 Anna Julia Pietrobon
Anna Julia Pietrobon Franciane Mouradian Emidio Teixeira
Franciane Mouradian Emidio Teixeira Maria Notomi Sato
Maria Notomi Sato
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 October 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.579220
This article is part of the Research Topic Immunosenescence and Clinical Consequences View all 10 articles
Old individuals are more susceptible to various infections due to immunological changes that occur during the aging process. These changes named collectively as “immunosenescence” include decreases in both the innate and adaptive immune responses in addition to the exacerbated production of inflammatory cytokines. This scenario of immunological dysfunction and its relationship with disease development in older people has been widely studied, especially in infections that can be fatal, such as influenza and, more recently, COVID-19. In the current scenario of SARS-CoV-2 infection, many mechanisms of disease pathogenesis in old individuals have been proposed. To better understand the dynamics of COVID-19 in this group, aspects related to immunological senescence must be well elucidated. In this article, we discuss the main mechanisms involved in immunosenescence and their possible correlations with the susceptibility of individuals of advanced age to SARS-CoV-2 infection and the more severe conditions of the disease.
Human history is marked by major epidemics, and viral respiratory infections have been major villains in this scenario. The 20th century was certainly marked by the devastating outbreak of the Spanish flu, caused by an influenza A virus of the H1N1 subtype (1). Currently, in the 21st century, coronaviruses appear to show their potential, with three epidemics in the past two decades.
Coronavirus epidemics include severe acute respiratory syndrome (SARS) coronavirus (CoV) (SARS-CoV-1), which occurred between 2002 and 2003, and Middle East respiratory syndrome (MERS)-CoV, which occurred in 2012 (2). Since December 2019, SARS-CoV-2, a new type of coronavirus, has caused respiratory infections ranging from mild to severe clinical conditions and death, and the disease caused by it has been called coronavirus disease 2019 (COVID-19) (3).
The current SARS-CoV-2 outbreak originated in the city of Wuhan in China (4), rapidly spread worldwide and was declared a pandemic by the World Health Organization. By September 20, 2020, COVID-19 had already infected more than 30 million people and caused over 950,000 deaths (5). In this global pandemic scenario, the United States and Brazil are the countries with the highest number of cases and deaths from COVID-19 (5). In Brazil, approximately 51% of SARS cases due to COVID-19 occur in old individuals (over 60 years of age), accounting for 73% of deaths (6).
In fact, it was observed that older people have a higher severity of the disease and are considered the main risk group for COVID-19 (4). This observation has been reinforced by SARS-CoV-2 infection in experimental models, where infected aged Syrian hamsters developed alveolar and perivascular edema (7). A greater severity in individuals of advanced age has also been reported in SARS-CoV-1 (8) and MERS (9). Interestingly, in addition to advanced age, male gender appears to be another risk factor for COVID-19 (10). Additionally, other conditions such as obesity, hypertension, and metabolic diseases are also risk factors for COVID-19 (11).
The following additional four coronaviruses circulate globally among the population: alpha (229E and NL63) and beta (OC43 and HKU1) CoVs. These coronaviruses generally cause mild infections of the upper respiratory tract similar to the common cold (12). However, there are reports of more severe respiratory diseases caused by OC43 and 229E, mainly in older individuals and individuals with chronic immune deficits (13).
Thus far, it is known that cytokine storm in the lungs may be among the immunological components involved in the pathogenesis of COVID-19 in the aged population. Although it has been suggested that alveolar macrophages from older individual have an anti-inflammatory profile, they can develop higher and uncontrolled responses of cellular activation and cytokine production after a pathogen insult and a lower ability to control tissue damage due to infection leaving the lung in a compromised state (14–16). In fact, at baseline state, the lungs of old individuals show increase in levels of complement and surfactant proteins and pro-inflammatory cytokines (15, 16). Interestingly, half of fatal cases of COVID-19 experience a cytokine storm, of which 82% are over the age of 60 (17).
Notably, a series of immunological changes occur with age, causing older individuals to develop immunosenescence (18). These factors can contribute to as pulmonary as systemic exacerbated inflammatory response in older individuals and seem to play a role in increasing susceptibility to respiratory infections.
In fact, a better knowledge of these mechanisms can contribute to the understanding of the infection dynamics in this scenario. Thus, here, we review the main factors related to the senescence of the innate and adaptive immune responses that can be responsible for both the severity and pathogenesis of COVID-19.
Coronaviruses have a positive single-stranded RNA genome of approximately 30 kb that forms the viral nucleocapsid with the nucleocapsid (N) protein. This structure is surrounded by an envelope formed of a lipid bilayer in which the spike (S) proteins, membrane (M) protein, and envelope (E) protein are inserted (19).
The coronavirus subfamily consists of four genera, i.e., α, β, γ, and δ coronaviruses, and the α and β genera are responsible for human infections (20, 21). Among the coronaviruses that infect humans, seven are known to cause diseases with flu-like symptoms, but SARS-CoV-1, MERS-CoV, and, more recently, SARS-CoV-2 have gained greater notoriety for their high transmission capacity and severe infections (22–24).
The transmission of coronaviruses occurs mainly through respiratory droplets and close contact between people. Once in the body, the viruses enter target cells when protein S binds to specific input receptors. SARS-CoV-2 S protein binds angiotensin-converting enzyme 2 (ACE2), which is present on the surface of several human cells (25). In addition, several studies have been suggesting that the MERS-CoV receptor, dipeptidyl peptidase 4 (DPP4), can also be used by SARS-CoV-2 during infection (26, 27). The interaction between SARS-CoV-2 and ACE2 recruits the transmembrane protease serine 2 (TMPRSS2), which promotes S protein priming and facilitates viral entry in the host cell (28). Other cellular proteins, such as the protease furin, can also promote SARS-CoV-2 S protein cleavage indicating their potential involvement in viral entrance (29). Once inside the cell, the envelope fuses with the endosomal membrane and releases the viral genome into the cytoplasm where replication and assembly of new viral particles occurs (30).
Coronavirus infection can affect the airways, causing cough, headache, and fever. In more severe cases, the infection can cause tissue damage, especially to the lung tissue, due to the high degree of inflammation generated to fight the virus, leading to the development of pneumonia and dyspnea, which can progress to death (31). Among patients with COVID-19, the highest incidence of severe cases occurs in individuals affected by comorbidities such as lung diseases, diabetes, and hypertension (32–34). Age also appears to be a risk factor for the disease, as worse outcomes and higher mortality rates are observed in older patients (35–37).
The aging process can be understood as a progressive and natural decrease in the biological functions of an organism (18). Despite its enormous plasticity and capacity for renewal, the immune system is also affected during the aging process. Since a functional immune response is essential for maintaining homeostasis and health, the immune aging process, called immunosenescence (Figure 1), contributes to the increased susceptibility to infections, cancers and autoimmune diseases (38–40).
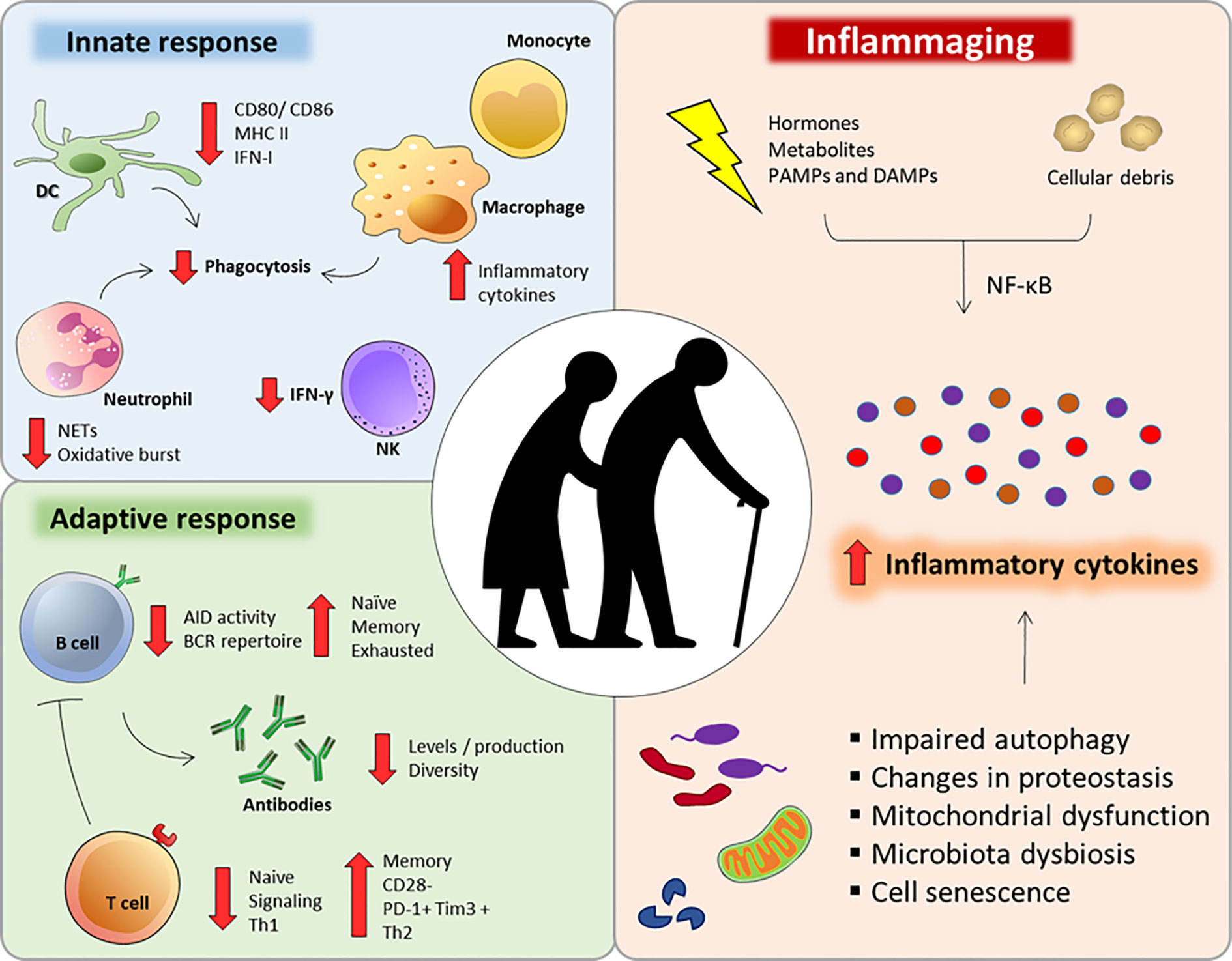
Figure 1 Major immunological alterations observed during immunosenescense. Aging interferes in a number of innate and adaptive immune cells aspects that can impair or compromise their function and response. Additionally, several factors can dysregulate intracellular homeostasis during aging, intensifying the secretion of inflammatory cytokines and chemokines (inflammaging).
A very striking feature of the immunosenescence process is a low-grade proinflammatory state, with an increase in serum inflammatory mediators, such as IL-6, IL-1RA, TNF-α, IL-1, and C-reactive protein (CRP) (41, 42). This low-grade inflammatory state named “inflammaging” is associated with the diminished ability to mount efficient immune responses during the aging process (42) (Figure 1).
Inflammaging is caused by a set of hormonal, metabolic and immune factors that constantly provide stimuli that are recognized by innate receptors, favoring an inflammatory environment (43). In addition, senescent cells commonly experience changes in their intracellular homeostasis, including telomeric perturbations and oxidative stress, leading to the activation of signaling pathways such as nuclear factor κB (NF-κB) and increased secretion of cytokines, chemokines, growth factors and lipids (44, 45). This condition in which senescent cells change their secretory phenotype is called the senescence-associated secretory phenotype (SASP) and is a potential contributor to inflammaging (46). The exacerbated inflammatory process associated with age may also be due to a failure to resolve inflammation since many regulatory factors are also deficient in older individuals (47–49).
The inflammatory stimuli that support the phenomenon of inflammaging can be triggered by several factors, including chronic infections and microbiota changes, which are going to be more detailed further in this text. However, sterile components naturally produced during cell cycle can also contribute to this phenomenon. Cellular debris resulting from the cell death process that occurs daily due to chemical and physical stresses as well as the accumulation of metabolic products and cellular catabolic products, such as lipofuscins and beta-amyloid proteins play a crucial role in inflammaging (50, 51). Under the physiological conditions of cell proliferation, such components are usually diluted between dividing cells. However, as the cell proliferation rate reaches its lowest levels due to aging, these molecules accumulate and can be recognized by pattern recognition receptors (PRRs) (52, 53).
In addition, infectious processes during aging can further accentuate the inflammatory condition by releasing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (54). During cytomegalovirus (CMV) infection, which infects 40–100% of the population worldwide (55), inflammatory mediators such as prostaglandin E2, IL-6 and TNF-α are released, highlighting the important contribution of this pathogen to inflammaging (56–58). However, a 10-year longitudinal study compared the impact of CMV infection on the serum levels of inflammatory cytokines in 249 individuals and showed that cytokine production in CMV-seropositive and CMV-seronegative individuals is similar (59).
Studies focusing on the current SARS-CoV-2 pandemic have already investigated the association between the pathogenesis of the disease and the inflammatory process. Regardless of the age group, patients affected by COVID-19 have higher plasma concentrations of inflammatory cytokines, such as TNF-α and IL-6, and the chemokines and molecules that activate cells, such as CXCL10, CCL2, CCL3, G-CSF, IL-2, IL-7, and IL-10 (24, 60). There is also a circulating increase in others well-known inflammatory markers, such as CRP, ferritin, D-dimer, and serum amyloid A (SAA) (61–64).
Additionally, in vitro SARS-CoV-1 studies have found that the viral cytopathic effect induces apoptosis in Vero E6 and HEK293 cells (65, 66) and that MERS-CoV promotes apoptosis in lung and kidney cells via Smad7 and FGF2 (67). A similar effect has also been observed in the HCoV virus (229E) in monocytic cells (68). These findings suggest that target cell apoptosis is a factor contributing to the tissue damage caused during in vivo infection. Potential DAMPs released during apoptosis can contribute to the local and systemic inflammatory response by activating PRRs, further aggravating the infection. Additionally, in silico studies have indicated that a strong protein-protein interaction exists between the viral S protein and TLR4, a PPR, suggesting that SARS-CoV-2 directly activates proinflammatory pathways (69).
One of the main intracellular pathways resulting from the activation of PRRs is NF-κB, which is the main pathway responsible for inducing the inflammatory response and the appearance of the SASP phenotype (70). DAMPs can also signal via the NLRP3 receptor, leading to the activation of the inflammasome pathway and secretion of the inflammatory cytokines IL-1β and IL-18 (71). Interestingly, serum IL-18 levels increase with age, indicating that the pathway strongly contributes to inflammaging (72). Higher levels of IL-18 were also observed in the serum of COVID-19 patients and were associated with disease severity and clinical outcome (73). Moreover, monocytes infected in vitro with SARS-CoV-2 presented the formation of NLRP3 puncta, and the same could be observed in mononuclear cells isolated from COVID-19 patients, indicating activation of the inflammasome pathway (73). In fact, NLRP3 inhibitors have already been proposed as potential drugs for the treatment of COVID-19 (74).
In addition, the autophagy pathway seems to be directly related to the development and progression of the inflammaging process. This pathway consists of specialized protein machinery that promotes the recycling of cellular content, generating nutrients and energy for maintaining homeostasis (75). Therefore, autophagy contributes to the elimination of the debris and products of cellular metabolism, preventing its recognition by PRRs and the consequent inflammation (76). However, it has been shown that there is a reduction in the activity of the autophagy pathway during aging (77). Additionally, deficiencies in other pathways that regulate proteostasis during aging, such as reduced proteasome activity, contribute to the accumulation of misfolded protein aggregates that can activate inflammatory pathways (78).
Preliminary studies in DAF2 mutant invertebrates, a longevity study model, indicate that silencing autophagy pathway genes reduces life expectancy in these organisms (79). Additionally, in a clinical trial, Mannick et al. (2018) demonstrated that enhancing the autophagy pathway using mTOR inhibitors reduces the incidence of infections in older individuals and promotes the expression of antiviral genes and a better response to vaccination against the influenza virus, corroborating the importance of the autophagy pathway in the immune response and fighting infections in individuals of advanced age (80).
Another consequence of reduced autophagy during aging is a lower rate of mitophagy, which leads to the accumulation of dysfunctional and damaged mitochondria, changes in the respiratory chain and the generation of reactive oxygen species (ROS) (81, 82). Oxidative phosphorylation products, such as ATP and ROS, induce an inflammatory response by activating the inflammasome pathway (83, 84). In an experimental model, it has been verified that the influenza virus induces the production of mitochondrial ROS, contributing to inflammation, higher viral titers and increased neutrophil infiltration in the airways and lungs (85). It has also been found that oxidative stress generated by H5N1 infection induces the formation of oxidized phospholipids, which activate the TLR4-TRIF pathway in pulmonary macrophages, inducing the inflammatory response (86). In fact, in the context of COVID-19, it was recently shown that mitoquinol and N-acetyl cysteine, two antioxidant drugs, prevented SARS-CoV-2 infection in human primary monocytes (87).
Additionally, mitochondrial lesions generated by stress lead to the release of DAMPs, such as mitochondrial DNA (mtDNA) rich in CpG motifs and bacterial DNA, and, therefore, can activate the inflammatory response via TLRs, NLRs and cGAS (88, 89). In this context, a positive correlation was found between the increase in mtDNA and proinflammatory cytokines such as TNF-α, IL-6 and CCL5 during aging (90).
Furthermore, it has been speculated that mitochondrial dysfunctions could be involved in the older population’s greater susceptibility to viral infections since the functioning of MAVS, a protein that assists the RIG-IRF-IFN cascade located in the mitochondrial membrane, depends on the integrity of the mitochondria and oxidative phosphorylation (91, 92). SARS-CoV-1 infection induces mitochondrial fission and MAVS degradation, suppressing the host’s antiviral response (93).
The intestinal microbiota can also play an important role in modulating the proinflammatory response during aging (94, 95). Over time, the composition and diversity of the microbiome changes, leading to dysbiosis in the host and a predominance of Th1-type responses (95, 96). Simultaneously, there is an increase in intestinal permeability with aging, favoring bacterial translocation and inflammaging (95). It has been observed that centenary individuals have a greater prevalence of opportunistic bacteria with proinflammatory characteristics in the intestinal microbiota and a reduced number of bacteria with anti-inflammatory properties. These data are strongly correlated with the serum levels of inflammatory cytokines such as IL-6 and IL-8, suggesting that the microbiota also contributes to the maintenance of inflammaging (97).
Some studies suggest that an exacerbated immune response is mainly responsible for the worsening of SARS-CoV-1 and MERS-CoV infections by contributing more to tissue damage than the actual infection, regardless of the age group (98). Regarding COVID-19, these effects do not seem to be any different. Excessive immune activation and production of proinflammatory cytokines are commonly observed in patients with COVID-19 (99). This exacerbated immune response involving high levels of cytokine release is known as cytokine storm syndrome. Although inflammatory responses are crucial for pathogen clearance, uncontrolled immune responses can be destructive by leading to systemic inflammation, vascular hyperpermeability, multiple organ failure and eventually death (100). In viral infections that reach the lungs, cytokine storm syndrome contributes to apoptosis in epithelial and endothelial cells, leading to fluid leakage in the lungs, the accumulation of leukocytes and tissue fibrosis (101), which, in turn, cause ARDS (102).
Considering the abovementioned aspects, it is possible that the inflammaging process favors the greater severity of COVID-19 in the aged population (Figure 2B). Although experimental reports are still scarce in the literature, several researchers have proposed that inflammaging could contribute to the more severe outcomes of COVID-19 in older patients (10, 103). In fact, Guaraldi and colleagues demonstrated that treatment with tocilizumab, a monoclonal anti-IL-6 receptor antibody, could attenuate COVID-19 severity in patients older than 60 years (104).
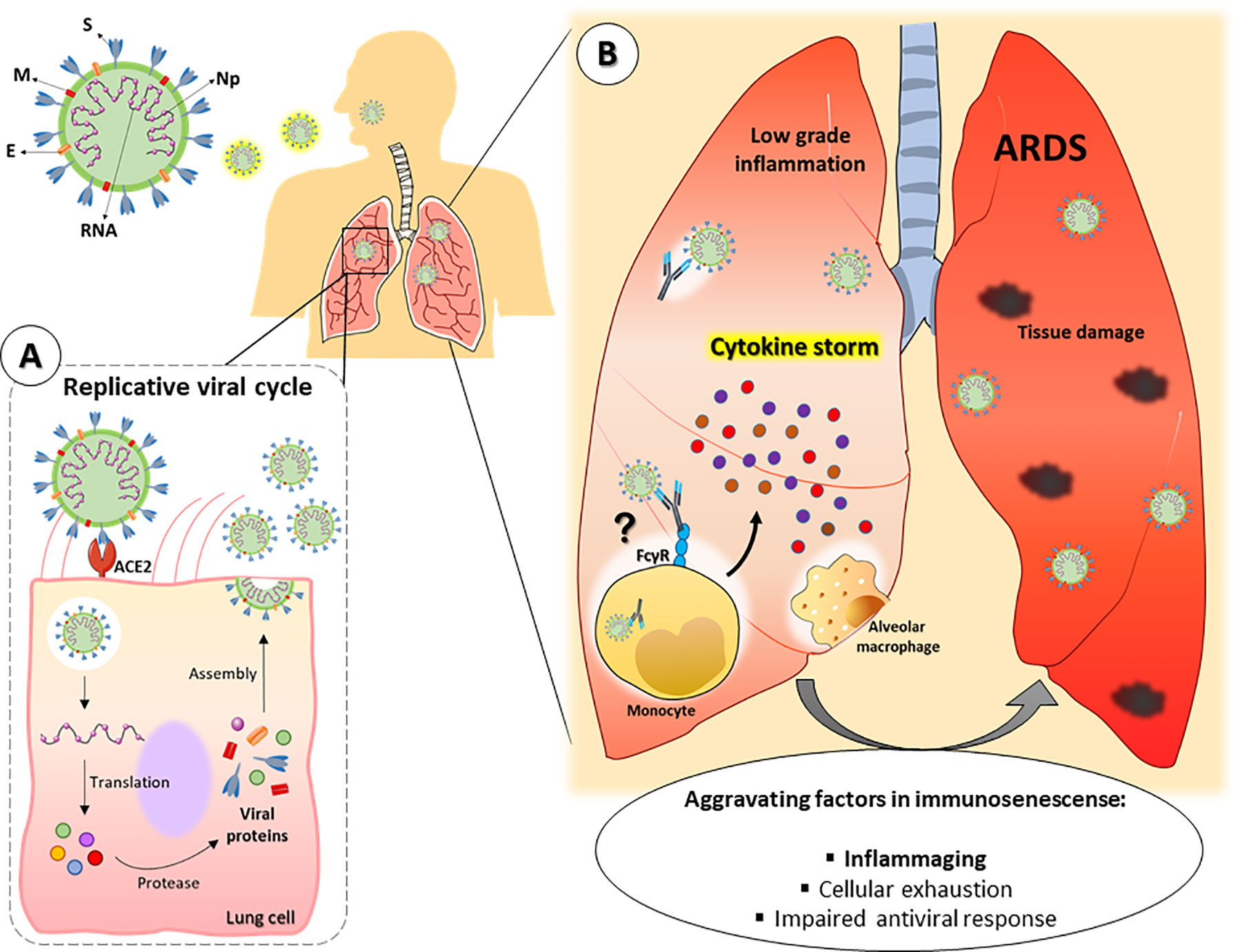
Figure 2 Hypothetical framework of SARS-CoV-2 pulmonary infection in old individuals. SARS-CoV-2 consists of single RNA strand and the following proteins: Spike (S), membrane (M), envelope (E) and nucleocapsid (Np). (A) After entering the organism, the virus infects lung cells by binding to the receptor angiotensin-converting enzyme 2 (ACE-2) and establishes its replicative cycle releasing new viral particles. (B) Older people have a constitutive low-grade proinflammatory state that, along with other peculiarities of the immunosenescence, can favor the cytokine storm syndrome, leading to a faster progression to ARDS and severe manifestations of COVID-19. In addition, tissue resident or lung-infiltrating immune cells (e.g., neutrophils, monocytes and alveolar macrophages) can contribute to disease severity either by dysfunctional responses associated to immunosenescence or by facilitating viral internalization through ADE. ARDS = Acute respiratory distress syndrome. ADE = Antibody-dependent enhancement.
The cells of the innate immune system can be quantitatively and qualitatively affected by the aging process. In the case of monocytes, there is a prevalence of nonclassical and intermediate subtypes associated with a lower phagocytic capacity (43). Monocytes from older individuals also secrete less IFN-α, IFN-γ, IL-1β, CCL20 and CCL8 when stimulated with adjuvants of the innate immune response (105), although some studies have suggested that these cells have a greater capacity to secrete proinflammatory cytokines under baseline conditions or after stimulation in older individuals (106–109). Recently, Zheng and colleagues reported an increase in the monocyte population in aged healthy adults, especially classical CD14 monocytes (110). Monocytes from aged individuals have higher expression of inflammatory genes, such as IL1B, TNF and CXCL8, and increased activation of the NF-κB, Toll-like receptor, inflammasome, and MAPK signaling pathways (110).
In the case of infection by SARS-CoV-2, there is a greater production of IL-6 and GM-CSF in the peripheral blood by CD14+ CD16+ monocytes (61). Additionally, in vitro infection of human monocytes with SARS-CoV-2 leads to the production of several inflammatory cytokines, such as IL-6, IL-1β, TNF-α, and IFN-I (87). Moreover, COVID-19 patients of advanced age have more monocytes than younger patients (110). These cells with an inflammatory profile can migrate into the lungs, contributing to the exacerbated inflammatory response and consequent tissue damage characteristic of the pathogenesis of the disease. In fact, because of their inflammatory properties, monocytes have been suggested to be among the main contributors to the disparate severity of COVID-19 in older patients (111).
The changes occurring in immunosenescence also affect antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages (112). In both cell populations, antigen presentation is compromised in the old population, possibly due to the lower expression of CD80, CD86 and MHC-II after exposure to a stimulus (113, 114) and a lower production of superoxide anion by macrophages after treatment with IFN-γ (115). In vitro studies investigating DCs derived from peripheral blood monocytes show that infection with MERS-CoV induces the expression of MHC-II and CD86 and promotes the production of IFN-γ, CXCL10, IL-12, and CCL5 (116). However, whether DCs from the older people respond similarly to infection by MERS or other coronaviruses is unclear.
In addition, SARS-CoV-1 is capable of infecting monocyte-derived DCs, rendering these cells producers of inflammatory cytokines, such as TNF-α and IL-6, and chemokines, such as CCL2, CCL3, CCL5 and CXCL10 (117). However, it was not possible to identify the production of antiviral cytokines, such as IFN-α, IFN-β, IFN-γ and IL-12p40, which may indicate a possible viral escape mechanism mediated by blocking these pathways. In fact, Hu et al. showed that the SARS-CoV-1 N protein interacts with TRIM25, preventing the generation of IFN-I via RIG-I (118). In COVID-19 patients, an imbalanced production of IFN-I has also been reported, and it seems to correlate with disease severity (119, 120). Moreover, SARS-CoV-2 infection elicits reduced expression of IFN-I and interferon-stimulated genes (121). In addition, pretreatment with IFN-α or IFN-β reduced SARS-CoV-2 titers in in vitro infection studies (122, 123). In fact, IFN-I administration has shown promising results in COVID-19 patient clinical trials (124). In a phase 2 study, the triple combination of lopinavir–ritonavir, ribavirin and interferon beta-1b was efficient in reducing symptoms, shortening the duration of infection and hospital stay in patients with mild to moderate COVID-19 (125).
Interestingly, in old individuals, the population of plasmacytoid DCs (pDCs), which is among the main mechanisms of fighting viral infections, is reduced and has less capacity for IFN-α secretion when stimulated with influenza virus due to the deficient expression of TLR7 and TLR9 (126–128). Complementarily, aged human monocytes have imbalanced IFN-I and IFN-III production in response to influenza infection due to defective induction of IFN transcription (129). Taken together, these findings suggest that a reduced IFN-I response in advanced age can contribute to poor clinical outcomes of COVID-19.
Another peculiarity that is possibly associated with the greater susceptibility of old individuals to viral infections is the reduced ability of DCs to perform cross-presentation due to dysfunction in mitochondrial activity and changes in the membrane potential of this organelle (130). These data indicate that the greater susceptibility of the advanced age population to infections may be associated with a lower functional capacity of phagocytes to eliminate pathogens and promote adequate activation of the adaptive immune response.
Aging also contributes to changes in alveolar macrophages (131, 132). During aging, there is a reduction in this cell population in the lungs, which is associated with the downregulation of pathways related to the cell cycle and upregulation of pathways associated with the inflammatory response (14). In fact, alveolar macrophages in animals with an advanced age are in a greater state of cellular activation, secrete more proinflammatory cytokines in response to a Mycobacterium tuberculosis stimulus and are refractory to an IFN-γ stimulus (16). Additionally, studies using murine models of influenza infection indicate that alveolar macrophages have a lower ability to control tissue damage due to infection (14). In addition, there is lower expression of the CD204 receptor, suggesting a reduced phagocytosis capacity of cellular debris that could contribute to increased tissue damage (14). In an experimental model of infection by coronavirus hepatitis virus type 1 (MHV-1), the depletion of alveolar macrophages contributes to the reduction in mortality and morbidity caused by the infection (133). In fact, SARS-CoV-2 infection in transgenic mice bearing human ACE2 leads to macrophage infiltration into the alveolar interstitium and alveolar cavities (134), and macrophage activation syndrome is associated with severe respiratory failure in COVID-19 patients (135), suggesting that this cell population plays a crucial role in the pathogenesis of the disease.
Neutrophils in old individuals are also affected by the immunosenescence process. During infections in older people, neutropenia may occur due to the lower proliferative capacity of neutrophil progenitor cells when stimulated by G-CSF (136). However, in SARS-CoV-1 infection, an increase in circulating neutrophils and an association between the infiltrates of this cell type in the lung and the severity of the injury have been observed (137). A similar scenario is observed in SARS-CoV-2 infection, where high neutrophil to lymphocyte ratio in peripheral blood have been reported in severely ill patients (138). Besides, lung infiltration of neutrophils was observed in autopsied COVID-19 patients, revealing capillary extravasation and neutrophilic mucositis (139). These findings indicate that neutrophils not only contribute to systemic inflammation in COVID-19 but also play a crucial role in local tissue damage.
Other characteristics of senescent neutrophils include lower microbicidal activity and a deficiency in the phagocytosis of opsonized bacteria, possibly due to a reduction in CD16 and the oxidative burst mediated by Fc-type receptors (140, 141). Some studies even suggest a deficiency in chemotaxis and release of ROS and neutrophil extracellular traps (NETs) in neutrophils in old individuals (140, 142, 143). However, there is a higher release of NETs in COVID-19 patients, and plasma from infected subjects induces NET release in neutrophils from healthy donors, indicating the participation of these cells in the immunopathogenesis of the disease (144).
The natural killer (NK) cell response is also compromised in old individuals. There is a prevalence of NK CD56dim cells, a cell population with high cytotoxic capacity and production of IFN-γ, and a decrease in NK CD56bright cells, which have a high capacity for cytokine and chemokine production (145, 146). NK cells produced in older people also produce less IFN-γ in the absence of stimulation, which helps to explain the greater susceptibility to viral infections during this stage of life (147). In an animal model of influenza infection, a decrease in NK cells in the lungs, with less capacity for IFN-γ production and degranulation, was observed (148, 149). Similarly, clinical observations of patients with COVID-19 revealed a significant reduction in this cell population during SARS-CoV-2 infection (99). In addition, Zheng et al. reported impaired NK function in severe COVID-19 patients, expressing higher levels of the NKG2A receptor, a cellular exhaustion marker, indicating impaired antiviral immunity (150). However, single-cell analysis of lung bronchoalveolar immune cells revealed a significant increase of NK cells in patients with COVID-19 when compared to healthy controls (151). In addition, in a senescent mice model of SARS-CoV-1 infection, NK cells have been shown to migrate to the lungs (152), indicating a possible contribution of these cells in coronaviruses infection pathogenesis.
Taken together, these findings lead us to propose that innate immune cell dysfunction linked to immunosenescence could be involved in the greater COVID pathogenesis in old individuals either by promoting a less efficient response for fighting the infection and/or favoring an exacerbated inflammatory response.
Changes due to aging are also present in the adaptive immune response and are associated with the functional impairment of T and B lymphocytes (153). The sum of these changes renders old people vulnerable to new emerging infectious diseases, as recently observed with SARS-CoV-2. The most prominent factor involves a decrease in the number of naïve cells because of thymic involution (154), an increase in memory/exhausted T cells and a reduction in B cell progenitors in the bone marrow (155). Consequently, these changes reflect the cumulative effect of previous and persistent infections in older individuals (156).
Initial studies involving patients with COVID-19 in China have observed decrease in peripheral lymphocytes was observed (24, 32). This lymphopenia was more prominent in the cases with more severe disease, and 42% of these patients were aged ≥65 years (32). SARS-CoV-1 patients also have been reported to have reduced circulating CD4+ and CD8+ T cells (157, 158). Indeed, in more severe cases of COVID-19, there is a reduction in CD8+ T cells (159), which could prevent an adequate cytotoxic response to fight the virus. Taken together, this profile has been proposed as a biomarker for diagnosis (160).
However, recent data from Arunachalam et al. evidences an increase in effector CD8+ T cells population in infected patients in comparison to health donors in an American and Chinese cohort (161). This could reflect the fact that COVID-19 has distinct effects in different population. In addition, the enhancement of effector T cells has been associated with recovery of SARS-CoV-2 infection (162, 163).
Whether the reduction in the number of T lymphocytes in old individuals could be a condition that predisposes such patients to more severe pathogenesis by COVID-19 remains unknown. However, analysis of immune cell sequencing showed that SARS-CoV-2 enhances T cell polarization from naïve to effector cells and that aging promotes the expression of SARS-CoV-2 susceptibility genes, mainly in T cells (110). In addition to lymphopenia, other age-related comorbidities are predictive of severe/critical cases and a high fatality rate during COVID-19 (37).
Individuals of advanced age have an increase in memory T cells with oligoclonal expansion and a decrease in the T cell receptor (TCR) repertoire (164, 165). These senescent T cells are mainly characterized not only by a low proliferative potential after activation but also by a shortening of telomeres and low telomerase activity, high production of ROS and constitutive activation of p38 MAP kinase, which once activated, blocks signaling via TCRs (166). Therefore, the inhibition of p38 MAPK could restore the proliferation and activation of telomerase in senescent T cells.
Phenotypically, senescent T lymphocytes can be identified by the expression of surface markers (CD28-, CD27-, CD57+ and CD45RA+) (167–169). In old individuals, the decrease of CD28 has been linked to persistent antigenic stimulation, and with each cycle of proliferation, its expression on the cell surface decreases (170). In addition, telomere shortening occurs, characterizing the process of replicative senescence in T lymphocytes (171).
CD28+CD27+ undifferentiated T cells have long telomeres, while highly differentiated or senescent CD28-CD27- cells have shortened telomeres (172). This phenotype (CD28-) is also observed in persistent human immunodeficiency virus (HIV), CMV infections and chronic inflammatory diseases such as rheumatoid arthritis (171). Under these conditions, a persistent antigenic stimulus occurs that leads to replicative senescence.
Several studies indicate that senescent T lymphocytes express the exhaustion molecules PD-1+ and Tim3+, a phenotype also observed in infections by lymphocytic choriomeningitis virus (LCMV), HIV and HCV (173). Exhausted cells have a low functional capacity, which could prevent the adequate cellular response to the virus, favoring viral escape and intensifying the pathogenesis of COVID-19 in old individuals. We base this hypothesis on studies showing that in SARS-CoV-2 infection, CD4+ and CD8+ T lymphocytes also have PD-1+ and Tim3+ expression, prominently in CD8+ T cells (159, 174).
In addition, changes in cytokine expression/secretion contribute to the development of immunological senescence. For example, IL-2 is decreased in old individuals, directly impacting the activation and proliferation of T cells (175, 176), which can lead to changes in the intensity and duration of the immune response and contribute to the immunosenescence process. In addition, senescent T cells also secrete high levels of the proinflammatory cytokines IFN-γ and TNF-α (166).
Regarding CD4+ helper T cells, the older people have a lower proportion of IFN-γ/IL-4 produced by memory CD4+ T cells, with increased Th2 cytokines and decreased Th1 cytokines, which may be a mechanism compensating for the increase in the proinflammatory state characteristic of the aging process (177). Interestingly, patients with severe COVID-19 (mean age of 61 years) also have decreased T-cell IFN-γ production (178). In addition, it has been shown that there is a lower frequency of memory CD4+ T cells producing IL-17 (179).
The functional impairment of the CD4+ T cell response contributes to the increase in pathology during influenza infection in old individuals (180). The same seems to be true for COVID-19 infections, since patients affected by the most severe form of the disease (mean age of 50 years) develop pathogenic Th1 lymphocytes that coexpress IFN-γ and GM-CSF and are associated with a hyperinflammatory response in the pathogenesis of the disease (61).
CD4+ T cells may also contribute to the production of cytokines in the cytokine storm, which is a main mechanism associated with the pathogenesis of COVID-19 in old individuals (181). In patients with severe COVID-19, CD4+ T cells express high levels of OX40 (159), a molecule involved in the production of cytokines by T cells (182).
However, an adequate balance between pro- and anti-inflammatory immune responses is essential for preserving health in old individuals. In fact, in severe cases of COVID-19, the evolution to acute respiratory distress syndrome (ARDS) and respiratory failure is a rapid process, which can occur before adaptive response establishment, emphasizing that excessive innate immunity (such as inflammaging) and inadequate regulatory responses may favor the evolution of the infection.
Regulatory T cells (Tregs) are potentially capable of suppressing the immune response and guaranteeing homeostasis (183). The number of naïve circulating Treg cells decreases while the number of memory Treg cells increases with age (184). Although both are suppressive, these different subtypes act at distinct sites in the body, according their expression of chemokine receptors. In addition increase in memory Treg cells is associated with a poor humoral response to influenza vaccination in older individuals (184). In mice, an increase in Treg cells at the expense of helper T cells has also been observed with age (185). Interestingly, patients with more severe COVID-19 present with fewer Treg cells than patients with less severe COVID-19 (186).
Immunosenescence studies are essential for understanding the greater susceptibility of older people to severe respiratory failure induced by viral infections. The presence of exhausted lymphocytes with a low functional capacity compromises the efficient antiviral cellular response, and changes in regulation favor the inflammatory status. These aspects appear to contribute to the severity of COVID-19 due to the cytokine storm.
Another important aspect of immunosenescence associated with the adaptive immune response concerns changes in B cells and the consequent failure of the humoral response. Memory B cells have a limited B cell receptor (BCR) repertoire, leading to a decrease in the humoral response to new antigens, with less efficient antibodies and less avidity (187).
A decrease in the ability to produce high-affinity antibodies in old individuals may result from defects in T cell signaling for the adequate activation of B cells, such as inadequate support mediated by T follicular helper cells (TFH) (188). Thus, many vaccines are ineffective in old individuals, rendeing them highly vulnerable to newly emerging pathogens, such as SARS and rapidly evolving viruses, such as influenza (189).
In an experimental model of influenza A infection, compared to young mice, aged mice showed a lower frequency of TFH cells and germinal center B cells, with reduced IgG titers but not IgM titers, but the IgM levels do not seem to depend on age (190). Thus, during the aging process, there may be some intrinsic impairment in B cells that compromises their functionality (191).
COVID-19 cohort studies show that seroconversion is observed on approximately the 10th day after symptom onset by increased IgM and IgG antibodies against the viral proteins N and S (60, 192). An age-dependent increase in the viral load (mean age of 66 years) was observed, but there was no correlation between age and the antibody levels. Interestingly, COVID-19 patients with associated comorbidities show lower levels of specific antibodies than COVID-19 patients without associated comorbidities (192).
A subset of B cells called age-associated B cells (ABCs) identified in mice has been closely related to the process of immunological senescence and minimally responds BCR and CD40 binding (193–195). ABCs have the potential to inhibit the growth of B cell precursors through the effects of TNF-α, inducing pro-B cell apoptosis (196, 197).
The transcription factor E47 is involved in the regulation of most B cell functions and is negatively regulated in splenic B cells in aged mice, promoting a reduction in the activation of activation-induced cytidine deaminase (AID) and class-switch recombination (198). In older humans, B cells have an age-dependent lower expression of E47 and AID, an associated decline in the number of memory B cells that have undergone class switching (IgG+ or IgA+) and an increase in naïve cells (IgG-/IgA-/CD27-) (199).
CD27 expression is related to somatically mutated B cell subsets (200, 201) and accordingly, CD27- and CD27+ B cells represent naïve and memory B cells, respectively. In fact, others studies also found higher number of naïve (CD27-) than memory (CD27+) B cells in individuals of advanced age (202–204).
Old people also have a reduction in the number of circulating B cells (205). In contrast, it has been observed that a double-negative (DN) B cell subtype (IgD-CD27-), which is the counterpart of ABCs in humans, is increased in the peripheral blood of older individuals (206). These DN B cells, also called late memory or exhausted cells, are associated with the failure to respond to the influenza vaccine in old individuals. DN B cells show SASP, with greater expression of proinflammatory cytokines (TNF, IL-6, and IL-8) and microRNAs associated with inflammation (miR 155/16/93) and are dependent on metabolic signaling via MAPK (207). These cells were increased in cases of chronic inflammation, such as HIV infection (208), and in systemic lupus erythematosus (209). For COVID-19, DN B cells are also significantly increased in severe patients (163, 210, 211) but is still unclear if this conditions is dependent on age.
In addition, recent studies have shown that seronegative healthy donors have SARS-CoV-2-specific CD4+ T cells, albeit at lower frequencies, which is indicative of cross-reactivity due to infection between circulating “common cold” coronaviruses (212–214). However, it is unknown whether the older individual could have previous cross-reactive antibodies to the new coronavirus.
In this context, a humoral immune response mechanism widely proposed to be associated with the severity of COVID-19 is related to the possible presence of a phenomenon called antibody-dependent enhancement (ADE) (215–217). ADE occurs when non-neutralizing antibodies generated in a previous viral infection bind Fcγ receptors (FcγR) present in host cells and promote viral internalization. This phenomenon has already been observed in dengue, yellow fever and HIV infection (218). In fact, ADE has also been demonstrated in other coronavirus infections, such as SARS-CoV-1 and MERS (215, 219). In COVID-19, ADE in phagocytes such as alveolar macrophages and lung-infiltrating monocytes could favor SARS-CoV-2 replication in the lung tissue (Figure 2B). In addition, the activation of these phagocytes through FcγR could contribute to the cytokine storm in these patients (220). Considering the decrease in the quality of antibody production in older individuals, it is reasonable to think that ADE could be involved in COVID-19 pathogenesis in advanced age patients.
As previously mentioned, different coronaviruses circulate among the population. Therefore, it is plausible that older people have been more exposed to these circulating viruses throughout their lives, thus generating a greater repertoire of antibodies, which could favor a more severe ADE-dependent COVID (Figure 2B). This hypothesis is reinforced since children show less susceptibility to SARS-CoV-2 infection (181) considering that their immune system is still developing and that they have had less time to be exposed to antigens. This hypothesis is also reinforced by the fact that some studies show rapid seroconversion to IgG in some patients with SARS-CoV-2 (221).
ADE can also occur when antibody concentrations decrease as a result of waning immunity, as observed by diluted antibodies for SARS-CoV-1 (219). Thus, high levels of antibodies can neutralize the virus, while subneutralizing concentrations could increase infection (222).
It is worth mentioning that highly neutralizing antibodies, such as those proposed to be generated by some SARS-CoV-2 candidate vaccines (223, 224) or those present in convalescent plasma used as treatment for some COVID-19 patients (225, 226), should not trigger ADE.
The IgG-mediated humoral response could also contribute to more severe pulmonary pathology. Compared to patients who recovered within the first 15 days after the onset of symptoms, the patients who died of SARS-CoV-1 had higher levels and faster development of neutralizing anti-S antibodies (227). In addition, in a nonhuman primate model, the previous presence of anti-S IgG antibodies resulted in more severe acute lung injury, with an increase in inflammatory cytokines (CCL2 and IL-8) and recruitment of monocytes/macrophages in the lung (228). These antibodies appear to promote activation via FcγR in these cells since their blockade reduced the inflammatory condition. The role of the virus-specific antibody response in lung injury in the pathogenesis of COVID-19 is still unknown.
The presence of immune complexes (ICs) worsens lung injury in viral infections by H1N1 influenza (229) and respiratory syncytial virus (230). Another severe lung disease has also been associated with IC deposition, which promotes not only FcγR-dependent cell activation but also complement system activation and consequent tissue damage (231). It is known that the aging process predisposes individuals to autoimmunity (232); however, whether the accumulation of ICs in old individuals is related to the severity of COVID-19 is unknown.
ICs have a high molecular weight, can be deposited in vessels and tissues, and can activate the complement system, thereby aggravating inflammation (233). In fact, the SARS-CoV-2 N protein has been shown to promote the activation of the complement system lectin pathway and aggravate lung injury in an animal model (234). In addition, these complement pathways were overactivated in the lungs of COVID-19 patients.
To date, no studies have proven that this senescent proinflammatory profile is dependent on B and T cells or other innate cell types and may in fact contribute to a more severe lung pathology in coronavirus-infected patients by increasing the inflammatory response and tissue injury.
Considering the clinical findings obtained thus far concerning SARS-CoV-2 infection and reports of diseases of a similar etiology, it is evident that the immunosenescence process, particularly the increased production of inflammatory cytokines resulting from inflammaging, plays a role in determining the prognosis of COVID-19 in old individuals. From an immunological perspective, the peculiarities of the immune system of older individuals may contribute to both the deficiency of effector mechanisms essential to fighting viral pathogens and the exacerbated inflammatory response, which can accelerate and intensify lung tissue damage. However, despite the strong evidence presented here, tests that accurately demonstrate the association between immunosenescence and the severity of COVID-19 are essential for assisting the search for treatments and the development of vaccines for this most affected age group.
AP and FT contributed equally to this work. All authors contributed to the article and approved the submitted version.
This work was supported by the Laboratório de Investigação Médica, Unidade 56, Department of Dermatology, School of Medicine, University of São Paulo, Brazil; Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP/n° 2018/18230-6 and no. 2017/18199-9).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ABC, Age-associated B cell; ACE2, Angiotensin-converting enzyme 2; ADE, Antibody-dependent enhancement; AID, Activation-induced cytidine deaminase; APC, Antigen-presenting cell; ARDS, Acute respiratory distress syndrome; ATP, Adenosine triphosphate; BCR, B cell receptor; CCL, CC chemokine ligand; CD, Cluster of differentiation; cGAS, Cyclic GMP-AMP synthase; CMV, Cytomegalovirus; CoV, Coronavirus; COVID-19, Coronavirus disease 2019; CpG, Cytosine-phosphate-Guanine; CRP, C-reactive protein; CXCL, C-X-C motif chemokine ligand; DAF2, Dauer formation-2; DAMP, Damage-associated molecular pattern; DC, Dendritic cell; DN, double-negative B cell; DNA, Deoxyribonucleic acid; DPP4, dipeptidyl peptidase 4; E47, E47 transcription factor; FGF2, Fibroblast growth factor 2; Fc, Fragment crystallizable; FcγR, Fc gamma receptors; G-CSF, Granulocyte colony-stimulating factor; GM-CSF, Granulocyte macrophage colony-stimulating factor; H1N1, Haemagglutinin-1 neuraminidase-1; H5N1, Haemagglutinin-5 neuraminidase-1; HCoV, Human coronavirus; HIV, Human immunodeficiency virus; IC, Immune complex; IFN-I, Interferon type I; Ig, Immunoglobulin; IL, Interleukin; IRF, Interferon regulatory factor; LCMV, Lymphocytic choriomeningitis virus; MAP, Mitogen-activated protein; MAPK, Mitogen-activated protein kinase; MAVS, Mitochondrial antiviral-signaling protein; MERS, Middle East respiratory syndrome; MHC, Major histocompatibility complex; MHV-1, Murine hepatitis virus type 1; mtDNA, Mitochondrial DNA; mTOR, Mammalian target of rapamycin; NET, Neutrophil extracellular trap; NF-κB, Nuclear factor kappa B; NK, Natural killer; NKG2A, CD94/NK group 2 member A; NLR, NOD-like receptor; NLRP3, NLR family pyrin domain containing 3; OX40, Tumor necrosis factor receptor superfamily, member 4 (TNFRSF4); PAMP, Pathogen-associated molecular pattern; PD-1, Programmed cell death 1; pDC, Plasmacytoid DC; PRR, Pattern recognition receptor; RIG, Retinoic acid-inducible gene; RNA, Ribonucleic acid; ROS, Reactive oxygen species; SAA, Serum Amyloid A; SARS, Severe acute respiratory syndrome; SARS-CoV-1, Severe acute respiratory syndrome coronavirus 1; SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; SASP, Senescence-associated secretory phenotype; Smad7, Mothers against decapentaplegic homolog 7; STING, Stimulator of interferon genes; TCR, T cell receptor; TFH, T follicular helper cell; Th, T helper cell; Tim3, T-cell immunoglobulin and mucin-domain containing-3; TLR, Toll like receptor; TMPRSS2, Transmembrane Protease Serine 2; TNF-α, Tumor necrosis factor alpha; Treg, Regulatory T cell; TRIF, TIR-domain-containing adapter-inducing interferon beta; TRIM, Tripartite motif.
ABC: Age-associated B cell
ACE2: Angiotensin-converting enzyme 2
ADE: Antibody-dependent enhancement
AID: Activation-induced cytidine deaminase
APC: Antigen-presenting cell
ARDS: Acute respiratory distress syndrome
ATP: Adenosine triphosphate
BCR: B cell receptor
CCL: CC chemokine ligand
CD: Cluster of differentiation
cGAS: Cyclic GMP-AMP synthase
CMV: Cytomegalovirus
CoV: Coronavirus
COVID-19: Coronavirus disease 2019
CpG: Cytosine-phosphate-Guanine
CRP: C-reactive protein
CXCL: C-X-C motif chemokine ligand
DAF2: Dauer formation-2
DAMP: Damage-associated molecular pattern
DC: Dendritic cell
DN: double-negative B cell
DNA: Deoxyribonucleic acid
DPP4: dipeptidyl peptidase 4
E47: E47 transcription factor
FGF2: Fibroblast growth factor 2
Fc: Fragment crystallizable
FcγR: Fc gamma receptors
G-CSF: Granulocyte colony-stimulating factor
GM-CSF: Granulocyte macrophage colony-stimulating factor
H1N1: Haemagglutinin-1 neuraminidase-1
H5N1: Haemagglutinin-5 neuraminidase-1
HCoV: Human coronavirus
HIV: Human immunodeficiency virus
IC: Immune complex
IFN-I: Interferon type I
Ig: Immunoglobulin
IL: Interleukin
IRF: Interferon regulatory factor
LCMV: Lymphocytic choriomeningitis virus
MAP: Mitogen-activated protein
MAPK: Mitogen-activated protein kinase
MAVS: Mitochondrial antiviral-signaling protein
MERS: Middle East respiratory syndrome
MHC: Major histocompatibility complex
MHV-1: Murine hepatitis virus type 1
mtDNA: Mitochondrial DNA
mTOR: Mammalian target of rapamycin
NET: Neutrophil extracellular trap
NF-κB: Nuclear factor kappa B
NK: Natural killer
NKG2A: CD94/NK group 2 member A
NLR: NOD-like receptor
NLRP3: NLR family pyrin domain containing 3
OX40: Tumor necrosis factor receptor superfamily, member 4 (TNFRSF4)
PAMP: Pathogen-associated molecular pattern
PD-1: Programmed cell death 1
pDC: Plasmacytoid DC
PRR: Pattern recognition receptor
RIG: Retinoic acid-inducible gene
RNA: Ribonucleic acid
ROS: Reactive oxygen species
SAA: Serum Amyloid A
SARS: Severe acute respiratory syndrome
SARS-CoV-1: Severe acute respiratory syndrome coronavirus 1
SARS-CoV-2: Severe acute respiratory syndrome coronavirus 2
SASP: Senescence-associated secretory phenotype
Smad7: Mothers against decapentaplegic homolog 7
STING: Stimulator of interferon genes
TCR: T cell receptor
TFH: T follicular helper cell
Th: T helper cell
Tim3: T-cell immunoglobulin and mucin-domain containing-3
TLR: Toll like receptor
TMPRSS2: Transmembrane Protease Serine 2
TNF-α: Tumor necrosis factor alpha
Treg: Regulatory T cell
TRIF: TIR-domain-containing adapter-inducing interferon beta
TRIM: Tripartite motif
1. Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Infect Dis (2007) 195(7):1018–28. doi: 10.1086/511989
2. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol (2016) 14(8):523–34. doi: 10.1038/nrmicro.2016.81
3. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
4. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome andDeath in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180(7):934–43. doi: 10.1001/jamainternmed.2020.0994
5. World Health Organization. World Health Organization. Coronavirus disease 2019 (COVID-19) WeeklyEpidemiological Update - 6. (2020). Available at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200921-weekly-epi-update-6.pdf?sfvrsn=d9cf9496_6.(Accessed October 10, 2020).
6. Ministério da Saúde do Brasil: Secretaria de Vigilância em Saúde. Boletim epidemiológico Especial - 30. Doença peloCoronavírus COVID-19. Semana Epidemiológica 36 (30/08 a 05/09). (2020). Available at: http://antigo.saude.gov.br/images/pdf/2020/September/09/Boletim-epidemiologico-COVID-30.pdf (Accessed October 10, 2020).
7. Osterrieder N, Bertzbach LD, Dietert K, Abdelgawad A, Vladimirova D, Kunec D, et al. Age-Dependent Progression of SARS-CoV-2 Infection in SyrianHamsters. Viruses (2020)12(7):779. doi: 10.3390/v12070779
8. Donnelly CA, Ghani AC, Leung GM, Hedley AJ, Fraser C, Riley S, et al. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet (2003) 361(9371):1761–6. doi: 10.1016/S0140-6736(03)13410-1
9. Nassar MS, Bakhrebah MA, Meo SA, Alsuabeyl MS, Zaher WA. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infection: epidemiology, pathogenesis and clinical characteristics. Eur Rev Med Pharmacol Sci (2018) 22(15):4956–61. doi: 10.26355/eurrev_201808_15635
10. Bonafè M, Prattichizzo F, Giuliani A, Storci G, Sabbatinelli J, Olivieri F. Inflamm-aging: Why older men are the most susceptible to SARS-CoV-2 complicated outcomes. Cytokine Growth Factor Rev (2020) 53:33–7. doi: 10.1016/j.cytogfr.2020.04.005
11. Alberca RW, Oliveira LM, Branco ACCC, Pereira NZ, Sato MN. Obesity as a risk factor for COVID-19: an overview. Crit Rev Food Sci Nutr (2020) 15:1–15. doi: 10.1080/10408398.2020.1775546
12. Frieman M, Baric R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol Mol Biol Rev (2008) 72(4):672–85. doi: 10.1128/MMBR.00015-08
13. Patrick DM, Petric M, Skowronski DM, Guasparini R, Booth TF, Krajden M, et al. An Outbreak of Human Coronavirus OC43 Infection and Serological Cross-reactivity with SARS Coronavirus. Can J Infect Dis Med Microbiol (2006) 17(6):330–6. doi: 10.1155/2006/152612
14. Wong CK, Smith CA, Sakamoto K, Kaminski N, Koff JL, Goldstein DR. Aging Impairs Alveolar Macrophage Phagocytosis and Increases Influenza-Induced Mortality in Mice. J Immunol (2017) 199(3):1060–8. doi: 10.4049/jimmunol.1700397
15. Kovacs EJ, Boe DM, Boule LA, Curtis BJ. Inflammaging and the Lung. Clin Geriatr Med (2017) 33(4):459–71. doi: 10.1016/j.cger.2017.06.002
16. Canan CH, Gokhale NS, Carruthers B, Lafuse WP, Schlesinger LS, Torrelles JB, et al. Characterization of lung inflammation and its impact on macrophage function in aging. J Leukoc Biol (2014) 96(3):473–80. doi: 10.1189/jlb.4A0214-093RR
17. Paranjpe I, Russak A, De Freitas JK, Lala A, Miotto R, Vaid A, et al. Clinical Characteristics of Hospitalized Covid-19 Patients in New York City. medRxiv (2020). doi: 10.1101/2020.04.19.20062117
18. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
19. Luk HKH, Li X, Fung J, Lau SKP, Woo PCY. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect Genet Evol (2019) 71:21–30. doi: 10.1016/j.meegid.2019.03.001
20. To KK, Hung IF, Chan JF, Yuen KY. From SARS coronavirus to novel animal and human coronaviruses. J Thorac Dis (2013) 5 Suppl 2:S103–8. doi: 10.3978/j.issn.2072-1439.2013.06.02
21. Xu J, Zhao S, Teng T, Abdalla AE, Zhu W, Xie L, et al. Systematic Comparison of Two Animal-to-Human Transmitted HumanCoronaviruses: SARS-CoV-2 and SARS-CoV. Viruses (2020) 12(2):244. doi: 10.3390/v12020244
22. Peiris JS, Yuen KY, Osterhaus AD, Stöhr K. The severe acute respiratory syndrome. N Engl J Med (2003) 349(25):2431–41. doi: 10.1056/NEJMra032498
23. Hijawi B, Abdallat M, Sayaydeh A, Alqasrawi S, Haddadin A, Jaarour N, et al. Novel coronavirus infections in Jordan, April 2012: epidemiological findings from a retrospective investigation. East Mediterr Health J (2013) 19 Suppl 1:S12–8. doi: 10.26719/2013.19.supp1.S12
24. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
25. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: anAnalysis Based on Decade-Long Structural Studies of SARS Coronavirus. J Virol (2020) 94(7):e00127-20. doi: 10.1128/JVI.00127-20
26. Li Y, Zhang Z, Yang L, Lian X, Xie Y, Li S, et al. The MERS-CoV Receptor DPP4 as a Candidate Binding Target of the SARS-CoV-2 Spike. iScience (2020) 23(8):101400. doi: 10.1016/j.isci.2020.101400
27. Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect (2020) 9(1):601–4. doi: 10.1080/22221751.2020.1739565
28. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e8. doi: 10.1016/j.cell.2020.02.052
29. Hoffmann M, Kleine-Weber H, Pöhlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol Cell (2020) 78(4):779–84.e5. doi: 10.1016/j.molcel.2020.04.022
30. Du L, He Y, Zhou Y, Liu S, Zheng BJ, Jiang S. The spike protein of SARS-CoV–a target for vaccine and therapeutic development. Nat Rev Microbiol (2009) 7(3):226–36. doi: 10.1038/nrmicro2090
31. Sahin AR, Erdogan A, Agaoglu PM, Dineri Y, Cakirci AY, Senel ME, et al. Novel Coronavirus (COVID-19) Outbreak:A Review of the Current Literature. EJMO (2020) 4(1):1–7. doi: 10.14744/ejmo.2020.12220
32. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Covid-19: Clinical Characteristics of Coronavirus Disease 2019 inChina. N Engl J Med (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
33. Jain V, Yuan J-M. Predictive symptoms and comorbidities for severe COVID-19 and intensive care unit admission: a systematic review and meta-analysis. Int J Public Health (2020) 65:533–46. doi: 10.1007/s00038-020-01390-7
34. Jain V, Yuan JM. Predictive symptoms and comorbidities for severe COVID-19 and intensive care unit admission: a systematic review and meta-analysis. Int J Public Health (2020) 65(5):533–46. doi: 10.1007/s00038-020-01390-7
35. Morley JE, Vellas B. Editorial: COVID-19 and Older Adults. J Nutr Health Aging (2020) 24(4):364–5. doi: 10.1007/s12603-020-1349-9
36. Liu K, Chen Y, Lin R, Han K. Clinical features of COVID-19 in elderly patients: A comparison with young and middle-aged patients. J Infect (2020) 80(6):e14–8. doi: 10.1016/j.jinf.2020.03.005
37. Wang L, He W, Yu X, Hu D, Bao M, Liu H, et al. Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on 4-week follow-up. J Infects (2020) 80(6):639–45. doi: 10.1016/j.jinf.2020.03.019
38. Goronzy JJ, Weyand CM. Understanding immunosenescence to improve responses to vaccines. Nat Immunol (2013) 14(5):428–36. doi: 10.1038/ni.2588
39. Falci C, Gianesin K, Sergi G, Giunco S, De Ronch I, Valpione S, et al. Immune senescence and cancer in elderly patients: results from an exploratory study. Exp Gerontol (2013) 48(12):1436–42. doi: 10.1016/j.exger.2013.09.011
40. Prelog M. Aging of the immune system: a risk factor for autoimmunity? Autoimmun Rev (2006) 5(2):136–9. doi: 10.1016/j.autrev.2005.09.008
41. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci (2014) 69 Suppl 1:S4–9. doi: 10.1093/gerona/glu057
42. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
43. Hearps AC, Martin GE, Angelovich TA, Cheng WJ, Maisa A, Landay AL, et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell (2012) 11(5):867–75. doi: 10.1111/j.1474-9726.2012.00851.x
44. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol (2009) 11(8):973–9. doi: 10.1038/ncb1909
45. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev (2011) 25(20):2125–36. doi: 10.1101/gad.17276711
46. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol (2008) 6(12):2853–68. doi: 10.1371/journal.pbio.0060301
47. Gigley JP, Khan IA. Plasmacytoid DC from aged mice down-regulate CD8 T cell responses by inhibiting cDC maturation after Encephalitozoon cuniculi infection. PloS One (2011) 6(6):e20838. doi: 10.1371/journal.pone.0020838
48. Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front Immunol (2019) 10:2247. doi: 10.3389/fimmu.2019.02247
49. Agrawal S, Gollapudi S, Gupta S, Agrawal A. Dendritic cells from the elderly display an intrinsic defect in the production of IL-10 in response to lithium chloride. Exp Gerontol (2013) 48(11):1285–92. doi: 10.1016/j.exger.2013.08.006
50. Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ (2016) 23(6):915–26. doi: 10.1038/cdd.2015.172
51. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol (2017) 8:1960. doi: 10.3389/fimmu.2017.01960
52. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
53. Lin L, Park S, Lakatta EG. RAGE signaling in inflammation and arterial aging. Front Biosci (Landmark Ed) (2009) 14:1403–13. doi: 10.2741/3315
54. Grignolio A, Mishto M, Faria AM, Garagnani P, Franceschi C, Tieri P. Towards a liquid self: how time, geography, and life experiences reshape the biological identity. Front Immunol (2014) 5:153. doi: 10.3389/fimmu.2014.00153
55. Tu W, Rao S. Mechanisms Underlying T Cell Immunosenescence: Aging and Cytomegalovirus Infection. Front Microbiol (2016) 7:2111. doi: 10.3389/fmicb.2016.02111
56. Yurochko AD, Hwang ES, Rasmussen L, Keay S, Pereira L, Huang ES. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J Virol (1997) 71(7):5051–9. doi: 10.1128/JVI.71.7.5051-5059.1997
57. Hummel M, Abecassis MM. A model for reactivation of CMV from latency. J Clin Virol (2002) 25 Suppl 2:S123–36. doi: 10.1016/S1386-6532(02)00088-4
58. Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Würzner R, et al. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol (2005) 79(6):3675–83. doi: 10.1128/JVI.79.6.3675-3683.2005
59. Bartlett DB, Firth CM, Phillips AC, Moss P, Baylis D, Syddall H, et al. The age-related increase in low-grade systemic inflammation (Inflammaging) is not driven by cytomegalovirus infection. Aging Cell (2012) 11(5):912–5. doi: 10.1111/j.1474-9726.2012.00849.x
60. Li B, Feng F, Yang G, Liu A, Yang N, Jiang Q, et al. Immunoglobulin G/M and Cytokines Detections in Continuous Sera from Patients with Novel Coronaviruses (2019-nCoV) Infection. (2020). doi: 10.2139/ssrn.3543609
61. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Y. qi R, et al. Aberrant pathogenic GM-CSF + T cells and inflammatory CD14 + CD16 +monocytes in severe pulmonary syndrome patients of a new coronavirus. biorxiv (2020). doi: 10.1101/2020.02.12.945576
62. Lin Z, Long F, Yang Y, Chen X, Xu L, Yang M. Serum ferritin as an independent risk factor for severity inCOVID-19 patients. J Infect (2020) 81(4):647–79. doi: 10.1016/j.jinf.2020.06.053
63. Huang I, Pranata R, Lim MA, Oehadian A, Alisjahbana B. C-reactive protein, procalcitonin, D-dimer, and ferritin in severe coronavirus disease-2019: a meta-analysis. Ther Adv Respir Dis (2020) 14:1753466620937175. doi: 10.1177/1753466620937175
64. Li H, Xiang X, Ren H, Xu L, Zhao L, Chen X, et al. Serum Amyloid A is a biomarker of severe Coronavirus Disease and poor prognosis. J Infect (2020) 80(6):646–55. doi: 10.1016/j.jinf.2020.03.035
65. Yan H, Xiao G, Zhang J, Hu Y, Yuan F, Cole DK, et al. SARS coronavirus induces apoptosis in Vero E6 cells. J Med Virol (2004) 73(3):323–31. doi: 10.1002/jmv.20094
66. Tan YX, Tan TH, Lee MJ, Tham PY, Gunalan V, Druce J, et al. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. J Virol (2007) 81(12):6346–55. doi: 10.1128/JVI.00090-07
67. Yeung ML, Yao Y, Jia L, Chan JF, Chan KH, Cheung KF, et al. MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat Microbiol (2016) 1:16004. doi: 10.1038/nmicrobiol.2016.4
68. Collins AR. In vitro detection of apoptosis in monocytes/macrophages infected with human coronavirus. Clin Diagn Lab Immunol (2002) 9(6):1392–5. doi: 10.1128/cdli.9.6.1392-1395.2002
69. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of theinteractions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol (2020) 10.1002/jmv.25987. doi: 10.1002/jmv.25987
70. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal (2012) 24(4):835–45. doi: 10.1016/j.cellsig.2011.12.006
71. Latz E, Duewell P. NLRP3 inflammasome activation in inflammaging. Semin Immunol (2018) 40:61–73. doi: 10.1016/j.smim.2018.09.001
72. Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab (2013) 18(4):519–32. doi: 10.1016/j.cmet.2013.09.010
73. Rodrigues TS, Sa KS, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasome activation in COVID-19 patients. medrvix (2020). doi: 10.1101/2020.08.05.20168872
74. Shah A. Novel Coronavirus-Induced NLRP3 Inflammasome Activation: A Potential Drug Target in the Treatment of COVID-19. Front Immunol (2020) 11:1021. doi: 10.3389/fimmu.2020.01021
75. Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol (2018) 20(5):521–7. doi: 10.1038/s41556-018-0092-5
76. Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell (2010) 140(6):798–804. doi: 10.1016/j.cell.2010.02.015
77. Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol (2018) 19(9):579–93. doi: 10.1038/s41580-018-0033-y
78. Cuanalo-Contreras K, Mukherjee A, Soto C. Role of protein misfolding and proteostasis deficiency in proteinmisfolding diseases and aging. Int J Cell Biol (2013) 2013:638083. doi: 10.1155/2013/638083
79. Meléndez A, Tallóczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science (2003) 301(5638):1387–91. doi: 10.1126/science.1087782
80. Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, et al. TORC1 inhibition enhances immune function and reduces infections inthe elderly. Sci Transl Med (2018) 10(449):eaaq1564. doi: 10.1126/scitranslmed.aaq1564
81. Harman D. Free radical theory of aging: Consequences of mitochondrial aging. Age (1983) 6:86–94. doi: 10.1007/BF02432509
82. Moreira OC, Estébanez B, Martínez-Florez S, de Paz JA, Cuevas MJ, González-Gallego J. Mitochondrial Function and Mitophagy in the Elderly: Effects of Exercise. Oxid Med Cell Longev (2017) 2017:2012798. doi: 10.1155/2017/2012798
83. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A (2009) 106(48):20388–93. doi: 10.1073/pnas.0908698106
84. Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol (2010) 40(3):616–9. doi: 10.1002/eji.200940168
85. To EE, Erlich JR, Liong F, Luong R, Liong S, Esaq F, et al. Mitochondrial Reactive Oxygen Species Contribute to Pathological Inflammation During Influenza A Virus Infection in Mice. Antioxid Redox Signal (2020) 32(13):929–42. doi: 10.1089/ars.2019.7727
86. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell (2008) 133(2):235–49. doi: 10.1016/j.cell.2008.02.043
87. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and MonocyteResponse through a HIF-1α/Glycolysis-Dependent Axis. CellMetab (2020) 32(3):437–46.e5. doi: 10.1016/j.cmet.2020.07.007
88. Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol (2004) 75(6):995–1000. doi: 10.1189/jlb.0703328
89. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339(6121):826–30. doi: 10.1126/science.1229963
90. Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur J Immunol (2014) 44(5):1552–62. doi: 10.1002/eji.201343921
91. McGuire PJ. Mitochondrial Dysfunction and the Aging ImmuneSystem. Biol (Basel) (2019) 8(2):26. doi: 10.3390/biology8020026
92. Koshiba T, Yasukawa K, Yanagi Y, Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal (2011) 4(158):ra7. doi: 10.1126/scisignal.2001147
93. Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol (2014) 193(6):3080–9. doi: 10.4049/jimmunol.1303196
94. Fransen F, van Beek AA, Borghuis T, Aidy SE, Hugenholtz F, van der Gaast-de Jongh C, et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front Immunol (2017) 8:1385. doi: 10.3389/fimmu.2017.01385
95. Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe (2017) 21(4):455–66.e4. doi: 10.1016/j.chom.2017.03.002
96. Carvalho FA, Aitken JD, Vijay-Kumar M, Gewirtz AT. Toll-like receptor-gut microbiota interactions: perturb at your own risk! Annu Rev Physiol (2012) 74:177–98. doi: 10.1146/annurev-physiol-020911-153330
97. Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, et al. Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PloS One (2010) 5(5):e10667. doi: 10.1371/journal.pone.0010667
98. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol (2017) 39(5):529–39. doi: 10.1007/s00281-017-0629-x
99. Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin Immunol (2020) 214:108393. doi: 10.1016/j.clim.2020.108393
100. D’Elia RV, Harrison K, Oyston PC, Lukaszewski RA, Clark GC. Targeting the “cytokine storm” for therapeutic benefit. Clin Vaccine Immunol (2013) 20(3):319–27. doi: 10.1128/CVI.00636-12
101. Avalos CR, Abreu CM, Queen SE, Li M, Price S, Shirk EN, et al. Brain Macrophages in Simian Immunodeficiency Virus-Infected,Antiretroviral-Suppressed Macaques: a Functional Latent Reservoir. MBio (2017) 8(4):e01186–17. doi: 10.1128/mBio.01186-17
102. Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev (2020) 53:25–32. doi: 10.1016/j.cytogfr.2020.05.003
103. Meftahi GH, Jangravi Z, Sahraei H, Bahari Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: the contribution of “inflame-aging”. Inflammation Res (2020) 69(9):825–39. doi: 10.1007/s00011-020-01372-8
104. Guaraldi G, Meschiari M, Cozzi-Lepri A, Milic J, Tonelli R, Menozzi M, et al. Tocilizumab in patients with severe COVID-19: a retrospective cohortstudy. Lancet Rheumatol (2020) 2(8):E474–84. doi: 10.1016/S2665-9913(20)30173-9
105. Metcalf TU, Wilkinson PA, Cameron MJ, Ghneim K, Chiang C, Wertheimer AM, et al. Human Monocyte Subsets Are Transcriptionally and Functionally Altered in Aging in Response to Pattern Recognition Receptor Agonists. J Immunol (2017) 199(4):1405–17. doi: 10.4049/jimmunol.1700148
106. Pinke KH, Calzavara B, Faria PF, do Nascimento MP, Venturini J, Lara VS. Proinflammatory profile of in vitro monocytes in the ageing is affected by lymphocytes presence. Immun Ageing (2013) 10(1):22. doi: 10.1186/1742-4933-10-22
107. Clark JA, Peterson TC. Cytokine production and aging: overproduction of IL-8 in elderly males in response to lipopolysaccharide. Mech Ageing Dev (1994) 77(2):127–39. doi: 10.1016/0047-6374(94)90020-5
108. Delpedro AD, Barjavel MJ, Mamdouh Z, Faure S, Bakouche O. Signal transduction in LPS-activated aged and young monocytes. J Interferon Cytokine Res (1998) 18(6):429–37. doi: 10.1089/jir.1998.18.429
109. Gabriel P, Cakman I, Rink L. Overproduction of monokines by leukocytes after stimulation with lipopolysaccharide in the elderly. Exp Gerontol (2002) 37(2-3):235–47. doi: 10.1016/s0531-5565(01)00189-9
110. Zheng Y, Liu X, Le W, Xie L, Li H, Wen W, et al. A human circulating immune cell landscape in aging andCOVID-19. Protein Cell (2020) 11(10):740–70. doi: 10.1007/s13238-020-00762-2
111. Pence BD. Severe COVID-19 and aging: are monocytes the key? Geroscience (2020) 42(4):1051–61. doi: 10.1007/s11357-020-00213-0
112. Linton PJ, Thoman ML. Immunosenescence in monocytes, macrophages, and dendritic cells: lessons learned from the lung and heart. Immunol Lett (2014) 162(1 Pt B):290–7. doi: 10.1016/j.imlet.2014.06.017
113. Li G, Smithey MJ, Rudd BD, Nikolich-Žugich J. Age-associated alterations in CD8α+ dendritic cells impair CD8 T-cell expansion in response to an intracellular bacterium. Aging Cell (2012) 11(6):968–77. doi: 10.1111/j.1474-9726.2012.00867.x
114. Herrero C, Marqués L, Lloberas J, Celada A. IFN-gamma-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J Clin Invest (2001) 107(4):485–93. doi: 10.1172/JCI11696
115. Yoon P, Keylock KT, Hartman ME, Freund GG, Woods JA. Macrophage hypo-responsiveness to interferon-gamma in aged mice is associated with impaired signaling through Jak-STAT. Mech Ageing Dev (2004) 125(2):137–43. doi: 10.1016/j.mad.2003.11.010
116. Chu H, Zhou J, Wong BH, Li C, Cheng ZS, Lin X, et al. Productive replication of Middle East respiratory syndromecoronavirus in monocyte-derived dendritic cells modulates innate immune response. Virology (2014) 454–455:197–205. doi: 10.1016/j.virol.2014.02.018
117. Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood (2005) 106(7):2366–74. doi: 10.1182/blood-2004-10-4166
118. Hu Y, Li W, Gao T, Cui Y, Jin Y, Li P, et al. The Severe Acute Respiratory Syndrome Coronavirus NucleocapsidInhibits Type I Interferon Production by Interfering with TRIM25-Mediated RIG-IUbiquitination. J Virol (2017) 91(8):e02143–16. doi: 10.1128/JVI.02143-16
119. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
120. Trouillet-Assant S, Viel S, Gaymard A, Pons S, Richard JC, Perret M, et al. Type I IFN immunoprofiling in COVID-19 patients. J Allergy Clin Immunol (2020) 146(1):206–208.e2. doi: 10.1016/j.jaci.2020.04.029
121. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
122. Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res (2020) 179:104811. doi: 10.1016/j.antiviral.2020.104811
123. Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R, Menachery VD. SARS-CoV-2 is sensitive to type I interferonpretreatment. bioRxiv (2020). doi: 10.1101/2020.03.07.982264
124. Davoudi-Monfared E, Rahmani H, Khalili H, Hajiabdolbaghi M, Salehi M, Abbasian L, et al. A Randomized Clinical Trial of the Efficacy and Safety of Interferonβ-1a in Treatment of Severe COVID-19. Antimicrob AgentsChemother (2020) 64(9):e01061–20. doi: 10.1128/AAC.01061-20
125. Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet (2020) 395(10238):1695–704. doi: 10.1016/S0140-6736(20)31042-4
126. Jing Y, Shaheen E, Drake RR, Chen N, Gravenstein S, Deng Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum Immunol (2009) 70(10):777–84. doi: 10.1016/j.humimm.2009.07.005
127. Pérez-Cabezas B, Naranjo-Gómez M, Fernández MA, Grífols JR, Pujol-Borrell R, Borràs FE. Reduced numbers of plasmacytoid dendritic cells in aged blood donors. Exp Gerontol (2007) 42(10):1033–8. doi: 10.1016/j.exger.2007.05.010
128. Splunter MV, Perdijk O, Fick-Brinkhof H, Floris-Vollenbroek EG, Meijer B, Brugman S, et al. Plasmacytoid dendritic cell and myeloid dendritic cell function in ageing: A comparison between elderly and young adult women. PloS One (2019) 14(12):e0225825. doi: 10.1371/journal.pone.0225825
129. Molony RD, Nguyen JT, Kong Y, Montgomery RR, Shaw AC, Iwasaki A. Aging impairs both primary and secondary RIG-I signaling forinterferon induction in human monocytes. Sci Signal (2017) 10(509):eaan2392. doi: 10.1126/scisignal.aan2392
130. Chougnet CA, Thacker R II, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells Is Associated with Mitochondrial Dysfunction. J Immunol (2015) 195(6):2624–32. doi: 10.4049/jimmunol.1501006
131. McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M, et al. The Aging Microenvironment Shapes Alveolar Macrophage Identity inAging. biorxiv (2019). doi: 10.1101/717033 Available at: https://www.biorxiv.org/content/10.1101/717033v2 (Accessed October 10, 2020).
132. van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, Leenen PJM. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol (2019) 40(2):113–27. doi: 10.1016/j.it.2018.12.007
133. Hartwig SM, Holman KM, Varga SM. Depletion of alveolar macrophages ameliorates virus-induced disease following a pulmonary coronavirus infection. PloS One (2014) 9(3):e90720. doi: 10.1371/journal.pone.0090720
134. Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenicmice. Nature (2020) 40(2):113–27. doi: 10.1038/s41586-020-2312-y
135. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex Immune Dysregulation in COVID-19 Patients with SevereRespiratory Failure. Cell Host Microbe (2020)27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009
136. Chatta GS, Andrews RG, Rodger E, Schrag M, Hammond WP, Dale DC. Hematopoietic progenitors and aging: alterations in granulocytic precursors and responsiveness to recombinant human G-CSF, GM-CSF, and IL-3. J Gerontol (1993) 48(5):M207–12. doi: 10.1093/geronj/48.5.m207
137. Wang YH, Lin AS, Chao TY, Lu SN, Liu JW, Chen SS, et al. A cluster of patients with severe acute respiratory syndrome in a chest ward in southern Taiwan. Intensive Care Med (2004) 30(6):1228–31. doi: 10.1007/s00134-004-2311-8
138. Fu J, Kong J, Wang W, Wu M, Yao L, Wang Z, et al. The clinical implication of dynamic neutrophil to lymphocyte ratio and D-dimer in COVID-19: A retrospective study in Suzhou China. Thromb Res (2020) 192:3–8. doi: 10.1016/j.thromres.2020.05.006
139. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: Neutrophil extracellulartraps. J Exp Med (2020) 217(6):e20200652. doi: 10.1084/jem.20200652
140. Wenisch C, Patruta S, Daxböck F, Krause R, Hörl W. Effect of age on human neutrophil function. J Leukoc Biol (2000) 67(1):40–5. doi: 10.1002/jlb.67.1.40
141. Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, et al. Senescence in innate immune responses: reduced neutrophil phagocyticcapacity and CD16 expression in elderly humans. J Leukoc Biol(2001) 70(6):881–6. doi: 10.1189/jlb.70.6.881
142. Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood (2014) 123(2):239–48. doi: 10.1182/blood-2013-08-519520
143. Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell (2014) 13(4):690–8. doi: 10.1111/acel.12222
144. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
145. Chidrawar SM, Khan N, Chan YL, Nayak L, Moss PA. Ageing is associated with a decline in peripheral blood CD56bright NK cells. Immun Ageing (2006) 3:10. doi: 10.1186/1742-4933-3-10
146. Campos C, Pera A, Sanchez-Correa B, Alonso C, Lopez-Fernandez I, Morgado S, et al. Effect of age and CMV on NK cell subpopulations. Exp Gerontol (2014) 54:130–7. doi: 10.1016/j.exger.2014.01.008
147. Le Garff-Tavernier M, Béziat V, Decocq J, Siguret V, Gandjbakhch F, Pautas E, et al. Human NK cells display major phenotypic and functional changes over the life span. Aging Cell (2010) 9(4):527–35. doi: 10.1111/j.1474-9726.2010.00584.x
148. Beli E, Clinthorne JF, Duriancik DM, Hwang I, Kim S, Gardner EM. Natural killer cell function is altered during the primary response of aged mice to influenza infection. Mech Ageing Dev (2011) 132(10):503–10. doi: 10.1016/j.mad.2011.08.005
149. Nogusa S, Ritz BW, Kassim SH, Jennings SR, Gardner EM. Characterization of age-related changes in natural killer cells during primary influenza infection in mice. Mech Ageing Dev (2008) 129(4):223–30. doi: 10.1016/j.mad.2008.01.003
150. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol (2020) 17(5):533–5. doi: 10.1038/s41423-020-0402-2
151. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
152. Chen J, Lau YF, Lamirande EW, Paddock CD, Bartlett JH, Zaki SR, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol (2010) 84(3):1289–301. doi: 10.1128/JVI.01281-09
153. Franceschi C, Bonafè M, Valensin S. Human immunosenescence: the prevailing of innate immunity, the failing of clonotypic immunity, and the filling of immunological space. Vaccine (2000) 18(16):1717–20. doi: 10.1016/S0264-410X(99)00513-7
154. Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol (2004) 5(2):133–9. doi: 10.1038/ni1033
155. Allman D, Miller JP. B cell development and receptor diversity during aging. Curr Opin Immunol (2005) 17(5):463–7. doi: 10.1016/j.coi.2005.07.002
156. Pawelec G, Akbar A, Caruso C, Solana R, Grubeck-Loebenstein B, Wikby A. Human immunosenescence: is it infectious? Immunol Rev (2005) 205:257–68. doi: 10.1111/j.0105-2896.2005.00271.x
157. Cui W, Fan Y, Wu W, Zhang F, Wang JY, Ni AP. Expression of lymphocytes and lymphocyte subsets in patients with severe acute respiratory syndrome. Clin Infect Dis (2003) 37(6):857–9. doi: 10.1086/378587
158. Li T, Qiu Z, Zhang L, Han Y, He W, Liu Z, et al. Significant changes of peripheral T lymphocyte subsets in patients with severe acute respiratory syndrome. J Infect Dis (2004) 189(4):648–51. doi: 10.1086/381535
159. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Y. qi R, et al. Pathogenic T cells and inflammatory monocytes incite inflammatorystorm in severe COVID-19 patients. Natl Sci Rev (2020)7(6): 998–1002. doi: 10.1093/nsr/nwaa041
160. Jiang M, Guo Y, Luo Q, Huang Z, Zhao R, Liu S, et al. T cell subset counts in peripheral blood can be used asdiscriminatory biomarkers for diagnosis and severity prediction of COVID-19. J Infect Dis (2020) 222(2):198–202. doi: 10.1093/infdis/jiaa252
161. Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science (2020) 369(6508):1210–20. doi: 10.1126/science.abc6261
162. Odak I, Barros-Martins J, Bošnjak B, Stahl K, David S, Wiesner O, et al. Reappearance of effector T cells is associated with recovery from COVID-19. EBioMedicine (2020) 57:102885. doi: 10.1016/j.ebiom.2020.102885
163. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive mapping of immune perturbations associated with severeCOVID-19. Sci Immunol (2020) 5(49):eabd7114. doi: 10.1126/sciimmunol.abd7114
164. Posnett DN, Yarilin D, Valiando JR, Li F, Liew FY, Weksler ME, et al. Oligoclonal expansions of antigen-specific CD8+ T cells in aged mice. Ann N Y Acad Sci (2003) 987:274–9. doi: 10.1111/j.1749-6632.2003.tb06061.x
165. Goronzy JJ, Lee WW, Weyand CM. Aging and T-cell diversity. Exp Gerontol (2007) 42(5):400–6. doi: 10.1016/j.exger.2006.11.016
166. Akbar AN, Henson SM, Lanna A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol (2016) 37(12):866–76. doi: 10.1016/j.it.2016.09.002
167. Bandrés E, Merino J, Vázquez B, Inogés S, Moreno C, Subirá ML, et al. The increase of IFN-gamma production through aging correlates with the expanded CD8(+high)CD28(-)CD57(+) subpopulation. Clin Immunol (2000) 96(3):230–5. doi: 10.1006/clim.2000.4894
168. Henson SM, Riddell NE, Akbar AN. Properties of end-stage human T cells defined by CD45RA re-expression. Curr Opin Immunol (2012) 24(4):476–81. doi: 10.1016/j.coi.2012.04.001
169. Onyema OO, Njemini R, Bautmans I, Renmans W, De Waele M, Mets T. Cellular aging and senescence characteristics of human T-lymphocytes. Biogerontology (2012) 13(2):169–81. doi: 10.1007/s10522-011-9366-z
170. Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev (2005) 205:158–69. doi: 10.1111/j.0105-2896.2005.00256.x
171. Chou J, RB E. T cell replicative senescence in human aging. Curr Pharm Des (2013) 19(9):1680–98. doi: 10.2174/138161213805219711
172. Plunkett FJ, Franzese O, Finney HM, Fletcher JM, Belaramani LL, Salmon M, et al. The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. J Immunol (2007) 178(12):7710–9. doi: 10.4049/jimmunol.178.12.7710
173. Lee KA, Shin KS, Kim GY, Song YC, Bae EA, Kim IK, et al. Characterization of age-associated exhausted CD8+ T cells defined by increased expression of Tim-3 and PD-1. Aging Cell (2016) 15(2):291–300. doi: 10.1111/acel.12435
174. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
175. Caruso C, Candore G, Cigna D, DiLorenzo G, Sireci G, Dieli F, et al. Cytokine production pathway in the elderly. Immunol Res (1996) 15(1):84–90. doi: 10.1007/BF02918286
176. Douziech N, Seres I, Larbi A, Szikszay E, Roy PM, Arcand M, et al. Modulation of human lymphocyte proliferative response with aging. Exp Gerontol (2002) 37(2-3):369–87. doi: 10.1016/s0531-5565(01)00204-2
177. Alberti S, Cevenini E, Ostan R, Capri M, Salvioli S, Bucci L, et al. Age-dependent modifications of Type 1 and Type 2 cytokines within virgin and memory CD4+ T cells in humans. Mech Ageing Dev (2006) 127(6):560–6. doi: 10.1016/j.mad.2006.01.014
178. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest (2020) 130(5):2620–9. doi: 10.1172/JCI137244
179. Lee JS, Lee WW, Kim SH, Kang Y, Lee N, Shin MS, et al. Age-associated alteration in naive and memory Th17 cell response in humans. Clin Immunol (2011) 140(1):84–91. doi: 10.1016/j.clim.2011.03.018
180. Lorenzo EC, Bartley JM, Haynes L. The impact of aging on CD4. Biogerontology (2018) 19(6):437–46. doi: 10.1007/s10522-018-9754-8
181. Abdulamir A, Hafidh R. The Possible Immunological Pathways for the VariableImmunopathogenesis of COVID-19 Infections among Healthy Adults, Elderly and Children. Electron J Gen Med (2020) 17(4):em202. doi: 10.29333/ejgm/7850
182. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev (2009) 229(1):173–91. doi: 10.1111/j.1600-065X.2009.00766.x
183. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10(7):490–500. doi: 10.1038/nri2785
184. van der Geest KS, Abdulahad WH, Tete SM, Lorencetti PG, Horst G, Bos NA, et al. Aging disturbs the balance between effector and regulatory CD4+ T cells. Exp Gerontol (2014) 60:190–6. doi: 10.1016/j.exger.2014.11.005
185. Pieren DKJ, Smits NAM, van de Garde MDB, Guichelaar T. Response kinetics reveal novel features of ageing in murine T cells. Sci Rep (2019) 9(1):5587. doi: 10.1038/s41598-019-42120-1
186. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan,China. Clin Infect Dis (2020) 71(15):762–8. doi: 10.1093/cid/ciaa248
187. Gibson KL, Wu YC, Barnett Y, Duggan O, Vaughan R, Kondeatis E, et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell (2009) 8(1):18–25. doi: 10.1111/j.1474-9726.2008.00443.x
188. Eaton SM, Burns EM, Kusser K, Randall TD, Haynes L. Age-related defects in CD4 T cell cognate helper function lead to reductions in humoral responses. J Exp Med (2004) 200(12):1613–22. doi: 10.1084/jem.20041395
189. Mosterín Höpping A, McElhaney J, Fonville JM, Powers DC, Beyer WEP, Smith DJ. The confounded effects of age and exposure history in response to influenza vaccination. Vaccine (2016) 34(4):540–6. doi: 10.1016/j.vaccine.2015.11.058
190. Swain SL, Kugler-Umana O, Kuang Y, Zhang W. The properties of the unique age-associated B cell subset reveal ashift in strategy of immune response with age. Cell Immunol (2017) 321:52–60. doi: 10.1016/j.cellimm.2017.05.009
191. Yang X, Stedra J, Cerny J. Relative contribution of T and B cells to hypermutation and selection of the antibody repertoire in germinal centers of aged mice. J Exp Med (1996) 183(3):959–70. doi: 10.1084/jem.183.3.959
192. To KK, Tsang OT, Leung WS, Tam AR, Wu TC, Lung DC, et al. Temporal profiles of viral load in posterior oropharyngeal salivasamples and serum antibody responses during infection by SARS-CoV-2: an observational cohortstudy. Lancet Infect Dis (2020) 20(5):565–74. doi: 10.1016/S1473-3099(20)30196-1
193. Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood (2011) 118(5):1294–304. doi: 10.1182/blood-2011-01-330530
194. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood (2011) 118(5):1305–15. doi: 10.1182/blood-2011-01-331462
195. Ma S, Wang C, Mao X, Hao Y. B Cell Dysfunction Associated With Aging and Autoimmune Diseases. Front Immunol (2019) 10:318. doi: 10.3389/fimmu.2019.00318
196. Frasca D, Romero M, Diaz A, Alter-Wolf S, Ratliff M, Landin AM, et al. A molecular mechanism for TNF-α-mediated downregulation of B cell responses. J Immunol (2012) 188(1):279–86. doi: 10.4049/jimmunol.1003964
197. Ratliff M, Alter S, Frasca D, Blomberg BB, Riley RL. In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell (2013) 12(2):303–11. doi: 10.1111/acel.12055
198. Frasca D, Van der Put E, Riley RL, Blomberg BB. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J Immunol (2004) 172(4):2155–62. doi: 10.4049/jimmunol.172.4.2155
199. Frasca D, Landin AM, Lechner SC, Ryan JG, Schwartz R, Riley RL, et al. Aging down-regulates the transcription factor E2A, activation-induced cytidine deaminase, and Ig class switch in human B cells. J Immunol (2008) 180(8):5283–90. doi: 10.4049/jimmunol.180.8.5283
200. Wu YB, Kipling B, Dunn-Walters DK. The relationship between CD27 negative and positive B cell populations in human peripheral blood. Front Immun (2011) 2:81. doi: 10.3389/fimmu.2011.00081
201. Klein U, Rajewsky K, Küppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med (1998) 188(9):1679–89. doi: 10.1084/jem.188.9.1679
202. Chong Y, Ikematsu H, Yamaji K, Nishimura M, Nabeshima S, Kashiwagi S, et al. CD27(+) (memory) B cell decrease and apoptosis-resistant CD27(-) (naive) B cell increase in aged humans: implications for age-related peripheral B cell developmental disturbances. Int Immunol (2005) 17:383–90. doi: 10.1093/intimm/dxh218
203. Frasca D, Diaz A, Romero M, Landin AM, Blomberg BB. Age effects on B cells and humoral immunity in humans. Ageing Res Rev (2011) 10(3):330–5. doi: 10.1016/j.arr.2010.08.004
204. Teixeira D, Ishimura ME, Longo-Maugeri IM, Lebrão ML, Duarte YAO, Bueno V. Biological Markers Changes at the Very Early Stage of Ageing (60-65Years). Is There a Gender-Related Effect? J Aging Sci (2015) 3:132. doi: 10.4172/2329-8847.1000132
205. Faria AM, de Moraes SM, de Freitas LH, Speziali E, Soares TF, Figueiredo-Neves SP, et al. Variation rhythms of lymphocyte subsets during healthy aging. Neuroimmunomodulation (2008) 15(4-6):365–79. doi: 10.1159/000156478
206. Colonna-Romano G, Bulati M, Aquino A, Pellicanò M, Vitello S, Lio D, et al. A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech Ageing Dev (2009) 130(10):681–90. doi: 10.1016/j.mad.2009.08.003
207. Frasca D, Diaz A, Romero M, Blomberg BB. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp Gerontol (2017) 87(Pt A):113–20. doi: 10.1016/j.exger.2016.12.001
208. Cagigi A, Du L, Dang LV, Grutzmeier S, Atlas A, Chiodi F, et al. CD27(-) B-cells produce class switched and somatically hyper-mutated antibodies during chronic HIV-1 infection. PloS One (2009) 4(5):e5427. doi: 10.1371/journal.pone.0005427
209. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol (2007) 178(10):6624–33. doi: 10.4049/jimmunol.178.10.6624
210. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep immune profiling of COVID-19 patients reveals distinctimmunotypes with therapeutic implications. Science (2020) 369(6508):eabc8511. doi: 10.1126/science.abc8511
211. Woodruff M, Ramonell R, Cashman K, Nguyen D, Ley A, Kyu S, et al. Critically ill SARS-CoV-2 patients display lupus-like hallmarks ofextrafollicular B cell activation. medRxiv (2020). doi: 10.1101/2020.04.29.20083717
212. Grifoni A, Weiskopf D, Ramirez S II, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell (2020) 181(7):1489–1501.e15. doi: 10.1016/j.cell.2020.05.015
213. Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature (2020). doi: 10.1038/s41586-020-2598-9
214. Weiskopf D, Schmitz KS, Raadsen MP, Grifoni A, Okba NMA, Endeman H, et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19patients with acute respiratory distress syndrome. Sci Immunol (2020) 5(48):eabd2071. doi: 10.1126/sciimmunol.abd2071
215. Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular Mechanism for Antibody-Dependent Enhancement ofCoronavirus Entry. J Virol (2020)94(5):e02015–19. doi: 10.1128/JVI.02015-19
216. Tetro JA. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect (2020) 22(2):72–3. doi: 10.1016/j.micinf.2020.02.006
217. Peron JPS, Nakaya H. Susceptibility of the Elderly to SARS-CoV-2 Infection: ACE-2 Overexpression, Shedding, and Antibody-dependent Enhancement (ADE). Clinics (Sao Paulo) (2020) 75:e1912. doi: 10.6061/clinics/2020/e1912
218. Tirado SM, Yoon KJ. Antibody-dependent enhancement of virus infection and disease. Viral Immunol (2003) 16(1):69–86. doi: 10.1089/088282403763635465
219. Wang SF, Tseng SP, Yen CH, Yang JY, Tsao CH, Shen CW, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem Biophys Res Commun (2014) 451(2):208–14. doi: 10.1016/j.bbrc.2014.07.090
220. Kuczera D, Assolini JP, Tomiotto-Pellissier F, Pavanelli WR, Silveira GF. Highlights for Dengue Immunopathogenesis: Antibody-Dependent Enhancement, Cytokine Storm, and Beyond. J Interferon Cytokine Res (2018) 38(2):69–80. doi: 10.1089/jir.2017.0037
221. Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med (2020) 26(6):845–8. doi: 10.1038/s41591-020-0897-1
222. Pitts PJ. Regulatory centaurs. Nat Biotechnol (2020) 38(7):788–9. doi: 10.1038/s41587-020-0589-x
223. Gao Q, Bao L, Mao H, Wang L, Xu K, Yang M, et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science (2020) 369(6499):77–81. doi: 10.1126/science.abc1932
224. van Doremalen N, Lambe T, Spencer A, Belij-Rammerstorfer S, Purushotham JN, Port JR, et al. ChAdOx1 nCoV-19 vaccination prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature (2020). doi: 10.1038/s41586-020-2608-y
225. Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 Critically Ill Patients With COVID-19 WithConvalescent Plasma. JAMA (2020) 323(16):1582–89. doi: 10.1001/jama.2020.4783
226. Roback JD, Guarner J. Convalescent Plasma to Treat COVID-19: Possibilities andChallenges. JAMA (2020) 323(16):1561–2. doi: 10.1001/jama.2020.4940
227. Zhang L, Zhang F, Yu W, He T, Yu J, Yi CE, et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J Med Virol (2006) 78(1):1–8. doi: 10.1002/jmv.20499
228. Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophageresponses during acute SARS-CoV infection. JCI Insight (2019) 4(4):e123158. doi: 10.1172/jci.insight.123158
229. Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med (2011) 17(2):195–9. doi: 10.1038/nm.2262
230. Polack FP, Teng MN, Collins PL, Prince GA, Exner M, Regele H, et al. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med (2002) 196(6):859–65. doi: 10.1084/jem.20020781
231. Ward PA, Fattahi F, Bosmann M. New Insights into Molecular Mechanisms of Immune Complex-Induced Injury in Lung. Front Immunol (2016) 7:86. doi: 10.3389/fimmu.2016.00086
232. Vadasz Z, Haj T, Kessel A, Toubi E. Age-related autoimmunity. BMC Med (2013) 11:94. doi: 10.1186/1741-7015-11-94
233. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol (2013) 33(6):479–92. doi: 10.1016/j.semnephrol.2013.08.001
Keywords: COVID-19, coronavirus, aging, immunosenescence, inflammaging, SARS-CoV-2
Citation: Pietrobon AJ, Teixeira FME and Sato MN (2020) I mmunosenescence and Inflammaging: Risk Factors of Severe COVID-19 in Older People. Front. Immunol. 11:579220. doi: 10.3389/fimmu.2020.579220
Received: 02 July 2020; Accepted: 29 September 2020;
Published: 27 October 2020.
Edited by:
Valquiria Bueno, Federal University of São Paulo, BrazilReviewed by:
Stefano Salvioli, University of Bologna, ItalyCopyright © 2020 Pietrobon, Teixeira and Sato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Notomi Sato, bWFyaXNhdG9AdXNwLmJy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.