
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 15 October 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.584048
 Yoshinobu Ikeno1†
Yoshinobu Ikeno1† Daiya Ohara2†
Daiya Ohara2† Yusuke Takeuchi2Hitomi Watanabe2Gen Kondoh2
Yusuke Takeuchi2Hitomi Watanabe2Gen Kondoh2 Kojiro Taura1Shinji Uemoto1
Kojiro Taura1Shinji Uemoto1 Keiji Hirota2*
Keiji Hirota2*Foxp3+ regulatory T (Treg) cells are pivotal in maintaining immunological self-tolerance and tissue homeostasis; however, it remains unclear how tissue Treg cells respond to liver injury and regulate chronic inflammation, which can cause liver fibrosis. We report here that hepatic Treg cells play a critical role in preventing liver pathology by suppressing inflammatory cellular immunity that can promote liver damage and fibrosis. Chronic liver inflammation induced by injections of carbon tetrachloride (CCl4) led to preferential expansion of hepatic Treg cells that prevented liver fibrosis. In contrast, depletion of Treg cells in the CCl4-induced liver fibrosis model exacerbated the severity of liver pathology. Treg depletion unleashed tissue cellular immunity and drove the activation and expansion of the pro-fibrotic IL-4-producing T helper 2 cells, as well as CCR2high Ly-6Chigh inflammatory monocytes/macrophages in the inflamed liver. Although Treg expression of amphiregulin plays a key role in tissue remodeling and repair in various inflammation models, amphiregulin from hepatic Treg cells, the largest producer among liver immune cells, was dispensable for maintaining liver homeostasis and preventing liver fibrosis during CCl4-induced chronic inflammation. Our results indicate that Treg cells control chronic liver inflammation and fibrosis by regulating the aberrant activation and functions of immune effector cells. Harnessing Treg functions, which effectively regulate tissue cellular immunity, may be a therapeutic strategy for preventing and treating liver fibrosis.
Liver fibrosis is characterized by the excessive accumulation of extracellular matrix proteins such as collagen, resulting from chronic liver inflammation mediated by infectious agents, excessive alcohol abuse, non-alcoholic steatohepatitis (NASH), and autoimmune liver diseases (1). It is widely appreciated that various types of inflammatory immune cells mediate chronic tissue inflammation in response to liver injury and the pathogenesis of liver fibrosis. For example, abundant inflammatory monocytes and macrophages are recruited to the inflamed liver tissue and are involved in the augmentation of liver fibrosis. The imbalance of IFN-γ-producing T helper (Th1) cells and IL-4-producing T helper (Th2) cells, which are the anti-fibrotic and pro-fibrotic Th subsets, respectively, also promotes liver fibrosis (2). Thus, activated innate and adaptive immune cells stimulate hepatic stellate cells to secrete extracellular matrix proteins, resulting in fibrotic changes in the liver that severely impair physiological liver functions. However, the mechanisms by which fibrotic tissue immune responses in the inflamed liver are negatively regulated remain unknown.
Foxp3+ regulatory T (Treg) cells play a pivotal role in maintaining immunological self-tolerance and tissue homeostasis by regulating inappropriate immune responses to self-organs and tissues (3). Under various pathogenic conditions in humans and animal models, Treg cells are recruited into the liver to regulate tissue inflammation and pathology (4–8). There is also accumulated evidence that tissue Treg cells actively engage in tissue remodeling and repair in stressed or damaged organs, for example, by secreting amphiregulin, a key molecule for tissue regeneration and repair in various inflammation models, including brain ischemia, muscle injury, and viral pneumonia (9–11). Thus, it is becoming increasingly clear that Treg cells have tissue-specific transcriptional signatures and functions to control tissue injury in a context-dependent manner. However, little is known about the mode of Treg actions regulating chronic liver inflammation and fibrosis.
Here, using a carbon tetrachloride (CCl4)-induced liver fibrosis model, we investigated the dynamics of cellular and molecular signatures in the inflamed liver regulated by Treg cells and Treg-specific amphiregulin.
C57BL/6J mice were purchased from CLEA Japan. Foxp3-DTR-eGFP (FDG) mice (12) and Foxp3YFP–Cre mice (13) were purchased from the Jackson Laboratory. Aregfl/fl mice were generated by inserting Loxp3 sites flanking exons 3 and 4 of the murine Areg gene using the CRISPR/Cas9 system. These mice were crossed with Foxp3YFP–Cre mice. All mice used had a C57BL/6J background and were maintained under specific-pathogen-free conditions. All animal experiments were approved by the Ethical Committee of Institute for Frontier Life and Medical Sciences and Graduate School of Medicine, Kyoto University. All relevant experiments were performed in accordance with the institutional guidelines.
Liver fibrosis was induced by intraperitoneal (i.p.) injections of CCl4 (0.5 μl/g body weight; Wako) diluted 1:4 in corn oil (Wako) twice a week for 6 weeks as described previously (14). In brief, CCl4 is converted into several radical forms by liver cytochrome P450 enzymes. The resulting radicals induce hepatotoxic damage and result in fibrosis. Mice were sacrificed 3 days after the final CCl4 injection. Liver fibrosis was also induced by feeding choline-deficient L-amino acid-defined (CDAA) diet for 8 weeks (Dyets Inc, Bethlehem, PA) (15). In brief, CDAA diet affects lipid metabolisms, resulting in steatohepatitis and liver fibrosis (16). For Treg-cell depletion experiments, FDG mice were injected i.p. with diphtheria toxin (DT; 1 μg in 100 μl PBS; Merck) every other day for 2 weeks (Figure 2A and Supplementary Figure 1A).
Serum alanine transaminase (ALT) levels were measured using an Infinity kit (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. Formalin-fixed frozen livers were stained with hematoxylin-eosin (Wako) or Sirius red (Wako) for histological analysis. The regions of the entire median lobe of the liver stained with Sirius red were extracted and quantified using Image J software (NIH). The mean percentage of the Sirius red-positive area was determined from four images of each mouse.
The collected livers were cut into small pieces and mashed through a 70 μm mesh filter. Leukocytes were then enriched by 37.5% Percoll (GE Healthcare) separation, followed by red blood cell lysis with RBC lysis buffer (Sigma-Aldrich). The resulting single-cell suspensions were incubated with anti-mouse CD16/32 antibody (2.4G2; BD Bioscience) and Fixable Viability Dye eFluorTM 780 (eBioscience). The cells were then stained using fluorescent conjugated antibodies against cell surface markers. For Foxp3 and amphiregulin staining, the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) was used in accordance with the manufacturer’s instructions. For intracellular cytokine staining, cells were re-stimulated with phorbol12-myristate 13 acetate (50 ng/ml; Sigma-Aldrich) and ionomycin (500 ng/ml; Sigma-Aldrich) in the presence of Brefeldin A (1 μg/ml; Merck) in IMDM (Sigma-Aldrich) containing 5% FBS (GIBCO), penicillin-streptomycin (Nacalai Tesque), 2-mercaptoethanol (GIBCO), GlutaMAX (GIBCO), sodium pyruvate (GIBCO), and MEM NEAA (GIBCO) for 4 h. Each sample was then fixed using 3.7% formaldehyde (Sigma-Aldrich), permeabilized with 0.1% NP-40 (Nacalai Tesque), and stained with antibodies against cytokines. The monoclonal antibodies used for flow cytometry analysis were as follows: anti-mouse CD3e (145-2C11), CD25 (PC61), Ly-6C (AL-21), IL-4 (11B11), Ki-67 (B56), PE-Streptavidin (all from BD Bioscience), anti-mouse CD4 (RM4-4), CD8a (53-6.7), CD11b (M1/70), CD45.2 (104), F4/80 (BM8), IL-33Ra (D1H9), Ly-6G (1A8), CCR2 (SA203G11), IL-17A (TC11-18H10.1) (all from Biolegend), anti-mouse Foxp3 (FJK-16s), and IFN-γ (XMG1.2) (both from eBioscience), and anti-mouse amphiregulin-biotin (BAF989; R&D Systems). All flow cytometry data were acquired using a BD FACSCantoII or BD FACS AriaSORP devise and analyzed using FlowJo software (Tree Star, Inc.).
Liver tissues were homogenized in PBS containing a cocktail of protease inhibitors using MagNALyzer (Roche). After centrifugation, IL-33 in the supernatant was measured using Mouse IL-33 ELISA Kit (Biolegend). The data was normalized by the original liver weight.
For extraction of total RNA from hepatic immune subsets, cells were directly sorted into 500 μl of TRIzol (Invitrogen) and were lysed by vortexing. For extraction of total RNA from liver tissues, the tissue was homogenized in 500 μl of Trizol using MagNALyzer. After RNA extraction, genomic DNA was removed using an RNeasy mini kit and RNase-Free DNase Set (QIAGEN). Purified RNA was reverse-transcribed with SuperScript VILO (Invitrogen) and the cDNA was used for quantitative PCR with Fast SYBR Green Master Mix (Applied Biosystems) on a LightCycler 480 (Roche). The expression levels of target genes were quantified after normalization to 18S rRNA expression. The sequence primers used for quantitative RT-PCR were as follows: aSMA-F: 5′CTGGGACGACATGGAAAAG3′, aSMA-R: 5′GTT CAGTGGTGCCTCTGTCA3′, Col1a1-F: 5′ACATGTTCAGCT TTGTGGACC3′, Col1a1-R: 5′TAGGCCATTGTGTATGCAGC3′, Areg-F: 5′GACTCACAGCGAGGATGACA3′, Areg-R: 5′GGCT TGGCAATGATTCAACT3′, 18s-F: 5′AGTCCCTGCCCTTTGT ACACA3′, and 18s-R: 5′CGATCCGAGGGCCTCACTA3′.
DNase-treated total RNA (1 μg) from liver tissues was used to construct a sequencing library using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB) and NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB) in accordance with the manufacturer’s instructions. The library DNA was sequenced using NextSeq 500 (Illumina). The sequenced reads were aligned to the UCSC mm10 mouse genome using Hisat2 (17) (version 2.1.0). The read counts of each gene were obtained using featureCounts (18) (version 1.6.4). Log2 fold change and adjusted p value of differential gene expression were calculated and statistically evaluated using DESeq2 (19) (version 1.20.0). Transcript per million (TPM) was calculated from read counts data and then each gene expression is normalized to Log10(TPM + 1). In Figure 4E, the expression of marker genes of inflammatory monocytes and macrophages are shown. For gene set enrichment analysis (GSEA) (Figure 4F), microarray gene expression datasets (E-MEXP-3177) (20) were analyzed to generate gene signatures of hepatic myeloid cell subsets using the limma package (21) (version 3.38.3) in R (version 3.5.1). Genes with an adjusted p value < 0.05 were ranked by log2 fold change. The top 200 up- and down-regulated genes were defined as hepatic Ly-6Chigh inflammatory monocyte/macrophage and Ly-6Clow macrophage gene signatures, respectively, and used to make gene set files. Then, ranked list files for GSEA were generated by using log2 fold change from DESeq2 analysis of CCl4-treated Treg-depleted DT(+) and DT(−) livers. These data files were processed with GSEA v3.0 software (22) and the resultant enrichment plots were shown.
Statistical analyses were performed using GraphPad PRISM7 (GraphPad Software). A two-tailed t-test or ANOVA followed by Tukey’s multiple comparisons test was used for statistical hypothesis testing. A p-value < 0.05 was considered statistically significant. Bar graphs are presented as mean ± standard deviation (SD). The sample sizes for all data are described in the figure legends.
To investigate the cellular dynamics of CD4+ T cells during liver inflammation, we induced chronic liver injury and fibrosis in an established procedure by injecting CCl4 for 6 weeks (Figure 1A). The kinetics of Foxp3+ Treg cells and effector Th subsets were then analyzed. The frequencies of Foxp3+ Treg cells were significantly increased in the livers of CCl4-treated mice, compared with those in the livers of vehicle-treated control mice, while the levels of Treg cells in the spleen were comparable between the two groups (Figures 1B,C). Consistently, the fraction of Ki67+ Treg cells from the CCl4-treated liver was significantly increased (Figure 1D). Notably, ST2 expression of hepatic Treg cells was higher than splenic Treg cells and after CCl4 injections, the frequencies of ST2+ Treg cells were significantly increased and correlated with protein levels of IL-33, suggesting that Treg expansion may be driven by IL-33 released from damaged hepatocytes (Figures 1E–G). In terms of effector Th subsets, IL-17A-producing T helper (Th17) cells were slightly increased in the CCl4-treated liver, whereas the proportions of Th1 and Th2 cells in the liver between the groups were unchanged (Figure 1H).
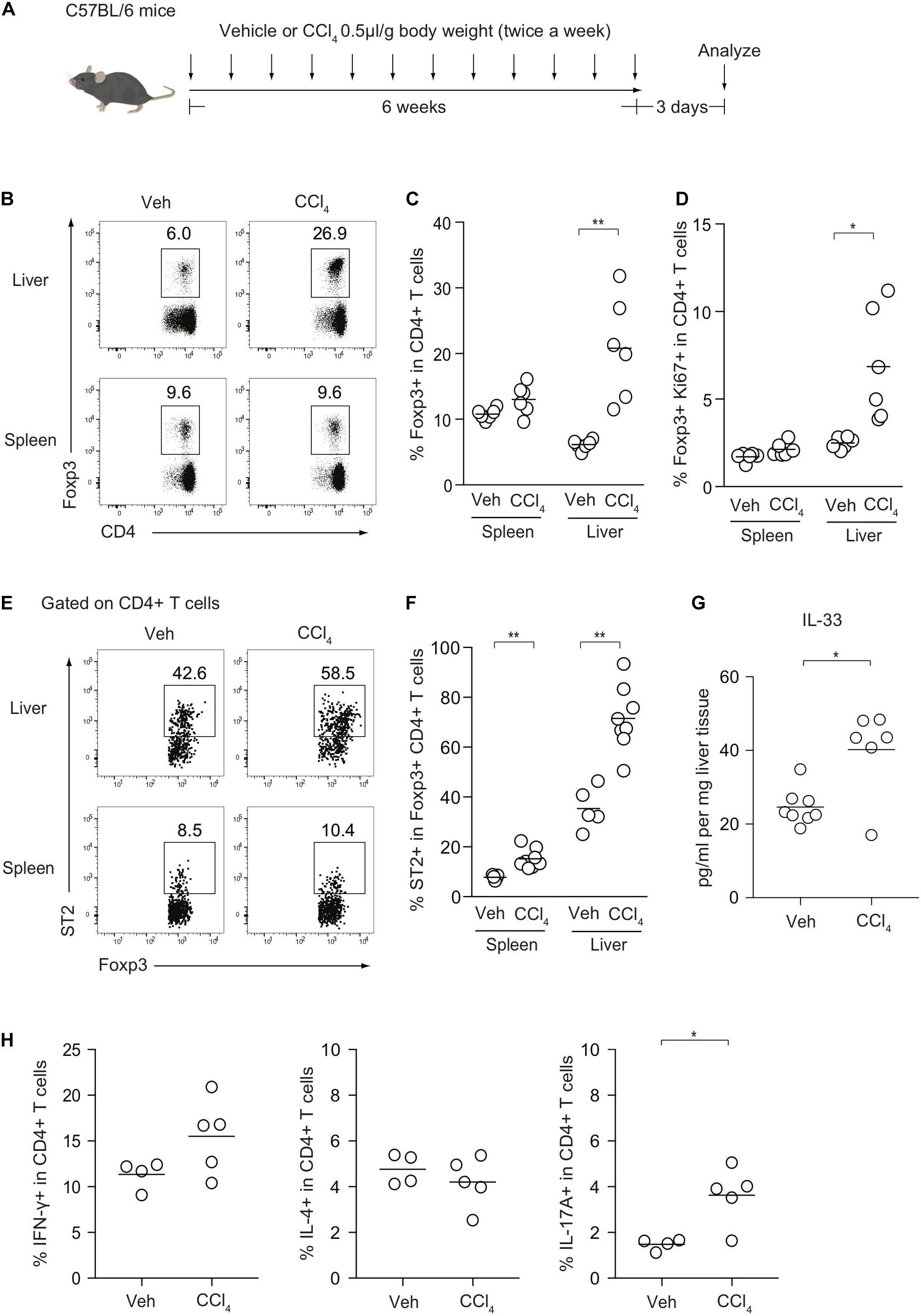
Figure 1. Kinetics of Foxp3+ Treg cells and effector Th subsets after CCl4-induced chronic liver injury. (A) Experimental design of CCl4-induced liver injury. (B) Flow cytometry analysis of hepatic and splenic CD4+ T cells for Foxp3 expression after intraperitoneal injections of either CCl4 or vehicle (Veh) as illustrated in panel (A). (C,D) Proportions of Foxp3+ or Ki67+ Foxp3+ in CD4+ T cells in the spleen or liver (n = 6). (E,F) Proportions of ST2+ in Foxp3+CD4+ Treg cells in the spleen or liver (n = 5–8). (G) Quantification of IL-33 protein by ELISA in CCl4- or vehicle-treated livers (n = 6–8). (H) Proportions of IFN-γ, IL-4, and IL-17-producing CD4+ T cells in the liver from CCl4- (n = 5) or Veh-treated mice (n = 4). Horizontal bars indicate the means in panels (C,D,F–H); *p < 0.05, **p < 0.01. Data are representative of three independent experiments in panels (B,C), and two independent experiments in panels (E–G) and pooled from two independent experiments in panels (D,H).
The observation of the preferential Treg expansion in damaged livers prompted us to examine the role of Foxp3+ Treg cells in the regulation of CCl4-induced liver inflammation and fibrosis. To this end, we removed Treg cells in the course of the development of the model using Foxp3-DTR-eGFP knock-in (FDG) mice in which Foxp3+ Treg cells are labeled as eGFP+ cells and can be specifically depleted by the administration of DT (Figure 2A). We confirmed that hepatic Treg cells in FDG mice were successfully depleted in both groups (Figures 2B,C). Using this system, we next evaluated if Treg depletion influenced the fibrotic regions of the liver as assessed by Sirius red staining and the serum levels of ALT, a marker of hepatocyte damage. Notably, the absence of Treg cells for the last 2 weeks over the course of CCl4 injections significantly increased the area of collagen deposition and the amounts of ALT in the sera compared with DT-untreated mice (Figures 2D–F). Although liver injury based on serum ALT levels could be exacerbated by Treg depletion alone, this acute inflammation for a limited period was not sufficient to induce liver fibrosis and the severe fibrosis required a combination of chronic inflammation by CCl4 and leukocyte infiltration in the liver by Treg ablation (Figures 2D–F). Consistent with collagen deposition in histology, the expression levels of the Col1al and Acta2 (α-SMA) markers for tissue fibrosis were significantly increased in the inflamed liver of CCl4- and DT-injected FDG mice (Figure 2G). We also observed similar findings with another liver fibrosis model driven by choline-deficient L-amino-defined (CDAA) diet. We fed this diet to FDG mice for 8 weeks and injected DT for the final two weeks to examine liver fibrosis (Supplementary Figure 1A). Similar to the CCl4 model, Treg cells significantly were increased in the liver by feeding the diet (Supplementary Figures 1B,C) and Treg depletion aggravated liver fibrosis assessed by liver fibrotic regions and expression levels of Col1a1 and Acta2 (α-SMA) (Supplementary Figures 1D,E). Taken together, these data indicated that hepatic Treg cells predominantly proliferate in response to CCl4-induced liver injury and suppress chronic inflammation as well as the progression of liver fibrosis.
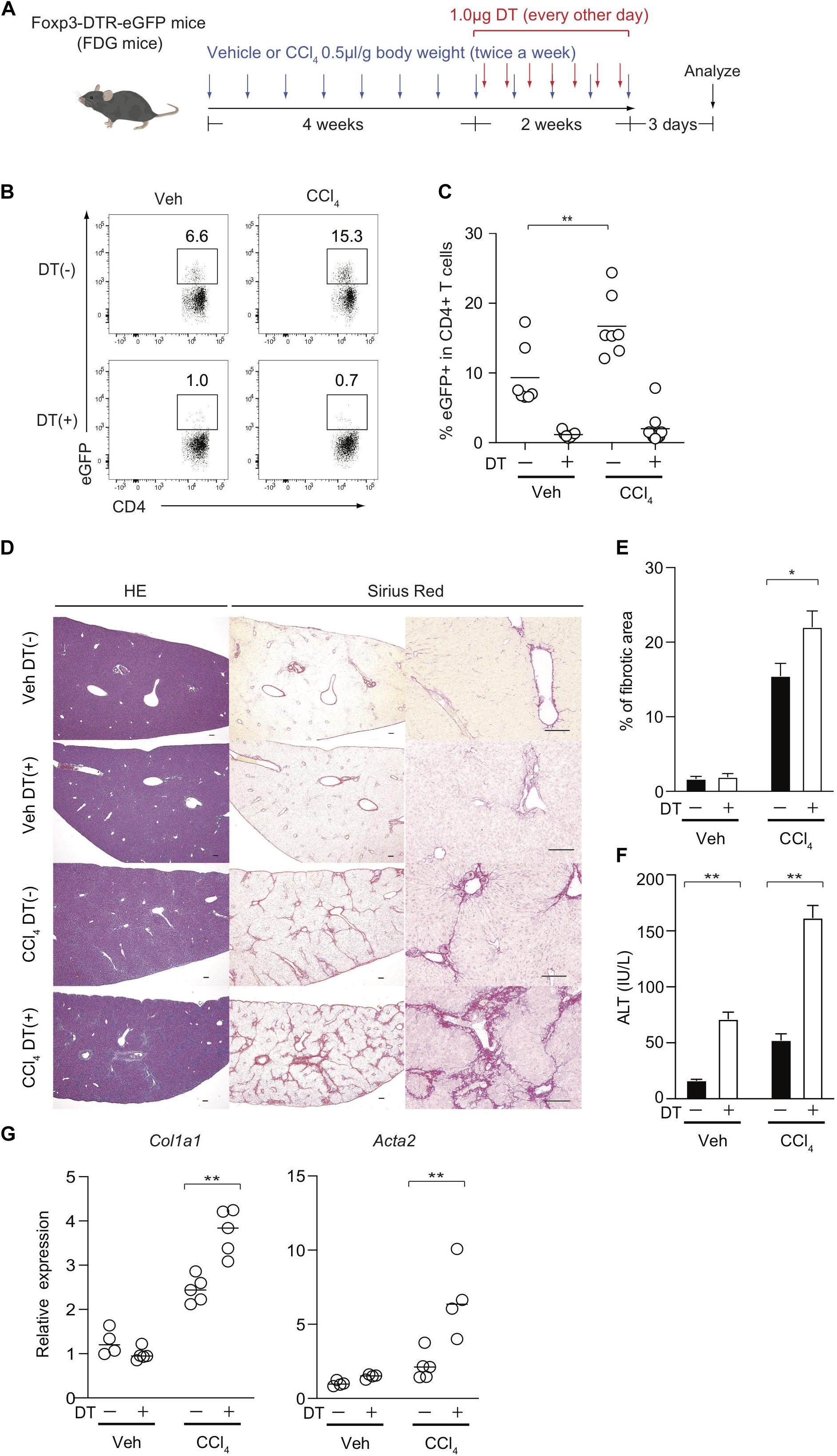
Figure 2. Treg-cell depletion aggravates CCl4-induced liver damage and fibrosis. (A) Experimental design of Treg-cell depletion during CCl4-induced liver injury. FDG mice were untreated or administered diphtheria toxin (DT) during the late phase of CCl4 and vehicle (Veh) treatment. (B,C) Proportions of eGFP+ in hepatic CD4+ T cells of FDG mice with DT(+) or without DT(−) as in panel (A). (D) Representative images of hematoxylin and eosin (H&E) and Sirius red (SR) staining of the liver of DT(−) and DT(+) FDG mice. Scale bars indicate 100 μm. (E) Quantification of fibrotic areas of the liver section evaluated by SR-positive area (n > 5). (F) Serum level of ALT of DT(−) and DT(+) FDG mice (n > 5). Vertical bars denote SD. Horizontal bars indicate the means in pannels (C,G). (G) Quantitative RT-PCR analysis for the expression of Col1a1 and Acta2 (α-SMA) in the liver tissue of DT(−) and DT(+) FDG mice (n = 4). Horizontal bars indicate the means; *p < 0.05, **p < 0.01. Data are representative of two independent experiments in panel (B) and are pooled in panel (C). Data are representative of three independent experiments in panels (E–G).
We next investigated the Treg-mediated molecular mechanisms underlying the prevention of liver inflammation and fibrosis in this model. Amphiregulin produced by immune subsets such as Treg cells and innate lymphoid cells has recently been highlighted as a key molecule for tissue regeneration and repair during acute and chronic inflammation (9–11, 23, 24). Among the immune cells in the CCl4-treated inflamed liver, which included CD4+ and CD8+ T cells, B220+ B cells, CD11b+ myeloid cells, NK1.1+ NK cells, and NK1.1+CD3+NKT cells, the Areg gene encoding amphiregulin was predominantly expressed by Treg cells (Figure 3A). To determine the specific effect of Treg-derived amphiregulin on CCl4-induced liver pathology, we generated Foxp3YFP–Cre Aregfl/fl mice in which the expression of amphiregulin from Treg cells was selectively deleted (Figures 3B,C). The absence of amphiregulin in Treg cells did not affect the proportions of hepatic Treg cells as compared with control Foxp3YFP–Cre Aregfl/wt mice (Figure 3D). We then treated Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice with vehicle or CCl4, and assessed gene expression profiles by q-PCR and RNA-seq, and liver fibrosis by staining with Sirius red. Despite the critical role of amphiregulin in various inflammation models, there were no significant changes in the expression of Col1al and Acta2 and Sirius red-positive fibrotic areas in the liver of Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice (Figures 3E,F). RNA-seq analysis of liver tissues from Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice six weeks after CCl4 treatment further revealed that the gene profiles between the two groups were almost identical with two minor genes with adjusted P value less than 0.05 in DESeq2 analysis. (Figure 3G). In addition, we collected liver tissue samples from Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice for RNA-seq analysis at three different time points before or during the course of CCl4 treatment (weeks 0, 2, and 6) and performed principal component analysis. No remarkable difference in the gene expression profiles, including markers for tissue fibrosis and T cell-mediated inflammation, were observed at any time point (Figure 3H and Table 1). These results demonstrated that the Treg-derived amphiregulin is dispensable for maintaining liver homeostasis and preventing CCl4-induced liver pathology.
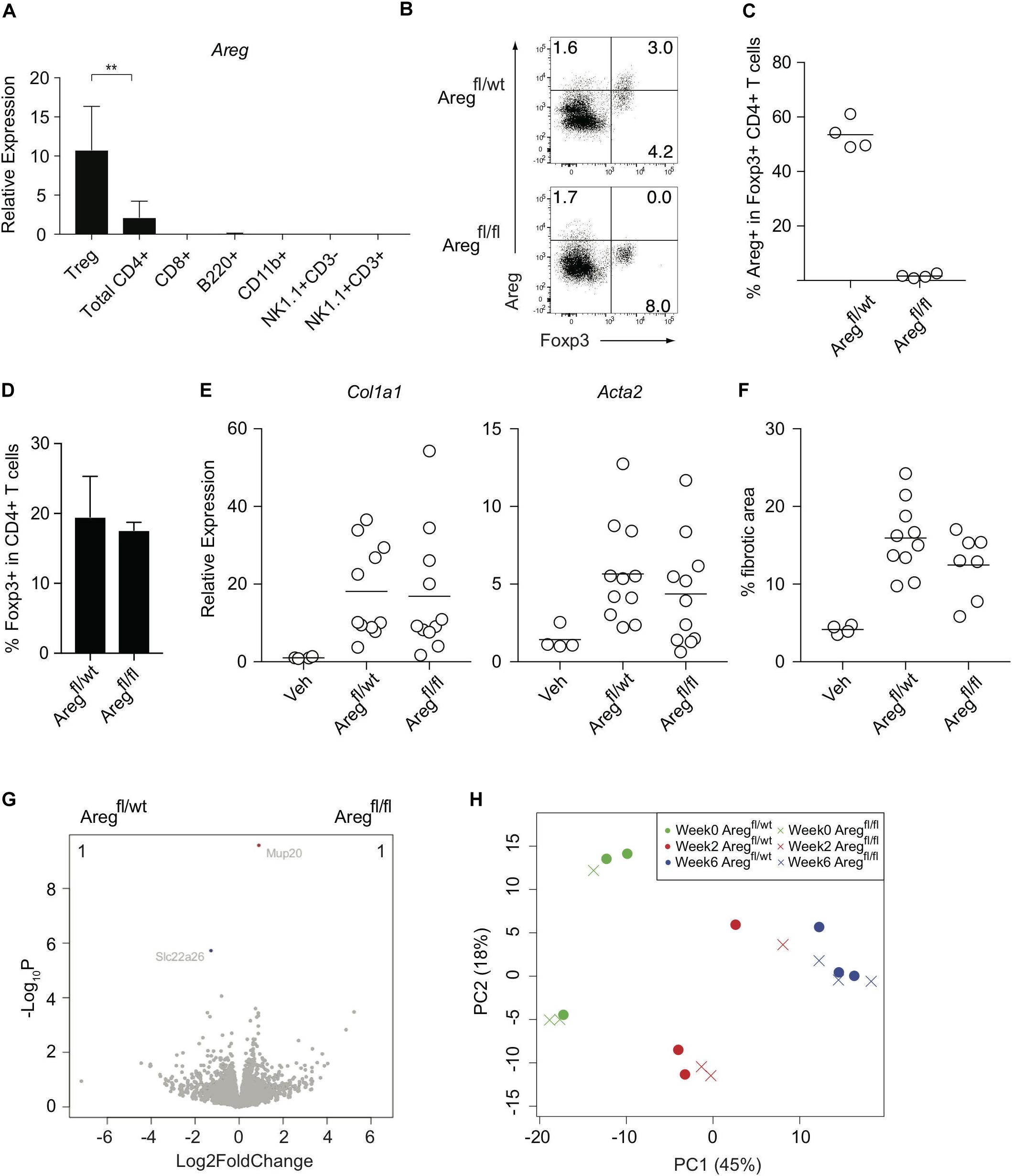
Figure 3. Treg-derived amphiregulin is dispensable for preventing CCl4-induced liver pathology. (A) Quantitative RT-PCR analysis of the amphiregulin gene (Areg) in CD4+ eGFP+ T (Treg) cells, CD4+ T cells, CD8+ T cells, B220+ B cells, CD11b+ myeloid cells, NK1.1+ CD3- NK cells, and NK1.1+CD3+ NKT cells isolated from the inflamed liver of FDG mice after the 6-week treatment with CCl4 (n = 2–7). (B) Intracellular Areg staining of hepatic CD4+ T cells from CCl4-treated Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice. (C) Proportions of Areg expression in hepatic Foxp3+CD4+ T cells as in panel (B) (n = 4 each). (D) Proportions of Foxp3 expression in hepatic CD4+ T cells in Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice. (E) Quantitative RT-PCR analysis for the expression of Col1a1 and Acta2 in the liver tissue after CCl4 treatment. (F) Proportions of fibrotic areas of the liver evaluated by Sirius red staining in CCl4-treated Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice or vehicle (Veh)-treated control mice. (G) RNA-seq analysis of whole liver tissues from Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice 6 weeks after CCl4 treatment. The volcano plots show genes with an adjusted P value < 0.05 in DESeq2 analysis between the groups. The genes upregulated in Foxp3YFP–Cre Aregfl/wt mice are shown in blue, and the genes downregulated are shown in red. The number of the genes is shown in the right and left top of the plots. (H) PCA analysis of liver RNA-seq data from the following six groups: Foxp3YFP–Cre Aregfl/wt or Foxp3YFP–Cre Aregfl/fl mice before CCl4 treatment (Week 0), or 2 weeks or 6 weeks after the treatment (Week 2) or (Week 6), respectively. Horizontal bars indicate the means in panels (C,E,F). Vertical bars denote SD; **p < 0.01. Data are representative of two independent experiments in panel (A). Data are pooled from two independent experiments in panels (E,F).
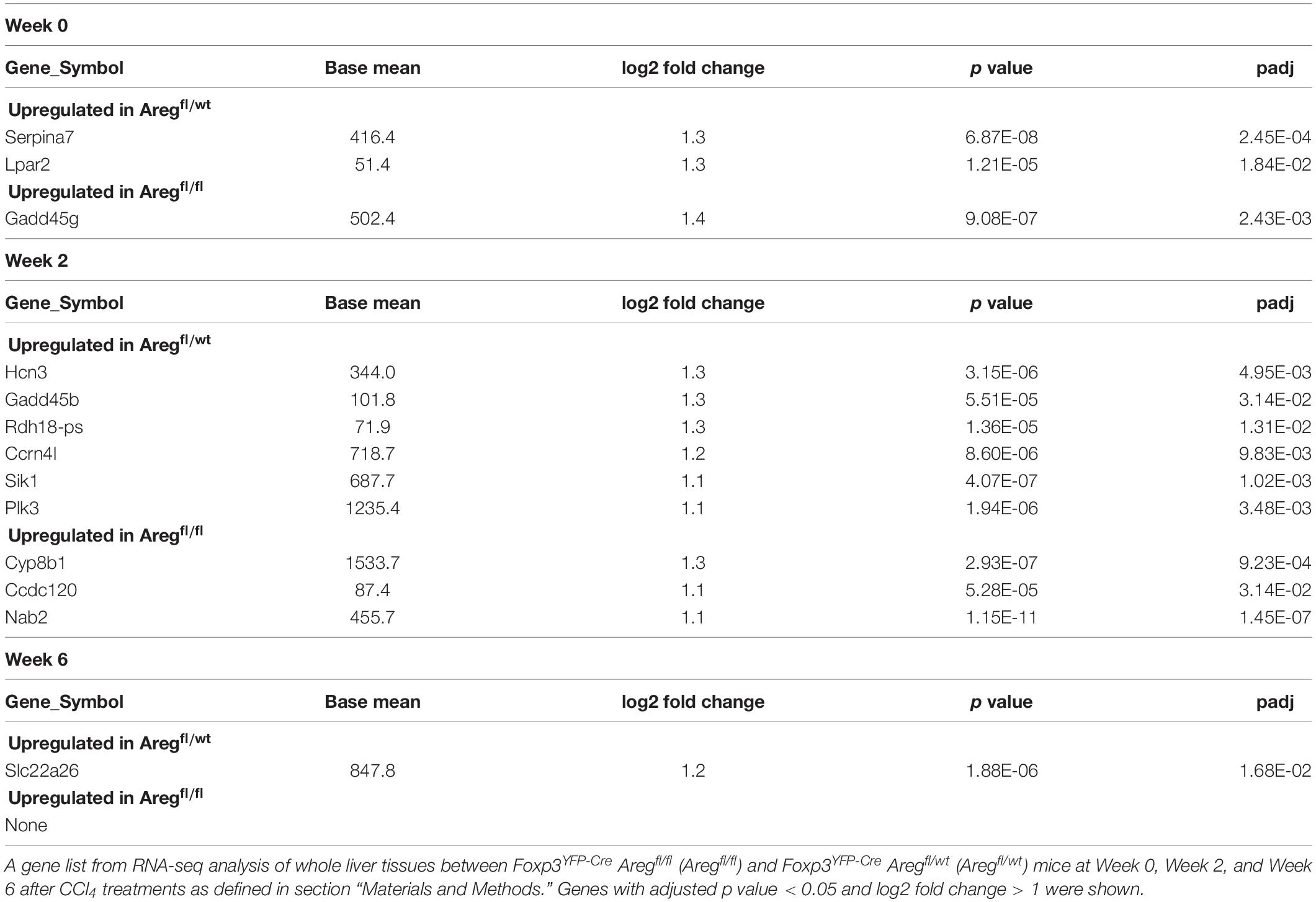
Table 1. Differentially expressed genes between Foxp3YFP–Cre Aregfl/wt and Foxp3YFP–Cre Aregfl/fl mice at the three time points.
Because Treg depletion exacerbated CCl4-induced liver damage and fibrosis (Figure 2), we assessed inflammatory cellular components in the inflamed liver from FDG mice with or without DT treatment. Although it is technically challenging to detect small numbers of antigen-specific immune responses induced by CCl4-mediated liver damages, the proportions of hepatic Th1 and Th2 cells, but not Th17 cells, in both vehicle- and CCl4-treated FDG mice, were dramatically increased upon DT administration (Figures 4A,B). The mediators involved in fibrogenesis are distinct from those involved in inflammation (25). In contrast to the type 2 cytokine IL-4 signaling as a pro-fibrotic mediator, IFN-γ has been characterized as an anti-fibrotic cytokine in the context of CCl4-mediated liver fibrosis (26, 27). Notably, the Th2/Th1 cell ratio, which is associated with tissue fibrosis, was specifically increased by the combination of CCl4 treatment and Treg depletion, but not by Treg depletion alone (Figure 4C). The proportions of CD8+ cytotoxic T cells in the CD45+ population also increased (Figure 4D). We next focused on Ly-6Chigh inflammatory monocytes/macrophages, which are also reported as a pro-fibrotic immune subset (20, 28), and compared the expression of relevant genes in the inflamed liver of CCl4-treated FDG mice with or without DT. Several cell surface markers for Ly-6Chigh inflammatory monocytes/macrophages, such as Ly6c2, Ccr2, Csf1r, and Sell, were significantly upregulated in Treg-depleted FDG mice (Figure 4E). In addition, GSEA revealed the significant enrichment of Ly-6Chigh inflammatory monocytes/macrophage signatures by Treg depletion. Ly-6Clow macrophage signatures, which are supposed to be an anti-fibrotic subset, were unchanged (Figure 4F). We also validated this finding by FACS analysis. The frequencies of Ly-6Chigh CCR2high inflammatory monocytes/macrophages were substantially expanded in the Treg-depleted fibrotic liver (Figures 4G,H). Collectively, these data indicated that Treg cells suppress the aberrant activation and expansion of the pro-fibrotic cellular immunity, especially Th2 cells and Ly-6Chigh CCR2high inflammatory monocytes/macrophages, during CCl4-induced liver inflammation, thereby inhibiting the progression of liver fibrosis.
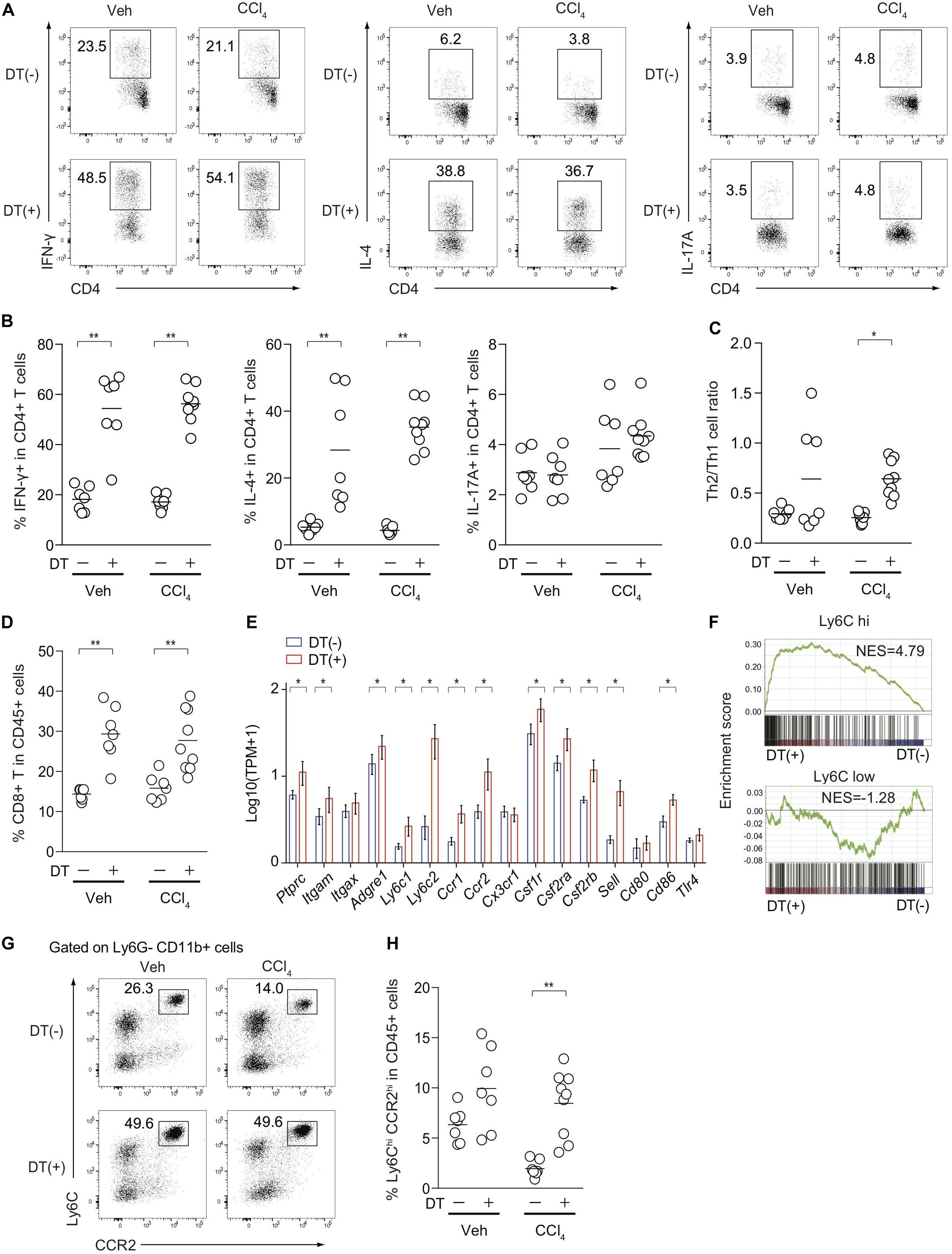
Figure 4. Treg-cell depletion in CCl4-treated FDG mice induces the activation and expansion of pro-fibrotic cellular immunity. (A,B) Proportions of IFN-γ, IL-4, and IL-17-producing hepatic CD4+ T cells from DT(−) or DT(+) FDG mice after vehicle (Veh) or CCl4 treatment. (C) Th2/Th1 cell ratios of hepatic CD4+ T cells from DT(−) or DT(+) FDG mice after Veh or CCl4 treatment. (D) Proportions of CD8a+ T cells in hepatic CD45.2+ cells after treatment. (E) Gene expression profiles of hepatic monocyte/macrophage associated genes from RNA-seq data of CCl4-treated DT(−) or DT(+) FDG mice (n = 3–4). Vertical bars denote SD. (F) Gene set enrichment analysis of liver RNA-seq data from DT(−) and DT(+) FDG mice with Ly-6Chigh- and Ly-6Clow-enriched signature as detailed in section “Materials and Methods.” (G,H) Proportions of Ly-6Chigh CCR2high cells in hepatic Ly-6G- CD11b+ cells (G) and CD45.2+ cells (H) from CCl4-treated DT(−) or DT(+) FDG mice. Horizontal bars indicate the means in panels (B–D,H); *p < 0.05, **p < 0.01. Data are representative of two independent experiments in panels (A,G). Data are pooled from two experiments in panels (B–D,H).
The current research focus seeks to understand tissue adaptation and organ-specific roles of Treg cells under physiological and pathological conditions. Accumulated evidence has indicated diverse immune suppression mechanisms of tissue Treg cells that operate in a cell-contact or secretory molecule-dependent manner (3). Moreover, recent findings have provided novel insight into Treg non-canonical functions in tissue regeneration and repair by the crosstalk between Treg and mesenchymal cells through amphiregulin in the inflamed brain, muscle, and lung (9–11). In this study, we demonstrate that Treg cells prevent liver pathology in a CCl4-induced liver fibrosis model by limiting the aberrant activation of pro-fibrotic cellular immunity, including Th2 cells and Ly-6Chigh CCR2high inflammatory monocytes/macrophages. Conversely, the tissue protective role of Treg-derived amphiregulin was limited in CCl4-induced liver inflammation and fibrosis, despite the predominant expression of amphiregulin in hepatic Treg cells.
Hepatic Treg cells sense various inflammatory cues to proliferate (29, 30) and regulate the pathogenesis of liver fibrosis in an acute disease model induced by bile duct ligation (29, 31). In addition, Foxp3+ human Treg cells expand in the fibrotic liver of hepatitis C virus infected patients (7). Although it was suggested that Treg cells might promote liver fibrosis based on Treg depletion experiments by anti-CD25 mAbs, the treatment of the antibody could also deplete pro-fibrotic effector T cells simultaneously, making the interpretation of the effect of the antibody on Treg or effector T cells difficult (8). In this study using FDG mice in which Foxp3+ Treg cells can specifically be depleted, we demonstrated that CCl4-induced liver inflammation and fibrosis also drive hepatic Foxp3+ Treg expansion, suggesting that common self-antigens or damage-associated molecular patterns (DAMPs) including IL-33 released from stressed or damaged hepatocytes are likely to stimulate Treg cells to proliferate and regulate excessive inflammation and liver pathology. Treg cells possess TCRs with high self-reactivity, which are key characteristics for mediating immunological self-tolerance (32, 33). Conversely, the experimental removal of Treg cells or Foxp3 mutations in mice and humans causes systemic autoimmune diseases, including liver inflammation (12, 34, 35). Thus, self-reactive Treg cells in the liver actively engage in liver homeostasis and prevent liver pathology resulting from hepatotoxic chemicals and immune activation. We found that Treg cells preferentially regulate cellular signatures during chronic liver inflammation, such as Th2 cells and Ly-6Chigh CCR2high inflammatory monocytes/macrophages, which were previously reported to produce the pro-fibrotic mediators IL-4 and TGF-β, respectively (28, 36, 37) and may be regulated by Treg-derived IL-10 (38). Group 2 innate lymphoid cells are also considered a pro-fibrotic population in the liver and promote liver fibrosis in an IL-33 dependent manner in the CCl4 model (39). These pro-fibrotic mediators activate hepatic stellate cells to secrete extracellular matrix proteins for the progression of liver fibrosis (40). However, it remains to be determined whether hepatic Treg cells directly control cellular immunity and stellate cells in situ, or whether Treg cells located in lymphoid organs like lymph nodes suppress the activation and trafficking of the effector cells before infiltrating into the liver.
Tissue Treg cells are capable of adapting to organ-specific environmental cues, including DAMPs, and subsequently acquire specific functions to control immune responses and tissue repair. In damaged tissue, IL-33, a well-known alarmin or DAMP, is abundantly released from necrotic cells and stimulates local Treg cells via signals from ST2, an IL-33 receptor, to produce amphiregulin, an epithelial growth factor receptor ligand (10). Recently, cell type-specific amphiregulin has been highlighted as a key molecule for tissue regeneration and repair in a context-dependent manner (9–11). Amphiregulin can also mediate protection of injured liver tissue by promoting hepatocyte proliferation (41, 42). Our RNA-seq data using Foxp3YFP–Cre Aregfl/fl mice clearly indicated that Treg-derived amphiregulin is not required to maintain liver homeostasis or to protect from liver inflammation and fibrosis through amphiregulin-mediated survival signals and cell growth, at least in CCl4-induced chronic liver injury, although Treg cells predominantly produce amphiregulin among immune cells in the liver. However, this finding does not necessarily exclude the possibility that hepatic Treg cells play a protective role in directly modulating regeneration of injured hepatocytes through unappreciated mechanisms. Further work is needed to explore this aspect of hepatic Treg non-canonical functions that can act on mesenchymal cells to prevent liver pathology during chronic inflammation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA010411.
The animal study was reviewed and approved by the Ethical Committee of Institute for Frontier Life and Medical Sciences and Graduate School of Medicine, Kyoto University.
YI, DO, and KH designed the study. YI, DO, YT, HW, GK, and KH performed the experiments. DO performed the statistical analysis and bioinformatics analysis of the RNA-seq data. KT and SU provided intellectual input. YI, DO, YT, and KH wrote the manuscript. KH supervised the project. All authors reviewed the manuscript.
This work was supported by JSPS Grant-in-Aid for Scientific Research (19H01026 to KH and 16H06233 to KH) and Takeda Science Foundation (KH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank T. Kondoh and Y. Sando for technical assistance with RNA sequencing. Illustration of C57BL/6 mice in Figures 1A, 2A and Supplementary Figure 1A was obtained from TogoPictureGallery (https://togotv.dbcls.jp/togopic.2014.60.html).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.584048/full#supplementary-material
2. Xu R, Zhang Z, Wang FS. Liver fibrosis: mechanisms of immune-mediated liver injury. Cell Mol Immunol. (2012) 9:296–301. doi: 10.1038/cmi.2011.53
3. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
4. Tedesco D, Thapa M, Gumber S, Elrod EJ, Rahman K, Ibegbu CC, et al. CD4+Foxp3+ T cells promote aberrant immunoglobulin G production and maintain CD8+ T-cell suppression during chronic liver disease. Hepatology. (2017) 65:661–77. doi: 10.1002/hep.28894
5. Gu L, Deng WS, Sun XF, Zhou H, Xu Q. Rapamycin ameliorates CCl4-induced liver fibrosis in mice through reciprocal regulation of the Th17/Treg cell balance. Mol Med Rep. (2016) 14:1153–61. doi: 10.3892/mmr.2016.5392
6. Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP, et al. Type 3 cytokines IL-17A and IL-22 drive TGF-β dependent liver fibrosis. Sci Immunol. (2018) 3:1–16. doi: 10.1126/sciimmunol.aar7754
7. Claassen MAA, de Knegt RJ, Tilanus HW, Janssen HLA, Boonstra A. Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C infected patients and limit the extent of fibrosis. J Hepatol. (2010) 52:315–21. doi: 10.1016/j.jhep.2009.12.013
8. Zhang X, Feng M, Liu X, Bai L, Kong M, Chen Y, et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl Res. (2016) 169:67–79.e2. doi: 10.1016/j.trsl.2015.10.008
9. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. (2019) 565:246–50. doi: 10.1038/s41586-018-0824-5
10. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. Distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
11. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
12. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. (2007) 8:191–7. doi: 10.1038/ni1428
13. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. (2008) 28:546–58. doi: 10.1016/j.immuni.2008.02.017
14. Scholten D, Trebicka J, Liedtke C, Weiskirchen R. The carbon tetrachloride model in mice. Lab Anim. (2015) 49:4–11. doi: 10.1177/0023677215571192
15. Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. (2009) 137:1467–77.e5. doi: 10.1053/j.gastro.2009.06.045
16. Ibrahim SH, Hirsova P, Malhi H, Gores GJ. Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame. Dig Dis Sci. (2016) 61:1325–36. doi: 10.1007/s10620-015-3977-1
17. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. (2019) 37:907–15. doi: 10.1038/s41587-019-0201-4
18. Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
19. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:1–21. doi: 10.1186/s13059-014-0550-8
20. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
21. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. (2015) 43:e47. doi: 10.1093/nar/gkv007
22. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
23. Jeffery HC, McDowell P, Lutz P, Wawman RE, Roberts S, Bagnall C, et al. Human intrahepatic ILC2 are IL-13positive amphiregulinpositive and their frequency correlates with model of end stage liver disease score. PLoS One. (2017) 12:e0188649. doi: 10.1371/journal.pone.0188649
24. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA. (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
25. Wynn TA. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol. (2004) 4:583–94. doi: 10.1038/nri1412
26. Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci USA. (1997) 94:10663–8. doi: 10.1073/pnas.94.20.10663
27. Weng HL, Cai WM, Liu RH. Animal experiment and clinical study of effect of gamma-interferon on hepatic fibrosis. World J Gastroenterol. (2001) 7:42–8. doi: 10.3748/wjg.v7.i1.42
28. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. (2009) 50:261–74. doi: 10.1002/hep.22950
29. Katz SC, Ryan K, Ahmed N, Plitas G, Chaudhry UI, Kingham TP, et al. Obstructive jaundice expands intrahepatic regulatory T cells, which impair liver T lymphocyte function but modulate liver cholestasis and fibrosis. J Immunol. (2011) 187:1150–6. doi: 10.4049/jimmunol.1004077
30. Popovic B, Golemac M, Podlech J, Zeleznjak J, Bilic-Zulle L, Lukic ML, et al. IL-33/ST2 pathway drives regulatory T cell dependent suppression of liver damage upon cytomegalovirus infection. PLoS Pathog. (2017) 13:e1006345. doi: 10.1371/journal.ppat.1006345
31. Roh YS, Park S, Lim CW, Kim B. Depletion of Foxp3+ regulatory T cells promotes profibrogenic milieu of cholestasis-induced liver injury. Dig Dis Sci. (2015) 60:2009–18. doi: 10.1007/s10620-014-3438-2
32. Corthay A. How do regulatory t cells work? Scand J Immunol. (2009) 70:326–36. doi: 10.1111/j.1365-3083.2009.02308.x
33. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
34. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. (2007) 204:57–63. doi: 10.1084/jem.20061852
35. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. (2016) 1417:5–22. doi: 10.1111/nyas.13011
36. Weng SY, Wang X, Vijayan S, Tang Y, Kim YO, Padberg K, et al. IL-4 receptor alpha signaling through macrophages differentially regulates liver fibrosis progression and reversal. EBioMedicine. (2018) 29:92–103. doi: 10.1016/j.ebiom.2018.01.028
37. Seki E, De Minicis S, Inokuchi S, Taura K, Miyai K, Van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. (2009) 50:185–97. doi: 10.1002/hep.22952
38. Louis H, Van Laethem JL, Wu W, Quertinmont E, Degraef C, Van Den Berg K, et al. Interleukin-10 controls neutrophilic infiltration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology. (1998) 28:1607–15. doi: 10.1002/hep.510280621
39. Mchedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. (2013) 39:357–71. doi: 10.1016/j.immuni.2013.07.018
40. Zhang CY, Yuan WG, He P, Lei JH, Wang CX. Liver fibrosis and hepatic stellate cells: etiology, pathological hallmarks and therapeutic targets. World J Gastroenterol. (2016) 22:10512–22. doi: 10.3748/wjg.v22.i48.10512
41. Berasain C, Avila MA. Amphiregulin. Semin Cell Dev Biol. (2014) 28:31–41. doi: 10.1016/J.SEMCDB.2014.01.005
Keywords: liver fibrosis, regulatory T cells, Foxp3, amphiregulin, liver inflammation
Citation: Ikeno Y, Ohara D, Takeuchi Y, Watanabe H, Kondoh G, Taura K, Uemoto S and Hirota K (2020) Foxp3+ Regulatory T Cells Inhibit CCl4-Induced Liver Inflammation and Fibrosis by Regulating Tissue Cellular Immunity. Front. Immunol. 11:584048. doi: 10.3389/fimmu.2020.584048
Received: 16 July 2020; Accepted: 09 September 2020;
Published: 15 October 2020.
Edited by:
Kiyoshi Hirahara, Chiba University, JapanReviewed by:
Chiaki Iwamura, Chiba University, JapanCopyright © 2020 Ikeno, Ohara, Takeuchi, Watanabe, Kondoh, Taura, Uemoto and Hirota. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keiji Hirota, aGtlaWppQGluZnJvbnQua3lvdG8tdS5hYy5qcA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.