 Tyler G. Normile
Tyler G. Normile Arielle M. Bryan
Arielle M. Bryan Maurizio Del Poeta
Maurizio Del Poeta- 1Department of Microbiology and Immunology, Stony Brook University, Stony Brook, NY, United States
- 2Ingenious Targeting Laboratory Incorporated, Ronkonkoma, NY, United States
- 3Division of Infectious Diseases, School of Medicine, Stony Brook University, Stony Brook, NY, United States
- 4Veterans Administration Medical Center, Northport, NY, United States
Cryptococcus species are environmental fungal pathogens and the causative agents of cryptococcosis. Infection occurs upon inhalation of infectious particles, which proliferate in the lung causing a primary infection. From this primary lung infection, fungal cells can eventually disseminate to other organs, particularly the brain, causing lethal meningoencephalitis. However, in most cases, the primary infection resolves with the formation of a lung granuloma. Upon severe immunodeficiency, dormant cryptococcal cells will start proliferating in the lung granuloma and eventually will disseminate to the brain. Many investigators have sought to study the protective host immune response to this pathogen in search of host parameters that keep the proliferation of cryptococcal cells under control. The majority of the work assimilates research carried out using the primary infection animal model, mainly because a reactivation model has been available only very recently. This review will focus on anti-cryptococcal immunity in both the primary and reactivation models. An understanding of the differences in host immunity between the primary and reactivation models will help to define the key host parameters that control the infections and are important for the research and development of new therapeutic and vaccine strategies against cryptococcosis.
Cryptococcus neoformans is an opportunistic fungal pathogen
Cryptococcus spp. are basidiomycetes ubiquitously found within the environment as basidiospores and budding yeast, most commonly in the soil, trees, and avian habitations (1, 2). Of clinical relevance, two main species, Cryptococcus neoformans and Cryptococcus gattii, are encapsulated fungal pathogens and the etiological agents of cryptococcosis, a life-threatening invasive fungal disease that infects humans via the respiratory tract (3, 4). Immunocompetent hosts can be infected but rarely succumb to infection. Conversely, under conditions where the host cannot immunologically control the initial pulmonary infection, such as during antiretroviral therapy in HIV/AIDS patients, chemotherapy in cancer patients, or immunosuppressive therapy in organ transplant patients, the yeast may uncontrollably proliferate and disseminate to the central nervous system resulting in meningoencephalitis (5–7).
The Cryptococcus neoformans/gattii species complex has a high degree of heterogeneity, and has been classified by numerous molecular typing techniques (8, 9). These genotypic classifications have greatly aided in the epidemiology and genetic diversity that exists within these species, however the serotype classification of these species will be most relevant to this review since our focus is on the host immune response to the yeast. There are five serotypes of Cryptococcus, with serotypes A, D, and AD belonging to C. neoformans and serotypes B and C belonging to C. gattii (9–11). These serotypes are based on differences in the arrangement of the glucuronoxylomannan (GXM) capsule surrounding the yeast, which is considered a major cryptococcal virulence factor (12, 13). The various serotypes result in prominent differences in both pathology and immune modulation through variations in pattern-recognition receptor ligation and immune modulation with host cells. In fact, the GXM capsule was shown to be a major deciding factor in determining pathogenicity of the various species of Cryptococcus (14). For instance, the capsule of Cryptococcus liquefaciens (a non-pathogenic species) does not exhibit the same microbial defenses against amoebas as does the pathogenic species capsule. As such, these serotype differences play distinct roles in modeling host susceptibility and geographical distribution from the clinical side.
Due to its worldwide distribution, it is widely accepted that human subjects are exposed to this fungus during early childhood (15, 16), and upon this primary infection, the host harbors fungal cells in lung granulomas (17–30). Perhaps the best evidence that supports this possibility is provided by studies showing that fungal strains from patients affected by cryptococcal meningoencephalitis are identical to those strains isolated earlier from the same asymptomatic patients (31–33). Other investigators have suggested that these findings were the result of patients being constantly re-exposed to the very same strain from the environment (34, 35). Although this assertion is in the realm of possibility, it does not take into consideration the enormous genetic variability of cryptococcal strains present in the environment (36–38). Due to the evidence of strain diversity mentioned above, it is our opinion that the chance of inhaling a genetically identical strain years apart is less probable than the reactivation of a prior infection. In light of this, the evidence together strongly suggests that primary infection, granuloma formation, and eventual reactivation of the dormant yeast cells upon immunosuppression reflect the stages of this disease.
Fungal Propagules: Spores vs. Yeast Cells as Infectious Particles
Pulmonary cryptococcosis begins upon inhalation of fungal particles, which can be either spores or/and yeast cells. The model organism, most commonly mice, receive these particles via intranasal or intra-tracheal challenge to recapitulate human infection (39). However, spores differ compared to yeast cells, and these differences may account for a different immune recognition and response to the infection (40).
Primarily, the spores of C. neoformans expose β-glucans, whereas encapsulated yeast cells expose the GXM capsule. β-glucans are strongly recognized by C-type lectin receptors (CLRs) on both resident and innate immune cells. The encapsulated yeast cells, however, weakly stimulate these same CLRs, as they ligate to TLR2, TLR4, CD14, and CD16 (41–44), resulting in different outcomes dependent on strain or host cell type.
Because of the pleiotropic effect of GXM and because GXM is a potent immunomodulator, yeast cells can induce a hyperinflammatory response that will eventually lead to the death of the host (particularly mice), or/and a non-protective type 2 immune response that will also be harmful to the host. Conversely, uptake of spores does not lead to a strong inflammation as spores can exist within macrophages for long periods of time without damaging them (13, 42, 45). Also, spores are less capable to stimulate adaptive immunity, which is well-known to be stimulated by GXM in encapsulated fungal cells through APCs, DCs, and macrophages (46–50). Thus, spores can evade host immunity much more efficiently than yeast cells, causing the development of a latent infection, and perhaps, they may represent a better model for studying the reactivation model. However, it is not known if spores are able to promote granuloma formation in mice and represents a topic for future investigation.
Animal Models for Studying Cryptococcosis
There have been several approaches utilized to investigate human cryptococcal infection in the laboratory. Whole-organism approaches and fungal mutants (24, 46, 51–56), in vivo antibody depletions and neutralizations (20, 56–61), and the assessment of fungal burden, host survival, and immune cell recruitment (present in nearly all C. neoformans studies) have been at the forefront of these modeling approaches with each uncovering notable findings as well as bringing limitations.
Invertebrate Models
Both invertebrate and vertebrate models exist in the literature for in vivo modeling of C. neoformans infection. This topic has been reviewed in the past (62), and this review will expand on the ample research that has been conducted since then. Invertebrate models include nematodes (Caenorhabditis elegans), amoebae (Acanthamoeba castellanii and Dictyostelium discoideum), and insects (Drosophila melanogaster and Galleria mellonella). The relatively low cost of maintenance, lesser ethical restrictions, ease of genetic manipulation, and short reproduction times makes invertebrates valuable tools in biomedical research. Research with invertebrate models has mainly addressed mapping signal transduction pathways for virulence during infection and the interactions of C. neoformans with innate phagocytic cells since invertebrates possess only the innate arm of the immune system (63). In light of invertebrate models, several studies have highlighted these organisms as practical tools.
First, Mylonakis et al. found that C. elegans can use non-pathogenic species of Cryptococcus (C. laurentii and C. kuetzingii) as food sources while C. neoformans resulted in death of the worms (64). In addition, the C. neoformans gene LAC1 was a virulence factor upregulated during infection of the worms, which confirmed prior mammalian studies as well as added to the growing belief that C. neoformans may have evolved into a pathogen during interactions with environmental predators such as C. elegans. From another group, C. elegans was used as part of a multi-host screen of C. neoformans mutants to assess previously unknown virulence factors (65). Several genes regulating lipid metabolism, chitin regulation, and melanin synthesis were discovered using this approach. These studies among others support C. elegans as a viable model for assessing C. neoformans infection in the lab.
Second, soil amoeba are reservoirs of C. neoformans, and resemble the way macrophages phagocytose microorganisms making them an important tool in C. neoformans research (17, 66, 67). A 2018 paper showed that D. discoideum phagocytosed C. neoformans but was unable to kill the yeast, a phenomenon similar to macrophages. However, Watkins et al. were able to ascertain that C. neoformans can either be expelled from the amoeba, or when the amoeba is pharmacologically blocked, C. neoformans can escape in a non-lytic manner (68), a recent phenomenon termed vomocytosis (69, 70). Ultimately, the amoeba model represents a novel approach for future studies on the cellular level, which has implications to how macrophages interact with C. neoformans in the host.
Finally, insects represent a valuable experimental tool for studying how innate phagocytes interact with the yeast cells as well as the effect of antimicrobial peptides on yeast viability (63, 71). G. mellonella can live at human body temperature and be infected with controlled doses of the yeast with minimal invasion. This is in contrast to the models above since C. elegans are inoculated onto an agar plate with colonies of C. neoformans and there is no way to regulate the infection dose and timing. This organism has been used primarily to assess virulence of C. neoformans (46, 65) and antifungal susceptibility (72). Altogether, G. mellonella represent one of the most versatile invertebrate tools for C. neoformans infection studies.
Vertebrate Models
Despite the progress that has been made utilizing invertebrate organisms, there are several limitations to their use as a model for cryptococcosis. One major limitation is the lack of an adaptive immune system, which has been repeatedly shown to be instrumental in host protection against C. neoformans. Secondly, the route of infection for invertebrates does not recapitulate the inhalation infection model in humans. Finally, a major immunological limitation of invertebrate organisms is that they lack differentiated phagocytes, such as macrophages and DCs and only have a generalized phagocyte cell type (73). However, these limitations can be overcome using vertebrate organisms as model hosts that offer a vast selection of tools. Primarily, the presence of an adaptive immune system and differentiated cell types, easy regulation of infection doses, and availability to infect via inhalation all offset the aforementioned invertebrate disadvantages. Additionally, the larger body sizes of these animals allow for more experimental manipulation to be carried out, such as endotracheal intubations, radiography and imaging, bronchoalveolar lavage, and cerebrospinal fluid collection (63), and the presence of organ systems allows for an infection model that closely resembles how humans become infected and succumb to CNS dissemination. As with all model hosts, each brings advantages as well as limitations, all of which will be briefly discussed below.
Several vertebrate organisms have been used in the modeling of C. neoformans infection, and some of these include zebrafish (Danio rerio), non-human primates, rabbits (Oryctolagus cuniculus), rats (Rattus rattus), and most commonly mice (Mus musculus). The zebrafish is a relatively newer host model for C. neoformans infection, although it has been established in other host-pathogen interactions and offers an exciting middle ground between the simplicity of invertebrate models and the organ system complexity found in mammals (74). Obvious advantages of this host include the optical transparency for live imaging where the yeast have been observed replicating in macrophages, genetic amenability for host mutational studies, and assessment of virulence for different strains of C. neoformans (75–77). However, certain elements must be considered when using this model. First, the authors reported that neutrophils did not accumulate at the site of infection or around infected macrophages in the zebrafish model (77). Although the role of neutrophils in mammalian infections is still uncertain, the early accumulation of neutrophils is a well-established immune signature for C. neoformans infections in mice and humans. Secondly, only three studies using zebrafish to model C. neoformans infection (and none using C. gattii) having been published to date, so more data are necessary for a thorough understanding of zebrafish-Cryptococcus interactions.
Non-human primates are an uncommon host for studying cryptococcosis. This is most likely due to the greater cost and housing requirements compared to other animal models (78, 79). Most studies in non-human primates were conducted decades ago, but a recent 2019 study performed a transcriptome analysis of cynomolgus monkeys (Macaca fascicularis) and mice using RNA-Seq during acute C. neoformans infections (80). The authors found that only about 20 percent of the differentially expressed genes were shared between these two hosts during infection, and they suggested monkeys could be a better model than mice at recapitulating the human response to C. neoformans. Although additional host data adds to the growing body of cryptococcal literature, this small sample size of monkeys (6 total monkeys; 3 control and 3 experimental) in one study does not justify the expense and challenge of using monkeys as hosts to C. neoformans.
Historically, rabbits have not been a commonly used host for studying cryptococcosis mainly because of the high cost to purchase and maintain, however the rabbit model closely mimics the human infection. Rabbits are naturally resistant to C. neoformans and they succumb to the fungus only when immunosuppressed (e.g., with corticosteroids) (81). The rabbit model is suitable for the introduction of fungal cells directly into and out of the subarachnoid space, allowing studies that address the fitness and the adaptation of fungal cells to the cerebrospinal fluid. This is a particular strength of this model because the most common clinical manifestation of cryptococcosis is meningitis. Understanding how fungal cellular pathways respond to this unique environment may provide important insights into the development of new therapeutic strategies targeting those specific pathways. Rabbits are also used to test the efficacy of new antifungals at the site of infection during cryptococcal meningitis (82–85). Thus, rabbits are a tractable alternative for studying cryptococcal meningitis.
Rats have been reported to develop chronic pulmonary cryptococcosis in the wild (86). Rat models of cryptococcal virulence and infection were more prevalent in the late 1980's, 1990's, and early 2000's (87–94), but have diminished over the years. In vivo and ex vivo studies using rats have mainly focused on C. neoformans interactions with alveolar macrophages (95–97). Similar to the rabbit model, rats also have the ability to form lung granulomas that efficiently contain C. neoformans within the granuloma until the animal becomes immunocompromised, which recapitulates the human pathophysiology [(87, 88, 92, 94)]. Rats are also a valuable model for C. gattii infections since they are naturally susceptible hosts and C. gattii infections are more common in immunocompetent humans (98–100). Although rats represent a well-defined host to cryptococcal infections, the limitations for this model are the cost and the lack of genetic knockout animals for studying the role of host parameters against cryptococcosis.
Notably, the mouse represents by far the most well-documented animal model to study host-pathogen interactions with C. neoformans. The mouse offers great flexibility for experimental studies with a large diversity in genetic knockout tools commercially available. In addition, the mouse has been well-characterized in biomedical research and ease of handling with numerous routes of infection. Nevertheless, discordance remains when distinguishing between the primary infection model and reactivation model mostly due to lack of tools used to study the reactivation model of infection in mice.
Differently from humans, rabbits, and rats (92, 94, 101–105), mice do not produce a granulomatous response against highly virulent C. neoformans strains, and they eventually succumb to the infection. Investigators have utilized less virulent strains, such as C. neoformans strain 52D, in which mice develop a persistent infection with a granulomatous response after intranasal infection (106–110). This model has allowed researchers to study chronic cryptococcosis and brain dissemination in mice, although there are several drawbacks to using less virulent strains that include induction of immunity not normally associated with highly virulent strains. Thus, due to a model that does not fully recapitulate human granuloma containment of the yeast, nearly all work has focused on the primary infection model of cryptococcosis.
However, our lab has developed a mouse model of cryptococcosis that recapitulates the human response to C. neoformans. In fact, we found that mice infected with an avirulent strain lacking the sphingolipid glucosylceramide (Δgcs1) leads to total containment of fungal cells in lung granulomas and no dissemination to the brain (111). However, when Tgε26 mice who are inherently immunocompromised (Tgε26 mice lack T and NK cells) are instranasally infected with this mutant, the mice do not form lung granulomas, the Δgcs1 mutant uncontrollably proliferates in the lung, disseminates to the brain, and results in complete death of these mice (20). Thus, this mutant mimics the physiopathology of the infection in humans with which cryptococcosis is mostly associated (112).
Although many animal models are available, the majority of immunological studies have been performed in mice, so the following sections of this review will focus on discussing key immunological findings in mice for both the primary infection model and reactivation model of cryptococcosis.
Modeling Cryptococcal Infections and What Has Been Learned
In the primary infection model to cryptococcosis, the host is exposed to the fungus for the first time upon inhalation of spores and/or desiccated yeast cells. It has been postulated that the primary infection happens very early on in life since there are data showing children having antibodies to the yeast in their blood [(16, 113)]. Protection against cryptococcosis is dependent on early host recognition, recruitment of proper cell types, and immunological control of yeast proliferation (114–116). The immune response to C. neoformans therefore can be temporally divided into three stages: (1) recognition of the inhaled particles by resident airway cells, (2) early recruitment of innate immunity, and (3) late recruitment of adaptive cell-mediated immunity (Figure 1). These stages collaboratively work in an orchestrated manner to control the pulmonary infection and deter extrapulmonary dissemination and are discussed in the following sections.
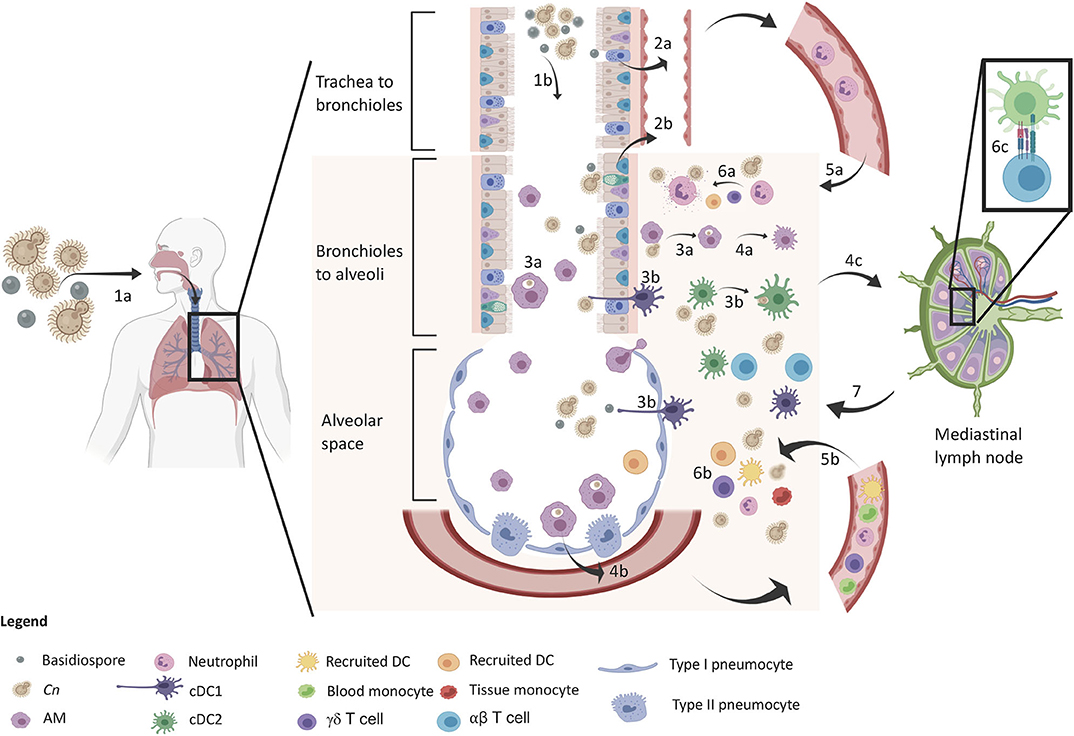
Figure 1. Protective immune response in the lung upon inhalation of C. neoformans. The sequence of events is denoted in the diagram. The yeast and basidiospores are inhaled (1a) and travel down the airways passing through the trachea to the bronchioles (1b) and further down to the alveolar spaces. Upper respiratory epithelia sense the spores (2a) and lower respiratory epithelium sense spores and encapsulated propagules (2b), which release IL-8 for early neutrophil recruitment. Macrophages (3a) and DCs (3b) phagocytose Cn in both the airways and lung tissue leading to M1 macrophage polarization (4a) and cytokine release (GM-CSF and MCP-1) for inflammatory cell recruitment (4b). DCs mature and migrate to the lung draining lymph node (4c) for induction of adaptive immunity. At this time, neutrophil (5a) and inflammatory cells (5b) infiltrate into the lung tissue for increased host defense against Cn. Neutrophils kill Cn via degranulation (6a) and a myriad of other defenses from other cells (6b) such as monocyte differentiation into recruited DCs that further amplify T cell induction (6c). Type 1/17 polarized T cells migrate back to the lung for adaptive immune control of infection leading to control/containment of the infection.
As previously mentioned, upon initial inhalation of C. neoformans, immunocompetent hosts generally control the infection, but the fungus is not always fully cleared from the lungs. In humans, C. neoformans can remain in a latent state of infection contained within lung granulomas, which are localized structures composed of several cell types that work in an coordinated manner to contain the pathogen (Figure 2B) (117). Using two strains of rats, it was shown that a type 1 immune response was needed to control pathogen replication and granuloma containment of C. neoformans, and that a type 2 immunity resulted in loss of fungal containment with the consequent exacerbation of the disease (88, 92). Although the host does not experience any pathology during latency, granuloma containment introduces a paradoxical situation: on the one hand these structures help to contain the infection, but on the other hand they also harbor the pathogen and provide a source of fungal replication once immunodeficiency occurs, such as upon HIV/AIDS-mediated lymphopenia (118, 119) or solid organ transplant recipients (4, 117). The immunological events associated with the reactivation model will be discussed following the primary model as well as current progress in a novel mouse model of cryptococcal granuloma containment.
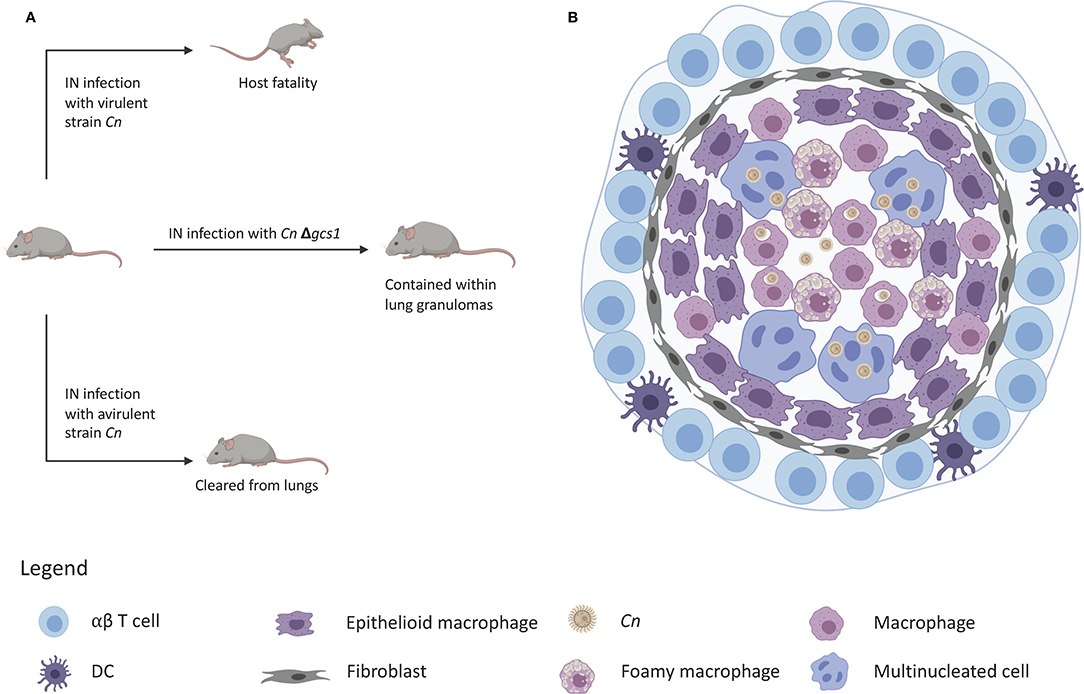
Figure 2. Granuloma containment in mice infected with C. neoformans△gcs1. (A) Mice are notorious for lacking the ability to form lung granulomas to contain C. neoformans infection. Infection with a virulent strain results in death and infection with an avirulent strain leads to clearance of the yeast from the lungs. Infection with C. neoformans△gcs1 results in granuloma containment of the yeast in the mouse lungs. (B) These granulomas exhibit a slightly necrotic center with macrophages, foamy cells, and giant multinucleated cells with internalized C. neoformans. These cells are surrounded by a closure of epithelioid cells and fibroblasts, followed by an outer ring of lymphocytes.
Primary Infection Model: Inhalation of the Fungal Particles
Airway Resident Cell Recognition of C. neoformans
In the absence of infection, an organism still has immune surveillance for early detection and response to foreign particles. This is especially exhibited at mucosal sites such as the intestines, skin, and the airways. Airway resident immune cells include the epithelial cells that line the airways, tissue-resident alveolar macrophages and dendritic cells, and non-canonical innate lymphocytes such as γδ T cells and innate lymphoid cells.
Epithelial cells
The inhalation of the fungal propagules, the first host cells to encounter these particles would naturally be the airway epithelium and resident airway immune cells. Airway epithelial cells serve as a barrier and immunological interface between the external and internal bodily environments (120–123). In depth studies on cryptococcal-epithelial interactions are lacking in relation to other cell types, but both the upper respiratory bronchial epithelia and lower alveolar epithelia have been shown to recognize, respond to, and internalize C. neoformans (124–127). Utilizing in vitro approaches, the human type II alveolar epithelial cell line, A549, several groups came to find that both GXM (128) and phospholipase B (124) were epithelial adherence factors. In fact, the adherence to lung epithelial cells (using A549 cell line) can be blocked by either using an antibody against the cryptococcal capsule (anti-GXM antibody, 18B7), or by using a Δplb1 mutant (lacking fungal phospholipase b). The cryptococcal capsule is clearly important for the adherence to A549 but its role is less clear because acapsular mutant (Δcap67) is still able to attach to A549 and this attachment was only inhibited in the presence of specific fungal mannoprotein (s) (e.g., MP84) (126). Upon adherence, internalization of C. neoformans by the airway epithelial will follow, and although the fungal and host factors regulating cryptococcal-epithelial interaction is still under investigation, one thing is clear: internalization of cryptococcal cells may cause death of the epithelial cells resulting in lung lesions and hyperinflammation (122).
Upon adherence, lung epithelial cells actively participate in the immunological response via secretion of specific cytokines. For instance, in vitro studies showed that when GXM binds to CD14 on A549 cells, they secrete IL-8 (41). IL-8 is a potent neutrophil recruitment chemokine, suggesting that lung epithelium may drive early recruitment of these early host effector cells. This paper was shortly followed by another study that used the BEAS-2B cell line that more closely mimics the upper bronchial epithelium, an area that would obviously be exposed to inhaled propagules first (127). This group also found that IL-8 was secreted in an NF-κB dependent manner in response to C. neoformans infection, but BEAS-2B cells only responded to an acapsular mutant and not the capsulated parent strain. It is possible that the different responses observed was due to the different cell types used (A549 vs. BEAS-2B), but another possibility is that upper bronchial epithelial cells respond more effectively to acapsular (or a thinner capsule) cryptococcal cells compared to epithelial cells found in the lower alveolar epithelium. It has been shown that cryptococcal particles have a very small or no capsule upon inhalation, and rapidly induce capsular growth as they proceed down the airway (42, 129–131). One final possibility stems from the limitations and drawbacks some researchers find in using the A549 cell line. Phenotypic studies of A549 cells showed they possess lower phospholipid content, fewer cytoplasmic lamellar bodies, and a higher autophogy rate compared to primary human type II alveolar cells (132, 133). Nonetheless, this evidence together with the cytokine data clearly suggests that the upper respiratory tract may sense less capsulated particles and respond by producing cytokines which recruit neutrophils early on, while differential responses may occur after capsule synthesis has been stimulated further down the airway.
Capsule synthesis consists in the production of a well-organized physical barrier containing glucuronic acid, xylose, and mannose (GXM) molecules connected through various glycosidic bonds. GXM strongly inhibits the protective host response to cryptococcal infection via initiation of a type 2 immunity (13, 42). Several groups have described the release of IL-33 in response to epithelial-cryptococcal interactions and the downstream effects of IL-33 through its receptor T1/ST2 (134, 135). IL-33 drives a type 2 immunity via the activation of type 2 innate lymphoid cells and promoting a Th2 T cell response in the lungs. These effects lead to an overabundance of IL-5 and IL-13 in the lung resulting in alternative M2 macrophage polarization and decreased epithelial barrier integrity promoting fungal cell persistence and penetration into the lung tissue and lung blood vessels. These negative facets may eventually lead to the dissemination of fungal cells from the lung to other organs through the bloodstream including the CNS. Interestingly, the source of IL-33 in the lungs was found to be from type II alveolar cells (135), the same source of IL-8 mentioned above. Specifically, IL-33 downregulates surface receptor expression for leukocyte extravasion, such as E-cadherin and ICAM-1, which decreases the recruited immune cell response. The protective (IL-8) and non-protective (IL-33) results together demonstrate the varied response to inhaled cryptococcal particles suggesting a strong dependence on strain, capsule size, and experimental conditions for host outcome.
Alveolar macrophages
In addition to the airway epithelial cells, CD11c+ airway resident immune cells such as alveolar macrophages (AM) and DCs have been reported to be the dominant cell populations in uninfected lungs (47, 136–139) and have been arguably the most widely studied cell population in the cryptococcal literature (Figure 1). These cells are found ubiquitously in the lower respiratory and alveolar spaces of the airway lumen and the underlying lamina propria below the epithelium. These pulmonary sentinels phagocytose debris, dead cell material, and inhaled particles found in the airways for removal as well as add to the early cytokine response in the lungs during the early phases of infection such as with C. neoformans.
AM have been suggested to play paradoxical roles during infection with C. neoformans (140, 141), so that the necessity of these cells to efficiently control C. neoformans lung infection has been strongly debated (142). On one hand, depletion of these cells (using anti-CD11c) prior to infection exhibited both increased lung fungal burden and the mortality rate in mouse models (50). On the other hand, C. neoformans is clearly capable of living within AMs (and other macrophages populations) unharmed shown by numerous studies (69, 143–146). It is important to note, however, that AMs are tissue resident macrophages in the lungs, but during an infection, a collection of macrophages consisting of AMs, interstitial macrophages, and monocyte-derived macrophages will populate the lungs. This collection, referred to in this review as pulmonary macrophages, needs to be set apart from studies looking at AMs alone.
Pulmonary macrophages have been shown to shift from an unpolarized state in uninfected animals to either classically activated M1 or alternatively activated M2 phenotypes depending on the lung cytokine microenvironment during infection (147–151). These polarization states dictate the effector functions of these cells. M1 macrophages are activated by IFN-γ and have been widely regarded as anti-cryptococcal in nature, while M2 macrophages become activated by IL-4 and are regarded as non-protective during infection with C. neoformans. M1 macrophages are regulated by STAT1 and are an early source of inflammatory cytokines and chemokines, such as IFN-γ, TNFα, IL-1, and IL-8, that drive the protective type 1 immune response against cryptococcosis (53, 137, 152–154). More specifically, M1 macrophages exhibit increased phagocytosis (147, 153), killing of C. neoformans via ROS/NOS production (53, 153), and aid the maturation of dendritic cells (DC) to activate adaptive cell-mediated immunity. Interestingly, these macrophages have also been reported as having innate memory in immunization studies (53). Through IFN-γ-primed macrophages from the IFN-γ-producing strain used in the Wormley lab, the authors observed an increased STAT1 binding to the promoter region and increased expression of antifungal gene expression. Thus, M1 polarized macrophages possess the ability to control C. neoformans, coordinate a protective response in the host, and may possess memory to future infections.
In contrast to anti-cryptococcal M1 macrophages, M2 polarized macrophages are regulated by STAT3, produce IL-5 and IL-13, and drive a type 2 immune response in the host, which is unable to contain the infection (147, 155). Because C. neoformans can survive and replicate within macrophages (13, 156, 157), M2 macrophages that have little to no fungicidal activity can harbor large numbers of yeast cells leading to dysfunctional macrophages (69, 145). Through either lytic or non-lytic (termed vomocytosis) exocytosis, cryptococcal cells may exit the macrophages either in the lungs or elsewhere if the macrophages have also left the lungs (70, 146, 158). This ability of C. neoformans to “hide” within macrophages protects the yeast against the harsh extracellular environment containing antimicrobial peptides and complement as well as killing by neutrophils, NK cells, and T cells (75). Additionally, the harsh environment of the phagosome may lead to the formation of titanized cryptococcal cells with consequential macrophage disruption and reduction in phagocytosis by other macrophages or/and extracellular killing of these titan cells (12, 159–162). Eventually, this leads to the persistence of the infection instead of containment or clearance.
These differential findings with AM in mice have also been observed with rat and human macrophages (94, 95, 97, 163–167). Early studies showed that C. neoformans was rapidly ingested by human AM, although no killing was observed, and the yeast were able to proliferate over the course of 6 h or 2 days (164, 166). The 1994 study also showed that human AM played a key role in antigen presentation to T cells. In rats, three studies from the same group in 1989 identified that rat AM phagocytose and effectively kill C. neoformans in the presence and absence of serum using in vitro assays (95–97). Taken together, the role of AM seems to be a variable response with respect to C. neoformans, especially when comparing in vitro and in vivo assays and between different model organisms.
Despite a large body of work studying alveolar macrophages in cryptococcosis models, several questions remain for future research in what determines M1 vs. M2 macrophage polarization. Indeed, serotype differences in the GXM capsule and certain cytokine profiles in the lung have been suggested to play a role in driving macrophage polarization (43, 44, 168, 169). Research on how other fungal factors and the lung microbiome influence the fate of AMs (and more generally pulmonary macrophages) during primary infection and reactivation is still greatly needed.
Resident dendritic cells
Lung resident DCs express CD11c and MHC-II markers and have been highly implicated in early defense against invading pathogens and have gained an increased appreciation in antifungal immunology over the last decade. DCs are innate phagocytes with a central role to bridge the innate and adaptive arms of the immune systems upon initial infection with a pathogen, including C. neoformans (47, 170). Phagocytosis of the yeast requires complement or antibody-mediated opsonization of the GXM capsule (116, 171), and the DCs kill C. neoformans through lysosomal degradation (47). Concurrently, DCs will upregulate co-stimulatory molecules such as CD80 and CD86 for antigen presentation to T cells. DCs are a diverse cell type with several subsets each possessing specialized functions and regarded as essential components of antifungal immunity including against C. neoformans. Three main subsets can be found within the lungs: CD103+ classic DCs (cDC1), CD11b+ cDC (cDC2), and plasmacytoid DCs (pDC) (47, 116, 172).
cDC1 cells are under the regulation of the Batf3 transcription factor (173) and are predominantly found within the airways and alveoli of the lungs (Figure 1). These cells sit at the epithelial interface and directly interact with the external microbial cells present in the airway lumen. These cells have been described to help drive a type 1 immune response via the release of IL-12 and stimulation of NK cells and ILC1s, recognize intracellular pathogens, and cross-present antigens to CD8+ T cells (173). cDC1 become activated in response to pathogen uptake and degradation and cytokine exposure (such as TNFα and GM-CSF), upregulate costimulatory molecules (CD40, CD80/86, MHC-II) and migratory receptors (CCR7). They can travel to the lung-draining lymph node via a CCL21 gradient to potentiate protective T cell-mediated immunity to cryptococcal infection (61, 174, 175).
cDC2 cells are under the regulation of the IRF4 transcription factor (176) and are also found within the lower respiratory tract and alveoli but deeper in the lamina propria tissue below the basement membrane of the epithelial cells (177). These cells have been shown to play a role for induction of both Th2 and Th17 CD4+ T cell-mediated immune responses depending on infectious antigen type and cytokine environment (174, 176) in a similar manner to cDC1 cells described above. While the Th2 induction mechanism by these cells is still unknown, the Th17 response is mediated through release of IL-23, IL-6, and TGF-β (all the cytokines needed for type 17 immune activation and sustainability). Additionally, cDC2 aid in inflammatory cell recruitment via secretion of IL-12, MCP-1/CCL2, MIP-1α/CCL3, MIP-1β/CCL4, and RANTES/CCL5 (172, 178, 179), and this subset has also been described to be essential for iBALT (inducible bronchus associated lymphoid tissue) maintenance with the release of CXCL12, CXCL13, and CXCL15 being the primary factors (178, 180). Overall, this subset exhibits highly diverse functions in the lung that help drive anti-cryptococcal immunity and containment.
Finally, pDCs have been studied far less in comparison to their classic DC counterparts and represent only a small percentage of the DCs in the lung. The main area of research for this cell type is in viral infections since they are the strongest producers of type I interferons (177, 181) but are also implicated in response against bacterial infections. However, pDC-related work in fungal immunity is relatively low with only one study for pDCs response to C. neoformans has been published to date. This study by Hole et al. (182) showed that pDCs exhibit direct killing ability toward C. neoformans via ROS production and this killing was dependent on the dectin-3 receptor. However, dectin-3 KO mice were not hypersusceptible to C. neoformans infection compared to WT mice, suggesting that although pDCs can kill the yeast, their killing ability though dectin-3 receptor is not essential for survival.
γδ T cells
γδ T cells and innate lymphoid cells (ILCs) have been often overlooked in anti-cryptococcal defense, although both cell types have been described to be critical players in mucosal immunity to other pathogens and diseases. γδ T cells express a non-canonical TCR bearing γ and δ subunits and can be found as both blood circulating and tissue-resident subsets. These cells are strongly defined by their ability to secrete IL-17 upon infection (183–185).
For the work associated with C. neoformans infection, there are two papers that report differing conclusions. In the first paper from 2004, Uezu et al. infected WT and TCRδ−/− KO mice (C57 background) with C. neoformans and assessed fungal burden and cytokine production between the two groups (186). The TCRδ−/− knockout mice cleared the infection quicker than WT mice and showed higher IFN-γ production by cells in the draining lymph nodes. These results suggest that γδ T cells hinder host protection to C. neoformans infection in this mouse model.
The second paper published 8 years later looked at the role of γδ T cells in mice (Balb/c) depleted of neutrophils (60). The authors found that γδ T cells were a significant source of IL-17A in the lungs during neutropenia. It was concluded that γδ T cells were a viable source of IL-17A in the absence of neutrophils and may be a significant source of leukocyte recruitment early on during infection with C. neoformans.
The differences between the role of γδ T cells in these studies may be attributed to the different cryptococcal strains, mouse genetic backgrounds, or experimental approaches. Although both groups used serotype A strains of C. neoformans, Uezu et al. used a clinical isolate in C57 mice while Wozniak et al. used an IFN-γ producing mutant derived from the H99 strain in Balb/c mice. C57 mice are genetically more susceptible to C. neoformans infection since they rapidly induce eosinophilia, whereas BALB/c mice are more resistant to C. neoformans (39). In addition, the H99-γ mutant induces strong immunity in mice, even protecting them against subsequent lethal challenges. Thus, the use of this protective strain may have skewed the host into a more beneficial outcome compared to a more virulent strain. Despite this caveat, the findings contribute key knowledge as IL-17 is a widely accepted anti-fungal immune response and γδ T cells respond early to pulmonary infection suggesting these cells could play an instrumental role early on during infection with C. neoformans.
Since reactivation of C. neoformans is a common occurrence associated with HIV/AIDS patients, it is worth noting that HIV progression downregulates the number of circulatory γδ T cells (187). Moreover, the antiretroviral therapy (ART) does not reconstitute these γδ T cells. Thus, stimulation or/and introduction of γδ T cells could be an effective immunotherapeutic approach to combat HIV and cryptococcosis.
Innate lymphoid cells
ILCs are one of the most recently emergent cell populations in immunology research. This widely heterogeneous group of cells reside at barrier regions, such as the skin and mucosal surfaces of the lung and intestines, and perform a wide variety of tasks including tissue remodeling, lymphoid tissue biogenesis, and early immunological responses to infection (183, 188–190). There are three major groupings of ILCs that are found in the lung parenchyma. Their nomenclature represents the type of immunity they provide and will be briefly mentioned for clarity. Type 1 ILCs that include natural killer (NK) cells and ILC1, which are defined by their expression of Tbet and secretion of IFN-γ in response to IL-12. Type 2 ILCs that are defined by expression of GATA3 and secrete IL-4, IL-5, IL-9, IL-13, and amphiregulin in response to IL-25 and IL-33. Type 3 ILCs that include lymphoid tissue inducer (LTi) cells and both NCR+ (CD335; NKp46) and NCR- subsets of ILC3, which are defined by expression of RORγt and secretion of IL-22 and IL-17 (as well as small amounts of IFN-γ) in response to IL-1β, IL-23, and IL-6 (188, 189, 191). Together, ILCs represent a group of cells that respond very quickly upon infection with great variety, which is dependent on the subset that becomes stimulated.
Innate lymphoid cell studies with regards to C. neoformans have been largely lacking until two recent studies have highlighted an important role for ILCs in the host response to C. neoformans. Prior to 2018, it was known that type 1 ILC NK cells were able to recognize, become activated in the presence of, and directly kill C. neoformans due to the activating receptor NKp30 in both mice (192, 193) and humans (194) but the fungal ligand responsible for this cytotoxicity was unknown. Additionally, NK cells were found to be defective in HIV-patients. Over two decades later, Li et al. came to find that the ligand to NKp30 was β-1,3-glucan using a variety of approaches with C. neoformans and Candida albicans (195). They found that type 1 ILC NK cells bind directly to β-1,3-glucan, increasing expression of perforin upon binding, and that the exogenous addition of β-1,3-glucan restored the anti-cryptococcal killing ability of NK cells in HIV-infected patients. This study provided exceptional information for an understudied ILC cell type that added to the mouse and human models of immunological protection.
Very recently, Kindermann et al. looked into the early response defining the protective type 1 immune protection against C. neoformans (196). Using RORα knockout mice (deficient in ILC2s), the group saw a downregulation of type 2 cytokines, IL-4, IL-5, and IL-13, decreased number of eosinophils, and significantly lower fungal burden compared to the WT C57 mouse control. This was accompanied by an increase in classical M1 macrophage activation, ultimately showing the host responded with a protective immune response. Lung histopathological analysis confirmed that there was reduced lung tissue damage associated with the increased type 1 immunity. These results further confirm the hypothesis that an ILC2 response may lead to a detrimental host outcome during cryptococcal infection (135). This study confirmed prior findings suggesting IL-33 preferentially increased ILC2 proliferation in the lung and downstream IL-13 release. Overall, these results suggest that ILC2s preferentially activate in response to highly virulent strains of C. neoformans during early infection timepoints to stimulate a non-protective, type 2 immune response.
Recruited Innate Immunity to C. neoformans
Inflammatory cell recruitment is essential for early control of C. neoformans infection (114, 197, 198). Neutrophils and dendritic cells have both been extensively studied and shown to play positive roles in the early host defense, while monocytes and eosinophils have been reported to play controversial or negative roles during infection with C. neoformans.
Neutrophils
Neutrophils are granulocytes originating in the bone marrow that are among the first inflammatory cells to respond to infection in great numbers and take part in a type 17 immune response (199–202). These cells exhibit a short lifespan and offer a myriad of defense mechanisms. Neutrophils have been widely implicated in anti-fungal defenses, especially against Candida albicans and Aspergillus fumigatus (136, 199, 203, 204), but since neutropenia is not a risk factor to developing cryptococcosis, the role of neutrophils has been sparsely explored in response to C. neoformans. However, the question remains if neutrophils are an important protective cell type even if the loss of them does not render host susceptibility.
Neutrophils have been suggested to play an important role for protection in humans once the infection has occurred. This defense stems from observations from several reports showing (i) dampened killing ability of macrophages and neutrophils during late stages of lymphopenia such as with AIDS progression (205, 206); (ii) impaired activation and effector functions of human neutrophils in response to TNF-α, IL-1β, and nitric oxide deficiency was observed in healthy patients who succumbed to pulmonary cryptococcosis (207); (iii) although neutropenia is not a risk factor for cryptococcosis, neutropenia is commonly observed in patients with HIV/AIDS (206, 208, 209). Taken together, these early studies strongly implicate human neutrophils as playing a role in anti-cryptococcal defense.
These data do not fully hold true in mice since murine neutrophils are known to be weaker than human neutrophils since they lack fully activated defensins (210). Neutrophils have been shown to exert a protective role during pulmonary infection caused by several microorganisms (211–213), but the role of neutrophils during C. neoformans infection remains unsettled in mice.
Neutrophils internalize C. neoformans, respond with the release of effector cytokines most notably IL-17A, kill the yeast cells via oxidative bursts and toxic cytoplasmic degranulation (214). Only a very limited amount of published work has addressed the role of neutrophils in animal models and depending on the animal model, strain of C. neoformans, and/or route of infection, the results seem to differ (50, 60, 215, 216). For example, mouse strains more resistant to C. neoformans (SJL/J) seem to benefit from the presence of neutrophils (216) but more susceptible strains (BALB/c) exhibited enhanced resistance upon neutrophil depletion (215). These studies were both conducted using the weaker C. neoformans strain D52. In addition, the BALB/c mice were depleted of neutrophils prior to infection but not continued throughout the course of infection. The number of neutrophils matched the control mice by day 7 post infection, so the role of neutrophils in the overall host outcome is not able to be formulated. Thus, it is obvious from these studies that the apparent different results in the literature are likely due to the use of different mouse models and most importantly the time frame of the induced neutropenia.
Aside from depletion studies, C. neoformans has also been reported in modulating the extracellular killing ability of neutrophils (217, 218). The work in these two studies implicates sphingomyelin synthase as a key enzyme in regulating the killing activity of neutrophils through the regulation of sphingomyelin production. Interestingly, the 2011 study found that neither the presence nor the size of the capsule did not influence the extracellular killing activity of these neutrophils although the GXM capsule is associated with immune modulation in many other cell types such as macrophages, monocytes, DCs, and T cells (217). The authors did find that melanized yeast cells completely abrogated the extracellular killing ability of these cells, and that live but not heat killed cells were necessary for this loss of extracellular killing activity. A key limitation in this study comes from using differentiated peripheral blood cell line, HL-60 cells, for the extracellular killing activity assays. These cells are human derived and possess increased killing ability over murine neutrophils but still derived from a propagated cell line.
Recruited dendritic cells
Recruited inflammatory DCs are monocyte-derived and termed monocyte-derived DCs (moDCs) (110, 116, 219). Overall, moDCs differ from resident DCs in phagocytosis and killing, cytokine release, and migration from the periphery to the nearest draining lymph nodes for activation of adaptive cell-mediated immunity (47, 198, 220–225). Upon arrival in the lung, moDCs downregulate the monocytic marker Ly6C and possess overlapping function with cDCs. The general consensus in the DC literature is less of a phenotypic classification compared to a functional classification, thus recruited DCs are believed to be an addition to the resident cDC defense arsenal rather than a functionally distinct defense (226), although moDCs have been described to be similar to cDC1 cells with a dependence on the transcription factors Irf8 and Batf3 for development, respond to IFN-γ, and express IL-2 and IL-12 (219).
In terms of anti-cryptococcal immunity, DCs have a wide repertoire of pattern recognition receptors (PRRs) that aid in recognition of fungal danger signals like capsular and cell wall components (via TLR2, TLR4, Dectin-1, and DC-SIGN), and fungal DNA upon yeast lysis (via TLR9). DCs respond with IL-2 and IL-23 production in the lung during inflammatory responses (47, 198, 219, 225, 227), thus aiding in the type 1 and 17 immune responses in the lung, respectively.
In the recent literature, the Wormley lab presented compelling evidence for the role of in vivo memory of DCs to C. neoformans (222). Although innate immunity has been widely believed to be devoid of immunological memory, the authors showed that DCs isolated from mice immunized with an IFN-γ producing H99 mutant strain (228) responded with a greatly increased type 1-based immune signature compared to naïve control mice 70 days after initial challenge. These DCs, when cultured ex vivo, responded with a significantly enhanced cytokine recall response of IFN-γ, IL-4, and IL-2 in response to C. neoformans components but not to C. albicans, Staphylococcus aureus, or LPS. These results not only uncover aspects of C. neoformans vaccination biology, but also greatly add to the growing body of literature that DCs are essential components to host defense against C. neoformans with a vast repertoire of anti-fungal capabilities.
Monocytes
Monocytes are mononuclear phagocytes that originate in the bone marrow and circulate through the blood with a variety of PRRs to enable detection of pathogens. Monocytes exist as both CCR2+ Ly6Chi in mice (CD14+ CD16- in humans) and CCR2lo Ly6Clo in mice (CD14lo and CD16- in humans). Ly6Chi are inflammatory monocytes that extravasate into infected tissues, while Ly6Clo are tissue patrolling monocytes that deal with tissue repair and homeostasis (229, 230). Monocytes respond very quickly in response to infection that succeeds neutrophils and are essentially precursors to macrophages and DCs (moDCs). Circulating monocytes have been characterized and functionally described as either mature or immature, which is highly dependent on the cytokine profile of the tissue they are infiltrating (231–236). In the presence of inflammation, mature monocytes express high levels of MHC-II and costimulatory markers such as CD80, CD86, and CD40, whereas immature monocytes express lower levels of these markers (169). However, the role of monocytes in response to C. neoformans infection has been debated, since the presence of monocytes can be detrimental or beneficial to the host depending on the infection model (50, 58, 237–239).
Both mature and immature monocytes can respond early on to C. neoformans infection. Both cells can phagocytose the fungus, however immature monocytes have decreased killing ability and harbor them back into the bloodstream across the blood brain barrier via the Trojan Horse model (240). For example, Charlier et al. intravenously infected mice with bone marrow-derived monocytes (BMDM) loaded with C. neoformans to demonstrate the existence of the Trojan horse model of crossing the blood brain barrier (BBB) (58). The authors found that BBB crossing was observed as early as 6 h post infection and a significant increase in the fungal burden in the spleen and kidney was observed with the yeast loaded BMDMs compared to unloaded BMDMs. These results suggest a role for monocyte trafficking of C. neoformans to the CNS via the Trojan horse model of infection. In addition to this, Heung and Hohl showed that CCR2+ Ly6Chi inflammatory monocytes respond early on in the lung to C. neoformans strain H99 and aid in fungal trafficking of the yeast from the lungs to the lung draining lymph node (237). Upon depletion of these cells using CCR2-DTR mice, a significant decrease in lung fungal burden and improved host survival were observed. This was in addition to lower numbers of ILC2s, Th2 lymphocytes, and M2 macrophages observed in the lungs of CCR2-DTR mice. These data suggest that CCR2+ monocytes may skew the lung immunity toward a non-protective type 2 immune response to the highly virulent H99 strain.
Conversely, three groups have reported a role for CCR2+ cells during C. neoformans infection. First, Traynor et al. infected both CCR2 KO and WT mice with C. neoformans strain 52D (less virulent serotype D) and reported that CCR2 KO mice had prolonged lung fungal burden (239). In addition, CCR2 KO mice had increased eosinophilia, increased leukocyte production of IL-4 and IL-5, and lack of a delayed-type hypersensitivity response, which all signify a type 2 immune response. Second, Osterholzer et al. showed that CCR2-deficient mice infected with C. neoformans strain 52D exhibited prolonged infection and a type 2 immune response compared to WT mice (238). These effects were attributed to impaired DC recruitment and protective T cell polarization. Third, Masso-Silva et al. compared the immune response between an infection with the WT H99 strain and a hypovirulent mutant, C. neoformans Δfbp1 (223). From flow cytometry and lung burden analyses, the authors concluded that CCR2+ monocytes differentiate into moDCs in response to the Δfbp1 mutant, and that CCR2 depleted mice were not protected due to an impaired T cell response in the lungs. Together, these studies suggest that CCR2-mediated recruitment of monocytes aid in fungal clearance of the less virulent C. neoformans 52D or the hypovirulent strain C. neoformans Δfbp1.
The collective conclusion from the above results in the mouse model point to monocytes being detrimental in response to the highly virulent serotype A C. neoformans H99, but protective in response to the weaker 52D strain. The GXM capsule of C. neoformans has been described as immunosuppressive (43) and has the ability to modulate macrophage polarization toward an M2 state (42, 44, 128, 241, 242). In addition to this, the five serotypes of Cryptococcus GXM capsule are differentially recognized by host innate immune cells, which suggests a possible variation in response to different serotypes. It is possible that an unknown GXM PAMP from serotype D strains drives monocytes into a protective maturation state, while serotype A strains either directly deter monocytes into an immature state or lack the necessary ligand needed to drive monocyte maturation.
Eosinophils
Eosinophils are another type of granulocyte (in addition to neutrophils) that may be recruited to the lungs during cryptococcal infection. Eosinophils are generally associated with allergic inflammation and are common effector cells against parasitic infections (243, 244), but these cells have been described as harmful in the host response to C. neoformans infection (135, 196, 239, 245, 246). Indeed, these studies (as well as others) have used eosinophil accumulation in the lungs as a marker of non-protective host immunity since these cells respond to increased levels of type 2 cytokines such as IL-5 and eotaxin.
In a recent study, Wiesner et al. utilized several approaches in surmising the lung lymphocyte-mediated recruitment of different granulocyte populations to the lung upon infection with the virulent C. neoformans strain KN99α in the C57BL/6 mice model (214). The authors used transgenic mouse models (STAT6 KO to ablate a Th2 response) and antibody depletions (anti-CD4 or anti-IL-5 to ameliorate these cells or cytokines, respectively) to determine the order of lymphocyte-mediated recruitment of granulocytes to the lungs went in order from Th2, Th17, Tc17, γδ T cells, and ILC2. In other words, Th2 CD4+ T cells from WT animals will preferentially recruit eosinophils via IL-5 in response to the virulent KN99α strain. When these cells were depleted, type 17 immune cells in order from Th17, Tc17, and γδ T cells recruited neutrophils to the lungs via IL-17A production. Not only did this study shed light upon a differential regulation of granulocyte recruitment in response to C. neoformans infection, but it also demonstrated a previously unstudied “pecking order” for lung cell recruitment in these mice that are notoriously susceptible to cryptococcosis. An interesting follow up to this study would involve using the less virulent serotype D strain in C57 mice as well as using both serotypes A and D in a more resistant mouse model such as CBA/J mice. This is an important concept because eosinophilia has been reported in human cryptococcosis cases (247) and the underlying cause of susceptibility of C57 mice to C. neoformans compared to other inbred strains such as CBA/J (39, 248, 249), which needs to be at the forefront of interpreting data across different strains of mice.
Eosinophils in rats exhibit different immunophenotypes than what is observed in mice (87, 250). In rats, eosinophils are part of the inflammatory response to C. neoformans. Garro et al. took an in vitro approach to studying the outcome of rat peritoneal eosinophils with opsonized C. neoformans (250). The authors found that eosinophils phagocytosed the yeast cells, upregulated MHC-I, MHC-II, and costimulatory molecules, and exhibited increased production of IL-12, TNFα, and IFN-γ. Additionally, these yeast-loaded eosinophils were able to induce CD4 and CD8 T cell proliferation and type 1 cytokine responses when cocultured together. Together these data suggest that eosinophils from rats possess anti-cryptococcal capabilities, act as antigen presenting cells, and promote a protective type 1 response.
Late Stage Adaptive Cell-Mediated Immunity to C. neoformans
Adaptive immune priming and recruitment to the lung is essential in host defense against C. neoformans. While B cells and antibody humoral responses show some beneficial aspects, T cell mediated immunity via CD4 and CD8 T cells provides optimal control and killing of the yeast cells. These cells express the canonical TCR with α and β subunits and require presentation of antigens on MHC molecules by APCs to become activated and recruited.
T Lymphocytes
T cells can be divided into CD4+ and CD8+ subsets, each possessing protective capabilities in the absence of the other during infection with C. neoformans (251–255). T cell-mediated immunity represents the most widely accepted potent host defense against C. neoformans infections, as well as the deepest downfall when a host is lymphopenic such as in HIV/AIDS patients. Indeed, T cells have been clinically implicated as major factor in the development of cryptococcal meningitis (115, 246, 256). These cells can aid in protection by either being directly cytotoxic or by secreting cytokines that aid in increased phagocyte uptake and anti-cryptococcal killing (252, 254, 257–260). Unlike the fluid polarization states of macrophages, once T cells polarize, they have more of a defined immune signature. Although differentiated T cell plasticity exists, type 1, 2, or 17 T cells (either CD4+ or CD8+) truly represent the subsets that pertain to anti-cryptococcal immunity and protection. These sub-sets are the most reported in the cryptococcal literature (expanded upon further down in this section), but a comprehensive review of this late-stage plasticity can be found in the following reviews (261–263).
The CD4+ T lymphocyte subsets Th1 and Th17 have been shown to be protective in response to infection with C. neoformans, with Th1 be more widely studied in the literature. Th1 T cells mature from naïve T cells in the presence of IFN-γ and are known to potently secrete IFN-γ and IL-2 as the major effector cytokines in response to IL-12 and IL-1β from lung macrophages to maintain the type 1 inflammatory response (258, 260, 264, 265). IFN-γ from Th1 T cells potently induces DC maturation for increased antigen presentation, classically activated M1 macrophages with increased reactive oxygen and nitrogen species production, and IgG2a/IgG3 B cell class switch recombination.
Th17 T cells are also necessary for anti-fungal defense and are defined by secretion of IL-17, which has a vast repertoire of inflammatory and tissue-specific responses. These cells mature from naïve T cells in the presence of TGF-β, IL-1, IL-23, and IL-6. IL-17 secretion from Th17 cells increases hematopoiesis and myelopoiesis, induce chemoattractants such as IL-8 and MCP-1 for neutrophil and monocyte recruitment, stimulate IgG2a/IgG3 B cell class switch recombination, and lead to downstream increases in prostaglandin E2, IL-6, nitric oxide, and IFN-γ production (120, 204, 224, 258, 266, 267). Th17 T cells also produce IL-22 in mucosal barrier tissues such as the gut and lung (268–270). Although Th22 T cells are a distinct subset, IL-22 producing Th17 cells are commonly observed in the mucosal barrier tissues. IL-22 increases the mucosal barrier integrity of these tissues, which in the case of pulmonary cryptococcosis has been suggested to prevent dissemination although this phenomenon has not been directly tested in vivo for C. neoformans.
Th2 T cells have been shown to be harmful during active cryptococcal infection. These cells mature from naïve T cells in the presence of IL-4 from alternatively activated macrophages and IL-33 lung epithelial and ILC2 cells (135, 243). These cells are seen in response to hosts that cannot control the early stages of infection when macrophages and DCs are polarized toward a type 2 state. These cells are associated with anti-parasitic defense via the secretion of IL-4, IL-5, and IL-13 and promote the recruitment of eosinophils. Altogether these cells lead to an anti-inflammatory response, M2 macrophage polarization, and persistence of fungal growth.
CD8+ T cells have also been described in antifungal defense but less so during infection with C. neoformans (251, 253, 267, 271–273). Analogously to the Th subsets of CD4+ T cells, the Tc subsets of CD8+ T cells have been described elsewhere (243, 251, 267) and follow the same immunological roles with Tc1, Tc17, and Tc2 mirroring Th1, Th17, and Th2, respectively. These Tc cell subsets have been reported have better direct killing ability of infected cells than the Th counterparts, however. Overall, the long-standing belief that CD4+ T cells are the sole driver of anti-cryptococcal immunity can be disregarded in our opinion since both CD8+ and CD4+ T cells possess potent anti-fungal capabilities during C. neoformans infection.
Summary of Immune Response Kinetics During Primary Infection
It is obvious that there is a well-orchestrated host response in the recognition, cytokine secretion, and temporal cell recruitment to immunological control of C. neoformans. When the fungal particles are inhaled into the lungs, epithelial and tissue resident cells recognize and take up the fungal particles (Figure 1). In response to this uptake, early cytokines such as IL-8 and IL-1 from the epithelial cells, TNFα and IFN-γ from macrophages, and IL-2, IL-12, and IL-23 from DCs induce an inflammatory state that promotes M1 macrophage polarization and chemokine secretion, such as GM-CSF and MCP-1. The collaborative effect of these early signals induces myelopoiesis and recruitment of neutrophils, monocytes, and DCs to the lung. In the presence of macrophage-derived IFN-γ and TNFα, DCs mature, phagocytose fungal particles, upregulate costimulatory molecules and migration markers, and migrate to the mLN for antigen presentation to naïve T cells. During this time, γδ T cells and ILCs may also play a role in the coordination the immune response, although their exact role is still unclear. As the innate immunity controls fungal proliferation, the adaptive immune system is recruited to the lung where type 1/17 T cells become potent sources of IFN-γ, IL-2, and IL-17 that activate the killing and/or containment of the yeast cells. Epithelial barrier integrity and antimicrobial peptide production are stimulated by IL-22 to deter extrapulmonary dissemination, phagocytic killing abilities are increased, and anti-fungal neutrophils are recruited and activated, leading to total elimination of fungal cells or/and containment of the yeast in a granuloma. Altogether, these cells work together synergistically for optimal host control of the infection.
Reactivation Model of a Latent Fungal Infection: Breakdown of Granuloma Containment
There are several noteworthy differences from the primary infection immune response previously described in mice and the response to reactivated C. neoformans. First, in the reactivation model, the fungal cells are already present in the lung tissue. Second, the host has already experienced a pulmonary cryptococcal infection, contained it, and has developed adaptive immunity against the yeast. Third, the host was able to control the initial infection during an immunocompetent state. Yet, if the host is experiencing reactivation of latent yeast cells, then some immunosuppressive event has led to this reactivation nullifying one or more host cell types that normally control the fungal replication.
With this said, the reactivation model is still one of the most understudied topics in regard to cryptococcosis mostly due to the lack of tools in model organisms. The mouse is by far the most popular animal model organism to study the primary infection model of cryptococcosis, yet the mouse not being able to form lung granulomas for containment of the fungus that recapitulate human granulomas results in the reactivation model being understudied (274). Mice have been previously mentioned to form a granulomatous response to moderately virulent C. neoformans strain 52D that forms a persistent infection. Although certain transgenic models have been shown to form granulomas, an immunocompetent mouse does not.
However, studies in our lab found that when mice are infected with a C. neoformans mutant strain in which the glucosylceramide synthase gene has been deleted (Δgcs1), they formed lung granulomas that closely resembled human lung granulomas (111). These granulomas are depicted by a necrotic center, a characteristic ring of foamy macrophages and multi-nucleated giant cells loaded with fungi or fungal debris (Figure 2). The macrophage ring is surrounded by an infiltration of lymphocytes, fibroblasts, and fibrotic tissue with collagen deposition. The resemblance of this mouse granuloma with the human lung granuloma (275–277) is simply striking.
In follow up studies with this mutant, the formation of these lung granulomas to contain C. neoformans Δgcs1 was found to be dependent on the host enzyme sphingosine kinase 1 (SK1) in the lungs (24, 274). Elevated levels of S1P, MCP-1, and TNFα in the bronchoalveolar lavage fluid were found to be significantly associated with granuloma containment. Interestingly, during the infection with Δgcs1, lung macrophages are a significant source of S1P, MCP-1, and TNFα and a highly regarded cell type in granuloma homeostasis. S1P is a widely studied immunological signaling molecule that directs leukocyte migration and function (278, 279), MCP-1/CCL2 is a pro-inflammatory cytokine that aids in recruitment of CCR2+ monocytes, memory T cells, and DCs, and TNFα is a pro-inflammatory cytokine that activates phagocytes and aids in DC maturation for induction of T cell-mediated immunity. Taken together, macrophages have been shown here to exert an early signaling cascade that is dependent on SK1 and drives the recruitment of innate and adaptive immunity to begin granuloma containment of C. neoformans Δgcs1.
Sphingosine phosphate receptors became an important function in granuloma reactivation since this family of five receptors are heterogeneously distributed on immune cells and respond to S1P gradients [for a detailed review of this topic please see (280, 281)].
Certain medications can cause immunosuppression and thus the prolonged administration of these medications is a predisposing factor for developing cryptococcosis. However, whether cryptococcosis develops as a result of the primary infection or reactivation is still controversial. One medication that causes a dramatic T cell depletion is FTY720 (Gilenya) used to treat multiple sclerosis (282).
Upon administration, FTY720 is rapidly phosphorylated into FTY720-P, becoming an analog of S1P and now able to bind S1P receptors 1, 3, 4, and 5. Other FTY720 derivatives, such as BAF312, is able to directly bind its receptors (1, 4, and 5) without being phosphorylated. Bryan et al. found that treatment with FTY720 but not BAF312 causes cryptococcosis in mice, and the infection is a result of a reactivation from the granuloma rather than a dissemination from the primary infection (283).
Very interestingly, FTY720 treated mice exhibited altered granuloma structure and impaired macrophage killing activity compared to BAF312 treated mice, despite the observation that both drugs caused similar lymphopenia. In addition, compared to BAF312, FTY720 not only disorganized the ring of macrophages at the granuloma site but these macrophages were mostly M2 polarized. These phenotypes are linked to the specific interaction of FTY720 to S1Pr3 (not targeted by BAF312), as macrophages lacking S1Pr3 also displayed poor phagocytosis and killing activity.
The observation that lymphopenia was present during both treatments clearly suggests that this immune condition is necessary but not sufficient to reactivate latent C. neoformans during FTY720 treatment (283).
It is important to mention that a limitation of this study is the use of C. neoformans Δgcs1 mutant. C. neoformans Δgcs1 mutant lacks the sphingolipid glucosylceramide. As a result, the mutant goes in cell cycle arrest in neutral/alkaline pH; it does not die when cells are exposed to these environments, rather it just cannot replicate (111). On the other hand, fungal glucosylceramides, including the species produced by C. neoformans, are highly immunogenic and mice develop antibodies against this fungal sphingolipid (284). It is possible that this antibody production in mice is counterproductive for the development of the granuloma, although antibody production also occurs in humans (285), whom are capable to develop lung granulomas. Due to its immunogenicity, fungal glucosylceramide may alter the initial lung innate immunity leading to the granuloma formation, in addition to have a notable effect on restricting fungal replication.
Nonetheless, the work using this cryptococcal mutant (30, 274) have laid the foundation for important animal studies in the reactivation of C. neoformans, such as the recent work by Bryan et al. (283) on the reactivation of the cryptococcal disease under certain drug immunodeficiency. The fact that macrophage function at the granuloma site must also be altered in addition to lymphopenia, it may explain why only a small fraction of subjects with AIDS develop cryptococcosis.
Conclusions and Future Perspectives
With the ever-increasing population of immunocompromised individuals, improvements in treatment and vaccinations are imperative for opportunistic infections, such as cryptococcosis. These improvements begin with structuring proper animal model systems to study the infection kinetics on the basic research side before being implemented into the clinical side. Work using the primary infection model purports invaluable information for protective anti-cryptococcal immunity and more research on certain host cell types, such as epithelial cells, γδ T cells, and ILCs, is warranted. This is especially important for investigators that look into protective immunity elicited by vaccination strains, since a thorough understanding of these models is at the forefront of transitioning from basic research into human clinical trials.
Also, the reactivation model of cryptococcosis has been widely understudied. The C. neoformans Δgcs1 mutant offers an invaluable tool that opens up new possibilities, but caution should be taken because the effect of glucosylceramide on the host immunity is not fully understood. Nonetheless, the observation that a drug (FTY720) used to treat reoccurring multiple sclerosis resulted in C. neoformans reactivation, has paved the way for a better understanding of the lung immunity required for both the formation and the containment of the lung granuloma.
Author Contributions
TN and MD contributed to the initial writing. TN, AB, and MD contributed to the editing and finalization of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants AI136934, AI116420, and AI125770, and by the Merit Review Grant I01BX002924 from the Veterans Affairs Program to MD. MD is a Burroughs Welcome Investigator in Infectious Diseases.
Conflict of Interest
MD is a Co-Founder and Chief Scientific Officer (CSO) of MicroRid Technologies Inc. AB is an employee of inGenious Targeting Laboratory Inc.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kozubowski L, Heitman J. Profiling a killer, the development of Cryptococcus neoformans. FEMS Microbiol Rev. (2012) 36:78–94. doi: 10.1111/j.1574-6976.2011.00286.x
2. Zhao Y, Lin J, Fan Y, Lin X. Life Cycle of Cryptococcus neoformans. Annu Rev Microbiol. (2019) 73:17–42. doi: 10.1146/annurev-micro-020518-120210
3. Maziarz EK, Perfect JR. Cryptococcosis. Infect Dis Clin North Am. (2016) 30:179–206. doi: 10.1016/j.idc.2015.10.006
4. Mourad A, Perfect JR. Present and future therapy of Cryptococcus infections. J Fungi. (2018) 4:79. doi: 10.3390/jof4030079
5. Goralska K, Blaszkowska J, Dzikowiec M. Neuroinfections caused by fungi. Infection. (2018) 46:443–59. doi: 10.1007/s15010-018-1152-2
6. Otto SBJ, George PE, Mercedes R, Nabukeera-Barungi N. Cryptococcal meningitis and immune reconstitution inflammatory syndrome in a pediatric patient with HIV after switching to second line antiretroviral therapy: a case report. BMC Infect Dis. (2020) 20:68. doi: 10.1186/s12879-020-4797-2
7. Wu X, Shen Y. Management of human immunodeficiency virus-associated cryptococcal meningitis: current status and future directions. Mycoses. (2019) 62:874–82. doi: 10.1111/myc.12977
8. Hagen F, Khayhan K, Theelen B, Kolecka A, Polacheck I, Sionov E, et al. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet Biol. (2015) 78:16–48. doi: 10.1016/j.fgb.2015.02.009
9. Kwon-Chung KJ, Fraser JA, Doering TL, Wang ZA, Janbon G, Idnurm A, et al. Cryptococcus neoformans and Cryptococcus gattii, the etiological agents of cryptococcosis. Cold Spring Harb Perspect Med. (2014) 4:a019760. doi: 10.1101/cshperspect.a019760
10. Desnos-Ollivier M, Patel S, Raoux-Barbot D, Heitman J, Dromer F, French Cryptococcosis Study G. Cryptococcosis serotypes impact outcome and provide evidence of Cryptococcus neoformans speciation. MBio. (2015) 6:e00311. doi: 10.1128/mBio.00311-15
11. Van Dyke MCC, Chaturvedi AK, Hardison SE, Leopold Wager CM, Castro-Lopez N, Hole CR, et al. Induction of broad-spectrum protective immunity against disparate Cryptococcus serotypes. Front Immunol. (2017) 8:1359. doi: 10.3389/fimmu.2017.01359
12. Probert M, Zhou X, Goodall M, Johnston SA, Bielska E, Ballou ER, et al. A glucuronoxylomannan epitope exhibits serotype-specific accessibility and redistributes towards the capsule surface during titanization of the fungal pathogen Cryptococcus neoformans. Infect Immun. (2019) 87:e00731-18. doi: 10.1128/IAI.00731-18
13. Zaragoza O. Basic principles of the virulence of Cryptococcus. Virulence. (2019) 10:490–501. doi: 10.1080/21505594.2019.1614383
14. Araujo Gde S, Fonseca FL, Pontes B, Torres A, Cordero RJ, Zancope-Oliveira RM, et al. Capsules from pathogenic and non-pathogenic Cryptococcus spp. manifest significant differences in structure and ability to protect against phagocytic cellsPLoS ONE. (2012) 7:e29561. doi: 10.1371/journal.pone.0029561
15. Abadi J, Pirofski L. Antibodies reactive with the cryptococcal capsular polysaccharide glucuronoxylomannan are present in sera from children with and without human immunodeficiency virus infection. J Infect Dis. (1999) 180:915–9.
16. Goldman DL, Khine H, Abadi J, Lindenberg DJ, Pirofski L, Niang R, et al. Serologic evidence for Cryptococcus neoformans infection in early childhood. Pediatrics. (2001) 107:E66. doi: 10.1542/peds.107.5.e66
17. Alvarez M, Casadevall A. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr Biol. (2006) 16:2161–5. doi: 10.1016/j.cub.2006.09.061
18. Feldmesser M, Tucker S, Casadevall A. Intracellular parasitism of macrophages by Cryptococcus neoformans. Trends Microbiol. (2001) 9:273–8. doi: 10.1016/s0966-842x(01)02035-2
19. Garcia J, Shea J, Alvarez-Vasquez F, Qureshi A, Luberto C, Voit EO, et al. Mathematical modeling of pathogenicity of Cryptococcus neoformans. Mol Syst Biol. (2008) 4:183–95. doi: 10.1038/msb.2008.17
20. Kechichian TB, Shea J, Del Poeta M. Depletion of alveolar macrophages decreases the dissemination of a glucosylceramide-deficient mutant of Cryptococcus neoformans in immunodeficient mice. Infect Immun. (2007) 75:4792–8. doi: 10.1128/IAI.00587-07
21. Khan MA, Jabeen R, Nasti TH, Mohammad O. Enhanced anticryptococcal activity of chloroquine in phosphatidylserine-containing liposomes in a murine model. J Antimicrob Chemother. (2005) 55:223–8. doi: 10.1093/jac/dkh522
22. Luberto C, Martinez-Marino B, Taraskiewicz D, Bolanos B, Chitano P, Toffaletti DL, et al. Identification of App1 as a regulator of phagocytosis and virulence of Cryptococcus neoformans. J. Clin. Invest. (2003) 112:1080–94. doi: 10.1172/JCI18309
23. Macura N, Zhang T, Casadevall A. Dependence of macrophage phagocytic efficacy on antibody concentration. Infect Immun. (2007) 75:1904–15. doi: 10.1128/IAI.01258-06
24. McQuiston T, Luberto C, Del Poeta M. Role of host sphingosine kinase 1 in the lung response against Cryptococcosis [Research Support, N.I.H., Extramural Research Support, U.S. Gov't, Non-P.H.S.]. Infect. Immun. (2010) 78:2342–52. doi: 10.1128/IAI.01140-09
25. Olszewski MA, Noverr MC, Chen GH, Toews GB, Cox GM, Perfect JR, et al. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am J Pathol. (2004) 164:1761–71. doi: 10.1016/S0002-9440(10)63734-0
26. Shea JM, Kechichian TB, Luberto C, Del Poeta M. The cryptococcal enzyme inositol phosphosphingolipid-phospholipase C confers resistance to the antifungal effects of macrophages and promotes fungal dissemination to the central nervous system [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't] Infect Immun. (2006) 74:5977–88. doi: 10.1128/IAI.00768-06
27. Steenbergen JN, Casadevall A. The origin and maintenance of virulence for the human pathogenic fungus Cryptococcus neoformans. Microbes Infect. (2003) 5:667–75. doi: 10.1016/s1286-4579(03)00092-3
28. Tucker SC, Casadevall A. Replication of Cryptococcus neoformans in macrophages is accompanied by phagosomal permeabilization and accumulation of vesicles containing polysaccharide in the cytoplasm. Proc Natl Acad Sci USA. (2002) 99:3165–70. doi: 10.1073/pnas.052702799
29. Wormley FL Jr, Cox GM, Perfect JR. Evaluation of host immune responses to pulmonary cryptococcosis using a temperature-sensitive Cryptococcus neoformans calcineurin A mutant strain. Microb Pathog. (2005) 38:113–23. doi: 10.1016/j.micpath.2004.12.007
30. McQuiston T, Luberto C, Del Poeta M. Role of sphingosine-1-phosphate (S1P) and S1P receptor 2 in the phagocytosis of Cryptococcus neoformans by alveolar macrophages. Microbiology. (2011) 157(Pt 5):1416–27. doi: 10.1099/mic.0.045989-0
31. Alanio A, Desnos-Ollivier M, Dromer F. Dynamics of Cryptococcus neoformans-macrophage interactions reveal that fungal background influences outcome during cryptococcal meningoencephalitis in humans. MBio. (2011) 2:e00158-11. doi: 10.1128/mBio.00158-11
32. Desnos-Ollivier M, Patel S, Spaulding AR, Charlier C, Garcia-Hermoso D, Nielsen K, et al. Mixed infections and in vivo evolution in the human fungal pathogen Cryptococcus neoformans. MBio. (2010) 1:e00091-10. doi: 10.1128/mBio.00091-10
33. Spitzer ED, Spitzer SG, Freundlich LF, Casadevall A. Persistence of initial infection in recurrent Cryptococcus neoformans meningitis. Lancet. (1993) 341:595–6.
34. McMullan BJ, Sorrell TC, Chen SC. Cryptococcus gattii infections: contemporary aspects of epidemiology, clinical manifestations and management of infection. Future Microbiol. (2013) 8:1613–31. doi: 10.2217/fmb.13.123
35. Sorrell TC, Chen SC-A, Phillips P, Marr KA. Clinical perspective on Cryptococcus neoformans and Cryptococcus gattii: implications for diagnosis and management. In: Heitman J, Kozel TR, Kwon-Chung KJ, Perfect J, Casadevall A, editors. Cryptococcus: From Human Pathogen to Model Yeast. Washington, DC: ASM (2011) p. 595–606.
36. Calo S, Billmyre RB, Heitman J. Generators of phenotypic diversity in the evolution of pathogenic microorganisms. PLoS Pathog. (2013) 9:e1003181. doi: 10.1371/journal.ppat.1003181
37. Danesi P, Firacative C, Cogliati M, Otranto D, Capelli G, Meyer W. Multilocus sequence typing (MLST) and M13 PCR fingerprinting revealed heterogeneity amongst Cryptococcus species obtained from Italian veterinary isolates. FEMS Yeast Res. (2014) 14:897–909. doi: 10.1111/1567-1364.12178
38. Escandon P, Sanchez A, Martinez M, Meyer W, Castaneda E. Molecular epidemiology of clinical and environmental isolates of the Cryptococcus neoformans species complex reveals a high genetic diversity and the presence of the molecular type VGII mating type a in Colombia. FEMS Yeast Res. (2006) 6:625–35. doi: 10.1111/j.1567-1364.2006.00055.x
39. Mukaremera L, McDonald TR, Nielsen JN, Molenaar CJ, Akampurira A, Schutz C, et al. The mouse inhalation model of Cryptococcus neoformans infection recapitulates strain virulence in humans and shows that closely related strains can possess differential virulence. Infect Immun. (2019) 87:e00046-19. doi: 10.1128/IAI.00046-19
40. Walsh NM, Botts MR, McDermott AJ, Ortiz SC, Wuthrich M, Klein B, et al. Infectious particle identity determines dissemination and disease outcome for the inhaled human fungal pathogen Cryptococcus. PLoS Pathog. (2019) 15:e1007777. doi: 10.1371/journal.ppat.1007777
41. Barbosa FM, Fonseca FL, Figueiredo RT, Bozza MT, Casadevall A, Nimrichter L, et al. Binding of glucuronoxylomannan to the CD14 receptor in human A549 alveolar cells induces interleukin-8 production. Clin Vaccine Immunol. (2007) 14:94–8. doi: 10.1128/CVI.00296-06
42. Decote-Ricardo D, LaRocque-de-Freitas IF, Rocha JDB, Nascimento DO, Nunes MP, Morrot A, et al. Immunomodulatory role of capsular polysaccharides constituents of Cryptococcus neoformans. Front Med. (2019) 6:129. doi: 10.3389/fmed.2019.00129
43. Monari C, Bistoni F, Vecchiarelli A. Glucuronoxylomannan exhibits potent immunosuppressive properties. FEMS Yeast Res. (2006) 6:537–42. doi: 10.1111/j.1567-1364.2006.00072.x
44. Monari C, Pericolini E, Bistoni G, Casadevall A, Kozel TR, Vecchiarelli A. Cryptococcus neoformans capsular glucuronoxylomannan induces expression of fas ligand in macrophages. J Immunol. (2005) 174:3461–8. doi: 10.4049/jimmunol.174.6.3461
45. Denham ST, Brown JCS. Mechanisms of pulmonary escape and dissemination by Cryptococcus neoformans. J Fungi. (2018) 4:25. doi: 10.3390/jof4010025
46. Colombo AC, Rella A, Normile T, Joffe LS, Tavares PM, de SAGR, et al. Cryptococcus neoformans glucuronoxylomannan and sterylglucoside are required for host protection in an animal vaccination model. MBio. (2019) 10:e02909-18. doi: 10.1128/mBio.02909-18
47. Nelson BN, Hawkins AN, Wozniak KL. Pulmonary macrophage and dendritic cell responses to Cryptococcus neoformans. Front Cell Infect Microbiol. (2020) 10:37. doi: 10.3389/fcimb.2020.00037
48. Normile TG, McEvoy K, Del Poeta M. Steryl glycosides in fungal pathogenesis: an understudied immunomodulatory adjuvant. J Fungi. (2020) 6:25. doi: 10.3390/jof6010025
49. Osterholzer JJ, Chen GH, Olszewski MA, Zhang YM, Curtis JL, Huffnagle GB, et al. Chemokine receptor 2-mediated accumulation of fungicidal exudate macrophages in mice that clear cryptococcal lung infection. Am J Pathol. (2011) 178:198–211. doi: 10.1016/j.ajpath.2010.11.006
50. Osterholzer JJ, Milam JE, Chen GH, Toews GB, Huffnagle GB, Olszewski MA. Role of dendritic cells and alveolar macrophages in regulating early host defense against pulmonary infection with Cryptococcus neoformans. Infect Immun. (2009) 77:3749–58. doi: 10.1128/IAI.00454-09
51. Campuzano A, Castro-Lopez N, Martinez AJ, Olszewski MA, Ganguly A, Leopold Wager C, et al. CARD9 is required for classical macrophage activation and the induction of protective immunity against pulmonary cryptococcosis. MBio. (2020) 11:e03005-19. doi: 10.1128/mBio.03005-19
52. Eastman AJ, He X, Qiu Y, Davis MJ, Vedula P, Lyons DM, et al. Cryptococcal heat shock protein 70 homolog Ssa1 contributes to pulmonary expansion of Cryptococcus neoformans during the afferent phase of the immune response by promoting macrophage M2 polarization. J Immunol. (2015) 194:5999–6010. doi: 10.4049/jimmunol.1402719
53. Leopold Wager CM, Hole CR, Campuzano A, Castro-Lopez N, Cai H, Caballero Van Dyke MC, et al. IFN-gamma immune priming of macrophages in vivo induces prolonged STAT1 binding and protection against Cryptococcus neoformans. PLoS Pathog. (2018) 14:e1007358. doi: 10.1371/journal.ppat.1007358
54. Rella A, Mor V, Farnoud AM, Singh A, Shamseddine AA, Ivanova E, et al. Role of Sterylglucosidase 1 (Sgl1) on the pathogenicity of Cryptococcus neoformans: potential applications for vaccine development. Front Microbiol. (2015) 6:836. doi: 10.3389/fmicb.2015.00836
55. Wang Y, Wang K, Masso-Silva JA, Rivera A, Xue C. A heat-killed cryptococcus mutant strain induces host protection against multiple invasive mycoses in a murine vaccine model. MBio. (2019) 10:02145-19. doi: 10.1128/mBio.02145-19
56. Wozniak KL, Hardison SE, Kolls JK, Wormley FL. Role of IL-17A on resolution of pulmonary C. neoformans infection. PLoS ONE. (2011) 6:e17204. doi: 10.1371/journal.pone.0017204
57. Bryan AM, Farnoud AM, Mor V, Del Poeta M. Macrophage cholesterol depletion and its effect on the phagocytosis of Cryptococcus neoformans. J Vis Exp. (2014) 94:52432. doi: 10.3791/52432
58. Charlier C, Nielsen K, Daou S, Brigitte M, Chretien F, Dromer F. Evidence of a role for monocytes in dissemination and brain invasion by Cryptococcus neoformans. Infect Immun. (2009) 77:120–7. doi: 10.1128/IAI.01065-08
59. Kawanishi N, Mizokami T, Niihara H, Yada K, Suzuki K. Macrophage depletion by clodronate liposome attenuates muscle injury and inflammation following exhaustive exercise. Biochem Biophys Rep. (2016) 5:146–51. doi: 10.1016/j.bbrep.2015.11.022
60. Wozniak KL, Kolls JK, Wormley FLJr. Depletion of neutrophils in a protective model of pulmonary cryptococcosis results in increased IL-17A production by gammadelta T cells. BMC Immunol. (2012) 13:65. doi: 10.1186/1471-2172-13-65
61. Xu J, Eastman AJ, Flaczyk A, Neal LM, Zhao G, Carolan J, et al. Disruption of early tumor necrosis factor alpha signaling prevents classical activation of dendritic cells in lung-associated lymph nodes and development of protective immunity against cryptococcal infection. MBio. (2016) 7:e00510-16. doi: 10.1128/mBio.00510-16
62. Carroll SF, Guillot L, Qureshi ST. Mammalian model hosts of cryptococcal infection. Comp Med. (2007) 57:9–17.
63. Sabiiti W, May RC, Pursall ER. Experimental models of cryptococcosis. Int J Microbiol. (2012) 2012:626745. doi: 10.1155/2012/626745
64. Mylonakis E, Ausubel FM, Perfect JR, Heitman J, Calderwood SB. Killing of Caenorhabditis elegans by Cryptococcus neoformans as a model of yeast pathogenesis. Proc Natl Acad Sci USA. (2002) 99:15675–80. doi: 10.1073/pnas.232568599
65. Desalermos A, Tan X, Rajamuthiah R, Arvanitis M, Wang Y, Li D, et al. A multi-host approach for the systematic analysis of virulence factors in Cryptococcus neoformans. J Infect Dis. (2015) 211:298–305. doi: 10.1093/infdis/jiu441
66. da Derengowski LS, Paes HC, Albuquerque P, Tavares AH, Fernandes L, Silva-Pereira I, et al. The transcriptional response of Cryptococcus neoformans to ingestion by Acanthamoeba castellanii and macrophages provides insights into the evolutionary adaptation to the mammalian host. Eukaryot Cell. (2013) 12:761–74. doi: 10.1128/EC.00073-13
67. Fu MS, Casadevall A. Divalent metal cations potentiate the predatory capacity of amoeba for Cryptococcus neoformans. Appl Environ Microbiol. (2018) 84:e01717-17. doi: 10.1128/AEM.01717-17
68. Watkins RA, Andrews A, Wynn C, Barisch C, King JS, Johnston SA. Cryptococcus neoformans escape from dictyostelium amoeba by both wash-mediated constitutive exocytosis and vomocytosis. Front Cell Infect Microbiol. (2018) 8:108. doi: 10.3389/fcimb.2018.00108
69. Cruz-Acuna M, Pacifici N, Lewis JS. Vomocytosis: too much booze, base, or calcium? MBio. (2019) 10:e02526-19. doi: 10.1128/mBio.02526-19
70. Seoane PI, May RC. Vomocytosis: what we know so far. Cell Microbiol. (2020) 22:e13145. doi: 10.1111/cmi.13145
71. Mylonakis E, Moreno R, El Khoury JB, Idnurm A, Heitman J, Calderwood SB, et al. Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesis. Infect Immun. (2005) 73:3842–50. doi: 10.1128/IAI.73.7.3842-3850.2005
72. Jemel S, Guillot J, Kallel K, Botterel F, Dannaoui E. Galleria mellonella for the evaluation of antifungal efficacy against medically important fungi, a narrative review. Microorganisms. (2020) 8:390. doi: 10.3390/microorganisms8030390
73. Melillo D, Marino R, Italiani P, Boraschi D. Innate immune memory in invertebrate metazoans: a critical appraisal. Front Immunol. (2018) 9:1915. doi: 10.3389/fimmu.2018.01915
74. Rosowski EE, Knox BP, Archambault LS, Huttenlocher A, Keller NP, Wheeler RT, et al. The zebrafish as a model host for invasive fungal infections. J Fungi. (2018) 4:136. doi: 10.3390/jof4040136
75. Bojarczuk A, Miller KA, Hotham R, Lewis A, Ogryzko NV, Kamuyango AA, et al. Cryptococcus neoformans intracellular proliferation and capsule size determines early macrophage control of infection. Sci Rep. (2016) 6:21489. doi: 10.1038/srep21489
76. Davis JM, Huang M, Botts MR, Hull CM, Huttenlocher A. A zebrafish model of cryptococcal infection reveals roles for macrophages, endothelial cells, and neutrophils in the establishment and control of sustained fungemia. Infect Immun. (2016) 84:3047–62. doi: 10.1128/IAI.00506-16
77. Tenor JL, Oehlers SH, Yang JL, Tobin DM, Perfect JR. live imaging of host-parasite interactions in a zebrafish infection model reveals cryptococcal determinants of virulence and central nervous system invasion. MBio. (2015) 6:e01425–e01415. doi: 10.1128/mBio.01425-15
78. Linares G, Daker RD. Cryptococcal dermotropism in the rhesus monkey. Mycopathol Mycol Appl. (1972) 46:17–32. doi: 10.1007/BF02051893
79. Pal M, Dube GD, Mehrotra BS. Pulmonary cryptococcosis in a rhesus monkey (Macaca mulatta). Mykosen. (1984) 27:309–12. doi: 10.1111/j.1439-0507.1984.tb02035.x
80. Li H, Li Y, Sun T, Du W, Li C, Suo C, et al. Unveil the transcriptional landscape at the Cryptococcus-host axis in mice and nonhuman primates. PLoS Negl Trop Dis. (2019) 13:e0007566. doi: 10.1371/journal.pntd.0007566
81. Perfect JR, Lang SD, Durack DT. Chronic cryptococcal meningitis: a new experimental model in rabbits. Am J Pathol. (1980) 101:177–94.
82. Kovanda LL, Giamberardino C, McEntee L, Toffaletti DL, Franke KS, Bartuska A, et al. Pharmacodynamics of isavuconazole in a rabbit model of cryptococcal meningoencephalitis. Antimicrob Agents Chemother. (2019) 63:e00546-19. doi: 10.1128/AAC.00546-19
83. Lestner J, McEntee L, Johnson A, Livermore J, Whalley S, Schwartz J, et al. Experimental models of short courses of liposomal amphotericin B for induction therapy for cryptococcal meningitis. Antimicrob Agents Chemother. (2017) 61:e00090-17. doi: 10.1128/AAC.00090-17
84. Nixon GL, McEntee L, Johnson A, Farrington N, Whalley S, Livermore J, et al. Repurposing and reformulation of the antiparasitic agent flubendazole for treatment of cryptococcal meningoencephalitis, a neglected fungal disease. Antimicrob Agents Chemother. (2018) 62:01909-17. doi: 10.1128/AAC.01909-17
85. Shaw KJ, Schell WA, Covel J, Duboc G, Giamberardino C, Kapoor M, et al. in vitro and in vivo evaluation of APX001A/APX001 and other Gwt1 inhibitors against cryptococcus. Antimicrob Agents Chemother. (2018) 62:e00523-18. doi: 10.1128/AAC.00523-18
86. Scrimgeour EM, Purohit RG. Chronic pulmonary cryptococcosis in a Rattus rattus from Rabaul, Papua New Guinea. Trans R Soc Trop Med Hyg. (1984) 78:827–8. doi: 10.1016/0035-9203(84)90034-8
87. Feldmesser M, Casadevall A, Kress Y, Spira G, Orlofsky A. Eosinophil-Cryptococcus neoformans interactions in vivo and in vitro. Infect Immun. (1997) 65:1899–907.
88. Kobayashi M, Ito M, Sano K, Koyama M. Granulomatous and cytokine responses to pulmonary Cryptococcus neoformans in two strains of rats. Mycopathologia. (2000) 151:121–30. doi: 10.1023/a:1017900604050
89. Masih DT, Sotomayor CE, Cervi LA, Riera CM, Rubinstein HR. Inhibition of I-A expression in rat peritoneal macrophages due to T-suppressor cells induced by Cryptococcus neoformans. J Med Vet Mycol. (1991) 29:125–8. doi: 10.1080/02681219180000211
90. Merkel GJ, Scofield BA, Rescorla FJ, Yang R, Grosfeld JL. Reduced recovery of a Cryptococcus neoformans adherence mutant from a rat model of cryptococcosis. Can J Microbiol. (1995) 41:428–32. doi: 10.1139/m95-057
91. Mizuochi T, Tentori L, Sharrow SO, Kruisbeek AM, Singer A. Differentiation of Ia-reactive CD8+ murine T cells does not require Ia engagement. Implications for the role of CD4 and CD8 accessory molecules in T cell differentiation. J Exp Med. (1988) 168:437–42. doi: 10.1084/jem.168.1.437
92. Shibuya K, Hirata A, Omuta J, Sugamata M, Katori S, Saito N, et al. Granuloma and cryptococcosis. J Infect Chemother. (2005) 11:115–22. doi: 10.1007/s10156-005-0387-x
93. Goldman D, Cho Y, Zhao M, Casadevall A, Lee SC. Expression of inducible nitric oxide synthase in rat pulmonary Cryptococcus neoformans granulomas. Am J Pathol. (1996) 148:1275–82.
94. Goldman DL, Casadevall A, Cho Y, Lee SC. Cryptococcus neoformans meningitis in the rat. Lab Invest. (1996) 75:759–70.
95. Bolanos B, Mitchell TG. Killing of Cryptococcus neoformans by rat alveolar macrophages. J Med Vet Mycol. (1989) 27:219–28.
96. Bolanos B, Mitchell TG. Phagocytosis and killing of Cryptococcus neoformans by rat alveolar macrophages in the absence of serum J Leukoc Biol. (1989) 46:521–8. doi: 10.1002/jlb.46.6.521
97. Bolanos B, Mitchell TG. Phagocytosis of Cryptococcus neoformans by rat alveolar macrophages. J Med Vet Mycol. (1989) 27:203–17.
98. Goulart L, Rosa e Silva LK, Chiapello L, Silveira C, Crestani J, Masih D, et al. Cryptococcus neoformans and Cryptococcus gattii genes preferentially expressed during rat macrophage infection. Med Mycol. (2010) 48:932–41. doi: 10.3109/13693781003677494
99. Krockenberger MB, Malik R, Ngamskulrungroj P, Trilles L, Escandon P, Dowd S, et al. Pathogenesis of pulmonary Cryptococcus gattii infection: a rat model. Mycopathologia. (2010) 170:315–30. doi: 10.1007/s11046-010-9328-z
100. Rosa RL, Berger M, Santi L, Driemeier D, Barros Terraciano P, Campos AR, et al. Proteomics of rat lungs infected by Cryptococcus gattii reveals a potential warburg-like effect. J Proteome Res. (2019) 18:3885–95. doi: 10.1021/acs.jproteome.9b00326
101. Chayakulkeeree M, Perfect JR. Cryptococcosis. Infect Dis Clin North Am. (2006) 20:507–44, v–vi. doi: 10.1016/j.idc.2006.07.001
102. Del Poeta M. Role of phagocytosis in the virulence of Cryptococcus neoformans. Eukaryot Cell. (2004) 3:1067–75. doi: 10.1128/EC.3.5.1067-1075.2004
103. Del Poeta M, Casadevall A. Ten challenges on Cryptococcus and cryptococcosis. Mycopathologia. (2011) 173:303–10. doi: 10.1007/s11046-011-9473-z
104. Goldman DL, Casadevall A. Cryptococcosis. In: Boros DL, editor. Granulomatous Infections and Inflammations: Cellular and Molecular Mechanisms. Washington, DC: ASM Press (2003) 84–116.
105. Goldman DL, Lee SC, Mednick AJ, Montella L, Casadevall A. Persistent Cryptococcus neoformans pulmonary infection in the rat is associated with intracellular parasitism, decreased inducible nitric oxide synthase expression, and altered antibody responsiveness to cryptococcal polysaccharide. Infect Immun. (2000) 68:832–8. doi: 10.1128/iai.68.2.832-838.2000
106. Leopold Wager CM, Hole CR, Wozniak KL, Olszewski MA, Wormley FL Jr. STAT1 signaling is essential for protection against Cryptococcus neoformans infection in mice. J Immunol. (2014) 193:4060–71. doi: 10.4049/jimmunol.1400318
107. Murdock BJ, Huffnagle GB, Olszewski MA, Osterholzer JJ. Interleukin-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and gamma interferon production. Infect Immun. (2014) 82:937–48. doi: 10.1128/IAI.01477-13
108. Roussey JA, Viglianti SP, Teitz-Tennenbaum S, Olszewski MA, Osterholzer JJ. Anti-PD-1 antibody treatment promotes clearance of persistent cryptococcal lung infection in mice. J Immunol. (2017) 199:3535–46. doi: 10.4049/jimmunol.1700840
109. Shourian M, Ralph B, Angers I, Sheppard DC, Qureshi ST. Contribution of IL-1RI signaling to protection against Cryptococcus neoformans 52D in a mouse model of infection. Front Immunol. (2017) 8:1987. doi: 10.3389/fimmu.2017.01987
110. Chen GH, Teitz-Tennenbaum S, Neal LM, Murdock BJ, Malachowski AN, Dils AJ, et al. Local GM-CSF-dependent differentiation and activation of pulmonary dendritic cells and macrophages protect against progressive cryptococcal lung infection in mice. J Immunol. (2016) 196:1810–21. doi: 10.4049/jimmunol.1501512
111. Rittershaus PC, Kechichian TB, Allegood J, Merrill AHJ, Hennig M, Luberto C, et al. Glucosylceramide is an essential regulator of pathogenicity of Cryptococcus neoformans. J Clin Invest. (2006) 116:1651–9. doi: 10.1172/JCI27890
112. Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS. (2009) 23:525–30. doi: 10.1097/QAD.0b013e328322ffac
113. Voelz K, May RC. Cryptococcal interactions with the host immune system. Eukaryot Cell. (2010) 9:835–46. doi: 10.1128/EC.00039-10
114. Campuzano A, Wormley FL. Innate immunity against cryptococcus, from recognition to elimination. J Fungi. (2018) 4:33. doi: 10.3390/jof4010033
115. Elsegeiny W, Marr KA, Williamson PR. Immunology of cryptococcal infections: developing a rational approach to patient therapy. Front Immunol. (2018) 9:651. doi: 10.3389/fimmu.2018.00651
116. Wozniak KL. Interactions of cryptococcus with dendritic cells. J Fungi. (2018) 4:36. doi: 10.3390/jof4010036
117. Brunet K, Alanio A, Lortholary O, Rammaert B. Reactivation of dormant/latent fungal infection. J Infect. (2018) 77:463–8. doi: 10.1016/j.jinf.2018.06.016
118. Mayer FL, Kronstad JW. Cryptococcus neoformans. Trends Microbiol. (2020) 28:163–4. doi: 10.1016/j.tim.2019.10.003
119. Shimizu H, Hara S, Nishioka H. Disseminated cryptococcosis with granuloma formation in idiopathic CD4 lymphocytopenia. J Infect Chemother. (2020) 26:257–60. doi: 10.1016/j.jiac.2019.07.006
120. Hernandez-Santos N, Wiesner DL, Fites JS, McDermott AJ, Warner T, Wuthrich M, et al. Lung epithelial cells coordinate innate lymphocytes and immunity against pulmonary fungal infection. Cell Host Microbe. (2018) 23:511–22 e515. doi: 10.1016/j.chom.2018.02.011
121. Leiva-Juarez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. (2018) 11:21–34. doi: 10.1038/mi.2017.71
122. Taylor-Smith LM. Cryptococcus-epithelial interactions. J Fungi. (2017) 3:53. doi: 10.3390/jof3040053
123. Hernandez-Santos N, Wiesner DL, Fites JS, McDermott AJ, Warner T, Wuthrich M, et al. Lung epithelial cells coordinate innate lymphocytes and immunity against pulmonary fungal infection. Cell Host Microbe. (2019) 25:630. doi: 10.1016/j.chom.2019.03.013
124. Ganendren R, Carter E, Sorrell T, Widmer F, Wright L. Phospholipase B activity enhances adhesion of Cryptococcus neoformans to a human lung epithelial cell line. Microbes Infect. (2006) 8:1006–15. doi: 10.1016/j.micinf.2005.10.018
125. Merkel GJ, Scofield BA. The in vitro interaction of Cryptococcus neoformans with human lung epithelial cells. FEMS Immunol Med Microbiol. (1997) 19:203–13. doi: 10.1111/j.1574-695X.1997.tb01089.x
126. Teixeira PA, Penha LL, Mendonca-Previato L, Previato JO. Mannoprotein MP84 mediates the adhesion of Cryptococcus neoformans to epithelial lung cells. Front Cell Infect Microbiol. (2014) 4:106. doi: 10.3389/fcimb.2014.00106
127. Guillot L, Carroll SF, Badawy M, Qureshi ST. Cryptococcus neoformans induces IL-8 secretion and CXCL1 expression by human bronchial epithelial cells. Respir Res. (2008) 9:9. doi: 10.1186/1465-9921-9-9
128. Barbosa FM, Fonseca FL, Holandino C, Alviano CS, Nimrichter L, Rodrigues ML. Glucuronoxylomannan-mediated interaction of Cryptococcus neoformans with human alveolar cells results in fungal internalization and host cell damage Microbes Infect. (2006) 8:493–502. doi: 10.1016/j.micinf.2005.07.027
129. Matsumoto Y, Azami S, Shiga H, Nagamachi T, Moriyama H, Yamashita Y, et al. Induction of signal transduction pathways related to the pathogenicity of Cryptococcus neoformans in the host environment. Drug Discov Ther. (2019) 13:177–82. doi: 10.5582/ddt.2019.01047
130. Trevijano-Contador N, de Oliveira HC, Garcia-Rodas R, Rossi SA, Llorente I, Zaballos A, et al. Cryptococcus neoformans can form titan-like cells in vitro in response to multiple signals. PLoS Pathog. (2018) 14:e1007007. doi: 10.1371/journal.ppat.1007007
131. Wang ZA, Li LX, Doering TL. Unraveling synthesis of the cryptococcal cell wall and capsule. Glycobiology. (2018) 28:719–30. doi: 10.1093/glycob/cwy030
132. Cooper JR, Abdullatif MB, Burnett EC, Kempsell KE, Conforti F, Tolley H, et al. Long term culture of the A549 cancer cell line promotes multilamellar body formation and differentiation towards an alveolar type II pneumocyte phenotype. PLoS ONE. (2016) 11:e0164438. doi: 10.1371/journal.pone.0164438
133. Swain RJ, Kemp SJ, Goldstraw P, Tetley TD, Stevens MM. Assessment of cell line models of primary human cells by Raman spectral phenotyping. Biophys J. (2010) 98:1703–11. doi: 10.1016/j.bpj.2009.12.4289
134. Alvarez F, Istomine R, Shourian M, Pavey N, Al-Aubodah TA, Qureshi S, et al. The alarmins IL-1 and IL-33 differentially regulate the functional specialisation of Foxp3(+) regulatory T cells during mucosal inflammation. Mucosal Immunol. (2019) 12:746–60. doi: 10.1038/s41385-019-0153-5
135. Piehler D, Eschke M, Schulze B, Protschka M, Muller U, Grahnert A, et al. The IL-33 receptor (ST2) regulates early IL-13 production in fungus-induced allergic airway inflammation. Mucosal Immunol. (2016) 9:937–49. doi: 10.1038/mi.2015.106
136. Espinosa V, Rivera A. First Line of defense: innate cell-mediated control of pulmonary aspergillosis. Front Microbiol. (2016) 7:272. doi: 10.3389/fmicb.2016.00272
137. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. (2013) 49:503–10. doi: 10.1165/rcmb.2013-0086MA
138. Vermaelen K, Pauwels R. Pulmonary dendritic cells. Am J Respir Crit Care Med. (2005) 172:530–51. doi: 10.1164/rccm.200410-1384SO
139. Wozniak KL, Vyas JM, Levitz SM. In vivo role of dendritic cells in a murine model of pulmonary cryptococcosis. Infect Immun. 2006) 74:3817–24. doi: 10.1128/IAI.00317-06
140. Kaufman-Francis K, Djordjevic JT, Juillard PG, Lev S, Desmarini D, Grau GER, et al. The early innate immune response to, and phagocyte-dependent entry of, Cryptococcus neoformans map to the perivascular space of cortical post-capillary venules in neurocryptococcosis. Am J Pathol. (2018) 188:1653–65. doi: 10.1016/j.ajpath.2018.03.015
141. McQuiston TJ, Williamson PR. Paradoxical roles of alveolar macrophages in the host response to Cryptococcus neoformans. J Infect Chemother. (2012) 18:1–9. doi: 10.1007/s10156-011-0306-2
142. Rudman J, Evans RJ, Johnston SA. Are macrophages the heroes or villains during cryptococcosis? Fungal Genet Biol. (2019) 132:103261. doi: 10.1016/j.fgb.2019.103261
143. De Leon-Rodriguez CM, Rossi DCP, Fu MS, Dragotakes Q, Coelho C, Guerrero Ros I, et al. The outcome of the Cryptococcus neoformans-macrophage interaction depends on phagolysosomal membrane integrity. J Immunol. (2018) 201:583–603. doi: 10.4049/jimmunol.1700958
144. Fu MS, Coelho C, De Leon-Rodriguez CM, Rossi DCP, Camacho E, Jung EH, et al. Cryptococcus neoformans urease affects the outcome of intracellular pathogenesis by modulating phagolysosomal pH. PLoS Pathog. (2018) 14:e1007144. doi: 10.1371/journal.ppat.1007144
145. Johnston SA, May RC. Cryptococcus interactions with macrophages: evasion and manipulation of the phagosome by a fungal pathogen. Cell Microbiol. (2013) 15:403–11. doi: 10.1111/cmi.12067
146. Smith LM, Dixon EF, May RC. The fungal pathogen Cryptococcus neoformans manipulates macrophage phagosome maturation. Cell Microbiol. (2015) 17:702–13. doi: 10.1111/cmi.12394
147. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio. (2013) 4:e00264–e00213. doi: 10.1128/mBio.00264-13
148. Leopold Wager CM, Wormley FL Jr. Classical versus alternative macrophage activation: the Ying and the Yang in host defense against pulmonary fungal infections. Mucosal Immunol. (2014) 7:1023–35. doi: 10.1038/mi.2014.65
149. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
150. Voelz K, Lammas DA, May RC. Cytokine signaling regulates the outcome of intracellular macrophage parasitism by Cryptococcus neoformans. Infect Immun. (2009) 77:3450–34. doi: 10.1128/IAI.00297-09
151. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. (2014) 26:192–7. doi: 10.1016/j.cellsig.2013.11.004
152. Draijer C, Penke LRK, Peters-Golden M. Distinctive effects of GM-CSF and M-CSF on proliferation and polarization of two major pulmonary macrophage populations. J Immunol. (2019) 202:2700–9. doi: 10.4049/jimmunol.1801387
153. Leopold Wager CM, Hole CR, Wozniak KL, Olszewski MA, Mueller M, Wormley FL Jr. STAT1 signaling within macrophages is required for antifungal activity against Cryptococcus neoformans. Infect Immun. (2015) 83:4513–27. doi: 10.1128/IAI.00935-15
154. Xu S, Shinohara ML. Tissue-resident macrophages in fungal infections. Front Immunol. (2017) 8:1798. doi: 10.3389/fimmu.2017.01798
155. Yin Z, Ma T, Lin Y, Lu X, Zhang C, Chen S, et al. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J Cell Biochem. (2018) 119:9419–32. doi: 10.1002/jcb.27259
156. Alanio A, Vernel-Pauillac F, Sturny-Leclere A, Dromer F. Cryptococcus neoformans host adaptation: toward biological evidence of dormancy. MBio. (2015) 6:e02580-14. doi: 10.1128/mBio.02580-14
157. Ballou ER, Johnston SA. The cause and effect of Cryptococcus interactions with the host. Curr Opin Microbiol. (2017) 40:88–94. doi: 10.1016/j.mib.2017.10.012
158. Garelnabi M, Taylor-Smith LM, Bielska E, Hall RA, Stones D, May RC. Quantifying donor-to-donor variation in macrophage responses to the human fungal pathogen Cryptococcus neoformans. PLoS ONE. (2018) 13:e0194615. doi: 10.1371/journal.pone.0194615
159. Crabtree JN, Okagaki LH, Wiesner DL, Strain AK, Nielsen JN, Nielsen K. Titan cell production enhances the virulence of Cryptococcus neoformans. Infect Immun. (2012) 80:3776–85. doi: 10.1128/IAI.00507-12
160. Mukaremera L, Lee KK, Wagener J, Wiesner DL, Gow NAR, Nielsen K. Titan cell production in Cryptococcus neoformans reshapes the cell wall and capsule composition during infection. Cell Surf. (2018) 1:15–24. doi: 10.1016/j.tcsw.2017.12.001
161. Okagaki LH, Nielsen K. Titan cells confer protection from phagocytosis in Cryptococcus neoformans infections. Eukaryot Cell. (2012) 11:820–6. doi: 10.1128/EC.00121-12
162. Zaragoza O, Nielsen K. Titan cells in Cryptococcus neoformans: cells with a giant impact. Curr Opin Microbiol. (2013) 16:409–13. doi: 10.1016/j.mib.2013.03.006
163. Billmyre RB, Applen Clancey S, Li LX, Doering TL, Heitman J. 5-fluorocytosine resistance is associated with hypermutation and alterations in capsule biosynthesis in Cryptococcus. Nat Commun. (2020) 11:127. doi: 10.1038/s41467-019-13890-z
164. Diamond RD, Bennett JE. Growth of Cryptococcus neoformans within human macrophages in vitro. Infect Immun. (1973) 7:231–6.
165. Miller MF, Mitchell TG. Killing of Cryptococcus neoformans strains by human neutrophils and monocytes. Infect Immun. (1991) 59:24–8.
166. Vecchiarelli A, Dottorini M, Pietrella D, Monari C, Retini C, Todisco T, et al. Role of human alveolar macrophages as antigen-presenting cells in Cryptococcus neoformans infection. Am J Respir Cell Mol Biol. (1994) 11:8.
167. Vogel DY, Glim JE, Stavenuiter AW, Breur M, Heijnen P, Amor S, et al. Human macrophage polarization in vitro: maturation and activation methods compared. Immunobiology. (2014) 219:695–703. doi: 10.1016/j.imbio.2014.05.002
168. Lupo P, Chang YC, Kelsall BL, Farber JM, Pietrella D, Vecchiarelli A, et al. The presence of capsule in Cryptococcus neoformans influences the gene expression profile in dendritic cells during interaction with the fungus. Infect Immun. (2008) 76:1581–9. doi: 10.1128/IAI.01184-07
169. Vecchiarelli A, Retini C, Pietrella D, Monari C, Kozel TR. T lymphocyte and monocyte interaction by CD40/CD40 ligand facilitates a lymphoproliferative response and killing of Cryptococcus neoformans in vitro. Eur J Immunol. (2000) 30:1385–93. doi: 10.1002/(SICI)1521-4141(200005)30:5<1385::AID-IMMU1385>3.0.CO
170. Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol. (2015) 16:36–44. doi: 10.1038/ni.3052
171. Hole CR, Bui H, Wormley FL Jr, Wozniak KL. Mechanisms of dendritic cell lysosomal killing of Cryptococcus. Sci Rep. (2012) 2:739. doi: 10.1038/srep00739
172. Schraml BU, Reis e Sousa C. Defining dendritic cells. Curr Opin Immunol. (2015) 32:13–20. doi: 10.1016/j.coi.2014.11.001
173. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
174. Bauman SK, Nichols KL, Murphy JW. Dendritic cells in the induction of protective and nonprotective anticryptococcal cell-mediated immune responses. J Immunol. (2000) 165:158–67. doi: 10.4049/jimmunol.165.1.158
175. Xu J, Flaczyk A, Neal LM, Fa Z, Eastman AJ, Malachowski AN, et al. Scavenger receptor MARCO orchestrates early defenses and contributes to fungal containment during cryptococcal infection. J Immunol. (2017) 198:3548–57. doi: 10.4049/jimmunol.1700057
176. Durai V, Murphy KM. Functions of murine dendritic cells. Immunity. (2016) 45:719–36. doi: 10.1016/j.immuni.2016.10.010
177. Kim TH, Lee HK. Differential roles of lung dendritic cell subsets against respiratory virus infection. Immune Netw. (2014) 14:128–37. doi: 10.4110/in.2014.14.3.128
178. Hwang JY, Randall TD, Silva-Sanchez A. Inducible bronchus-associated lymphoid tissue: taming inflammation in the lung. Front Immunol. (2016) 7:258. doi: 10.3389/fimmu.2016.00258
179. Whitney PG, Bar E, Osorio F, Rogers NC, Schraml BU, Deddouche S, et al. Syk signaling in dendritic cells orchestrates innate resistance to systemic fungal infection. PLoS Pathog. (2014) 10:e1004276. doi: 10.1371/journal.ppat.1004276
180. GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D, et al. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med. (2009) 206:2339–49. doi: 10.1084/jem.20090410
181. Smit JJ, Lindell DM, Boon L, Kool M, Lambrecht BN, Lukacs NW. The balance between plasmacytoid DC versus conventional DC determines pulmonary immunity to virus infections. PLoS ONE. (2008) 3:e1720. doi: 10.1371/journal.pone.0001720
182. Hole CR, Leopold Wager CM, Mendiola AS, Wozniak KL, Campuzano A, Lin X, et al. Antifungal activity of plasmacytoid dendritic cells against Cryptococcus neoformans in vitro requires expression of dectin-3 (CLEC4D) and reactive oxygen species. Infect Immun. (2016) 84:2493–504. doi: 10.1128/IAI.00103-16
183. Borger JG, Lau M, Hibbs ML. The influence of innate lymphoid cells and unconventional T cells in chronic inflammatory lung disease. Front Immunol. (2019) 10:1597. doi: 10.3389/fimmu.2019.01597
184. Khairallah C, Chu TH, Sheridan BS. Tissue adaptations of memory and tissue-resident gamma delta T cells. Front Immunol. (2018) 9:2636. doi: 10.3389/fimmu.2018.02636
185. Zhao Y, Lin L, Xiao Z, Li M, Wu X, Li W, et al. Protective role of gammadelta T cells in different pathogen infections and its potential clinical application. J Immunol Res. (2018) 2018:5081634. doi: 10.1155/2018/5081634
186. Uezu K, Kawakami K, Miyagi K, Kinjo Y, Kinjo T, Ishikawa H, et al. Accumulation of gammadelta T cells in the lungs and their regulatory roles in Th1 response and host defense against pulmonary infection with Cryptococcus neoformans. J Immunol. (2004) 172:7629–34. doi: 10.4049/jimmunol.172.12.7629
187. Juno JA, Eriksson EM. gammadelta T-cell responses during HIV infection and antiretroviral therapy. Clin Transl Immunol. (2019) 8:e01069. doi: 10.1002/cti2.1069
188. Barlow JL, McKenzie ANJ. Innate lymphoid cells of the lung. Annu Rev Physiol. (2019) 81:429–52. doi: 10.1146/annurev-physiol-020518-114630
189. Jacquelot N, Luong K, Seillet C. Physiological regulation of innate lymphoid cells. Front Immunol. (2019) 10:405. doi: 10.3389/fimmu.2019.00405
190. Seo GY, Giles DA, Kronenberg M. The role of innate lymphoid cells in response to microbes at mucosal surfaces. Mucosal Immunol. (2020) 13:399–412. doi: 10.1038/s41385-020-0265-y
191. Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. (2009) 31:15–23. doi: 10.1016/j.immuni.2009.06.008
192. Hidore MR, Mislan TW, Murphy JW. Responses of murine natural killer cells to binding of the fungal target Cryptococcus neoformans. Infect Immun. (1991) 59:1489–99.
193. Murphy JW, Hidore MR, Nabavi N. Binding interactions of murine natural killer cells with the fungal target Cryptococcus neoformans. Infect Immun. (1991) 59:1476–88.
194. Levitz SM, Dupont MP, Smail EH. Direct activity of human T lymphocytes and natural killer cells against Cryptococcus neoformans. Infect Immun. (1994) 62:194–202.
195. Li SS, Ogbomo H, Mansour MK, Xiang RF, Szabo L, Munro F, et al. Identification of the fungal ligand triggering cytotoxic PRR-mediated NK cell killing of Cryptococcus and Candida. Nat Commun. (2018) 9:751. doi: 10.1038/s41467-018-03014-4
196. Kindermann M, Knipfer L, Obermeyer S, Muller U, Alber G, Bogdan C, et al. Group 2 innate lymphoid cells (ILC2) suppress beneficial type 1 immune responses during pulmonary cryptococcosis. Front Immunol. (2020) 11:209. doi: 10.3389/fimmu.2020.00209
197. Garelnabi M, May RC. Variability in innate host immune responses to cryptococcosis. Mem Inst Oswaldo Cruz. (2018) 113:e180060. doi: 10.1590/0074-02760180060
198. Hunniger K, Kurzai O. Phagocytes as central players in the defence against invasive fungal infection. Semin Cell Dev Biol. (2019) 89:3–15. doi: 10.1016/j.semcdb.2018.03.021
199. Gazendam RP, van de Geer A, Roos D, van den Berg TK, Kuijpers TW. How neutrophils kill fungi. Immunol Rev. (2016) 273:299–311. doi: 10.1111/imr.12454
200. Selders GS, Fetz AE, Radic MZ, Bowlin GL. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen Biomater. (2017) 4:55–68. doi: 10.1093/rb/rbw041
201. Urban CF, Nett JE. Neutrophil extracellular traps in fungal infection. Semin Cell Dev Biol. (2019) 89:47–57. doi: 10.1016/j.semcdb.2018.03.020
202. Yang X, Wang H, Hu F, Chen X, Zhang M. Nonlytic exocytosis of Cryptococcus neoformans from neutrophils in the brain vasculature. Cell Commun Signal. (2019) 17:117. doi: 10.1186/s12964-019-0429-0
203. Taylor PR, Roy S, Leal SM Jr, Sun Y, Howell SJ, Cobb BA, et al. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORgammat and dectin-2. Nat Immunol. (2014) 15:143–51. doi: 10.1038/ni.2797
204. Li Z, Lu G, Meng G. Pathogenic fungal infection in the lung. Front Immunol. (2019) 10:1524. doi: 10.3389/fimmu.2019.01524
205. Dobmeyer TS, Raffel B, Dobmeyer JM, Findhammer S, Klein SA, Kabelitz D, et al. Decreased function of monocytes and granulocytes during HIV-1 infection correlates with CD4 cell counts. Eur J Med Res. (1995) 1:9–15.
206. Pugliese A, Vidotto V, Beltramo T, Torre D. Phagocytic activity in human immunodeficiency virus type 1 infection. Clin Diagn Lab Immunol. (2005) 12:889–95. doi: 10.1128/CDLI.12.8.889-895.2005
207. Marroni M, Pericolini E, Cenci E, Bistoni F, Vecchiarelli A. Functional defect of natural immune system in an apparent immunocompetent patient with pulmonary cryptococcosis. J Infect. (2007) 54:e5–8. doi: 10.1016/j.jinf.2006.03.018
208. Malhotra P, Shah SS, Kaplan M, McGowan JP. Cryptococcal fungemia in a neutropenic patient with AIDS while receiving caspofungin. J Infect. (2005) 51:e181–3. doi: 10.1016/j.jinf.2005.01.002
209. Munoz JF, Salmen S, Berrueta LR, Carlos MP, Cova JA, Donis JH, et al. Effect of human immunodeficiency virus type 1 on intracellular activation and superoxide production by neutrophils. J Infect Dis. (1999) 180:206–10. doi: 10.1086/314821
211. Fulurija A, Ashman RB, Papadimitriou JM. Neutrophil depletion increases susceptibility to systemic and vaginal candidiasis in mice, and reveals differences between brain and kidney in mechanisms of host resistance. Microbiology. (1996) 142 (Pt 12):3487–96.
212. Pedrosa J, Saunders BM, Appelberg R, Orme IM, Silva MT, Cooper AM. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect Immun. (2000) 68:577–83. doi: 10.1128/iai.68.2.577-583.2000
213. Seiler P, Aichele P, Raupach B, Odermatt B, Steinhoff U, Kaufmann SH. Rapid neutrophil response controls fast-replicating intracellular bacteria but not slow-replicating Mycobacterium tuberculosis. J Infect Dis. (2000) 181:671–80. doi: 10.1086/315278
214. Wiesner DL, Smith KD, Kashem SW, Bohjanen PR, Nielsen K. Different lymphocyte populations direct dichotomous eosinophil or neutrophil responses to pulmonary Cryptococcus infection. J Immunol. (2017) 198:1627–37. doi: 10.4049/jimmunol.1600821
215. Mednick AJ, Feldmesser M, Rivera J, Casadevall A. Neutropenia alters lung cytokine production in mice and reduces their susceptibility to pulmonary cryptococcosis. Eur J Immunol. (2003) 33:1744–53. doi: 10.1002/eji.200323626
216. Guillot L, Carroll SF, Homer R, Qureshi ST. Enhanced innate immune responsiveness to pulmonary Cryptococcus neoformans infection is associated with resistance to progressive infection. Infect Immun. (2008) 76:4745–56. doi: 10.1128/IAI.00341-08
217. Qureshi A, Grey A, Rose KL, Schey KL, Del Poeta M. Cryptococcus neoformans modulates extracellular killing by neutrophils. Front Microbiol. (2011) 2:193. doi: 10.3389/fmicb.2011.00193
218. Qureshi A, Subathra M, Grey A, Schey K, Del Poeta M, Luberto C. Role of sphingomyelin synthase in controlling the antimicrobial activity of neutrophils against Cryptococcus neoformans. PLoS ONE. (2010) 5:e15587. doi: 10.1371/journal.pone.0015587
219. Gardner A, Ruffell B. moDCs, less problems. Immunity. (2018) 48:6–8. doi: 10.1016/j.immuni.2017.12.017
220. Fites JS, Gui M, Kernien JF, Negoro P, Dagher Z, Sykes DB, et al. An unappreciated role for neutrophil-DC hybrids in immunity to invasive fungal infections. PLoS Pathog. (2018) 14:e1007073. doi: 10.1371/journal.ppat.1007073
221. Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, et al. Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity. (2017) 46:504–15. doi: 10.1016/j.immuni.2017.02.019
222. Hole CR, Wager CML, Castro-Lopez N, Campuzano A, Cai H, Wozniak KL, et al. Induction of memory-like dendritic cell responses in vivo. Nat Commun. (2019) 10:2955. doi: 10.1038/s41467-019-10486-5
223. Masso-Silva J, Espinosa V, Liu TB, Wang Y, Xue C, Rivera A. The F-box protein Fbp1 shapes the immunogenic potential of Cryptococcus neoformans. MBio. (2018) 9:e01828-17. doi: 10.1128/mBio.01828-17
224. Mukaremera L, Nielsen K. Adaptive immunity to Cryptococcus neoformans infections. J Fungi. (2017) 3:63. doi: 10.3390/jof3040064
225. Patin EC, Thompson A, Orr SJ. Pattern recognition receptors in fungal immunity. Semin Cell Dev Biol. (2019) 89:24–33. doi: 10.1016/j.semcdb.2018.03.003
226. Cook PC, MacDonald AS. Dendritic cells in lung immunopathology. Semin Immunopathol. (2016) 38:449–60. doi: 10.1007/s00281-016-0571-3
227. Shourian M, Qureshi ST. Resistance and tolerance to cryptococcal infection: an intricate balance that controls the development of disease. Front Immunol. (2019) 10:66. doi: 10.3389/fimmu.2019.00066
228. Wormley FLJr, Perfect JR, Steele C, Cox GM. Protection against cryptococcosis by using a murine gamma interferon-producing Cryptococcus neoformans strain. Infect Immun. (2007) 75:1453–62. doi: 10.1128/IAI.00274-06
229. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/s1074-7613(03)00174-2
230. Yrlid U, Jenkins CD, MacPherson GG. Relationships between distinct blood monocyte subsets and migrating intestinal lymph dendritic cells in vivo under steady-state conditions. J Immunol. (2006) 176:4155–62. doi: 10.4049/jimmunol.176.7.4155
231. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
232. Gower RM, Wu H, Foster GA, Devaraj S, Jialal I, Ballantyne CM, et al. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. (2011) 31:160–6. doi: 10.1161/ATVBAHA.110.215434
233. Huaux F, Lo Re S, Giordano G, Uwambayinema F, Devosse R, Yakoub Y, et al. IL-1alpha induces CD11b(low) alveolar macrophage proliferation and maturation during granuloma formation. J Pathol. (2015) 235:698–709. doi: 10.1002/path.4487
234. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. (2008) 26:421–52. doi: 10.1146/annurev.immunol.26.021607.090326
235. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
236. Zhang L, Wang CC. Inflammatory response of macrophages in infection. Hepatobiliary Pancreat Dis Int. (2014) 13:138–52. doi: 10.1016/s1499-3872(14)60024-2
237. Heung LJ, Hohl TM. Inflammatory monocytes are detrimental to the host immune response during acute infection with Cryptococcus neoformans. PLoS Pathog. (2019) 15:e1007627. doi: 10.1371/journal.ppat.1007627
238. Osterholzer JJ, Curtis JL, Polak T, Ames T, Chen GH, McDonald R, et al. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J Immunol. (2008) 181:610–20. doi: 10.4049/jimmunol.181.1.610
239. Traynor TR, Kuziel WA, Toews GB, Huffnagle GB. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol. (2000) 164:2021–7. doi: 10.4049/jimmunol.164.4.2021
240. Sorrell TC, Juillard PG, Djordjevic JT, Kaufman-Francis K, Dietmann A, Milonig A, et al. Cryptococcal transmigration across a model brain blood-barrier: evidence of the Trojan horse mechanism and differences between Cryptococcus neoformans var. grubii strain H99 and Cryptococcus gattii strain R265. Microbes Infect. (2016) 18:57–67. doi: 10.1016/j.micinf.2015.08.017
241. Fonseca FL, Nohara LL, Cordero RJ, Frases S, Casadevall A, Almeida IC, et al. Immunomodulatory effects of serotype B glucuronoxylomannan from Cryptococcus gattii correlate with polysaccharide diameter. Infect Immun. (2010) 78:3861–70. doi: 10.1128/IAI.00111-10
242. Scriven JE, Graham LM, Schutz C, Scriba TJ, Wilkinson KA, Wilkinson RJ, et al. A glucuronoxylomannan-associated immune signature, characterized by monocyte deactivation and an increased interleukin 10 level, is a predictor of death in cryptococcal meningitis. J Infect Dis. (2016) 213:1725–34. doi: 10.1093/infdis/jiw007
243. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
244. Resnick MB, Weller PF. Mechanisms of eosinophil recruitment. Am J Respir Cell Mol Biol. (1993) 8:349–55. doi: 10.1165/ajrcmb/8.4.349
245. Holmer SM, Evans KS, Asfaw YG, Saini D, Schell WA, Ledford JG, et al. Impact of surfactant protein D, interleukin-5, and eosinophilia on Cryptococcosis. Infect Immun. (2014) 82:683–93. doi: 10.1128/IAI.00855-13
246. Zhang Y, Wang F, Tompkins KC, McNamara A, Jain AV, Moore BB, et al. Robust Th1 and Th17 immunity supports pulmonary clearance but cannot prevent systemic dissemination of highly virulent Cryptococcus neoformans H99. Am J Pathol. (2009) 175:2489–500. doi: 10.2353/ajpath.2009.090530
247. Yokoyama T, Kadowaki M, Yoshida M, Suzuki K, Komori M, Iwanaga T. Disseminated cryptococcosis with marked eosinophilia in a postpartum woman. Intern Med. (2018) 57:135–9. doi: 10.2169/internalmedicine.9247-17
248. Coelho C, Camacho E, Salas A, Alanio A, Casadevall A. Intranasal inoculation of Cryptococcus neoformans in mice produces nasal infection with rapid brain dissemination. mSphere. (2019) 4:e00483–19. doi: 10.1128/mSphere.00483-19
249. Tao L, Reese TA. Making mouse models that reflect human immune responses. Trends Immunol. (2017) 38:181–93. doi: 10.1016/j.it.2016.12.007
250. Garro AP, Chiapello LS, Baronetti JL, Masih DT. Rat eosinophils stimulate the expansion of Cryptococcus neoformans-specific CD4(+) and CD8(+) T cells with a T-helper 1 profile. Immunology. (2011) 132:174–87. doi: 10.1111/j.1365-2567.2010.03351.x
251. Flores-Santibanez F, Cuadra B, Fernandez D, Rosemblatt MV, Nunez S, Cruz P, et al. In vitro-generated Tc17 cells present a memory phenotype and serve as a reservoir of Tc1 cells in vivo. Front Immunol. (2018) 9:209. doi: 10.3389/fimmu.2018.00209
252. Martin MD, Badovinac VP. Defining memory CD8 T cell. Front Immunol. (2018) 9:2692. doi: 10.3389/fimmu.2018.02692
253. Srenathan U, Steel K, Taams LS. IL-17+ CD8+ T cells: differentiation, phenotype and role in inflammatory disease. Immunol Lett. (2016) 178:20–6. doi: 10.1016/j.imlet.2016.05.001
254. Wilk MM, Mills KHG. CD4 TRM cells following infection and immunization: implications for more effective vaccine design. Front Immunol. (2018) 9:1860. doi: 10.3389/fimmu.2018.01860
255. Nanjappa SG, Heninger E, Wuthrich M, Sullivan T, Klein B. Protective antifungal memory CD8(+) T cells are maintained in the absence of CD4(+) T cell help and cognate antigen in mice. J Clin Invest. (2012) 122:987–99. doi: 10.1172/JCI58762
256. Flaczyk A, Duerr CU, Shourian M, Lafferty EI, Fritz JH, Qureshi ST. IL-33 signaling regulates innate and adaptive immunity to Cryptococcus neoformans. J Immunol. (2013) 191:2503–13. doi: 10.4049/jimmunol.1300426
257. Gratz IK, Campbell DJ. Resident memory T cells show that it is never too late to change your ways. Nat Immunol. (2020) 21:359–60. doi: 10.1038/s41590-020-0637-1
258. McDermott AJ, Klein BS. Helper T-cell responses and pulmonary fungal infections. Immunology. (2018) 155:155–63. doi: 10.1111/imm.12953
259. Ogongo P, Porterfield JZ, Leslie A. Lung tissue resident memory T-cells in the immune response to Mycobacterium tuberculosis. Front Immunol. (2019) 10:992. doi: 10.3389/fimmu.2019.00992
260. Schreiner D, King CG. CD4+ Memory T cells at home in the tissue: mechanisms for health and disease. Front Immunol. (2018) 9:2394. doi: 10.3389/fimmu.2018.02394
261. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. (2009) 30:92–107. doi: 10.1016/j.immuni.2008.11.005
262. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. (2010) 11:674–80. doi: 10.1038/ni.1899
263. O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells Science. (2010) 327:1098–102. doi: 10.1126/science.1178334
264. Barinov A, Galgano A, Krenn G, Tanchot C, Vasseur F, Rocha B. CD4/CD8/Dendritic cell complexes in the spleen: CD8+ T cells can directly bind CD4+ T cells and modulate their response. PLoS ONE. (2017) 12:e0180644. doi: 10.1371/journal.pone.0180644
265. Neal LM, Xing E, Xu J, Kolbe JL, Osterholzer JJ, Segal BM, et al. CD4(+) T cells orchestrate lethal immune pathology despite fungal clearance during Cryptococcus neoformans meningoencephalitis. MBio. (2017) 8:e01415-17. doi: 10.1128/mBio.01415-17
266. Martinez-Lopez M, Iborra S, Conde-Garrosa R, Mastrangelo A, Danne C, Mann ER, et al. Microbiota sensing by mincle-syk axis in dendritic cells regulates interleukin-17 and−22 production and promotes intestinal barrier integrity. Immunity. (2019) 50:446–61 e449. doi: 10.1016/j.immuni.2018.12.020
267. Nanjappa SG, McDermott AJ, Fites JS, Galles K, Wuthrich M, Deepe GSJr, et al. Antifungal Tc17 cells are durable and stable, persisting as long-lasting vaccine memory without plasticity towards IFNgamma cells. PLoS Pathog. (2017) 13:e1006356. doi: 10.1371/journal.ppat.1006356
268. Broquet A, Jacqueline C, Davieau M, Besbes A, Roquilly A, Martin J, et al. Interleukin-22 level is negatively correlated with neutrophil recruitment in the lungs in a Pseudomonas aeruginosa pneumonia model. Sci Rep. (2017) 7:11010. doi: 10.1038/s41598-017-11518-0
269. Ronacher K, Sinha R, Cestari M. IL-22: an underestimated player in natural resistance to tuberculosis? Front Immunol. (2018) 9:2209. doi: 10.3389/fimmu.2018.02209
270. Valeri M, Raffatellu M. Cytokines IL-17 and IL-22 in the host response to infection. Pathog Dis. (2016) 74:ftw111. doi: 10.1093/femspd/ftw111
271. Liang Y, Pan HF, Ye DQ. Tc17 Cells in immunity and systemic autoimmunity. Int Rev Immunol. (2015) 34:318–31. doi: 10.3109/08830185.2014.954698
272. Chen HW, Tsai JP, Yao TY, Hsieh CL, Chen IH, Liu SJ. TGF-beta and IL-21 cooperatively stimulate activated CD8(+) T cells to differentiate into Tc17 cells. Immunol Lett. (2016) 174:23–7. doi: 10.1016/j.imlet.2016.04.006
273. Nanjappa SG, Heninger E, Wuthrich M, Gasper DJ, Klein BS. Tc17 cells mediate vaccine immunity against lethal fungal pneumonia in immune deficient hosts lacking CD4+ T cells. PLoS Pathog. (2012) 8:e1002771. doi: 10.1371/journal.ppat.1002771
274. Farnoud AM, Bryan AM, Kechichian T, Luberto C, Del Poeta M. The granuloma response controlling cryptococcosis in mice depends on the sphingosine kinase 1-sphingosine 1-phosphate pathway Infect Immun. (2015) 83:2705–13. doi: 10.1128/IAI.00056-15
275. Yanagawa N, Sakai F, Takemura T, Ishikawa S, Takaki Y, Hishima T, et al. Pulmonary cryptococcosis in rheumatoid arthritis (RA) patients: comparison of imaging characteristics among RA, acquired immunodeficiency syndrome, and immunocompetent patients. Eur J Radiol. (2013) 82:2035–42. doi: 10.1016/j.ejrad.2013.07.014
276. Yu JQ, Tang KJ, Xu BL, Xie CM, Light RW. Pulmonary cryptococcosis in non-AIDS patients. Braz J Infect Dis. (2012) 16:531–9. doi: 10.1016/j.bjid.2012.07.004
277. Zeng Y, Wu X, Yi X, Luo B, Zhu M, Rui W, et al. Clinicopathologic and ultrastructural study of non-HIV-related primary pulmonary cryptococcosis in China: report of 43 cases. Ultrastruct Pathol. (2011) 35:19–25. doi: 10.3109/01913123.2010.521293
278. Baeyens A, Fang V, Chen C, Schwab SR. Exit strategies: S1P signaling and T cell migration. Trends Immunol. (2015) 36:778–87. doi: 10.1016/j.it.2015.10.005
279. Matloubian M, Lo CG, Cinamon G, Lesneski M, Xu Y, Brinkman V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. (2004) 427:355–60. doi: 10.1038/nature02284
280. Bryan AM, Del Poeta M. Sphingosine-1-phosphate receptors and innate immunity. Cell Microbiol. (2018) 20:e12836. doi: 10.1111/cmi.12836
281. Hla T. Sphingosine 1-phosphate receptors. Prostaglandins Other Lipid Mediat. (2001) 64:135–42. doi: 10.1016/s0090-6980(01)00109-5
282. Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. (2006) 355:1124–40. doi: 10.1056/NEJMoa052643
283. Bryan AM, You JK, McQuiston T, Lazzarini C, Qiu Z, Sheridan BS, et al. FTY720 reactivates cryptococcal granulomas in mice through S1P receptor 3 on macrophages. J Clin Invest. (2020) 130:1–16. doi: 10.1172/JCI136068
284. Mor V, Farnoud AM, Singh A, Rella A, Tanno H, Ishii K, et al. Glucosylceramide administration as a vaccination strategy in mouse models of cryptococcosis. PLoS ONE. (2016) 11:e0153853. doi: 10.1371/journal.pone.0153853
Keywords: cryptococcosis, infection models, primary infection, granuloma, latent infection, immunodeficiency, host immune response
Citation: Normile TG, Bryan AM and Del Poeta M (2020) Animal Models of Cryptococcus neoformans in Identifying Immune Parameters Associated With Primary Infection and Reactivation of Latent Infection. Front. Immunol. 11:581750. doi: 10.3389/fimmu.2020.581750
Received: 09 July 2020; Accepted: 12 August 2020;
Published: 15 September 2020.
Edited by:
Paul Laszlo Bollyky, Stanford University, United StatesReviewed by:
Chaoyang Xue, Rutgers University, Newark, United StatesKaren L. Wozniak, Oklahoma State University, United States
Copyright © 2020 Normile, Bryan and Del Poeta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Del Poeta, bWF1cml6aW8uZGVscG9ldGFAc3Rvbnlicm9vay5lZHU=; Tyler G. Normile, dHlsZXIubm9ybWlsZUBzdG9ueWJyb29rLmVkdQ==