 Portia Gough
Portia Gough Ian A. Myles
Ian A. Myles- Epithelial Therapeutics Unit, National Institute of Allergy and Infectious Disease, National Institutes of Health, Bethesda, MD, United States
Since its discovery in 1975, TNFα has been a subject of intense study as it plays significant roles in both immunity and cancer. Such attention is well deserved as TNFα is unique in its engagement of pleiotropic signaling via its two receptors: TNFR1 and TNFR2. Extensive research has yielded mechanistic insights into how a single cytokine can provoke a disparate range of cellular responses, from proliferation and survival to apoptosis and necrosis. Understanding the intracellular signaling pathways induced by this single cytokine via its two receptors is key to further revelation of its exact functions in the many disease states and immune responses in which it plays a role. In this review, we describe the signaling complexes formed by TNFR1 and TNFR2 that lead to each potential cellular response, namely, canonical and non-canonical NF-κB activation, apoptosis and necrosis. This is followed by a discussion of data from in vivo mouse and human studies to examine the differential impacts of TNFR1 versus TNFR2 signaling.
Introduction
Tumor Necrosis Factor alpha (TNFα) is a central mediator in the immunologic processes of infection control, autoimmunity, allergic disease, as well as the anti-neoplastic activity for which it was named. While too often categorized as simply “pro-inflammatory”, modern research has identified the complex and pleotropic nature of TNFα signaling. However, many gaps in knowledge persist in differentiating the signaling through the two main TNFα receptors (TNFR1 and TNFR2) or how the balance between them may influence downstream consequences. Activation of signaling by TNFα through TNFR1 and TNFR2 initiates a variety of potential outcomes, including cell proliferation, gene activation or cell death. Mediating this variety of cellular responses from just two receptors requires complex control of signal transduction within the cell.
The particular response of a given cell to activation by TNFα is determined by receptor expression and intracellular conditions, such as ubiquitination of the signaling complex and the availability of caspases. Herein, we attempt to summarize the difference in intracellular signaling downstream from TNFR1 versus TNFR2 as well as their specific biologic consequences. In addition, we collate the animal model and human clinical studies which independently assess TNFR activity. Overall, we identify that a greater understanding of this pathway may offer opportunities for novel therapeutic targeting as well as treatment optimization for current TNFα inhibition approaches.
TNFR Cellular Expression
TNFα receptor expression is central to the cellular response to TNFα and varies by cell type. TNFR1 is a death receptor, as its structure includes a death domain (DD), that is constitutively expressed on most cell types and is activated by TNFα in either its membrane-bound (mTNFα) or soluble (sTNFα) forms (1–3). Following activation by binding TNFα, intracellular signaling via TNFR1 is initiated via its DD (4).
In contrast, TNFR2 is limited in both its expression and its activation. Expression of TNFR2 is restricted to particular cell types, including, endothelial cells, fibroblasts and subsets of neurons and immune cells (myeloid cells, T- and B-cell subsets) (5, 6). Among the peripheral immune cells, both the percentage of cells expressing, and the number of receptors per cell, is much greater for TNFR2 than TNFR1 (7). TNFR2 is only fully activated by mTNFα, and this receptor lacks the DD that is central to intracellular signaling by TNFR1 (8). The selectivity of ligand, binding sTNFα and/or mTNFα, adds a layer to the regulation of responses that result from activation of TNFR1 or TNFR2. There is a structural basis for preferential binding of mTNFα by TNFR2 based on its rigid proline-rich stalk region, which alters the organization of TNFR2 monomers in the absence of ligand in a manner that inhibits their ability to bind sTNFα (9). Thus, the mechanism of activated pathways and cellular responses differ between the two TNFα receptors.
Further adding to the complexity of TNFR signaling is the production of soluble TNFR1 or TNFR2 during cellular activation and the potential for reverse signaling through binding mTNFα. TNFα or other stimuli (e.g., formyl methionine-leucine-phenylalanine (fMLP), lipopolysaccharide (LPS), GM-CSF) induce proteolytic cleavage of TNFRs by the metalloproteinase TNFα converting enzyme (TACE; also called ADAM17) (10, 11). Soluble receptors can also be generated by alternative splicing of mRNA to produce TNFR2 that lacks the membrane-spanning region (12). These soluble receptors retain their ability to bind TNFα, albeit with lower affinity than their membrane-bound counterparts, thereby reducing the availability of TNFα for receptors that remain membrane-bound (12, 13). When mTNFα is bound by either TNFR1 or TNFR2, reverse signaling can occur. In this case, once TNFR binds mTNFα, it is phosphorylated within the ligand-bearing cell, resulting in NFκB activation (a pathway described in more detail below) (14, 15). Receptor expression and receptor shedding and reverse signaling through membrane-bound ligand provide extracellular regulation for this signaling pathway.
TNFR1 Activation: Formation of the Core Signaling Complex
When activated by binding TNFα, a core signaling complex is constructed on the cytoplasmic tail of TNFR1. The first step in this process is the trimerization of TNFR1, initiated by contact with TNFα (16). Once TNFR1 forms a trimer, the DD is able to recruit TNFR1-associated death domain (TRADD) (4). TRADD acts as a scaffold in the TNFR1 signaling complex, directing all downstream signaling events as it recruits TNF receptor-associated factor (TRAF) 2, or TRAF5, and receptor-interacting serine/threonine-protein kinase 1 (RIPK1) (17, 18). TRAF2 then provides a platform for recruitment of cellular inhibitor of apoptosis protein (cIAP) 1 and cIAP2 (19, 20) (Figure 1; Supplemental Figure 1).
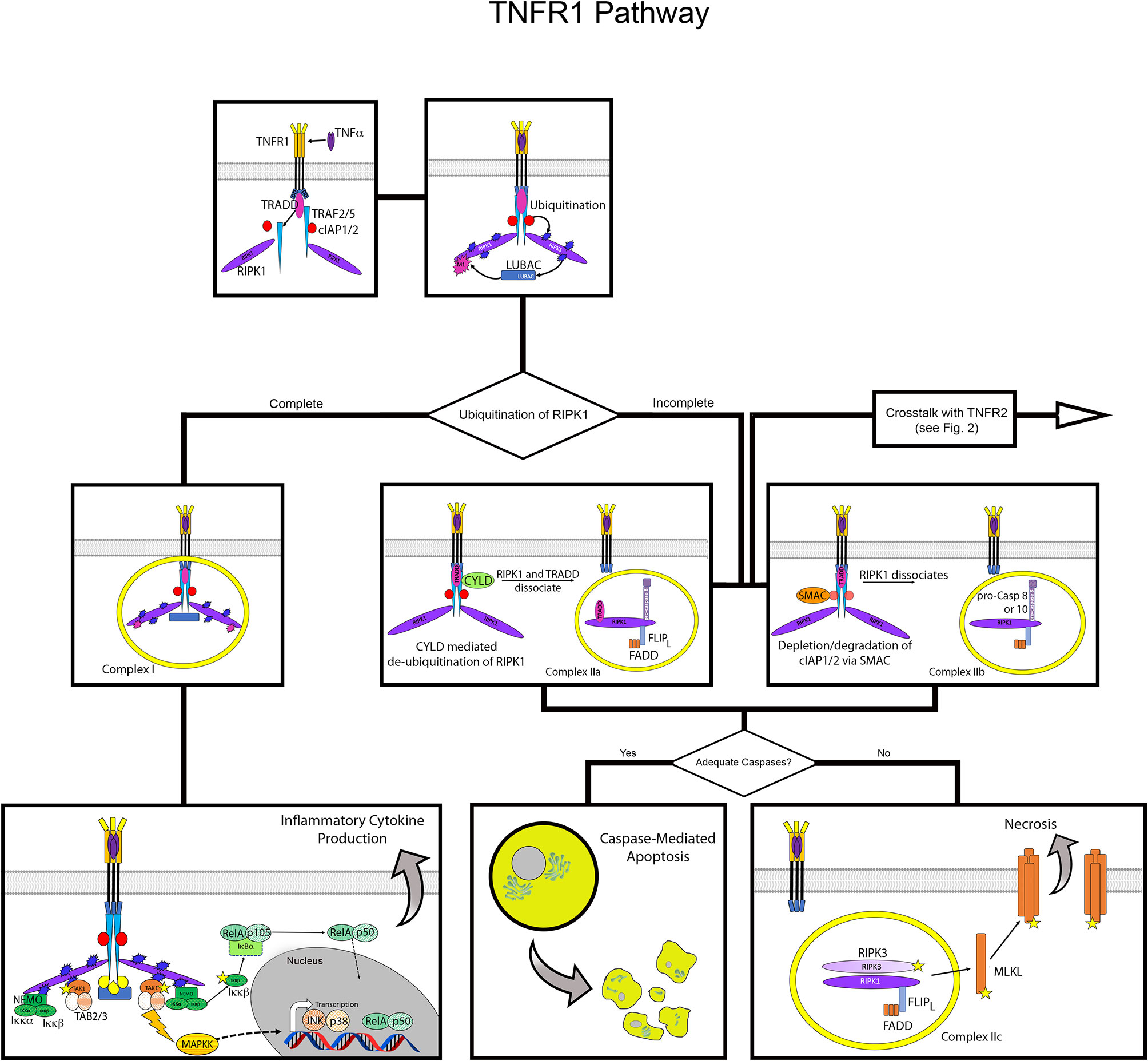
Figure 1 Overview of TNFR1 activation pathway. Flow diagram for TNFR1 activation contrasting the outcomes of inflammatory cytokine production versus cell death. The top panels depict formation of the core signaling complex with recruitment and assembly of TRAF2/5, cIAP 1/2 and RIPK1 at the death domain of TNFR1, and subsequent recruitment of LUBAC. The middle and lower panels show the divergence of potential pathways from formation of the core complex. On the left, complete ubiquitination of RIPK1 results in formation of Complex I, which leads to recruitment of NEMO and TAK1 that activate NFκB and JNK, respectively. On the right, incomplete ubiquitination of RIPK1 leads to formation of complex IIa or IIb, with assembly of FLIPL, FADD and pro-caspase 8 or 10, leading apoptosis via activation of the latter. Below this, the formation of Complex IIc in the absence of sufficient caspases leads to necroptosis via activation of MLKL via the necrosome formed by assembly of RIPK1 and RIPK3. Stars indicate phosphorylation, blue bursts represent ubiquitination, purple bursts represent M1 ubiquitination.
The assembly of TRADD, TRAF2 (or TRAF5), RIPK1, and cIAP1/2 forms the core signaling complex of activated TNFR1. From here, the cellular response mediated by TNFR1 is determined by the ubiquitination of RIPK1 and the availability of caspase molecules (21, 22). The TRAF proteins and cIAP1/2 are ubiquitin E3 enzymes that add ubiquitin chains to RIPK1 at multiple locations (20, 23). These ubiquitin chains act as scaffolds for additional factors that lead to the formation of Complex I, which activates NF-κB, JNK, and p38 pathways to induce cytokine signaling and cell survival (20). If the addition of these ubiquitin chains is disrupted, various versions of Complex II can form, leading to apoptotic or necrotic cell death. The details of the formation of these complexes are discussed below.
Complex I: Activation of NF-κB, JNK, and p38 Pathways Leads to Gene Activation and Survival
The formation of Complex I is stabilized by linear ubiquitination of RIPK1, which allows the recruitment of additional signaling factors (24). Addition of K63, K11, and K48 poly-ubiquitin chains by cIAP1/2 recruits a multimeric complex known as linear ubiquitin chain assembly complex (LUBAC) (25, 26). Complex I is fully activated by attachment of an M1 poly-ubiquitin chain to RIPK1 by LUBAC, completing the formation of the scaffolding network that recruits additional mediators for gene activation (25).
Activation of multiple pathways, specifically NF-κB, JNK, and p38, by Complex I is achieved by parallel assembly of proteins, recruited via their ubiquitin-binding domains, to activate TGFβ-activated kinase 1 (TAK1) and inhibitor of IκB kinase (IKK). TAK1 is activated by its recruitment with TAK1 and MAP3K7-binding protein (TAB) 2 and TAB3, with TAB2 and TAB3 binding to poly-ubiquitin chains (27). Alongside the recruitment and activation of TAK1, the IKK complex is recruited by assembly of IKKα, IKKβ and NF-κB essential modulator (NEMO), via the ubiquitin-binding domain of the latter (28–30). Once in proximity to each other, TAK1 activates the IKK complex via phosphorylation of IKKβ (27, 28).
TAK1 also activates mitogen-activated protein kinase kinases (MAPKKs), which activate JUN NH2-terminal kinase (JNK) and p38 pathways (21, 31). The NEMO-dependent activation of the IKK complex results in canonical NF-κB activation, freeing the RelA/p105 heterodimer to undergo proteolytic processing to RelA/p52 and subsequent nuclear translocation (30, 32). Thus, formation of Complex I initiates signaling that induces inflammatory gene activation and cell survival via activation of multiple transcription factors. The particular genes induced by TNFα vary widely by cell type, with as many as >5000 genes modulated by the various transcription factors in a highly dynamic manner (33–36). If Complex I does not form upon stimulation of signaling through TNFR1, the pathways for cell survival are not initiated and different versions of Complex II form to mediate cell death via apoptosis or necroptosis (37).
Complexes IIa and IIb: Apoptosis via Caspase 8
Returning to the principle that full ubiquitination of RIPK1 is necessary for the formation of Complex I, conversely, the formation of Complex II is dependent on incomplete ubiquitination of RIPK1 (20, 38). When RIPK1 is not fully ubiquitinated, it dissociates from the signaling complex and apoptotic signaling via Complex IIa or IIb is initiated (39). There are multiple ubiquitin-modifying enzymes that can act on RIPK1 to facilitate this process.
Formation of Complex IIa is initiated when RIPK1 is de-ubiquitinated by cylindromatosis (CYLD). CYLD provides a negative feedback loop for NF-κB activation, as its expression is induced by NF-κB (40, 41). When CYLD associates with TRAF2, it removes the K63 and M1 poly-ubiquitin chains from RIPK1, thereby allowing RIPK1 to dissociate from the TNFR1 complex (40, 41). Once released into the cytosol, RIPK1 forms Complex IIa by assembling with TRADD, Fas-associated death domain (FADD), FLICE-like inhibitory protein (FLIPL), pro-caspase 8 (39). The assembly of these proteins converts pro-caspase 8 to its active form, caspase 8, initiating the apoptosis pathway.
Incomplete ubiquitination of RIPK1 can also be caused by depletion or degradation of cIAP1/2, which results in the absence or reduction of K63 poly-ubiquitin chains added to RIPK1, leading to the formation of Complex IIb (42, 43). Regulation of cIAP1/2 is mediated, in part, by second mitochondria-derived activator caspase (SMAC) (44). The interaction with SMAC causes auto-ubiquitination of cIAPs, leading to their degradation (45). Due to its destabilization in the core signaling complex by the resultant absence of K63-linked ubiquitination, RIPK1 dissociates from TNFR1 as in the start of Complex IIa. It again forms a complex in the cytosol with FADD, FLIPL, and pro-caspase 8. Thus, Complex IIb includes the same components as Complex IIa, except it lacks TRADD (21, 31). This assembly also serves to activate pro-caspase 8 to caspase 8, resulting in cell death by apoptosis (39). Activation of caspase 10 has also been linked to Complex IIa and IIb formation (37).
Complex IIc: Necroptosis in the Absence of Caspase Activity
After activation of caspase 8 in Complex IIa and IIb, RIPK1 and RIPK3 are degraded by the activated caspase in the cytosol (46, 47). However, if there are not sufficient caspases available to perform this inactivation, necroptosis will occur via the activity of RIPK1 and RIPK3. The depletion of caspase 8 to induce necroptosis has mainly been established and studied using targeted inhibition of caspase 8 by pharmacological agents such as Z-VAD-FMK (48, 49). This process has also been observed in cells expressing the caspase inhibitor SPI-2, resulting from vaccinia virus infection, which sensitizes cells to TNFα-induced necroptosis in a RIPK1-dependent manner (50, 51). Once caspase 8 is inhibited or depleted, RIPK1 and RIPK3 assemble together in an amyloid-like structure to form the necrosome, and RIPK3 activates mixed-lineage kinase domain-like protein (MLKL) (52–54). Once activated, MLKL oligomerizes and translocates to the plasma membrane where it binds phosphoinositides to cause cell lysis (53).
TNFR2 Activation: Canonical and Non-Canonical NF-κB Pathways
Canonical NF-κB activation is defined by the requirement for NEMO-dependent IKK activation, which frees the RelA/p50 NF-κB heterodimer for nuclear translocation. Thus, NEMO-independent IKK activation is the defining feature of non-canonical NF-κB activation; instead, this pathway involves the accumulation of NF-κB inducing kinase (NIK) to activate IKKα, leading to nuclear translocation of the RelB/p52 heterodimer (32, 33). Although TNFR2 signaling can result in canonical NF-κB activation, this is most often a result of TNFR1 activity (21) (Figure 2; Supplemental Figure 1).
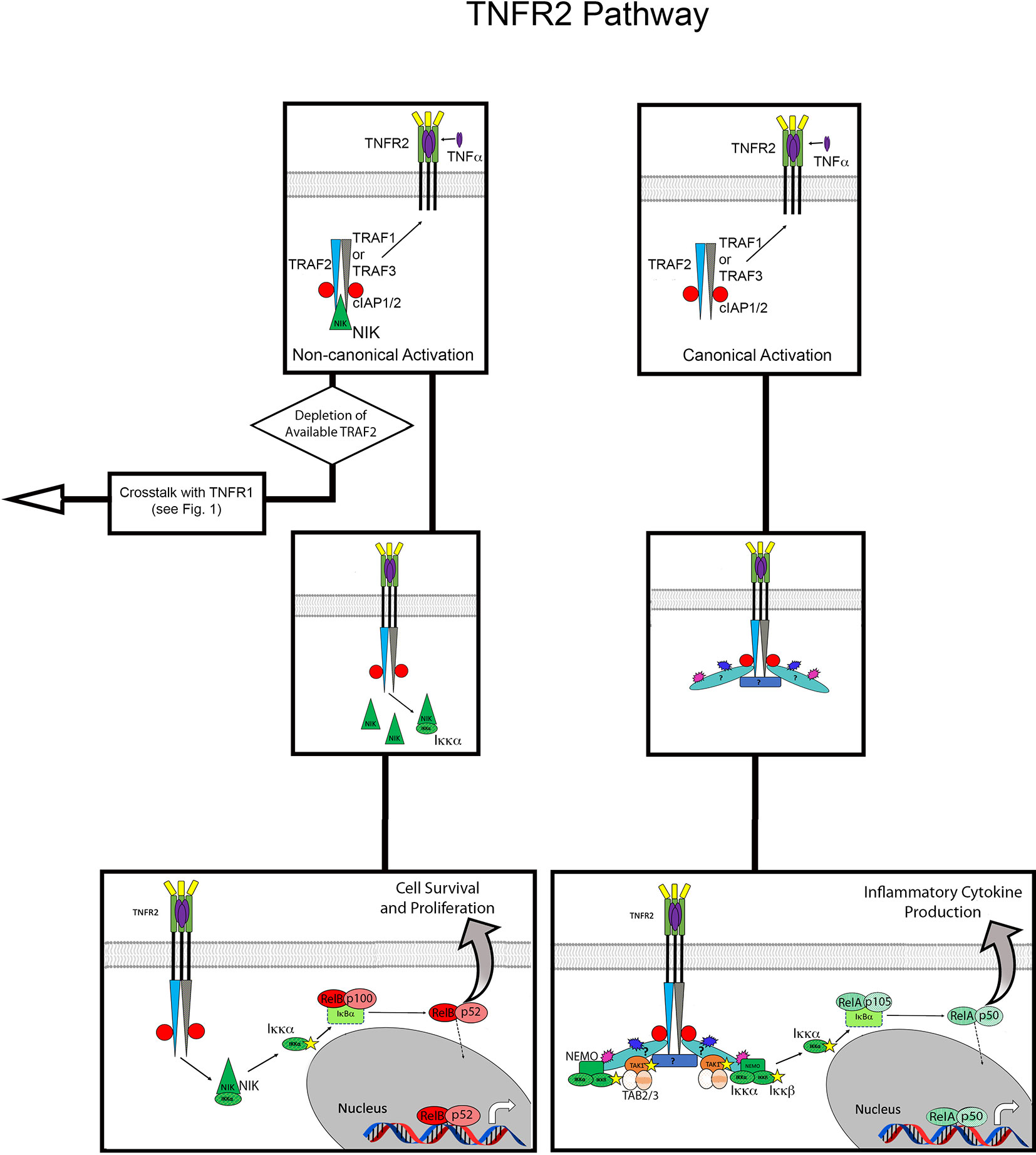
Figure 2 Overview of TNFR2 activation pathway. Flow diagram for TNFR2 activation contrasting the non-canonical versus canonical pathways. The top panels show recruitment of TRAF proteins, cIAP1/2 and NIK to TNFR2 upon binding ligand (mTNFα). On the left, this assembly leads to non-canonical NFκB activation via accumulation of NIK. On the right, the pathway of canonical NFκB activation is shown, the details of which are unknown but presumably result from K63 and M1 polyubiquitin chains mediating the recruitment of TAK1 and NEMO. Stars indicate phosphorylation, blue bursts represent ubiquitination, purple burst represent M1 ubiquitination. Question mark in canonical pathways indicate the target of ubiquitination is unknown.
Activation of TNFR2, which lacks the DD that characterizes signaling by TNFR1, most frequently results in cell proliferation and survival. After binding mTNFα, TNFR2 forms a trimer and directly recruits TRAF2 and TRAF1 or TRAF3, albeit with lower affinity than TNFR1-TRADD (55–57). TRAF2 is central to assembly of the TNFR2 signaling complex, and also recruits cIAP1/2 (19, 20, 58). Although the exact targets remain unknown, it has been demonstrated that the TNFR2 signaling complex includes K63 and M1 poly-ubiquitin chains (59). This suggests a mechanism of canonical NF-κB activation similar to that observed in the TNFR1 signaling complex. Indeed, it has been shown that activation of this pathway by TNFR2 also relies on TAK1 and IKKβ, and the K63 and M1 poly-ubiquitin chains bring these kinases into close proximity with each other (59, 60).
While canonical NF-κB activation tends to be rapid and result in the expression of pro-inflammatory genes, non-canonical NF-κB pathways activate in a sustained manner, on a slower timescale, and promote cell survival and proliferation. This alternative NF-κB pathway utilizes p100, which is produced from the canonical pathway, usually via activation of TNFR1 signaling (61). Again, a defining feature of non-canonical NF-κB activation is its induction by NIK (62). In its basal state, NIK is in an inhibitory complex with TRAF 2/3 and cIAP 1/2. When TRAF 2/3 is recruited to TNFR2, the inhibitory complex of NIK is disrupted (63). Activated NIK then accumulates and activates IKKα, allowing p100 to be proteolytically processed to p52, thereby generating the active NF-κB p52/RelB heterodimer (64). Expression of p100 and RelB is potentiated by activation of the canonical NF-κB pathway by TNFR1, which then feeds into the non-canonical activation induced by TNFR2 and results in cell survival and proliferation (61). This signaling synergy between TNFR1 and TNFR2 is an example of crosstalk between the receptors, a type of interaction that is also central to cell death mediated by TNFR2.
Cell Death via TNFR2
As stated in the previous section, TRAF2 is central to the cellular responses generated from the TNFR2 signaling complex. Activation of TNFR2 influences formation of TNFR1 signaling complexes by depleting available TRAF2 for Complex I, as expression levels of TNFR2 can be up to 10-fold higher on cells that express both receptors (65) (Figure 2; Supplemental Figure 1). Additionally, TRAF2 can be degraded by its interaction with cIAP1/2. This depletion and degradation of TRAF2 has two major consequences: promoting the non-canonical NF-κB pathway, described above, and inhibiting canonical NF-κB activation via TNFR1 Complex I (61, 64). The reduction of TRAF2 inhibits recruitment of cIAP1/2 to the TNFR1 signaling complex, thereby allowing formation of Complex IIa, IIb or IIc. Thus, cell death mediated by TNFR2 is a result of crosstalk, where activity of TNFR2 indirectly influences the signaling complexes that can form at TNFR1. In TNFR signaling, cell survival, proliferation and death are a matter of quantitative balance between TNFR1 and TNFR2 and the key components of their respective signaling complexes.
TNFR Influence on Epithelial and Endothelial Cells
Epithelial-to-mesenchymal transition (EMT) is a pathway heavily mediated by TNFR in endo- and epithelial cells. EMT is an essential process for normal tissue repair; epithelial cells take on a migratory mesenchymal phenotype via a complex modulation of adhesion proteins and intracellular filaments (66, 67). During wound healing, polarization of cells on the leading edge of the damaged tissue activates EMT-related pathways to fill the cellular gaps through a combination of migration and proliferation (67, 68). Subsequent mesenchymal-to-epithelial transition (MET) re-epithelializes the damaged area (67, 68). While both receptors are involved in the inflammatory and anti-infection responses (69), TNFR2 is central for EMT and cell proliferation (70). The potential for differing impacts of TNFR activation in MET is currently unknown. TNFα-related EMT can be further modified by phospholipid exposure (71, 72) as well as co-stimulation of Toll-Like Receptors (TLR) and neurotransmitters like nicotine and serotonin (73–78).
Furthermore, the consistent, direct contact between neighboring epithelial cells heightens the potential influence of mTNFα. While genetic deletion of TACE/ADAM17 does not appear to impact bacterial responses in peripheral immune cells (79), TACE/ADAM17 knockout mice develop a microbiome-dependent, eczematous phenotype (80, 81). Endothelial-to-mesenchymal transition (EndoMT) is also a process of developing a migratory phenotype important in vascularization. Although EndoMT is known to be influenced by TNFα (82, 83), differential impacts of the receptors of membrane bound status of TNFα remain unelucidated.
Differential TNFR Activity in Mouse Models
Table 1 details all identified mouse models in which TNFR1 and TNFR2 were assessed independently. In brief, neurologic models have demonstrated even more complex effects of selective TNFR deletion; in EAE, TNFR2 is protective on microglia but deleterious on monocytes and macrophages whereas TNFR1 appears harmful in all contexts due to reducing the blood brain barrier function against cell infiltration into the CNS (84, 105, 117). In mouse models of pain, deletion of TNFR1 lead to reduction of mechanical pain as well as NMDA activation of lamina II neurons (86, 87, 134). Early-phase hyperalgesia responses to heat after exposure to complete Freud’s adjuvant (CFA) showed dual dependence on TNFR1 and TNFR2, whereas late phase reactions were TNFR1 exclusive (86). CFA injection into mouse footpads led to an increase in TNFR2 mRNA in the spinal cord (86).
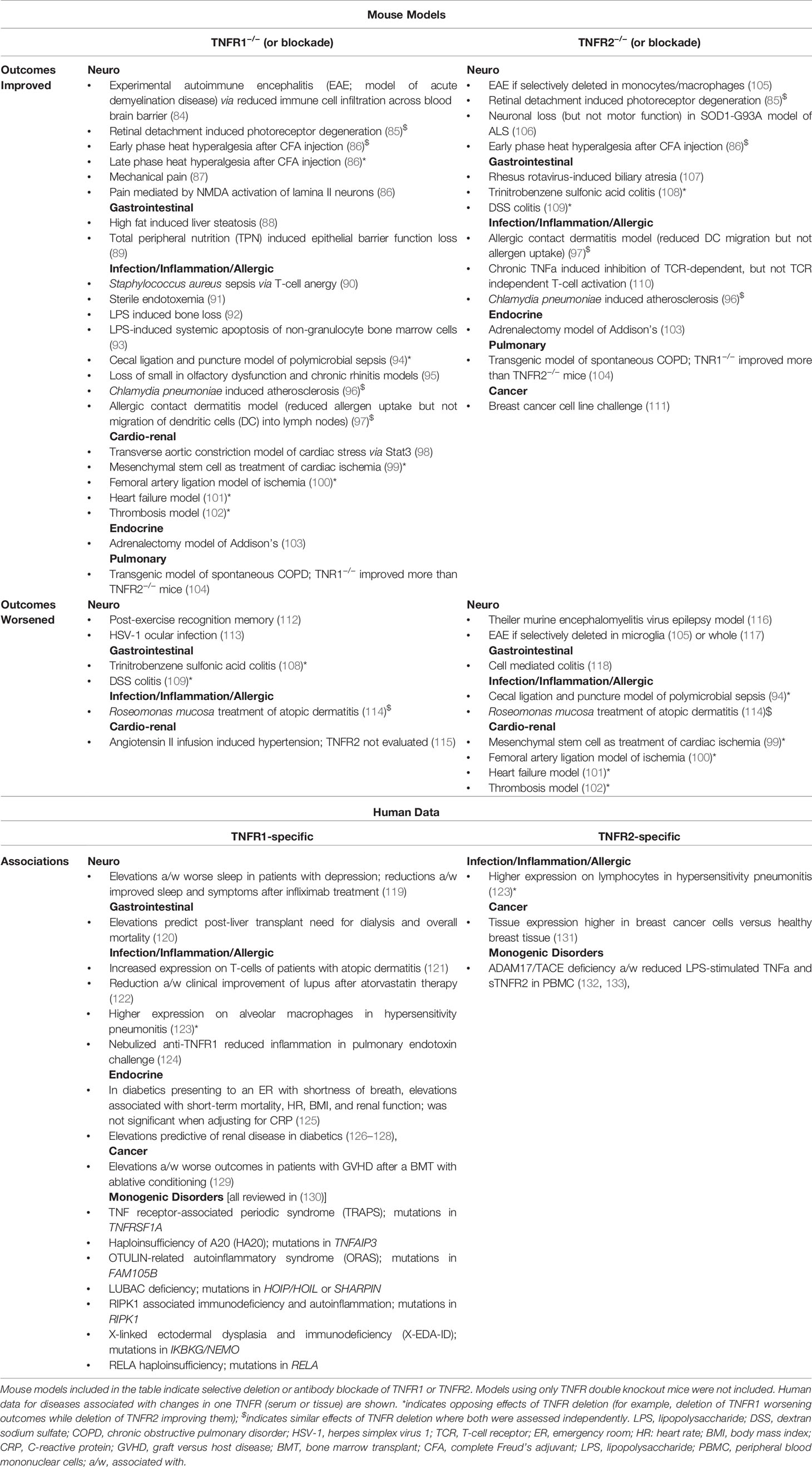
Table 1 Summary of mouse model and clinical association data for TNFR1 versus TNFR2.
Gastrointestinal models identified TNFRdko and TNFR1−/− mice have worse outcomes during DSS colitis (135), however selective deletion of TNFR2 improves outcomes (109). While TNFR2 deletion improved DSS colitis (109), TNFR2 is protective in cell-mediated colitis models, potentially through its role in fostering the expansion and stability of regulatory T cells (Tregs) (118) and vitamin D dependent tolerogenic dendritic cells (136, 137).
Opposite, but similarly discordant effects were seen in thrombosis models where TNFR2−/− mice display worse outcomes and TNFR1−/− mice were protected (102). Additional cardio-renal models including heart failure and ischemia also displayed opposing influence of TNFR1 versus TNFR2 (99–101). Models of adrenal insufficiency (103), chronic obstructive pulmonary disease (COPD) (104), and breast cancer (111) each identified a pathogenic role for TNFR2.
The lone infection model differentiating TNFR impacts uncovered in our search demonstrated TNFR2-mediated protection and TNFR1-mediated harm during polymicrobial sepsis resulting from cecal ligation and perforation (94). However, the importance of TNFR2 in wound repair may confound any infection-control conclusions of this surgical model. Meanwhile, LPS mediated effects on infection and inflammation appear to operate through TNFR1 (91–93), while off target effects of inflammation (such as allergic sensitization and Chlamydia induced atherosclerosis) were mediated by TNFR2 (96, 97). Both allergic rhinitis-associated and neurogenic olfactory dysfunction are mediated by TNFR1 (95). Our recent publication demonstrated that the modeled treatment efficacy of topical, commensal Roseomonas mucosa for atopic dermatitis operates through TNFR2, however both receptors are essential for modeled therapeutic benefit (114). Consistent with prior literature, the TNFR2-mediated effects also demonstrated a role for neurologic and innate signaling; TNFR2 potentiation by optimal TLR5 and nAChR activation were essential to modeled treatment outcomes (114). Therefore, while in vivo evidence is limited, the therapeutic potential of selective TNFR blockade remains an under-elucidated topic.
Viral TNF Blockade
Several examples of virus encoded TNFα-binding proteins have been described in the literature (138). T2 have been identified in the leporipovirus myxoma virus (M-T2) and the Shope fibroma virus (S-T2) (139, 140). While extracellular M-T2 inhibition is species-limited to rabbits, S-T2 can also inactivate human TNFα. However, in the intracellular form, M-T2 can inhibit TNFR-1–mediated cell death by directly binding to human TNFR1 (141). The poxvirus produces several TNFR decoy receptors: CrmB and CrmD bind TNFα as well as leukotriene alpha (142, 143); CrmC and CrmE exclusively bind TNFα (142, 144, 145). Additionally, the YLDV and tanapox viruses produce the decoy receptor 2L, an major histocompatibility complex I (MHC-I) analogous protein which also competitively binds TNFa (146). Other examples of direct viral influence over the TNFα pathway include the SARS-CoV1-S protein, which activates TACE to cause increased shedding of ACE2 and increased solubilization of TNFα (147). These viral TNFα modulation strategies present intriguing opportunities for therapeutic development.
Pharmacologic TNF Blockade
The predominant TNF blockade strategy is through antibody-mediated inhibition: chimeric (infliximab); fully humanized (adalimumab and golimumab); and variable binding regions fused to a Peg moiety (certolizumab-pegol). Alternatively, etanercept is a dimer of recombinant TNFR binding regions fused to the constant region of a human IgG1 antibody (148). Although these medications have been revolutionary in the treatment of autoimmune and autoinflammatory diseases, their results do not inform TNFR actions given that they bind the TNF ligand and thus preclude signaling through both receptors (148).
Pentoxifylline is an oral medication which inhibits the release of TNFα. While the mechanism remains to be elucidated, the proposed mechanism is via influence over phosphodiesterase (PDE) and cyclic (cAMP) (149), further implicating lipid metabolism in the control of TNFα (71, 72). Of note, current TNFα inhibition strategies are unlikely to block the impacts of mTNFα. Given the greater binding affinity of the current anti-TNF agents for sTNFα (150, 151), tissue-level analysis may be needed to determine if meaningful neutralization of mTNFα can be achieved by any current anti-TNFα therapy.
Although TNF blockade is now a routine part of clinical medicine, details of differential impacts of TNFR signaling in humans have larger gaps in knowledge than murine models. As stated, all current anti-TNF treatments are non-selective, and thus, only a paucity of clinical evidence for selective blockade exists (Table 1). While several anti-TNFR1 antibodies have been used in vitro (137), only one anti-TNFR1 antibody has been studied in humans; a nebulized formulation reduced inflammation and endothelial injury in healthy subjects that underwent an inhalational LPS challenge (124). The drugs thalidomide and cyclophosphamide have reportedly TNFR2-specific inhibitory effects, but confirmatory evidence is thus far limited to the effects on Treg differentiation; and, the side effect profile for thalidomide does not favor clinical use outside of cancer treatment (137).
Soluble and Tissue TNFR Expression in Inflammation
While interventional evidence is lacking, several publications demonstrate disease associations with differential TNFR effects, particularly for serum levels of sTNFR (119–123, 125–129, 131). Intriguingly, the concentrations of both sTNFR1 and sTNFR2 follow circadian rhythms that run about 1 h ahead of the well-established cortisol fluctuations (152). Thus, time of day may be an important confounder for clinical studies that aim to compare sTNFR levels in disease. However, a far more concerning confounder is the possibility of acute phase reactions. Several examples of treatment responsive, equivalent elevations in both sTNFR1 and sTFNR2 have been described in tuberculosis (153), multiple sclerosis (154), schizophrenia (155), Alzheimer’s disease (156), hyperlipidemia (157), severe burns (158), and after dietary challenge with transfat (159). A study evaluating predictors of infarction in heart disease may provide the most illustrative example for the possibility of sTNFR1 and sTNFR2 being general inflammatory markers; elevations of both sTNFR1 and sTNFR2 were highly predictive of future cardiac events but lost statistical significance when adjusted for levels of the more commonly deployed inflammatory marker CRP (160). Similarly, tissue levels of both TNFR1 and TNFR2 are elevated in psoriasis and decrease with successful anti-TNFα treatment (161). However, not all patients respond to anti-TNFα treatment and the risk of infectious complications must be considered whenever TNF-inhibitors are prescribed (162).
Differential Soluble and Tissue TNFR Associations in Disease
While sTNFR1 and sTNFR2 may simultaneously track with general inflammation, a unique differential association is found in obesity, where elevations are seen in serum TNFα but not sTNFR1 or sTNFR2 (163). More distinguishing associations between disease and TNFR1 have been reported compared to TNFR2, however several studies failed to assess sTNFR2 levels (164, 165). Overall, the most robust clinical association for sTNFR1 is for predicting future renal disease in diabetics (126–128). Elevated sTNFR1 (but not sTNFR2) was associated with worse outcomes when presenting to an emergency room with shortness of breath, but also lost statistical significance when adjusting for CRP (125). Associations unique to sTNFR1 were also seen with treatment-responsive lupus (122), sleep disruption in depression (119), worse outcomes in GVHD after ablative-conditioned BMT (129), and mortality and dialysis needs for post-liver transplant patients (120). Two studies have assessed the expression of TNFR1 versus TNFR2 on immune cells (121, 123), compared to healthy controls: patients with atopic dermatitis have increased expression of TFNR1 on their T-cells (121); patients with hypersensitivity pneumonitis have elevated TNFR1 expression on alveolar macrophages but elevated TNFR2 on peripheral lymphocytes (123). Furthermore, human cell culture models challenged with R. mucosa confirmed the importance of the TNFR2 related pathways identified in our mouse models (114).
Monogenic Disorders of TNFR Signaling
Although many gaps in knowledge for common diseases, monogenic disorders related to TNFR signaling can provide valuable insights. The most demonstrative of these disorders in TNF receptor-associated periodic syndrome (TRAPS) resulting from autosomal dominant mutations in TNFSF1A. Although the exact mechanism is unclear, aberrant TNFR1 signaling, intracellular accumulation of misfolded proteins, and constitutive immune activation have been implicated in the pathogenesis (166). Patients most commonly experience severe abdominal pain, arthralgias, and myalgias (167). Although the anti-IL-1 agent anakinra has become the preferred treatment in TRAPS, etanercept has been used (130). Multiple other monogenic autoinflammatory syndromes are associated with the intracellular complexes downstream of TNFR1, including haploinsufficiency of A20 (HA20), LUBAC deficiency, RIPK1 deficiency, OTULIN-related autoinflammatory syndrome (ORAS), RELA haploinsufficiency, and X-linked ectodermal dysplasia and immunodeficiency (X-EDA-ID); each of these has been expertly detailed in a recent review (130).
In 2011, a syndrome including inflammatory skin disease and recurrent infections was linked to autosomal recessive mutations in ADAM17/TACE in two siblings. One sibling died at age 12 of parvovirus B19-associated myocarditis. However, while the other was shown to have exaggerated production of IL-1β and IL-6 in response to lipopolysaccharide (LPS) challenge, he was described as living a “relatively normal life” despite continued recurrent infection (132). Subsequently, another patient with ADAM17/TACE mutations resulting in inflammatory skin disease, recurrent infections, and fatal sepsis was identified (133). Studies into LPS responses in the patient’s peripheral blood mononuclear cells (PBMC) revealed reduced production of TNFα and sTNFR2 (133).
Discussion
Data from cell cultures, animal models, and humans strongly suggests the balance between TNFR1 and TNFR2 activity has meaningful impacts on health and disease. Unfortunately, mouse models which independently assess TNFR influence are uncommon in the literature and often lack confirmatory validation between research groups. Similarly, clinical studies that independently assess sTNFR levels are typically small and lack tissue-level verification. Clinical utility of anti-TNF treatments should not preclude investigation into differential impacts of TNFRs, given that more targeted blockade could lead to further optimization of clinical benefit, or a reduction in side effects.
The current literature on the importance of the TNFRs suggests actionable needs for future research to assess sTNFR1 and sTNFR2 during studies of diseases in which TNFα is known to play a role. Ideally, this research could be bolstered by tissue staining for receptor expression, mTNFα concentration, and/or transcription factor post-translational modifications. Furthermore, functional assessments such as the scratch assay should be considered wherever a role for TNFα-modulation of epithelial or endothelial cellular activity is suspected.
However, a notable portion of the differences in downstream signaling through TNFR1 versus TNFR2 is neither transcriptionally regulated nor governed directly by receptor expression. Thus, research into TNFα-modulated diseases using transcript- or proteomics approaches may require direct assessment of the ubiquitination and phosphorylation of signal transducers of the TNFR pathway. Similarly, investigations into disease states or experimental conditions that reveal involvement of pathways that modulate TNFR activity, such as lipid metabolism or neurotransmitter signaling, may be enhanced by directed inquiry into possible TNFR-dependent impacts.
Author Contributions
PG and IM contributed equally to writing the manuscript and editing the text. IM made the figures and table, with the assistance of PG. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Intramural Research Program of the NIH and NIAID.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.585880/full#supplementary-material
References
1. Naude PJ, den Boer JA, Luiten PG, Eisel UL. Tumor necrosis factor receptor cross-talk. FEBS J (2011) 278:888–98. doi: 10.1111/j.1742-4658.2011.08017.x
2. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature (1997) 385:729–33. doi: 10.1038/385729a0
3. Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature (1997) 385:733–6. doi: 10.1038/385733a0
4. Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell (1995) 81:495–504. doi: 10.1016/0092-8674(95)90070-5
5. Medler J, Wajant H. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin Ther Targets (2019) 23:295–307. doi: 10.1080/14728222.2019.1586886
6. Borghi A, Verstrepen L, Beyaert R. TRAF2 multitasking in TNF receptor-induced signaling to NF-kappaB, MAP kinases and cell death. Biochem Pharmacol (2016) 116:1–10. doi: 10.1016/j.bcp.2016.03.009
7. Sennikov S, Alshevskaya A, Zhukova J, Belomestnova I, Karaulov A, Evsegneeva I, et al. Co-expression of membrane-bound TNF-alpha type 1 and 2 receptors differ in the subsets of immunocompetent cells. Immunol Lett (2019) 207:1–5. doi: 10.1016/j.imlet.2019.01.005
8. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell (1995) 83:793–802. doi: 10.1016/0092-8674(95)90192-2
9. Richter C, Messerschmidt S, Holeiter G, Tepperink J, Osswald S, Zappe A, et al. The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol Cell Biol (2012) 32:2515–29. doi: 10.1128/MCB.06458-11
10. Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. An essential role for ectodomain shedding in mammalian development. Science (1998) 282:1281–4. doi: 10.1126/science.282.5392.1281
11. Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, et al. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem (2000) 275:14608–14. doi: 10.1074/jbc.275.19.14608
12. Lainez B, Fernandez-Real JM, Romero X, Esplugues E, Canete JD, Ricart W, et al. Identification and characterization of a novel spliced variant that encodes human soluble tumor necrosis factor receptor 2. Int Immunol (2004) 16:169–77. doi: 10.1093/intimm/dxh014
13. Van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL, Lowry SF. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor alpha in vitro and in vivo. Proc Natl Acad Sci USA (1992) 89:4845–9. doi: 10.1073/pnas.89.11.4845
14. Zhang H, Yan D, Shi X, Liang H, Pang Y, Qin N, et al. Transmembrane TNF-alpha mediates “forward” and “reverse” signaling, inducing cell death or survival via the NF-kappaB pathway in Raji Burkitt lymphoma cells. J Leukoc Biol (2008) 84:789–97. doi: 10.1189/jlb.0208078
15. Zhang M, Wang J, Jia L, Huang J, He C, Hu F, et al. Transmembrane TNF-alpha promotes activation-induced cell death by forward and reverse signaling. Oncotarget (2017) 8:63799–812. doi: 10.18632/oncotarget.19124
16. Banner DW, D’Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, et al. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell (1993) 73:431–45. doi: 10.1016/0092-8674(93)90132-A
17. Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell (1996) 84:299–308. doi: 10.1016/S0092-8674(00)80984-8
18. Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity (1996) 4:387–96. doi: 10.1016/S1074-7613(00)80252-6
19. Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc Natl Acad Sci USA (1996) 93:13973–8. doi: 10.1073/pnas.93.24.13973
20. Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J Biol Chem (2009) 284:35906–15. doi: 10.1074/jbc.M109.072256
21. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol Rev (2019) 99:115–60. doi: 10.1152/physrev.00045.2017
22. Walczak H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol Rev (2011) 244:9–28. doi: 10.1111/j.1600-065X.2011.01066.x
23. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell (2008) 30:689–700. doi: 10.1016/j.molcel.2008.05.014
24. Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell (2009) 36:831–44. doi: 10.1016/j.molcel.2009.10.013
25. Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature (2011) 471:591–6. doi: 10.1038/nature09816
26. Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael-Tomasevic A, Phu L, et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J (2010) 29:4198–209. doi: 10.1038/emboj.2010.300
27. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
28. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell (2009) 136:1098–109. doi: 10.1016/j.cell.2009.03.007
29. Lo YC, Lin SC, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, et al. Structural basis for recognition of diubiquitins by NEMO. Mol Cell (2009) 33:602–15. doi: 10.1016/j.molcel.2009.01.012
30. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell (2006) 22:245–57. doi: 10.1016/j.molcel.2006.03.026
31. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol (2015) 15:362–74. doi: 10.1038/nri3834
32. Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol (2010) 2:a000158. doi: 10.1101/cshperspect.a000158
33. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
34. Lee A, Qiao Y, Grigoriev G, Chen J, Park-Min KH, Park SH, et al. Tumor necrosis factor alpha induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum (2013) 65:928–38. doi: 10.1002/art.37853
35. Sun L, Yang G, Zaidi M, Iqbal J. TNF-induced gene expression oscillates in time. Biochem Biophys Res Commun (2008) 371:900–5. doi: 10.1016/j.bbrc.2008.03.114
36. Zimmermann M, Aguilera FB, Castellucci M, Rossato M, Costa S, Lunardi C, et al. Chromatin remodelling and autocrine TNFalpha are required for optimal interleukin-6 expression in activated human neutrophils. Nat Commun (2015) 6:6061. doi: 10.1038/ncomms7061
37. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell (2003) 114:181–90. doi: 10.1016/S0092-8674(03)00521-X
38. Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem (2006) 281:13636–43. doi: 10.1074/jbc.M600620200
39. Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell (2008) 133:693–703. doi: 10.1016/j.cell.2008.03.036
40. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature (2003) 424:793–6. doi: 10.1038/nature01803
41. Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature (2003) 424:801–5. doi: 10.1038/nature01802
42. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell (2011) 43:432–48. doi: 10.1016/j.molcel.2011.06.006
43. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell (2007) 131:669–81. doi: 10.1016/j.cell.2007.10.030
44. Fulda S. Smac mimetics as IAP antagonists. Semin Cell Dev Biol (2015) 39:132–8. doi: 10.1016/j.semcdb.2014.12.005
45. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell (2000) 102:33–42. doi: 10.1016/S0092-8674(00)00008-8
46. Newton K, Manning G. Necroptosis and Inflammation. Annu Rev Biochem (2016) 85:743–63. doi: 10.1146/annurev-biochem-060815-014830
47. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature (2011) 471:363–7. doi: 10.1038/nature09852
48. Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol (2000) 151:1247–56. doi: 10.1083/jcb.151.6.1247
49. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (2009) 325:332–6. doi: 10.1126/science.1172308
50. Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem (2003) 278:51613–21. doi: 10.1074/jbc.M305633200
51. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell (2009) 137:1112–23. doi: 10.1016/j.cell.2009.05.037
52. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell (2012) 150:339–50. doi: 10.1016/j.cell.2012.06.019
53. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
54. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
55. Grech AP, Gardam S, Chan T, Quinn R, Gonzales R, Basten A, et al. Tumor necrosis factor receptor 2 (TNFR2) signaling is negatively regulated by a novel, carboxyl-terminal TNFR-associated factor 2 (TRAF2)-binding site. J Biol Chem (2005) 280:31572–81. doi: 10.1074/jbc.M504849200
56. Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, Nakagawa S, et al. Solution of the structure of the TNF-TNFR2 complex. Sci Signal (2010) 3:ra83. doi: 10.1126/scisignal.2000954
57. Park YC, Ye H, Hsia C, Segal D, Rich RL, Liou HC, et al. A novel mechanism of TRAF signaling revealed by structural and functional analyses of the TRADD-TRAF2 interaction. Cell (2000) 101:777–87. doi: 10.1016/S0092-8674(00)80889-2
58. Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell (2010) 38:101–13. doi: 10.1016/j.molcel.2010.03.009
59. Borghi A, Haegman M, Fischer R, Carpentier I, Bertrand MJM, Libert C, et al. The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-kappaB signaling. Biochem Pharmacol (2018) 153:292–8. doi: 10.1016/j.bcp.2018.01.039
60. Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci USA (2013) 110:15247–52. doi: 10.1073/pnas.1314715110
61. Rauert H, Wicovsky A, Muller N, Siegmund D, Spindler V, Waschke J, et al. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J Biol Chem (2010) 285:7394–404. doi: 10.1074/jbc.M109.037341
62. Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res (2011) 21:71–85. doi: 10.1038/cr.2010.177
63. Hostager BS, Bishop GA. CD40-Mediated Activation of the NF-kappaB2 Pathway. Front Immunol (2013) 4:376. doi: 10.3389/fimmu.2013.00376
64. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol (2017) 17:545–58. doi: 10.1038/nri.2017.52
65. Wajant H, Siegmund D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front Cell Dev Biol (2019) 7:91. doi: 10.3389/fcell.2019.00091
66. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15:178–96. doi: 10.1038/nrm3758
67. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119:1420–8. doi: 10.1172/JCI39104
68. Das V, Bhattacharya S, Chikkaputtaiah C, Hazra S, Pal M. The basics of epithelial-mesenchymal transition (EMT): A study from a structure, dynamics, and functional perspective. J Cell Physiol (2019) 234(9):14535–55. doi: 10.1002/jcp.28160
69. Sedger LM, McDermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev (2014) 25:453–72. doi: 10.1016/j.cytogfr.2014.07.016
70. Markopoulos GS, Roupakia E, Marcu KB, Kolettas E. Epigenetic Regulation of Inflammatory Cytokine-Induced Epithelial-To-Mesenchymal Cell Transition and Cancer Stem Cell Generation. Cells (2019) 8:1143. doi: 10.3390/cells8101143
71. Treede I, Braun A, Jeliaskova P, Giese T, Fullekrug J, Griffiths G, et al. TNF-alpha-induced up-regulation of pro-inflammatory cytokines is reduced by phosphatidylcholine in intestinal epithelial cells. BMC Gastroenterol (2009) 9:53. doi: 10.1186/1471-230X-9-53
72. Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, et al. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci U S A (2011) 108:1903–7. doi: 10.1073/pnas.1019267108
73. Thagia I, Shaw EJ, Smith E, Else KJ, Rigby RJ. Intestinal epithelial suppressor of cytokine signaling 3 enhances microbial-induced inflammatory tumor necrosis factor-alpha, contributing to epithelial barrier dysfunction. Am J Physiol Gastrointest Liver Physiol (2015) 308:G25–31. doi: 10.1152/ajpgi.00214.2014
74. Chen PC, Lee WY, Ling HH, Cheng CH, Chen KC, Lin CW. Activation of fibroblasts by nicotine promotes the epithelial-mesenchymal transition and motility of breast cancer cells. J Cell Physiol (2018) 233:4972–80. doi: 10.1002/jcp.26334
75. Curtis BJ, Radek KA. Cholinergic regulation of keratinocyte innate immunity and permeability barrier integrity: new perspectives in epidermal immunity and disease. J Invest Dermatol (2012) 132:28–42. doi: 10.1038/jid.2011.264
76. Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, et al. Expression and Function of the Cholinergic System in Immune Cells. Front Immunol (2017) 8:1085. doi: 10.3389/fimmu.2017.01085
77. Uchida M, Oyanagi E, Kawanishi N, Iemitsu M, Miyachi M, Kremenik MJ, et al. Exhaustive exercise increases the TNF-alpha production in response to flagellin via the upregulation of toll-like receptor 5 in the large intestine in mice. Immunol Lett (2014) 158:151–8. doi: 10.1016/j.imlet.2013.12.021
78. Sadiq A, Shah A, Jeschke MG, Belo C, Qasim Hayat M, Murad S, et al. The Role of Serotonin during Skin Healing in Post-Thermal Injury. Int J Mol Sci (2018) 19:1034. doi: 10.3390/ijms19041034
79. Link MA, Lucke K, Schmid J, Schumacher V, Eden T, Rose-John S, et al. The role of ADAM17 in the T-cell response against bacterial pathogens. PloS One (2017) 12:e0184320. doi: 10.1371/journal.pone.0184320
80. Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity (2015) 42:756–66. doi: 10.1016/j.immuni.2015.03.014
81. Woodring T, Kobayashi T, Kim D, Nagao K. ADAM17-Deficient Mice Model the Transcriptional Signature of Human Atopic Dermatitis. J Invest Dermatol (2018) 138:2283–6. doi: 10.1016/j.jid.2018.04.021
82. Yoshimatsu Y, Wakabayashi I, Kimuro S, Takahashi N, Takahashi K, Kobayashi M, et al. TNF-alpha enhances TGF-beta-induced endothelial-to-mesenchymal transition via TGF-beta signal augmentation. Cancer Sci (2020) 111(7):2385–99. doi: 10.1111/cas.14455
83. Dmitrieva NI, Walts AD, Nguyen DP, Grubb A, Zhang X, Wang X, et al. Impaired angiogenesis and extracellular matrix metabolism in autosomal-dominant hyper-IgE syndrome. J Clin Invest (2020) 130(8):4167–81. doi: 10.1172/JCI135490
84. Williams SK, Fairless R, Maier O, Liermann PC, Pichi K, Fischer R, et al. Anti-TNFR1 targeting in humanized mice ameliorates disease in a model of multiple sclerosis. Sci Rep (2018) 8:13628. doi: 10.1038/s41598-018-31957-7
85. Nakazawa T, Kayama M, Ryu M, Kunikata H, Watanabe R, Yasuda M, et al. Tumor necrosis factor-alpha mediates photoreceptor death in a rodent model of retinal detachment. Invest Ophthalmol Vis Sci (2011) 52:1384–91. doi: 10.1167/iovs.10-6509
86. Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain (2011) 152:419–27. doi: 10.1016/j.pain.2010.11.014
87. Jin X, Gereau RWT. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci (2006) 26:246–55. doi: 10.1523/JNEUROSCI.3858-05.2006
88. Wandrer F, Liebig S, Marhenke S, Vogel A, John K, Manns MP, et al. TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis (2020) 11:212. doi: 10.1038/s41419-020-2411-6
89. Feng Y, Teitelbaum DH. Tumour necrosis factor–induced loss of intestinal barrier function requires TNFR1 and TNFR2 signalling in a mouse model of total parenteral nutrition. J Physiol (2013) 591:3709–23. doi: 10.1113/jphysiol.2013.253518
90. Ledo C, Gonzalez CD, Poncini CV, Mollerach M, Gomez MI. TNFR1 Signaling Contributes to T Cell Anergy During Staphylococcus aureus Sepsis. Front Cell Infect Microbiol (2018) 8:259. doi: 10.3389/fcimb.2018.00259
91. Vandewalle J, Steeland S, Van Ryckeghem S, Eggermont M, Van Wonterghem E, Vandenbroucke RE, et al. A Study of Cecal Ligation and Puncture-Induced Sepsis in Tissue-Specific Tumor Necrosis Factor Receptor 1-Deficient Mice. Front Immunol (2019) 10:2574. doi: 10.3389/fimmu.2019.02574
92. Espirito Santo AI, Ersek A, Freidin A, Feldmann M, Stoop AA, Horwood NJ. Selective inhibition of TNFR1 reduces osteoclast numbers and is differentiated from anti-TNF in a LPS-driven model of inflammatory bone loss. Biochem Biophys Res Commun (2015) 464:1145–50. doi: 10.1016/j.bbrc.2015.07.094
93. Kotani J, Avallone NJ, Lin E, Goshima M, Lowry SF, Calvano SE. Tumor necrosis factor receptor regulation of bone marrow cell apoptosis during endotoxin-induced systemic inflammation. Shock (2006) 25:464–71. doi: 10.1097/01.shk.0000209544.22048.02
94. Ebach DR, Riehl TE, Stenson WF. Opposing effects of tumor necrosis factor receptor 1 and 2 in sepsis due to cecal ligation and puncture. Shock (2005) 23:311–8. doi: 10.1097/01.shk.0000157301.87051.77
95. Sousa Garcia D, Chen M, Smith AK, Lazarini PR, Lane AP. Role of the type I tumor necrosis factor receptor in inflammation-associated olfactory dysfunction. Int Forum Allergy Rhinol (2017) 7:160–8. doi: 10.1002/alr.21855
96. Zafiratos MT, Cottrell JT, Manam S, Henderson KK, Ramsey KH, Murthy AK. Tumor necrosis factor receptor superfamily members 1a and 1b contribute to exacerbation of atherosclerosis by Chlamydia pneumoniae in mice. Microbes Infect (2019) 21:104–8. doi: 10.1016/j.micinf.2018.09.003
97. Becke FM, Hehlgans T, Brockhoff G, Mannel DN. Development of allergic contact dermatitis requires activation of both tumor necrosis factor-receptors. Eur Cytokine Netw (2001) 12:45–50.
98. Nan J, Hu H, Sun Y, Zhu L, Wang Y, Zhong Z, et al. TNFR2 Stimulation Promotes Mitochondrial Fusion via Stat3- and NF-kB-Dependent Activation of OPA1 Expression. Circ Res (2017) 121:392–410. doi: 10.1161/CIRCRESAHA.117.311143
99. Kelly ML, Wang M, Crisostomo PR, Abarbanell AM, Herrmann JL, Weil BR, et al. TNF receptor 2, not TNF receptor 1, enhances mesenchymal stem cell-mediated cardiac protection following acute ischemia. Shock (2010) 33:602–7. doi: 10.1097/SHK.0b013e3181cc0913
100. Luo D, Luo Y, He Y, Zhang H, Zhang R, Li X, et al. Differential functions of tumor necrosis factor receptor 1 and 2 signaling in ischemia-mediated arteriogenesis and angiogenesis. Am J Pathol (2006) 169:1886–98. doi: 10.2353/ajpath.2006.060603
101. Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation (2004) 109:1892–7. doi: 10.1161/01.CIR.0000124227.00670.AB
102. Pircher J, Merkle M, Wornle M, Ribeiro A, Czermak T, Stampnik Y, et al. Prothrombotic effects of tumor necrosis factor alpha in vivo are amplified by the absence of TNF-alpha receptor subtype 1 and require TNF-alpha receptor subtype 2. Arthritis Res Ther (2012) 14:R225. doi: 10.1186/ar4064
103. Koniaris LG, Wand G, Wright TM. TNF mediates a murine model of Addison’s crisis. Shock (2001) 15:29–34. doi: 10.1097/00024382-200115010-00005
104. Fujita M, Ouchi H, Ikegame S, Harada E, Matsumoto T, Uchino J, et al. Critical role of tumor necrosis factor receptor 1 in the pathogenesis of pulmonary emphysema in mice. Int J Chron Obstruct Pulmon Dis (2016) 11:1705–12. doi: 10.2147/COPD.S108919
105. Gao H, Danzi MC, Choi CS, Taherian M, Dalby-Hansen C, Ellman DG, et al. Opposing Functions of Microglial and Macrophagic TNFR2 in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Cell Rep (2017) 18:198–212. doi: 10.1016/j.celrep.2016.11.083
106. Tortarolo M, Vallarola A, Lidonnici D, Battaglia E, Gensano F, Spaltro G, et al. Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J Neurochem (2015) 135:109–24. doi: 10.1111/jnc.13154
107. Shivakumar P, Mizuochi T, Mourya R, Gutta S, Yang L, Luo Z, et al. Preferential TNFalpha signaling via TNFR2 regulates epithelial injury and duct obstruction in experimental biliary atresia. JCI Insight (2017) 2:e88747. doi: 10.1172/jci.insight.88747
108. Ebach DR, Newberry R, Stenson WF. Differential role of tumor necrosis factor receptors in TNBS colitis. Inflammation Bowel Dis (2005) 11:533–40. doi: 10.1097/01.MIB.0000163698.34592.30
109. Wang K, Han G, Dou Y, Wang Y, Liu G, Wang R, et al. Opposite role of tumor necrosis factor receptors in dextran sulfate sodium-induced colitis in mice. PloS One (2012) 7:e52924. doi: 10.1371/journal.pone.0052924
110. Aspalter RM, Wolf HM, Eibl MM. Chronic TNF-alpha exposure impairs TCR-signaling via TNF-RII but not TNF-RI. Cell Immunol (2005) 237:55–67. doi: 10.1016/j.cellimm.2005.10.001
111. Li JQ, Xue H, Zhou L, Dong LH, Wei DP, Li H. Mechanism of fatty acid synthase in drug tolerance related to epithelial-mesenchymal transition of breast cancer. Asian Pac J Cancer Prev (2014) 15:7617–23. doi: 10.7314/APJCP.2014.15.18.7617
112. Morgan JA, Singhal G, Corrigan F, Jaehne EJ, Jawahar MC, Baune BT. TNF signalling via the TNF receptors mediates the effects of exercise on cognition-like behaviours. Behav Brain Res (2018) 353:74–82. doi: 10.1016/j.bbr.2018.06.036
113. Mohankrishnan A, Parmar R, Bhurani V, Dalai SK. Lack of TNF-alpha signaling through p55 makes the mice more susceptible to acute infection but does not alter state of latency and reactivation of HSV-1. Virus Res (2018) 244:1–5. doi: 10.1016/j.virusres.2017.11.004
114. Myles IA, Castillo CR, Barbian KD, Kanakabandi K, Virtaneva K, Fitzmeyer E, et al. Therapeutic Responses to Roseomonas mucosa in Atopic Dermatitis Involve Lipid-Mediated TNF-related Epithelial Repair. Sci Trans Med (2020) 12(560):eaaz8631. doi: 10.1126/scitranslmed.aaz8631
115. Chen CC, Pedraza PL, Hao S, Stier CT, Ferreri NR. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am J Physiol Renal Physiol (2010) 299:F1141–50. doi: 10.1152/ajprenal.00344.2010
116. Patel DC, Wallis G, Dahle EJ, McElroy PB, Thomson KE, Tesi RJ, et al. Hippocampal TNFalpha Signaling Contributes to Seizure Generation in an Infection-Induced Mouse Model of Limbic Epilepsy. eNeuro (2017) 4:ENEURO.0105–17.2017. doi: 10.1523/ENEURO.0105-17.2017
117. Madsen PM, Desu HL, de Rivero Vaccari JP, Florimon Y, Ellman DG, Keane RW, et al. Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav Immun (2020) 84:132–46. doi: 10.1016/j.bbi.2019.11.017
118. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype inflammatory environment. J Immunol (2013) 190:1076–84. doi: 10.4049/jimmunol.1202659
119. Weinberger JF, Raison CL, Rye DB, Montague AR, Woolwine BJ, Felger JC, et al. Inhibition of tumor necrosis factor improves sleep continuity in patients with treatment resistant depression and high inflammation. Brain Behav Immun (2015) 47:193–200. doi: 10.1016/j.bbi.2014.12.016
120. Morais BS, Teixeira AL, Maciel JF, Lima AA, Barbosa IG, Sanches MD. Soluble tumor necrosis factor receptors as predictors of 1-year mortality and renal dysfunction after liver transplantation. Transpl Immunol (2016) 35:29–33. doi: 10.1016/j.trim.2016.01.006
121. Lopatnikova JA, Alshevskaya AA, Krugleeva OL, Nepomnyschih VM, Gladkikh VS, Lukinov VL, et al. Expression of TNFalpha Receptors on Immunocompetent Cells Is Increased in Atopic Dermatitis. Int Arch Allergy Immunol (2017) 174:151–60. doi: 10.1159/000481135
122. Ferreira GA, Teixeira AL, Calderaro DC, Sato EI. Atorvastatin reduced soluble receptors of TNF-alpha in systemic lupus erythematosus. Clin Exp Rheumatol (2016) 34:42–8.
123. Chen B, Tong Z, Ye Q, Nakamura S, Costabel U, Guzman J. Expression of tumour necrosis factor receptors by bronchoalveolar cells in hypersensitivity pneumonitis. Eur Respir J (2005) 25:1039–43. doi: 10.1183/09031936.05.00084704
124. Proudfoot A, Bayliffe A, O’Kane CM, Wright T, Serone A, Bareille PJ, et al. Novel anti-tumour necrosis factor receptor-1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax (2018) 73:723–30. doi: 10.1136/thoraxjnl-2017-210305
125. Wandell P, Carlsson AC, Larsson A, Melander O, Wessman T, Arnlov J, et al. TNFR1 is associated with short-term mortality in patients with diabetes and acute dyspnea seeking care at the emergency department. Acta Diabetol (2020) 57(10):1145–50. doi: 10.1007/s00592-020-01527-3
126. Fernandez-Juarez G, Villacorta Perez J, Luno Fernandez JL, Martinez-Martinez E, Cachofeiro V, Barrio Lucia V, et al. High levels of circulating TNFR1 increase the risk of all-cause mortality and progression of renal disease in type 2 diabetic nephropathy. Nephrol (Carlton) (2017) 22:354–60. doi: 10.1111/nep.12781
127. Kamei N, Yamashita M, Nishizaki Y, Yanagisawa N, Nojiri S, Tanaka K, et al. Association between circulating tumor necrosis factor-related biomarkers and estimated glomerular filtration rate in type 2 diabetes. Sci Rep (2018) 8:15302. doi: 10.1038/s41598-018-33590-w
128. Vendrell J, Broch M, Fernandez-Real JM, Gutierrez C, Simon I, Megia A, et al. Tumour necrosis factor receptors (TNFRs) in Type 2 diabetes. Analysis of soluble plasma fractions and genetic variations of TNFR2 gene in a case-control study. Diabetes Med (2005) 22:387–92. doi: 10.1111/j.1464-5491.2004.01392.x
129. Choi SW, Stiff P, Cooke K, Ferrara JL, Braun T, Kitko C, et al. TNF-inhibition with etanercept for graft-versus-host disease prevention in high-risk HCT: lower TNFR1 levels correlate with better outcomes. Biol Blood Marrow Transplant (2012) 18:1525–32. doi: 10.1016/j.bbmt.2012.03.013
130. Jarosz-Griffiths HH, Holbrook J, Lara-Reyna S, McDermott MF. TNF receptor signalling in autoinflammatory diseases. Int Immunol (2019) 31:639–48. doi: 10.1093/intimm/dxz024
131. Ghods A, Ghaderi A, Shariat M, Talei AR, Mehdipour F. TNFR2 but not TNFR1 is the main TNFR expressed by B and T lymphocytes in breast cancer draining lymph nodes. Immunol Lett (2019) 209:36–44. doi: 10.1016/j.imlet.2019.03.013
132. Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med (2011) 365:1502–8. doi: 10.1056/NEJMoa1100721
133. Tsukerman P, Eisenstein EM, Chavkin M, Schmiedel D, Wong E, Werner M, et al. Cytokine secretion and NK cell activity in human ADAM17 deficiency. Oncotarget (2015) 6:44151–60. doi: 10.18632/oncotarget.6629
134. Baral P, Udit S, Chiu IM. Pain and immunity: implications for host defence. Nat Rev Immunol (2019) 19:433–47. doi: 10.1038/s41577-019-0147-2
135. Bradford EM, Ryu SH, Singh AP, Lee G, Goretsky T, Sinh P, et al. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J Immunol (2017) 199:1886–97. doi: 10.4049/jimmunol.1601066
136. Kleijwegt FS, Laban S, Duinkerken G, Joosten AM, Zaldumbide A, Nikolic T, et al. Critical role for TNF in the induction of human antigen-specific regulatory T cells by tolerogenic dendritic cells. J Immunol (2010) 185:1412–8. doi: 10.4049/jimmunol.1000560
137. Zou H, Li R, Hu H, Hu Y, Chen X. Modulation of Regulatory T Cell Activity by TNF Receptor Type II-Targeting Pharmacological Agents. Front Immunol (2018) 9:594. doi: 10.3389/fimmu.2018.00594
138. Felix J, Savvides SN. Mechanisms of immunomodulation by mammalian and viral decoy receptors: insights from structures. Nat Rev Immunol (2017) 17:112–29. doi: 10.1038/nri.2016.134
139. Smith CA, Davis T, Wignall JM, Din WS, Farrah T, Upton C, et al. T2 open reading frame from the Shope fibroma virus encodes a soluble form of the TNF receptor. Biochem Biophys Res Commun (1991) 176:335–42. doi: 10.1016/0006-291X(91)90929-2
140. Schreiber M, McFadden G. The myxoma virus TNF-receptor homologue (T2) inhibits tumor necrosis factor-alpha in a species-specific fashion. Virology (1994) 204:692–705. doi: 10.1006/viro.1994.1585
141. Sedger LM, Osvath SR, Xu XM, Li G, Chan FK, Barrett JW, et al. Poxvirus tumor necrosis factor receptor (TNFR)-like T2 proteins contain a conserved preligand assembly domain that inhibits cellular TNFR1-induced cell death. J Virol (2006) 80:9300–9. doi: 10.1128/JVI.02449-05
142. Graham SC, Bahar MW, Abrescia NG, Smith GL, Stuart DI, Grimes JM. Structure of CrmE, a virus-encoded tumour necrosis factor receptor. J Mol Biol (2007) 372:660–71. doi: 10.1016/j.jmb.2007.06.082
143. Pontejo SM, Alejo A, Alcami A. Comparative Biochemical and Functional Analysis of Viral and Human Secreted Tumor Necrosis Factor (TNF) Decoy Receptors. J Biol Chem (2015) 290:15973–84. doi: 10.1074/jbc.M115.650119
144. Smith CA, Hu FQ, Smith TD, Richards CL, Smolak P, Goodwin RG, et al. Cowpox virus genome encodes a second soluble homologue of cellular TNF receptors, distinct from CrmB, that binds TNF but not LT alpha. Virology (1996) 223:132–47. doi: 10.1006/viro.1996.0462
145. Saraiva M, Alcami A. CrmE, a novel soluble tumor necrosis factor receptor encoded by poxviruses. J Virol (2001) 75:226–33. doi: 10.1128/JVI.75.1.226-233.2001
146. Yang Z, West A, Bjorkman PJ. Crystal structure of TNFalpha complexed with a poxvirus MHC-related TNF binding protein. Nat Struct Mol Biol (2009) 16:1189–91. doi: 10.1038/nsmb.1683
147. Haga S, Nagata N, Okamura T, Yamamoto N, Sata T, Yamamoto N, et al. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antiviral Res (2010) 85:551–5. doi: 10.1016/j.antiviral.2009.12.001
148. Gerriets V, Bansal P, Goyal A, Khaddour K. Tumor Necrosis Factor (TNF) Inhibitors. Treasure Island (FL): StatPearls Publishing (2020).
149. Genoves P, Garcia D, Cejalvo D, Martin A, Zaragoza C, Toledo AH, et al. Pentoxifylline in liver ischemia and reperfusion. J Invest Surg (2014) 27:114–24. doi: 10.3109/08941939.2013.835454
150. Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatol (Oxford) (2010) 49:1215–28. doi: 10.1093/rheumatology/keq031
151. Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol (2009) 131:308–16. doi: 10.1016/j.clim.2009.01.002
152. Haack M, Pollmacher T, Mullington JM. Diurnal and sleep-wake dependent variations of soluble TNF- and IL-2 receptors in healthy volunteers. Brain Behav Immun (2004) 18:361–7. doi: 10.1016/j.bbi.2003.12.009
153. Alessandri AL, Souza AL, Oliveira SC, Macedo GC, Teixeira MM, Teixeira AL. Concentrations of CXCL8, CXCL9 and sTNFR1 in plasma of patients with pulmonary tuberculosis undergoing treatment. Inflammation Res (2006) 55:528–33. doi: 10.1007/s00011-006-5136-9
154. Laske C, Oschmann P, Tofighi J, Kuehne SB, Diehl H, Bregenzer T, et al. Induction of sTNF-R1 and sTNF-R2 by interferon beta-1b in correlation with clinical and MRI activity. Acta Neurol Scand (2001) 103:105–13. doi: 10.1034/j.1600-0404.2001.103002105.x
155. Coelho FM, Reis HJ, Nicolato R, Romano-Silva MA, Teixeira MM, Bauer ME, et al. Increased serum levels of inflammatory markers in chronic institutionalized patients with schizophrenia. Neuroimmunomodulation (2008) 15:140–4. doi: 10.1159/000148197
156. Jiang H, Hampel H, Prvulovic D, Wallin A, Blennow K, Li R, et al. Elevated CSF levels of TACE activity and soluble TNF receptors in subjects with mild cognitive impairment and patients with Alzheimer’s disease. Mol Neurodegener (2011) 6:69. doi: 10.1186/1750-1326-6-69
157. Fernandez-Real JM, Gutierrez C, Ricart W, Castineira MJ, Vendrell J, Richart C. Plasma levels of the soluble fraction of tumor necrosis factor receptors 1 and 2 are independent determinants of plasma cholesterol and LDL-cholesterol concentrations in healthy subjects. Atherosclerosis (1999) 146:321–7. doi: 10.1016/S0021-9150(99)00156-2
158. Yamada Y, Endo S, Inada K, Nakae H, Nasu W, Taniguchi S, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptor I, II levels in patients with severe burns. Burns (2000) 26:239–44. doi: 10.1016/S0305-4179(99)00137-0
159. Bendsen NT, Stender S, Szecsi PB, Pedersen SB, Basu S, Hellgren LI, et al. Effect of industrially produced trans fat on markers of systemic inflammation: evidence from a randomized trial in women. J Lipid Res (2011) 52:1821–8. doi: 10.1194/jlr.M014738
160. Carlsson AC, Ruge T, Kjoller E, Hilden J, Kolmos HJ, Sajadieh A, et al. 10-Year Associations Between Tumor Necrosis Factor Receptors 1 and 2 and Cardiovascular Events in Patients With Stable Coronary Heart Disease: A CLARICOR (Effect of Clarithromycin on Mortality and Morbidity in Patients With Ischemic Heart Disease) Trial Substudy. J Am Heart Assoc (2018) 7. doi: 10.1161/JAHA.117.008299
161. Caldarola G, De Simone C, Carbone A, Tulli A, Amerio P, Feliciani C. TNFalpha and its receptors in psoriatic skin, before and after treatment with etanercept. Int J Immunopathol Pharmacol (2009) 22:961–6. doi: 10.1177/039463200902200411
162. Minozzi S, Bonovas S, Lytras T, Pecoraro V, Gonzalez-Lorenzo M, Bastiampillai AJ, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Saf (2016) 15:11–34. doi: 10.1080/14740338.2016.1240783
163. Olszanecka-Glinianowicz M, Zahorska-Markiewicz B, Janowska J, Zurakowski A. Serum concentrations of nitric oxide, tumor necrosis factor (TNF)-alpha and TNF soluble receptors in women with overweight and obesity. Metabolism (2004) 53:1268–73. doi: 10.1016/j.metabol.2004.07.001
164. Ribeiro-Samora GA, Rabelo LA, Ferreira ACC, Favero M, Guedes GS, Pereira LSM, et al. Inflammation and oxidative stress in heart failure: effects of exercise intensity and duration. Braz J Med Biol Res (2017) 50:e6393. doi: 10.1590/1414-431x20176393
165. Coelho FM, Narciso FM, Oliveira DM, Pereira DS, Teixeira AL, Teixeira MM, et al. sTNFR-1 is an early inflammatory marker in community versus institutionalized elderly women. Inflammation Res (2010) 59:129–34. doi: 10.1007/s00011-009-0079-6
166. Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF Receptor-Associated Periodic Syndrome (TRAPS): Current Perspectives. Int J Mol Sci (2020) 21:3263. doi: 10.3390/ijms21093263
167. Gaggiano C, Vitale A, Obici L, Merlini G, Soriano A, Viapiana O, et al. Clinical Features at Onset and Genetic Characterization of Pediatric and Adult Patients with TNF-alpha Receptor-Associated Periodic Syndrome (TRAPS): A Series of 80 Cases from the AIDA Network. Mediators Inflammation (2020) 2020:8562485. doi: 10.1155/2020/8562485
Keywords: epithelial to mesenchymal transition, NF-kappa B, signaling/signaling pathways, TNF, TNF receptor, TNF blockade
Citation: Gough P and Myles IA (2020) Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 11:585880. doi: 10.3389/fimmu.2020.585880
Received: 21 July 2020; Accepted: 02 November 2020;
Published: 25 November 2020.
Edited by:
Annalisa Del Prete, University of Brescia, ItalyReviewed by:
Aristine Cheng, National Taiwan University, TaiwanSamuel Lara-Reyna, University of Birmingham, United Kingdom
Copyright © 2020 Gough and Myles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Portia Gough, cG9ydGlhLmdvdWdoQG5paC5nb3Y=