 Daniella Bianchi Reis Insuela1†
Daniella Bianchi Reis Insuela1† Maximiliano Ruben Ferrero1†
Maximiliano Ruben Ferrero1† Diego de Sá Coutinho1
Diego de Sá Coutinho1 Marco Aurélio Martins1
Marco Aurélio Martins1 Vinicius Frias Carvalho1,2*
Vinicius Frias Carvalho1,2*- 1Laboratory of Inflammation, Oswaldo Cruz Institute, Oswaldo Cruz Foundation (FIOCRUZ), Rio de Janeiro, Brazil
- 2Laboratory of Inflammation, National Institute of Science and Technology on Neuroimmunomodulation (INCT-NIM), Rio de Janeiro, Brazil
Asthma represents one of the leading chronic diseases worldwide and causes a high global burden of death and disability. In asthmatic patients, the exacerbation and chronification of the inflammatory response are often related to a failure in the resolution phase of inflammation. We reviewed the role of the main arachidonic acid (AA) specialized pro-resolving mediators (SPMs) in the resolution of chronic lung inflammation of asthmatics. AA is metabolized by two classes of enzymes, cyclooxygenases (COX), which produce prostaglandins (PGs) and thromboxanes, and lypoxygenases (LOX), which form leukotrienes and lipoxins (LXs). In asthma, two primary pro-resolving derived mediators from COXs are PGE2 and the cyclopentenone prostaglandin15-Deoxy-Delta-12,14-PGJ2 (15d-PGJ2) while from LOXs are the LXA4 and LXB4. In different models of asthma, PGE2, 15d-PGJ2, and LXs reduced lung inflammation and remodeling. Furthermore, these SPMs inhibited chemotaxis and function of several inflammatory cells involved in asthma pathogenesis, such as eosinophils, and presented an antiremodeling effect in airway epithelial, smooth muscle cells and fibroblasts in vitro. In addition, PGE2, 15d-PGJ2, and LXs are all able to induce macrophage reprogramming to an alternative M2 pro-resolving phenotype in vitro and in vivo. Although PGE2 and LXA4 showed some beneficial effects in asthmatic patients, there are limitations to their clinical use, since PGE2 caused side effects, while LXA4 presented low stability. Therefore, despite the strong evidence that these AA-derived SPMs induce resolution of both inflammatory response and tissue remodeling in asthma, safer and more stable analogs must be developed for further clinical investigation of their application in asthma treatment.
Introduction
Asthma is a high prevalence chronic inflammatory pulmonary disease, the respiratory symptoms of which include cough, wheezing, shortness of breath, and chest tightness which leads to elevated morbidity, mortality, and disease social and economic costs (1–3). Pulmonary inflammation is the hallmark of asthma, which is driven by a Th2 immune reponse to inhaled allergens, and associated with infiltration of the bronchial mucosa with eosinophils, CD4+ T cells, macrophages and, in exacerbations and severe cases, neutrophils (4, 5). Macrophages are classified into classical (M1) or alternative activation (M2a, M2b, M2c, or M2d subtypes). During allergic asthma, under exposure to Th2 cytokines (IL-4 and IL-13), macrophages are reprogrammed to M2a profile and perform diverse functions ranging between protective and pathogenic roles (6–9). Airway remodelling is another key feature of asthma pathogenesis and can precede the development of inflammation (10). It is characterized by mucous gland and airway smooth muscle (ASM) cells hyperplasia and/or hypertrophy, deposition of extracellular matrix (EM) proteins, and myofibroblast proliferation, leading to the thickening and occlusions of airways (11). In severe asthmatic patients, a failure in pro-resolving pathways extends the pro-inflammatory mechanisms, resulting in a chronic inflammation, which is associated with a major cause of admission to the intensive care unit and high mortality rates (12, 13). Lipid mediators, such as those originated by arachidonic acid (AA), are key factors of the resolution of inflammation, once they orchestrate the clearance of pro-inflammatory cells and signals promoting tissue restoration (13, 14). In this review, we discussed the impact of AA-derived specialized pro-resolving mediators (SPMs) in the resolution of inflammation and remodeling in asthma.
Resolution of Inflammation
The resolution of inflammation is an active and controlled process that reduces inflammation through the elimination of danger signals, leading to the restoration of tissue homeostasis and preventing the progression towards an uncontrolled chronic inflammatory state. Thus, catabolization and antagonization of pro-inflammatory mediators, a decrease in leukocyte numbers at inflammatory sites, and tissue repair are key events in the resolution process (13). It is notewothy that different from classical anti‐inflammatory molecules, the SPMs modulate the end of the inflammatory response, without promoting unwanted immunosuppression (15). During the resolution phase, leukocyte apoptosis and metabolization of intracellular inflammatory signals lead to the clearance of inflammatory cells by specialized phagocytes. Together, these events promote the end of the acute inflammatory response and initiate tissue repair and healing (12, 16).
Endogenous mediators that actively participate in the resolution process include lipids (i.e., lipoxins, resolvins, maresin, and protectins), peptides (i.e. alpha-melanocortin-stimulating hormone and chemerin), proteins (i.e., annexin A1, Galectin-1, TGF-β and IL-10), and nucleotides (i.e. adenosine and inosine) (17–19). They promote cessation of polymorphonuclear infiltration into the inflamed tissue, reprogramming of macrophages and TCD4+ cells to M2 and T regulatory phenotypes respectively, sequestration and counter-regulation of pro-inflammatory mediators, apoptosis of polymorphonuclear cells with subsequent phagocytosis by M2 macrophages, and tissue repair (20–22).
Among the SPMs, the lipid mediators activate many aspects of the resolution process (23). These endogenous mediators are biosynthesized in local inflamed tissue microenvironments, and can control the magnitude/duration of the inflammatory response as well as the timing of tissue restoration (17). They primarily come from the metabolism of polyunsaturated fatty acids, such as AA, docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), and docosapentaenoic acid (DPA) (15). Interestingly, several AA-derived mediators have consistently presented pro-resolving and tissue protecting activities in asthma (24, 25).
SPMs Derived From Cyclooxygenases
Cyclooxygenases (COX), especially COX-2 isoform, play a pivotal role in the conversion of AA into different pro-inflammatory lipid mediators, including prostaglandins (PG) and thromboxanes (26). Despite the clear ligation of COX-2 activity with the development of the inflammatory response, it has also been proved that the inhibition of this enzyme impairs leukocyte clearance, indicating that some COX-2 derived mediators possess pro-resolving action. This occurs mainly due to the ability of COX-2 to metabolize EPA into resolvins, which are one of the main classes of SPMs (27). In asthma, beyond resolvins, the COX-2 activity also culminates in the formation of other important SPMs, such as PGE2 and 15-Deoxy-Delta-12,14-PGJ2 (15d-PGJ2), a metabolite of PGD2 (28).
PGE2 is synthesized by three distinct enzymes, microsomal PGE synthase-1 (mPGES-1), mPGES-2, and cytosolic PGES (cPGES), which use PGH2 as substrate. The actions of PGE2 are mediated by four distinct 7TM receptors (EP1–EP4) (29). Although PGE2 is a pro-inflammatory mediator, several works have shown that this lipid presents pro-resolving actions in some contexts (30). So, what determines when PGE2 presents pro-resolving effects? There are three major factors, not mutually exclusive: i) time: the kinetics of PGE2 release can separate its pro-inflammatory and pro-resolving effects due to the presence of different targets (31, 32); ii) context: eg. PGE2 can inhibit ERK activation and MMP-1 secretion by gastric epithelial cells in the presence of cytokines, however, in their absence, PGE2 does the opposite (31, 33, 34); iii) concentration: eg. very low PGE2 concentrations inhibit chondrocyte-dependent collagen cleavage in osteoarthritis cartilage, while higher concentrations enhance it (35).
IL-4 and IL-13, essential cytokines in the asthma pathogenesis (4), suppressed PGE2 production by dendritic cells through reduction of COX-2 and mPGES-1 expression (36). Also, asthmatic patients presented an inverse correlation between the sputum levels of PGE2 and eosinophil numbers (37, 38), suggesting that PGE2 may reduce airway eosinophilia in these patients. Inhaled PGE2 markedly inhibits the early and late bronchoconstrictor response to an allergen in asthmatic patients (39); however, these effects may be related only to the PGE2-induced ASM relaxation (40). Nevertheless, COX-1 knock out (KO) and EP2KO mice that were ovalbumin (OVA)-sensitized and challenged showed increased eosinophilia and Th2 cytokines levels in the lungs and bronchoalveolar lavage (BAL), respectively, compared to wild-type (WT) mice (41, 42). Besides, treatment with PGE2 inhibited the house dust mite (HDM)-induced lung eosinophilia (43), and OVA-provoked accumulation of eosinophils and Th2 cytokines in the BAL (42), probably because PGE2 can inhibit β2 integrin and L-selectin function with a consequent reduction in eosinophil migration (44, 45). Furthermore, prior studies of our group showed that PGE2 derived from eosinophils induced an early resolution of allergic pleural edema (25, 46).
Until now, there has been no agreement on the effects of PGE2 on the differentiation of naïve T cells to Th1, Th2, or Th17 (47); however, type 2 innate lymphoid cells (ILC2), that emerged in the literature as novel Th2 cytokine-producing cells, strongly express both EP2 and EP4. PGE2 inhibited proliferation, activation, and release of cytokines by ILC2 (48, 49). Besides, alveolar macrophages from asthmatics presented a reduction in the EP2 expression (50) and PGE2 generation, in parallel with decreased efferocytosis of apoptotic cells (51). PGE2 is a well-known inductor of M2 macrophage reprogramming (52). Furthermore, PGE2 induced IL-10 production by macrophages in vitro, and the adoptive transfer of those PGE2‐treated macrophages led to fewer infiltrating eosinophils, macrophages, activated TCD4+, and regulatory T lymphocytes in lungs of HDM‐exposed mice (43).
In lung fibroblasts, there is an inverse relationship between COX-2 and mPGES-1 expression and the number of allergen challenges, resulting in a reduction in PGE2 production by those cells (53). Besides, mPGES-1 KO mice showed an augmentation of allergen-induced vascular smooth muscle cell numbers and thickness of intrapulmonary vessels (54). PGE2 also inhibited fibroblast migration, proliferation, collagen deposition, and myofibroblast differentiation in the lung (55). In vitro, PGE2 decreased the expression of tenacin C and fibronectin by human fibroblast and ASM cells (56), reduced the proliferation of ASM cells derived from asthmatic patients (57), and upregulated the expression of the anti-inflammatory protein tristetraprolin in human ASM cells (58). Prior investigations of our group revealed that the instillation of glucagon induced a high production of PGE2 into the lungs (59). Also, we reported that a non-selective COX inhibitor decreased the inhibitory effect of glucagon on OVA-induced collagen deposition in the lungs (60), suggesting that the anti-remodeling effect of glucagon depends on PGE2 production. Interestingly, inhaled PGE2 showed bronchodilator capacity in small clinical trials with asthmatic patients (61, 62) (Table 1).
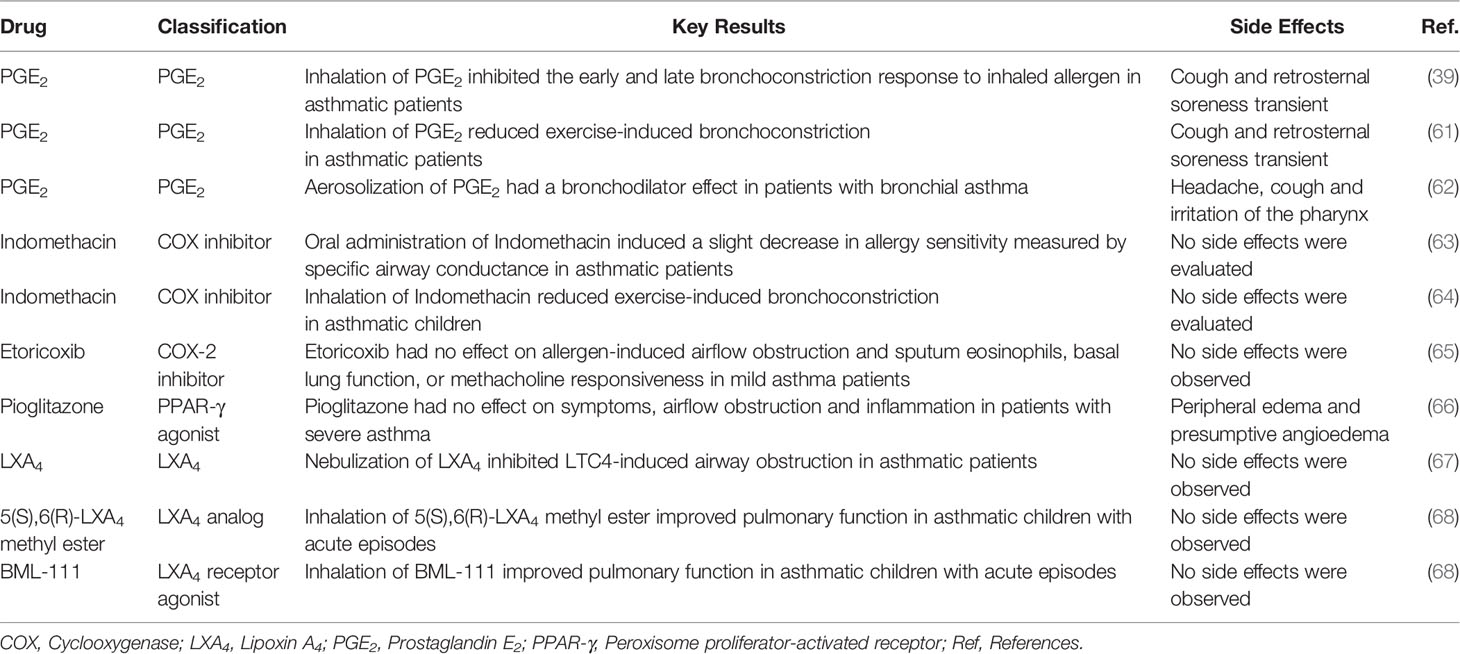
Table 1 Summary of clinical studies using mediators related to arachidonic acid metabolism pathways in asthma.
Despite the possible benefits of PGE2 in asthma, non-selective COX inhibitors improved specific airway conductance and airway constriction of asthmatics (63, 64), suggesting that COX-derivatives may play a role in the development or worsening of asthma. Nevertheless, patients with mild allergic asthma treated with specific COX-2 inhibitors did not present an effect on lung function and eosinophil accumulation in the sputum (65) (Table 1). Although PGE2 acts directly in the resolution of inflammation, it can also drive a pro-inflammatory response in human fibroblast and ASM cells (56). Furthermore, PGE2 apparently desensitized β2 adrenergic receptors during asthma exacerbation triggered by Rhinovirus infection (69). A high dose of PGE2 can also induce airway contraction in asthmatic patients, probably through activating different receptors (70), and cough by activation of EP3 receptor (71). As the most of pro-resolving actions of PGE2 are related to the activation of EP2, the development of selective agonists of this receptor can be a good strategy to be consider for treating asthma in the future.
15d-PGJ2 is formed spontaneously by a series of dehydration of PGD2 (72), and it is produced abundantly in the inflamed site, making it important in the resolution of the inflammation (73). Most of the pro-resolving actions of 15d-PGJ2 depend on the peroxisome proliferator-activated receptor-gamma (PPARγ) activation, but some of its effects are independent of this receptor (74). In asthmatic patients, there is a reduction in the PPARγ expression in BAL cells (75). Furthermore, polymorphism of the PPARG gene may be related to an increased risk of asthma development (76). Activation of PPARγ by synthetic agonists reduced the levels of Th2 cytokines and inhibited AHR, the influx of eosinophils and structural changes in the airway wall in murine OVA-challenge models of asthma (77, 78). Together, these data indicate that the reduction in PPARγ expression by inflammatory cells in asthmatic patients may be one of the mechanisms that contribute to the development of chronic asthma.
In a model of carrageenin-induced pleurisy, 48h after the provocation, when mononuclear cells dominate the reaction up to the resolution, there was an immense increase in COX-2 protein expression and 15d-PGJ2 levels coincident with inflammatory resolution and associated with minimal exudate PGE2 levels. In this model, the use of both nonselective or selective COX-2 inhibitors, 24h after carrageenin challenge, increased the number of inflammatory cells and exudate volume in parallel to a reduction in the 15d-PGJ2 levels. In addition, 15d-PGJ2 reversed the selective-COX-2 inhibitor-induced rise in cell number and exudate volume, indicating that the production of 15d-PGJ2 is important to the resolution in this model (79). The pro-resolving effect of 15d-PGJ2 was related to an induction of apoptosis of inflammatory cells (80). Besides, 15d-PGJ2 also regulates the balance of cytokines and chemokines that control leukocyte trafficking during acute inflammation, promotes M2 macrophage differentiation, as well as the efflux of macrophage to draining lymphatics, facilitating the resolution of inflammation (81). This pro-resolving effect of 15d-PGJ2 may be dependent on PPARγ, once IL-4-induced PPARγ activity becomes indispensible for M2 activation (82, 83).
In an OVA-induced asthma model, KO mice for PGD synthase (H-PGDS), an enzyme that catalyzes PGH2 into PGD2, showed accelerated chronic allergic lung eosinophil inflammation in parallel to an increase in the local levels of TNFα and eotaxin-1. Furthermore, the exogenous administration of 15d-PGJ2 decreased the excessive eosinophilic infiltration and TNFα and eotaxin-1 levels noted in those mice (84). Furthermore, the activation of PPARγ reduced OVA-induced eosinophilia and IL-4, IL-5, and IL-6 levels in the lungs of mice (85). We previously showed that interventional treatment with 15d-PGJ2 inhibited both OVA- and HDM-induced eosinophils accumulation and IL-5 and IL-13 levels in the lungs (86). The pro-resolving effect of 15d-PGJ2 on lung eosinophilia is probably related to its ability to block the traffic and induce apoptosis of these granulocytes (87). The inhibitory effect of 15d-PGJ2 on eosinophil migration is possibly dependent on PPARγ, once the activation of this receptor by synthetic agonists inhibits chemotaxis of eosinophils (85). However, the pro-apoptotic effect of 15d-PGJ2 is independent of PPARγ (87). 15d-PGJ2 also inhibited T lymphocyte proliferation in a mechanism probably dependent on PPARγ, as it is mimicked by PPARγ synthetic agonists (88, 89).
We previously demonstrated that interventional treatment with 15d-PGJ2 reversed structural changes related to airway remodeling, including epithelial thickening, mucus exacerbation, and EM deposition, in both OVA and HDM murine models of asthma (86). These antiremodeling effects of 15d-PGJ2 may be related to its ability to reduce differentiation of fibroblasts into myofibroblasts, the proliferation of myofibroblasts (90), and fibroblast growth factor-induced human ASM cell proliferation (91). Although PPARγ agonists are extremely promising to asthma therapy, unfortunately severe asthmatic patients treated with pioglitazone did not present with an improvement in asthma features and showed significant side effects (66) (Table 1).
SPMs Derived From Lipoxygenases
5- Lipoxygenase (LOX) and 15-LOX are the main LOXs involved in the metabolization of AA (92), resulting in the formation leukotrienes (LTs) and lipoxins (LXs). While LTs are recognized to exert broad proinflammatory effects, LXs present pro-resolving actions (93). Endogenously, LXs are typically produced by three main pathways. In one route, LXA4 and LXB4 are produced by 5-LOX (94), and in other by 12-LOX (95). It is described that aspirin treatment can also promote the synthesis of LXs epimers denominated aspirin-triggered lipoxins (ATLs), including 15-epimeric (epi)-LXA4 and 15-epi-LXB4 (94). LXA4 and ATLs act primarily on a 7TMN receptor denominated ALXR (96). ALXR is expressed in several tissues, including lungs, and different cell types such as leukocytes, fibroblasts, and bronchial epithelial cells. LXA4 can also activate the aryl hydrocarbon receptor, and both LXA4 and ATLs are antagonists of the cysteinyl leukotriene receptor 1 (97). Nevertheless, the LXB4 receptor has not yet been identified (98).
The failure in the generation and action of LXs is associated with more severe airway inflammation (99). Indeed, severely asthmatic patients presented a reduction of LXA4 levels in BAL fluid, sputum, and whole blood compared to moderately asthmatic individuals. This reduction in LXA4 concentrations observed in severe asthma was associated with a higher degree of airway obstruction (24). Eosinophils from the blood of asthmatic patients presented a decreased ALXR expression compared to those obtained from healthy individuals (100). Furthermore, transgenic mice that overexpress ALXR showed a reduction in OVA-induced eosinophilia in the BAL and lung tissue (101). We previously showed that 15-epi-LXA4 analogs inhibited OVA-induced pleural eosinophil influx by reducting local eotaxin and IL-5 generation (102). We also noted that 15-epi-LXA4 analogs accelerate the drainage of OVA-induced pleural edema (25). In human eosinophils, LXA4 inhibited chemotaxis toward chemoattractants (103), and granulocyte-macrophage colony-stimulating factor-induced IL-13 and eotaxin release in vitro (104). In spite of inhibiting eosinophil migration, 15-epi-LXA4 is a potent chemoattractant to monocytes in vitro (105) and restored the balance between M2 and M1 populations into the lungs in a murine model of pulmonary damage induced by bleomycin (106). Furthermore, LXA4 stimulates macrophage efferocytosis of apoptotic polymorphonuclear cells and cellular debris (107).
Among the ILC family, natural killer (NK) cells and ILC2s are important in the control and exacerbation of asthma, respectively. NK cell depletion induced a persistent allergic airway inflammation in association with reduction of the LXA4 levels in the BAL (108). LXA4 enhanced activated NK cells-induced eosinophil apoptosis through ALXR activation (109). Meanwhile, the blood and sputum of patients with severe asthma presented elevated numbers of ILC2 compared to mild asthmatics, which was related to persistent airway eosinophilia (110). LXA4 inhibited both PGD2- and IL-25 plus IL-33-induced IL-13 release by ILC2 in vitro in a mechanism dependent on ALXR activation (109).
In vitro, LXA4 and 15-epi-LXA4 reduced IL-8 secretion induced by serum amyloid A in a human alveolar A549 cell line (111). Also, activation of ALXR by LXA4 increased basal proliferation and wound repair of human airway epithelial cells (112). In a murine model of asthma caused by OVA, airway epithelial cells presented with an increased expression of ALXR (101) and LXB4 reduced mucus production (113). ASM hypertrophy and hyperplasia, as well as accumulation of muscle cells in the subepithelial layer, are some of the changes observed in asthma remodeling. It was shown that LXA4 reduced both LTE4- and IL-13–primed ASM migration toward platelet-derived growth factor in vitro (114). Another critical pathological feature of airway remodeling in asthma is the EM deposition in the peribronchiolar area. It is noteworthy that both fibroblasts and myofibroblasts can express ALXR (115). Moreover, LXA4 inhibited connective tissue growth factor-induced human lung fibroblast proliferation in vitro (115) and blocked TGF-β-triggered increase in α-smooth muscle actin expression and collagen release by human myofibroblasts in vitro (116). Besides, treatment with 15-epi-LXA4 reversed bleomycin-promoted fibrosis and lung damage in mice (106). Altogether, these data suggest a potential role of LXs in the resolution of the airway and peribronchiolar remodeling observed in asthmatics.
Due to the possible therapeutic application of LXA4, some clinical trials using this LX, its analogues, or LXA4 receptor agonist BML-111 were administered in asthmatic patients. The nebulization of LXA4 reduced LTC4-induced bronchoconstriction (67); however, the rapid inactivation and significant instability to exposure to light and acids of LXA4 (117) make its clinical use difficult. Furthermore, the inhalation of LXA4 analog or BML-111, which is more potent and stable than LXA4 itself (118), improved the lung function (68) (Table 1). Interestingly, both LXA4 analog and BML-111 were well tolerated and presented no side effects (68).
Conclusion
This mini-review presents several aspects of the pro-resolving effects of COX- and LOX-derivative mediators in asthma (Figure 1), addressing their efficacy and current limitations for clinical use. Nevertheless, the review presents several strong pieces of evidence that support the development of new drugs based on analogs of PGE2, 15d-PGJ2, and LXs with better physical-chemical properties, allowing greater stability and superior selectivity for specific receptors. Moreover, new analogs of AA-derived SPMs could also improve efficiency and reduce the required dose of glucocorticoid, the latter often leading to adverse effects and steroid-refractoriness, despite being the best asthma treatment so far.
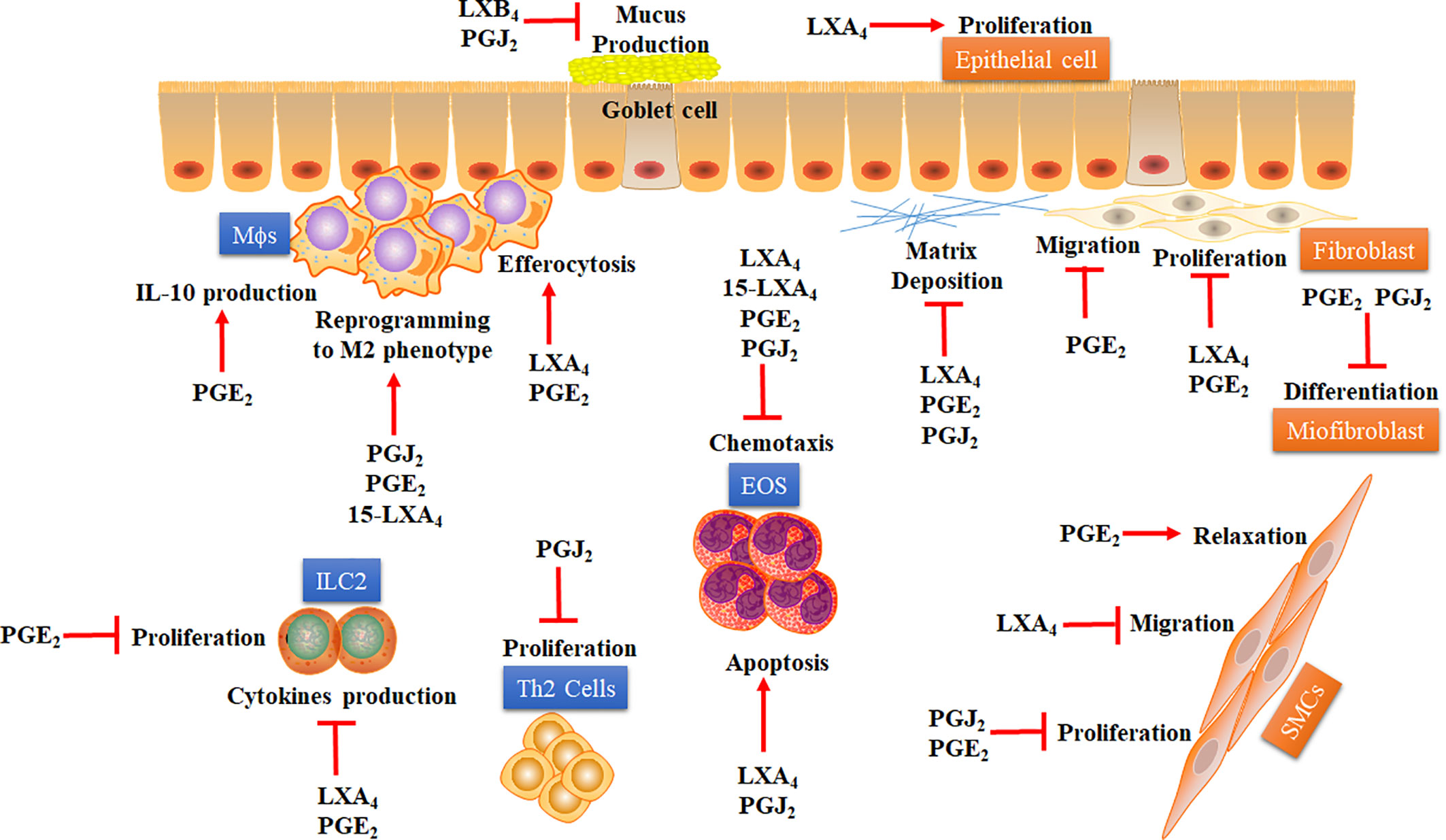
Figure 1 Proposed mechanisms whereby COX- and LOX-derived lipid mediators may accelerate the resolution of lung inflammation in asthma. Some COX- and LOX-derived lipid mediators, including PGE2, 15dPGJ2, LXA4, and LXB4, have demonstrated several pro-resolving actions over immune cells (blue squares) and structural cells (orange squares) involved in asthma. Pro-resolving effects of COX- and LOX-derived lipid mediators are: i) inhibition of EOS chemotaxis and stimulation of apoptosis on those cells; ii) inhibition of ILC-2 proliferation and cytokine production; iii) inhibition of Th2 lymphocytes proliferation; iv) stimulation of efferocytosis and IL-10 production by MΦs; v) induction of macrophage reprogramming to alternative M2 phenotype. Besides, these SPMs derived from COX and LOX present some important antiremodeling effects in asthma, like: i) inhibition of mucus production by globet cells and stimulation of airway epithelial cells proliferation; ii) inhibition of proliferation and migration of SMCs and stimulation of relaxation of these cells; iii) inhibition of proliferation, migration, and extracellular matrix deposition by fibroblasts; iv) inhibition of fibroblast differentiation into myofibroblasts. EOS: Eosinophil. ILC-2: Type-2 innate lymphoid cells. LXA4: Lipoxin A4. 15-LXA4: 15-epimeric (epi)-LXA4. LXB4: Lipoxin B4. MΦs: Macrophages. M2: M2 macrophage phenotype. PGE2: Prostaglandin E2. PGJ2: 15-Deoxy-Delta-12,14-PGJ2. SMCs: Smooth muscle cells. Th2: Type-2 CD4+ T helper. The arrow represents stimulation while the flat arrow represents inhibition.
Author Contributions
DI, MF, and DC contributed to the conception and design of the study, wrote the manuscript, discussed the content, and contributed to the manuscript revision. MM discussed the content and contributed to the manuscript revision. VC contributed to the conception and design of the study, wrote the manuscript, discussed the content, and contributed to the manuscript revision. All authors reviewed and/or edited the manuscript prior submission. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by PrInt Fiocruz-CAPES Program N° 01/2020.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank PrInt Fiocruz-CAPES Program; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Programa Fiocruz de Fomento a Inovação (INOVA-FIOCRUZ), and Fundação Carlos Chagas de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) for support.
Abbreviations
AA, arachidonic acid; AHR, airway hyperreactivity; ASM, airway smooth muscle; ATLs, aspirin-triggered-lipoxins; BAL, bronchoalveolar lavage; COX, cyclooxygenase; cPGES, cytosolic PGE synthase; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; 15d-PGJ2, 15-deoxy-delta-12,14-PGJ2; EM, extracellular matrix; EPA, eicosapentaenoic acid; Epi, epimeric; HDM, house dust mite; H-PGDS, PGD synthase; ILC2, type 2 innate lymphoid cells; KO, knock out; LOXs, lipoxygenases; LTs, leukotrienes; LXs, lipoxins; mPGES-1, microsomal PGE synthase-1; NK, natural killer; OVA, ovalbumin; PG, prostaglandin; PPAR-γ, peroxisome proliferator-activated receptor; SPM, specialized pro-resolving mediator; WT, wild-type.
References
1. Perez-Garcia J, Espuela-Ortiz A, Lorenzo-Diaz F, Pino-Yanes M. Pharmacogenetics of Pediatric Asthma: Current Perspectives. Pharmgenom Pers Med (2020) 13:89–103. doi: 10.2147/PGPM.S201276
2. Ramadan WH, Sarkis A, Aderian SS, Milane A. Asthma and COPD Patients’ Perception of Appropriate Metered-Dose Inhaler Technique. Dose Response (2020) 18(2):1559325820917832–. doi: 10.1177/1559325820917832
3. Hough KP, Curtiss ML, Blain TJ, Liu R-M, Trevor J, Deshane JS, et al. Airway Remodeling in Asthma. Front Med (Lausanne) (2020) 7:191. doi: 10.3389/fmed.2020.00191
4. Barnes PJ. Cellular and molecular mechanisms of asthma and COPD. Clin Sci (Lond) (2017) 131(13):1541–58. doi: 10.1042/CS20160487
5. Lahousse L, Bahmer T, Cuevas-Ocana S, Flajolet P, Mathioudakis AG, McDonnell M, et al. ERS International Congress, Madrid, 2019: highlights from the Airway Diseases, Asthma and COPD Assembly. ERJ Open Res (2020) 6(1):1–14. doi: 10.1183/23120541.00341-2019
6. Girodet PO, Nguyen D, Mancini JD, Hundal M, Zhou X, Israel E, et al. Alternative Macrophage Activation Is Increased in Asthma. Am J Respir Cell Mol Biol (2016) 55(4):467–75. doi: 10.1165/rcmb.2015-0295OC
7. Abdelaziz MH, Abdelwahab SF, Wan J, Cai W, Huixuan W, Jianjun C, et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J Transl Med (2020) 18(1):58. doi: 10.1186/s12967-020-02251-w
8. Moreira AP, Cavassani KA, Hullinger R, Rosada RS, Fong DJ, Murray L, et al. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J Allergy Clin Immunol (2010) 126(4):712–21 e7. doi: 10.1016/j.jaci.2010.06.010
9. van der Veen TA, de Groot LES, Melgert BN. The different faces of the macrophage in asthma. Curr Opin Pulm Med (2020) 26(1):62–8. doi: 10.1097/MCP.0000000000000647
10. Lezmi G, Gosset P, Deschildre A, Abou-Taam R, Mahut B, Beydon N, et al. Airway Remodeling in Preschool Children with Severe Recurrent Wheeze. Am J Respir Crit Care Med (2015) 192(2):164–71. doi: 10.1164/rccm.201411-1958OC
11. Lloyd CM, Robinson DS. Allergen-induced airway remodelling. Eur Respir J (2007) 29(5):1020–32. doi: 10.1183/09031936.00150305
12. Barnig C, Bezema T, Calder PC, Charloux A, Frossard N, Garssen J, et al. Activation of Resolution Pathways to Prevent and Fight Chronic Inflammation: Lessons From Asthma and Inflammatory Bowel Disease. Front Immunol (2019) 10:1699. doi: 10.3389/fimmu.2019.01699
13. Schett G, Neurath MF. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun (2018) 9(1):3261. doi: 10.1038/s41467-018-05800-6
14. Kytikova O, Novgorodtseva T, Denisenko Y, Antonyuk M, Gvozdenko T. Pro-Resolving Lipid Mediators in the Pathophysiology of Asthma. Med (Kaunas) (2019) 55(6):1–12. doi: 10.3390/medicina55060284
15. Romano M, Patruno S, Pomilio A, Recchiuti A. Proresolving Lipid Mediators and Receptors in Stem Cell Biology: Concise Review. Stem Cells Transl Med (2019) 8(10):992–8. doi: 10.1002/sctm.19-0078
16. Rahtes A, Geng S, Lee C, Li L. Cellular and molecular mechanisms involved in the resolution of innate leukocyte inflammation. J Leukoc Biol (2018) 104(3):535–41. doi: 10.1002/JLB.3MA0218-070R
17. Fredman G, Tabas I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am J Pathol (2017) 187(6):1211–21. doi: 10.1016/j.ajpath.2017.01.018
18. Cash JL, Norling LV, Perretti M. Resolution of inflammation: targeting GPCRs that interact with lipids and peptides. Drug Discovery Today (2014) 19(8):1186–92. doi: 10.1016/j.drudis.2014.06.023
19. Cash JL, Bass MD, Campbell J, Barnes M, Kubes P, Martin P. Resolution Mediator Chemerin15 Reprograms the Wound Microenvironment to Promote Repair and Reduce Scarring. Curr Biol (2014) 24(12):1435. doi: 10.1016/j.cub.2014.06.010
20. Sendama W. The effect of ageing on the resolution of inflammation. Ageing Res Rev (2020) 57:101000. doi: 10.1016/j.arr.2019.101000
21. Schett G. Resolution of inflammation in arthritis. Semin Immunopathol (2019) 41(6):675–9. doi: 10.1007/s00281-019-00768-x
22. Recchiuti A, Mattoscio D, Isopi E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front Pharmacol (2019) 10:252:252. doi: 10.3389/fphar.2019.00252
23. Gilroy DW, Bishop-Bailey D. Lipid mediators in immune regulation and resolution. Br J Pharmacol (2019) 176(8):1009–23. doi: 10.1111/bph.14587
24. Kazani S, Planaguma A, Ono E, Bonini M, Zahid M, Marigowda G, et al. Exhaled breath condensate eicosanoid levels associate with asthma and its severity. J Allergy Clin Immunol (2013) 132(3):547–53. doi: 10.1016/j.jaci.2013.01.058
25. Bandeira-Melo C, Serra MF, Diaz BL, Cordeiro RS, Silva PM, Lenzi HL, et al. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats: relationship with concurrent eosinophilia. J Immunol (2000) 164(2):1029–36. doi: 10.4049/jimmunol.164.2.1029
26. Lee K, Lee SH, Kim TH. The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy. Int J Mol Sci (2020) 21(5):1–26. doi: 10.3390/ijms21051851
27. Kohli P, Levy BD. Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol (2009) 158(4):960–71. doi: 10.1111/j.1476-5381.2009.00290.x
28. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol (2016) 7:160:160. doi: 10.3389/fimmu.2016.00160
29. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449
30. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol (2001) 2(7):612–9. doi: 10.1038/89759
31. Pillinger MH, Dinsell V, Apsel B, Tolani SN, Marjanovic N, Chan ES, et al. Regulation of metalloproteinases and NF-kappaB activation in rabbit synovial fibroblasts via E prostaglandins and Erk: contrasting effects of nabumetone and 6MNA. Br J Pharmacol (2004) 142(6):973–82. doi: 10.1038/sj.bjp.0705864
32. Gomez PF, Pillinger MH, Attur M, Marjanovic N, Dave M, Park J, et al. Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-kappaB subunits (p65, p50) in stimulated rheumatoid synovial fibroblasts. J Immunol (2005) 175(10):6924–30. doi: 10.4049/jimmunol.175.10.6924
33. Pillinger MH, Rosenthal PB, Tolani SN, Apsel B, Dinsell V, Greenberg J, et al. Cyclooxygenase-2-derived E prostaglandins down-regulate matrix metalloproteinase-1 expression in fibroblast-like synoviocytes via inhibition of extracellular signal-regulated kinase activation. J Immunol (2003) 171(11):6080–9. doi: 10.4049/jimmunol.171.11.6080
34. Pillinger MH, Marjanovic N, Kim SY, Scher JU, Izmirly P, Tolani S, et al. Matrix metalloproteinase secretion by gastric epithelial cells is regulated by E prostaglandins and MAPKs. J Biol Chem (2005) 280(11):9973–9. doi: 10.1074/jbc.M413522200
35. Tchetina EV, Di Battista JA, Zukor DJ, Antoniou J, Poole AR. Prostaglandin PGE2 at very low concentrations suppresses collagen cleavage in cultured human osteoarthritic articular cartilage: this involves a decrease in expression of proinflammatory genes, collagenases and COL10A1, a gene linked to chondrocyte hypertrophy. Arthritis Res Ther (2007) 9(4):R75. doi: 10.1186/ar2273
36. Cho W, Kim Y, Jeoung DI, Kim YM, Choe J. IL-4 and IL-13 suppress prostaglandins production in human follicular dendritic cells by repressing COX-2 and mPGES-1 expression through JAK1 and STAT6. Mol Immunol (2011) 48(6-7):966–72. doi: 10.1016/j.molimm.2011.01.007
37. Aggarwal S, Moodley YP, Thompson PJ, Misso NL. Prostaglandin E2 and cysteinyl leukotriene concentrations in sputum: association with asthma severity and eosinophilic inflammation. Clin Exp Allergy (2010) 40(1):85–93. doi: 10.1111/j.1365-2222.2009.03386.x
38. Pavord ID, Ward R, Woltmann G, Wardlaw AJ, Sheller JR, Dworski R. Induced sputum eicosanoid concentrations in asthma. Am J Respir Crit Care Med (1999) 160(6):1905–9. doi: 10.1164/ajrccm.160.6.9903114
39. Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am Rev Respir Dis (1993) 148(1):87–90. doi: 10.1164/ajrccm/148.1.87
40. Buckley J, Birrell MA, Maher SA, Nials AT, Clarke DL, Belvisi MG. EP4 receptor as a new target for bronchodilator therapy. Thorax (2011) 66(12):1029–35. doi: 10.1136/thx.2010.158568
41. Carey MA, Germolec DR, Bradbury JA, Gooch RA, Moorman MP, Flake GP, et al. Accentuated T helper type 2 airway response after allergen challenge in cyclooxygenase-1-/- but not cyclooxygenase-2-/- mice. Am J Respir Crit Care Med (2003) 167(11):1509–15. doi: 10.1164/rccm.200211-1383OC
42. Zaslona Z, Okunishi K, Bourdonnay E, Domingo-Gonzalez R, Moore BB, Lukacs NW, et al. Prostaglandin E(2) suppresses allergic sensitization and lung inflammation by targeting the E prostanoid 2 receptor on T cells. J Allergy Clin Immunol (2014) 133(2):379–87. doi: 10.1016/j.jaci.2013.07.037
43. Draijer C, Boorsma CE, Reker-Smit C, Post E, Poelstra K, Melgert BN. PGE2-treated macrophages inhibit development of allergic lung inflammation in mice. J Leukoc Biol (2016) 100(1):95–102. doi: 10.1189/jlb.3MAB1115-505R
44. Sturm EM, Schratl P, Schuligoi R, Konya V, Sturm GJ, Lippe IT, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J Immunol (2008) 181(10):7273–83. doi: 10.4049/jimmunol.181.10.7273
45. Konya V, Philipose S, Balint Z, Olschewski A, Marsche G, Sturm EM, et al. Interaction of eosinophils with endothelial cells is modulated by prostaglandin EP4 receptors. Eur J Immunol (2011) 41(8):2379–89. doi: 10.1002/eji.201141460
46. Bandeira-Melo C, Singh Y, Cordeiro RS, Silva PM, Martins MA. Involvement of prostaglandins in the down-regulation of allergic plasma leakage observed in rats undergoing pleural eosinophilia. Br J Pharmacol (1996) 118(8):2192–8. doi: 10.1111/j.1476-5381.1996.tb15662.x
47. Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim Biophys Acta (2015) 1851(4):414–21. doi: 10.1016/j.bbalip.2014.07.008
48. Maric J, Ravindran A, Mazzurana L, Bjorklund AK, Van Acker A, Rao A, et al. Prostaglandin E2 suppresses human group 2 innate lymphoid cell function. J Allergy Clin Immunol (2018) 141(5):1761–73 e6. doi: 10.1016/j.jaci.2017.09.050
49. Zhou Y, Wang W, Zhao C, Wang Y, Wu H, Sun X, et al. Prostaglandin E2 Inhibits Group 2 Innate Lymphoid Cell Activation and Allergic Airway Inflammation Through E-Prostanoid 4-Cyclic Adenosine Monophosphate Signaling. Front Immunol (2018) 9:501:501. doi: 10.3389/fimmu.2018.00501
50. Corrigan CJ, Napoli RL, Meng Q, Fang C, Wu H, Tochiki K, et al. Reduced expression of the prostaglandin E2 receptor E-prostanoid 2 on bronchial mucosal leukocytes in patients with aspirin-sensitive asthma. J Allergy Clin Immunol (2012) 129(6):1636–46. doi: 10.1016/j.jaci.2012.02.007
51. Huynh ML, Malcolm KC, Kotaru C, Tilstra JA, Westcott JY, Fadok VA, et al. Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med (2005) 172(8):972–9. doi: 10.1164/rccm.200501-035OC
52. Wang Z, Brandt S, Medeiros A, Wang S, Wu H, Dent A, et al. MicroRNA 21 is a homeostatic regulator of macrophage polarization and prevents prostaglandin E2-mediated M2 generation. PloS One (2015) 10(2):e0115855. doi: 10.1371/journal.pone.0115855
53. Stumm CL, Wettlaufer SH, Jancar S, Peters-Golden M. Airway remodeling in murine asthma correlates with a defect in PGE2 synthesis by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol (2011) 301(5):L636–44. doi: 10.1152/ajplung.00158.2011
54. Lundequist A, Nallamshetty SN, Xing W, Feng C, Laidlaw TM, Uematsu S, et al. Prostaglandin E(2) exerts homeostatic regulation of pulmonary vascular remodeling in allergic airway inflammation. J Immunol (2010) 184(1):433–41. doi: 10.4049/jimmunol.0902835
55. Huang S, Wettlaufer SH, Hogaboam C, Aronoff DM, Peters-Golden M. Prostaglandin E(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am J Physiol Lung Cell Mol Physiol (2007) 292(2):L405–13. doi: 10.1152/ajplung.00232.2006
56. Van Ly D, Burgess JK, Brock TG, Lee TH, Black JL, Oliver BG. Prostaglandins but not leukotrienes alter extracellular matrix protein deposition and cytokine release in primary human airway smooth muscle cells and fibroblasts. Am J Physiol Lung Cell Mol Physiol (2012) 303: (3):L239–50. doi: 10.1152/ajplung.00097.2012
57. Burgess JK, Ge Q, Boustany S, Black JL, Johnson PR. Increased sensitivity of asthmatic airway smooth muscle cells to prostaglandin E2 might be mediated by increased numbers of E-prostanoid receptors. J Allergy Clin Immunol (2004) 113(5):876–81. doi: 10.1016/j.jaci.2004.02.029
58. Bradbury P, Patel BS, Cidem A, Nader CP, Oliver BG, Ammit AJ. Prostaglandin E2, but not cAMP nor beta2-agonists, induce tristetraprolin (TTP) in human airway smooth muscle cells. Inflammation Res (2019) 68(5):369–77. doi: 10.1007/s00011-019-01224-0
59. Insuela DB, Daleprane JB, Coelho LP, Silva AR, e Silva PM, MA M, et al. Glucagon induces airway smooth muscle relaxation by nitric oxide and prostaglandin E(2). J Endocrinol (2015) 225(3):205–17. doi: 10.1530/JOE-14-0648
60. Insuela DBR, Azevedo CT, Coutinho DS, Magalhaes NS, Ferrero MR, Ferreira TPT, et al. Glucagon reduces airway hyperreactivity, inflammation, and remodeling induced by ovalbumin. Sci Rep (2019) 9(1):6478. doi: 10.1038/s41598-019-42981-6
61. Melillo E, Woolley KL, Manning PJ, Watson RM, O’Byrne PM. Effect of inhaled PGE2 on exercise-induced bronchoconstriction in asthmatic subjects. Am J Respir Crit Care Med (1994) 149(5):1138–41. doi: 10.1164/ajrccm.149.5.8173753
62. Kawakami Y, Uchiyama K, Irie T, Murao M. Evaluation of aerosols of prostaglandins E1 and E2 as bronchodilators. Eur J Clin Pharmacol (1973) 6(2):127–32. doi: 10.1007/BF00562439
63. Fish JE, Ankin MG, Adkinson NF Jr., Peterman VI. Indomethacin modification of immediate-type immunologic airway responses in allergic asthmatic and non-asthmatic subjects: evidence for altered arachidonic acid metabolism in asthma. Am Rev Respir Dis (1981) 123(6):609–14. doi: 10.1164/arrd.1981.123.6.609
64. Shimizu T, Mochizuki H, Shigeta M, Morikawa A. Effect of inhaled indomethacin on exercise-induced bronchoconstriction in children with asthma. Am J Respir Crit Care Med (1997) 155(1):170–3. doi: 10.1164/ajrccm.155.1.9001307
65. Daham K, James A, Balgoma D, Kupczyk M, Billing B, Lindeberg A, et al. Effects of selective COX-2 inhibition on allergen-induced bronchoconstriction and airway inflammation in asthma. J Allergy Clin Immunol (2014) 134(2):306–13. doi: 10.1016/j.jaci.2013.12.002
66. Kaler M, Barochia AV, Weir NA, Cuento RA, Stylianou M, Roth MJ, et al. A randomized, placebo-controlled, double-blinded, crossover trial of pioglitazone for severe asthma. J Allergy Clin Immunol (2017) 140(6):1716–8. doi: 10.1016/j.jaci.2017.05.033
67. Christie PE, Spur BW, Lee TH. The effects of lipoxin A4 on airway responses in asthmatic subjects. Am Rev Respir Dis (1992) 145(6):1281–4. doi: 10.1164/ajrccm/145.6.1281
68. Kong X, Wu SH, Zhang L, Chen XQ. Pilot application of lipoxin A4 analog and lipoxin A4 receptor agonist in asthmatic children with acute episodes. Exp Ther Med (2017) 14(3):2284–90. doi: 10.3892/etm.2017.4787
69. Van Ly D, Faiz A, Jenkins C, Crossett B, Black JL, McParland B, et al. Characterising the mechanism of airway smooth muscle beta2 adrenoceptor desensitization by rhinovirus infected bronchial epithelial cells. PloS One (2013) 8(2):e56058. doi: 10.1371/journal.pone.0056058
70. Safholm J, Manson ML, Bood J, Delin I, Orre AC, Bergman P, et al. Prostaglandin E2 inhibits mast cell-dependent bronchoconstriction in human small airways through the E prostanoid subtype 2 receptor. J Allergy Clin Immunol (2015) 136(5):1232–9 e1. doi: 10.1016/j.jaci.2015.04.002
71. Maher SA, Birrell MA, Belvisi MG. Prostaglandin E2 mediates cough via the EP3 receptor: implications for future disease therapy. Am J Respir Crit Care Med (2009) 180(10):923–8. doi: 10.1164/rccm.200903-0388OC
72. Fitzpatrick FA, Wynalda MA. Albumin-catalyzed metabolism of prostaglandin D2. Identification of products formed in vitro. J Biol Chem (1983) 258(19):11713–8.
73. Surh YJ, Na HK, Park JM, Lee HN, Kim W, Yoon IS, et al. 15-Deoxy-Delta(1)(2),(1)(4)-prostaglandin J(2), an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem Pharmacol (2011) 82(10):1335–51. doi: 10.1016/j.bcp.2011.07.100
74. Li J, Guo C, Wu J. 15-Deoxy–(12,14)-Prostaglandin J2 (15d-PGJ2), an Endogenous Ligand of PPAR-gamma: Function and Mechanism. PPAR Res (2019) 2019:7242030. doi: 10.1155/2019/7242030
75. Kobayashi M, Thomassen MJ, Rambasek T, Bonfield TL, Raychaudhuri B, Malur A, et al. An inverse relationship between peroxisome proliferator-activated receptor gamma and allergic airway inflammation in an allergen challenge model. Ann Allergy Asthma Immunol (2005) 95(5):468–73. doi: 10.1016/S1081-1206(10)61173-8
76. Oh SH, Park SM, Lee YH, Cha JY, Lee JY, Shin EK, et al. Association of peroxisome proliferator-activated receptor-gamma gene polymorphisms with the development of asthma. Respir Med (2009) 103(7):1020–4. doi: 10.1016/j.rmed.2009.01.015
77. Honda K, Marquillies P, Capron M, Dombrowicz D. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol (2004) 113(5):882–8. doi: 10.1016/j.jaci.2004.02.036
78. Hammad H, de Heer HJ, Soullie T, Angeli V, Trottein F, Hoogsteden HC, et al. Activation of peroxisome proliferator-activated receptor-gamma in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am J Pathol (2004) 164(1):263–71. doi: 10.1016/s0002-9440(10)63116-1
79. Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med (1999) 5(6):698–701. doi: 10.1038/9550
80. Gilroy DW, Colville-Nash PR, McMaster S, Sawatzky DA, Willoughby DA, Lawrence T. Inducible cyclooxygenase-derived 15-deoxy(Delta)12-14PGJ2 brings about acute inflammatory resolution in rat pleurisy by inducing neutrophil and macrophage apoptosis. FASEB J (2003) 17(15):2269–71. doi: 10.1096/fj.02-1162fje
81. Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, et al. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc Natl Acad Sci USA (2007) 104(52):20979–84. doi: 10.1073/pnas.0707394104
82. Marathe C, Bradley MN, Hong C, Chao L, Wilpitz D, Salazar J, et al. Preserved glucose tolerance in high-fat-fed C57BL/6 mice transplanted with PPARgamma-/-, PPARdelta-/-, PPARgammadelta-/-, or LXRalphabeta-/- bone marrow. J Lipid Res (2009) 50(2):214–24. doi: 10.1194/jlr.M800189-JLR200
83. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity (2010) 33(5):699–712. doi: 10.1016/j.immuni.2010.11.009
84. Maehara T, Nakamura T, Maeda S, Aritake K, Nakamura M, Murata T. Epithelial cell-derived prostaglandin D2 inhibits chronic allergic lung inflammation in mice. FASEB J (2019) 33(7):8202–10. doi: 10.1096/fj.201802817R
85. Woerly G, Honda K, Loyens M, Papin JP, Auwerx J, Staels B, et al. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J Exp Med (2003) 198(3):411–21. doi: 10.1084/jem.20021384
86. Coutinho DS, Anjos-Valotta EA, do Nascimento C, Pires ALA, Napimoga MH, Carvalho VF, et al. 15-Deoxy-Delta-12,14-Prostaglandin J2 Inhibits Lung Inflammation and Remodeling in Distinct Murine Models of Asthma. Front Immunol (2017) 8:740:740. doi: 10.3389/fimmu.2017.00740
87. Ward C, Dransfield I, Murray J, Farrow SN, Haslett C, Rossi AG. Prostaglandin D2 and its metabolites induce caspase-dependent granulocyte apoptosis that is mediated via inhibition of I kappa B alpha degradation using a peroxisome proliferator-activated receptor-gamma-independent mechanism. J Immunol (2002) 168(12):6232–43. doi: 10.4049/jimmunol.168.12.6232
88. Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol (2000) 164(3):1364–71. doi: 10.4049/jimmunol.164.3.1364
89. Nencioni A, Lauber K, Grunebach F, Van Parijs L, Denzlinger C, Wesselborg S, et al. Cyclopentenone prostaglandins induce lymphocyte apoptosis by activating the mitochondrial apoptosis pathway independent of external death receptor signaling. J Immunol (2003) 171(10):5148–56. doi: 10.4049/jimmunol.171.10.5148
90. Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, et al. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol (2005) 288(6):L1146–53. doi: 10.1152/ajplung.00383.2004
91. Ward JE, Gould H, Harris T, Bonacci JV, Stewart AG. PPAR gamma ligands, 15-deoxy-delta12,14-prostaglandin J2 and rosiglitazone regulate human cultured airway smooth muscle proliferation through different mechanisms. Br J Pharmacol (2004) 141(3):517–25. doi: 10.1038/sj.bjp.0705630
92. Ikei KN, Yeung J, Apopa PL, Ceja J, Vesci J, Holman TR, et al. Investigations of human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation. J Lipid Res (2012) 53(12):2546–59. doi: 10.1194/jlr.M026385
93. Macdonald LJ, Boddy SC, Denison FC, Sales KJ, Jabbour HN. A role for lipoxin A(4) as an anti-inflammatory mediator in the human endometrium. Reproduction (2011) 142(2):345–52. doi: 10.1530/REP-11-0021
94. Maderna P, Godson C. Lipoxins: resolutionary road. Br J Pharmacol (2009) 158(4):947–59. doi: 10.1111/j.1476-5381.2009.00386.x
95. Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol (2014) 76:467–92. doi: 10.1146/annurev-physiol-021113-170408
96. Machado FS, Johndrow JE, Esper L, Dias A, Bafica A, Serhan CN, et al. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med (2006) 12(3):330–4. doi: 10.1038/nm1355
97. Martini AC, Forner S, Bento AF, Rae GA. Neuroprotective effects of lipoxin A4 in central nervous system pathologies. BioMed Res Int (2014) 2014:316204. doi: 10.1155/2014/316204
98. Kim C, Livne-Bar I, Gronert K, Sivak JM. Fair-Weather Friends: Evidence of Lipoxin Dysregulation in Neurodegeneration. Mol Nutr Food Res (2020) 64(4):e1801076. doi: 10.1002/mnfr.201801076
99. Haworth O, Levy BD. Endogenous lipid mediators in the resolution of airway inflammation. Eur Respir J (2007) 30(5):980–92. doi: 10.1183/09031936.00005807
100. Planaguma A, Kazani S, Marigowda G, Haworth O, Mariani TJ, Israel E, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med (2008) 178(6):574–82. doi: 10.1164/rccm.200801-061OC
101. Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, et al. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat Med (2002) 8(9):1018–23. doi: 10.1038/nm748
102. Bandeira-Melo C, Bozza PT, Diaz BL, Cordeiro RS, Jose PJ, Martins MA, et al. Cutting edge: lipoxin (LX) A4 and aspirin-triggered 15-epi-LXA4 block allergen-induced eosinophil trafficking. J Immunol (2000) 164(5):2267–71. doi: 10.4049/jimmunol.164.5.2267
103. Soyombo O, Spur BW, Lee TH. Effects of lipoxin A4 on chemotaxis and degranulation of human eosinophils stimulated by platelet-activating factor and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Allergy (1994) 49(4):230–4. doi: 10.1111/j.1398-9995.1994.tb02654.x
104. Starosta V, Pazdrak K, Boldogh I, Svider T, Kurosky A. Lipoxin A4 counterregulates GM-CSF signaling in eosinophilic granulocytes. J Immunol (2008) 181(12):8688–99. doi: 10.4049/jimmunol.181.12.8688
105. Simoes RL, Fierro IM. Involvement of the Rho-kinase/myosin light chain kinase pathway on human monocyte chemotaxis induced by ATL-1, an aspirin-triggered lipoxin A4 synthetic analog. J Immunol (2005) 175(3):1843–50. doi: 10.4049/jimmunol.175.3.1843
106. Guilherme RF, Xisto DG, Kunkel SL, Freire-de-Lima CG, Rocco PR, Neves JS, et al. Pulmonary antifibrotic mechanisms aspirin-triggered lipoxin A(4) synthetic analog. Am J Respir Cell Mol Biol (2013) 49(6):1029–37. doi: 10.1165/rcmb.2012-0462OC
107. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510(7503):92–101. doi: 10.1038/nature13479
108. Haworth O, Cernadas M, Levy BD. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J Immunol (2011) 186(11):6129–35. doi: 10.4049/jimmunol.1004007
109. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med (2013) 5(174):174ra26. doi: 10.1126/scitranslmed.3004812
110. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O’Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol (2016) 137(1):75–86 e8. doi: 10.1016/j.jaci.2015.05.037
111. Bozinovski S, Uddin M, Vlahos R, Thompson M, McQualter JL, Merritt AS, et al. Serum amyloid A opposes lipoxin A(4) to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc Natl Acad Sci U.S.A. (2012) 109(3):935–40. doi: 10.1073/pnas.1109382109
112. Bonnans C, Fukunaga K, Levy MA, Levy BD. Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. Am J Pathol (2006) 168(4):1064–72. doi: 10.2353/ajpath.2006.051056
113. Karra L, Haworth O, Priluck R, Levy BD, Levi-Schaffer F. Lipoxin B(4) promotes the resolution of allergic inflammation in the upper and lower airways of mice. Mucosal Immunol (2015) 8(4):852–62. doi: 10.1038/mi.2014.116
114. Parameswaran K, Radford K, Fanat A, Stephen J, Bonnans C, Levy BD, et al. Modulation of human airway smooth muscle migration by lipid mediators and Th-2 cytokines. Am J Respir Cell Mol Biol (2007) 37(2):240–7. doi: 10.1165/rcmb.2006-0172OC
115. Wu SH, Wu XH, Lu C, Dong L, Chen ZQ. Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am J Respir Cell Mol Biol (2006) 34(1):65–72. doi: 10.1165/rcmb.2005-0184OC
116. Roach KM, Feghali-Bostwick CA, Amrani Y, Bradding P. Lipoxin A4 Attenuates Constitutive and TGF-beta1-Dependent Profibrotic Activity in Human Lung Myofibroblasts. J Immunol (2015) 195(6):2852–60. doi: 10.4049/jimmunol.1500936
117. Kim H, Park SH, Han SY, Lee YS, Cho J, Kim JM. LXA4-FPR2 signaling regulates radiation-induced pulmonary fibrosis via crosstalk with TGF-beta/Smad signaling. Cell Death Dis (2020) 11(8):653. doi: 10.1038/s41419-020-02846-7
Keywords: asthma, lipoxins, PGE2, 15d-PGJ2, resolution
Citation: Insuela DBR, Ferrero MR, Coutinho DS, Martins MA and Carvalho VF (2020) Could Arachidonic Acid-Derived Pro-Resolving Mediators Be a New Therapeutic Strategy for Asthma Therapy? Front. Immunol. 11:580598. doi: 10.3389/fimmu.2020.580598
Received: 06 July 2020; Accepted: 20 October 2020;
Published: 09 December 2020.
Edited by:
Danuta Radzioch, McGill University, CanadaReviewed by:
Vanessa Pinho, Federal University of Minas Gerais, BrazilBrian Gregory George Oliver, University of Technology Sydney, Australia
Copyright © 2020 Insuela, Ferrero, Coutinho, Martins and Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinicius Frias Carvalho, dmluaWNpdXNmcmlhc0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work