 Scott R. Barnum
Scott R. Barnum Doryen Bubeck2
Doryen Bubeck2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 November 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.585108
This article is part of the Research Topic Perforins and Cholesterol-Dependent Cytolysins in Immunity and Pathogenesis View all 18 articles
The soluble membrane attack complex (sMAC, a.k.a., sC5b-9 or TCC) is generated on activation of complement and contains the complement proteins C5b, C6, C7, C8, C9 together with the regulatory proteins clusterin and/or vitronectin. sMAC is a member of the MACPF/cholesterol-dependent-cytolysin superfamily of pore-forming molecules that insert into lipid bilayers and disrupt cellular integrity and function. sMAC is a unique complement activation macromolecule as it is comprised of several different subunits. To date no complement-mediated function has been identified for sMAC. sMAC is present in blood and other body fluids under homeostatic conditions and there is abundant evidence documenting changes in sMAC levels during infection, autoimmune disease and trauma. Despite decades of scientific interest in sMAC, the mechanisms regulating its formation in healthy individuals and its biological functions in both health and disease remain poorly understood. Here, we review the structural differences between sMAC and its membrane counterpart, MAC, and examine sMAC immunobiology with respect to its presence in body fluids in health and disease. Finally, we discuss the diagnostic potential of sMAC for diagnostic and prognostic applications and potential utility as a companion diagnostic.
The complement system is the most complex of the immunological and hematological pathways in human biology. Composed of ~50 proteins, four activation pathways (classical, lectin, alternative, and extrinsic) and a terminal lytic pathway, it is an important part of both innate and adaptive immune responses (1–3). Complement-mediated immune effector functions include chemoattraction of immune cells, activation of leukocytes, platelets and essentially all cell types proximal to complement activation, opsonization of invading pathogens, enhancement of the acute-phase response, lysis of susceptible pathogens and modulation of lymphocyte-mediated immune responses (1, 2, 4–6). Complement also serves to help in controlling T and B cell activation and function, stem cells and developmental processes, modulate basic cellular processes in intracellular sensing and cellular metabolism as it relates to immune responses (7–15), synaptic pruning (16, 17), modulation of the circadian clock (18), and possible contributions to schizophrenia (19, 20). Effector functions mediated by complement are driven by the proteolytic generation of activation fragments that either 1) bind to receptors expressed on both immune and non-immune cells, or 2) covalently attach to cell surfaces adjacent to sites of complement activation (1–3). These activities are tightly controlled by more than a dozen fluid-phase and membrane-bound regulatory molecules whose function is to keep complement activation in proportion to the amount of activator present and to limit damage to host tissues (1, 21, 22). Additional non-canonical roles of complement are discussed in a series published in Seminars in Immunology (23).
Activation of complement liberates more functional polypeptide fragments of various molecular species than any other immunological or hematological pathway. For example, activation of factor B releases Bb, a serine protease and key component of the alternative pathway C3 and C5 convertases, and Ba, a small polypeptide composed of three shushi domains with no known biological function. In contrast, cleavage of C3 and C4 generates C3a and C4a, respectively, which are small (~10 kDa) fragments that possess a wide range of functions including chemoattraction, antimicrobial activity, and modulation of T cell responses [reviewed in (6, 24)]. In addition, C3 and C4 cleavage produces multiple polypeptides from the larger ‘b’ fragments which are equally diverse in function (1, 2, 25). Enzymatic activity of complement serine proteases is responsible for production of at least a dozen activation fragments. The soluble membrane attack complex (sMAC) is an exception to the production of functionally-active polypeptide fragments. Generated by activation of the complement pathways, the formation of sMAC in the fluid-phase starts with the cleavage of C5 by C5 convertases, to C5a and C5b (Figure 1A). The addition of C6, C7, C8, C9 to C5b forms a basic MAC structure, which associates with the regulatory proteins clusterin and/or vitronectin, to form a soluble MAC complex inhibited from inserting into lipid bilayers (26–33). sMAC may have one to three C9 molecules and can bind one to two clusterin or vitronectin molecules, or a combination of clusterin and vitronectin molecules (Figure 1B). Thus, sMAC is not a single molecular species, but a family of closely related multi-molecular complexes. Based on this stoichiometry, at least fifteen different sMAC complexes are possible. Since each of the protein subunits in sMAC have polymorphic variants (34–43), there are many sMAC variants at the population level (similar to polymorphism at the population level for MHC molecules). The biological roles of these sMAC species in homeostatic conditions and disease pathophysiology are undefined. In contrast, studies in recent years have demonstrated that the MAC contributes to intracellular signaling, inflammation, and other functions (44–47).
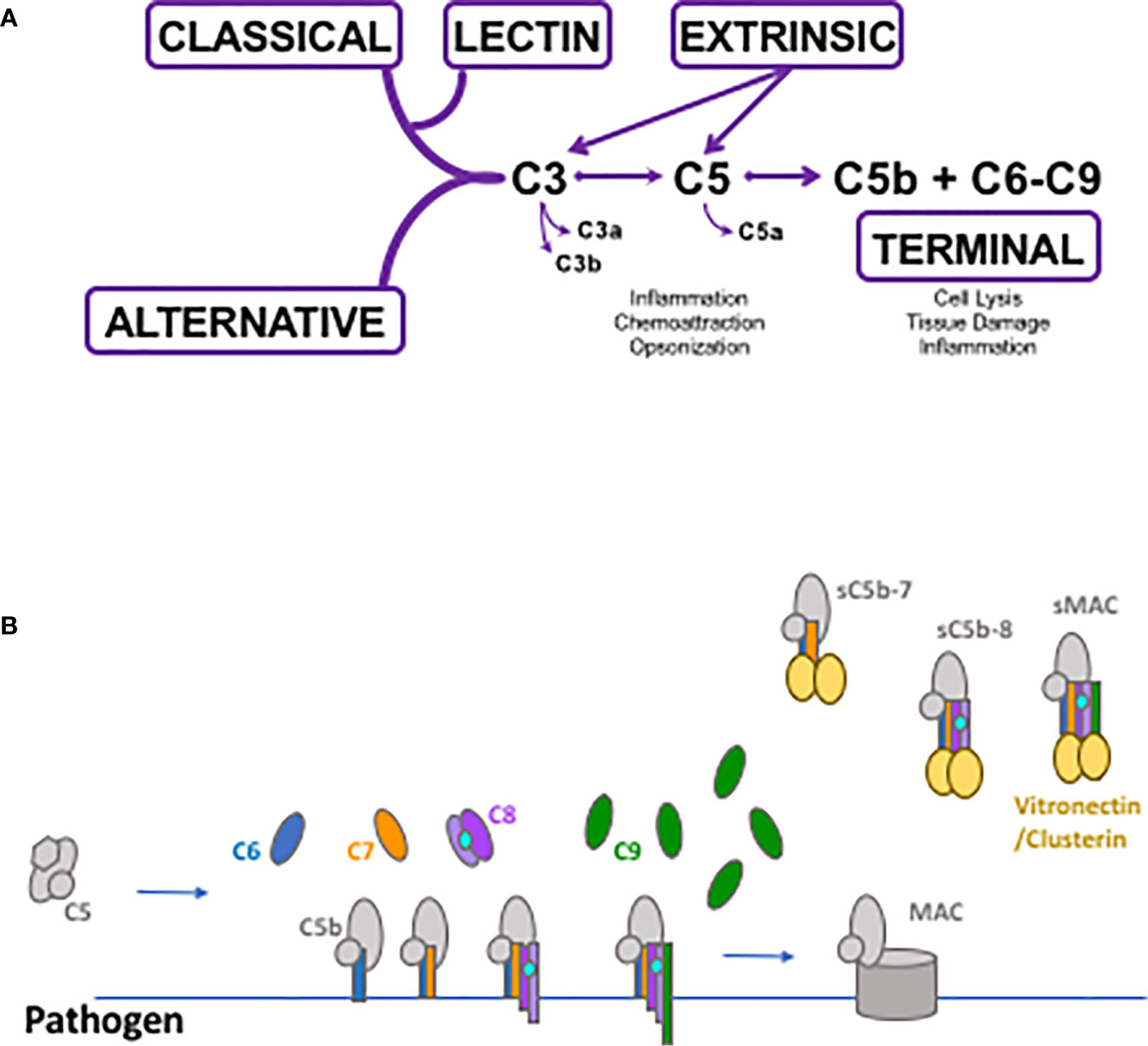
Figure 1 Schematic depicting complement activation and soluble membrane attack complex (sMAC) and MAC formation. (A) The classical, lectin and alternative pathways generate C3 and C5 convertases that cleave C3 and C5 into their biologically active fragments. Direct cleavage of C3 and C5 occurs through the extrinsic protease pathway which utilizes several enzymes of the coagulation system such as activated thrombin and plasmin and others. Activation through any of the pathways can generate C5b which initiates the formation of MAC and sMAC through the terminal pathway. (B) Schematic of MAC formation on a pathogen surface. Generation of C5b as a result of complement activation allows the non-covalent association of C6 through C9 and the production of the pore-forming membrane attack complex. Simultaneously with MAC formation, C5b in the fluid-phase can associate with C6 through C9 forming soluble intermediates leading to sMAC generation. All of the soluble intermediates and sMAC associate with vitronectin and/or clusterin preventing their insertion into pathogen or human cell membranes.
Several intermediates leading to the formation of sMAC and MAC have been well characterized biochemically (Table 1). Cryo electron microscopy structures of sMAC (33) and MAC (33, 52, 53) suggests a similar overall arrangement of complement proteins within the complex (Figure 2A). In both complexes, C5b serves as a structural scaffold that organizes C6, C7, C8 and C9 into an arc through their pore-forming membrane attack complex perforin (MACPF) domains. During MAC formation, the core MACPF domains of C6, C7 C8 and C9 undergo a dramatic structural rearrangement in which two helical bundles unfurl to form a pair of β-hairpins that insert into the lipid bilayer. While it remains unclear from the low-resolution sMAC structure if these transmembrane-hairpins domains have unfurled, both complexes are of a similar length suggesting that at least some of sMAC β-hairpins maybe extended (Figure 2B). Negative stain electron microscopy images of vitronectin-labeled sMAC suggest that chaperones may bind to exposed hydrophobic hairpins (29), however, the molecular details of how clusterin and vitronectin prevent membrane insertion of sMAC are still unresolved. In MAC, the helix-to-hairpin transition of membrane-interacting residues exposes a charged surface of the MACPF. Charge complementarity between MACPF-MACPF interfaces is one of main factors that determines the direction of MAC assembly, and likely plays a similar role in formation of sMAC (53). The non-pore forming domains of complement proteins act as regulatory auxiliary modules, preventing the premature release of transmembrane β-hairpins during MAC assembly. How these regulatory domains are oriented in sMAC remains to be seen. High resolution structures of inhibited MAC complexes will be necessary to understand how regulators, such as vitronectin and clusterin, block membrane association of MAC.
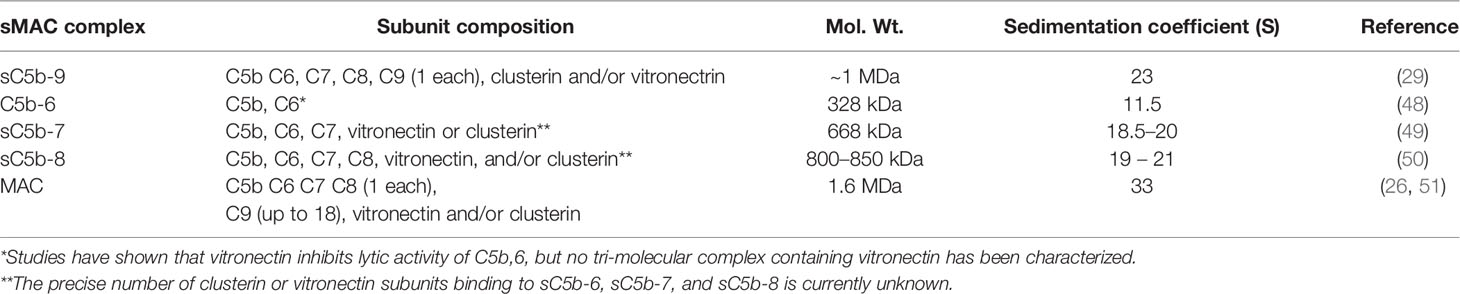
Table 1 Physicochemical parameters of soluble membrane attack complex (sMAC) and related complexes.
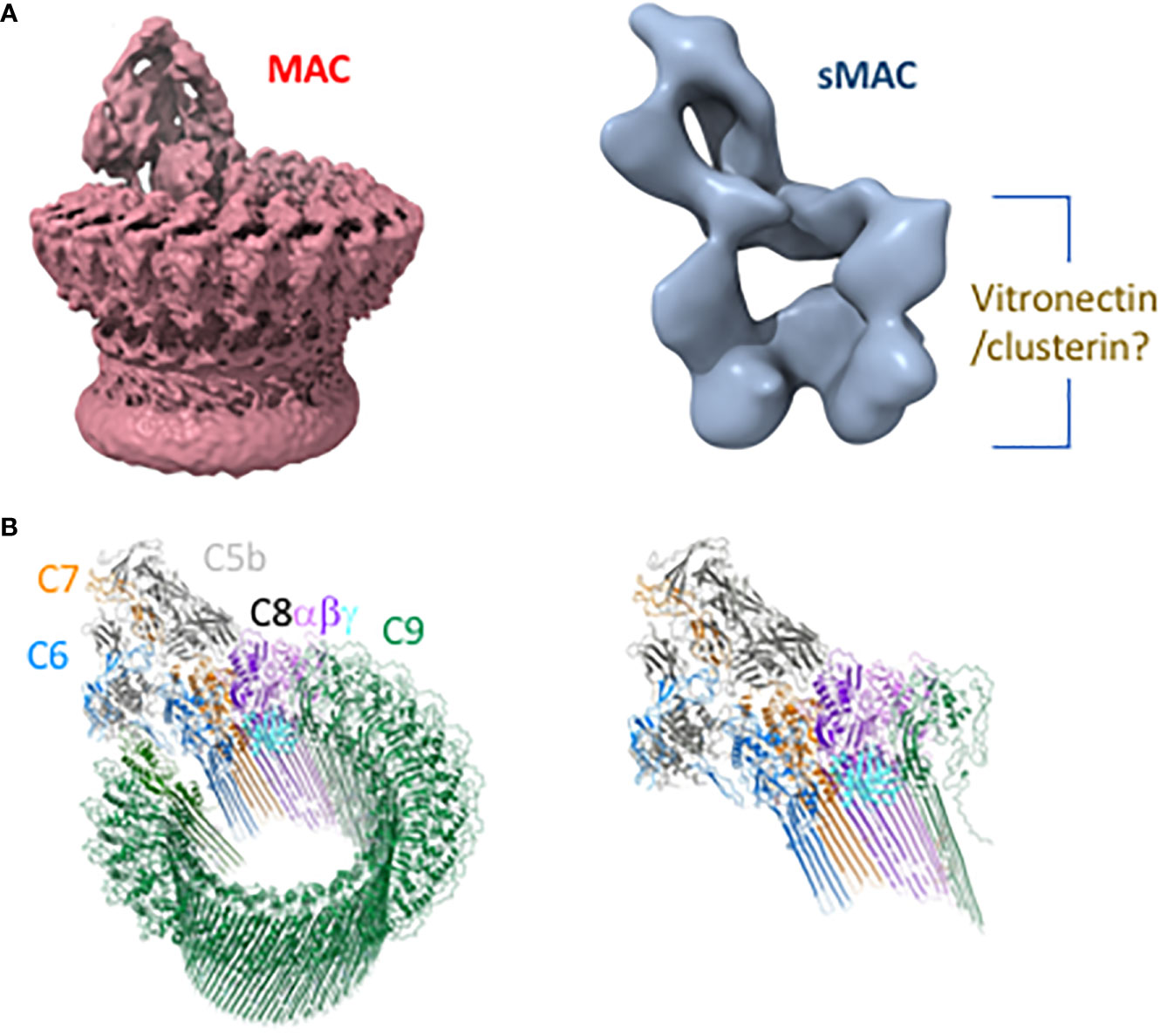
Figure 2 Structures of membrane attack complex (MAC) and soluble MAC (sMAC). (A) CryoEM reconstruction of MAC at 4.9 Å resolution (EMD-0110) (53) shows a ring-like arrangement of complement proteins that comprise MAC (left). By contrast, the cryoEM reconstruction of sMAC at 24 Å resolution (EMD-1991) (33) reveals that the ring is stopped short by chaperones vitronectin and clusterin to form an arc (right). Vitronectin and clusterin may act to cap the arc and/or bind exposed hydrophobic residues of unfurled beta-hairpins. (B) The left depicts the MAC (PDB-6H04) as a ribbon diagram in which complement proteins are colored: C5b (gray), C6 (blue), C7 (orange), C8β (dark purple), C8α (light purple), C8γ (cyan), and C9 oligomer (green). On the right is a single copy of each protein that may fill the arc of density depicted in the low-resolution sMAC structure.
sMAC was first quantitated in plasma in healthy individuals almost 50 years ago (54). Numerous studies since that time have documented the presence of sMAC in most body fluids as discussed below. Although sMAC is present in many of these fluids, the mechanism(s) that generate tissue-specific, basal sMAC levels have received little attention. The continuous activation of complement at low levels through C3 tickover, first described in the early 1970s (55, 56), likely contributes to sMAC generation. In addition to tickover, it is well established that the coagulation system, like complement, is continuously active at a basal level, and thus basal activation of the coagulation and fibrinolytic systems may also contribute to sMAC generation (57–61). This mechanism of complement activation is known as the extrinsic pathway, and it bypasses convertase formation and directly activates C3 and C5 [reviewed in (62, 63)]. The relative contribution of each of these mechanisms and any others that may be involved in basal sMAC generation remains to be established. Once complement is activated however, the high plasma concentration of vitronectin (200–400 μg/ml, (64) and clusterin [150–540 μg/ml, (65)] relative to C9 [~50–60 μg/ml, (66)] suggests formation of sMAC is favored, particularly since both regulatory proteins are elevated in concentration in the acute phase response (67, 68). Mathematical modeling supports this possibility revealing that sMAC is generated rapidly on activation of the classical and alternative pathways reaching peak concentration within 15 min (69). In contrast, MAC production and deposition on pathogens surfaces is characterized by a lag phase of ~20 min, followed by rapid production and deposition that peaks after 50 min (70).
Timing is important for the formation of functional MAC pores and could tip the balance to sMAC production. Rapid over-activation of the complement terminal pathway may overwhelm the C5 convertase. In addition to the proteolytic cleavage of C5, the C5 convertase also plays an important role in orienting MAC assembly precursors at the membrane (71). Improperly inserted precursors could then be scavenged by fluid-phase vitronectin and clusterin, to produce sMAC. On a bacterial surface, convertase generated C5b6 must rapidly recruit C7 to form functional MAC (72). If there is a delay in availability of C7, the inert C5b7 complex could be scavenged by clusterin and vitronectin. Indeed, C5b7 is the first MAC intermediate to bind these two chaperones.
Although sMAC was detected in plasma several decades ago (54), the first quantitative assays based on sMAC neo-epitopes were not developed until the mid-1980’s (73, 74). sMAC levels of plasma or serum were frequently measured with in-house assays using serum activated zymosan or inulin as a standard control. These standards and assays were not well characterized and interpretation of the results were complicated by variable assay sensitivity, sample handling, and sample storage (73–77) as highlighted in recent study by Yang and colleagues (78). The International Complement Standardization Committee has since defined an activation standard for quantitation of complement activation products, including sMAC, termed International complement standard #2 (79). International complement standard #2 was derived from healthy donor-derived pooled serum activated with heat-aggregated IgG and zymosan. The utility of this standard (defined as complement activation units, CAU) is limited by variability in pooled serum between different donor cohorts and the reagents used to activate complement. The relationship of CAU to standard measures of protein concentration remains poorly defined. However, immunoassays using purified sMAC as a reagent for generating quantitative standard curves overcomes these limitations.
A recent study using a sMAC ELISA (Quidel, Corp., San Diego, CA) reported mean plasma and serum levels of sMAC of 121 ± 3.7 ng/ml (n=199) and 175 ± 8.1 ng/ml (n=49) respectively, in healthy adult donors (80). Plasma sMAC levels were similar between African-Americans and Caucasians and between males and females. Interestingly, plasma sMAC levels in individuals above 50 years of age were significantly higher than those in their 40’s and younger. The levels of sMAC in neonates have been measured in cord blood plasma samples obtained immediately after birth and were markedly lower than adult levels (81). This study, and a number of others, have shown that terminal pathway proteins comprising the MAC (C6-C9) are significantly lower in pre-term and full term infants, as are proteins of the classical and alterative pathways [reviewed in (82)]. In addition, a recent study determined that the blood levels of C9 in children less than one year of age were significantly lower compared to adults, and adult levels were reached between two and eighteen years of age (83). These studies indicate that sMAC levels in are lower in children, in part, because the concentration of proteins that compose the MAC are lower. Nonetheless, sMAC levels in children increase during infection, and activation of complement in neonatal serum by cobra venom factor also increased sMAC levels (81). Additional studies to determine sMAC blood levels in healthy adults and children are warranted to determine the value of sMAC as a diagnostic and prognostic tool in disease settings.
The sMAC in plasma and serum has been measured in many clinical settings including infectious and autoimmune disease, transplantation, trauma, and complement deficiencies (Table 2). The level of sMAC increases in these conditions in a disease-dependent fashion. However, an encompassing generalization regarding the magnitude and kinetics of the responses is not possible due to the variability in assay types used to quantify sMAC, and the baseline differences of sMAC concentration between serum and plasma. For this reason, we have not included the level of sMAC for the diseases and conditions listed in Tables 2–4. In vivo studies in rabbits have demonstrated that sMAC is eliminated with a half-life of 30–50 min (170), but no half-life studies have been reported for human sMAC. It is clear the diagnostic and prognostic value of sMAC in blood requires assay and sample handling standardization, particularly as complement therapeutics move into the clinical treatment repertoire (171).

Table 2 Soluble membrane attack complex (sMAC) changes in blood, plasma, and serum in various clinical conditions.
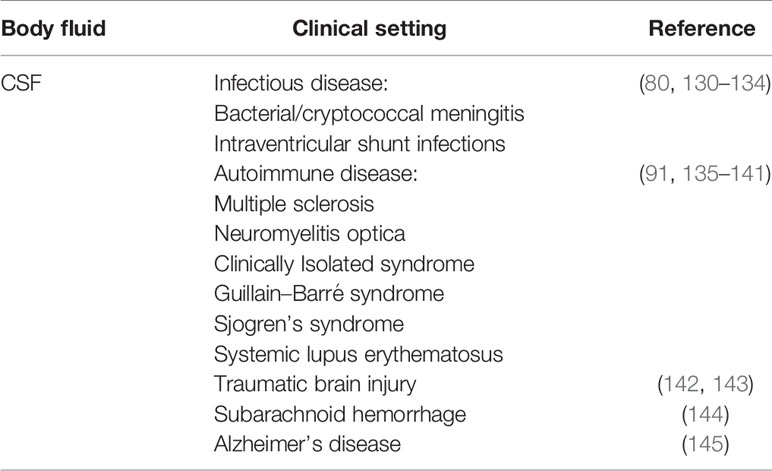
Table 3 Soluble membrane attack complex (sMAC) changes in cerebrospinal fluid (CSF) in various clinical conditions.
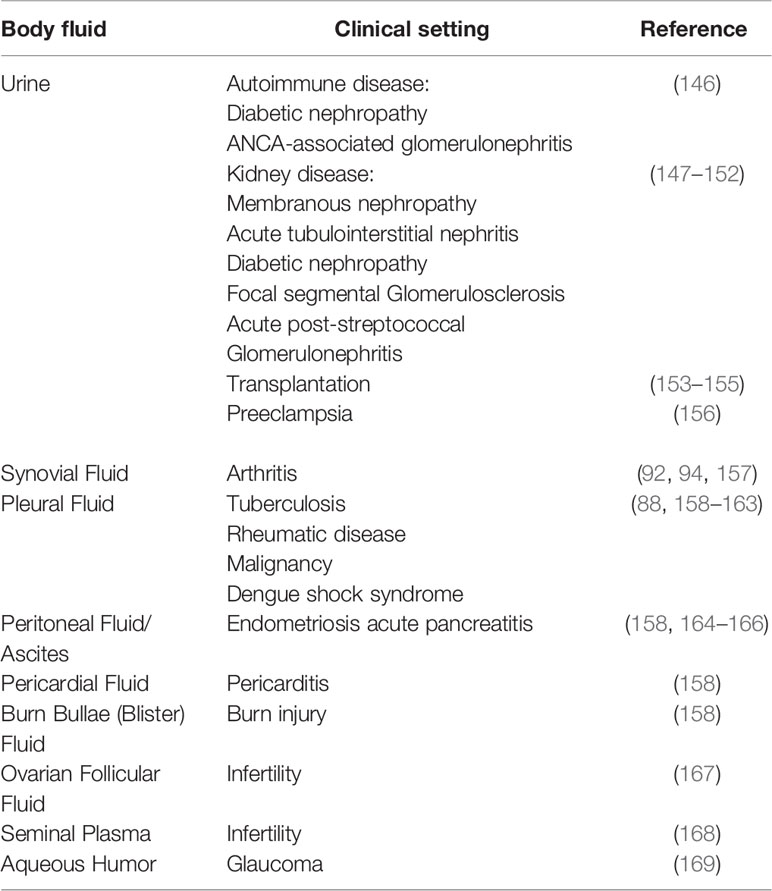
Table 4 Mucosal and synovial soluble membrane attack complex (sMAC) changes in various clinical conditions.
The normal range of sMAC concentration in cerebrospinal fluid (CSF) of healthy individuals has not been established, in part, because of the clinical risk and discomfort surrounding procuring CSF via lumbar puncture. As a result, normal levels of sMAC in CSF have frequently been derived from cohorts with “other neurological diseases” or from patients who underwent lumbar puncture as a part of standard clinical care and had negative bacterial cultures. In most studies using the Quidel sC5b-9 ELISA, CSF sMAC levels range from undetectable to the low nanogram/ml range (10–20 ng/ml) (130, 142, 144, 172, 173). Studies analyzing the sMAC CSF/serum quotient using Reiber–Felgenhauer nomograms of IgM suggest that sMAC is intrathecally produced rather than diffusing across the blood brain barrier (BBB) as has been shown for C9 (173, 174). There are exceptions to this low normal range. For example, Aly and colleagues reported mean levels of sMAC CSF to be ~50 ng/ml in neonates with hypoxic-ischemic encephalopathy (175). The reasons for this higher level are unclear, but may be due to developmentally-reduced integrity of the BBB shortly after birth, to the elevated level of plasma proteins found in neonatal CSF compared to adults, or to the transport of plasma proteins across choroid plexus epithelial cells in fetal and neonatal brain [reviewed in (176)]. It is also possible that CSF sMAC in neonates is generated as a result of complement-mediated synaptic pruning (16) during neurodevelopment, which is subsequently cleared postnatally. Other studies have reported sMAC concentrations in control groups range from high ng/ml to low μg/ml levels (131, 135, 136). Although non-standardized, in-house sMAC assays were used many of these studies, one likely reason for the high sMAC levels in the control groups was the inclusion of patients with tumors, Huntington’s disease, stroke, seizure disorder, cerebellar degeneration, progressive supranuclear palsy, or undetermined infections, which are, at least in part, inflammatory in nature.
Despite the contrasting reports on the levels of CSF sMAC in healthy individuals, it is clear the levels increase in a number of pathological conditions. Table 3 lists a number of neurological diseases in which sMAC increases relative to levels in other neurological diseases. In bacterial meningitis and shunt infections, sMAC levels have been reported to increase compared to uninfected controls (130, 132–134). In shunt infections, the increase in sMAC was remarkably high (over 100-fold) compared to control CSF (130). Similar dramatic changes in sMAC levels have been reported for traumatic brain injury (as high as 1,800-fold) and subarachnoid hemorrhage (~200-fold) compared to control CSF (142–144). sMAC concentration of this magnitude in CSF suggests its production is derived through multiple mechanisms and sources including: 1) increased intrathecal complement production and activation, 2) blood-derived sMAC leaking across a compromised BBB, and 3) in situ generation of sMAC at injury site(s). Interestingly, admixture experiments using human CSF and serum demonstrated that sMAC could be generated in a dose-dependent fashion (up to 5-fold over CSF alone) (144). However, there are cases where sMAC levels are not elevated in infectious or other pathological conditions. For example, in viral and fungal infections, CSF sMAC levels do not increase or increase minimally (131). In idiopathic normal pressure hydrocephalus, a disorder characterized by faulty CSF mechanical dynamics and associated neurodegeneration and inflammation (177), median sMAC levels were a low ~13 ng/ml (173). The reason for the differences in sMAC levels in these latter pathological conditions is unclear, but if verified by additional studies, they could provide differential diagnostic opportunities.
In central nervous system, autoimmune diseases such as multiple sclerosis (MS), Guillain-Barre syndrome, Sjogren’s syndrome and systemic lupus erythematosus, there have been reports of increased levels of sMAC (91, 135–138). However, other MS studies have reported no increases in sMAC (139, 178). Studies also present conflicting findings for sMAC levels in neuromyelitis optica (91, 139, 178). Clinically isolated syndrome, a neurodegenerative disease reminiscent of MS (179), has also been examined for changes in CSF sMAC levels. Although sMAC has been detected, the levels do not appear to increase in a clinically meaningful way, but the number of studies is limited (140, 141). There are several other central nervous system diseases where the MAC contributes to disease pathogenesis and, by extension, sMAC levels may change during the course of disease progression. These include epilepsy (180), Parkinson’s disease (181) amyotrophic lateral sclerosis (182), Alzheimer’s disease (183), various psychiatric conditions (20, 184) and possibly autoimmune encephalitis (185, 186). The inconsistencies noted in some of the above-mentioned studies most likely stem from the use of different sMAC assays, differential sample handling and storage, and the rarity of healthy patient CSF as a negative control. Going forward it would be important to agree on a standard assay for quantitating sMAC and to adopt standardized protocols for handling CSF samples such as that employed by the BioMS-eu network (187, 188).
Most complement proteins are too large to be excreted in urine. Even factor D, the smallest of the complement proteins (~ 24 Kd), does not pass through the tubular epithelium unless there is a kidney defect (189, 190). With a molecular weight approaching 1 MDa (29), studies have suggested that urinary sMAC is most likely locally generated rather than transported from blood into urine (147, 148, 153, 191). A number of factors may contribute to local sMAC generation in the kidney including high levels of proteinuria, cellular debris, urinary ammonia, and low urinary pH (153). In healthy individuals, sMAC is generally undetectable in urine regardless of the type of assay employed. The kidney appears particularly susceptible to complement-mediated damage for a variety of reasons [reviewed in (192)]. Not surprisingly then, nearly all studies examining for sMAC in urine are derived from patients with a variety of acute and chronic kidney diseases or post-kidney transplantation (147–155) (Table 4). A number of urinary biomarkers have been identified for acute kidney injury including kidney injury molecule-1 (KIM-1), IL-18, and others (193). Recent studies suggest that sMAC urinary levels are diagnostic for interstitial inflammation in acute kidney injury associated with nephritis (150) and severe preeclampsia (156) particularly in combination with KIM-1.
Surprisingly there is little known regarding sMAC and urinary tract infections (UTI). It has been shown that C3 promotes colonization of the upper urinary tract by E. coli and that C3- and C4-deficent mice develop fewer renal infections (194). Furthermore, in animal studies, C5a appears to exacerbate UTI through enhancing inflammation and recruitment of leukocytes as C5aR-deficient mice had less renal injury and reduced bacterial load compared to wild type mice (195). These studies indicate that complement contributes to UTI, at least for some pathogens, and that the terminal pathway could be involved since C5a is generated. It would be worth determining baseline levels of sMAC (once assays with higher sensitivity have been developed) in the urine of healthy individuals and comparing it to the levels in UTI patients. sMAC might be an easy biomarker to monitor in UTI, especially in chronic pyelonephritis.
In addition to blood, CSF, and urine, sMAC is found in synovial, pleural, pericardial and peritoneal fluid under conditions of infection, malignancy, or autoimmune disease (listed in Table 4). These fluids are routinely collected for diagnostic purposes, primarily to identify bacterial or viral infections, as well as other medical conditions (196–201). Less commonly analyzed is blister fluid from burn patients. Blister fluid is receiving more attention as a possible diagnostic tool based on recent biochemical and proteomic studies [reviewed in (202, 203)]. sMAC and other complement activation proteins have been detected in blister fluid, however their diagnostic utility remains to be determined (158). Complement components are present in male and female reproductive systems and play a role in both fertility and infertility (204, 205). The presence of sMAC in ovarian follicular fluid and seminal plasma not only indicates complement activation, but suggests possible complement-mediated contributions to infertility (204). This remains an understudied topic and is worth pursuing given the general worldwide decline in fertility (206). sMAC has also been detected in aqueous humor of patients with exfoliating glaucoma (169), but not in patients with neovascular age-related macular degeneration (207). sMAC may be present in other body fluids such as tears, nasopharynx secretions, intestinal secretions, and gingival crevicular fluid, but these have not yet been reported. Support for this possibility comes from studies demonstrating the presence of C5a in normal tears and aqueous humor from patients with cataracts, glaucoma, anterior uveitis, or gingival crevicular fluid (208–210).
Changes in the blood levels of either complement proteins or complement functional activity have served as a valuable diagnostic tool for autoimmune diseases, syndromes, and complement deficiencies for over 60 years [initially reviewed in (211)]. Since then our understanding of the complement system and its relationship to the pathophysiology of infectious and autoimmune disease has increased significantly, and most clinical laboratories routinely run at least some complement-related diagnostic assays (212–214). In addition, commercial diagnostic laboratories offer an extensive array of assays to quantitate blood levels of many complement proteins, measure overall complement function, assess pathway- and protein-specific function and identify auto-antibodies to complement proteins. Identifying complement genetic mutations that contribute to syndromes such as hereditary angioedema, hemolytic uremic syndrome, and rare variants that contribute to deficiency or dysfunction [reviewed in (215)], is now offered by some diagnostic laboratories. The value of complement diagnostics will continue to grow as understanding of the role of complement in autoimmune, infectious, psychiatric diseases, and malignancies expands in the coming years (17, 216–218).
Although sMAC levels in blood, CSF, and other body fluids have been studied as a possible biomarker for diseases and inflammatory conditions (Tables 2–4), those studies have not translated into common use of sMAC as a clinical diagnostic tool. The literature provides numerous examples of the utility of sMAC as a diagnostic biomarker, but the lack of comprehensive reviews on this topic may be one contributing factor to the under-appreciation of its potential. There is no evidence to suggest that intermediates on the way to sMAC formation (sC5b-7 and sC5b-8) have any diagnostic value and there are currently no assays to specifically measure these MAC-related complexes. The advent of complement therapeutics may, however, be a game-changer for sMAC as a diagnostic tool. The anti-C5 antibody eculizumab prevents MAC and sMAC formation by blocking the cleavage of C5 into C5a and C5b, thereby inhibiting the terminal pathway (219). Initially used for treatment of patients with paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, eculizumab has more recently been used in the management of myasthenia gravis, antibody-mediated graft rejection, neuromyelitis optica, and other conditions (220). Several studies have demonstrated that sMAC levels correlate well with eculizumab dosing further indicating that sMAC may be a useful biomarker for monitoring dosing and also aid in developing personalized patient treatment plans. This would usher in a new era in complement diagnostics particularly if patients could measure sMAC (and/or other complement fragments) at home and relay the information directly to their physician or clinic. This could include patients being treated for paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome (106, 221, 222), age-related macular degeneration (107), glomerulonephritis (223), hematopoietic stem cell transplantation (transplant associated thrombotic microangiopathy) (224), thrombotic thrombocytopenia purpura (108, 225), and acute post-infectious glomerulonephritis (226). sMAC monitoring may also have diagnostic value in anti-TNF-α treatment of spondylarthropathies (227), indicating the diagnostic value of sMAC exists beyond complement-specific therapeutics. By extension, sMAC may have diagnostic value in monitoring treatment in rheumatoid, psoriatic arthritis, and other autoimmune diseases given the findings in spondylarthropathies. Complement therapeutic drugs that target the terminal pathway directly or that inhibit the alternative pathway [through which most MAC/sMAC is generated (228)] are currently in development, and it may be beneficial to use sMAC as a biomarker in companion diagnostics to monitor drug efficacy and help manage patient dosing.
The terminal complement pathway gives rise to the MAC and multiple sMAC isoforms. Although multiple immunological roles have been identified for the MAC, little is known regarding the immunobiology of sMAC and intermediates generated during the formation of sMAC. There is, however, a large body of preclinical and clinical studies suggesting that sMAC may be a valuable diagnostic tool in multiple disease settings. In order to fully appreciate the diagnostic potential of sMAC, a number of points should be addressed going forward. These include:
● Assay standardization for quantitating sMAC to allow comparison between datasets and disease settings
● Sample handling and storage standardization to maximize sample stability
● Increased reliance on true healthy controls instead of “non-inflammatory” or “other disease” control sample sets
● Studies to determine basal sMAC fluid levels across multiple demographics
In addition to formalized standardization, there is still much we do not know regarding sMAC with respect to basic physiology and biology. For instance, does sMAC containing vitronectin mediate unknown complement functions or contribute to hematological or cancer-related functions? The multi-functional roles of clusterin and vitronectin may provide insight into sMAC immunobiology, including identification of receptors used in the course of sMAC turnover. These would further aid in the use of sMAC as a biomarker for disease.
Conception and design was by TS and SB. Writing, review, and revision of the manuscript was performed by TS, DB, and SB. All authors contributed to the article and approved the submitted version.
This work was supported in part by a grant from the National Institutes of Health (R43-AI132038) to TS and SB. This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 864751 to DB).
SB and TS are co-founders of CNine Biosolutions, LLC, a biotech company developing complement diagnostics and co-inventors on United States patents # 10,535,004 and 10,630,774 and European Union patent # 3137908, both entitled, Methods and Compositions for Diagnosis and Treatment of Meningitis.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Philip Stahel for critical reading of the manuscript.
1. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Merle NS, Church SE, Noe R, Halbwachs-Mecarell L, Roumenina LT. Complement System Part II: Role in Immunity. Front Immunol (2015) 6:257. doi: 10.3389/fimmu.2015.00257?
3. The Complement Factsbook. 2nd ed. Barnum SR, Schein TN eds. London: Academic Press (2018). p. 480.
4. Freeley S, Kemper C, Le Friec G. The “ins and outs” of complement-driven immune responses. Immunol Rev (2016) 274(1):16–32. doi: 10.1111/imr.12472
5. Erdei A, Sandor N, Macsik-Valent B, Lukacsi S, Kremlitzka M, Bajtay Z. The versatile functions of complement C3-derived ligands. Immunol Rev (2016) 274(1):127–40. doi: 10.1111/imr.12498
6. West EE, Kolev M, Kemper C. Complement and the Regulation of T Cell Responses. Annu Rev Immunol (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
7. Hajishengallis G, Lambris JD. More than complementing Tolls: complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol Rev (2016) 274(1):233–44. doi: 10.1111/imr.12467
8. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol (2017) 18(12):1288–98. doi: 10.1038/ni.3858
9. Hawksworth OA, Coulthard LG, Mantovani S. Complement in stem cells and development. Semin Immunol (2018) 37:74–84. doi: 10.1016/j.smim.2018.02.009
10. Tam JC, Bidgood SR, McEwan WA. Intracellular sensing of complement C3 activates cell autonomous immunity. Science (2014) 345(6201):1256070. doi: 10.1126/science.1256070
11. Barbu A, Hamad OA, Lind L, Ekdahl KN, Nilsson B. The role of complement factor C3 in lipid metabolism. Mol Immunol (2015) 67(1):101–7. doi: 10.1016/j.molimm.2015.02.027
12. Hess C, Kemper C. Complement-mediated regulation of metabolism and basic cellular process. Immunity (2016) 45(2):240–54. doi: 10.1016/j.immuni.2016.08.003
13. Liszewski MK, Elvington M, Kulkarni HS, Atkinson JP. Complement’s hidden arsenal: New insights and novel functions inside the cell. Mol Immunol (2017) 84:2–9. doi: 10.1016/j.molimm.2017.01.004
14. Arbore G, Kemper C, Kolev M. Intracellular complement - the complosome - in immune cell regulation. Mol Immunol (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
15. Kolev M, Kemper C. Keeping It All Going-Complement Meets Metabolism. Front Immunol (2017) 8:1. doi: 10.3389/fimmu.2017.00001
16. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci (2012) 35:369–89. doi: 10.1146/annurev-neuro-061010-113810
17. Presumey J, Bialas AR, Carroll MC. Complement System in Neural Synapse Elimination in Development and Disease. Adv Immunol (2017) 135:53–79. doi: 10.1016/bs.ai.2017.06.004
18. Shivshankar P, Fekry B, Eckel-Mahan K, Wetsel RA. Circadian clock and complement immune system - complementary control of physiology and pathology? Front Cell Infect Microbiol (2020). doi: 10.3389/fcimb.2020.00418
19. Laskaris L, Zalesky A, Weickert CS, Di Biase MA, Chana G, Baune BT. Investigation of peripheral complement factors across stages of psychosis. Schizophr Res (2019) 204:30–7. doi: 10.1016/j.schres.2018.11.035
20. Kopczynska M, Zelek W, Touchard S, Gaughran F, Di Forti M, Mondelli V. Complement system biomarkers in first episode psychosis. Schizophr Res (2019) 204:16–22. doi: 10.1016/j.schres.2017.12.012
21. Schatz-Jakobsen JA, Pedersen DV, Andersen GR. Structural insight into proteolytic activation and regulation of the complement system. Immunol Rev (2016) 274(1):59–73. doi: 10.1111/imr.12465
22. Cserhalmi M, Papp A, Brandus B, Uzonyi B, Jozsi M. Regulation of regulators: Role of the complement factor H-related proteins. Semin Immunol (2019) 45:101341. doi: 10.1016/j.smim.2019.101341
23. Kemper C, Kohl J. Back to the future - non-canonical functions of complement. Semin Immunol (2018) 37:1–3. doi: 10.1016/j.smim.2018.05.002
24. Barnum SR. C4a: An Anaphylatoxin in Name Only. J Innate Immun (2015) 7(4):333–9. doi: 10.1159/000371423
25. Barnum SR. Complement: A primer for the coming therapeutic revolution. Pharmacol Ther (2017) 172:63–72. doi: 10.1016/j.pharmthera.2016.11.014
26. Podack ER, Esser AF, Biesecker G, Muller-Eberhard HJ. Membrane attack complex of complement: a structural analysis of its assembly. J Exp Med (1980) 151(2):301–13. doi: 10.1084/jem.151.2.301
27. Bhakdi S, Tranum-Jensen J. Terminal membrane C5b-9 complex of human complement: transition from an amphiphilic to a hydrophilic state through binding of the S protein from serum. J Cell Biol (1982) 94(3):755–9. doi: 10.1083/jcb.94.3.755
28. Murphy BF, Kirszbaum L, Walker ID, d’Apice AJ. SP-40,40, a newly identified normal human serum protein found in the SC5b-9 complex of complement and in the immune deposits in glomerulonephritis. J Clin Invest (1988) 81(6):1858–64. doi: 10.1172/JCI113531
29. Preissner KP, Podack ER, Muller-Eberhard HJ. SC5b-7, SC5b-8 and SC5b-9 complexes of complement: ultrastructure and localization of the S-protein (vitronectin) within the macromolecules. Eur J Immunol (1989) 19(1):69–75. doi: 10.1002/eji.1830190112
30. Milis L, Morris CA, Sheehan MC, Charlesworth JA, Pussell BA. Vitronectin-mediated inhibition of complement: evidence for different binding sites for C5b-7 and C9. Clin Exp Immunol (1993) 92(1):114–9. doi: 10.1111/j.1365-2249.1993.tb05956.x
31. Tschopp J, Chonn A, Hertig S, French LE. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement C7, C8 beta, and the b domain of C9. J Immunol (1993) 151(4):2159–65.
32. McDonald JF, Nelsestuen GL. Potent inhibition of terminal complement assembly by clusterin: characterization of its impact on C9 polymerization. Biochemistry (1997) 36(24):7464–73. doi: 10.1021/bi962895r
33. Hadders MA, Bubeck D, Roversi P, Hakobyan S, Forneris F, Morgan BP, et al. Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep (2012) 1(3):200–7. doi: 10.1016/j.celrep.2012.02.003
34. Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med (2014) 370(7):632–9. doi: 10.1056/NEJMoa1311084
35. Giles JL, Choy E, van den Berg C, Morgan BP, Harris CL. Response to Comment on “Functional Analysis of a Complement Polymorphism (rs17611) Associated with Rheumatoid Arthritis”. J Immunol (2015) 195(1):4. doi: 10.4049/jimmunol.1500822
36. Fernie BA, Wurzner R, Unsworth DJ, Tuxworth RI, Hobart MJ. DNA polymorphisms of the complement C6 and C7 genes. Ann Hum Genet (1995) 59(Pt 2):163–81. doi: 10.1111/j.1469-1809.1995.tb00739.x
37. Fernie BA, Hobart MJ. Complement C7 deficiency: seven further molecular defects and their associated marker haplotypes. Hum Genet (1998) 103(4):513–9. doi: 10.1007/s004390050859
38. Rittner C, Stradmann-Bellinghausen B. Human complement C81 (C8 A) polymorphism: detection and segregation of new variants. Hum Genet (1993) 92(4):413–6. doi: 10.1007/BF01247347
39. Dewald G, Hemmer S, Nothen MM. Human complement component C8. Molecular basis of the beta-chain polymorphism. FEBS Lett (1994) 340(3):211–5. doi: 10.1016/0014-5793(94)80140-1
40. Dewald G, Cichon S, Bryant SP, Hemmer S, Nothen MM, Spurr NK. The human complement C8G gene, a member of the lipocalin gene family: polymorphisms and mapping to chromosome 9q34.3. Ann Hum Genet (1996) 60(4):281–91. doi: 10.1111/j.1469-1809.1996.tb01192.x
41. Bae JS, Pasaje CF, Park BL, Cheong HS, Kim JH, Park TJ, et al. Genetic analysis of complement component 9 (C9) polymorphisms with clearance of hepatitis B virus infection. Dig Dis Sci (2011) 56(9):2735–41. doi: 10.1007/s10620-011-1657-3
42. Kremlitzka M, Geerlings MJ, de Jong S, Bakker B, Nilsson SC, Fauser S, et al. Functional analyses of rare genetic variants in complement component C9 identified in patients with age-related macular degeneration. Hum Mol Genet (2018) 27(15):2678–88. doi: 10.1093/hmg/ddy178
43. Bumiller-Bini V, Cipolla GA, de Almeida RC, Petzl-Erler ML, Augusto DG, Boldt A. Sparking Fire Under the Skin? Answers From the Association of Complement Genes With Pemphigus Foliaceus. Front Immunol (2018) 9:695. doi: 10.3389/fimmu.2018.00695
44. Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology (2016) 221(6):747–51. doi: 10.1016/j.imbio.2015.04.006
45. Morgan BP, Walters D, Serna M, Bubeck D. Terminal complexes of the complement system: new structural insights and their relevance to function. Immunol Rev (2016) 274(1):141–51. doi: 10.1111/imr.12461
46. Triantafilou M, Hughes TR, Morgan BP, Triantafilou K. Complementing the inflammasome. Immunology (2016) 147(2):152–64. doi: 10.1111/imm.12556
47. Morgan BP, Boyd C, Bubeck D. Molecular cell biology of complement membrane attack. Semin Cell Dev Biol (2017) 72:124–32. doi: 10.1016/j.semcdb.2017.06.009
48. Podack ER, Kolb WP, Muller-Eberhard HJ. The C5b-6 complex: formation, isolation, and inhibition of its activity by lipoprotein and the S-protein of human serum. J Immunol (1978) 120(6):1841–8.
49. Podack ER, Kolb WP, Muller-Eberhard HJ. The SC5b-7 complex: formation, isolation, properties, and subunit composition. J Immunol (1977) 119(6):2024–9.
50. Bhakdi S, Roth M. Fluid-phase SC5b-8 complex of human complement: generation and isolation from serum. J Immunol (1981) 127(2):576–80.
51. Tschopp J, Engel A, Podack ER. Molecular weight of poly(C9). 12 to 18 C9 molecules form the transmembrane channel of complement. J Biol Chem (1984) 259(3):1922–8.
52. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun (2016) 7:10587. doi: 10.1038/ncomms10587
53. Menny A, Serna M, Boyd CM, Gardner S, Joseph AP, Morgan BP, et al. CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat Commun (2018) 9(1):5316. doi: 10.1038/s41467-018-07653-5
54. Kolb WP, Muller-Eberhard HJ. The membrane attack mechanism of complement. Verification of a stable C5-9 complex in free solution. J Exp Med (1973) 138(2):438–51. doi: 10.1084/jem.138.2.438
55. Abramson N, Lachmann PJ, Rosen FS, Jandl JH. Deficiency of C3 inactivator in man. J Immunol (1971) 107(1):19–27.
56. Alper CA, Abramson N, Johnston RB Jr., Jandl JH, Rosen FS. Studies in vivo and in vitro on an abnormality in the metabolism of C3 in a patient with increased susceptibility to infection. J Clin Invest (1970) 49(11):1975–85. doi: 10.1172/JCI106417
57. Nossel HL, Ti M, Kaplan KL, Spanondis K, Soland T, Butler VP Jr. The generation of fibrinopeptide A in clinical blood samples: evidence for thrombin activity. J Clin Invest (1976) 58(5):1136–44. doi: 10.1172/JCI108566
58. Nossel HL, Yudelman I, Canfield RE, Butler VP Jr., Spanondis K, Wilner GD, et al. Measurement of fibrinopeptide A in human blood. J Clin Invest (1974) 54(1):43–53. doi: 10.1172/JCI107749
59. Bauer KA, Rosenberger RD. The pathophysiology of the prethrombotic state in humans: Insights gained from studies using markers of hemostatic system activation. Blood (1987) 70:343–50. doi: 10.1182/blood.V70.2.343.bloodjournal702343
60. Bauer KA, Kass BL, ten Cate H, Hawiger JJ. Factor IX is activated in vivo by the tissue factor mechanism. Blood (1990) 76:731–6. doi: 10.1182/blood.V76.4.731.bloodjournal764731
61. Bauer KA, Kass BL, ten Cate H, Bednarek MA, Hawiger JJ, Rosenberger RD. Detection of factor X activation in humans. Blood (1989) 74(2007-2015). doi: 10.1182/blood.V74.6.2007.2007
62. Conway EM. Complement-coagulation connections. Coagulation Fibrinol (2018) 29:243–51. doi: 10.1097/MBC.0000000000000720
63. D’zik S. Complement and coagulation: Cross talk through time. Transfusion Med Rev (2019) 33:199–206. doi: 10.1016/j.tmrv.2019.08.004
64. Izumi M, Yamada KM, Hayashi M. Vitronectin exists in two structurally and functionally distinct forms in human plasma. Biochim Biophys Acta (1989) 990(2):101–8. doi: 10.1016/S0304-4165(89)80019-4
65. Trougakos IP, Poulakou M, Stathatos M, Chalikia A, Melidonis A, Gonos ES. Serum levels of the senescence biomarker clusterin/apolipoprotein J increase significantly in diabetes type II and during development of coronary heart disease or at myocardial infarction. Exp Gerontol (2002) 37(10-11):1175–87. doi: 10.1016/S0531-5565(02)00139-0
66. Biesecker G, Muller-Eberhard HJ. The ninth component of human complement: purification and physicochemical characterization. J Immunol (1980) 124(3):1291–6.
67. Falgarone G, Chiocchia G. Chapter 8: Clusterin: A multifacet protein at the crossroad of inflammation and autoimmunity. Adv Cancer Res (2009) 104:139–70. doi: 10.1016/S0065-230X(09)04008-1
68. Seiffert D. Constitutive and regulated expression of vitronectin. Histol Histopathol (1997) 12(3):787–97.
69. Zewde N, Morikis D. A computational model for the evaluation of complement system regulation under homeostasis, disease, and drug intervention. PLoS One (2018) 13(6):e0198644. doi: 10.1371/journal.pone.0198644
70. Zewde N, Gorham RD Jr., Dorado A, Morikis D. Quantitative Modeling of the Alternative Pathway of the Complement System. PLoS One (2016) 11(3):e0152337. doi: 10.1371/journal.pone.0152337
71. Heesterbeek DA, Bardoel BW, Parsons ES, Bennett I, Ruyken M, Doorduijn DJ, et al. Bacterial killing by complement requires membrane attack complex formation via surface-bound C5 convertases. EMBO J (2019) 38(4). doi: 10.15252/embj.201899852
72. Doorduijn DJ, Bardoel BW, Heesterbeek DAC, Ruyken M, Benn G, Parsons ES, et al. Bacterial killing by complement requires direct anchoring of membrane attack complex precursor C5b-7. PLoS Pathog (2020) 16(6):e1008606. doi: 10.1371/journal.ppat.1008606
73. Mollnes TE, Lea T, Froland SS, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand J Immunol (1985) 22(2):197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x
74. Mollnes TE, Lea T, Harboe M, Tschopp J. Monoclonal antibodies recognizing a neoantigen of poly(C9) detect the human terminal complement complex in tissue and plasma. Scand J Immunol (1985) 22(2):183–95. doi: 10.1111/j.1365-3083.1985.tb01870.x
75. Bhakdi S, Muhly M. A simple immunoradiometric assay for the terminal SC5b-9 complex of human complement. J Immunol Methods (1983) 57(1-3):283–9. doi: 10.1016/0022-1759(83)90088-1
76. Hugo F, Kramer S, Bhakdi S. Sensitive ELISA for quantitating the terminal membrane C5b-9 and fluid-phase SC5b-9 complex of human complement. J Immunol Methods (1987) 99(2):243–51. doi: 10.1016/0022-1759(87)90134-7
77. Haahr-Pedersen S, Bjerre M, Flyvbjerg A, Mogelvang R, Dominquez H, Hansen TK, et al. Level of complement activity predicts cardiac dysfunction after acute myocardial infarction treated with primary percutaneous coronary intervention. J Invasive Cardiol (2009) 21(1):13–9.
78. Yang S, McGookey M, Wang Y, Cataland SR, Wu HM. Effect of blood sampling, processing, and storage on the measurement of complement activation biomarkers. Am J Clin Pathol (2015) 143(4):558–65. doi: 10.1309/AJCPXPD7ZQXNTIAL
79. Bergseth G, Kirschfink M, Giclas PC, Nilsson B, Mollnes TE. An international serum standard for application in assays to detect human complement activation products. Mol Immunol (2013) 56(3):232–9. doi: 10.1016/j.molimm.2013.05.221
80. Schein TN, Blackburn TE, Heath SL, Barnum SR. Plasma levels of soluble membrane attack complex are elevated despite viral suppression in HIV patients with poor immune reconstitution. Clin Exp Immunol (2019) 198(3):359–66. doi: 10.1111/cei.13366
81. Hogasen AK, Overlie I, Hansen TW, Abrahamsen TG, Finne PH, Hogasen K. The analysis of the complement activation product SC5 b-9 is applicable in neonates in spite of their profound C9 deficiency. J Perinat Med (2000) 28(1):39–48. doi: 10.1515/JPM.2000.006
82. McGreal EP, Hearne K, Spiller OB. Off to a slow start: under-development of the complement system in term newborns is more substantial following premature birth. Immunobiology (2012) 217(2):176–86. doi: 10.1016/j.imbio.2011.07.027
83. Willems E, Alkema W, Keizer-Garritsen J, Suppers A, van der Flier M, Philipsen R, et al. Biosynthetic homeostasis and resilience of the complement system in health and infectious disease. EBioMedicine (2019) 45:303–13. doi: 10.1016/j.ebiom.2019.06.008
84. Tjernberg AR, Woksepp H, Sandholm K, Johansson M, Dahle C, Ludvigsson JF, et al. Celiac disease and complement activation in response to Streptococcus pneumoniae. Eur J Pediatr (2020) 179(1):133–40. doi: 10.1007/s00431-019-03490-w
85. Lin RY, Astiz ME, Saxon JC, Saha DC, Rackow EC. Alterations in C3, C4, factor B and related metabolites in septic shock. Clin Immunol Immunopathol (1993) 69:136–42. doi: 10.1006/clin.1993.1161
86. Brandtzaeg P, Mollnes TE, Kierulf P. Complement activation and endotoxin levels in systemic meningococcal disease. J Infect Dis (1989) 160(1):58–65. doi: 10.1093/infdis/160.1.58
87. Segura-Cervantes E, Mancilla-Ramirez J, Gonzalez-Canudas J, Alba E, Santillan-Ballesteros R, Morales-Barquet D, et al. Inflammatory Response in Preterm and Very Preterm Newborns with Sepsis. Mediators Inflamm (2016) 2016:6740827. doi: 10.1155/2016/6740827
88. Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S. Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J Infect Dis (2006) 193(8):1078–88. doi: 10.1086/500949
89. Schein TN, Barnum SR. “Role of complement in cerebral malaria”. In: Stoute JA, editor. Complement Activation in Malaria Immunity and Pathogenesis. Berlin: Springer-Verlag (2018). p. 65–90.
90. Taylor RP, Stoute JA, Lindorfer MA. “Mechanisms of complement activation in malaria”. In: Stoute JA, editor. Compement Activation in Malaria Immunity and Pathogenesis. Berlin: Springer-Verlag (2018). p. 31–49.
91. Wang H, Wang K, Wang C, Qui W, Lu Z, Hu X, et al. Increased soluble C5b-9 in CSF of neuromyelitis optica. Scand J Immunol (2014) 79:127–30. doi: 10.1111/sji.12132
92. Morgan BP, Daniels RH, Williams BD. Measurement of terminal complement complexes in rheumatoid arthritis. Clin Exp Immunol (1988) 73(3):473–8.
93. Dalmasso AP, Falk RJ, Raij L. The pathobiology of the terminal complement complexes. Complement Inflammation (1989) 6(1):36–48. doi: 10.1159/000463070
94. Rus V, Malide D, Bolosiu HD, Parasca I, Dutu AL. Levels of SC5b–9 complement complex in plasma and synovial fluid of patients with rheumatic disease. Med Internet (1990) 28(4):305–10.
95. Buyon JP, Tamerius J, Belmont HM, Abramson SB. Assessment of disease activity and impending flare in patients with systemic lupus erythematosus. Comparison of the use of complement split products and conventional measurements of complement. Arthritis Rheum (1992) 35(9):1028–37. doi: 10.1002/art.1780350907
96. Horigome I, Seino J, Sudo K, Kinoshita Y, Saito T, Yoshinaga K. Terminal complement complex in plasma from patients with systemic lupus erythematosus and other glomerular diseases. Clin Exp Immunol (1987) 70(2):417–24.
97. Mollnes TE, Haga HJ, Brun JG, Nielsen EW, Sjoholm A, Sturfeldt G, et al. Complement activation in patients with systemic lupus erythematosus without nephritis. Rheumatol (Oxford) (1999) 38(10):933–40. doi: 10.1093/rheumatology/38.10.933
98. Nagy G, Li DY, Chen M, Zhao MH. Usefulness of detection of complement activation products in evaluating SLE activity. Lupus (2000) 9(1):19–25. doi: 10.1177/096120330000900105
99. Nytrova P, Potlukova E, Kemlink D, Woodhall M, Horakova D, Waters P, et al. Complement activation in patients with neuromyelitis optica. J Neuroimmunol (2014) 274(1-2):185–91. doi: 10.1016/j.jneuroim.2014.07.001
100. Hakobyan S, Luppe S, Evans DR, Harding K, Loveless S, Robertson NP, et al. Plasma complement biomarkers distinguish multiple sclerosis and neuromyelitis optica spectrum disorder. Mult Scler (2017) 23(7):946–55. doi: 10.1177/1352458516669002
101. Miao D, Li DY, Chen M, Zhao MH. Platelets are activated in ANCA-associated vasculitis via thrombin-PARs pathway and can activate the alternative complement pathway. Arthritis Res Ther (2017) 19(1):252. doi: 10.1186/s13075-017-1458-y
102. Kim MY, Guerra MM, Kaplowitz E, Laskin CA, Petri M, Branch DW, et al. Complement activation predicts adverse pregnancy outcome in patients with systemic lupus erythematosus and/or antiphospholipid antibodies. Ann Rheum Dis (2018) 77(4):549–55. doi: 10.1136/annrheumdis-2017-212224
103. Wu EY, McInnis EA, Boyer-Suavet S, Mendoza CE, Aybar LT, Kennedy KB, et al. Measuring Circulating Complement Activation Products in Myeloperoxidase- and Proteinase 3-Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol (2019) 71(11):1894–903. doi: 10.1002/art.41011
104. Barohn RJ, Brey RL. Soluble terminal complement components in human myasthenia gravis. Clin Neurol Neurosurg (1993) 95(4):285–90. doi: 10.1016/0303-8467(93)90103-N
105. Donadelli R, Pulieri P, Piras R, Iatropoulos P, Valoti E, Benigni A, et al. Unraveling the Molecular Mechanisms Underlying Complement Dysregulation by Nephritic Factors in C3G and IC-MPGN. Front Immunol (2018) 9:2329. doi: 10.3389/fimmu.2018.02329
106. Wehling C, Amon O, Bommer M, Hoppe B, Kentouche K, Schalk G, et al. Monitoring of complement activation biomarkers and eculizumab in complement-mediated renal disorders. Clin Exp Immunol (2017) 187(2):304–15. doi: 10.1111/cei.12890
107. Tian Y, Kijlstra A, van der Veen RL, Makridaki M, Murray IJ, Berendschot TT. Lutein supplementation leads to decreased soluble complement membrane attack complex sC5b-9 plasma levels. Acta Ophthalmol (2015) 93(2):141–5. doi: 10.1111/aos.12535
108. Bitzan M, Hammad RM, Bonnefoy A, Al Dhaheri WS Vezina C, Rivard GE. Acquired thrombotic thrombocytopenic purpura with isolated CFHR3/1 deletion-rapid remission following complement blockade. Pediatr Nephrol (2018) 33(8):1437–42. doi: 10.1007/s00467-018-3957-8
109. Omine M, Kinoshita T, Nakakuma H, Maciejewski JP, Parker CJ, Socie G. Paroxysmal nocturnal hemoglobinuria. Int J Hematol (2005) 82(5):417–21. doi: 10.1532/IJH97.05140
110. Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood (2005) 106(12):3699–709. doi: 10.1182/blood-2005-04-1717
111. Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood (2014) 124(11):1715–26. doi: 10.1182/blood-2014-02-558296
112. Scholl HP, Charbel I, Walier M, Janzer S, Pollok-Kopp B, Borncke F, et al. Systemic complement activation in age-related macular degeneration. PLoS One (2008) 3(7):e2593. doi: 10.1371/journal.pone.0002593
113. Vallhonrat H, Williams WW, Dec GW, Keck S, Schoenfeld D, Cosimi AB, et al. Complement activation products in plasma after heart transplantation in humans. Transplantation (2001) 71(9):1308–11. doi: 10.1097/00007890-200105150-00022
114. Vallhonrat H, Williams WW, Cosimi AB, Tolkoff-Rubin N, Ginns LC, Wain JC, et al. In vivo generation of C4d, Bb, iC3b, and SC5b-9 after OKT3 administration in kidney and lung transplant recipients. Transplantation (1999) 67(2):253–8. doi: 10.1097/00007890-199901270-00011
115. Vallhonrat H, Swinford RD, Ingelfinger JR, Williams WW, Ryan DP, Tolkoff-Rubin N, et al. Rapid activation of the alternative pathway of complement by extracorporeal membrane oxygenation. ASAIO J (1999) 45(1):113–4. doi: 10.1097/00002480-199901000-00025
116. Horvath O, Kallay K, Csuka D, Mezo B, Sinkovits G, Kassa C, et al. Early Increase in Complement Terminal Pathway Activation Marker sC5b-9 Is Predictive for the Development of Thrombotic Microangiopathy after Stem Cell Transplantation. Biol Blood Marrow Transplant (2018) 24(5):989–96. doi: 10.1016/j.bbmt.2018.01.009
117. Roumenina LT, Bartolucci P, Pirenne F. The role of Complement in Post-Transfusion Hemolysis and Hyperhemolysis Reaction. Transfus Med Rev (2019) 33(4):225–30. doi: 10.1016/j.tmrv.2019.09.007
118. Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood (2014) 124(4):645–53. doi: 10.1182/blood-2014-03-564997
119. Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, et al. Early complementopathy after multiple injuries in humans. Shock (2012) 37(4):348–54. doi: 10.1097/SHK.0b013e3182471795
120. Li Y, Zhao Q, Liu B, Dixon A, Cancio L, Dubick M, et al. Early complementopathy predicts the outcomes of patients with trauma. Trauma Surg Acute Care Open (2019) 4(1):e000217. doi: 10.1136/tsaco-2018-000217
121. Deppisch R, Schmitt V, Bommer J, Hansch GM, Ritz E. Fluid phase generation of terminal complement complex as a novel index of bioincompatibility. Kidney Int (1990) 37(2):696–706. doi: 10.1038/ki.1990.36
122. Mizuno M, Suzuki Y, Higashide K, Sei Y, Iguchi D, Sakata F, et al. High Levels of Soluble C5b-9 Complex in Dialysis Fluid May Predict Poor Prognosis in Peritonitis in Peritoneal Dialysis Patients. PLoS One (2017) 12(1):e0169111. doi: 10.1371/journal.pone.0169111
123. Poppelaars F, Faria B, Gaya da Costa M, Franssen CFM, van Son WJ, Berger SP, et al. The Complement System in Dialysis: A Forgotten Story? Front Immunol (2018) 9:71. doi: 10.3389/fimmu.2018.00071
124. Faria B, Gaya da Costa M, Poppelaars F, Franssen CFM, Pestana M, Berger SP, et al. Administration of Intravenous Iron Formulations Induces Complement Activation in-vivo. Front Immunol (2019) 10:1885. doi: 10.3389/fimmu.2019.01885
125. Lines SW, Richardson VR, Thomas B, Dunn EJ, Wright MJ, Carter AM. Complement and Cardiovascular Disease–The Missing Link in Haemodialysis Patients. Nephron (2016) 132(1):5–14. doi: 10.1159/000442426
126. Yasuda M, Takeuchi K, Hiruma M, Iida H, Tahara A, Itagane H, et al. The complement system in ischemic heart disease. Circulation (1990) 81(1):156–63. doi: 10.1161/01.CIR.81.1.156
127. Trendelenburg M, Stallone F, Pershyna K, Eisenhut T, Twerenbold R, Wildi K, et al. Complement activation products in acute heart failure: Potential role in pathophysiology, responses to treatment and impacts on long-term survival. Eur Heart J Acute Cardiovasc Care (2018) 7(4):348–57. doi: 10.1177/2048872617694674
128. Orrem HL, Nilsson PH, Pischke SE, Grindheim G, Garred P, Seljeflot I, et al. Acute heart failure following myocardial infarction: complement activation correlates with the severity of heart failure in patients developing cardiogenic shock. ESC Heart Fail (2018) 5(3):292–301. doi: 10.1002/ehf2.12266
129. Akcan U, Karabulut S, Ismail Kucukali C, Cakir S, Tuzun E. Bipolar disorder patients display reduced serum complement levels and elevated peripheral blood complement expression levels. Acta Neuropsychiatr (2018) 30(2):70–8. doi: 10.1017/neu.2017.10
130. Ramos TN, Arynchyna AA, Blackburn TE, Barnum SR, Johnston JM. Soluble membrane attack complex is diagnostic for intraventricular shunt infection in children. JCI Insight (2016) 1(10):e87919. doi: 10.1172/jci.insight.87919
131. Shen L, Zheng J, Wang Y, Zhu M, Zhu H, Cheng Q, et al. Increased activity of the complement system in cerebrospinal fluid of the patients with Non-HIV cryptococcal meningitis. BMC Infect Dis (2017) 17. doi: 10.1186/s12879-016-2107-9
132. Woehrl B, Brouwer MC, Murr C, Heckenberg SG, Baas F, Pfister HW, et al. Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis. J Clin Invest (2011) 121(10):3943–53. doi: 10.1172/JCI57522
133. Brouwer MC, Baas F, van der Ende A, van de Beek D. Genetic variation and cerebrospinal fluid levels of mannose binding lectin in pneumococcal meningitis patients. PLoS One (2013) 8(5):e65151. doi: 10.1371/journal.pone.0065151
134. Mook-Kanamori BB, Brouwer MC, Geldhoff M, Ende AV, van de Beek D. Cerebrospinal fluid complement activation in patients with pneumococcal and meningococcal meningitis. J Infect (2014) 68(6):542–7. doi: 10.1016/j.jinf.2013.12.016
135. Sanders ME, Koski CL, Robbins D, Shin ML, Frank MM, Joiner KA. Activated terminal complement in cerebrospinal fluid in Guillain-Barre syndrome and multiple sclerosis. J Immunol (1986) 136(12):4456–9.
136. Sanders ME, Alexander EL, Koski CL, Frank MM, Joiner KA. Detection of activated terminal complement (C5b-9) in cerebrospinal fluid from patients with central nervous system involvement of primary Sjogren’s syndrome or systemic lupus erythematosus. J Immunol (1987) 138(7):2095–9.
137. Sellebjerg F, Jaliashvili I, Christiansen M, Garred P. Intrathecal activation of the complement system and disability in multiple sclerosis. J Neurol Sci (1998) 157(2):168–74. doi: 10.1016/S0022-510X(98)00086-0
138. Jonsen A, Bengtsson AA, Nived O, Ryberg B, Truedsson L, Ronnblom L, et al. The heterogeneity of neuropsychiatric systemic lupus erythematosus is reflected in lack of association with cerebrospinal fluid cytokine profiles. Lupus (2003) 12(11):846–50. doi: 10.1191/0961203303lu472sr
139. Zelek WM, Fathalla D, Morgan A, Touchard S, Loveless S, Tallantyre E, et al. Cerebrospinal fluid complement system biomarkers in demyelinating disease. Mult Scler (2019) 1352458519887905. doi: 10.1177/1352458519887905
140. Ingram G, Hakobyan S, Hirst CL, Harris CL, Loveless S, Mitchell JP, et al. Systemic complement profiling in multiple sclerosis as a biomarker of disease state. Mult Scler (2012) 18(10):1401–11. doi: 10.1177/1352458512438238
141. Hakansson I, Vrethem M, Dahle C, Ekdahl KN. Complement activation in cerebrospinal fluid in clinically isolated syndrome and early stages of relapsing remitting multiple sclerosis. J Neuroimmunol (2020) 340:577147. doi: 10.1016/j.jneuroim.2020.577147
142. Stahel PF, Morganti-Kossmann MC, Perez D, Redaelli C, Gloor B, Trentz O, et al. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma (2001) 18(8):773–81. doi: 10.1089/089771501316919139
143. Parry J, Hwang J, Stahel CF, Henderson C, Nadeau J, Stacey S, et al. Soluble terminal complement activation fragment sC5b-9: a new serum biomarker for traumatic brain injury? Eur J Trauma Emerg Surg (2020). doi: 10.1007/s00068-020-01407-z
144. Lindsberg PJ, Ohman J, Lehto T, Wuorimaa T, et al. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann Neurol (1996) 40(4):587–96. doi: 10.1002/ana.410400408
145. Loeffler DA, Brickman CM, Juneau PL, Perry MF, Pomara N, Lewitt PA. Cerebrospinal fluid C3a increases with age, but does not increase further in Alzheimer’s disease. Neurobiol Aging (1997) 18(5):555–7. doi: 10.1016/S0197-4580(97)00110-3
146. Pelletier K, Bonnefoy A, Chapdelaine H, Pichette V, Lejars M, Madore F, et al. Clinical Value of Complement Activation Biomarkers in Overt Diabetic Nephropathy. Kidney Int Rep (2019) 4(6):797–805. doi: 10.1016/j.ekir.2019.03.004
147. Matsell DG, Wyatt RJ, Gaber LW. Terminal complement complexes in acute poststreptococcal glomerulonephritis. Pediatr Nephrol (1994) 8(6):671–6. doi: 10.1007/BF00869086
148. Kusunoki Y, Akutsu Y, Itami N, Tochimaru H, Nagata Y, Takekoshi Y, et al. Urinary excretion of terminal complement complexes in glomerular disease. Nephron (1991) 59(1):27–32. doi: 10.1159/000186513
149. Zhang MF, Huang J, Zhang YM, Qu Z, Wang X, Wang F, et al. Complement activation products in the circulation and urine of primary membranous nephropathy. BMC Nephrol (2019) 20(1):313. doi: 10.1186/s12882-019-1509-5
150. Zhao WT, Huang JW, Sun PP, Su T, Tang JW, Wang SX, et al. Diagnostic roles of urinary kidney injury molecule 1 and soluble C5b-9 in acute tubulointerstitial nephritis. Am J Physiol Renal Physiol (2019) 317(3):F584–92. doi: 10.1152/ajprenal.00176.2019
151. Huang H, Li D, Huang X, Wang Y, Wang S, Wang X, et al. Association of Complement and Inflammatory Biomarkers with Diabetic Nephropathy. Ann Clin Lab Sci (2019) 49(4):488–95.
152. Thurman JM, Wong M, Renner B, Frazer-Abel A, Giclas PC, Joy MS, et al. Complement Activation in Patients with Focal Segmental Glomerulosclerosis. PLoS One (2015) 10(9):e0136558. doi: 10.1371/journal.pone.0136558
153. van der Pol P, de Vries DK, van Gijlswijk DJ, van Anken GE, Schlagwein N, Daha MR, et al. Pitfalls in urinary complement measurements. Transpl Immunol (2012) 27(1):55–8. doi: 10.1016/j.trim.2012.06.001
154. Biglarnia AR, Huber-Lang M, Mohlin C, Ekdahl KN, Nilsson B, et al. The multifaceted role of complement in kidney transplantation. Nat Rev Nephrol (2018) 14(12):767–81. doi: 10.1038/s41581-018-0071-x
155. Lammerts RGM, Eisenga MF, Alyami M, Daha MR, Seelen MA, Pol RA, et al. Urinary Properdin and sC5b-9 Are Independently Associated With Increased Risk for Graft Failure in Renal Transplant Recipients. Front Immunol (2019) 10:2511. doi: 10.3389/fimmu.2019.02511
156. Burwick RM, Easter SR, Dawood HY, Yamamoto HS, Fichorova RN, Feinberg BB, et al. Complement activation and kidney injury molecule-1-associated proximal tubule injury in severe preeclampsia. Hypertension (2014) 64(4):833–8. doi: 10.1161/HYPERTENSIONAHA.114.03456
157. Brodeur JP, Ruddy S, Schwartz LB, Moxley G. Synovial fluid levels of complement SC5b-9 and fragment Bb are elevated in patients with rheumatoid arthritis. Arthritis Rheum (1991) 34(12):1531–7. doi: 10.1002/art.1780341209
158. Bengtsson A, Bengtson JP, Rydenhag A, Roxvall L, Heideman M. Accumulation of anaphylatoxins and terminal complement complexes in inflammatory fluids. J Intern Med (1990) 228(2):173–6. doi: 10.1111/j.1365-2796.1990.tb00212.x
159. Hara N, Abe M, Inuzuka S, Kawarada Y, Shigematsu N. Pleural SC5b-9 in differential diagnosis of tuberculous, malignant, and other effusions. Chest (1992) 102(4):1060–4. doi: 10.1378/chest.102.4.1060
160. Salomaa ER, Viander M, Saaresranta T, Terho EO. Complement components and their activation products in pleural fluid. Chest (1998) 114(3):723–30. doi: 10.1378/chest.114.3.723
161. Porcel JM, Vives M, Gazquez I, Vicente de Vera MC, Perez B, Rubio M. Usefulness of pleural complement activation products in differentiating tuberculosis and malignant effusions. Int J Tuberc Lung Dis (2000) 4(1):76–82.
162. Vives M, Porcel JM, Gazquez I, Perez B, Rubio M. Pleural SC5b-9: a test for identifying complicated parapneumonic effusions. Respiration (2000) 67(4):433–8. doi: 10.1159/000029543
163. Porcel JM. Pleural fluid tests to identify complicated parapneumonic effusions. Curr Opin Pulm Med (2010) 16(4):357–61. doi: 10.1097/MCP.0b013e328338a108
164. Kabut J, Kondera-Anasz Z, Sikora J, Mielczarek-Palacz A. Levels of complement components iC3b, C3c, C4, and SC5b-9 in peritoneal fluid and serum of infertile women with endometriosis. Fertil Steril (2007) 88(5):1298–303. doi: 10.1016/j.fertnstert.2006.12.061
165. Bjerre M, Holland-Fischer P, Gronbaek H, Frystyk J, Hansen TK, Vilstrup H, et al. Soluble membrane attack complex in ascites in patients with liver cirrhosis without infections. World J Hepatol (2010) 2(6):221–5. doi: 10.4254/wjh.v2.i6.221
166. Shigemoto E, Mizuno M, Suzuki Y, Kobayashi K, Sakata F, Kariya T, et al. Increase of Eosinophil in Dialysate During Induction of Peritoneal Dialysis. Perit Dial Int (2019) 39(1):90–2. doi: 10.3747/pdi.2017.00205
167. D’Cruz OJ, Haas GG Jr., Lambert H. Evaluation of antisperm complement-dependent immune mediators in human ovarian follicular fluid. J Immunol (1990) 144(10):3841–8.
168. D’Cruz OJ, Haas GG Jr. Lack of complement activation in the seminal plasma of men with antisperm antibodies associated in vivo on their sperm. Am J Reprod Immunol (1990) 24(2):51–7. doi: 10.1111/j.1600-0897.1990.tb01038.x
169. Doudevski I, Rostagno A, Cowman M, Liebmann J, Ritch R, Ghiso J. Clusterin and complement activation in exfoliation glaucoma. Invest Ophthalmol Vis Sci (2014) 55(4):2491–9. doi: 10.1167/iovs.13-12941
170. Hugo F, Berstecher C, Kramer S, Fassbender W, Bhakdi S. In vivo clearance studies of the terminal fluid-phase complement complex in rabbits. Clin Exp Immunol (1989) 77(1):112–6.
171. Zelek WM, Xie L, Morgan BP, Harris CL. Compendium of current complement therapeutics. Mol Immunol (2019) 114:341–52. doi: 10.1016/j.molimm.2019.07.030
172. Anderson AM, Schein TN, Kalapila A, Lai L, Waldrop-Valverde D, Moore RC, et al. Soluble membrane attcak complex in the blood and cerebrospinal fluid of HIV-infected individuals, relationship to HIV RNA, and comparison with HIV negatives. J Neuroimmunol (2017) 311:35–9. doi: 10.1016/j.jneuroim.2017.07.014
173. Seele J, Kirschfink M, Djukic M, Lange P, Gossner J, Bunkowski S, et al. Cisterno-lumbar gradient of complement fractions in geriatric patients with suspected normal pressure hydrocephalus. Clin Chim Acta (2018) 486:1–7. doi: 10.1016/j.cca.2018.07.008
174. Broadwell RD, Sofroniew MV. Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol (1993) 120(2):245–63. doi: 10.1006/exnr.1993.1059
175. Aly H, Khashaba MT, Nada A, Hasanen BM, McCarter R, Schultz SJ, et al. The role of complement in neurodevelopment impairment following neonatal hypoxic-ischemic encephalopathy. Am J Perinatol (2009) 26:659–65. doi: 10.1055/s-0029-1220793
176. Saunders NR, Dziegielewska KM, Mollgard K, Habgood MD. Physiology and molecular biology of barrier mechanisms in the fetal and neonatal brain. J Physiol (2018) 596(23):5723–56. doi: 10.1113/JP275376
177. Brautigam K, Vakis A, Tsitsipanis C. Pathogenesis of idiopathic Normal Pressure Hydrocephalus: A review of knowledge. J Clin Neurosci (2019) 61:10–3. doi: 10.1016/j.jocn.2018.10.147
178. Tuzun E, Kurtuncu M, Turkoglu R, Icoz S, Pehlivan M, Birisik O, et al. Enhanced complement consumption in neuromyelitis optica and Behcet’s disease patients. J Neuroimmunol (2011) 233(1-2):211–5. doi: 10.1016/j.jneuroim.2010.11.010
179. Miller DH, Chard DT, Ciccarelli O. Clinically isolated syndromes. Lancet Neurol (2020) 11:157–69. doi: 10.1016/S1474-4422(11)70274-5
180. Buckingham SC, Ramos TN, Barnum SR. Complement C5-deficient mice are protected from seizures in experimental cerebral malaria. Epilepsia (2014) 55(12):e139–42. doi: 10.1111/epi.12858
181. Loeffler DA, Camp DM, Conant SB. Complement activation in the Parkinson’s disease substantia nigra: an immunocytochemical study. J Neuroinflamm (2006) 3:29. doi: 10.1186/1742-2094-3-29
182. Kjaeldgaard AL, Pilely K, Olsen KS, Pedersen SW, Lauritsen AO, Moller K, et al. Amyotrophic lateral sclerosis: The complement and inflammatory hypothesis. Mol Immunol (2018) 102:14–25. doi: 10.1016/j.molimm.2018.06.007
183. Kolev MV, Ruseva MM, Harris CL, Morgan BP, Donev RM. Implication of complement system and its regulators in Alzheimer’s disease. Curr Neuropharmacol (2009) 7(1):1–8. doi: 10.2174/157015909787602805
184. Ishii T, Hattori K, Miyakawa T, Watanabe K, Hidese S, Sasayama D, et al. Increased cerebrospinal fluid complement C5 levels in major depressive disorder and schizophrenia. Biochem Biophys Res Commun (2018) 497(2):683–8. doi: 10.1016/j.bbrc.2018.02.131
185. Dalmau J, Graus F. Antibody-Mediated Encephalitis. N Engl J Med (2018) 378(9):840–51. doi: 10.1056/NEJMra1708712
186. Lungen EM, Maier V, Venhoff N, Salzer U, Dersch R, Berger B, et al. Systemic Lupus Erythematosus With Isolated Psychiatric Symptoms and Antinuclear Antibody Detection in the Cerebrospinal Fluid. Front Psychiatry (2019) 10:226. doi: 10.3389/fpsyt.2019.00226
187. Teunissen CE, Petzold A, Bennett JL, Berven FS, Brundin L, Comabella M, et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology (2009) 73(22):1914–22. doi: 10.1212/WNL.0b013e3181c47cc2
188. Teunissen CE, Tumani H, Engelborghs S, Mollenhauer B. Biobanking of CSF: international standardization to optimize biomarker development. Clin Biochem (2014) 47(4-5):288–92. doi: 10.1016/j.clinbiochem.2013.12.024
189. Volanakis JE, Barnum SR, Kilpatrick JM. Purification and properties of human factor D. Methods Enzymol (1993) 223:82–97. doi: 10.1016/0076-6879(93)23039-P
190. Volanakis JE, Barnum SR, Kilpatrick JM. Renal filtration and catabolism of complement protein D. N Engl J Med (1985) 312(7):395–9. doi: 10.1056/NEJM198502143120702
191. Thurman JM, Nester CM. All Things Complement. Clin J Am Soc Nephrol (2016) 11(10):1856–66. doi: 10.2215/CJN.01710216
192. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol (2016) 12(7):383–401. doi: 10.1038/nrneph.2016.70
193. Desanti De Oliveira B, Xu K, Shen TH, Callahan M, Kiryluk K, D’Agati VD, et al. Molecular nephrology: types of acute tubular injury. Nat Rev Nephrol (2019) 15(10):599–612. doi: 10.1038/s41581-019-0184-x
194. Springall T, Sheerin NS, Abe K, Holers VM, Wan H, Sacks SH. Epithelial secretion of C3 promotes colonization of the upper urinary tract by Escherichia coli. Nat Med (2001) 7(7):801–6. doi: 10.1038/89923
195. Choudhry N, Li K, KY Wu, Song Y, Farrar CA, et al. The complement factor 5a receptor 1 has a pathogenic role in chronic inflammation and renal fibrosis in a murine model of chronic pyelonephritis. Kidney Int (2016) 90(3):540–54. doi: 10.1016/j.kint.2016.04.023
196. Oliviero F, Galozzi P, Ramonda R, de Oliveira FL, Schiavon F, Scanu A, et al. Unusual Findings in Synovial Fluid Analysis: A Review. Ann Clin Lab Sci (2017) 47(3):253–9.
197. Seidman AJ, Limaiem F. “Synovial Fluid Analysis”. In: StatPearls. (Treasure Island (FL): Stat Pearls Publishing (2020).
198. Mercer RM, Corcoran JP, Porcel JM, Rahman NM, Psallidas I. Interpreting pleural fluid results. Clin Med (Lond) (2019) 19(3):213–7. doi: 10.7861/clinmedicine.19-3-213
199. Hou W, Sanyal AJ. Ascites: diagnosis and management. Med Clin North Am (2009) 93(4):801–17, vii. doi: 10.1016/j.mcna.2009.03.007
200. Oey RC, van Buuren HR, de Man RA. The diagnostic work-up in patients with ascites: current guidelines and future prospects. Neth J Med (2016) 74(8):330–5.
201. Trindade F, Vitorino R, Leite-Moreira A, Falcao-Pires I. Pericardial fluid: an underrated molecular library of heart conditions and a potential vehicle for cardiac therapy. Basic Res Cardiol (2019) 114(2):10. doi: 10.1007/s00395-019-0716-3
202. Widgerow AD, King K, Tocco-Tussardi I, Banyard DA, Chiang R, Awad A, et al. The burn wound exudate-an under-utilized resource. Burns (2015) 41(1):11–7. doi: 10.1016/j.burns.2014.06.002
203. Zang T, Broszczak DA, Broadbent JA, Cuttle L, Lu H, Parker TJ. The biochemistry of blister fluid from pediatric burn injuries: proteomics and metabolomics aspects. Expert Rev Proteomics (2016) 13(1):35–53. doi: 10.1586/14789450.2016.1122528
204. Rooney IA, Oglesby TJ, Atkinson JP. Complement in human reproduction: activation and control. Immunol Res (1993) 12(3):276–94. doi: 10.1007/BF02918258
205. Harris CL, Mizuno M, Morgan BP. Complement and complement regulators in the male reproductive system. Mol Immunol (2006) 43(1-2):57–67. doi: 10.1016/j.molimm.2005.06.026
206. Vander Borght M, Wyns C. Fertility and infertility: Definition and epidemiology. Clin Biochem (2018) 62:2–10. doi: 10.1016/j.clinbiochem.2018.03.012
207. Schick T, Steinhauer M, Aslanidis A, Altay L, Karlstetter M, Langmann T, et al. Local complement activation in aqueous humor in patients with age-related macular degeneration. Eye (Lond) (2017) 31(5):810–3. doi: 10.1038/eye.2016.328
208. Mondino BJ, Sumner H. Anaphylatoxin levels in human aqueous humor. Invest Ophthalmol Vis Sci (1986) 27(8):1288–92.
209. Gallenkamp J, Spanier G, Worle E, Englbrecht M, Kirschfink M, Greslechner R, et al. A novel multiplex detection array revealed systemic complement activation in oral squamous cell carcinoma. Oncotarget (2018) 9(3):3001–13. doi: 10.18632/oncotarget.22963
210. Hajishengallis G, Kajikawa T, Hajishengallis E, Maekawa T, Reis ES, Mastellos DC, et al. Complement-Dependent Mechanisms and Interventions in Periodontal Disease. Front Immunol (2019) 10:406. doi: 10.3389/fimmu.2019.00406
211. Schur PH, Austen KF. Complement in human disease. Annu Rev Med (1968) 19:1–24. doi: 10.1146/annurev.me.19.020168.000245
212. Lopez-Lera A, Corvillo F, Nozal P, Regueiro JR, Sanchez-Corral P, Lopez-Trascasa M. Complement as a diagnostic tool in immunopathology. Semin Cell Dev Biol (2019) 85:86–97. doi: 10.1016/j.semcdb.2017.12.017
213. Ekdahl KN, Persson B, Mohlin C, Sandholm K, Skattum L, Nilsson B. Interpretation of Serological Complement Biomarkers in Disease. Front Immunol (2018) 9:2237. doi: 10.3389/fimmu.2018.02237
214. Prohaszka Z, Kirschfink M, Frazer-Abel A. Complement analysis in the era of targeted therapeutics. Mol Immunol (2018) 102:84–8. doi: 10.1016/j.molimm.2018.06.001
215. Goicoechea de Jorge E, Lopez Lera A, Bayarri-Olmos R, Yebenes H, Lopez-Trascasa M, Rodriguez de Cordoba S. Common and rare genetic variants of complement components in human disease. Mol Immunol (2018) 102:42–57. doi: 10.1016/j.molimm.2018.06.011
216. Nimgaonkar VL, Prasad KM, Chowdari KV, Severance EG, Yolken RH. The complement system: a gateway to gene-environment interactions in schizophrenia pathogenesis. Mol Psychiatry (2017) 22(11):1554–61. doi: 10.1038/mp.2017.151
217. Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest (2017) 127(3):780–9. doi: 10.1172/JCI90962
218. Mamidi S, Hone S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology (2017) 222(1):45–54. doi: 10.1016/j.imbio.2015.11.008
219. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol (2007) 25(11):1256–64. doi: 10.1038/nbt1344
220. Patriquin CJ, Kuo KHM. Eculizumab and Beyond: The Past, Present, and Future of Complement Therapeutics. Transfus Med Rev (2019) 33(4):256–65. doi: 10.1016/j.tmrv.2019.09.004
221. Cofiell R, Kukreja A, Bedard K, Yan Y, Mickle AP Ogawa M, et al. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood (2015) 125(21):3253–62. doi: 10.1182/blood-2014-09-600411
222. Subias Hidalgo M, Martin Merinero H, Lopez A, Anter J, Garcia SP, Ataulfo Gonzalez-Fernandez F, et al. Extravascular hemolysis and complement consumption in Paroxysmal Nocturnal Hemoglobinuria patients undergoing eculizumab treatment. Immunobiology (2017) 222(2):363–71. doi: 10.1016/j.imbio.2016.09.002
223. Ruggenenti P, Daina E, Gennarini A, Carrara C, Gamba S, Noris M, et al. C5 Convertase Blockade in Membranoproliferative Glomerulonephritis: A Single-Arm Clinical Trial. Am J Kidney Dis (2019) 74(2):224–38. doi: 10.1053/j.ajkd.2018.12.046
224. Jodele S, Fukuda T, Mizuno K, Vinks AA, Laskin BL, Goebel J, et al. Variable Eculizumab Clearance Requires Pharmacodynamic Monitoring to Optimize Therapy for Thrombotic Microangiopathy after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant (2016) 22(2):307–15. doi: 10.1016/j.bbmt.2015.10.002
225. Pecoraro C, Ferretti AV, Rurali E, Galbusera M, Noris M, Remuzzi G. Treatment of Congenital Thrombotic Thrombocytopenic Purpura With Eculizumab. Am J Kidney Dis (2015) 66(6):1067–70. doi: 10.1053/j.ajkd.2015.06.032
226. Chehade H, Rotman S, Fremeaux-Bacchi V, Aubert V, Sadallah S, Sifaki L, et al. Blockade of C5 in Severe Acute Postinfectious Glomerulonephritis Associated With Anti-Factor H Autoantibody. Am J Kidney Dis (2016) 68(6):944–8. doi: 10.1053/j.ajkd.2016.06.026
227. Hokstad I, Deyab G, Wang Fagerland M, Lyberg T, Hjeltnes G, Forre O, et al. Tumor necrosis factor inhibitors are associated with reduced complement activation in spondylarthropathies: An observational study. PLoS One (2019) 14(7):e0220079. doi: 10.1371/journal.pone.0220079
Keywords: complement, soluble membrane attack complex, sC5b-9, cholesterol-dependent cytolysins, MAC, diagnostics, sMAC
Citation: Barnum SR, Bubeck D and Schein TN (2020) Soluble Membrane Attack Complex: Biochemistry and Immunobiology. Front. Immunol. 11:585108. doi: 10.3389/fimmu.2020.585108
Received: 19 July 2020; Accepted: 14 October 2020;
Published: 10 November 2020.
Edited by:
Robert John Crispin Gilbert, University of Oxford, United KingdomReviewed by:
Marjetka Podobnik, National Institute of Chemistry Slovenia, SloveniaCopyright © 2020 Barnum, Bubeck and Schein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott R. Barnum, c2Jhcm51bUBjbmluZWJpby5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.