 Ronald G. Gill
Ronald G. Gill Adam L. Burrack
Adam L. Burrack- 1Departments of Surgery and Immunology and Microbiology, University of Colorado Denver, Aurora, CO, United States
- 2Department of Microbiology and Immunology, University of Minnesota Medical School, Minneapolis, MN, United States
Memory T lymphocytes constitute a significant problem in tissue and organ transplantation due their contribution to early rejection and their relative resistance to tolerance-promoting therapies. Memory cells generated by environmental antigen exposure, as with T cells in general, harbor a high frequency of T cell receptors (TCR) spontaneously cross-reacting with allogeneic major histocompatibility complex (MHC) molecules. This phenomenon, known as ‘heterologous’ immunity, is thought to be a key barrier to transplant tolerance induction since such memory cells can potentially react directly with essentially any prospective allograft. In this review, we describe two additional concepts that expand this commonly held view of how memory cells contribute to transplant immunity and tolerance disruption. Firstly, autoimmunity is an additional response that can comprise an endogenously generated form of heterologous alloimmunity. However, unlike heterologous immunity generated as a byproduct of indiscriminate antigen sensitization, autoimmunity can generate T cells that have the unusual potential to interact with the graft either through the recognition of graft-bearing autoantigens or by their cross-reactive (heterologous) alloimmune specificity to MHC molecules. Moreover, we describe an additional pathway, independent of significant heterologous immunity, whereby immune memory to vaccine- or pathogen-induced antigens also may impair tolerance induction. This latter form of immune recognition indirectly disrupts tolerance by the licensing of naïve alloreactive T cells by vaccine/pathogen directed memory cells recognizing the same antigen-presenting cell in vivo. Thus, there appear to be recognition pathways beyond typical heterologous immunity through which memory T cells can directly or indirectly impact allograft immunity and tolerance.
Introduction
Memory T cells constitute a formidable obstacle both for preventing early graft rejection and for the eventual induction of allograft tolerance. For example, memory CD8 T cells can trigger early aggressive rejection of cardiac allograft rejection in mice (1). Importantly, memory T cells are also relatively resistant to tolerance-promoting therapies (2–4). An important property of memory cells thought to be especially relevant for impairing allograft survival is their strong ‘heterologous’ reactivity to allogeneic MHC molecules. The concept of heterologous immunity originated by the observation that humoral or cellular immunity to one pathogen could impart reactivity to a secondary, unrelated pathogen (5). This property is found in memory T cells generated in response to one virus that that can cross-react with a new unrelated viral infection (6, 7). This term has been borrowed by the transplantation field to describe a specific feature of memory T cells that imparts a high degree of cross-reactivity to allogeneic MHC molecules (8, 9). This phenomenon is almost certainly due to the high intrinsic bias of the TCR repertoire for MHC recognition (10, 11). Thus, simply by chance, any polyclonal antigen-specific T cell response would be expected to contain a significant subpopulation of allogeneic MHC-reactive T cells. This concept is strikingly illustrated by the findings from Amir et al. showing that nearly half of CD4 and CD8 virus-specific human T cell clones cross-reacted to at least one allogeneic HLA allele (12). A high degree of cross-reactivity to alloantigens by pathogen-induced T cells has also been demonstrated in mouse infection models (13–15).
Autoimmunity as an Endogenous Source of Heterologous Allograft Immunity
There has been interest in the transplant field for how alloimmunity may initiate nascent autoimmunity that can impact the graft. This has been especially evident in chronic allograft reactivity in lung transplantation (16, 17). However, the converse may also be true; pre-existing autoimmunity may be a source of potential alloimmunity in the form of heterologous immunity. We have had a long-standing interest in islet transplantation using the non-obese diabetic (NOD) model of spontaneous autoimmune Type 1 diabetes. NOD mice have a multi-factorial predisposition for developing diabetes due to T and B cell dependent islet beta cell-specific autoimmunity (18–20). Importantly, diseased NOD mice destroy syngeneic (NOD) pancreas (21) or isolated islet (22) transplants through a process of recurrent disease, a phenomenon that also occured in non-immunosuppressed Type 1 patients receiving a partial pancreas transplant from a non-diseased identical twin (23). Moreover, NOD mice also show a strong response to islet allografts (22, 24, 25). As such, the NOD mouse model is highly useful for studying islet transplant autoimmunity and alloimmunity, including potential heterologous immunity, in the setting of Type 1 diabetes.
Based on the discussion above, the autoimmune T cell repertoire, like any polyclonal T cell population, would be expected to have a high degree of cross-reactivity to allogeneic MHC molecules. Interestingly, a survey of established autoreactive (islet antigen-specific) T cell clones derived from NOD mice revealed that over one third cross-reacted to one or more of three allogeneic MHC haplotypes (26), a result conceptually similar to what had been found previously for human virus-specific T cell clones (12). Based on this concept, we interrogated the TCR specificity of T cells infiltrating MHC-unrelated islet allografts grafted into spontaneously diabetic NOD mice. Consistent with results from screening autoreactive T cell clones, TCRs from islet allografts were profoundly enriched with dual autoreactive/alloreactive specificities (26). Thus, autoimmunity can be a source endogenously generated heterologous immunity contributing to allograft rejection.
Heterologous Autoreactive T Cells With Alloreactivity: One or Two TCRs?
While the simultaneous reactivity of individual T cells for both self-MHC-restricted cognate antigens and allogeneic MHC molecules has been apparent for many years (27), it is not always clear whether this is due to a single TCR α/β pair or due to two separate TCRs on a given cell. There is ample reason to posit that autoreactive T cells demonstrating additional alloreactivity could be due to the contribution of two separate TCRs. A significant percentage (estimated to be roughly between 1-8%) of mature mouse (28–30) and human (31) peripheral T cells express two TCRs, presumably due to a substantial frequency of developing T cells expressing two functional TCR α chains (32). Moreover, dual TCR-expressing T cells indeed have a high frequency of an alloreactive second receptor (33, 34), and these can play an important role in triggering graft-versus-host disease in mice (33). It is conceivable, then, that autoreactive T cells demonstrating cross-reactive alloreactivity could be the result of two separate TCR specificities on the same cells.
Conversely, when studying a dual TCR-expressing T cell clone with both self-MHC-restricted peptide specify (OVA) and alloreactvity, Malissen et al. found that only one the two TCR α/β pairs imparted this dual reactivity (35). This demonstrates that a single TCR can possess combined nominal antigen plus cross-reactive allo-specificity. This concept was supported by studies involving high-throughput sequencing of a large repertoire of TCR transcripts from T cells targeting islet allografts in spontaneously diabetic NOD mice (26). Importantly, screening the antigen reactivity of highly expressed TCRs indicated that single TCR α/β pairs conferred simultaneous dual autoantigen/alloantigen (MHC) reactivity (26). Thus, the predominant heterologous immunity identified by this approach could be accounted for by single autoreactive TCRs with clear cross-reactivity to allogeneic MHC molecules. Of course, these finds do not preclude the potential of heterologous alloimmunity emerging from autoreactive T cells being the result of a second TCR. However, results to date suggest that the most frequent source of simultaneous autoreactive/alloreactive T cells in islet transplants in the setting of autoimmunity is the result of a single, cross-reactive TCR.
Conventional Antigen-Stimulated Versus Autoimmune Heterologous Immunity: A ‘Trojan Horse’ Model of Allograft Immunity
One potentially key difference between memory T cells generated by past antigen challenge and ongoing autoimmunity may simply be in the activation state of antigen-experienced T cells in these two scenarios. In fact, memory T cells may not be completely resistant to tolerance induction (2). For example, naïve mice can be tolerized to tissue and organ transplants despite bearing a degree of memory T cells generated by environmental antigen exposure. If this is the case, then the impact of immune memory on allograft rejection and tolerance may be related in part to the pre-transplant burden of pre-existing alloreactive T cell memory (8, 36). However, the activation state of memory cells may also impact their potential to be tolerized. In most cases, one would expect memory cells from past antigen exposure to be in a more quiescent state of central memory (2). However, the autoimmune T cell pool may be experiencing persistent activation/re-activation in the host, including those cells expressing cross-reactive alloimmunity. This means that the alloimmune component found in autoimmune disease may already be in a heightened activation state and potentially more challenging to tolerize. While NOD mice have a variety of tolerance defects (37, 38) the presence of alloreactvity found within the smoldering autoreactive repertoire may contribute to the dramatic resistance of NOD mice to allograft tolerance, even toward tissues/organs for which they have no apparent autoimmunity (37). It will be most interesting to test this concept in future studies.
In addition, there is a second and more unusual property of heterologous (alloreactive) autoreactive T cells that may make them especially virulent as mediators of islet rejection. In the conventional view of heterologous immunity, antigen-experienced memory cells contribute to allograft immunity and tolerance resistance due to their chance cross-reactivity to the graft, unrelated to the specificity of the original stimulating antigen. However, in the case of autoimmunity, heterologous T cells have the potential to interact with graft through two qualitatively distinct recognition pathways simultaneously (Figure 1). One route of islet graft interaction can be through the recognition of self MHC-restricted islet autoantigens acquired from the transplant and processed and presented by host antigen-presenting cells (APCs). We previously found that monoclonal BDC2.5 TCR transgenic CD4 T cells without allogeneic cross-reactivity could nevertheless recognize allograft-derived autoantigens processed by host APCs and destroy islet allografts though this type of indirect autoantigen recognition (39). Thus, this autoreactive specificity alone was sufficient to trigger allograft rejection. However, since polyclonal autoreactive T cells targeting the islet graft also contain cross-reactive, alloreactive T cells (26), some of these cells can also directly recognize the native allogeneic MHC expressed by the graft. An example of this phenomenon is a CD4 TCR (9860-A3B3) isolated from an MHC mismatched C3H (H-2k) islet graft in NOD mice. This TCR recognizes an islet-associated Chromogranin A peptide presented by the NOD MHC class II I-Ag7 while also directly recognizing allogeneic I-Ak expressed by the donor (26). Thus, this unusual situation could represent a sort of ‘Trojan Horse’ phenomenon in the islet graft in which the influx of T cells responding to autoantigens also ferries in a cohort of heterologous alloreactive T cells that can directly engage the allograft MHC. This simultaneous graft recognition through either auto- or allo- specificities could account for the accelerated response to MHC unrelated islet allografts in NOD mice despite the lack of intentional prior alloantigen exposure in these mice (24, 25). This property of heterologous immunity within autoimmune T cells could potentially be a general dilemma in controlling allograft rejection in the setting of autoimmune disease.
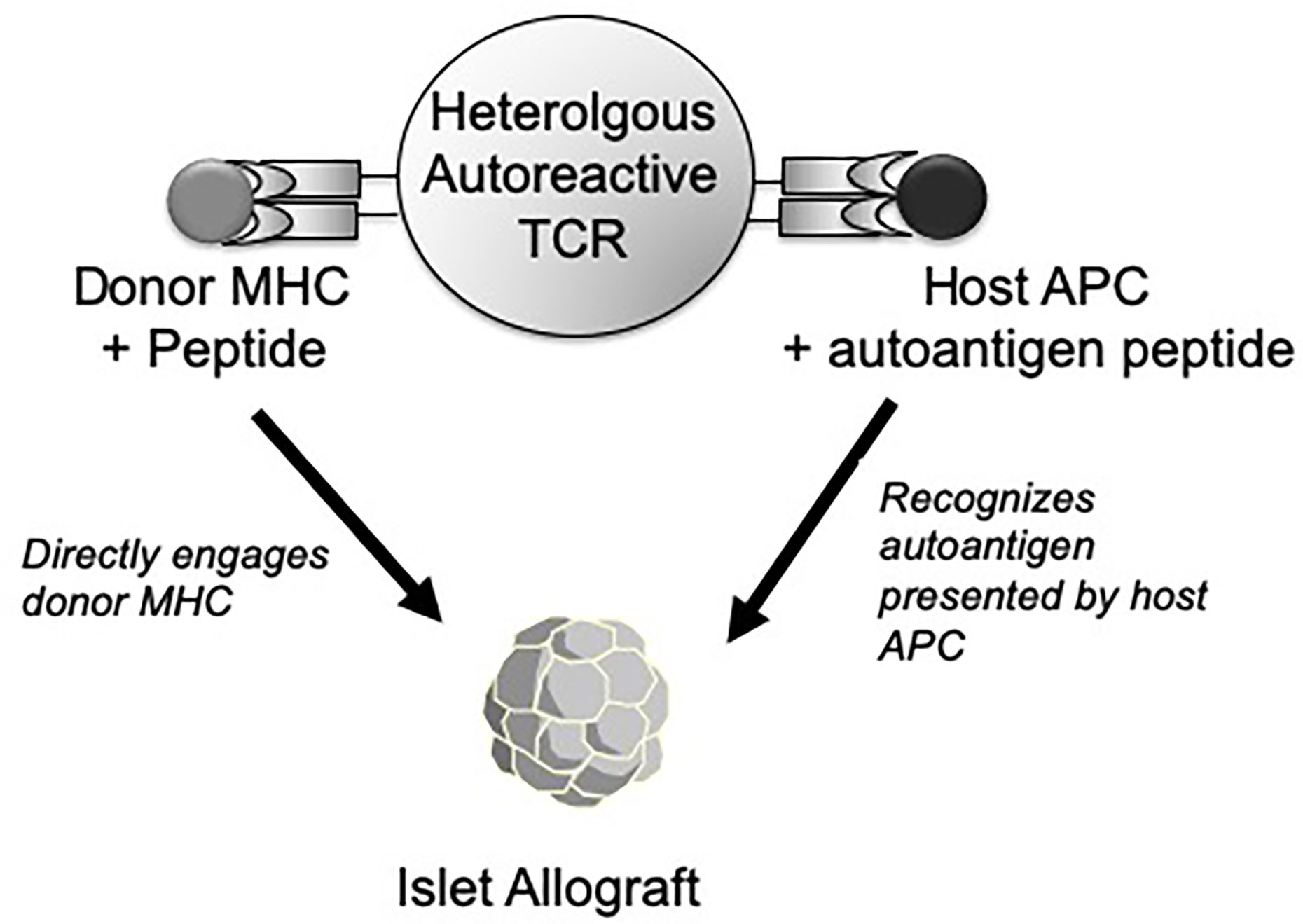
Figure 1 Depiction of an autoreactive (islet-specific) T cell with a TCR with both autoantigen specificity and cross-reactivity to allogenic MHC molecules. In response to an islet allograft, this type of heterologous TCR can recognize a host MHC-restricted, graft-derived autoantigen peptide presented by host APC (right side). Alternatively, the same TCR may also directly recognize a native donor MHC molecule plus an unidentified peptide expressed by the graft (left side). As such, the same T cell has the potential to interact with the islet graft through either a host MHC-restricted (autoreactive) or donor MHC-restricted (alloreactive) recognition pathway.
An Additional and Less Apparent Route of Tolerance Blockade by Memory T Cells
A major ongoing goal in transplantation is to transition from chronic non-specific immunosuppression towards the induction of antigen (donor)-specific tolerance. To this point, this discussion has centered on heterologous immunity by memory T cells to allogeneic MHC molecules themselves (either from prior antigen exposure or via autoimmunity) as a key problem in transplant immunity and tolerance. As such, the importance of assessing pre-existing humoral or cellular immunity to donor MHC has been a major focus of screening efforts in transplantation (40–42). While such efforts are clearly warranted, there are potentially alternative routes whereby T cell memory could impair tolerance induction without a requirement for substantial heterologous immunity to the donor MHC (43). Unfortunately, the metagenome of both organ donors and recipients encode a variety of non-self antigens, such as those derived from microbiota (44, 45) or from latent infections such as CMV and EBV (46–49) that are clearly associated with impaired allograft outcomes in clinical transplantation. It is clear that the activation of anti-viral immunity can abrogate allograft tolerance (50, 51), possibly by the induction of inflammation that itself may non-specifically impair tolerance induction (52, 53).
One could assume that much of the impairment of tolerance induced by recipient responses to donor-associated pathogens is related to either heterologous immunity generated during the pathogen response and/or to the associated inflammation. We propose another more provocative form of host immunity to donor-derived non-MHC antigens that also could impair tolerance induction. This problem of donor-derived, non-self, non-MHC antigens has arguably been under-represented in most small animal studies. This being the case, we developed a model system in which the donor expressed a non-self transgenic antigen (OVA) to which the host was immune via vaccination (43), a scenario that could have relevance to clinical transplantation in which vaccination might protect from a donor-derived pathogen (54). Tolerance was induced using a common approach of administering a pre-transplant donor-specific transfusion (DST) in the form of donor spleen cells plus costimulation blockade (55, 56). Interestingly, host anti-OVA vaccination alone was innocuous, generating negligible anti-donor heterologous alloimmunity and had no impact on tolerance induction to wild-type allografts. Even peri-transplant re-activation of host anti-OVA reactivity did not impair tolerance induction. However, treatment with an OVA-expressing allogeneic DST in an OVA-immune recipient profoundly abrogated tolerance induction, even if the subsequent allograft did not express OVA (43).
Tolerance Disruption of Naïve T Cells by Memory T Cells via Linked Antigen Presentation
A key feature of this admittedly contrived system was that that the alloantigen and non-self (OVA) antigen had to be presented on the same APC in order to disrupt tolerance (Figure 2). That is, ‘linked’ recognition of the vaccine-directed antigen and the alloantigen was required for tolerance blockade (43). This scenario illustrates the potential for an alternative route whereby memory cells may impact the microenvironment during initial tolerance induction at the level of antigen presentation, not via donor MHC recognition, but rather through the recognition of another non-MHC antigen introduced by donor cells. Currently, probably the most recognizable concept involving T cells influencing one another via recognition of the same APC is that of ‘linked suppression’ in which putative regulatory T cells inhibits the function of another uncommitted T cell through interacting with the same APC (57). However, the concept of linked recognition leading to cell activation is actually considerably older. The original description of ‘linked’ antigen recognition referred to the observation of the carrier-hapten phenomenon in which the ‘helper’ determinant for antigen formation required physical linkage between the ‘helper’ determinant and the antibody specificity (58). This concept was later adapted to refer to the finding that helper T cells for the generation of cytotoxic T cells required recognition of the same APC in vivo (59). Three seminal studies later found that the basis of such CD4 T cell help for CD8 T cells was in the form of CD40:CD154 interactions with the APC resulting in the licensing of such APCs to activate other T cells (60–62). We had proposed that such T-T cell collaboration could be bi-directional in that CD4 and CD8 T cells could potentially influence one another through linked recognition of the APC (63). This latter concept could explain how memory CD8 T cells could disrupt T cell tolerance and promote allograft rejection instead (64). Of course, while this model of tolerance disruption required memory CD8 T cells (43), there is clear evidence that both CD4 (65–67) and CD8 (64, 65, 68) T cells can be involved in tolerance blockade in pre-clinical models. However, it is usually unclear how specific memory T cell subsets actually impair tolerance induction.
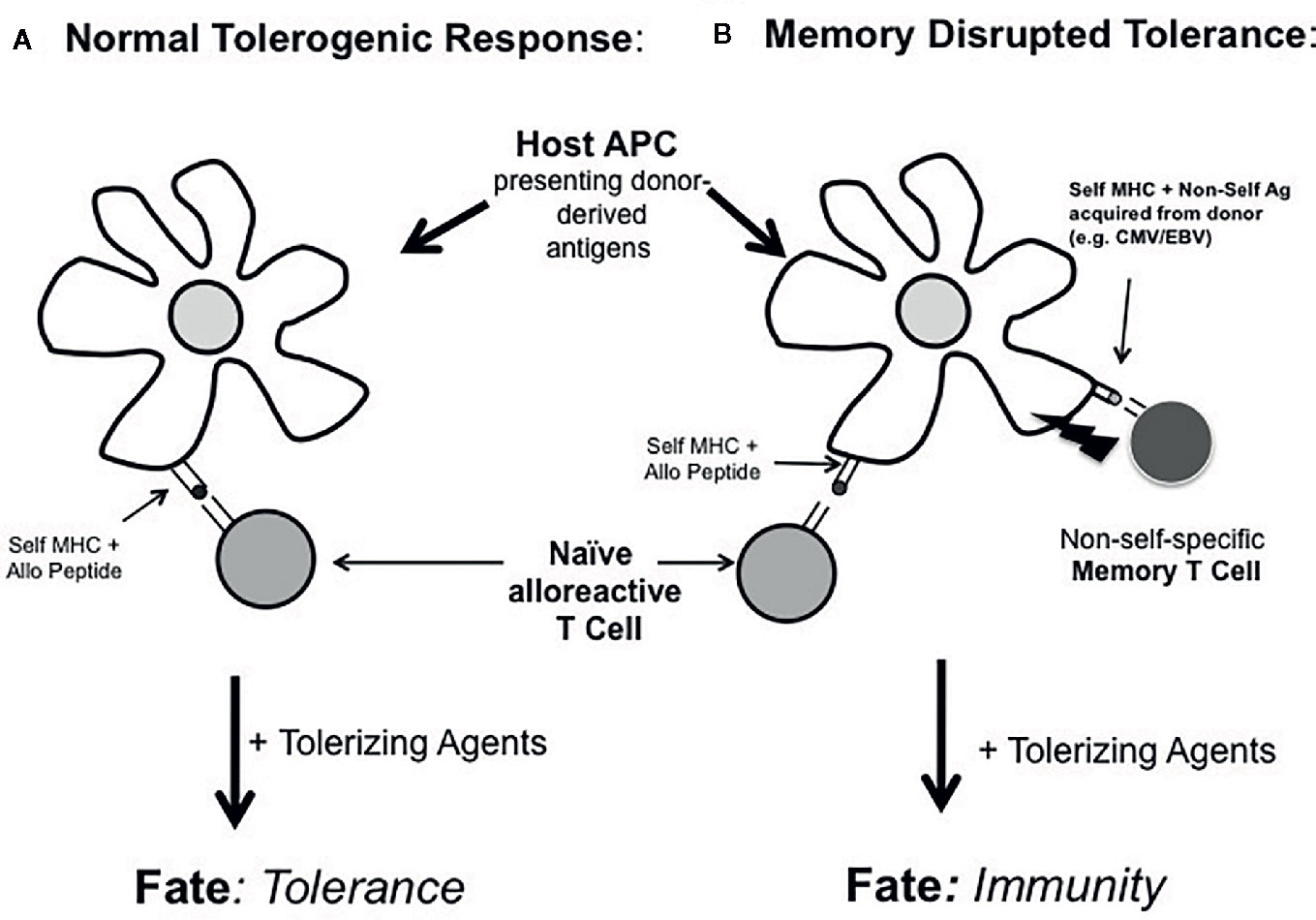
Figure 2 Working model of tolerance blockade by linked recognition of alloantigens and non-MHC donor antigens. In (A) host naïve T cells responding to donor antigens acquired by host APCs are amendable to tolerance induction by tolerance-promoting agents. In (B), if these same APCs also acquire other donor-derived antigens to which the host has pre-existing immunity (e.g., to non-self pathogen-derived antigens or autoantigens), tolerance is disrupted at the level of the APC. In this case, the fate of such uncommitted alloreactive T cells is diverted from tolerance to immunity.
In what situation might this type of memory cell reactivity be important in transplantation? In the setting of autoimmunity or c donor pathogen infections such as CMV and EBV (46–49), the host could be immune to donor-derived, non-MHC antigens without obvious pre-transplant anti-donor MHC immunity. However, depending on the tissue distribution of autoantigens or donor pathogen-derived antigens, memory cells for these antigens could disrupt tolerance induction by diverting the naïve T cells recognizing the same APC from a tolerized fate to an effector phenotype (Figure 2). Because the existing host T cell memory to donor-derived, non-self antigens was self-MHC restricted in this linked recognition model (65), we would propose that the ‘indirect’ pathway of alloantigen recognition by host APCs was chiefly involved in disrupting tolerance. This could contrast sharply with how heterologous memory T cells (i.e., T cells with direct donor MHC reactivity) disrupt tolerance. Such donor MHC cross-reactive T cells may influence tolerance through the ‘direct’ pathway of antigen recognition. Future studies are needed to define the specific cellular interactions required by memory T cells to impair tolerance. Moreover, this tolerance blockade could occur without significant evidence of conventional heterologous anti-donor MHC reactivity. If this alternate and less apparent route of memory T cell tolerance blockade is significant, it implies that assessing pre-transplant anti-donor MHC reactivity alone may not be sufficient to predict the potential success of tolerance-promoting therapies. It may also be important to more carefully assess the presence of donor-derived pathogens and the corresponding host immunity to these antigens or to autoantigens.
Concluding Remarks
The role of memory T cells for providing resistance to allograft tolerance induction is well established. Moreover, the high degree of heterologous anti-donor MHC alloreactivity found within memory T cell populations is rightly considered a major potential source of tolerance disruption. By this view, the implied paradigm is that memory cells behave essentially as directly allo-sensitized cells that are resistant to regulation. However, there are other routes of memory cell specificity that expand and perhaps complicate this straightforward view (summarized in Table 1). For example, autoimmunity may constitute a form of ongoing memory T cell generation and heterologous alloreactivity that does not require exogenous antigen exposure. Also, heterologous autoreactive T cells have the unusual potential for recognizing autoantigen-expressing allografts through autoreactive and alloreactive specificities simultaneously. Alternatively, memory T cells can potentially subvert tolerance induction by recognizing donor-derived, non-MHC antigens (such as autoantigens or from pathogens) co-presented on APCs with conventional alloantigens resulting in the disruption of tolerance by naïve alloreactive T cells. Importantly, this latter form of antigen recognition could impair tolerance even in the absence of significant anti-donor MHC reactivity. The relative significance of this latter route of tolerance blockade by memory T cells requires further clarification. Unfortunately, the clinical transplantation field currently relies on chronic non-specific immunosuppression to maintain graft survival and has not yet progressed to the point of using defined therapeutics to induce allograft tolerance in prospective trials. As such, it is challenging to determine the degree to which these or other potential pathways of tolerance impairment by immune memory pose significant barriers to achieving transplantation tolerance in humans.

Table 1 Characteristics of differing pathways whereby memory T cells impair allograft tolerance.
Author Contributions
AB and RG wrote and edited the review article. RG created the model figures and AB created Table 1. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants RO1 DK115745 and UC4 DK104223 (RG) and by RO1 DK099187 (AB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Su CA, Iida S, Abe T, Fairchild RL. Endogenous memory CD8 T cells directly mediate cardiac allograft rejection. Am J Transplant (2014) 14:568–79. doi: 10.1111/ajt.12605
2. Lakkis FG, Sayegh MH. Memory T cells: a hurdle to immunologic tolerance. J Am Soc Nephrol (2003) 14:2402–10. doi: 10.1097/01.ASN.0000085020.78117.70
3. Yang J, Brook MO, Carvalho-Gaspar M, Zhang J, Ramon HE, Sayegh MH, et al. Allograft rejection mediated by memory T cells is resistant to regulation. Proc Natl Acad Sci USA (2007) 104:19954–9. doi: 10.1073/pnas.0704397104
4. Ford ML, Larsen CP. Transplantation tolerance: memories that haunt us. Sci Transl Med (2011) 3:86ps22. doi: 10.1126/scitranslmed.3002504
5. Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol (2002) 2:417–26. doi: 10.1038/nri820
6. Selin LK, Cornberg M, Brehm MA, Kim SK, Calcagno C, Ghersi D, et al. CD8 memory T cells: cross-reactivity and heterologous immunity. Semin Immunol (2004) 16:335–47. doi: 10.1016/j.smim.2004.08.014
7. Welsh RM, Che JW, Brehm MA, Selin LK. Heterologous immunity between viruses. Immunol Rev (2010) 235:244–66. doi: 10.1111/j.0105-2896.2010.00897.x
8. Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest (2003) 111:1887–95. doi: 10.1172/JCI200317477
9. Selin LK, Brehm MA, Naumov YN, Cornberg M, Kim SK, Clute SC, et al. Memory of mice and men: CD8+ T-cell cross-reactivity and heterologous immunity. Immunol Rev (2006) 211:164–81. doi: 10.1111/j.0105-2896.2006.00394.x
10. Zerrahn J, Held W, Raulet DH. The MHC reactivity of the T cell repertoire prior to positive and negative selection. Cell (1997) 88:627–36. doi: 10.1016/S0092-8674(00)81905-4
11. Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C, et al. How the T cell repertoire becomes peptide and MHC specific. Cell (2005) 122:247–60. doi: 10.1016/j.cell.2005.05.013
12. Amir AL, D’Orsogna LJ, Roelen DL, van Loenen MM, Hagedoorn RS, de Boer R, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood (2010) 115:3146–57. doi: 10.1182/blood-2009-07-234906
13. Yang H, Welsh RM. Induction of alloreactive cytotoxic T cells by acute virus infection of mice. J Immunol (1986) 136:1186–93.
14. Pantenburg B, Heinzel F, Das L, Heeger PS, Valujskikh A. T cells primed by Leishmania major infection cross-react with alloantigens and alter the course of allograft rejection. J Immunol (2002) 169:8686–3693. doi: 10.4049/jimmunol.169.7.3686
15. Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsch RM. Direct visualization of cross-reactive effector and memory allo-specific CD8 T cells generated in response to virl infections. J Immunol (2003) 170:4077–86. doi: 10.4049/jimmunol.170.8.4077
16. Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, et al. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant (2011) 30:624–31. doi: 10.1016/j.healun.2011.01.708
17. Vittal R, Fan L, Greenspan DS, Mickler EA, Gopalakrishnan B, Gu H, et al. IL-17 induces type V collagen overexpression and EMT via TGF-β-dependent pathways in obliterative bronchiolitis. Am J Physiol Lung Cell Mol Physiol (2013) 304:L401–14. doi: 10.1152/ajplung.00080.2012
18. Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity (1997) 7:727–38. doi: 10.1016/S1074-7613(00)80392-1
19. Leiter EH. NOD mice and related strains: Origin, husbandry, and biology. In: NOD Mice and Related Strains: Research Applications in Diabetes, AIDS, Cancer, and Other Diseases. Leiter EH, Atkinson MA, editors. R.G. Landes, Austin (1998). pp. 1–35.
20. Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med (1999) 5:601–4. doi: 10.1038/9442
21. Terada M, Lennartz K, Mullen Y. Allogeneic and syngeneic pancreas transplantation in non-obese diabetic mice. Transplant Proc (1987) 19:960.
22. Wang Y, Pontesilli O, Gill RG, La Rosa FG, Lafferty KJ. The role of CD4+ and CD8+ T cells in the destruction of islet grafts by spontaneously diabetic mice. Proc Natl Acad Sci USA (1991) 88:527–31. doi: 10.1073/pnas.88.2.527
23. Sutherland DER, Sibley R, Xu XZ, Michael A, Srikanta S, Taub F, et al. Twin-to-twin pancreas transplantation: Reversal and reenactment of the pathogenesis of type I diabetes. Transplant Assoc Am Physicians (1984) 97:80–7.
24. Okitsu T, Bartlett ST, Hadley GA, Drachenberg CB, Farney AC. Recurrent autoimmunity accelerates destruction of minor and major histoincompatible islet grafts in non-obese diabetic (NOD) mice. Am J Transplant (2001) 1:138–45. doi: 10.1034/j.1600-6143.2001.10207.x
25. Makhlouf L, Kishimoto K, Smith RN, Abdi R, Koulmanda M, Winn HJ, et al. The role of autoimmunity in islet allograft destruction: major histocompatibility complex class II matching is necessary for autoimmune destruction of allogeneic islet transplantation after T-cell costimulatory blockade. Diabetes (2002) 51:3202–10. doi: 10.2337/diabetes.51.11.3202
26. Burrack AL, Landry LG, Siebert J, Coulombe M, Gill RG, Nakayama M. Simultaneous Recognition of Allogeneic MHC and Cognate Autoantigen by Autoreactive T Cells in Transplant Rejection. J Immunol (2018) 200:1504–12. doi: 10.4049/jimmunol.1700856
27. Webb SR, Sprent J. T cells with multiple specifities. Intern Rev Immunol (1986) 1:151–82. doi: 10.3109/08830188609056605
28. Hardardottir F, Baron JL, Janeway CA Jr. T cells with two functional antigen-specific receptors. Proc Natl Acad Sci USA (1995) 92:354–8. doi: 10.1073/pnas.92.2.354
29. Alam SM, Gascoigne NR. Posttranslational regulation of TCR Valpha allelic exclusion during T cell differentiation. J Immunol (1998) 160:3883–90.
30. He X, Janeway CA, Levine M, Robinson E, Preston-Hurlburt P, Viret C, et al. Dual receptor T cells extend the immune repertoire for foreign antigens. Nat Immunol (2002) 3:127–34. doi: 10.1038/ni751
31. Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A. Expression of two T cell receptor alpha chains: dual receptor T cells. Science (1993) 262:422–4. doi: 10.1126/science.8211163
32. Malissen M, Trucy J, Jouvin-Marche E, Cazenave PA, Scollay R, Malissen B. Regulation of TCR alpha and beta gene allelic exclusion during T-cell development. Immunol Today (1992) 13:315–22. doi: 10.1016/0167-5699(92)90044-8
33. Morris GP, Allen PM. Cutting Edge: Highly alloreactive dual TCR T cells play a dominant role in graft-versus-host disease. J Immunol (2009) 182:6639–43. doi: 10.4049/jimmunol.0900638
34. Balakrishnan A, Morris GP. The highly alloreactive nature of dual TCR T cells. Curr Opin Organ Transplant (2016) 21:22–8. doi: 10.1097/MOT.0000000000000261
35. Malissen M, Trucy J, Letourneur F, Rebaï N, Dunn DE, Fitch FW, et al. A T cell clone expresses two T cell receptor alpha genes but uses one alpha beta heterodimer for allorecognition and self MHC-restricted antigen recognition. Cell (1988) 55:49–59. doi: 10.1016/0092-8674(88)90008-6
36. Heeger PS, Greenspan NS, Kuhlenschmidt S, Dejelo C, Hricik DE, Schulak JA, et al. Pretransplant frequency of donor-specific, IFN-g-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of postransplant rejection episodes. J Immunol (1999) 163:2267–75.
37. Markees TG, Serreze DV, Phillips NE, Sorli CH, Gordon EJ, Shultz LD, et al. NOD mice have a generalized defect in their response to transplantation tolerance induction. Diabetes (1999) 48:967–74. doi: 10.2337/diabetes.48.5.967
38. Pearson T, Markees TG, Serreze DV, Pierce MA, Marron MP, Wicker LS, et al. Genetic disassociation of autoimmunity and resistance to costimulation blockade-induced transplantation tolerance in nonobese diabetic mice. J Immunol (2003) 171:185–95. doi: 10.4049/jimmunol.171.1.185
39. Kupfer TM, Crawford ML, Pham K, Gill RG. MHC-mismatched islet allografts are vulnerable to autoimmune recognition in vivo. J Immunol (2005) 175:2309–16. doi: 10.4049/jimmunol.175.4.2309
40. Augustine JJ, Siu DS, Clemente MJ, Schulak JA, Heeger PS. Pre-transplant IFN-gamma ELISPOTs are associated with post-transplant renal function in African American renal transplant recipients. Am J Transplant (2005) 5:1971–5. doi: 10.1111/j.1600-6143.2005.00958.x
41. Tambur AR, Campbell P, Claas FH, Feng S, Gebel HM, Jackson AM, et al. Sensitization in Transplantation: Assessment of Risk (STAR) 2017 Working Group Meeting Report. Am J Transplant (2018) 18:1604–14. doi: 10.1111/ajt.14752
42. Tambur AR, Campbell P, Chong AS, Feng S, Ford ML, Gebel H, et al. Sensitization in Transplantation: Assessment of Risk (STAR) 2019 Working Group Meeting Report. Am J Transplant (2020) 20:2652–68. doi: 10.1111/ajt.15937
43. Nelsen MK, Beard KS, Plenter RJ, Kedl RM, Clambey ET, Gill RG. Disruption of Transplant Tolerance by an ‘Incognito’ Form of CD8+ T Cell-Dependent Memory. Am J Transplant (2017) 17:1742–53. doi: 10.1111/ajt.14194
44. Alegre ML, Mannon RB, Mannon PJ. The microbiota, the immune system and the allograft. Am J Transplant (2014) 14:1236–48. doi: 10.1111/ajt.12760
45. Lei YM, Chen L, Wang Y, Stefka AT, Molinero LL, Theriault B, et al. The composition of the microbiota modulates allograft rejection. J Clin Invest (2016) 126:2736–44. doi: 10.1172/JCI85295
46. Martin JM, Danziger-Isakov LA. Cytomegalovirus risk, prevention, and management in pediatric solid organ transplantation. Pediatr Transplant (2011) 15:229–36. doi: 10.1111/j.1399-3046.2010.01454.x
47. Gala-Lopez BL, Senior PA, Koh A, Kashkoush SM, Kawahara T, Kin T, et al. Late cytomegalovirus transmission and impact of T-depletion in clinical islet transplantation. Am J Transplant (2011) 11:2708–14. doi: 10.1111/j.1600-6143.2011.03724.x
48. Zamora MR. DNA viruses (CMV, EBV, and the herpesviruses). Semin Respir Crit Care Med (2011) 32:454–70. doi: 10.1055/s-0031-1283285
49. Kovacs CS Jr., Koval CE, van Duin D, de Morais AG, Gonzalez BE, Avery RK, et al. Selecting suitable solid organ transplant donors: Reducing the risk of donor-transmitted infections. World J Transplant (2014) 4:43–56. doi: 10.5500/wjt.v4.i2.43
50. Welsh RM, Markees TG, Woda BA, Daniels KA, Brehm MA, Mordes JP, et al. Virus-induced abrogation of transplantation tolerance induced by donor-specific transfusion and anti-CD154 antibody. J Virol (2000) 74:2210–8. doi: 10.1128/JVI.74.5.2210-2218.2000
51. Forman D, Welsh RM, Markees TG, Woda BA, Mordes JP, Rossini AA, et al. Viral abrogation of stem cell transplantation tolerance causes graft rejection and host death by different mechanisms. J Immunol (2002) 168:6047–56. doi: 10.4049/jimmunol.168.12.6047
52. Chen L, Wang T, Zhou P, Ma L, Yin D, Shen J, et al. TLR engagement prevents transplantation tolerance. Am J Transplant (2006) 6:2282–91. doi: 10.1111/j.1600-6143.2006.01489.x
53. Thornley TB, Brehm MA, Markees TG, Shultz LD, Mordes JP, Welsh RM, et al. TLR agonists abrogate costimulation blockade-induced prologation of skin allografts. J Immunol (2006) 176:1561–70. doi: 10.4049/jimmunol.176.3.1561
54. Streblow DN, Hwee YK, Kreklywich CN, Andoh T, Denton M, Smith P, et al. Rat Cytomegalovirus Vaccine Prevents Accelerated Chronic Rejection in CMV-Naive Recipients of Infected Donor Allograft Hearts. Am J Transplant (2015) 15:1805–16. doi: 10.1111/ajt.13188
55. Parker DC, Greiner DL, Phillips NE, Appel MC, Steele AW, Durie FH, et al. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA (1995) 92:9560–4. doi: 10.1073/pnas.92.21.9560
56. Markees TG, Phillips NE, Noelle RJ, Shultz LD, Mordes JP, Greiner DL, et al. Prolonged survival of mouse skin allografts in recipients treated with donor splenocytes and antibody to CD40 ligand. Transplantation (1997) 64:329–35. doi: 10.1097/00007890-199707270-00026
57. Honey K, Cobbold SP, Waldmann H. CD40 ligand blockade induces CD4+ T cell tolerance and linked suppression. J Immunol (1999) 163:4805–10.
58. Rajewsky K, Schirrmacher V, Nase S, Jerne NK. The requirement of more than one antigenic determinant for immunogenicity. J Exp Med (1969) 129:1131–43. doi: 10.1084/jem.129.6.1131
59. Cassell D, Forman J. Linked recognition of helper and cytotoxic antigenic determinants for the generation of cytotoxic T lymphocytes. Ann N Y Acad Sci (1988) 532:51–60. doi: 10.1111/j.1749-6632.1988.tb36325.x
60. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature (1998) 393:474–8. doi: 10.1038/30989
61. Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature (1998) 393:480–3. doi: 10.1038/31002
62. Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature (1998) 393:478–80. doi: 10.1038/30996
63. Gill RG. T-cell-T-cell collaboration in allograft responses. Curr Opin Immuno (1993) 5:782–7. doi: 10.1016/0952-7915(93)90137-H
64. Nelsen MK, Beard KS, Plenter RJ, Kedl RM, Clambey ET, Gill RG. Disruption of Transplant Tolerance by an “Incognito” Form of CD8 T Cell-Dependent Memory. Am J Transplant (2017) 17:1742–53. doi: 10.1111/ajt.14194
65. Valujskikh A, Pantenburg B, Heeger PS. Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant (2002) 2:501–9. doi: 10.1034/j.1600-6143.2002.20603.x
66. Gorbacheva V, Fan R, Fairchild RL, Baldwin WM, Valujskikh A. Memory CD4 T Cells Induce Antibody-Mediated Rejection of Renal Allografts. J Am Soc Nephrol (2016) 27:3299–307. doi: 10.1681/ASN.2015080848
67. Cortes-Cerisuelo M, Laurie SJ, Mathews DV, Winterberg PD, Larsen CP, Adams AB, et al. Increased Pretransplant Frequency of CD28(+) CD4(+) T(EM) Predicts Belatacept-Resistant Rejection in Human Renal Transplant Recipients. Am J Transplant (2017) 17:2350–62. doi: 10.1111/ajt.14350
68. Mathews DV, Wakwe WC, Kim SC, Lowe MC, Breeden C, Roberts ME, et al. Belatacept-Resistant Rejection Is Associated With CD28(+) Memory CD8 T Cells. Am J Transplant (2017) 17:2285–99. doi: 10.1111/ajt.14349
69. Adams AG, Pearson TC, Larsen CP. Heterologous immunity: An overlloked barrier to tolerance. Immunol Rev (2003) 196:147–60. doi: 10.1046/j.1600-065X.2003.00082.x
70. Merritt E, Londoño MC, Childs K, Whitehouse G, Kodela E, Sánchez-Fueyo A, et al. On the impact of hepatitis C virus and heterologous immunity on alloimmune responses following liver transplantation. Am J Transplant (2020). doi: 10.1111/ajt.16134
71. Dangi A, Yu S, Lee FT, Burnette M, Wang JJ, Kanwar YS, et al. Murine cytomegalovirus dissemination but not reactivation in donor-positive/recipient-negative allogeneic kidney transplantation can be effectively prevented by transplant immune tolerance. Kidney Int (2020) 98:147–58. doi: 10.1016/j.kint.2020.01.034
72. Danziger-Isakov L, Cherkassky L, Siegel H, McManamon M, Kramer K, Budev M, et al. Effects of influenza immunization on humoral and cellular alloreactivity in humans. Transplantation (2010) 89:838–44. doi: 10.1097/TP.0b013e3181ca56f8
Keywords: immune memory, autoimmunity, tolerance, transplantation, infection, vaccination
Citation: Gill RG and Burrack AL (2020) Diverse Routes of Allograft Tolerance Disruption by Memory T Cells. Front. Immunol. 11:580483. doi: 10.3389/fimmu.2020.580483
Received: 06 July 2020; Accepted: 24 September 2020;
Published: 08 October 2020.
Edited by:
Xunrong Luo, Duke University, United StatesReviewed by:
Anna Valujskikh, Cleveland Clinic, United StatesXian Chang Li, Houston Methodist Hospital, United States
Copyright © 2020 Gill and Burrack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronald G. Gill, cm9uYWxkLmdpbGxAY3VhbnNjaHV0ei5lZHU=