cells: Role of Tumor-Derived TGF-β. J Immunol (2007) 178(5):2883–92. doi: 10.4049/jimmunol.178.5.2883
42. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The Central Role of CD4 + T Cells in the Antitumor Immune Response. J Exp Med (2002) 188(12):2357–68. doi: 10.1084/jem.188.12.2357
43. Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol (1998) 10(5):588–94. doi: 10.1016/S0952-7915(98)80228-8
44. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4 + T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med (2010) 207(3):637–50. doi: 10.1084/jem.20091918
45. McKinstry KK, Strutt TM, Swain SL. Regulation of CD4+ T-cell contraction during pathogen challenge. Immunol Rev (2010) 236:110–24. doi: 10.1111/j.1600-065X.2010.00921.x
46. Taylor JJ, Jenkins MK. CD4+ memory T cell survival. Curr Opin Immunol (2011) 23(3):319–23. doi: 10.1016/j.coi.2011.03.010
47. Strutt TM, McKinstry KK, Kuang Y, Bradley LM, Swain SL. Memory CD4+ T-cell-mediated protection depends on secondary effectors that are distinct from and superior to primary effectors. Proc Natl Acad Sci (2012) 109(38):E2551–60. doi: 10.1073/pnas.1205894109
48. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Pillars Article : Two Subsets of Memory T Lymphocytes with Distinct Homing Potentials and Effector Functions . Nature. Nature (1999) 192(3):840–4. doi: 10.1038/35005534
49. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur J Immunol (2013) 43(11):2797–809. doi: 10.1002/eji.201343751
50. Lanna A, Gomes DCO, Muller-Durovic B, McDonnell T, Escors D, Gilroy DW, et al. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat Immunol (2017) 18(3):354–63. doi: 10.1038/ni.3665
51. Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol (2014) 15(10):965–72. doi: 10.1038/ni.2981
52. Amyes E, Hatton C, Montamat-Sicotte D, Gudgeon N, Rickinson AB, McMichael AJ, et al. Characterization of the CD4 + T Cell Response to Epstein-Barr Virus during Primary and Persistent Infection. J Exp Med (2003) 198(6):903–11. doi: 10.1084/jem.20022058
53. Gato-Cañas M, Zuazo M, Arasanz H, Ibañez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep (2017) 20(8):1818–29. doi: 10.1016/j.celrep.2017.07.075
54. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Nicholas Haining W, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med (2017) 214(4):895–904. doi: 10.1084/jem.20160801
55. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25(2):214–21. doi: 10.1016/j.coi.2012.12.003
56. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
57. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy-naïve melanoma patients and expand significantly during anti-PD-1 treatment. Clin Cancer Res (2018) 24(13):3036–3045. doi: 10.1158/1078-0432.CCR-17-2257
58. Kim KH, Cho J, Ku BM, Koh J, Sun JM, Lee SH, et al. The first-week proliferative response of peripheral blood PD-1 þ CD8 þ T cells predicts the response to Anti-PD-1 therapy in solid tumors. Clin Cancer Res (2019) 25(7):2144–2154. doi: 10.1158/1078-0432.CCR-18-1449
59. Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1–targeted therapy in lung cancer patients. Proc Natl Acad Sci (2017) 114:4993–8. doi: 10.1073/pnas.1705327114
60. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature (2017) 545(7652):60–5. doi: 10.1038/nature22079
61. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1 + PD-1 + CD8 + T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity (2019) 50(1):195–211.e10. doi: 10.1016/j.immuni.2018.12.021
62. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8 + T cells. EMBO Mol Med (2011) 3(10):581–92. doi: 10.1002/emmm.201100165
63. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8 + T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
64. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8 + T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330
65. Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin JX, et al. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc Natl Acad Sci USA (2019) 116(28):14113–8. doi: 10.1073/pnas.1903520116
66. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Sci (80- ) (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
67. Valpione S, Galvani E, Tweedy J, Mundra PA, Banyard A, Middlehurst P, et al. Immune awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer (2020) 1(2):210–21. doi: 10.1038/s43018-019-0022-x
68. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell (2017) 168(3):487–502.e15. doi: 10.1016/j.cell.2016.12.022
69. Markowitz GJ, Havel LS, Crowley MJP, Ban Y, Lee SB, Thalappillil JS, et al. Immune reprogramming via PD-1 inhibition enhances early-stage lung cancer survival. JCI Insight (2018) 3(13):e96836. doi: 10.1172/jci.insight.96836
70. Moreno BH, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol Res (2016) 4(10):845–57. doi: 10.1158/2326-6066.CIR-16-0060
71. Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature (2015) 520:692–6. doi: 10.1038/nature14426
72. Knocke S, Fleischmann-Mundt B, Saborowski M, Manns MP, Kühnel F, Wirth TC, et al. Tailored Tumor Immunogenicity Reveals Regulation of CD4 and CD8 T Cell Responses against Cancer. Cell Rep (2016) 17(9):2234–46. doi: 10.1016/j.celrep.2016.10.086
73. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547:222–6. doi: 10.1038/nature23003
74. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature (2019) 574(7780):696–701. doi: 10.1038/s41586-019-1671-8
75. Zuazo M, Arasanz H, Fernández-Hinojal G, García-Granda MJ, Gato M, Bocanegra A, et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol Med (2019) 11(7):e10293. doi: 10.15252/emmm.201910293
76. Kagamu H, Kitano S, Yamaguchi O, Yoshimura K, Horimoto K, Kitazawa M, et al. CD4 + T-cell Immunity in the Peripheral Blood Correlates with Response to Anti-PD-1 Therapy. Cancer Immunol Res (2019) 8(3):334–44. doi: 10.1158/2326-6066.cir-19-0574
77. Nagasaki J, Togashi Y, Sugawara T, Itami M, Yamauchi N, Yuda J, et al. The critical role of CD4+ T cells in PD-1 blockade against MHC-II–expressing tumors such as classic Hodgkin lymphoma. Blood Adv (2020) 4(17):4069–82. doi: 10.1182/bloodadvances.2020002098
79. Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, et al. Non-conventional Inhibitory CD4+Foxp3–PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell (2018) 34(4):691. doi: 10.1016/j.ccell.2018.05.009
80. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Sci (80- ) (2018) 362(6411):eaar3593. doi: 10.1126/science.aar3593
81. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Sci (80- ) (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
82. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med (2018) 24(7):994–1004. doi: 10.1038/s41591-018-0057-z
83. Prat A, Navarro A, Paré L, Reguart N, Galván P, Pascual T, et al. Immune-related gene expression profiling after PD-1 blockade in non–small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res (2017) 77(13):3540–50. doi: 10.1158/0008-5472.CAN-16-3556
84. Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med (2018) 24:144–53. doi: 10.1038/nm.4466
85. Subrahmanyam PB, Dong Z, Gusenleitner D, Giobbie-Hurder A, Severgnini M, Zhou J, et al. Distinct predictive biomarker candidates for response to anti-CTLA-4 and anti-PD-1 immunotherapy in melanoma patients. J Immunother Cancer (2018) 6:18. doi: 10.1186/s40425-018-0328-8
86. Arrieta O, Montes-Servín E. MA14.02 Evaluation of PD1/PDL1 Expression on Peripheral Blood Cells Subpopulations in Patients with Non-Small Cell Lung Cancer. J Thorac Oncol (2017) 12:S422–3. doi: 10.1016/j.jtho.2016.11.488
87. Manjarrez-Orduño N, Menard LC, Kansal S, Fischer P, Kakrecha B, Jiang C, et al. Circulating T cell subpopulations correlate with immune responses at the tumor site and clinical response to PD1 inhibition in non-small cell lung cancer. Front Immunol (2018) 12:S422–S423. doi: 10.3389/fimmu.2018.01613
88. Olugbile S, Kiyotani K, Inoue H, Park J-H, Hoffman P, Szeto L, et al. P3.02c-058 In-Depth Molecular Characterization of T Cell Clonal Expansion Induced by Anti-PD1 Therapy in NSCLC. J Thorac Oncol (2017) 30(1):13–22. doi: 10.1016/j.jtho.2016.11.1853
89. Takeuchi Y, Tanemura A, Tada Y, Katayama I, Kumanogoh A, Nishikawa H. Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int Immunol (2018) 9:1613. doi: 10.1093/intimm/dxx073
90. Tanizaki J, Haratani K, Hayashi H, Chiba Y, Nakamura Y, Yonesaka K, et al. Peripheral Blood Biomarkers Associated with Clinical Outcome in Non–Small Cell Lung Cancer Patients Treated with Nivolumab. J Thorac Oncol (2018) 13:97–105. doi: 10.1016/j.jtho.2017.10.030
91. Bagley SJ, Kothari S, Aggarwal C, Bauml JM, Alley EW, Evans TL, et al. Pretreatment neutrophil-to-lymphocyte ratio as a marker of outcomes in nivolumab-treated patients with advanced non-small-cell lung cancer. Lung Cancer (2017) 106:1–7. doi: 10.1016/j.lungcan.2017.01.013
92. Bilen MA, Dutcher GMA, Liu Y, Ravindranathan D, Kissick HT, Carthon BC, et al. Association Between Pretreatment Neutrophil-to-Lymphocyte Ratio and Outcome of Patients With Metastatic Renal-Cell Carcinoma Treated With Nivolumab. Clin Genitourin Cancer (2018) 16(3):e563–75. doi: 10.1016/j.clgc.2017.12.015
93. Jiang T, Qiao M, Zhao C, Li X, Gao G, Su C, et al. Pretreatment neutrophil-to-lymphocyte ratio is associated with outcome of advanced-stage cancer patients treated with immunotherapy: a meta-analysis. Cancer Immunol Immunother (2018) 67(5):713–27. doi: 10.1007/s00262-018-2126-z
94. Mezquita L, Auclin E, Ferrara R, Charrier M, Remon J, Planchard D, et al. Association of the lung immune prognostic index with immune checkpoint inhibitor outcomes in patients with advanced non-small cell lung cancer. JAMA Oncol (2018) 4:351–7. doi: 10.1001/jamaoncol.2017.4771
95. Bocanegra A, Fernandez-Hinojal G, Zuazo-Ibarra M, Arasanz H, Garcia-Granda MJ, Hernandez C, et al. PD-L1 expression in systemic immune cell populations as a potential predictive biomarker of responses to PD-L1/PD-1 blockade therapy in lung cancer. Int J Mol Sci (2019) 20:1–13. doi: 10.3390/ijms20071631
96. Arasanz H, Zuazo M, Bocanegra A, Gato M, Martínez-Aguillo M, Morilla I, et al. Early detection of hyperprogressive disease in non-small cell lung cancer by monitoring of systemic T cell dynamics. Cancers (Basel) (2020) 12(2):344. doi: 10.3390/cancers12020344
Keywords: CD4, immune checkpoint inhibitor, PD-L1, PD-1, biomarker
Citation: Zuazo M, Arasanz H, Bocanegra A, Fernandez G, Chocarro L, Vera R, Kochan G and Escors D (2020) Systemic CD4 Immunity as a Key Contributor to PD-L1/PD-1 Blockade Immunotherapy Efficacy . Front. Immunol. 11:586907. doi: 10.3389/fimmu.2020.586907
Received: 24 July 2020; Accepted: 30 October 2020;
Published: 30 November 2020.
Edited by:
José Manuel González-Navajas, University General Hospital of Alicante, SpainReviewed by:
Umaimainthan Palendira, The University of Sydney, AustraliaVijayakumar Velu, Emory University, United States
Copyright © 2020 Zuazo, Arasanz, Bocanegra, Fernandez, Chocarro, Vera, Kochan and Escors. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grazyna Kochan, Z3JhenluYS5rb2NoYW5AbmF2YXJyYS5lcw==; Ruth Vera, cnV0aC52ZXJhLmdhcmNpYUBuYXZhcnJhLmVz; David Escors, ZGVzY29yc21AbmF2YXJyYS5lcw==