 Xuewei Ding
Xuewei Ding Wei Xiang
Wei Xiang Xiaojie He
Xiaojie He- 1Institute of Pediatrics, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Laboratory of Pediatric Nephrology, Institute of Pediatrics, The Second Xiangya Hospital, Central South University, Changsha, China
- 3Key Laboratory of Tropical Translational Medicine of Ministry of Education, Hainan Medical University, NHC Key Laboratory of Control of Tropical diseases (Hainan Medical University), Haikou, China
Systemic lupus erythematosus (SLE) is a multi-system autoimmune disease including the cardiovascular system. Atherosclerosis is the most common cardiovascular complication of SLE and a significant risk factor for morbidity and mortality. Vascular damage/protection mechanism in SLE patients is out of balance, caused by the cascade reaction among oxidative stress, proinflammatory cytokines, Neutrophil Extracellular Traps, activation of B cells and autoantibodies and abnormal T cells. As a precursor cell repairing vascular endothelium, endothelial progenitor cells (EPCs) belong to the protective mechanism and show the reduced number and impaired function in SLE. However, the pathological mechanism of EPCs dysfunction in SLE remains ill-defined. This paper reviews the latest SLE epidemiology and pathogenesis, discusses the changes in the number and function of EPCs in SLE, expounds the role of EPCs in SLE atherosclerosis, and provides new guidance and theoretical basis for exploring novel targets for SLE treatment.
Introduction
SLE is an immune complex-mediated autoimmune disease involving multiple systems. Its prevalence and incidence rate can be as high as 241/100,000 per year and 23.2/100,000 per year, and the rate of premature death is 2–3 times that of healthy people (1). Since 2000, the prevalence rate of adult SLE in women has been 30–150/100,000, and the incidence rate is 2.2–23.1/100,000 per year (2). SLE is also an autoimmune disease characterized by cardiovascular disease (CVD). A multicenter study found that a quarter of the nearly 10,000 deaths from SLE were caused by CVD (3). Current studies have demonstrated that the inherent factors of SLE are independent risk factors for the premature occurrence of atherosclerosis in SLE patients (4). With the improvement of the diagnosis and treatment, the early mortality of SLE patients has been dramatically reduced. However, atherosclerosis is still one of the leading causes of death of late SLE patients. It is of considerable significance to explore the natural course and mechanism of SLE combined with atherosclerosis, find useful therapeutic targets, provide evidence for clinical intervention, and delay the death of SLE.
Vascular endothelial dysfunction is the starting point in SLE atherosclerosis. Endothelial progenitor cells (EPCs) are closely related to vascular endothelial function. Therefore, the relationship between atherosclerosis and EPCs in SLE is a research direction worth exploring. However, in recent decades, there are few studies on the relationship between atherosclerosis and EPCs in SLE, and the results are controversial. This paper analyzes the changes in the number and function of EPCs in SLE and reviews the potential role of EPCs in SLE atherosclerosis.
Mechanism of Atherosclerosis in SLE
Arteriosclerosis is a series of aggregation events of leukocytes and vascular smooth muscle cells (VSMCs) in intima triggered by endothelial dysfunction and lipoprotein retention, resulting in fibrous plaques. Then fibrous plaques rupture, followed by thrombosis. This process requires the immune response’s help (5, 6). The abnormal immune response driven by SLE enhances vascular injury mechanism and weaken repair mechanism, breaking vascular dynamic balance which determines the occurrence of CVD (Figure 1).
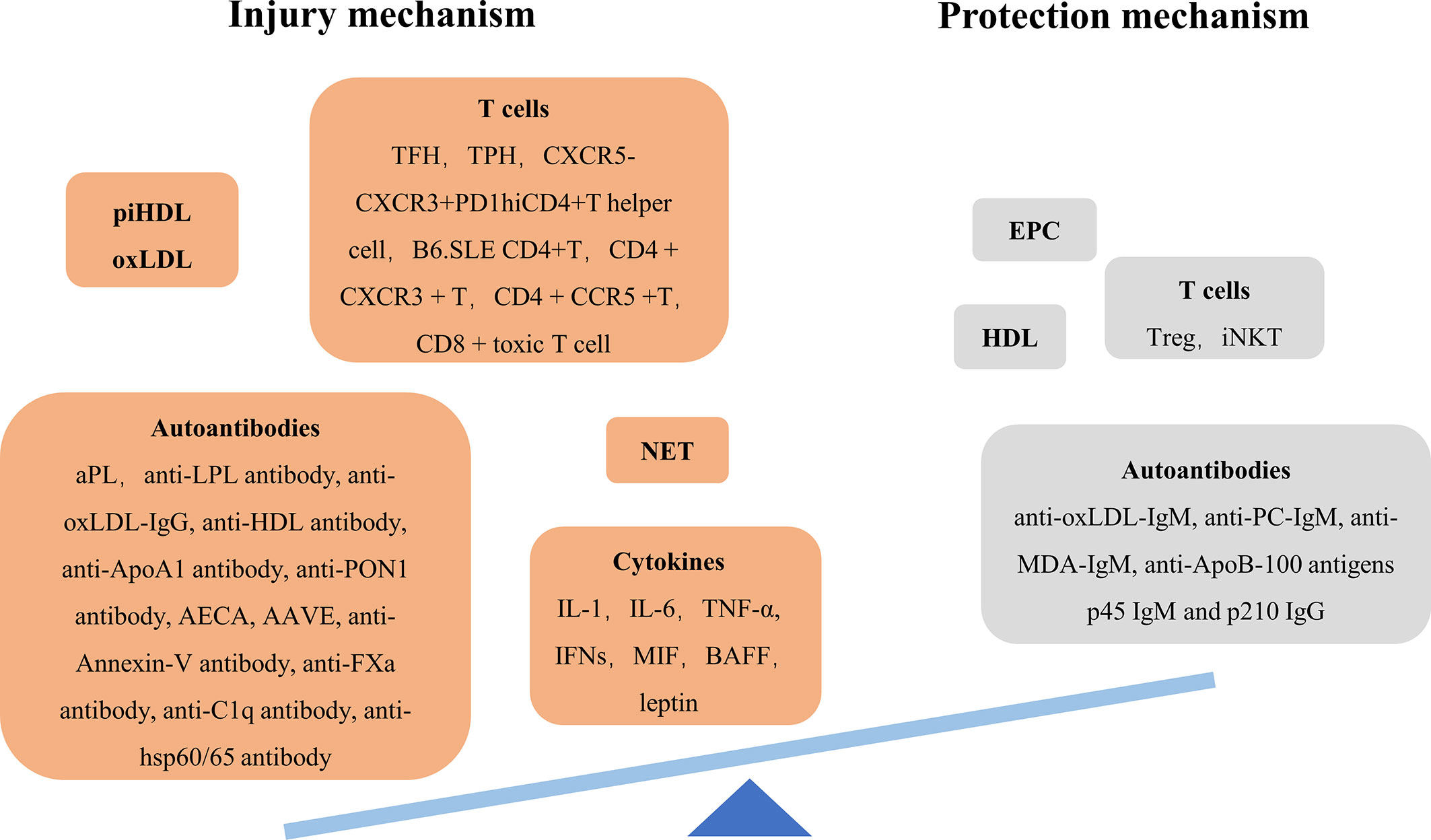
Figure 1 Imbalance of injury/protection mechanism of SLE arteriosclerosis. AAVE anti-vascular endothelial-cadherin antibody AECA anti-endothelial cell antibody aPL antiphospholipid antibody BAFF B cell-activating factor EPC endothelial progenitor cell iNKT invariant natural killer T cell LPL lipoprotein lipase MDA malondialdehyde; MIF, macrophage migration inhibitory factor; NET, neutrophil extracellular trap; PC, choline phosphate; TFH, follicular helper T cell; TPF, peripheral helper T cell.
Oxidative Stress
Mitochondrial dysfunctions, abnormal bioenergetics/immunometabolism and telomere/telomerase disequilibrium endowed SLE patients with intense oxidative stress (7). Among the three main targets of oxidative stress, oxidized lipids—oxLDL and proinflammatory HDL (piHDL)—play a prominent role in accelerating SLE atherosclerosis (8). OxLDL participates in many stages of atherosclerosis, from endothelial dysfunction to plaque rupture (6, 9). Normal HDL plays a role in protecting atherosclerosis by promoting cholesterol outflow, inhibiting vascular inflammation and scavenging oxidizing substances. However, lupus-altered HDL shifts from a normal anti-inflammatory state to a proinflammatory state, causing atherosclerosis (10). Increased piHDL weakens the ability to prevent LDL oxidation (8).
Cytokines
Cytokines, the primary regulators of immune responses, regulate and coordinate multiple stages of atherosclerosis. There is a cascade reaction between these proinflammatory cytokines in accelerating SLE atherosclerosis (Figure 2).
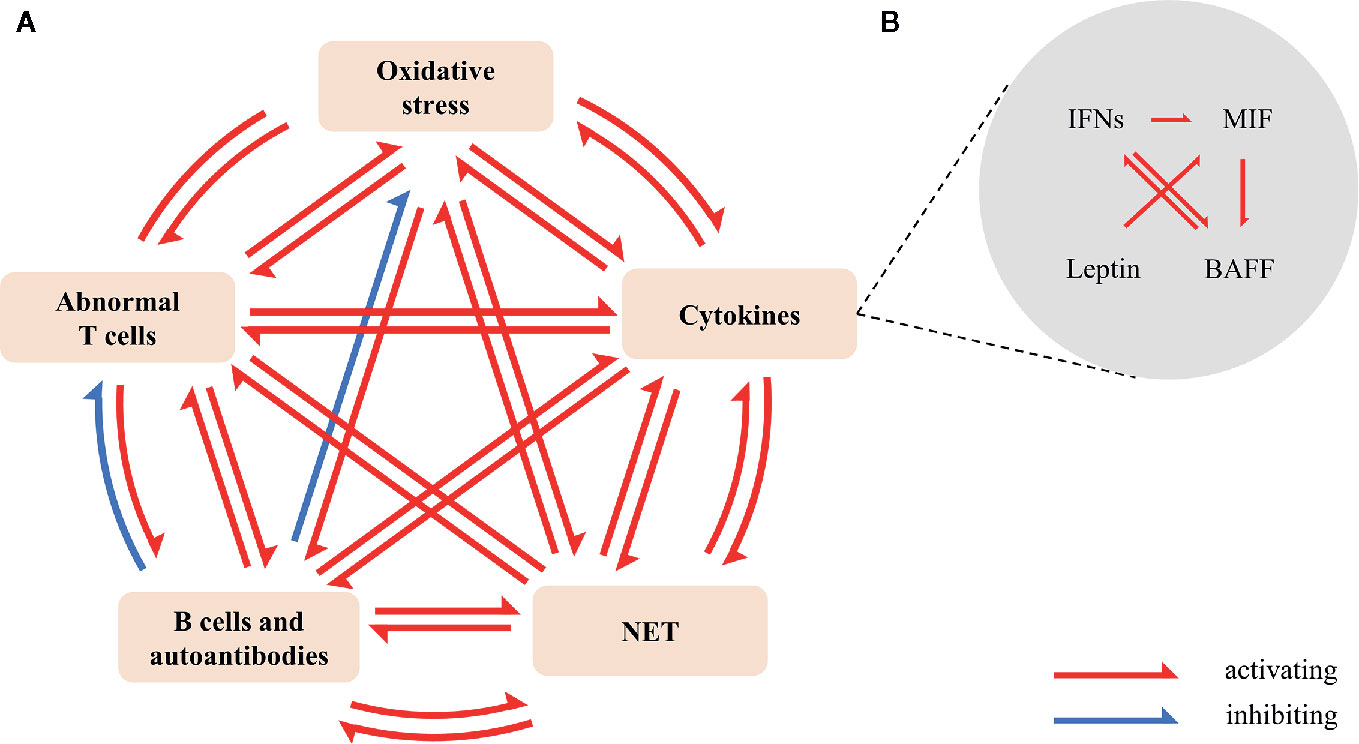
Figure 2 Cross-talk between oxidative stress, cytokines, NETs, activation of B cells and autoantibodies, and abnormal T cells in SLE. (A) Oxidative stress promotes the production of IFN-I (11), NETs (12), autoantibodies (13), and the imbalance of Th17/Treg (14). IFN-α and IFN-γ promote lipids oxidative modification (15, 16); BAFF promotes the production of autoantibodies (17), the release of NETs (18) and the activation of T cells (19); leptin promotes the production of autoantibodies, the release of NET and the imbalance of Th17/Treg (20, 21). NET encourages oxidation HDL (22), the expression of IFN-α (23) and IL-1β (24), and activates NET-specific memory B cells to proliferate and secrete polyclonal IgG (25). Overactive T cells increase ROS (26) and cytokines, especially IFN-γ; TFH (27, 28), CXCR5-CXCR3+PD1hiCD4+T helper cell (29), and peripheral helper T cell (TPH) (30) promote the differentiation of B cells and the production of antibodies. SLE-related autoantibodies and immune complexes induce the release of NET (31); anti-ApoA1-IgG guides the expression of cytokines (32). Anti-PC-IgM increases Tregs (33); anti-PC-IgM and anti-MDA-IgM reduce oxidative stress (34). (B) IFN-I (35, 36) and IFN-II (37) induce the expression and mobilization of BAFF. BAFF promotes the activation of B cells by IFN (38). Moreover, IFN-I encourages the production of MIF (39). MIF/CD74 signal regulates BAFF (40, 41). Leptin enhances MIF-induced inflammation (42). Besides, IFNs, MIF and leptin strengthen the expression of chemokine, adhesion molecule, TNF- α and ILs. BAFF, B cell-activating factor; MDA, malondialdehyde; MIF, macrophage migration inhibitory factor; NET, neutrophil extracellular trap; PC, choline phosphate; ROS, reactive oxygen species; TFH, follicular helper T cell; TPF, peripheral helper T cell.
IFNs are divided into three classes: IFN-I (IFN-α, IFN-β, IFN-δ, IFN-ϵ, IFN-κ, IFN-τ, IFN-ω), IFN-II (IFN-γ), IFN-III (IFN-λ1, IFN-λ2, IFN-λ3). IFNs participated in the whole process of atherosclerosis, especially IFN-I (15, 43–45). For example, IFN-α and IFN-γ promote lipoproteins’ oxidation (15, 16). IFN-α promotes endothelial dysfunction by accelerating endothelial cells (ECs) apoptosis and damaging EPCs, one of the vascular repair mechanisms (15, 46–53). IFN-α enhances the expression of chemokine and adhesion molecules without leukocytes adhesion (53); while IFN-γ can regulate the attraction and adhesion of leukocytes (54). IFN-α induces the up-regulation of SR-A expression in monocytes/macrophages, then promoting the lipid uptake and the formation of macrophage-derived foam cells (55); IFN-γ not only up-regulates SR-A, but also up-regulate ACAT1 (56) and inhibit specific anti-atherosclerotic MSRN proteins (APOE and C3) in macrophages (57) to reduce cholesterol efflux. IFN-α prevents smooth muscle progenitor cell (SMPC) from maturation which could give rise to macrophages and eventually foam cells (58); IFN-γ enhances VSMCs’ proliferation and migration (56). IFN-α and IFN-γ induce VSMC and macrophages apoptosis in atherosclerotic plaques, contributing to plaque instability (59–61). Moreover, IFN-α inhibits the expression of type I collagen gene COL1A1 in VSMCs (62) and induces the synthesis of TNF-α, IL-12 and MMP-9 (63); while IFN-γ inhibits the expression of type I collagen gene COL1A2 in VSMCs (64) and induces the synthesis of MMP-1, MMP-2 and MMP-9 (56). Besides, IFN-α forms an IFN-α-platelet-CD154-CD40 forward feedback loop to promote thrombosis (65, 66).
Macrophage migration inhibitory factor (MIF) is an inflammatory and chemokine-like cytokine and an upstream regulator of innate immunity. MIF enhances LDL uptake (67), recruits monocytes and T cells (68–70), migrates VSMCs (71), resulting in plaques. MIF also increases the expression of MMP-1 and MMP-9, inducing plaques rupture (72, 73).
B-Cell Activating Factor (BAFF) is a critical factor in B cell maturation, survival and function, and an independent factor in accelerating SLE atherosclerosis (17). BAFF/BAFF-R axis supports pathogenic B cells producing pathogenic anti-IgG-oxLDL antibodies (74, 75), which is over-activated in SLE (76). The co-expression of BAFF/TNFSF13B and APRIL/TNFSF13 in the plaque lymphocytes and macrophages up-regulate FURIN, the primary Proprotein convertase subtilisin/Kexin (PCSK), which inactivates lipases and regulates inflammation in atherosclerosis (19). And BAFF weakens EPCs’ function and promotes EPCs’ apoptosis (77).
As an immunopotentiator (78), leptin significantly increases the risk of atherosclerosis in SLE patients (79). And the serum leptin level ≥ 34ng/dL was significantly correlated with carotid plaque (79). Leptin induces oxidative stress, increases MCP-1, TNF-α, IL-6 and endothelin-1, accompanied by the expression of other EC adhesion molecules, MMPs and VEGF, which damages VSMCs and ECs (80). And leptin promotes the secretion of atherosclerotic factor (42, 81). Besides, leptin promotes the production of autoantibodies, increases the release of NET and imbalance of Th17/Treg in SLE (20).
Neutrophil Extracellular Traps (NETs)
NET is a unique form of neutrophils death, characterized by the extrusion of chromatin and a driver of SLE atherosclerosis (82–87). NETs damages ECs. They promote vascular leakage and endothelial-to-mesenchymal transition through the degradation of VE-cadherin and the activation of β-catenin signaling (87); they induce EC death through the activation of endothelial MMP-2 (88). NETs also kill VSMCs (89). Moreover, NETs mediate HDL’s oxidation, interfering with cholesterol outflow (22). NETs induce the secretion of IFN-α (23) and IL-1β (24). Serine proteases from NETs degrade tissue factor pathway inhibitor (TFPI) (90) and promote FXII (91) that activate coagulation cascade and thrombosis.
The Activation of B Cells and Autoantibodies
B cells mainly affect atherosclerosis by producing autoantibodies: B1 cells secrete protective natural IgM and IgA antibodies, whereas B2 cells produce pathogenic IgG antibodies. And the tendency of overactive B cells to produce pathogenic IgG antibodies is a potential risk factor for lupus-associated atherosclerosis (17). In particular, antiphospholipid antibodies (aPL) have been identified as independent predictors of atherosclerotic plaque progression in SLE (92, 93).
Anti-HDL-IgG induces LDL to enter the ECs, which is a major contributor to atherosclerosis in SLE. Recently, Kurien BT et al. found that SLE RNP and anti-Ro/LaRNP antibodies probably increase the level of anti-oxLDL antibodies (94). Anti-HDL antibody, anti-ApoA1 antibody and anti-PON1 antibody probably have a common atherogenic pathway—they unbalance PON-1/MPO, which enhances lipids oxidative modification and interferes with HDL’s anti-inflammation (95–97). Besides, anti-ApoA1-IgG has two pathways that induce atherosclerosis in a TLR2/TLR4/CD14-dependent manner: it activates transcriptional nuclear factor NF-kB to guide the expression of inflammatory factors; it provides an alternative (or a concomitant) signal to PI3K in an Src-dependent pathway, activates L-type Ca2+ channels and potassium/calcium exchangers, resulting in the depolarization of myocardial plasma membrane (32). Anti-FXa-IgG unbalances hemostasis and thrombosis by inhibiting the FXa enzyme (98) and promotes endothelial dysfunction by enhancing FXa-PAR-mediated Ca2+ signal transduction (99). Recent studies have found that IgA-AECA is involved in SLE endothelial damage by recognizing the membrane proteins of ECs (100). Anti-C1q antibody plays a role in atherosclerosis by reducing C1q’s level and protective effects (101, 102), which polarizes macrophages towards an M2-like anti-inflammatory phenotype (103) and improves macrophages’ survival and excretion (104).
There are potential protective autoantibodies in SLE patients, such as anti-oxLDL-IgM, anti-ApoB100 antibodies, anti-choline phosphate (PC) antibodies and anti-malondialdehyde (MDA) antibodies. The first three have a synergistic effect: they reduce the level of oxLDL, the uptake of oxLDL, and the formation of foam cells (105–107). And Anti-PC-IgM increases Tregs in SLE and atherosclerosis, reduces IL-17 and TNF-α, and makes dendritic cells (DCs) immature (33). The combined application of anti-PC-IgM and anti-MDA-IgM has a doubly preventive impact on atherosclerosis (34). However, SLE patients showed a low level of protective autoantibodies (34, 107). Some dietary and metabolic factors may be responsible for the low levels of anti-PC-IgM and anti-MDA-IgM (108).
SLE increases the risk of CVD by promoting pathogenic autoantibodies and inhibiting potential protective autoantibodies.
The Abnormal T Cells
Abnormal T cell subsets are considered to be an essential factor leading to endothelial dysfunction and CVD in SLE patients. Tregs are protective T cells in atherogenesis, inhibiting atherogenic T cell subsets and inflammation. And Treg/Th17 imbalance is common in SLE, becoming a risk factor for atherosclerosis (109). In human circulation, atherosclerosis’s severity is not directly related to the number of Tregs (110) but is closely related to the dysfunction of Tregs (111). During atherosclerosis, most Treg lost Foxp3 expression and its immunosuppressive function, then transform into follicular helper T cell (TFH) (112), which is used to stimulate the formation of germinal center (GC) and the selection of high-affinity B cells in GC (27). TFH has also been shown to accelerate atherosclerosis, although not necessarily by inducing the production of pathogenic IgG (112, 113). Besides, CD4+T cells in peripheral blood of SLE patients highly express CCR5 and CXCR3 promoting the migration of inflammatory T cells to the arterial wall in a chemokine-dependent way (114, 115). In particular, CCR5 is the critical factor for CD4+T cells homing to atherosclerotic plaques (116).
A recent study has shown that Invariant natural killer T (iNKT) in SLE patients has an anti-atherosclerotic phenotype which induces macrophages to polarize into anti-inflammatory and anti-atherosclerotic M2 phenotype (117). The protection is triggered in early atherosclerosis but is lost or submerged in the development of clinical atherosclerosis (117).
Oxidative stress, cytokines, NETs, activation of B cells and autoantibodies, and abnormal T cells in SLE interact with each other, amplifying their pro-atherogenic effects (Figure 2). As a result, the dynamic vascular homeostasis is broken in SLE patients, characterized by enhanced injury mechanism and weakened protection mechanism. Subclinical atherosclerosis in SLE accelerates, even in environments with low disease activity (92).
The Role of EPCs in Arteriosclerosis
Atherosclerosis is a manifestation of the imbalance between vascular injury and protection mechanisms, especially in endothelial dysfunction. EPCs are the primary protection mechanism for endothelial dysfunction, which promote angiogenesis and maintains endothelial integrity with a series of reactions. But the situation of this protection mechanism in SLE is not optimistic.
Classification, Immunophenotype, and Physiology of EPCs
Scientists have reached a consensus that EPCs isolated by cell culture are distinguished into two different groups: myeloid angiogenic cells (MACs), used to identify early EPCs (118); endothelial colony forming cells (ECFCs), used to identify late EPCs (119). They promote vascular repair through different mechanisms (120). ECFCs, considered to be real EPCs, can differentiate into ECs promoting vascular repair and neovascularization (121), with the immunophenotype positive for CD31, CD105, CD146, and negative for CD45, CD14 (120). MACs can’t become ECs but secretes angiogenic cytokines to promote angiogenesis through a paracrine mechanism (122), with the immunophenotype positive for CD45, CD14, CD31, and negative for CD146, CD133, and Tie2 (120) (Table 1).
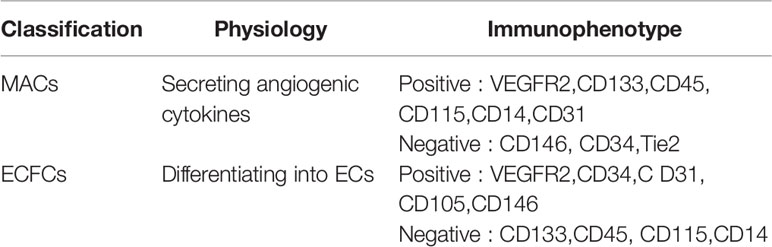
Table 1 Classification, immunophenotype and physiology of EPCs.
The Role of EPCs in Vascular Repair
After the injury, vascular repair occurs by accelerating the replacement of ECs. Re-endothelialization is a self-repairing process that maintains vascular endothelial protection after injury, including the proliferation and migration of adjacent intact ECs, resident EPCs and recruited EPCs. EPCs provide an endogenous repair mechanism to counteract persistent cell damage induced by risk factors. Scientists suggested EPCs are a useful tool for the treatment of endothelial injury in regenerative cardiovascular medicine (123–126). Thus, EPCs have been studied as biomarkers for the diagnosis and prognosis of CVD (127–129).
ECs
Healthy ECs protect atherosclerosis by promoting vasodilation, antioxidant and anti-inflammatory and inhibiting leukocyte adhesion and migration, and smooth muscle cell proliferation and migration. Remarkably, ECs can repair themselves. VEGF activates Cdc-42 and Rac1, mediates the formation of filamentous pseudopodia and plate pseudopodia, leading to EC migration (130). SDF-1 activates GPCR-dependent p110γPI3K, increases the expression of FoxM1 in ECs, participates in the transcriptional regulation of cell cycle progression genes, promoting the proliferation of ECs (131). Also, FoxM1 promotes re-adhesion between ECs through transcriptional control of β-catenin (132). When cells exfoliate after injury, surrounding ECs proliferate and migrate to coverage the basement membrane. However, mature ECs have limited ability to replace damaged ECs. Compared to ECs, EPCs show a higher proliferation potential, thus can serve as an additional source of ECs.
EPCs
EPCs could differentiation into ECs. EPCs locate at the site of vascular injury, restore endothelial integrity and participate in neovascularization. The process of re-endothelialization includes mobilization, chemotaxis, homing, proliferation and differentiation (Figure 3). Early EPCs release growth factors, adhesion molecules and chemokines to promote the proliferation, survival and migration of late EPCs; late EPCs directly participate in the formation of endothelium (133). EPCs also release exocrine bodies to respond to injured ECs (134, 135).
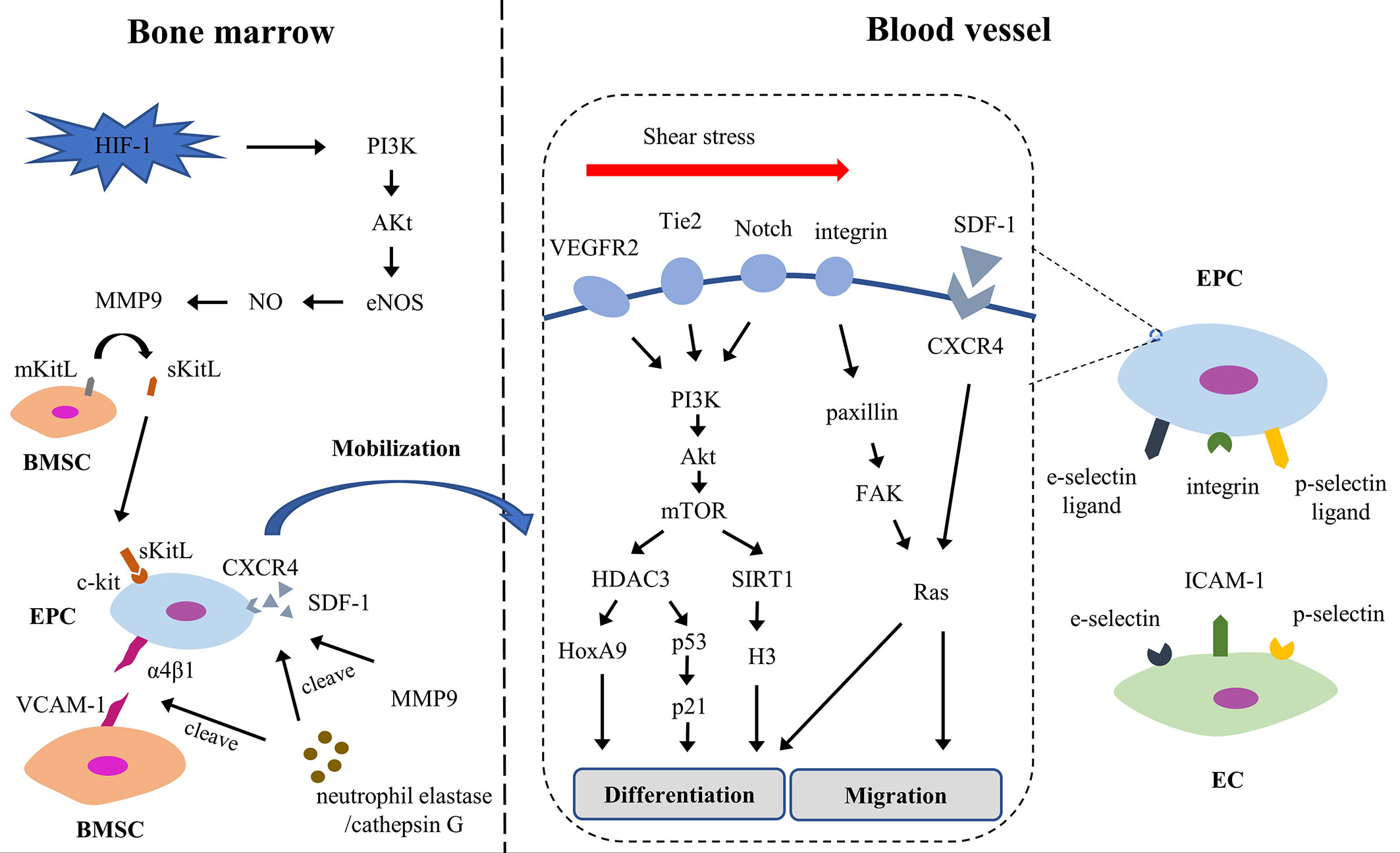
Figure 3 The process of EPC re-endothelialization. Akt, Protein kinase B; BMSC, bone marrow stromal cell; CXCR4, chemokine (C-X-C motif) receptor 4; EC, endothelial cell; EPC, endothelial progenitor cell; eNOS, endothelial nitric oxide synthase; FAK, focal adhesion kinase; HDAC3, histone deacetylase 3; HIF-1, hypoxia-inducible factor 1; HoxA9, Homeobox A9; ICAM-1, intercellular adhesion molecule-1; mKitL, membrane-bound form of Kit ligand; MMP9, matrix metalloproteinase-9; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol-3-kinase; SDF-1, stromal cell-derived factor-1; sKitL, soluble Kit-ligand; VCAM-1, vascular cell adhesion molecule-1; VEGFR2, vascular endothelial growth factor receptor-2.
Mobilization
The mobilization is the first step and is strictly regulated. EPCs are mainly seen in the bone marrow and in an inactive state which bind to bone marrow stromal cells (BMSCs) through the interaction of integrins (α4β1 and β3) and VCAM-1 (136). Under hypoxia, hypoxia-inducible factor 1 (HIF-1) rapidly increases, then weakens the interaction between EPCs and BMSC through NO and MMP-9 in PI3K/AKt/eNOS-dependent manner. Moreover, neutrophil elastase and cathepsin G prevent EPCs from combining with BMSCs by cutting integrin and VCAM-1; and they cooperated with MMP9 to degrade SDF-1 in peripheral blood matrix niches forming a high SDF-1 concentration gradient. Under the synergistic action of elastase, cathepsin G and MMPs, EPCs are driven into the peripheral circulation (137).
Homing
After entering the peripheral circulation from the bone marrow, EPCs are summoned and stay at the site of endothelial injury in the tissue. This process involves multi-step cascade adhesion and signaling events, including chemotaxis, involvement, adhesion and migration (138). The SDF-1/CXCR4 axis regulates the downstream signal Rac, changes the polarity and cytoskeleton of the cells, maintains the motor state of the transitional cells, and navigates the EPCs to the target organ (139). Meanwhile, integrin, p-selectin ligand and e-selectin ligand expressed on EPCs interact with p-selectin, e-selectin and ICAM-1 expressed on activated ECs, supporting EPCs adhesion and migration to ECs (140, 141). Some studies have shown that SDF-1 increases the expression of e-selectin in microvascular ECs and then increases the adhesion of EC-EPC (142).
Differentiation
On the way to the target organ, EPCs begin to differentiate into ECs. During differentiation, cytokines and shear stress trigger a series of events, which cause EPCs to acquire some phenotypic characteristics of ECs. Shear stress supports the differentiation and proliferation of EPCs via VEGFR2, Tie2, Notch, and β1/3 integrin signaling (143). It stabilizes and activates histone deacetylase 3 (HDAC3) through the VEGFR2/Tie2/Notch/PI3K/Akt/mTOR pathway, which in turn deacetylated p53, leading to increased cell cycle arrest protein p21 and endothelial markers (144). The homeobox transcription factor HoxA9 contributes to HDAC-mediated differentiation (145). Histone deacetylase SIRT1, another downstream factor of shear stress/PI3K/Akt pathway, is overexpressed in EPCs and decreases histone H3 acetylation, upregulating endothelial markers (146). Beside, integrins β1 and β3, also overexpressed, enhance the expression of endothelial markers via paxillin/FAK/RAS/ERK pathway (147–149).
Mobilized EPCs enter into the peripheral blood and build a cell pool, repairing the endothelium by forming a patch at the site of intimal injury. EPCs represent negative feedback in intravascular homeostasis. The number and function of EPCs are regulated by the same molecular pathway, so the decrease of EPCs number is related to weakened function, and the increase of EPCs number is related to enhanced function.
Changes in the Number and Function of EPCs in SLE
There are 15 research articles about the number and function of SLE EPCs by searching “(Endothelial Progenitor Cells) AND (Lupus Erythematosus, Systemic)” in PubMed, which have shown inconsistent results (Table 2). Most of the results on the quantitative studies of SLE EPCs have shown a low level. Four studies have shown different results. The difference in the detection, quantification and identification of EPCs and the active phase of SLE might explain the quantitative differences. Studies on the qualitative of SLE EPCs also showed different results. Ablin JN et al. shown enhanced adhesion of SLE EPCs (156), while the others shown weakened proliferation/migration/adhesion/differentiation (46–49, 77, 150, 153, 154, 157–159). The different adhesion test and quantification seems to be the reason.
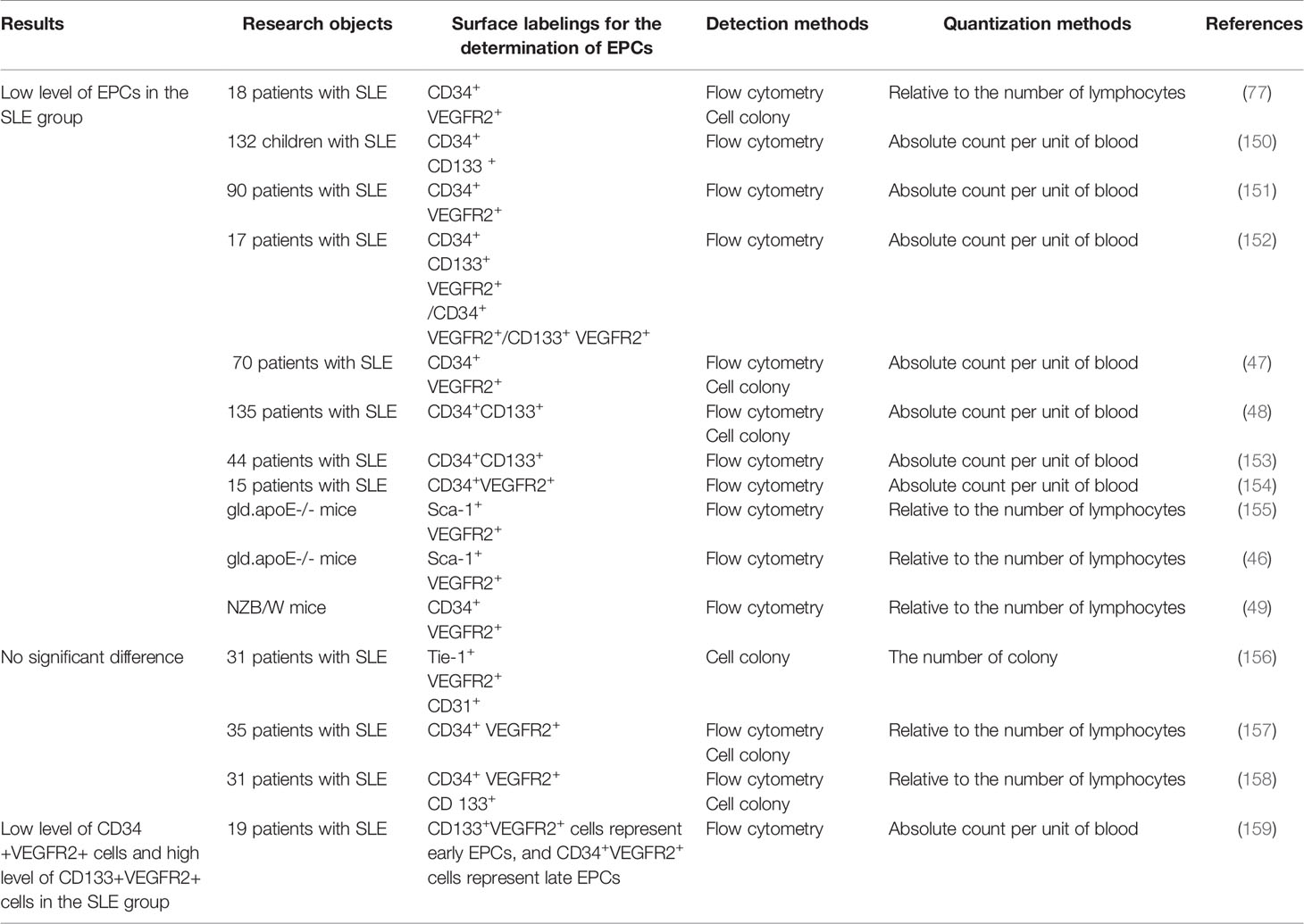
Table 2 Quantitative analysis of circulating EPCs between SLE and healthy control.
Causes of Reduced Number and Impaired Function of EPCs in SLE
Although the results are controversial, we believe that SLE EPCs show a trend of reduced number and impaired function. The risk factors (IFN-I, BAFF, OPG, IL-10, IL-18) and protective factors (Tang) both exist in SLE. The reduced number and impaired function of SLE EPCs seem to be the result of the game between the two sides.
There is no doubt that IFN-I accelerates SLE atherosclerosis, whether in the initiation or development of the disease (15, 52). The adult and mouse models’ researches conclude that IFN-I accelerating SLE atherosclerosis by interfering with EPCs (15, 46–49, 51, 52, 160). Like adult-onset SLE, childhood-onset SLE also shown reduced number and impaired function of EPCs (150). But there was no significant correlation between IFN-I activity and childhood-onset SLE subclinical atherosclerosis and endothelial function (150). We need a longitudinal assessment in the future to assess whether vascular damage in childhood-onset SLE is related to IFN-I. Inflammatory body activation is a key downstream pathway leading to vascular abnormalities. The interaction between IFN-I and inflammatory factors mediates reduced number and impaired function of SLE EPCs. IFN-α down-regulates IL-1β and VEGF (52) and up-regulates IL-18 and its activator caspase-1 (51)— IL-1β promotes the differentiation of EPCs (52); IL-18 inhibits the differentiation of EPCs (51). IL10 inhibits EC differentiation and enhances IFN-α-mediated EPC dysfunction (50). OPG plays a pathogenic role in atherosclerosis. OPG binds to syndecan 4, the receptor of OPG on EPC, then induces oxidative stress, causing apoptosis of EPC (151). Spinelli FR et al. has observed that BAFF receptors are expressed in both EPC and EC, and mediated the apoptosis of EPC (77). The addition of BAFF inhibitor—belimumab—restored the quantity and quality of EPCs in vivo and in vitro, which further proved this point (77).
Tang, a specific T cell group expressing CD3, CD31 and CXCR4, promotes early EPCs differentiation and activates locally resident ECs (161). And the percentage of circulating Tang increased in SLE patients (162–164). However, the chronic inflammatory environment of SLE accelerates autoimmune aging. Aging Tang (CD28null-Tang) is not protective but cytotoxic, secreting inflammatory mediators and releasing cytolytic molecules from intracellular particles to induce EC damage and accelerates atherosclerosis in most SLE patients (165). And the frequency of CD28null-Tang increased in SLE patients with traditional CVD risk factors and active diseases (165).
Therefore, we speculate that Tang activates the vascular endothelial protective mechanism in the early SLE. With the progress of the disease, the chronic inflammatory environment of SLE not only accelerates the aging of Tang but also enriches a variety of risk factors for EPCs, which leads to the dysfunction of EPC in SLE patients.
The Role of IFN-I in the Injury of EPCs in SLE
The Immune Mechanism of IFN-I Production in SLE
The IFN-I system in SLE is chronically active. pDCs (plasmacytoid pre-dendritic cells) are the primary source, which have high levels of interferon regulatory factor (IRF) 7, facilitating rapid and large-scale IFN-α generation (166). Up-regulated interferon-induced genes such as MX1, ISG54, and ISG56 and transcription factors of interferon pathway such as IRF5, IRF7, IRAK1, TREX1, STAT4, and PTPN22 mediate abnormal immune responses and the production of ICs, resulting in abnormal activation of pDCs (167). And other immune cells such as neutrophils, NK cells, T cells, B cells and platelets enhance IFN-I production by IC-stimulated pDCs; IFN-I, in turn, stimulates the activation of these immune cells, forming a self-magnifying pathogenic loop (65, 66, 168–173).
During exploring the signaling pathway, the increased exposure of nuclear contents to corresponding nucleic acid biosensors is the critical risk factors. Under normal physiological conditions, self DNA/RNA exists in different cell compartments and is isolated from the nucleic acid biosensor in the cytoplasm. Due to the insufficient clearance of apoptotic/necrotic cells, SLE patients are rich in endogenous free DNA/RNA, which form ICs with anti-DNA/RNA antibodies (174). Exogenous microbial DNA/RNA also induce autoimmune response (175–177). Exposed RNA and DNA stimulate the relevant nucleic acid biosensor in the form of ICs. DNA biosensors are divided into two types: endosomal membrane receptors and intracellular receptors (178). TLR9 is the only known DNA biosensor based on endosomes, which is mainly expressed in pDCs. The DNA ICs are absorbed and transported into the endosome through the Fcγ RIIa in pDCs, activating TLR9-MyD88-IRF7 pathway (166). Moreover, TLR9 can bind to the curli-DNA complex, composed of bacterial DNA and amyloid protein curli—a component of bacterial biofilms (175, 176). Compared with TLR9, cytoplasmic DNA biosensors are widely expressed in mammalian cells. Thirteen cytoplasmic DNA biosensors have been found so far and cGAS is the most important cytoplasmic DNA biosensor (178). cGAS binds to cytoplasmic DNA to produce cGAMP, which then activates ER-resident STING protein. The activated STING is transported from the endoplasmic reticulum to the ER-resident Golgi apparatus and recruits TBK1 to enter the endosome. TBK1 activates IRF3 and IRF7, leading to the expression of IFN-I (179). Major RNA biosensors include TLR7 and RIG-I/MDA5. TLR7 also belongs to the endosomal membrane receptor, activated by single-stranded RNA. The U1snRNA induces PDCs to produce IFN-α through Fcγ RIIa-TLR7-MyD88-IRF7 pathway in SLE patients (180, 181). RIG-I/MDA5 signal is mainly used to deal with viral infections. After recognizing viral double-stranded RNA, intracellular RNA helicases (RIG-I and MDA5) undergo conformational changes to induce MAVS, and activates IRF3/7 through TRAF6/3, resulting in the secretion of IFN-I (182). Recent studies have shown that RIG-I/MDA5 signal may reduce the degradation capacity of insoluble virus-like aggregates, inducing a continuous increase of IFN-I (177).
The Pathways of IFN-I Damaging EPCs
IFN-I is one of the causes of impaired EPCs, but the specific mechanism remains to be elucidated. IFN-I damages EPCs in two ways: direct toxicity and indirect toxicity (Figure 4).
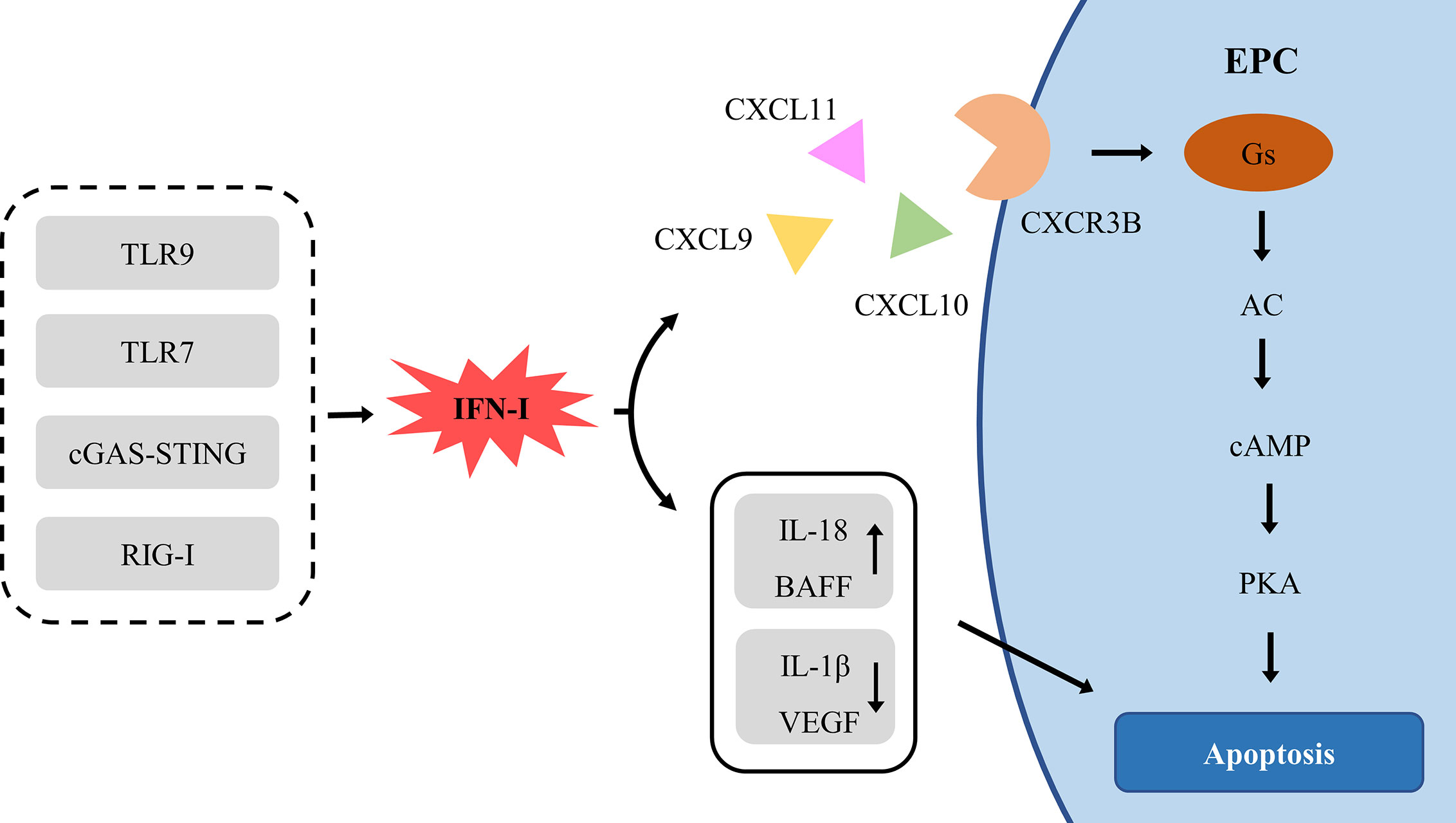
Figure 4 The signal pathway of IFN-Idamaging EPC. AC, adenylyl cyclase; BAFF, B cell-activating factor; cAMP, cyclic adenosine monophosphate; cGAS, cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase; CXCL, chemokine (C-X-C Motif) ligand; CXCR, chemokine (C-X-C motif) receptor; EPC, endothelial progenitor cell; PKA, Protein kinase A; RIG-I, retinoic acid-inducible gene I; TLR, Toll-like receptor; VEGF, vascular endothelial growth factor.
IFN-I actively induces the production of ELR-negative CXC chemokines CXCL9, CXCL10 and CXCL11, which mediate angiostasis through the receptor CXCR3 (183). CXCR3 exists in three different splice variants, CXCR3A, CXCR3B, and CXCR3-alt (184). CXCR3A recruits leukocytes, especially in Th1 lymphocytes (185). CXCR3-alt has a higher affinity for CXCL11, but its role in angiogenesis remains to be determined (186). Conversely, CXCR3B, expressed in ECs, is the main angiostatic variant of CXCR3 and is the primary angiostatic receptor for CXCL9, CXCL10, and CXCL11, inducing anti-proliferation and anti-migration (187–189). CXCR3A and CXCR3B differ for 52 amino acids at the NH2 end and couple different types of G proteins, triggering different signal transduction pathways, CXCR3A-Gi-PI3K-MAPK and CXCR3B-Gs-AC-cAMP-PKA (187, 190). The coupling of CXCR3B with Gs results in the selective activation of adenylyl cyclase (AC) and a consequent increase of intracellular cAMP levels (187). Up-regulation of cAMP in ECs directly activates PKA, inducing apoptosis (191).
Moreover, IFN-I enhances the toxicity of ILs and BAFF, which are EPC risk factor as well. IFN-I interacts with inflammatory factor ILs to damage EPC synergistically. IL-10 enhances the effect of IFN-α on SLE EPC (50). IFN-I down-regulates angiogenic molecules IL-1β and VEGF (52) and up-regulates IL-18 and its activator caspase-1 (51), enhancing the anti-angiogenic effect. There was a positive correlation between the levels of IFN-I and BAFF in SLE (192). IFN-I induces the expression and mobilization of BAFF in SLE monocytes and neutrophils (35, 36). The expression of BAFF is directly induced by IFN-I through IRF1 and IRF2 (36). IFN-α stimulates the secretion of IL-17, then IL-17 and BAFF promote the survival and differentiation of B cells and production of autoantibodies, which enhances IFN by pDCs, forming a closed vicious circle (192).
Therefore, IFN-I has direct and indirect toxic effects on EPC, resulting in endothelial dysfunction, which starts atherosclerosis in SLE. It is proved once again that IFN-I plays a central pathogenic role in SLE CVD.
Conclusion
Long-term activation of IFN-I system in SLE induces the expression of CXCL9/10/11, activating CXCR3B-Gs-AC-cAMP-PKA signal pathway to promote the dysfunction of ECs and EPCs; and CXCR3A-Gi-PI3K-MAPK signaling pathway to recruit leukocytes into the inflammatory site. Besides, IFN-I enhances the toxicity of other EPCs dysfunction factors, indirectly accelerating arteriosclerosis. Overexpression of IFN-I through the activation of TLR7/9 signal decreases the number and function of EPCs and increases atherosclerotic lesions in SLE patients (46), suggesting that targeted therapy of cGAS and RIG-I signal pathway may have a potential therapeutic effect on SLE atherosclerosis. Targeted therapy of the IFN-I system has a potential therapeutic effect on early atherosclerosis in SLE patients.
Author Contributions
XD did the literature search and drafted the article. WX and XH gave insight. XH revised the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (No. 61562021 and No. 81560275, No. 81960885, No. 81260139, No. 81060073, No. 30560161), Hainan Major Science and Technology Projects (ZDKJ2039010), Hainan Association for academic excellence Youth Science and Technology Innovation Program (201515), Hainan special projects of Social Development (ZDYF2018103 and 2015SF39).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thanked Caiqiong Liu and Dahai Yuan for reviewing and revising this paper.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.581385/full#supplementary-material
References
1. Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatol (Oxford) (2017) 56:1945–61. doi: 10.1093/rheumatology/kex260
2. Durcan L, O’Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet (2019) 393:2332–43. doi: 10.1016/S0140-6736(19)30237-5
3. Croca S, Rahman A. Atherosclerosis in systemic lupus erythematosus. Best Pract Res Clin Rheumatol (2017) 31:364–72. doi: 10.1016/j.berh.2017.09.012
4. Giannelou M, Mavragani CP. Cardiovascular disease in systemic lupus erythematosus: A comprehensive update. J Autoimmun (2017) 82:1–12. doi: 10.1016/j.jaut.2017.05.008
5. Miteva K, Madonna R, De Caterina R, Van Linthout S. Innate and adaptive immunity in atherosclerosis. Vascul Pharmacol (2018) 107:67–77. doi: 10.1016/j.vph.2018.04.006
6. Wu MY, Li CJ, Hou MF, Chu PY. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int J Mol Sci (2017) 18. doi: 10.3390/ijms18102034
7. Tsai CY, Shen CY, Liao HT, Li KJ, Lee HT, Lu CS, et al. Molecular and Cellular Bases of Immunosenescence, Inflammation, and Cardiovascular Complications Mimicking “Inflammaging” in Patients with Systemic Lupus Erythematosus. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20163878
8. Park JK, Kim JY, Moon JY, Ahn EY, Lee EY, Lee EB, et al. Altered lipoproteins in patients with systemic lupus erythematosus are associated with augmented oxidative stress: a potential role in atherosclerosis. Arthritis Res Ther (2016) 18:306. doi: 10.1186/s13075-016-1204-x
9. Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D, et al. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front Physiol (2017) 8:600. doi: 10.3389/fphys.2017.00600
10. Kim SY, Yu M, Morin EE, Kang J, Kaplan MJ, Schwendeman A. High-Density Lipoprotein in Lupus: Disease Biomarkers and Potential Therapeutic Strategy. Arthritis Rheumatol (2020) 72:20–30. doi: 10.1002/art.41059
11. Buskiewicz IA, Montgomery T, Yasewicz EC, Huber SA, Murphy MP, Hartley RC, et al. Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal (2016) 9:a115. doi: 10.1126/scisignal.aaf1933
12. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
13. Lightfoot YL, Blanco LP, Kaplan MJ. Metabolic abnormalities and oxidative stress in lupus. Curr Opin Rheumatol (2017) 29:442–9. doi: 10.1097/BOR.0000000000000413
14. Yang J, Yang X, Zou H, Li M. Oxidative Stress and Treg and Th17 Dysfunction in Systemic Lupus Erythematosus. Oxid Med Cell Longev (2016) 2016:2526174. doi: 10.1155/2016/2526174
15. Thacker SG, Zhao W, Smith CK, Luo W, Wang H, Vivekanandan-Giri A, et al. Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum (2012) 64:2975–85. doi: 10.1002/art.34504
16. Casbon AJ, Long ME, Dunn KW, Allen LA, Dinauer MC. Effects of IFN-gamma on intracellular trafficking and activity of macrophage NADPH oxidase flavocytochrome b558. J Leukoc Biol (2012) 92:869–82. doi: 10.1189/jlb.0512244
17. Theodorou E, Nezos A, Antypa E, Ioakeimidis D, Koutsilieris M, Tektonidou M, et al. B-cell activating factor and related genetic variants in lupus related atherosclerosis. J Autoimmun (2018) 92:87–92. doi: 10.1016/j.jaut.2018.05.002
18. Jeremic I, Djuric O, Nikolic M, Vlajnic M, Nikolic A, Radojkovic D, et al. Neutrophil extracellular traps-associated markers are elevated in patients with systemic lupus erythematosus. Rheumatol Int (2019) 39:1849–57. doi: 10.1007/s00296-019-04426-1
19. Turpeinen H, Raitoharju E, Oksanen A, Oksala N, Levula M, Lyytikainen LP, et al. Proprotein convertases in human atherosclerotic plaques: the overexpression of FURIN and its substrate cytokines BAFF and APRIL. Atherosclerosis (2011) 219:799–806. doi: 10.1016/j.atherosclerosis.2011.08.011
20. Yuan Q, Chen H, Li X, Wei J. Leptin: an unappreciated key player in SLE. Clin Rheumatol (2020) 39:305–17. doi: 10.1007/s10067-019-04831-8
21. Yu Y, Fu S, Zhang X, Wang L, Zhao L, Wan W, et al. Leptin facilitates the differentiation of Th17 cells from MRL/Mp-Fas lpr lupus mice by activating NLRP3 inflammasome. Innate Immun (2020) 26:294–300. doi: 10.1177/1753425919886643
22. Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol (2014) 66:2532–44. doi: 10.1002/art.38703
23. Doring Y, Manthey HD, Drechsler M, Lievens D, Megens RT, Soehnlein O, et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation (2012) 125:1673–83. doi: 10.1161/CIRCULATIONAHA.111.046755
24. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (2015) 349:316–20. doi: 10.1126/science.aaa8064
25. Gestermann N, Di Domizio J, Lande R, Demaria O, Frasca L, Feldmeyer L, et al. Netting Neutrophils Activate Autoreactive B Cells in Lupus. J Immunol (2018) 200:3364–71. doi: 10.4049/jimmunol.1700778
26. Sharabi A, Tsokos GC. T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nat Rev Rheumatol (2020) 16:100–12. doi: 10.1038/s41584-019-0356-x
27. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
28. Mountz JD, Hsu HC, Ballesteros-Tato A. Dysregulation of T Follicular Helper Cells in Lupus. J Immunol (2019) 202:1649–58. doi: 10.4049/jimmunol.1801150
29. Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, et al. A CD4(+) T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med (2019) 25:75–81. doi: 10.1038/s41591-018-0254-9
30. Makiyama A, Chiba A, Noto D, Murayama G, Yamaji K, Tamura N, et al. Expanded circulating peripheral helper T cells in systemic lupus erythematosus: association with disease activity and B cell differentiation. Rheumatol (Oxford) (2019) 58:1861–9. doi: 10.1093/rheumatology/kez077
31. Granger V, Peyneau M, Chollet-Martin S, de Chaisemartin L. Neutrophil Extracellular Traps in Autoimmunity and Allergy: Immune Complexes at Work. Front Immunol (2019) 10:2824. doi: 10.3389/fimmu.2019.02824
32. Chistiakov DA, Orekhov AN, Bobryshev YV. ApoA1 and ApoA1-specific self-antibodies in cardiovascular disease. Lab Invest (2016) 96:708–18. doi: 10.1038/labinvest.2016.56
33. Sun J, Lundstrom SL, Zhang B, Zubarev RA, Steuer J, Gillgren P, et al. IgM antibodies against phosphorylcholine promote polarization of T regulatory cells from patients with atherosclerotic plaques, systemic lupus erythematosus and healthy donors. Atherosclerosis (2018) 268:36–48. doi: 10.1016/j.atherosclerosis.2017.11.010
34. Rahman M, Sing S, Golabkesh Z, Fiskesund R, Gustafsson T, Jogestrand T, et al. IgM antibodies against malondialdehyde and phosphorylcholine are together strong protection markers for atherosclerosis in systemic lupus erythematosus: Regulation and underlying mechanisms. Clin Immunol (2016) 166-167:27–37. doi: 10.1016/j.clim.2016.04.007
35. Lopez P, Scheel-Toellner D, Rodriguez-Carrio J, Caminal-Montero L, Gordon C, Suarez A. Interferon-alpha-induced B-lymphocyte stimulator expression and mobilization inhealthy and systemic lupus erthymatosus monocytes. Rheumatol (Oxford) (2014) 53:2249–58. doi: 10.1093/rheumatology/keu249
36. Sjostrand M, Johansson A, Aqrawi L, Olsson T, Wahren-Herlenius M, Espinosa A. The Expression of BAFF Is Controlled by IRF Transcription Factors. J Immunol (2016) 196:91–6. doi: 10.4049/jimmunol.1501061
37. Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol (2008) 181:2211–9. doi: 10.4049/jimmunol.181.3.2211
38. Dong G, Yang Y, Li X, Yao X, Zhu Y, Zhang H, et al. Granulocytic myeloid-derived suppressor cells contribute to IFN-I signaling activation of B cells and disease progression through the lncRNA NEAT1-BAFF axis in systemic lupus erythematosus. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165554. doi: 10.1016/j.bbadis.2019.165554
39. Feng X, Chen W, Xiao L, Gu F, Huang J, Tsao BP, et al. Artesunate inhibits type I interferon-induced production of macrophage migration inhibitory factor in patients with systemic lupus erythematosus. Lupus (2017) 26:62–72. doi: 10.1177/0961203316651738
40. Lapter S, Ben-David H, Sharabi A, Zinger H, Telerman A, Gordin M, et al. A role for the B-cell CD74/macrophage migration inhibitory factor pathway in the immunomodulation of systemic lupus erythematosus by a therapeutic tolerogenic peptide. Immunology (2011) 132:87–95. doi: 10.1111/j.1365-2567.2010.03342.x
41. Schmitz C, Noels H, El BO, Straussfeld E, Megens R, Sternkopf M, et al. Mif-deficiency favors an atheroprotective autoantibody phenotype in atherosclerosis. FASEB J (2018) 32:4428–43. doi: 10.1096/fj.201800058R
42. Murata T, Asanuma K, Ara N, Iijima K, Hatta W, Hamada S, et al. Leptin Aggravates Reflux Esophagitis by Increasing Tissue Levels of Macrophage Migration Inhibitory Factor in Rats. Tohoku J Exp Med (2018) 245:45–53. doi: 10.1620/tjem.245.45
43. Lewandowski LB, Kaplan MJ. Update on cardiovascular disease in lupus. Curr Opin Rheumatol (2016) 28:468–76. doi: 10.1097/BOR.0000000000000307
44. de Winther MP. The Plot Thickens Further for Type I Interferons in Atherosclerotic Disease. Arterioscler Thromb Vasc Biol (2016) 36:217–8. doi: 10.1161/ATVBAHA.115.305464
45. Boshuizen MC, de Winther MP. Interferons as Essential Modulators of Atherosclerosis. Arterioscler Thromb Vasc Biol (2015) 35:1579–88. doi: 10.1073/pnas.0807841105
46. Geng L, Wang S, Li X, Wang D, Chen H, Chen J, et al. Association between Type I interferon and depletion and dysfunction of endothelial progenitor cells in C57BL/6 mice deficient in both apolipoprotein E and Fas ligand. Curr Res Transl Med (2018) 66:71–82. doi: 10.1016/j.retram.2018.02.002
47. Lee PY, Li Y, Richards HB, Chan FS, Zhuang H, Narain S, et al. Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum (2007) 56:3759–69. doi: 10.1002/art.23035
48. Denny MF, Thacker S, Mehta H, Somers EC, Dodick T, Barrat FJ, et al. Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood (2007) 110:2907–15. doi: 10.1182/blood-2007-05-089086
49. Thacker SG, Duquaine D, Park J, Kaplan MJ. Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelial dysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus (2010) 19:288–99. doi: 10.1177/0961203309353773
50. Cates AM, Holden VI, Myers EM, Smith CK, Kaplan MJ, Kahlenberg JM. Interleukin 10 hampers endothelial cell differentiation and enhances the effects of interferon alpha on lupus endothelial cell progenitors. Rheumatol (Oxford) (2015) 54:1114–23. doi: 10.1093/rheumatology/keu431
51. Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol (2011) 187:6143–56. doi: 10.4049/jimmunol.1101284
52. Thacker SG, Berthier CC, Mattinzoli D, Rastaldi MP, Kretzler M, Kaplan MJ. The detrimental effects of IFN-alpha on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J Immunol (2010) 185:4457–69. doi: 10.4049/jimmunol.1001782
53. Buie JJ, Renaud LL, Muise-Helmericks R, Oates JC. IFN-alpha Negatively Regulates the Expression of Endothelial Nitric Oxide Synthase and Nitric Oxide Production: Implications for Systemic Lupus Erythematosus. J Immunol (2017) 199:1979–88. doi: 10.4049/jimmunol.1600108
54. Chmielewski S, Olejnik A, Sikorski K, Pelisek J, Blaszczyk K, Aoqui C, et al. STAT1-dependent signal integration between IFNgamma and TLR4 in vascular cells reflect pro-atherogenic responses in human atherosclerosis. PloS One (2014) 9:e113318. doi: 10.1371/journal.pone.0113318
55. Li J, Fu Q, Cui H, Qu B, Pan W, Shen N, et al. Interferon-alpha priming promotes lipid uptake and macrophage-derived foam cell formation: a novel link between interferon-alpha and atherosclerosis in lupus. Arthritis Rheum (2011) 63:492–502. doi: 10.1002/art.30165
56. Yu XH, Zhang J, Zheng XL, Yang YH, Tang CK. Interferon-gamma in foam cell formation and progression of atherosclerosis. Clin Chim Acta (2015) 441:33–43. doi: 10.1016/j.cca.2014.12.007
57. Reardon CA, Lingaraju A, Schoenfelt KQ, Zhou G, Cui C, Jacobs-El H, et al. Obesity and Insulin Resistance Promote Atherosclerosis through an IFNgamma-Regulated Macrophage Protein Network. Cell Rep (2018) 23:3021–30. doi: 10.1016/j.celrep.2018.05.010
58. Diao Y, Mohandas R, Lee P, Liu Z, Sautina L, Mu W, et al. Effects of Long-Term Type I Interferon on the Arterial Wall and Smooth Muscle Progenitor Cells Differentiation. Arterioscler Thromb Vasc Biol (2016) 36:266–73. doi: 10.1161/ATVBAHA.115.306767
59. Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation (2006) 114:2482–9. doi: 10.1161/CIRCULATIONAHA.106.642801
60. Rosner D, Stoneman V, Littlewood T, McCarthy N, Figg N, Wang Y, et al. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am J Pathol (2006) 168:2054–63. doi: 10.2353/ajpath.2006.050473
61. Inagaki Y, Yamagishi S, Amano S, Okamoto T, Koga K, Makita Z. Interferon-gamma-induced apoptosis and activation of THP-1 macrophages. Life Sci (2002) 71:2499–508. doi: 10.1016/S0024-3205(02)02042-8
62. Zhang CY, Qu B, Ye P, Li J, Bao CD. Vulnerability of atherosclerotic plaques is associated with type I interferon in a murine model of lupus and atherosclerosis. Genet Mol Res (2015) 14:14871–81. doi: 10.4238/2015.November.18.52
63. Niessner A, Shin MS, Pryshchep O, Goronzy JJ, Chaikof EL, Weyand CM. Synergistic proinflammatory effects of the antiviral cytokine interferon-alpha and Toll-like receptor 4 ligands in the atherosclerotic plaque. Circulation (2007) 116:2043–52. 10.1161/CIRCULATIONAHA.107.697789
64. Weng X, Cheng X, Wu X, Xu H, Fang M, Xu Y. Sin3Bmediates collagen type I gene repression by interferon gamma in vascular smooth muscle cells. Biochem Biophys Res Commun (2014) 447:263–70. doi: 10.1016/j.bbrc.2014.03.140
65. Lood C, Amisten S, Gullstrand B, Jonsen A, Allhorn M, Truedsson L, et al. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: up-regulation of the type I interferon system is strongly associated with vascular disease. Blood (2010) 116:1951–7. doi: 10.1182/blood-2010-03-274605
66. Duffau P, Seneschal J, Nicco C, Richez C, Lazaro E, Douchet I, et al. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med (2010) 2:47r–63r. doi: 10.1126/scitranslmed.3001001
67. Domschke G, Linden F, Pawig L, Hafner A, Akhavanpoor M, Reymann J, et al. Systematic RNA-interference in primary human monocyte-derived macrophages: A high-throughput platform to study foam cell formation. Sci Rep (2018) 8:10516. doi: 10.1038/s41598-018-28790-3
68. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007) 13:587–96. doi: 10.1038/nm1567
69. Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, et al. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol (2006) 177:8072–9. doi: 10.4049/jimmunol.177.11.8072
70. Amin MA, Haas CS, Zhu K, Mansfield PJ, Kim MJ, Lackowski NP, et al. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFkappaB. Blood (2006) 107:2252–61. doi: 10.1182/blood-2005-05-2011
71. Schrans-Stassen BH, Lue H, Sonnemans DG, Bernhagen J, Post MJ. Stimulation of vascular smooth muscle cell migration by macrophage migration inhibitory factor. Antioxid Redox Signal (2005) 7:1211–6. doi: 10.1089/ars.2005.7.1211
72. Kong YZ, Huang XR, Ouyang X, Tan JJ, Fingerle-Rowson G, Bacher M, et al. Evidence for vascular macrophage migration inhibitory factor in destabilization of human atherosclerotic plaques. Cardiovasc Res (2005) 65:272–82. doi: 10.1016/j.cardiores.2004.09.020
73. Kong YZ, Yu X, Tang JJ, Ouyang X, Huang XR, Fingerle-Rowson G, et al. Macrophage migration inhibitory factor induces MMP-9 expression: implications for destabilization of human atherosclerotic plaques. Atherosclerosis (2005) 178:207–15. doi: 10.1016/j.atherosclerosis.2004.08.030
74. Scholz JL, Crowley JE, Tomayko MM, Steinel N, O’Neill PJ, Quinn WR, et al. BLyS inhibition eliminates primary B cells but leaves natural and acquired humoral immunity intact. Proc Natl Acad Sci USA (2008) 105:15517–22. doi: 10.1073/pnas.0807841105
75. Schweighoffer E, Tybulewicz VL. Signalling for B cell survival. Curr Opin Cell Biol (2018) 51:8–14. doi: 10.1016/j.ceb.2017.10.002
76. Duan JH, Jiang Y, Mu H, Tang ZQ. Expression of BAFF and BR3 in patients with systemic lupus erythematosus. Braz J Med Biol Res (2016) 49. doi: 10.1590/1414-431X20154853
77. Spinelli FR, Barbati C, Cecarelli F, Morello F, Colasanti T, Vomero M, et al. B lymphocyte stimulator modulates number and function of endothelial progenitor cells in systemic lupus erythematosus. Arthritis Res Ther (2019) 21:245. doi: 10.1186/s13075-019-2015-7
78. Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gomez-Reino JJ, et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol (2017) 13:100–9. doi: 10.1038/nrrheum.2016.209
79. McMahon M, Skaggs BJ, Grossman JM, Sahakian L, Fitzgerald J, Wong WK, et al. A panel of biomarkers is associated with increased risk of the presence and progression of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheumatol (2014) 66:130–9. doi: 10.1002/art.38204
80. Adya R, Tan BK, Randeva HS. Differential effects of leptin and adiponectin in endothelial angiogenesis. J Diabetes Res (2015) 2015:648239. doi: 10.1155/2015/648239
81. Naylor C, Petri WJ. Leptin Regulation of Immune Responses. Trends Mol Med (2016) 22:88–98. doi: 10.1016/j.molmed.2015.12.001
82. Barnado A, Crofford LJ, Oates JC. At the Bedside: Neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J Leukoc Biol (2016) 99:265–78. doi: 10.1189/jlb.5BT0615-234R
83. Grayson PC, Kaplan MJ. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J Leukoc Biol (2016) 99:253–64. doi: 10.1189/jlb.5BT0615-247R
84. Moore S, Juo HH, Nielsen CT, Tyden H, Bengtsson AA, Lood C. Neutrophil extracellular traps identify patients at risk of increased disease activity and cardiovascular comorbidity in systemic lupus erythematosus. J Rheumatol (2019). doi: 10.3899/jrheum.190875
85. O’Neil LJ, Kaplan MJ, Carmona-Rivera C. The Role of Neutrophils and Neutrophil Extracellular Traps in Vascular Damage in Systemic Lupus Erythematosus. J Clin Med (2019) 8. doi: 10.3390/jcm8091325
86. Goel RR, Kaplan MJ. Deadliest catch: neutrophil extracellular traps in autoimmunity. Curr Opin Rheumatol (2020) 32:64–70. doi: 10.1097/BOR.0000000000000667
87. Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler Thromb Vasc Biol (2017) 37:1371–9. doi: 10.1161/ATVBAHA.117.309002
88. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis (2015) 74:1417–24. doi: 10.1136/annrheumdis-2013-204837
89. Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature (2019) 569:236–40.
90. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med (2010) 16:887–96. doi: 10.1038/nm.2184
91. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209:819–35. doi: 10.1084/jem.20112322
92. Kravvariti E, Konstantonis G, Sfikakis PP, Tektonidou MG. Progression of subclinical atherosclerosis in systemic lupus erythematosus versus rheumatoid arthritis: the impact of low disease activity. Rheumatol (Oxford) (2018) 57:2158–66. doi: 10.1093/rheumatology/key233
93. Perez-Sanchez C, Barbarroja N, Messineo S, Ruiz-Limon P, Rodriguez-Ariza A, Jimenez-Gomez Y, et al. Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann Rheum Dis (2015) 74:1441–9. doi: 10.1136/annrheumdis-2013-204600
94. Kurien BT, Fesmire J, Anderson CJ, Scofield RH. Anti-Ro and Concomitant Anti-La Autoantibodies Strongly Associated With Anti-oxLDL or Anti-Phospholipid Antibody in Systemic Lupus Erythematosus. J Clin Rheumatol (2016) 22:418–25. doi: 10.1097/RHU.0000000000000429
95. Kiss E, Seres I, Tarr T, Kocsis Z, Szegedi G, Paragh G. Reduced paraoxonase1 activity is a risk for atherosclerosis in patients with systemic lupus erythematosus. Ann N Y Acad Sci (2007) 1108:83–91. doi: 10.1196/annals.1422.009
96. Lopez P, Rodriguez-Carrio J, Martinez-Zapico A, Perez-Alvarez AI, Lopez-Mejias R, Benavente L, et al. Serum Levels of Anti-PON1 and Anti-HDL Antibodies as Potential Biomarkers of Premature Atherosclerosis in Systemic Lupus Erythematosus. Thromb Haemost (2017) 117:2194–206. doi: 10.1160/TH17-03-0221
97. Batuca JR, Ames PR, Isenberg DA, Alves JD. Antibodies toward high-density lipoprotein components inhibit paraoxonase activity in patients with systemic lupus erythematosus. Ann N Y Acad Sci (2007) 1108:137–46. doi: 10.1196/annals.1422.016
98. Artim-Esen B, Pericleous C, Mackie I, Ripoll VM, Latchman D, Isenberg D, et al. Anti-factor Xa antibodies in patients with antiphospholipid syndrome and their effects upon coagulation assays. Arthritis Res Ther (2015) 17:47. doi: 10.1186/s13075-015-0568-7
99. Artim-Esen B, Smoktunowicz N, McDonnell T, Ripoll VM, Pericleous C, Mackie I, et al. Factor Xa Mediates Calcium Flux in Endothelial Cells and is Potentiated by Igg From Patients With Lupus and/or Antiphospholipid Syndrome. Sci Rep (2017) 7:10788. doi: 10.1038/s41598-017-11315-9
100. Kondo A, Takahashi K, Mizuno T, Kato A, Hirano D, Yamamoto N, et al. The Level of IgA Antibodies to Endothelial Cells Correlates with Histological Evidence of Disease Activity in Patients with Lupus Nephritis. PloS One (2016) 11:e163085. doi: 10.1371/journal.pone.0163085
101. Thanei S, Vanhecke D, Trendelenburg M. Anti-C1q autoantibodies from systemic lupus erythematosus patients activate the complement system via both the classical and lectin pathways. Clin Immunol (2015) 160:180–7. doi: 10.1016/j.clim.2015.06.014
102. Thanei S, Trendelenburg M. Anti-C1q Autoantibodies from Systemic Lupus Erythematosus Patients Induce a Proinflammatory Phenotype in Macrophages. J Immunol (2016) 196:2063–74. doi: 10.4049/jimmunol.1501659
103. Ho MM, Manughian-Peter A, Spivia WR, Taylor A, Fraser DA. Macrophage molecular signaling and inflammatory responses during ingestion of atherogenic lipoproteins are modulated by complement protein C1q. Atherosclerosis (2016) 253:38–46. doi: 10.1016/j.atherosclerosis.2016.08.019
104. Pulanco MC, Cosman J, Ho MM, Huynh J, Fing K, Turcu J, et al. Complement Protein C1q Enhances Macrophage Foam Cell Survival and Efferocytosis. J Immunol (2017) 198:472–80. doi: 10.4049/jimmunol.1601445
105. de Faire U, Frostegard J. Natural antibodies against phosphorylcholine in cardiovascular disease. Ann N Y Acad Sci (2009) 1173:292–300. doi: 10.1111/j.1749-6632.2009.04748.x
106. Caligiuri G, Khallou-Laschet J, Vandaele M, Gaston AT, Delignat S, Mandet C, et al. Phosphorylcholine-targeting immunization reduces atherosclerosis. J Am Coll Cardiol (2007) 50:540–6. doi: 10.1016/j.jacc.2006.11.054
107. Svenungsson E, Engelbertsen D, Wigren M, Gustafsson JT, Gunnarsson I, Elvin K, et al. Decreased levels of autoantibodies against apolipoprotein B-100 antigens are associated with cardiovascular disease in systemic lupus erythematosus. Clin Exp Immunol (2015) 181:417–26. doi: 10.4049/jimmunol.1801150
108. Lourdudoss C, Ajeganova S, Frostegard J. Association between dietary and metabolic factors and IgM antibodies to phosphorylcholine and malondialdehyde in patients with systemic lupus erythematosus and population-based matched controls. Clin Exp Rheumatol (2018) 36:428–33.
109. Zhu M, Mo H, Li D, Luo X, Zhang L. Th17/Treg imbalance induced by increased incidence of atherosclerosis in patients with systemic lupus erythematosus (SLE). Clin Rheumatol (2013) 32:1045–52. doi: 10.1007/s10067-013-2237-z
110. Garcia-Carrasco M, Soto-Santillan P, Mendoza-Pinto C, Gonzalez-Ramirez R, Lopez-Carmona AL, Munguia-Realpozo P, et al. The Role of Circulating Regulatory T Cell Levels on Subclinical Atherosclerosis and Cardiovascular Risk Factors in Women with Systemic Lupus Erythematosus. Mediators Inflammation (2018) 2018:3271572. doi: 10.1155/2018/3271572
111. Wilhelm AJ, Rhoads JP, Wade NS, Major AS. Dysregulated CD4+ T cells from SLE-susceptible mice are sufficient to accelerate atherosclerosis in LDLr-/- mice. Ann Rheum Dis (2015) 74:778–85. doi: 10.1136/annrheumdis-2013-203759
112. Gaddis DE, Padgett LE, Wu R, McSkimming C, Romines V, Taylor AM, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun (2018) 9:1095. doi: 10.1038/s41467-018-03493-5
113. Nus M, Sage AP, Lu Y, Masters L, Lam B, Newland S, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med (2017) 23:601–10. doi: 10.1038/nm.4315
114. Baragetti A, Ramirez GA, Magnoni M, Garlaschelli K, Grigore L, Berteotti M, et al. Disease trends over time and CD4(+)CCR5(+) T-cells expansion predict carotid atherosclerosis development in patients with systemic lupus erythematosus. Nutr Metab Cardiovasc Dis (2018) 28:53–63. doi: 10.1016/j.numecd.2017.09.001
115. Clement M, Charles N, Escoubet B, Guedj K, Chauveheid MP, Caligiuri G, et al. CD4+CXCR3+ T cells and plasmacytoid dendritic cells drive accelerated atherosclerosis associated with systemic lupus erythematosus. J Autoimmun (2015) 63:59–67. doi: 10.1016/j.jaut.2015.07.001
116. Li J, McArdle S, Gholami A, Kimura T, Wolf D, Gerhardt T, et al. CCR5+T-bet+FoxP3+ Effector CD4 T Cells Drive Atherosclerosis. Circ Res (2016) 118:1540–52. doi: 10.1161/CIRCRESAHA.116.308648
117. Smith E, Croca S, Waddington KE, Sofat R, Griffin M, Nicolaides A, et al. Cross-talk between iNKT cells and monocytes triggers an atheroprotective immune response in SLE patients with asymptomatic plaque. Sci Immunol (2016) 1. doi: 10.1126/sciimmunol.aah4081
118. Vaughan EE, O’Brien T. Isolation of circulating angiogenic cells. Methods Mol Biol (2012) 916:351–6. doi: 10.1007/978-1-61779-980-8_25
119. Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest (2000) 105:71–7. doi: 10.1172/JCI8071
120. Medina RJ, Barber CL, Sabatier F, Dignat-George F, Melero-Martin JM, Khosrotehrani K, et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl Med (2017) 6:1316–20. doi: 10.1002/sctm.16-0360
121. Tasev D, Koolwijk P, van Hinsbergh VW. Therapeutic Potential of Human-Derived Endothelial Colony-Forming Cells in Animal Models. Tissue Eng Part B Rev (2016) 22:371–82. doi: 10.1089/ten.teb.2016.0050
122. Medina RJ, O’Neill CL, O’Doherty TM, Knott H, Guduric-Fuchs J, Gardiner TA, et al. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol Med (2011) 17:1045–55. doi: 10.2119/molmed.2011.00129
123. Chong MS, Ng WK, Chan JK. Concise Review: Endothelial Progenitor Cells in Regenerative Medicine: Applications and Challenges. Stem Cells Transl Med (2016) 5:530–8. doi: 10.5966/sctm.2015-0227
124. Takizawa S, Nagata E, Nakayama T, Masuda H, Asahara T. Recent Progress in Endothelial Progenitor Cell Culture Systems: Potential for Stroke Therapy. Neurol Med Chir (Tokyo) (2016) 56:302–9. doi: 10.2176/nmc.ra.2016-0027
125. Kamei N, Atesok K, Ochi M. The Use of Endothelial Progenitor Cells for the Regeneration of Musculoskeletal and Neural Tissues. Stem Cells Int (2017) 2017:1960804. doi: 10.1155/2017/1960804
126. Bianconi V, Sahebkar A, Kovanen P, Bagaglia F, Ricciuti B, Calabro P, et al. Endothelial and cardiac progenitor cells for cardiovascular repair: A controversial paradigm in cell therapy. Pharmacol Ther (2018) 181:156–68. doi: 10.1016/j.pharmthera.2017.08.004
127. Lee PS, Poh KK. Endothelial progenitor cells in cardiovascular diseases. World J Stem Cells (2014) 6:355–66. doi: 10.4252/wjsc.v6.i3.355
128. Balistreri CR, Buffa S, Pisano C, Lio D, Ruvolo G, Mazzesi G. Are Endothelial Progenitor Cells the Real Solution for Cardiovascular Diseases? Focus on Controversies and Perspectives. BioMed Res Int (2015) 2015:835934. doi: 10.1155/2015/835934
129. Madonna R, De Caterina R. Circulating endothelial progenitor cells: Do they live up to their name? Vascul Pharmacol (2015) 67-69:2–5.doi: 10.1016/j.vph.2015.02.018
130. Hasan SS, Siekmann AF. The same but different: signaling pathways in control of endothelial cell migration. Curr Opin Cell Biol (2015) 36:86–92. doi: 10.1016/j.ceb.2015.07.009
131. Huang X, Dai Z, Cai L, Sun K, Cho J, Albertine KH, et al. Endothelial p110gammaPI3K Mediates Endothelial Regeneration and Vascular Repair After Inflammatory Vascular Injury. Circulation (2016) 133:1093–103. doi: 10.1161/CIRCULATIONAHA.115.020918
132. Zhao YD, Huang X, Yi F, Dai Z, Qian Z, Tiruppathi C, et al. Endothelial FoxM1 mediates bone marrow progenitor cell-induced vascular repair and resolution of inflammation following inflammatory lung injury. Stem Cells (2014) 32:1855–64. doi: 10.1002/stem.1690
133. Hou Y, Li C. Stem/Progenitor Cells and Their Therapeutic Application in Cardiovascular Disease. Front Cell Dev Biol (2018) 6:139.doi: 10.3389/fcell.2018.00139
134. Kong J, Wang F, Zhang J, Cui Y, Pan L, Zhang W, et al. Exosomes of Endothelial Progenitor Cells Inhibit Neointima Formation After Carotid Artery Injury. J Surg Res (2018) 232:398–407. doi: 10.1016/j.jss.2018.06.066
135. Zhou Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Chang E, et al. Exosomes from Endothelial Progenitor Cells Improve the Outcome of a Murine Model of Sepsis. Mol Ther (2018) 26:1375–84. doi: 10.1016/j.ymthe.2018.02.020
136. de la Puente P, Muz B, Azab F, Azab AK. Cell trafficking of endothelial progenitor cells in tumorprogression. Clin Cancer Res (2013) 19:3360–8. doi: 10.1158/1078-0432.CCR-13-0462
137. Sun R, Huang J, Sun B. Mobilization of endothelial progenitor cells in sepsis. Inflammation Res (2020) 69:1–9. doi: 10.1007/s00011-019-01299-9
138. Williams PA, Silva EA. The Role of Synthetic Extracellular Matrices in Endothelial Progenitor Cell Homing for Treatment of Vascular Disease. Ann BioMed Eng (2015) 43:2301–13. doi: 10.1007/s10439-015-1400-x
139. Shen L, Gao Y, Qian J, Sun A, Ge J. Anovel mechanism for endothelial progenitor cells homing: The SDF-1/CXCR4-Rac pathway may regulate endothelial progenitor cells homing through cellular polarization. Med Hypotheses (2011) 76:256–8. doi: 10.1016/j.mehy.2010.10.014
140. Avci-Adali M, Ziemer G, Wendel HP. Induction of EPC homing on biofunctionalized vascular grafts for rapid in vivo self-endothelialization–a review of current strategies. Biotechnol Adv (2010) 28:119–29. doi: 10.1016/j.biotechadv.2009.10.005
141. Chavakis E, Aicher A, Heeschen C, Sasaki K, Kaiser R, El MN, et al. Role of beta2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J Exp Med (2005) 201:63–72. doi: 10.1084/jem.20041402
142. Liu ZJ, Tian R, An W, Zhuge Y, Li Y, Shao H, et al. Identification of E-selectin as a novel target for the regulation of postnatal neovascularization: implications for diabetic wound healing. Ann Surg (2010) 252:625–34.
143. Kutikhin AG, Sinitsky MY, Yuzhalin AE, Velikanova EA. Shear stress: An essential driver of endothelial progenitor cells. J Mol Cell Cardiol (2018) 118:46–69. doi: 10.1016/j.yjmcc.2018.03.007
144. Zeng L, Xiao Q, Margariti A, Zhang Z, Zampetaki A, Patel S, et al. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J Cell Biol (2006) 174:1059–69. doi: 10.1083/jcb.200605113
145. Rössig L, Urbich C, Brühl T, Dernbach E, Heeschen C, Chavakis E, et al. Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J Exp Med (2005) 201:1825–35. doi: 10.1084/jem.20042097
146. Cheng BB, Yan ZQ, Yao QP, Shen BR, Wang JY, Gao LZ, et al. Association of SIRT1 expression with shear stress induced endothelial progenitor cell differentiation. J Cell Biochem (2012) 113:3663–71. doi: 10.1002/jcb.24239
147. Cheng M, Guan X, Li H, Cui X, Zhang X, Li X, et al. Shear stress regulates late EPC differentiation via mechanosensitive molecule-mediated cytoskeletal rearrangement. PloS One (2013) 8:e67675. doi: 10.1371/journal.pone.0067675
148. Obi S, Masuda H, Shizuno T, Sato A, Yamamoto K, Ando J, et al. Fluid shear stress induces differentiation of circulating phenotype endothelial progenitor cells. Am J Physiol Cell Physiol (2012) 303:C595–606. doi: 10.1152/ajpcell.00133.2012
149. Cui X, Zhang X, Guan X, Li H, Li X, Lu H, et al. Shear stress augments the endothelial cell differentiation marker expression in late EPCs by upregulating integrins. Biochem Biophys Res Commun (2012) 425:419–25. doi: 10.1016/j.bbrc.2012.07.115
150. Mohan S, Barsalou J, Bradley TJ, Slorach C, Reynolds JA, Hasni S, et al. Endothelial progenitor cell phenotype and function are impaired in childhood-onset systemic lupus erythematosus. Arthritis Rheumatol (2015) 67:2257–62. doi: 10.1002/art.39149
151. Kim JY, Park YJ, Kim KJ, Choi JJ, Kim WU, Cho CS. Osteoprotegerin causes apoptosis of endothelial progenitor cells by induction of oxidative stress. Arthritis Rheum (2013) 65:2172–82. doi: 10.1002/art.37997
152. Baker JF, Zhang L, Imadojemu S, Sharpe A, Patil S, Moore JS, et al. Circulating endothelial progenitor cells are reduced in SLE in the absence of coronary artery calcification. Rheumatol Int (2012) 32:997–1002. doi: 10.1007/s00296-010-1730-9
153. Moonen JR, de Leeuw K, van Seijen XJ, Kallenberg CG, van Luyn MJ, Bijl M, et al. Reduced number and impaired function of circulating progenitor cells in patients with systemic lupus erythematosus. Arthritis Res Ther (2007) 9:R84. doi: 10.1186/ar2283
154. Westerweel PE, Luijten RK, Hoefer IE, Koomans HA, Derksen RH, Verhaar MC. Haematopoietic and endothelial progenitor cells are deficient in quiescent systemic lupus erythematosus. Ann Rheum Dis (2007) 66:865–70. doi: 10.1136/ard.2006.065631
155. Yao G, Qi J, Zhang Z, Huang S, Geng L, Li W, et al. Endothelial cell injury is involved in atherosclerosis and lupus symptoms in gld.apoE(-) (/) (-) mice. Int J Rheum Dis (2019) 22:488–96. doi: 10.1111/1756-185X.13458
156. Ablin JN, Boguslavski V, Aloush V, Elkayam O, Paran D, Levartovski D, et al. Enhanced adhesive properties of endothelial progenitor cells (EPCs) in patients with SLE. Rheumatol Int (2011) 31:773–8. doi: 10.1007/s00296-010-1377-6
157. Deng XL, Li XX, Liu XY, Sun L, Liu R. Comparative study on circulating endothelial progenitor cells in systemic lupus erythematosus patients at active stage. Rheumatol Int (2010) 30:1429–36. doi: 10.1007/s00296-009-1156-4
158. Grisar J, Steiner CW, Bonelli M, Karonitsch T, Schwarzinger I, Weigel G, et al. Systemic lupus erythematosus patients exhibit functional deficiencies of endothelial progenitor cells. Rheumatol (Oxford) (2008) 47:1476–83. doi: 10.1093/rheumatology/ken286
159. Ebner P, Picard F, Richter J, Darrelmann E, Schneider M, Strauer BE, et al. Accumulation of VEGFR-2+/CD133+ cells and decreased number and impaired functionality of CD34+/VEGFR-2+ cells in patients with SLE. Rheumatol (Oxford) (2010) 49:63–72.doi: 10.1093/rheumatology/kep335
160. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
161. Hur J, Yang HM, Yoon CH, Lee CS, Park KW, Kim JH, et al. Identification of a novel role of T cells in postnatal vasculogenesis: characterization of endothelial progenitor cell colonies. Circulation (2007) 116:1671–82. doi: 10.1161/CIRCULATIONAHA.107.694778
162. Cavazzana I, Piantoni S, Sciatti E, Fredi M, Taraborelli M, Bonadei I, et al. Relationship between endothelial dysfunction, videocapillaroscopy and circulating CD3+CD31+CXCR4+ lymphocytes in systemic lupus erythematosus without cardiovascular risk factors. Lupus (2019) 28:210–6. doi: 10.1177/0961203318821161
163. Zhao P, Miao J, Zhang K, Lv M, Han Q, Zhu P. Circulating Angiogenic T Cells Are Increased in Lupus Nephritis Patients. Med Sci Monit (2018) 24:5384–90. doi: 10.12659/MSM.908406
164. Miao J, Qiu F, Li T, Zhao P, Zhang K, Lv M, et al. Circulating Angiogenic T Cells and Their Subpopulations in Patients with Systemic Lupus Erythematosus. Mediators Inflammation (2016) 2016:2842143. doi: 10.1155/2016/2842143
165. Lopez P, Rodriguez-Carrio J, Martinez-Zapico A, Caminal-Montero L, Suarez A. Senescent profile of angiogenic T cells from systemic lupus erythematosus patients. J Leukoc Biol (2016) 99:405–12. doi: 10.1189/jlb.5HI0215-042R
166. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol (2015) 15:471–85. doi: 10.1038/nri3865
167. Luo S, Wang Y, Zhao M, Lu Q. Theimportant roles of type I interferon and interferon-inducible genes in systemic lupus erythematosus. Int Immunopharmacol (2016) 40:542–9. doi: 10.1016/j.intimp.2016.10.012
168. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3:20r–73r. doi: 10.1126/scitranslmed.3001201
169. Leonard D, Eloranta ML, Hagberg N, Berggren O, Tandre K, Alm G, et al. Activated T cells enhance interferon-alpha production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes. Ann Rheum Dis (2016) 75:1728–34. doi: 10.1136/annrheumdis-2015-208055
170. Hagberg N, Berggren O, Leonard D, Weber G, Bryceson YT, Alm GV, et al. IFN-alpha production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1beta and LFA-1. J Immunol (2011) 186:5085–94. doi: 10.4049/jimmunol.1003349
171. Berggren O, Hagberg N, Weber G, Alm GV, Ronnblom L, Eloranta ML. B lymphocytes enhance interferon-alpha production by plasmacytoid dendritic cells. Arthritis Rheum (2012) 64:3409–19. doi: 10.1002/art.34599
172. Ward JM, Ratliff ML, Dozmorov MG, Wiley G, Guthridge JM, Gaffney PM, et al. Human effector B lymphocytes express ARID3a and secrete interferon alpha. J Autoimmun (2016) 75:130–40. doi: 10.1016/j.jaut.2016.08.003
173. Domeier PP, Chodisetti SB, Schell SL, Kawasawa YI, Fasnacht MJ, Soni C, et al. B-Cell-Intrinsic Type 1 Interferon Signaling Is Crucial for Loss of Tolerance and the Development of Autoreactive B Cells. Cell Rep (2018) 24:406–18. doi: 10.1016/j.celrep.2018.06.046
174. O’Neill LA. Immunology. Sensing the dark side of DNA. Science (2013) 339:763–4. doi: 10.1126/science.1234724
175. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity (2015) 42:1171–84. doi: 10.1016/j.immuni.2015.06.002
176. Tursi SA, Lee EY, Medeiros NJ, Lee MH, Nicastro LK, Buttaro B, et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PloS Pathog (2017) 13:e1006315. doi: 10.1371/journal.ppat.1006315
177. Shao WH, Shu DH, Zhen Y, Hilliard B, Priest SO, Cesaroni M, et al. Prion-like Aggregation of Mitochondrial Antiviral Signaling Protein in Lupus Patients Is Associated With Increased Levels of Type I Interferon. Arthritis Rheumatol (2016) 68:2697–707. doi: 10.1002/art.39733
178. Lou H, Pickering MC. Extracellular DNA and autoimmune diseases. Cell Mol Immunol (2018) 15:746–55. doi: 10.1038/cmi.2017.136
179. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol (2014) 35:88–93. doi: 10.1016/j.it.2013.10.010
180. Savarese E, Chae OW, Trowitzsch S, Weber G, Kastner B, Akira S, et al. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood (2006) 107:3229–34. doi: 10.1182/blood-2005-07-2650
181. Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med (2005) 202:1575–85. doi: 10.1084/jem.20051696
182. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol (2011) 11:389–402. doi: 10.1038/nri2975
183. Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev (2015) 26:311–27. doi: 10.1016/j.cytogfr.2014.11.009
184. Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol (2008) 28:1928–36. doi: 10.1161/ATVBAHA.108.162925
185. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res (2011) 317:620–31. doi: 10.1016/j.yexcr.2010.12.017
186. Ehlert JE, Addison CA, Burdick MD, Kunkel SL, Strieter RM. Identification and partial characterization of a variant of human CXCR3 generated by posttranscriptional exon skipping. J Immunol (2004) 173:6234–40. doi: 10.4049/jimmunol.173.10.6234
187. Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med (2003) 197:1537–49. doi: 10.1084/jem.20021897
188. Romagnani P, Annunziato F, Lasagni L, Lazzeri E, Beltrame C, Francalanci M, et al. Cell cycle-dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. J Clin Invest (2001) 107:53–63. doi: 10.1172/JCI9775
189. Salcedo R, Resau JH, Halverson D, Hudson EA, Dambach M, Powell D, et al. Differential expression and responsiveness of chemokine receptors (CXCR1-3) by human microvascular endothelial cells and umbilical vein endothelial cells. FASEB J (2000) 14:2055–64. doi: 10.1096/fj.99-0963com
190. Datta D, Contreras AG, Grimm M, Waaga-Gasser AM, Briscoe DM, Pal S. Calcineurin inhibitors modulate CXCR3 splice variant expression and mediate renal cancer progression. J Am Soc Nephrol (2008) 19:2437–46. doi: 10.1681/ASN.2008040394
191. Kim S, Bakre M, Yin H, Varner JA. Inhibition of endothelial cell survival and angiogenesis by protein kinase A. J Clin Invest (2002) 110:933–41. doi: 10.1172/JCI0214268
Keywords: atherosclerosis, endothelial cell, endothelial progenitor cell, pathogenesis, systemic lupus erythematosus, IFN-I
Citation: Ding X, Xiang W and He X (2020) IFN-I Mediates Dysfunction of Endothelial Progenitor Cells in Atherosclerosis of Systemic Lupus Erythematosus. Front. Immunol. 11:581385. doi: 10.3389/fimmu.2020.581385
Received: 08 July 2020; Accepted: 14 October 2020;
Published: 11 November 2020.
Edited by:
Joshua Daniel Ooi, Monash University, AustraliaReviewed by:
Genhong Yao, Nanjing Drum Tower Hospital, ChinaMicaela Fredi, Civil Hospital of Brescia, Italy
Copyright © 2020 Ding, Xiang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojie He, aGV4ajcxNTBAMTYzLmNvbQ==