 Ingrid Kazue Mizuno Watanabe1,2*
Ingrid Kazue Mizuno Watanabe1,2* Magaiver Andrade-Silva2
Magaiver Andrade-Silva2 Orestes Foresto-Neto1
Orestes Foresto-Neto1 Raphael José Ferreira Felizardo1
Raphael José Ferreira Felizardo1 Marco Aurélio Costa Matheus2
Marco Aurélio Costa Matheus2 Reinaldo Correa Silva1
Reinaldo Correa Silva1 Marcos Antônio Cenedeze2
Marcos Antônio Cenedeze2 Tâmisa Seeko Bandeira Honda1Luiz Augusto Buoro Perandini1Rildo Aparecido Volpini3
Tâmisa Seeko Bandeira Honda1Luiz Augusto Buoro Perandini1Rildo Aparecido Volpini3 Alvaro Pacheco-Silva2,4
Alvaro Pacheco-Silva2,4 Niels Olsen Saraiva Câmara1,2
Niels Olsen Saraiva Câmara1,2- 1Department of Immunology, Institute of Biomedical Science, University of São Paulo, São Paulo, Brazil
- 2Nephrology Division, Department of Medicine, Federal University of São Paulo, São Paulo, Brazil
- 3Department of Nephrology, University of São Paulo, School of Medicine, São Paulo, Brazil
- 4Kidney Transplant Unit, Hospital Israelita Albert Einstein, São Paulo, Brazil
Increasing evidence shows the essential participation of gut microbiota in human health and diseases by shaping local and systemic immunity. Despite an accumulating body of studies showing that chronic kidney disease (CKD) is closely associated with disturbances in the composition of gut microbiota, it remains unclear the importance of gut microbiota in the onset and development of CKD. For the purpose of untangling the role of gut microbiota in CKD, gut microbiota was depleted with a pool of broad-spectrum antibiotics in mice submitted to unilateral ureteral obstruction (UUO). Depletion of gut microbiota significantly decreased levels of proinflammatory cytokines and fibrosis markers, attenuating renal injury. Additionally, to study whether the pathogenic role of gut microbiota is dependent of microbial-host crosstalk, we generated mice lacking Myd88 (myeloid differentiation primary response gene 8) expression in intestinal epithelial cells (IECs) and performed UUO. The absence of Myd88 in IECs prevented a bacterial burden in mesenteric lymph nodes as observed in WT mice after UUO and led to lower expression of proinflammatory cytokines and chemokines, reducing deposition of type I collagen and, ultimately, attenuating renal damage. Therefore, our results suggest that the presence of gut microbiota is crucial for the development of CKD and may be dependent of Myd88 signaling in IECs, which appears to be essential to maturation of immune cells intimately involved in aggravation of inflammatory scenarios.
Introduction
Chronic kidney disease (CKD) has been considered as a global public health problem. In 2017, CKD was ranked as the 12th leading cause of death and the number of cases reached almost 700 million worldwide (1). The onset and progression of CKD involve a complex interplay among metabolic, neuroendocrine and immunologic events, which generates a high risk clinical phenotype (2). In the past decades, other players have been included in this scenario, including the gut and its indigenous residents so-called the gut microbiota.
Despite the existence of a large variety of causative factors in the onset and progression of CKD, accumulating evidence has continuously linked local and systemic outcomes from kidney damage to dysbiosis. Kidney failure provokes accumulation of uremic toxins, which, in turn, can impair the integrity of intestinal barrier and disturb the composition and functionality of gut microbiota (dysbiosis) enhancing toxin production and inflammation in CKD. Accumulation of toxins originated from the gut microbiota in CKD, such as p-cresol sulfate and TMAO, has been linked to the progression of kidney function (3, 4). Profound disturbances in gut microbiota may also alter the levels of microbial metabolites with benefits to the host metabolism, such as short-chain fatty acids (SCFAs), which participate in the modulation of inflammatory responses in models of kidney injury (5–7).
Alterations in the microbial composition and functionality can influence the maintenance of intestinal barrier. In homeostatic conditions, intestinal epithelial cells (IECs) and immune cells “feel and react” to microbial stimuli orchestrating a symphony of immune responses and reinforcing the intestinal barrier to keep a balance between tolerance and pathogenic immunity. The integrity of intestinal barrier critically relies on the fitness of IECs, which are central players of a microbial-dependent strategy for intestinal repair and homeostasis. IECs sense microbial-associated molecular patterns (MAMPs), through pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and NOD-like receptors (NLRs), promoting proliferation and differentiation of epithelial cells, reinforcement of tight junctions (8), and secretion of IgA and antimicrobial peptides (AMPs) in the lumen (9).
The dysfunction of intestinal barrier has been observed in a variety of diseases, including type 1 diabetes mellitus, inflammatory bowel disease and coeliac disease (10). In the context of CKD, uremia has been strongly associated with disturbances in tight junction dynamics. Experimental studies observed a pronounced depletion of components of intestinal epithelial tight junction in uremic animals (11). Moreover, Varizi et al. observed that in vitro treatment with urea remarkably reduced the expression of claudin-1, occludin and ZO-1 in IECs, which was accompanied by lower transepithelial electrical resistance (12). Impaired intestinal barrier not only aggravates local inflammation but could also contribute to systemic inflammation. Indeed, circulating endotoxemia and bacterial-derived DNA fragments were found in CKD patients (13, 14). Furthermore, the blood levels of endotoxins seem to be correlated with CKD stage (14).
Together, evidence points to the importance of gut microbiota and intestinal barrier fitness in the pathogenesis of CKD. Therefore, the purpose of this study was to evaluate the role of gut microbiota in the onset and development of CKD and whether the influence of gut microbiota was dependent of the microbial-host crosstalk, specifically of the Myd88 signaling in IECs.
Material and Methods
Mice
All animal procedures described in this study were approved by the Ethics Committee in Animal Research of the Federal University of São Paulo (Protocol number: 7562120416) and all the animal study methods were performed according to the Ethical Principles in Animal Research. Animals were held under temperature-controlled conditions on a 12-h light cycle with access to food and water ad libitum.
Gut microbiota was depleted in 6-week-old C57Bl/6 mice by oral administration of broad-spectrum antibiotics (ABX) [1 g/L ampicillin, 1 g/L metronidazole, 1 g/L neomycin and 0.5 g/L vancomycin (Sigma-Aldrich)] via autoclaved drinking water with 1% (wt/vol) glucose for 4 weeks, as previously described (15). During ABX treatment, bacterial load in the feces was assessed once in a week. Briefly, fresh fecal pellets were resuspended in sterile PBS, and an aliquot of the suspension was inoculated in 5 ml of Luria-Bertani (LB) broth and kept at 37°C for 3 days. Bacteria concentration in the culture was estimated through the optical density at 600 nm. After 3 weeks of ABX treatment, irreversible unilateral ureteral obstruction (UUO) was performed in control and ABX mice, as previously described (16).
Selective deletion of Myd88 in IECs (Myd88ΔIEC) was obtained through the intercross of B6.129P2-MyD88 tm1Defr/J (Myd88fl/fl) and B6N.Cg-Tg (Vil1-Cre)997Gum/J strains. After confirmation of Myd88 deletion in isolated IECs, mice were submitted to UUO. After 7 days of UUO, mice were euthanized, and samples [blood, colon, liver, mesenteric lymph node (MLN) and kidney] were collected for further analysis.
Pelvic Biochemical Urinary Parameters
Total protein and creatinine in urine were quantified using commercial kits according to instructions (Labtest). Urinary albumin content was estimated using dye-binding technique. Briefly, under reducing conditions, urine samples were resolved by 10% SDS-polyacrylamide gel electrophoresis. Gels were stained with 0.1% Coomassie blue R-250 solution for 2 h and destained overnight. Albumin concentration was determined according to the intensity of the bands using a positive loading control (bovine serum albumin, 0.2 mg/ml) using ImageJ software.
Cytokines Quantification
Serum levels of IL-6 were quantified using available commercial kit (R&D Systems) according to manufacturer’s instructions. Quantification of cytokines and chemokines in kidney and colon samples were performed using Bio-Plex Pro mouse cytokine, chemokine, and growth factor assay (Bio-Rad) according to manufacturer’s instructions. Briefly, aliquots of tissue homogenate were incubated with premixed “color-coded” magnetic beads in a shaker at room temperature for 30 min. After washing steps, incubation with biotinylated detection antibody and streptavidin PE conjugate were performed subsequently, allowing measurement of a specific cytokine later in the laser detection step. Cytokine concentrations were determined using standards curves and results were expressed as pg/mg of total protein.
Immunohistochemistry
Formalin-fixed tissue was embedded in paraffin using standard procedure and histological sections were immunostained by standard avidin-biotin-peroxidase methodology (Vectastain ABC Kit, Vector Laboratories) using diaminobenzidine (DAB) as the chromogen. Profibrotic and cell proliferation status were assessed using antibodies anti-type I collagen (1:200, Abcam), anti-fibroblast-specific protein 1 (FSP-1) (1:800, Dako) and anti-KI-67 (1:1,000, Abcam), respectively. Digital microphotographs of renal cortex were obtained at 40-fold magnification using a Carl Zeiss Axioskop 40 microscope (Carl Zeiss Microscopy) and AxioCam MRc5 digital camera (Carl Zeiss Microscopy). The area stained positive for type I collagen, FSP-1 and KI-67 was quantified as percentage of total cross-sectional area using Axiovision software (Carl Zeiss Microscopy).
Fluorescence In Situ Hybridization
Segments of distal colon containing feces were harvested and fixed in methacarn solution at 4°C for 24 h and embedded in paraffin using standard procedure. Briefly, deparaffinized sections were incubated with Eubacteria probe (AF488-conjugated, Sigma-Aldrich) in a humid chamber at 50°C overnight. After washing step, sections were incubated with lectin from Ulex europaeus (TRITC-conjugated, Sigma-Aldrich) at room temperature for 2 h. After mounting step with Vectashield (Vector laboratories), images were obtained using confocal microscope at a magnification of 40×.
Detection of Bacterial DNA in Organ Samples
DNA from MLN and liver samples (collected in sterile conditions) was extracted using DNeasy Blood & Tissue Kit (Qiagen) according to manufacturer’s instructions. All reactions were carried out with 20 ng of DNA through Sybr system using primers designed for bacterial 16S gene and for mouse genomic DNA (Table 1). Quantification of bacterial 16S rDNA was normalized using mouse genomic DNA (pIgR genomic region) as internal control and calculated using the ΔΔCT-method. Results were expressed as an n-fold difference in relation to the expression of matched controls.

Table 1 Primer sequences used for qPCR.
Gene Expression
Total RNA from colon and kidney samples were extracted with Trizol® (Invitrogen), and cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression of proinflammatory cytokines and profibrotic markers were performed based on Sybr system using designed primers listed in Table 1. Quantification of target gene expression was normalized using HPRT or GAPDH as internal controls and calculated using the ΔΔCT-method. Results were expressed as an n-fold difference in relation to the expression of matched controls. Analyses were performed using QuantStudio™ Design & Analysis Software v1.5.0 (Applied Biosystems).
Western Blotting
Kidney samples were homogenized in ice-cold RIPA buffer containing protease inhibitors cocktail (Roche Applied Science). Aliquots of 20 µg of protein were subjected to electrophoresis under reducing conditions. Proteins transferred to nitrocellulose membranes were probed overnight with mouse monoclonal anti-α smooth muscle actin (α-SMA) antibody (1:1,000, Dako), and, sequentially, incubated with appropriate secondary antibody. The bands were detected by chemiluminescence (Clarity Western ECL Substrate, Bio-Rad) according to the manufacturers’ recommendations. Results were normalized relative to α-tubulin expression.
Statistics
Results were expressed as mean ± SE. Student t test was performed to compare two groups, and one-way ANOVA with Tukey’s post hoc test was performed to compare more than three groups using GraphPad Prism 5.0 (GraphPad Software). Statistical significance was set at p ≤ 0.05.
Results
Depletion of Gut Microbiota Protects Against UUO Injury
Treatment with ABX was effective in lowering gut bacterial load and in modulating gene expression of some components of intestinal barrier (Figure S1). Depletion of gut microbiota was followed by upregulation of occludin and ZO-1, which are considered key elements of tight junctions. CKD has been implicated in disruption of intestinal epithelial tight junctions, which has been associated with local and systemic inflammation (Figure S1). UUO led to increased gene expression of proinflammatory cytokines, such as IL-1b and IL-6, in the colon (Figure 1), which was prevented by depletion of gut microbiota. Systemic inflammatory status was also greatly influenced by UUO, as indicated by a 7-fold increase of serum levels of IL-6 in untreated group (Figure 1G), which was partially prevented by ABX treatment.
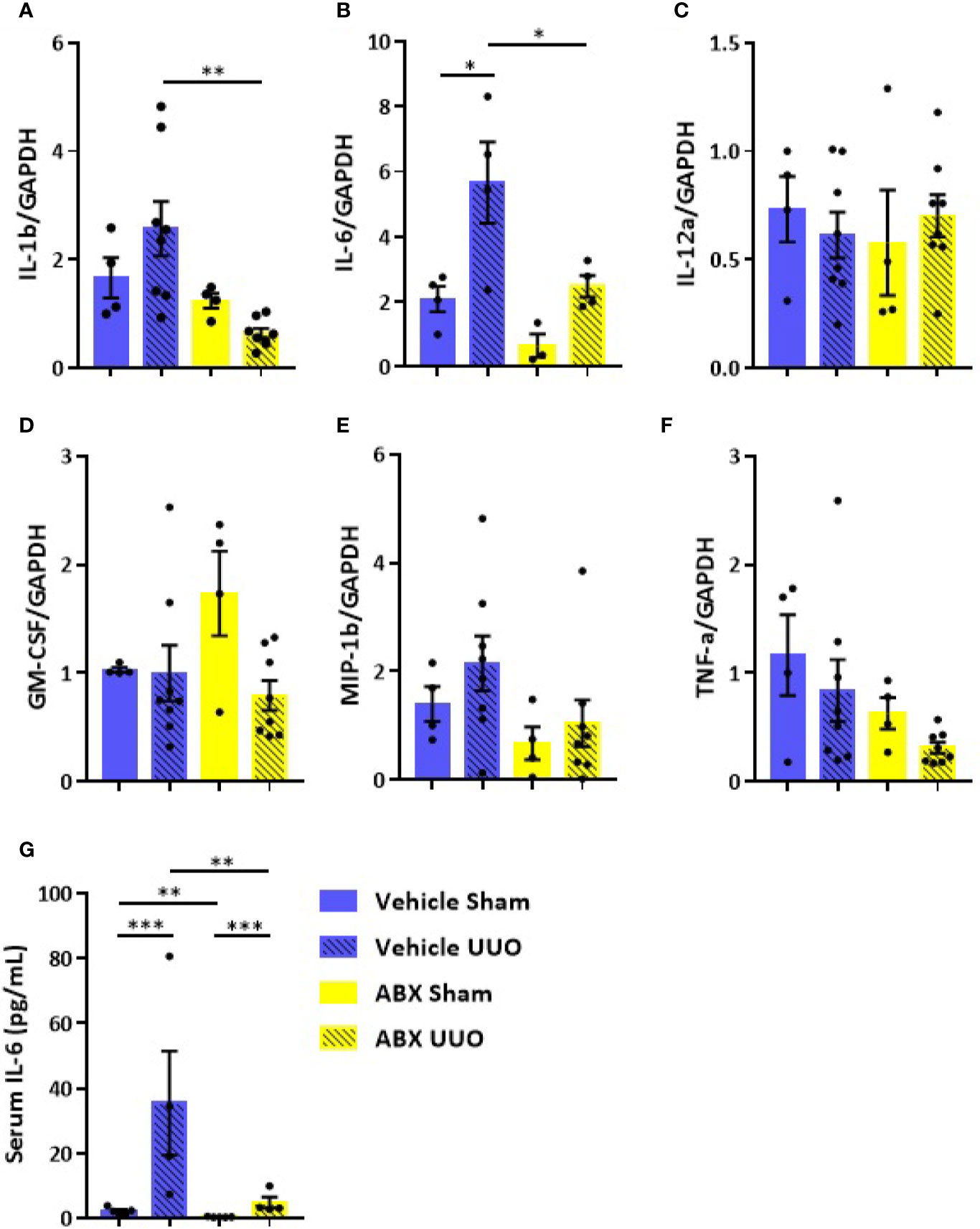
Figure 1 Impact of depleted gut microbiota on the colon and systemic inflammation in CKD. Gene expression of (A) IL-1b, (B) IL-6, (C) IL-12a, (D) GM-CSF, (E) MIP-1β, and (F) TNF-α in the colon of Vehicle and ABX-treated mice submitted or not to UUO (N = 4 to 8). (G) Serum levels of IL-6 of Vehicle and ABX-treated mice submitted or not to UUO (N = 4). *p < 0.05; **p < 0.01; ***p < 0.001.
The systemic inflammation observed after UUO was followed by increased gene expression of proinflammatory cytokines in the kidney of untreated mice. On the other hand, ABX-treated group showed strikingly lower expression of cytokines, such as IL-1b, IL-4, and IL-6, and chemokines (GM-CSF and MIP-1β) after UUO (Figure 2). In addition to that, depletion of gut microbiota modulated the expression of markers of tubulointerstitial fibrosis. Decreased gene expression of profibrotic markers, such as fibronectin and type I collagen, was observed in the kidney of ABX-treated mice (Figures 2H, I). We also observed lower protein expression of indicators of kidney injury, including α-SMA and type I collagen (Figure 3). In addition, depletion of gut microbiota affected the expression of markers of cell activation, such as FSP-1 and KI-67. ABX treatment led to lower quantity of FSP-1 positive cells in the renal intestitium, and decreased number of KI-67 positive cells, including renal tubular cells and interstitial cell populations (Figure 3B). This protection against UUO injury was followed by better pelvic urinary parameters. Abx-treated mice presented lower levels of protein in pelvic urine after UUO compared with untreated group (Figure 3B). Taken together, our results indicated that the proinflammatory and profibrotic scenarios caused by UUO are dependent of the presence of gut microbiota.
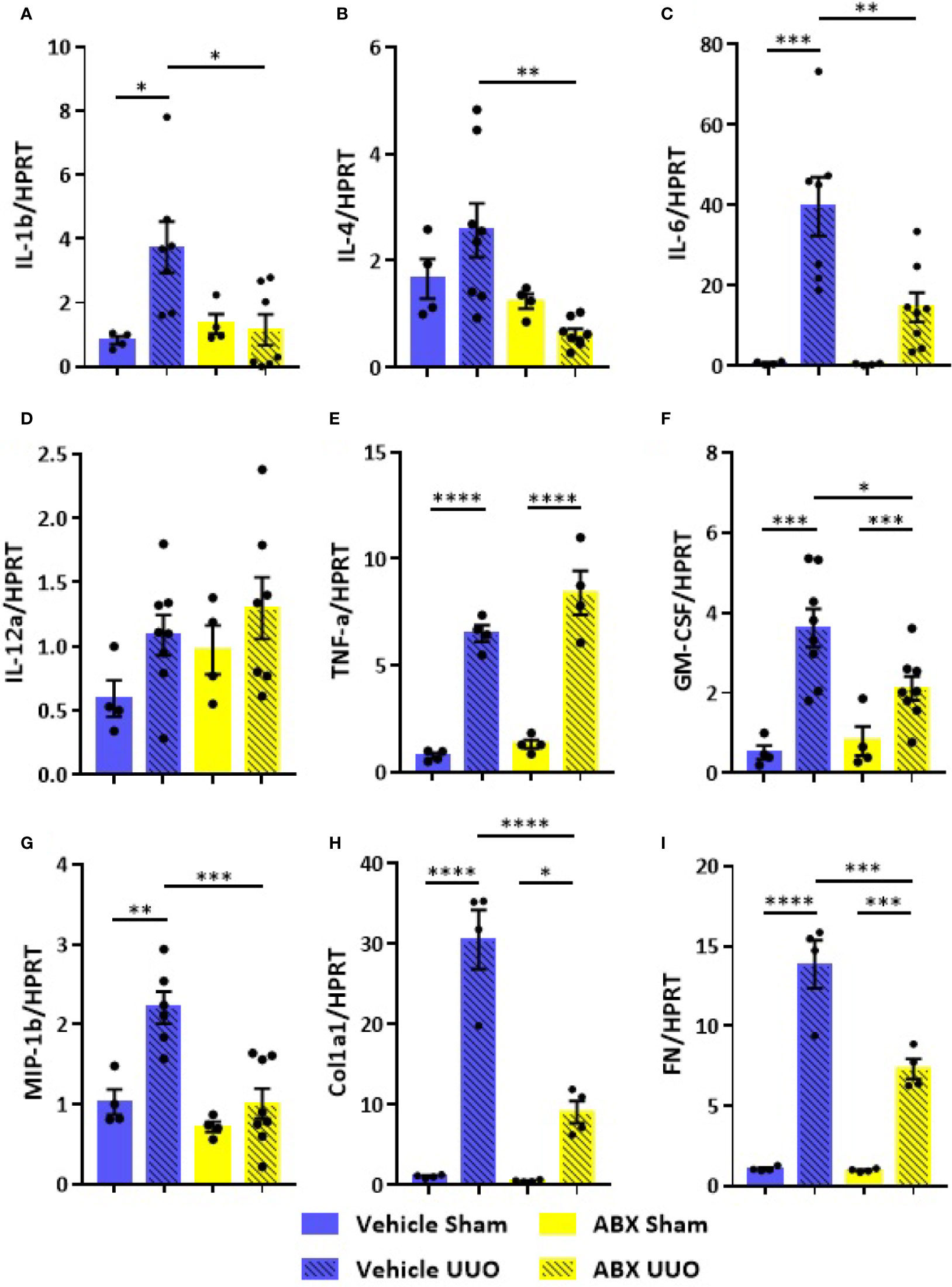
Figure 2 Impact of depleted gut microbiota on the kidney in CKD. Gene expression of (A) IL-1b, (B) IL-4, (C) IL-6, (D) IL-12a, (E) TNF-α, (F) GM-CSF, (G) MIP-1β, (H) Col1a1, and (I) fibronectin (FN) in the kidney of Vehicle and ABX-treated mice submitted or not to UUO (N = 4 to 8). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
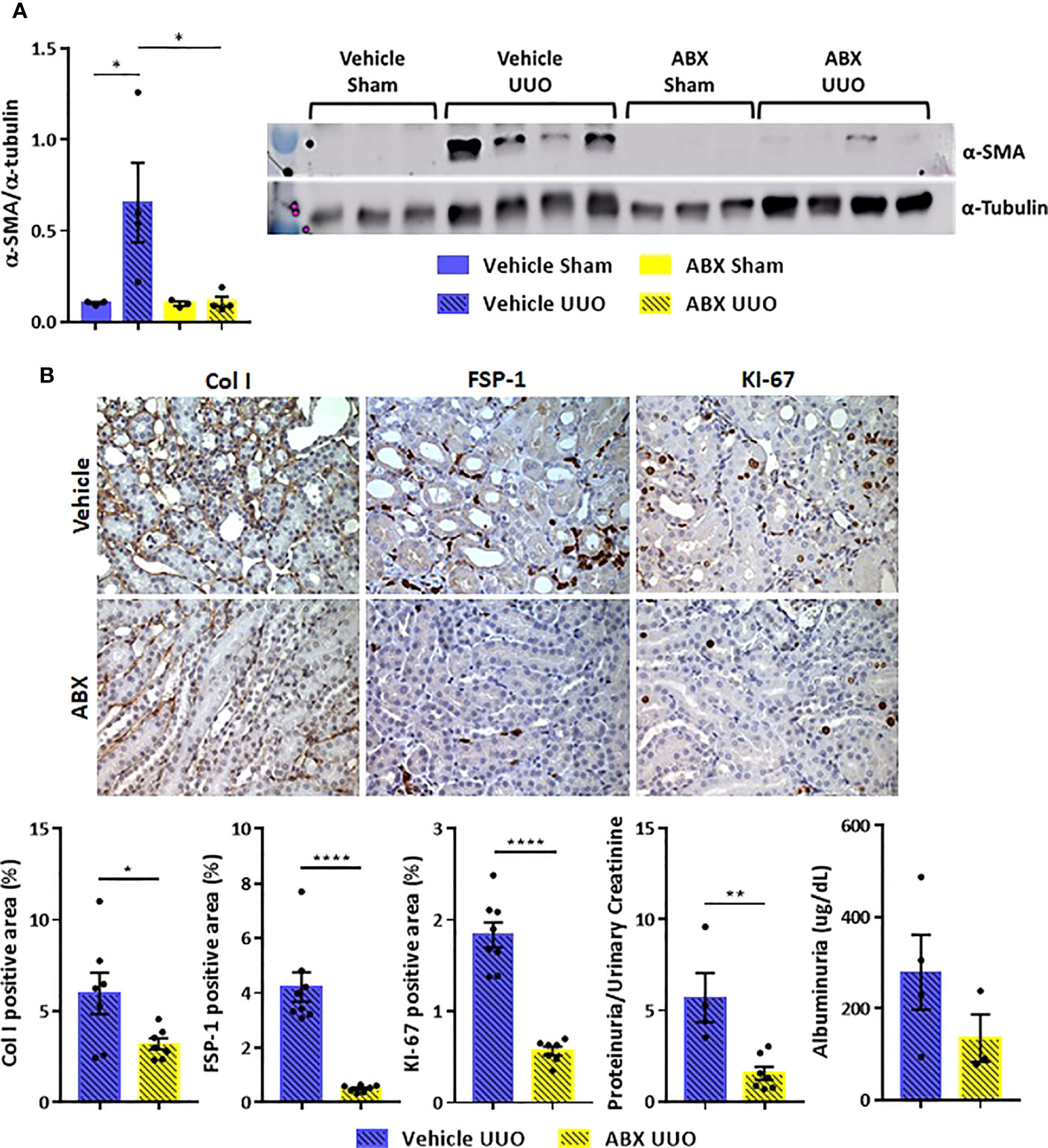
Figure 3 (A) Protein expression of α-SMA in the kidney of Vehicle and ABX-treated mice submitted or not to UUO (N = 3 to 4) by Western blotting. (B) Representative images and positive area (%) of immunohistochemical staining for type I collagen (Col I), FSP-1 and KI-67 in the kidney of Vehicle and ABX-treated mice submitted to UUO (N = 7 to 8); concentration of total protein normalized by creatinine in pelvic urine (N = 4 to 7); estimation of albuminuria in pelvic urine collected from Vehicle and ABX-treated mice submitted to UUO (N = 3 to 4). *p < 0.05; **p < 0.01; ****p < 0.0001.
Myd88 Signaling in IECs Participates in UUO Injury
On the next step of the study, we decided to examine whether this UUO-generated inflammation would be dependent on Myd88 signaling in IECs. For this purpose, after confirmation of Myd88 deletion in IEC isolated from intestine (Figure S2), Myd88ΔIEC mice were submitted to UUO and expression of proinflammatory cytokines was evaluated aiming to verify the participation of IEC-specific Myd88 signaling in local and systemic inflammation. Analysis of colon samples of Myd88ΔIEC mice, similar to untreated WT mice, revealed that UUO led to a massive bacterial invasion in the mucus layer (Figure 4A). Surprisingly, a higher bacterial burden was only observed in MLN of untreated WT mice after UUO (Figure 4B). In addition, the lack of Myd88 in IECs decreased levels of IL-1b, IL-12p40, and IL-17A in the colon after UUO, while levels of TNF-α and colony stimulating factors, such as GM-CSF and MIP-1β, were unaltered in both strains (Figure 5). Kidney assessment revealed that Myd88ΔIEC mice submitted to UUO presented lower levels of IL-4, IL-12p40, IL-17A, GM-CSF, and MIP-1β compared to Myd88fl/fl mice (Figure 6). Besides the attenuation of the proinflammatory scenario, the selective deletion of Myd88 also influenced the expression of profibrotic markers. Myd88ΔIEC mice submitted to UUO presented lower expression of Col1a1 and fibronectin compared to Myd88flox mice followed by decreased deposition of type I collagen (Figure 7). Renal benefits also included the observation of better pelvic urinary parameters (Figure 7). Low urinary levels of total protein and albumin were observed in Myd88ΔIEC mice.
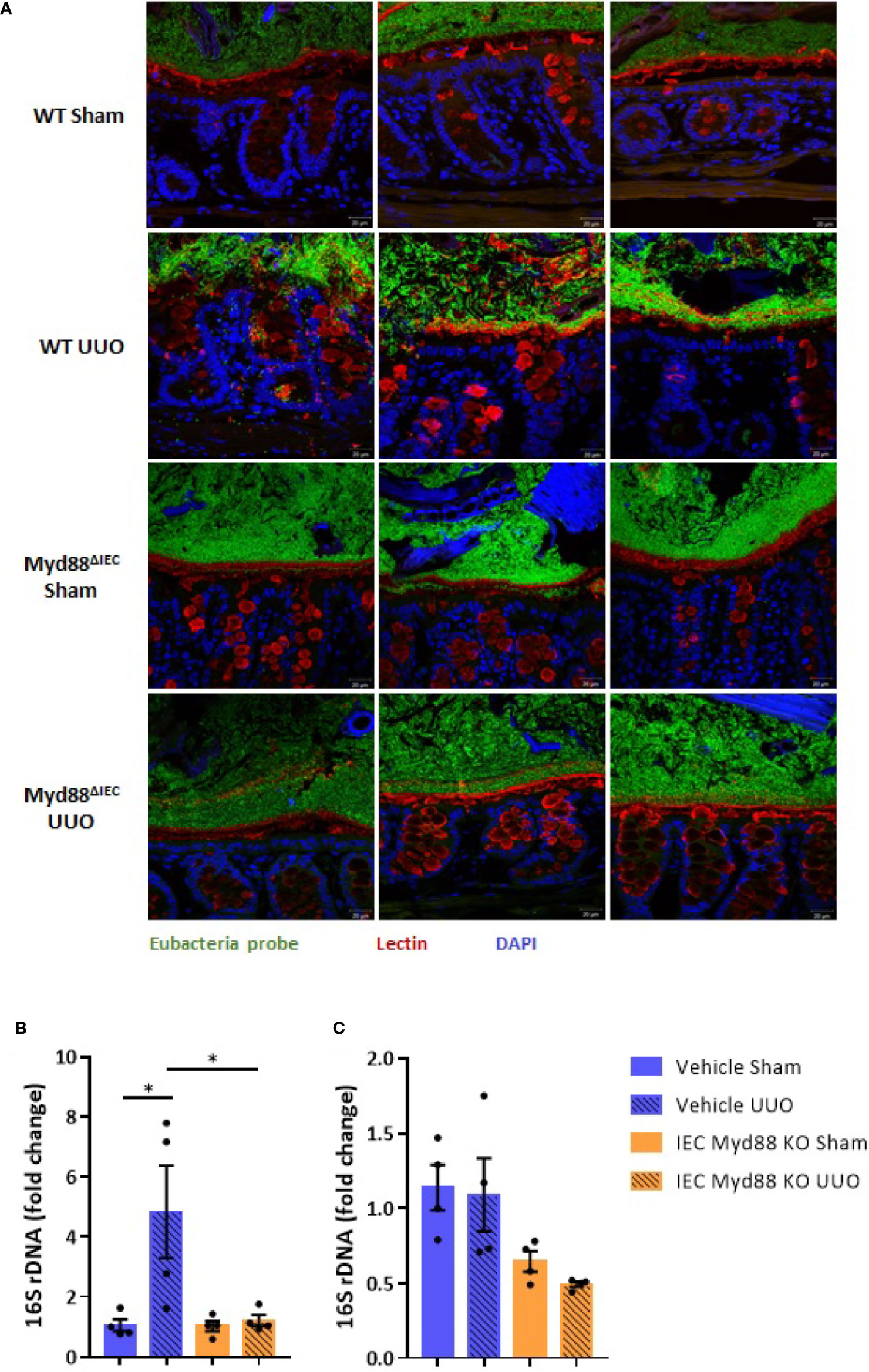
Figure 4 (A) Fluorescence in situ hybridization (FISH) analysis of the total bacteria in the colon. Visualization of colon fragments containing feces labeled with probe for Eubacteria (green) and FITC-conjugated lectin (red) for mucus (x40 fold magnification). (B) Quantification of bacterial DNA in the mesenteric lymph node (MLN) of untreated wild type (WT) and Myd88ΔIEC mice submitted or not to UUO based on gene expression of 16S rDNA (N = 4). (C) Quantification of bacterial DNA in the liver of untreated wild type (WT) and Myd88ΔIEC mice submitted or not to UUO based on gene expression of 16S rDNA (N = 4). *p < 0.05.
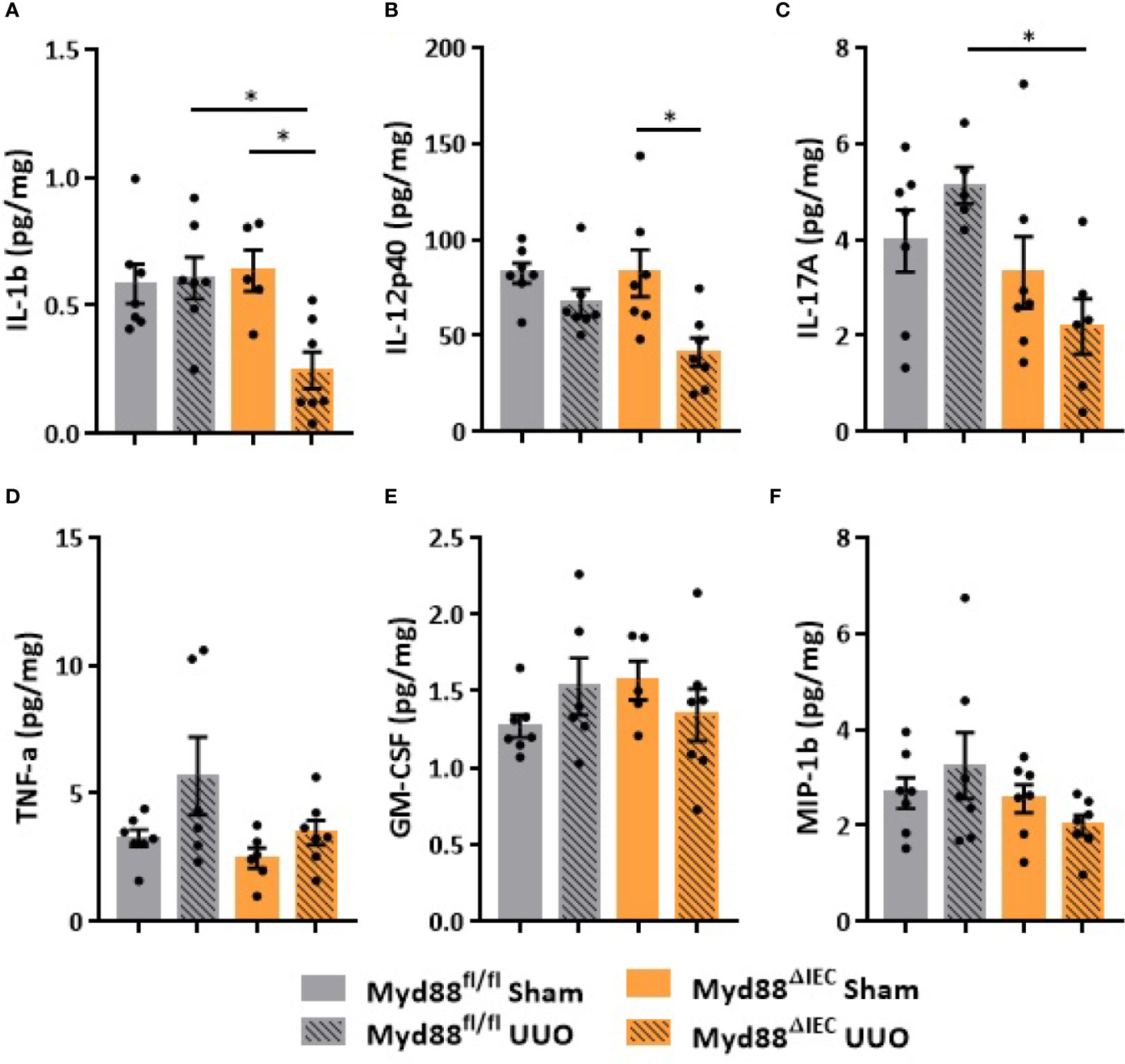
Figure 5 Effect of Myd88 signaling in IECs on the colon in CKD. Concentration of (A) IL-1b, (B) IL-12p40, (C) IL-17A, (D) TNF-α, (E) GM-CSF, and (F) MIP-1β in the colon of Myd88fl/fl and Myd88ΔIEC mice submitted or not to UUO determined by multiplex cytokine assay (N = 5 to 7). *p < 0.05.
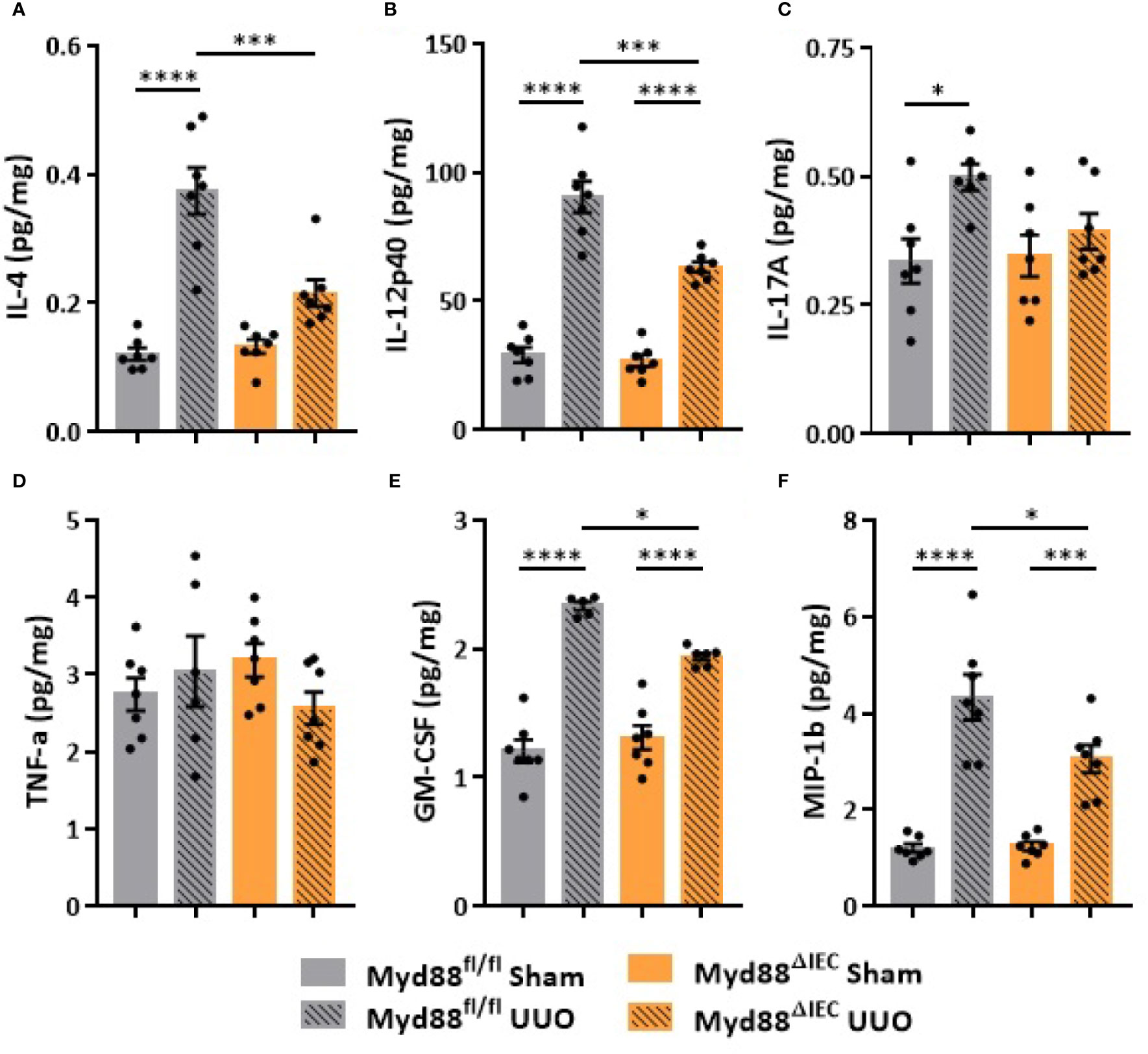
Figure 6 Effect of Myd88 signaling in IECs on the inflammatory profile of the kidney in CKD. Concentration of (A) IL-4, (B) IL-12p40, (C) IL-17A, (D) TNF-α, (E) GM-CSF, and (F) MIP-1β in the kidney of Myd88fl/fl and Myd88ΔIEC mice submitted or not to UUO determined by multiplex cytokine assay (N = 5 to 7). *p < 0.05; ***p < 0.001; ****p < 0.0001.
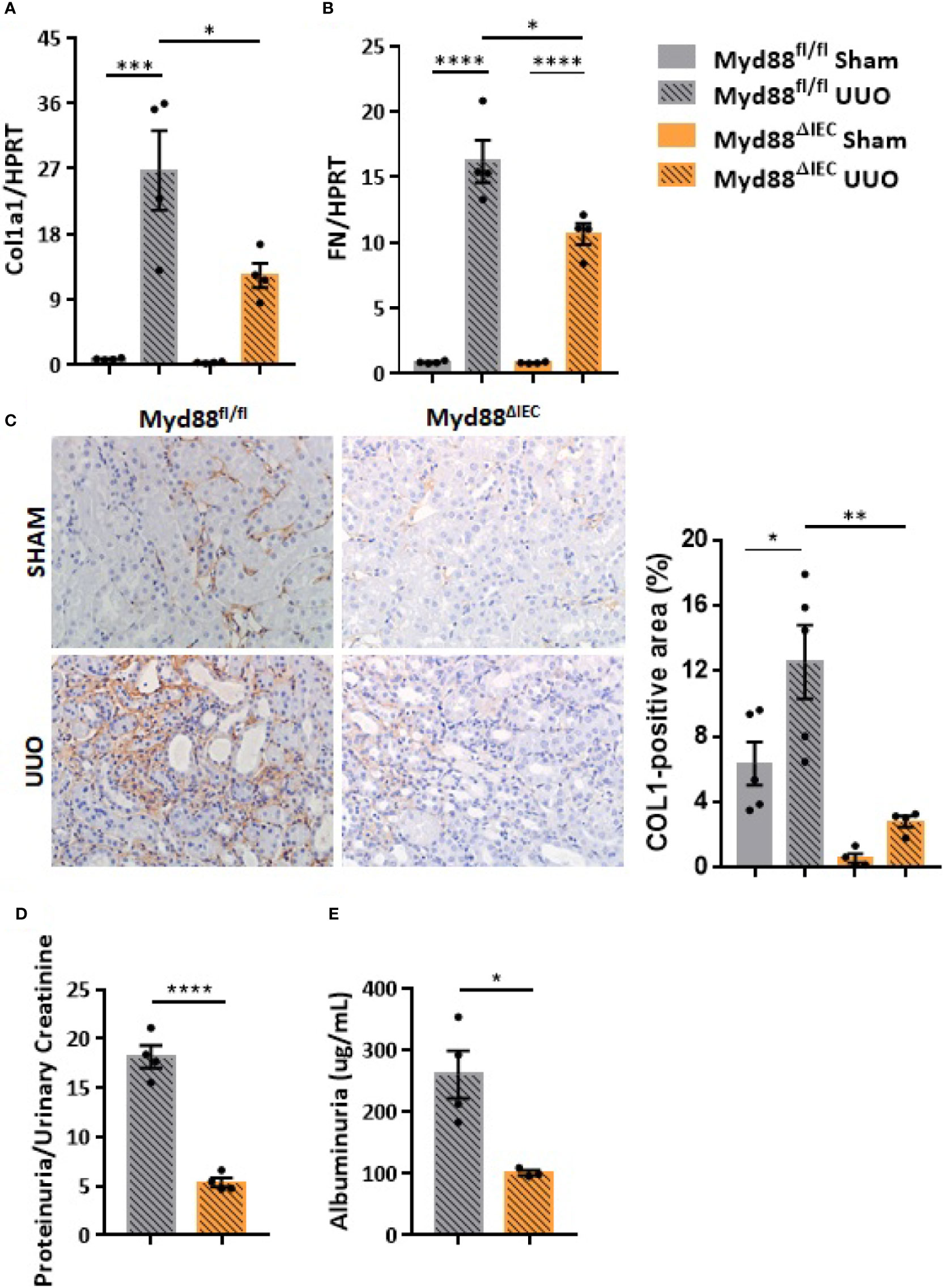
Figure 7 Effect of Myd88 signaling in IECs on the kidney remodeling in CKD. Gene expression of (A) Col1a1 and (B) fibronectin (FN) in the kidney of Myd88fl/fl and Myd88ΔIEC mice submitted or not to UUO (N = 4). (C) Representative images and positive area (%) of immunohistochemical staining for type I collagen (Col I) in the kidney of Myd88fl/fl (N = 5) and Myd88ΔIEC mice (N = 4) submitted or not to UUO. (D) Concentration of total protein normalized by creatinine in pelvic urine (N = 4). (E) Estimation of albuminuria in pelvic urine collected from Myd88fl/fl (N = 4) and Myd88ΔIEC mice (N = 3) submitted to UUO. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Discussion
In the last decades, important advances have shown the essential role of gut microbiota in host physiology, including modulation of host metabolism, central nervous system activity and, especially, immune system. Several studies have shown that gut microbiota may dictate how host immune system would respond to inflammatory hits (17–19). Despite the increasing data supporting the association of CKD and dysbiosis, little is known whether alteration in the gut microbiota would influence the onset and development of CKD. Therefore, in this study we evaluated the effect of depletion of gut microbiota and the role of Myd88 signaling in IECs during UUO injury. Changes in the gut microbiota induced by ABX treatment were followed by upregulation of occludin and ZO-1, which are considered critical components of tight junctions. Substantial data have correlated the expression of both components with epithelial barrier properties. Suppression of occludin or ZO-1 in epithelial monolayer culture alters the dynamic behavior of tight junction by increasing leak pathway flux (20, 21). Moreover, overexpression of occludin limited intestinal barrier disruption in an experimental TNF-induced diarrhea (22). Despite our results may be an indicative of a pre-reinforcement of tight junctions, there is not a consensus whether ABX treatment impairs the intestinal barrier integrity. Tulstrup et al. did not observe alterations in the gene expression of ZO-1, occludin, claudin-1 suggesting that ABX effect is dependent on ABX class (23).
Depletion of gut microbiota abrogated inflammation induced by UUO in the colon. The presence of gut microbiota seems to be crucial to trigger local UUO-induced inflammation and intestinal barrier disruption. Untreated mice showed high expression of proinflammatory cytokines, such as IL-1b and IL-6, in the colon along with a massive presence of bacteria in the mucus layer. This bacterial burden was not limited to the mucus layer since we detected a higher bacterial load in the MLN after UUO. In physiological conditions, the intestinal immune system is taught by the gut microbiota to tolerate luminal antigens, thus limiting host immune responses. However, in dysbiosis conditions, such as the one generated by UUO, a higher trafficking of microbial antigens via CX3CR1hi mononuclear phagocytes can occur aiming at compartmentalizing the intestinal immune response to avoid inflammation (24). Indeed, we did not detect alterations in bacterial load in the liver after UUO, which supports the idea of MLN as a key immune inductive site. Taken together, these observations indicate that depletion of gut microbiota protects against UUO intestinal injury probably by lowering the load of bacteria, which could play a pathogenic role when the intestinal barrier is disrupted. This scenario is aggravated by increased serum levels of IL-6, which support the idea of a sustained inflammatory status generated by CKD.
Beneficial effects of depleted gut microbiota reach the kidney reinforcing the importance of kidney-gut axis in CKD. ABX-treated mice presented lower expression of proinflammatory cytokines after UUO compared to untreated mice. Moreover, depletion of gut microbiota downregulated the expression of several profibrotic markers, such as type I collagen, fibronectin and α-SMA, alleviating tubulointerstitial fibrosis, which is a main histological hallmark of CKD. Moreover, ABX treatment also reduced the expression of cell activation markers, such as FSP-1 and KI-67. During tubular injury, increased FSP-1 expression allows the tracking of cellular behavior associated with matrix production, including cell phenotypic transition, migration, and invasiveness. Evidences indicate that the origin of kidney fibroblasts includes perivascular and resident fibroblasts, pericytes (25), bone marrow derived fibrocytes (26), and transformed epithelial cells (27). Increased expression of KI-67 as observed in untreated mice is an indicative of proliferation of tubular epithelial cells and fibroblasts. All the aforementioned renal improvement fostered the preservation of kidney structure highlighting the idea that, despite the benefits generated by gut microbiota in physiological conditions, the bacterial community can feed inflammatory states generated by other hits.
The importance of IECs in maintenance of intestinal barrier is clearly highlighted in infectious diseases and pathologies with host immune genetic susceptibility. Of great relevance to intestinal homeostasis, Myd88 is a crucial player in the interface between host and microbes and essential to host resistance to infection (28, 29). Deletion of Myd88 decreases AMP secretion, such as RegIIIγ (30) and RELMβ (28) and impairs epithelial cell turnover and repair (31) leading to intestinal barrier dysfunction in experimental infectious colitis. Therefore, in the next step of the study, we decided to evaluate the impact of Myd88 signaling in IECs in the onset and development of CKD. The lack of Myd88 signaling led to lower levels of cytokines in the colon, such as IL-1b, IL-12p40, and IL-17A, in the CKD condition. This significant reduction indicates that, at least locally, Myd88 signaling is essential to the production of critical cytokines involved in the maintenance of intestinal barrier, probably by decreased signaling after PAMP sensing. Unlike in other organs, in the colon IL-1b has a differential role and appears to influence epithelial proliferation promoting healing and tissue repair (32). IL-17A also promotes epithelial barrier integrity by regulating the cellular localization of occludin (33) and regulates antimicrobial activity at mucosal surface (34). On the other hand, IL-17-producing cells can be pathogenic in intestinal diseases, when co-expressed with IL-23, demonstrating a dual role for this cytokine in gut homeostasis and disease (35). Despite the downregulation of these critical cytokines, we did not observe differences in the pattern of bacterial invasion in the mucus layer between WT and Myd88ΔIEC mice. Apparently, activation of myeloid cells via Myd88 is necessary for the development of chronic intestinal inflammation, whereas epithelial Myd88 signaling is essential for host survival (36). This cell-specific role of Myd88 could explain why we did not observe any evidence of intestinal inflammation in the scenario of IEC-deletion of Myd88. On the other hand, unexpectedly, the lack of Myd88 signaling in IECs prevented a bacterial burden in MLN after UUO, indicating that TLR-mediated epithelial recognition of luminal bacteria is required for bacterial trafficking, probably via CX3CR1hi mononuclear phagocytes, aiming at induction of local immune response to avoid systemic inflammation (24).
The lack of Myd88 in IECs also modulated inflammation in the kidney supporting the importance of kidney-gut axis in CKD. Myd88ΔIEC mice presented lower levels of proinflammatory cytokines including IL-4, IL-12p40, and IL-17A, and reduced renal concentration of macrophage-stimulating factors, such as GM-CSF and MIP-1β. GM-CSF has been implicated in the crosstalk between cell tubular injury and cell immune infiltration involved in the sustained tubular injury and progressive interstitial fibrosis at the time of AKI-to-CKD transition (37). Indeed, the lack of Myd88 in IECs modulated the gene expression of profibrotic markers and abrogated the increase of type I collagen after UUO, indicating a substantial prevention of tubulointerstitial fibrosis. The attenuation of inflammation and fibrosis reflected in urinary parameters, once we observed lower levels of protein and albumin in pelvic urine of Myd88ΔIEC mice, supporting the idea that Myd88 signaling in IECs participates in UUO injury. Here, we found that IEC-specific Myd88 deletion alleviated kidney injury indicating that, besides gut microbiota, innate signaling plays an important role in the microbiota-gut-kidney axis in CKD. IECs are capable to sense the microbial presence through a variety of antigen receptors expressed on their apical surface, leading to activation of innate and adaptative immune responses (38). Therefore, we can speculate that the beneficial effects of Myd88 deletion after UUO may be due to an attenuation of intestinal inflammation similarly observed in a model of intestinal injury induced by ischemia/reperfusion. Mühlbauer et al. reported that impaired epithelial Myd88 signaling decreased neutrophil infiltration, reduced binding of IgA to neoantingens, as well as diminished complement activation (39). Myd88 signaling in IECs may affect the maturation of renal resident macrophages and of bone marrow monocytes recruited during inflammatory conditions. Indeed, Emal et al. reported the critical role of gut microbiota in keeping maturation/supply status of renal macrophages in an ischemia-reperfusion injury (40), supporting the importance of gut microbiota-host interaction in modulating the systemic immune system and ultimately inflammatory conditions. Several studies, especially using germ-free mice, demonstrated the importance of microbiota and host interactions in priming immunity (19, 41, 42), which could have deleterious effects in inflammatory states, such as kidney injury.
In summary, we showed that depletion of gut microbiota protects against UUO injury supporting the idea that gut microbiota and host interactions can be detrimental in inflammatory pathologies, such as CKD. Moreover, the renal benefits generated by Myd88 deletion in IECs indicate that the deleterious effects of gut microbiota in such conditions may be dependent of host innate signaling.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Ethics Committee in Animal Research of the Federal University of São Paulo.
Author Contributions
IW and NC conceived and initiated the study. IW, MA-S, OF-N, and NC planned the experiments. IW, MA-S, OF-N, and RF performed the experiments. MM, RC, MC, TH, LP, and RV gave technical assistance. IW, MA-S, OF-N, and NC analyzed the data. IW, AP-S, and NC wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, São Paulo, Brazil) (grant numbers 2014/50833-1 and 2017/05264-7), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, financial code 001). This work was also supported under the International Collaboration Research Funding from FAPESP and The Netherlands Organization for Scientific Research (NWO, The Netherlands, Grant number 2019/19435-3). This work was also a result of collaboration between NC and Ivan Cruz Moura at Institute Imagine, Paris, under the support of CAPES Cofecub Program (Grant number 19/954).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Ivan Cruz Moura for the financial and scientific contribution for this work. Unfortunately, Ivan Cruz Moura passed away in 2019.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.578623/full#supplementary-material
References
1. Collaboration GBDCKD. Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (2020) 395(10225):709–33. doi: 10.1016/S0140-6736(20)30045-3
2. Zoccali C, Vanholder R, Massy ZA, Ortiz A, Sarafidis P, Dekker FW, et al. The systemic nature of CKD. Nat Rev Nephrol (2017) 13(6):344–58. doi: 10.1038/nrneph.2017.52
3. Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol JASN (2014) 25(9):1897–907. doi: 10.1681/ASN.2013101062
4. Mueller DM, Allenspach M, Othman A, Saely CH, Muendlein A, Vonbank A, et al. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis (2015) 243(2):638–44. doi: 10.1016/j.atherosclerosis.2015.10.091
5. Machado RA, Constantino Lde S, Tomasi CD, Rojas HA, Vuolo FS, Vitto MF, et al. Sodium butyrate decreases the activation of NF-kappaB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast-induced nephropathy. Nephrol Dialysis Transplant Off Publ Eur Dialysis Transplant Assoc Eur Renal Assoc (2012) 27(8):3136–40. doi: 10.1093/ndt/gfr807
6. Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J Am Soc Nephrol JASN (2015) 26(8):1877–88. doi: 10.1681/ASN.2014030288
7. Al-Harbi NO, Nadeem A, Ahmad SF, Alotaibi MR, AlAsmari AF, Alanazi WA, et al. Short chain fatty acid, acetate ameliorates sepsis-induced acute kidney injury by inhibition of NADPH oxidase signaling in T cells. Int Immunopharmacol (2018) 58:24–31. doi: 10.1016/j.intimp.2018.02.023
8. Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr (2011) 141(5):769–76. doi: 10.3945/jn.110.135657
9. Muniz LR, Knosp C, Yeretssian G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front Immunol (2012) 3:310. doi: 10.3389/fimmu.2012.00310
10. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol (2017) 14(1):9–21. doi: 10.1038/nrgastro.2016.169
11. Vaziri ND, Yuan J, Rahimi A, Ni Z, Said H, Subramanian VS. Disintegration of colonic epithelial tight junction in uremia: a likely cause of CKD-associated inflammation. Nephrol Dialysis Transplant Off Publ Eur Dialysis Transplant Assoc Eur Renal Assoc (2012) 27(7):2686–93. doi: 10.1093/ndt/gfr624
12. Vaziri ND, Yuan J, Norris K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol (2013) 37(1):1–6. doi: 10.1159/000345969
13. Bossola M, Sanguinetti M, Scribano D, Zuppi C, Giungi S, Luciani G, et al. Circulating bacterial-derived DNA fragments and markers of inflammation in chronic hemodialysis patients. Clin J Am Soc Nephrol CJASN (2009) 4(2):379–85. doi: 10.2215/CJN.03490708
14. McIntyre CW, Harrison LE, Eldehni MT, Jefferies HJ, Szeto CC, John SG, et al. Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol CJASN (2011) 6(1):133–41. doi: 10.2215/CJN.04610510
15. Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut (2016) 65(4):575–83. doi: 10.1136/gutjnl-2015-309728
16. Hesketh EE, Vernon MA, Ding P, Clay S, Borthwick G, Conway B, et al. A murine model of irreversible and reversible unilateral ureteric obstruction. J Visualized Experiments JoVE (2014) 94(e52559):1–6. doi: 10.3791/52559
17. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity (2012) 37(1):158–70. doi: 10.1016/j.immuni.2012.04.011
18. Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell (2017) 171(5):1015–28.e13. doi: 10.1016/j.cell.2017.09.016
19. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res (2020) 30(6):492–506. doi: 10.1038/s41422-020-0332-7
20. Al-Sadi R, Khatib K, Guo S, Ye D, Youssef M, Ma T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointestinal Liver Physiol (2011) 300(6):G1054–64. doi: 10.1152/ajpgi.00055.2011
21. Van Itallie CM, Fanning AS, Bridges A, Anderson JM. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell (2009) 20(17):3930–40. doi: 10.1091/mbc.E09-04-0320
22. Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR,2, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol (2010) 189(1):111–26. doi: 10.1083/jcb.200902153
23. Tulstrup MV, Christensen EG, Carvalho V, Linninge C, Ahrne S, Hojberg O, et al. Antibiotic Treatment Affects Intestinal Permeability and Gut Microbial Composition in Wistar Rats Dependent on Antibiotic Class. PloS One (2015) 10(12):e0144854. doi: 10.1371/journal.pone.0144854
24. Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, et al. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature (2013) 494(7435):116–20. doi: 10.1038/nature11809
25. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol (2008) 173(6):1617–27. doi: 10.2353/ajpath.2008.080433
26. Li J, Deane JA, Campanale NV, Bertram JF, Ricardo SD. The contribution of bone marrow-derived cells to the development of renal interstitial fibrosis. Stem Cells (2007) 25(3):697–706. doi: 10.1634/stemcells.2006-0133
27. Zeisberg M, Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci J Virtual Library (2008) 13:6991–8. doi: 10.2741/3204
28. Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, et al. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infection Immun (2014) 82(9):3753–63. doi: 10.1128/IAI.02045-14
29. Friedrich C, Mamareli P, Thiemann S, Kruse F, Wang Z, Holzmann B, et al. MyD88 signaling in dendritic cells and the intestinal epithelium controls immunity against intestinal infection with C. rodentium. PloS Pathog (2017) 13(5):e1006357. doi: 10.1371/journal.ppat.1006357
30. Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med (2007) 204(8):1891–900. doi: 10.1084/jem.20070563
31. Gibson DL, Ma C, Bergstrom KS, Huang JT, Man C, Vallance BA. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol (2008) 10(3):618–31. doi: 10.1111/j.1462-5822.2007.01071.x
32. Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, et al. Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut (2014) 63(4):598–609. doi: 10.1136/gutjnl-2012-303329
33. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity (2015) 43(4):727–38. doi: 10.1016/j.immuni.2015.09.003
34. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol (2017) 18(6):612–21. doi: 10.1038/ni.3742
35. Fuss IJ. IL-17: intestinal effector or protector? Nature (2011) 4(4):366–7. doi: 10.1038/mi.2011.24
36. Asquith MJ, Boulard O, Powrie F, Maloy KJ. Pathogenic and protective roles of MyD88 in leukocytes and epithelial cells in mouse models of inflammatory bowel disease. Gastroenterology (2010) 139(2):519–29.e1-2. doi: 10.1053/j.gastro.2010.04.045
37. Xu L, Sharkey D, Cantley LG. Tubular GM-CSF Promotes Late MCP-1/CCR2-Mediated Fibrosis and Inflammation after Ischemia/Reperfusion Injury. J Am Soc Nephrol JASN (2019) 30(10):1825–40. doi: 10.1681/ASN.2019010068
38. Campeau JL, Salim SY, Albert EJ, Hotte N, Madsen KL. Intestinal epithelial cells modulate antigen-presenting cell responses to bacterial DNA. Infection Immun (2012) 80(8):2632–44. doi: 10.1128/IAI.00288-12
39. Muhlbauer M, Perez-Chanona E, Jobin C. Epithelial cell-specific MyD88 signaling mediates ischemia/reperfusion-induced intestinal injury independent of microbial status. Inflammatory Bowel Dis (2013) 19(13):2857–66. doi: 10.1097/01.MIB.0000435445.96933.37
40. Emal D, Rampanelli E, Stroo I, Butter LM, Teske GJ, Claessen N, et al. Depletion of Gut Microbiota Protects against Renal Ischemia-Reperfusion Injury. J Am Soc Nephrol JASN (2017) 28(5):1450–61. doi: 10.1681/ASN.2016030255
41. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol (2009) 9(5):313–23. doi: 10.1038/nri2515
Keywords: chronic kidney disease, gut microbiota, Myd88, inflammation, fibrosis
Citation: Watanabe IKM, Andrade-Silva M, Foresto-Neto O, Felizardo RJF, Matheus MAC, Silva RC, Cenedeze MA, Honda TSB, Perandini LAB, Volpini RA, Pacheco-Silva A and Câmara NOS (2020) Gut Microbiota and Intestinal Epithelial Myd88 Signaling Are Crucial for Renal Injury in UUO Mice. Front. Immunol. 11:578623. doi: 10.3389/fimmu.2020.578623
Received: 30 June 2020; Accepted: 09 November 2020;
Published: 22 December 2020.
Edited by:
Gislane Lelis Vilela de Oliveira, São Paulo State University, BrazilReviewed by:
Hee-Seong Jang, University of Nebraska Medical Center, United StatesZhengxiang He, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2020 Watanabe, Andrade-Silva, Foresto-Neto, Felizardo, Matheus, Silva, Cenedeze, Honda, Perandini, Volpini, Pacheco-Silva and Câmara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ingrid Kazue Mizuno Watanabe, aW5ncmlkd2F0YW5hYmVAZ21haWwuY29t