 Claire E. McQuitty
Claire E. McQuitty Roger Williams1,2
Roger Williams1,2 Shilpa Chokshi
Shilpa Chokshi Luca Urbani
Luca Urbani- 1Institute of Hepatology, Foundation for Liver Research, London, United Kingdom
- 2Faculty of Life Sciences & Medicine, King’s College London, London, United Kingdom
Chronic liver disease when accompanied by underlying fibrosis, is characterized by an accumulation of extracellular matrix (ECM) proteins and chronic inflammation. Although traditionally considered as a passive and largely architectural structure, the ECM is now being recognized as a source of potent damage-associated molecular pattern (DAMP)s with immune-active peptides and domains. In parallel, the ECM anchors a range of cytokines, chemokines and growth factors, all of which are capable of modulating immune responses. A growing body of evidence shows that ECM proteins themselves are capable of modulating immunity either directly via ligation with immune cell receptors including integrins and TLRs, or indirectly through release of immunoactive molecules such as cytokines which are stored within the ECM structure. Notably, ECM deposition and remodeling during injury and fibrosis can result in release or formation of ECM-DAMPs within the tissue, which can promote local inflammatory immune response and chemotactic immune cell recruitment and inflammation. It is well described that the ECM and immune response are interlinked and mutually participate in driving fibrosis, although their precise interactions in the context of chronic liver disease are poorly understood. This review aims to describe the known pro-/anti-inflammatory and fibrogenic properties of ECM proteins and DAMPs, with particular reference to the immunomodulatory properties of the ECM in the context of chronic liver disease. Finally, we discuss the importance of developing novel biotechnological platforms based on decellularized ECM-scaffolds, which provide opportunities to directly explore liver ECM-immune cell interactions in greater detail.
Introduction
The extracellular matrix (ECM) constitutes a complex network of proteins which make up the overall architecture of an organ. It acts as a structural support, imparts mechano-elastic properties and delivers environmental cues to influence cellular proliferation, survival, shape, migration and differentiation and plays an important role in modulating tissue homeostasis, remodeling and regeneration (1–3). The ECM network can be broadly divided into different families of proteins based on their structure: collagens, elastin, cell-adhesion glycoproteins, glycosaminoglycans (GAGs), proteoglycans, and matricellular proteins. GAGs are long, unbranched chains of repeating disaccharide units; proteoglycans are formed by GAGs which are bound to core proteins. Matricellular proteins are non-structural ECM-associated proteins with regulatory activity that are released and/or incorporated into the ECM particularly during development and in response to injury (4). The ECM also binds a large number of ECM-associated molecules including growth factors, cytokines, and chemokines (5), which can bind to numerous ECM proteins (5), discussed in detail in the section “ECM protein association with growth factors, cytokines and chemokines”.
The ECM is ubiquitous in all tissues throughout the body; however, the proportions, specific composition and organization of ECM proteins differs organ-to-organ, resulting in a unique landscape which provides a set of properties integral to the organs’ function. Berger et al., highlighted this in a recent study using proteomics to compare the ECM of decellularized porcine pancreas, small intestine, and lung. The study showed clear differences in the ECM signature between decellularized organs and show that organ specific ECM landscapes drive spontaneously differentiating human induced pluripotent stem cells (hiPSCs) toward different lineages in an organ- dependent manner, elegantly revealing that the ECM possesses organ-specific biological cues (6).
In this review, we highlight the importance of the intricate ECM-immune cell interactions in the liver and during liver fibrosis and describe the known immunomodulatory activities of individual ECM molecules and fragments in chronic liver disease (CLD)s.
ECM in the Healthy Liver
In the healthy liver, the ECM comprises up to 10% of the total volume (7) and is largely restricted to the portal tracts, sinusoidal walls and the central vein (8).
An in-depth proteomics analysis of decellularized human liver ECM by Naba et al., identified more than 150 distinct ECM proteins which make up the healthy human liver matrisome (9). Among these are: (1) a large and diverse number of collagens, including transmembrane-, network-forming-, fibrillar and fibril-associated collagens; (2) 44 ECM glycoproteins, the most abundant of which include fibrinogens and fibronectins; (3) 11 proteoglycans, including the small leucine-rich repeat proteoglycans: biglycan, decorin, and lumican; several understudied proteins: proteoglycan-2, -3, and -4, and versican; (4) a large number of proteins associated with the formation of elastic fibres; and (5) the matricellular proteins – periostin, tenascin-C and -X. Further details regarding these ECM proteins found in the healthy human liver matrisome can be found in a recent review by Arteel et al. (10).
In homeostasis, the composition of the ECM is constantly fine-tuned by a series of synthesis and degradation events. Hepatocytes, liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs) and Kupffer cells (KCs) are all capable of synthesising ECM proteins (11), as well as matrix metalloproteinase (MMP)s and their inhibitors, tissue-inhibitors of MMPs (TIMPs) which can degrade most ECM proteins (12) and contribute to ECM remodeling and turnover in homeostasis and disease. MMPs are considered major contributors to ECM remodeling. They are secreted as latent zymogens, requiring activation, and are marginally expressed in the healthy liver, but expressed immediately after hepatic injury and thought to be involved in fibrosis resolution (12). Indeed, several rodent models of CLD show enhanced MMP expression linked to amelioration of fibrosis. For example, Kim et al. show that in a mouse model of CCl4 induced liver fibrosis, enhanced MMP-13 expression promoted recovery from liver fibrosis (13). Similarly, Siller-Lopez and colleagues showed that treatment with human MMP-8 showed amelioration of liver fibrosis in a CCl4 treated bile-duct ligation (BDL) rat model of liver fibrosis (14).
Careful management of ECM remodeling is required for tissue regeneration and wound healing and is crucial for maintaining homeostasis. Indeed, if the balance of synthesis and degradation is perturbed, e.g., following injury or insult, the liver can progress towards increased scar formation/fibrosis, during which ECM synthesis greatly outweighs degradation resulting in excessive accumulation of ECM proteins. HSCs are central participants in this fibrogenic processes. Local inflammatory signals can activate quiescent HSCs to a highly proliferative phenotype driving their transdifferentiation into myofibroblasts, which characteristically increase ECM secretion, this process underlies the development and progression of liver fibrosis (15). The mechanisms of HSC activation are complex and multi-factorial and can be characterized as: “initiation”, in which increased susceptibility to a range of extracellular signals promotes expression of growth factor receptors, modulation of growth factor signaling and transdifferentiation to a contractile and fibrogenic phenotype; and “perpetuation”, in which a series of positive feed-back loops from the ECM amplify this activated phenotype. Indeed, HSCs can interact with ECM proteins via αvβ3 integrin which binds to (arginine- glycine- aspartic acid) RGD domains on ECM proteins. αvβ3 integrin expression is upregulated during liver fibrosis development and progression (16) and ECM-αvβ3 interactions have been suggested to support HSC proliferation (17). Initiation and perpetuation of HSC activation is modulated by complex cross-talk between liver cells, infiltrating immune cells and the ECM (18, 19). Finally, the ECM directly influences cell behaviour, including immune cells which can interact with ECM proteins through cell surface receptors such as integrins which activate intracellular signaling pathways (2), cell migration (1) and proliferation (4).
In summary, the interplay between the ECM, liver cells and immune cells helps maintain a balance between injury and repair. Perturbation of this equilibrium, for example by toxic injuries such as alcohol, can drive liver fibrogenesis which is characterized by progressive accumulation of ECM proteins coupled with chronic inflammation.
ECM in Chronic Liver Disease
The progression of CLDs, including alcoholic liver disease (ALD), non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), and Hepatitis B and C (HBV/HCV), is associated with the development of fibrosis. In these diseases, active remodeling of the ECM proteins, drives dramatic changes in the ECM landscape and the release of ECM-associated bioactive molecules (e.g., growth factors and cytokines). In addition, fragmented ECM proteins, generated upon remodeling or during injury, are recognized by immune cells as damage-associated molecular patterns (DAMPs), termed ECM-DAMPs.
Generally, liver fibrosis involves increased ECM protein synthesis and deposition, particularly of fibrillar collagens type I, which can be 8-fold higher than in healthy liver (20), collagen type III, fibronectin, and laminin (21) which are deposited in the Space of Disse, forming a dense matrix. If unresolved, a proportion of patients with fibrosis can progress to cirrhosis and/or hepatocellular carcinoma (HCC), the rate of which is associated with the underlying aetiology (22, 23). The reasons for these differences are not well understood but interestingly the stage of fibrosis and the causative aetiology seem to differ in the composition and organization of ECM components. This is detailed in Table 1. For example, fibrotic stage-specific changes in ECM composition during chronic HCV infection correlates with progressive accumulation of collagens I and II, followed by elastin over expression (28). Similarly, Daneshgar et al. reported upregulation of MMPs 23B and 28 and versican, in decellularized human liver samples with increasing grades of fibrosis and cirrhosis (30). Aetiology specific differences have also been observed. Mazza and colleagues report increased collagen 10A, 5A, fibulin 5, and fibronectin in cirrhotic liver from patients with HCC (31). Whereas, advanced ALD is associated with increased deposition of collagen type 1, upregulated synthesis of osteopontin (OPN) and increased synthesis of cellular fibronectin (cFN). To date, ECM accumulation in NASH has not specifically characterized.

Table 1 Disease-specific changes in the extracellular matrix (ECM).
Much of this information is available thanks to ongoing advances in proteomics analysis and the development of online platforms, such as MatrisomeDB (32), which allows comparison between healthy liver ECM and diseased liver ECM from different aetiologies. However, currently available proteomics data on human liver ECM is relatively limited, and it is difficult to draw conclusions on which ECM proteins are upregulated or downregulated in different liver diseases and during disease progression and understand their biological context.
Inflammation in Chronic Liver Disease
Inflammation is central to the pathogenesis and progression of CLD (33). In general, pro-inflammatory immune responses are thought to drive progressive CLD through bystander cell damage and activation of fibrogenic and hepatocarcinogenic pathways. Anti-inflammatory responses may counter these injurious pro-inflammatory responses and slow pathogenesis, but excessive anti-inflammatory responses may inadvertently impair immune-mediated wound healing and induce a state of immune insufficiency leading to a vulnerability to bacterial infection and increase the risk of hepatic decompenzation, as seen in patients with liver cirrhosis (34). A number of immune cells contribute to liver inflammation in CLD, including resident macrophages (KCs), infiltrating monocyte-derived macrophages, T cells, dendritic cell (DC)s and neutrophils. These immune cells, and especially macrophages, activate HSCs, and chronic activation drives progressive fibrosis and ECM remodeling (35, 36). The causative mechanisms responsible for the development of non-resolving and chronic inflammation varies dependant on disease aetiology and is appraised in detail in these reviews (35, 37). For example, ALD inflammation is primarily driven through loss of barrier integrity in the gut and excessive translocation of gut-derived bacterial pathogen-associated molecular pattern (PAMP)s, such as lipopolysaccharide (LPS), and bacterial PAMPs (22) which trigger release of pro-inflammatory cytokines from KCs (22). This in turn drives recruitment of infiltrating neutrophils, a prominent feature of alcoholic hepatitis (37). Inflammation in NAFLD is initially thought to be driven by lipotoxicity (accumulation of toxic lipids in hepatocytes). Lipotoxicity promotes chronic inflammation through activation of KCs and recruitment of infiltrating monocytes, resulting in induction of pro-inflammatory intracellular pathways, such as transforming growth factor (TGF)-β1 (38, 39) promoting HSC activation and driving progressive fibrosis. However, in later stages increased bacterial translocation also contributes to hepatic and systemic inflammation. The concomitant presence of infiltrating monocyte-derived macrophages and KC activation is not exclusive to NASH and is seen in progressive liver fibrosis underlying other CLDs, including HBV and HCV, where viral infection is followed by rapid recruitment of pro-inflammatory monocytes and T-cells into the liver (40). Chronic infection with HBV and HCV drives both inflammatory and anti-viral suppressive immune responses and generally leads to progressive fibrosis and ECM remodeling.
Although multiple mediators of inflammation have been described in CLD, including cytokines, chemokines, DAMPs, and PAMPs, the immunomodulatory role of the ECM has largely been ignored and in particular the ability of the ECM network to modulate immune responses through mechanotransduction, and changes in organ stiffness is not well described.
ECM Stiffness and Inflammation
Remodeling and accumulation of ECM during fibrosis results in gradual liver stiffening. This is not only one of the results of the fibrogenic process, but also an important promoter of the progression of liver fibrosis itself. Central to the mechanosensing regulation of hepatic cells are the Hippo-related transcriptional coactivators Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) (41). In patients with organ fibrosis, a feed-forward cycle of matrix stiffness, myofibroblast activation and proliferation, and ECM accumulation perpetuates fibrosis progression (42), and YAP/TAZ mechanosignaling represents a core pathway of this cycle. The regulation and biological effects of YAP/TAZ signaling are cell type specific (43). In liver fibrosis, YAP is activated in HSCs in response to matrix stiffening, ultimately resulting in the translocation of YAP/TAZ from the cytoplasm to the nucleus and inducing the expression of profibrotic genes and increasing expression of alpha- smooth muscle actin (α-SMA) and excessive ECM deposition (44, 45). In hepatocytes, YAP/TAZ activity was increased in chronic liver injury in mice, and YAP-expressing hepatocytes activated the expression of pro-fibrogenic proteins (collagen I, TIMP-1, platelet-derived growth factor (PDGF) -c, TGF-β2) and pro-inflammatory factors (tumor necrosis factor (TNF)-α, Interleukin (IL) -1β), and stimulated the expansion of myofibroblasts and macrophages (46). This was confirmed in experiments with YAP and YAP/TAZ knockout (KO) animals, where hepatocytes exhibited limited myofibroblast expansion, inflammation, and decreased fibrosis after CCl4 injury.
Macrophages can sense changes in stiffness through mechano-transduction signaling and respond by regulating Toll-like receptor (TLR)-mediated inflammation. Macrophages grown on softer substrates induce stronger TLR4 and TLR9 signaling and release higher amounts of the strong pro-inflammatory cytokine TNF-α (47). These events are clearly influenced by growth surface stiffness, as inhibition of mechanotransduction signaling enhances TLR signaling, which increases TNF-α production and prolongs activation of TLR downstream kinases p38 and extracellular signal-regulated kinase (ERK)1/2. ECM deposition that occurs in hepatic fibrosis drastically increases tissue stiffness, indicating that these TLR-mediated events could be involved in modulating inflammatory microenvironments in CLD.
Active ECM remodeling and accumulation underpins liver stiffening in fibrosis, and results in release and/or activation of a range bioactive molecules harboured in the ECM. These molecules can directly and indirectly modulate immune responses during CLD.
ECM Protein Association With Growth Factors, Cytokines, and Chemokines
ECM proteins anchor a number of growth factors, cytokines, and chemokines, creating a reservoir of bio-active, immunomodulatory molecules. The release and availability of these molecules is regulated by ECM proteins in response to injury, wound-healing, and tissue regeneration, creating a local inflammatory milieu which can drive recruitment of immune cells. In turn, recruited immune cells can actively remodel the ECM network, both by secreting and digesting ECM proteins, further contributing to the release of ECM-bound molecules.
One such ECM-bound molecule, is TGF-β1. TGF-β1 is stored in the ECM and responds to perturbations in the microenvironment to ensure ECM homeostasis (48, 49). This is coordinated through a tightly controlled mechanism of deposition into and release from the ECM. Secreted TGF-β1 is held inactive through its association with a complex of proteins termed the large latency complex (LLC). The LLC, in turn, is anchored to fibrillar ECM proteins (fibronectin, fibrillins, fibulins) (48) by latent- TGF-β- binding proteins (LTBP)s, creating a reservoir of inactive TGF-β1 within the ECM. Active TGF-β1 can be released from the LLC by either proteolytic degradation or mechanical deformation of the complex. Cells bearing αν integrins bind the latency associated protein (LAP) within the ECM- anchored LLC. This generates a pulling force causing a conformational change in the LLC that frees active TGF-β1 (50, 51, 52). The integrin-dependent mechanical release of TGF-β1, explains why latent TGF-β1 can only be activated if bound to the ECM. Mechanical resistance by the ECM ensures that when force is applied to LAP, it will produce the conformational change needed to free TGF-β1. Consequently, increasing ECM stiffness has a direct effect on TGF-β1 activation. Disorganized ECM, typical of remodeling tissues, does not provide enough resistance to induce release of TGF-β1, while stiffer ECM, typical of advanced fibrosis, is expected to ensure sufficient resistance for latent TGF-β1 activation by cell pulling (48).
Most hepatic cells are sensitive to TGF-β1 and in fibrogenesis excessive TGF-β1 activates HSCs inducing their transdifferentiation into myofibroblasts, which further deposit ECM proteins. TGF-β1 also contributes to ECM deposition by amplifying hepatocyte death (53). Furthermore, timely release of TGF-β1 in response to changes in ECM stiffness during liver fibrosis can drive both pro-inflammatory and inhibitory immune responses. For example, TGF-β1 is a key mediator of the terminal differentiation of regulatory T cells (Tregs), important negative regulators of the inflammatory process in liver fibrosis. Studies have also shown that TGF-β1 is immunosuppressive through induction of macrophages, impairment of DC and natural killer (NK) cell activity (52, 54, 55, 56).
One way in which TGF-β1 drives immune responses is thorough the establishment of paracrine senescence in hepatic cells. This describes the transmission of senescence properties from senescent cells to otherwise normal cells in acute and chronic liver injuries (57). Senescence is considered as a multistep, dynamic cellular process in response to endogenous and exogenous stress, such as oncogene activation, DNA damage or other stress-mediated signals, which induces cells to enter a permanent cell cycle arrest. Senescent cells with cell cycle arrest undergo chromatin remodeling, present senescence-associated secretory phenotype (SASP), change morphology and gain other characteristics of a complete senescence phenotype. In the liver, cellular senescence may cause loss of regenerative capability affecting the function and tissue renewal (58). The SASP is a dynamic pro-inflammatory response that activates and reinforces the senescent phenotype in the surrounding cells and modulates fibrosis (57). Most senescent cells express multiple cytokines such as IL-8, chemokine ligand (CCL)-2 (monocyte chemoattractant protein (MCP)-1), CCL7 (MCP-3), CCL8 (MCP-2), CCL13 (MCP-4), CCL3, etc (59, 60). The most characterized SASP components include multiple pro-inflammatory cytokines: IL-1β, IL-6 and TGF-β1. Thus, TGF-β1 is a critical element for the establishment of paracrine senescence through the SASP. TGF-β1 induces and maintains paracrine senescence through a mechanism that generates reactive oxygen species (ROS) and DNA damage response (DDR) (61).
Senescence has been reported in hepatocytes in different liver diseases such as cirrhosis (62, 63) in cholangiocytes during primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC) (64, 65, 66); and in HSCs in fibrosis (67). Specifically, acute liver injury was shown to induce senescence predominantly in hepatocytes in vivo, and this was amplified and transmitted between hepatocytes in a feedback loop TGF-β1-dependent produced by macrophages (68). In a model of biliary disease, senescence was specifically induced in cholangiocytes, with evident alterations in the cellular and signaling microenvironment, recruitment of myofibroblasts and macrophages causing collagen deposition, TGF-β1 production and induction of senescence in surrounding cholangiocytes and hepatocytes. The induction of senescence in cholangiocytes also results in the establishment of paracrine senescence in the liver parenchyma through TGF-β1-dependent mechanisms (64, 69).
Numerous ECM proteins in the liver can interact with TGF-β1 and regulate its local availability and activation, which in turn can modulate immune cell responses. For example, Thrombospondin-1 (TSP-1) is an endogenous activator of TGF-β1 during tissue repair and remodeling, causing release of active TGF-β1 from the LLC through a non-proteolytic mechanism (52, 70, 71). The ability to regulate local TGF-β1 release provides TSP-1 certain immunomodulatory properties (49, 72). TSP-1 null mice have a lower TGF-β1 activity that reduces T helper cell (Th)17 and IL-17 levels (73, 74). TSP-1 is implicated in the activation of TGF-β1 by human NK cells (75) and also seems to be responsible for CD36-dependent activation of TGF-β2 by murine antigen presenting cells, a growth factor important for induction of Foxp3+Tregs (76).
Biglycan and decorin have been shown to inhibit TGF-β1 activity. Biglycan inhibits the activity of TGF-β1 in vitro (77) and in vivo, as demonstrated by experiments in biglycan-deficient mice that showed elevated levels of active TGF-β1 in plasma (78), and decorin is reported to bind to and inhibit multiple growth factors and growth factor receptors including TGF-β family members (79).
In liver fibrosis, TGF-β1 availability is regulated by fibronectin (FN), and directly affects collagen fibrillogenesis in response to liver injury (80). Similarly, increasing local availability of TGF-β1 by tenascin-C promotes fibrogenesis through activation of fibroblasts to produce ECM (81). Importantly, Tenascin-C is upregulated in patients with cirrhosis, HCC and chronic hepatitis C (82, 83, 84, 85). Extracellular matrix protein 1 (ECM-1) is reported to downregulate TGF-β1 activity. ECM-1 is produced in the liver mainly by hepatocytes and is downregulated upon liver damage. This event directly induces release of activated TGF-β1 from its latent complex. Furthermore, ECM-1 inhibits latent TGF-β1 activation during homeostasis preventing spontaneous fibrogenesis. In fact, ECM-1 supplementation can reverse fibrosis when ECM-1-knockout mice are exposed to liver damage (86).
Other ECM proteins which are reported to bind TGF-β include fibrillin-1, plasma fibronectin (pFN) (48), heparan sulphate (HS), and agrin which can bind to both TGF-β1 and TGF-β2 (87) and regulate local gradients and growth factor activity (88, 89).
Many ECM proteins bind a range of growth factors and are summarized in Table 2. Some examples are represented by biglycan, which interacts with TNF-β (103), bone morphogenetic proteins (BMPs) BMP-2, BMP-4, BMP-6 (104, 105), and Wnt-1-induced secreted protein 1 (WISP1) (106). Decorin sequesters PDGF before it can bind to receptors on target cells, thus blocking its activity (107). Similarly, Decorin binds to and neutralizes connective tissue growth factor (CTGF) (108), Low density lipoprotein receptor related protein 1 (LRP1) and WSP1 (106). Fibrillin-1 binds to BMPs, integrins responsible for cell-matrix communication, and other growth and differentiation factors (100–102, 109). pFN binds to vascular endothelial growth factor (VEGF), BMP-1, hepatocyte growth factor (HGF) fibroblast growth factor (FGF)-2, PDGF, and latent TGF-β1 (48). Finally, tenascin-C binds to the FGF family, VEGF, PDGF, and insulin growth factor-binding proteins (IGF-BPs) (95, 96). A detailed account of the specific interactions can be found in this review (4).
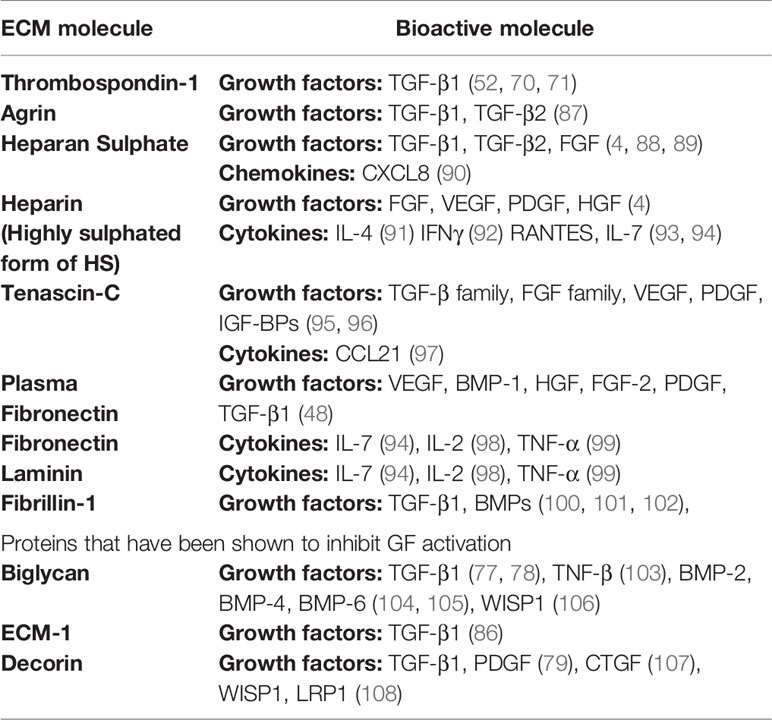
Table 2 Growth factors, chemokines, and cytokines that bind to specific extracellular matrix (ECM) proteins.
The ECM also harbours a large number of cytokines and chemokines secreted, for example, by immune cells infiltrating to sites of damage or infection. Many ECM proteins have affinity for these molecules creating a chemoattractant and immune-modulatory gradient further attracting and activating incoming immune cells. For example, heparan sulphate binds IL-4 (91),which primarily acts on B cells; interferon (IFN)γ (92), which acts on monocytes, T cells, DCs, and NK cells; Regulated on Activation Normal T cell Expressed and Secreted (RANTES) (93), which acts on memory T cells, eosinophils, basophils, NK cells, and DCs; and IL-7 (94). Similarly, IL-7 and IL-2 bind fibronectin (94), laminin, and collagen IV; while TNF-α (98), which acts on T cells, B cells, monocytes, endothelial cells and fibroblasts binds with FN (110) and laminin (99). Finally, tenascin-C binds to CCL21 enforcing an immune-suppressive lymphoid stroma, as shown in a model of oral squamous cell carcinoma (97). By inducing CCL21 and binding to it, tenascin-C instructed lymphoid stroma to recruit T regulatory cells and express high levels of anti-inflammatory cytokines.
Immunomodulatory Properties of the ECM
In addition to influencing immune responses through the release and modulation of bioactive molecules, ECM components can directly modulate immune responses. The prevalence, topological organization and molecular structure of ECM proteins changes during tissue remodeling and wound repair, as well as during disease development, progression and inflammation (111, 112). Changes in the ECM occur in both healthy and diseased tissues, and matrix molecules are able to create microenvironmental niches that affect intracellular signaling pathways critical for the regulation of local cells and immune cells. Many ECM proteins harbour bioactive domains which act as direct ligands on a large number of immune cell receptors (113, 114). ECM ligand-immune cell interactions drive both pro- and anti-inflammatory immune responses and are involved in maintaining homeostasis but also in establishing pathways of pathogenesis. For example, collagen is a high affinity ligand for leukocyte associated immune receptor (LAIR)-1 expressed on most immune cells including T cells, B cell, NK cells, monocytes, macrophages, monocyte-derived DCs, mast cells, eosinophils, and basophils (115). Collagen-LAIR-1 association promotes immune suppressive phenotype essentially holding the immune response in check and contributing to homeostasis. Similarly, differential expression of LAIR-1 by immune cells or interruption of collagen-LAIR-1 binding by the action of soluble receptor LAIR-2 (115), may be involved in driving disease processes. Indeed, Martinez-Esparza et al., found a reduction of LAIR-1 expression on macrophages in the liver of patients with liver cirrhosis but an increase LAIR-1 expression in circulating monocytes in the blood of cirrhosis patients (116). The authors suggest that upregulation of collagen in the cirrhotic liver may serve to inhibit LAIR-1 expression on liver macrophages, suggesting a role for collagen-LAIR-1 interaction in liver cirrhosis.
In addition, newly synthetized or fragmented ECM proteins, generated upon remodeling or during injury, are recognized by immune cells as DAMPs, or in this case, ECM-DAMPs (117, 118). ECM-DAMPs engage directly with multiple immune cells, including macrophages (119–122), monocytes (123), dendritic cells, and T cells (124), as well as with endothelial cells (3, 41), and modulate a wide range of pro- and anti-inflammatory responses, which will be discussed in the following sections. These endogenous glycoproteins, peptides, proteoglycans and GAGs support infiltrating immune cells and proliferating tissue resident cells to orchestrate sterile inflammation, for example by modulating TGF-β and IL-1β signaling (117, 125–128). ECM-DAMPs engage with multiple pattern recognition receptors (PRRs), which recognize and induce activation of cells of the innate and adaptive immune system such as monocytes, macrophages and T cells, and non-immune cells like endothelial cells. Among the PRRs identified as able to bind ECM-DAMPs, TLR2, and TLR4 play a preponderant role as effectors of their immunomodulatory properties (129). A summary of interactions between TLR4 on immune cells and ECM proteins/ECM-DAMPs and consequent immune response is shown in Figure 1. ECM-DAMPs promote both pro- and anti-inflammatory immune responses, suggesting that the matrix proteins are actively involved in fine tuning the inflammatory processes (117, 130–132).
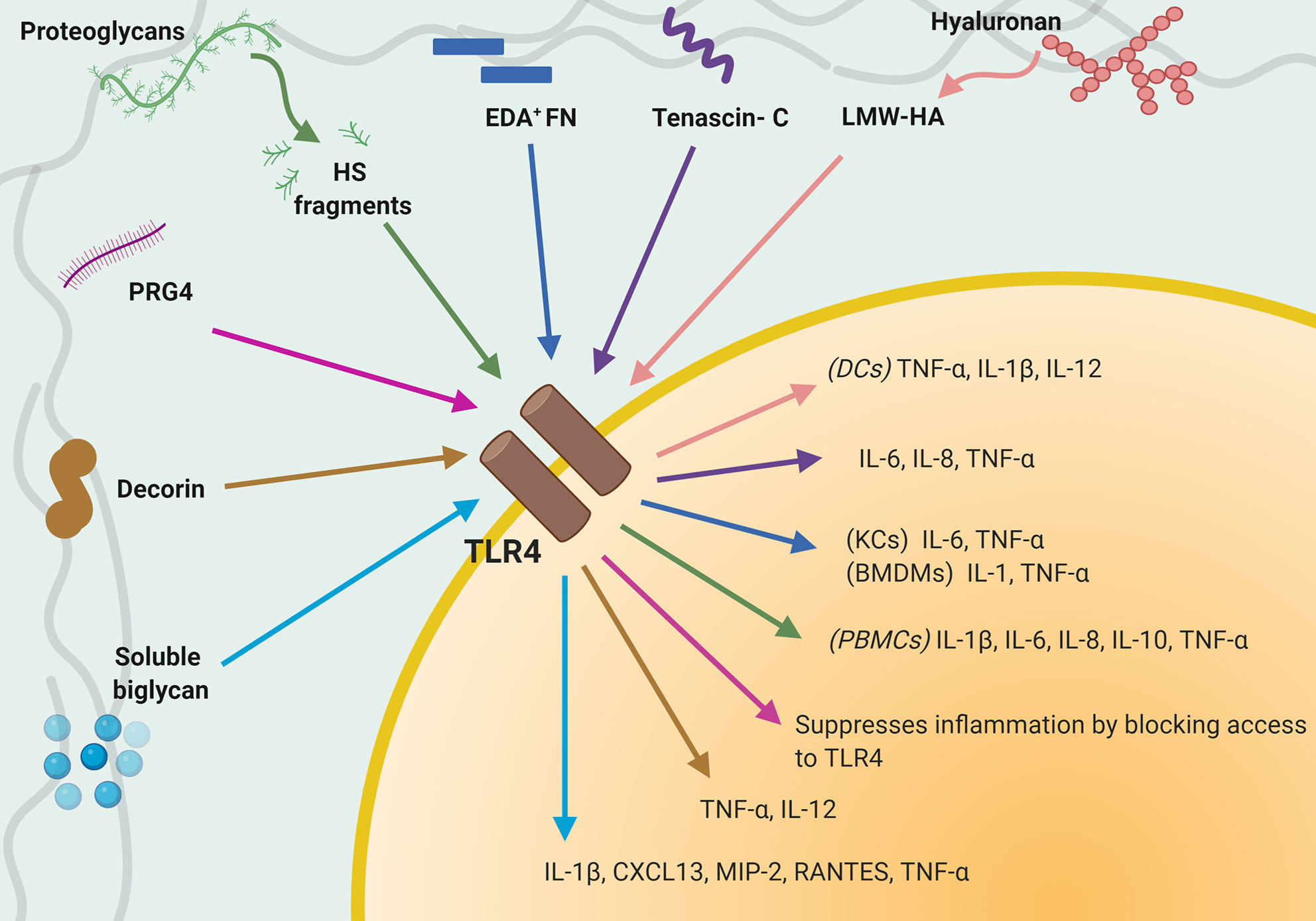
Figure 1 Interaction between TLR4 expressed on immune cells and extracellular matrix (ECM) proteins/ECM-damage-associated molecular patterns (DAMPs). Schematic that summaries known interactions between TLR4 and individual ECM-DAMPs or ECM proteins and consequent immune responses.
This evidence demonstrates that the ECM is a biologically active participant in perpetuating inflammation and suggests an important role in the progression of liver fibrosis and inflammatory disease. However, there is a lack of knowledge of specific ECM-immune interactions involved in CLD. Exploring the role of immunomodulatory ECM proteins in CLD could be key to understanding the inflammatory responses upon liver injury, providing new biomarkers to identify disease stage, predict disease progression, and develop new anti-inflammatory or immunomodulatory drugs. Some of these proteins have been studied for their immunomodulatory properties in the healthy liver, in liver cancer and in CLD, for example the role of periostin and TSP-1 in NAFLD and NASH. Other ECM components have been marginally studied in the liver but have been identified as key players in other inflammatory environments, such as the recruitment and induction of cytokine synthesis in macrophages by decorin and versican. Individual ECM molecules and their known pro- and anti-inflammatory role in inflammatory diseases are reported in the next sections. Some ECM components have been briefly studied in the liver but have been identified as key players in other inflammatory environments.
Over 150 ECM proteins have been identified in the human liver (9, 30). In this review we highlight 20 liver ECM proteins, selected for their implication in CLD and that have been shown to modulate immune responses. For clarity, we have grouped these proteins as “pro-inflammatory” – those with predominantly pro-inflammatory actions (versican, TSP-1, agrin, cysteine-rich protein 61, lumican, periostin, heparan sulphate, low molecular weight-hyaluronic acid, biglycan, elastin, and secreted protein, acidic and rich in cysteine); “anti-inflammatory” - those with predominantly anti-inflammatory actions (ECM-1, high molecular weight-hyaluronic acid and proteoglycan-4); and “dual acting”- those which have distinct pro- and anti-inflammatory properties (tenascins, OPN, collagen, decorin, and fibronectin). Further, where possible, proteins are organized as those which drive immune responses through direct ligand-immune cell interactions, followed by proteins which are recognized as ECM-DAMPs (Figure 2). Each protein is discussed for its specific immune-modulatory properties and the known implications in CLD. This is summarized in Table 3.
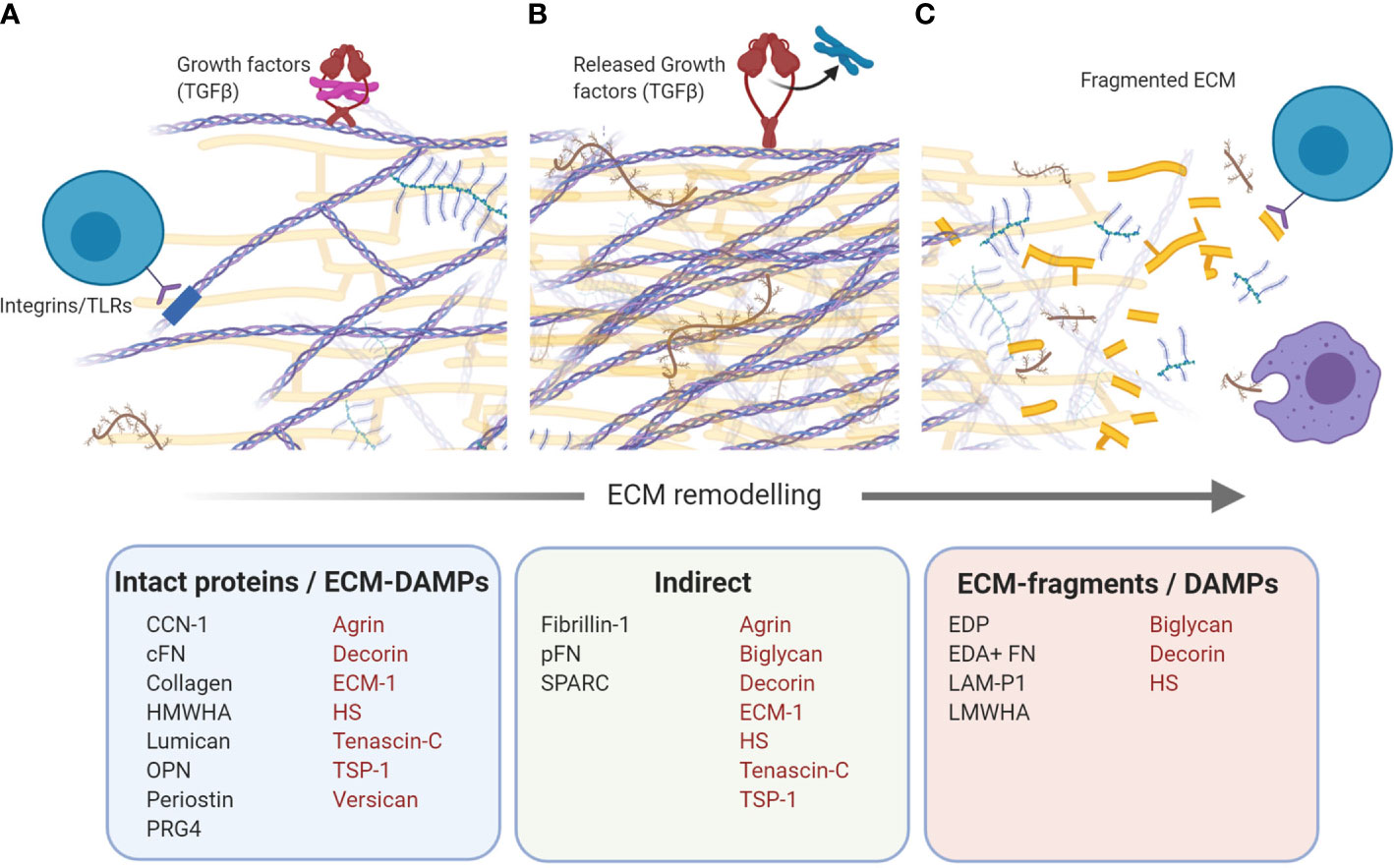
Figure 2 Immunomodulatory extracellular matrix (ECM) proteins. Schematic that shows ECM proteins with direct and indirect modulation of immune responses. (A) Intact proteins/ECM-damage-associated molecular patterns (DAMPs). Bioactive domains on numerous ECM proteins act as direct ligands for a wide number of immune cell receptors, including toll-like receptors (TLRs) and integrins, and drive a range of inflammatory and immune-suppressive responses. (B) Indirect. Numerous ECM proteins can bind growth factors, harbouring them in the ECM. Growth factors can be released upon ECM remodeling, creating a local inflammatory milieu which drives recruitment and polarization of infiltrating and resident immune cells. (C) ECM-fragments/DAMPs. Active remodeling of the ECM during fibrosis, results in the generation of ECM protein fragments which can be recognized by immune cells as DAMPs. Proteins with more than one mechanism of immunomodulation are indicated in red. Schematic was created using BioRender (https://app.biorender.com).
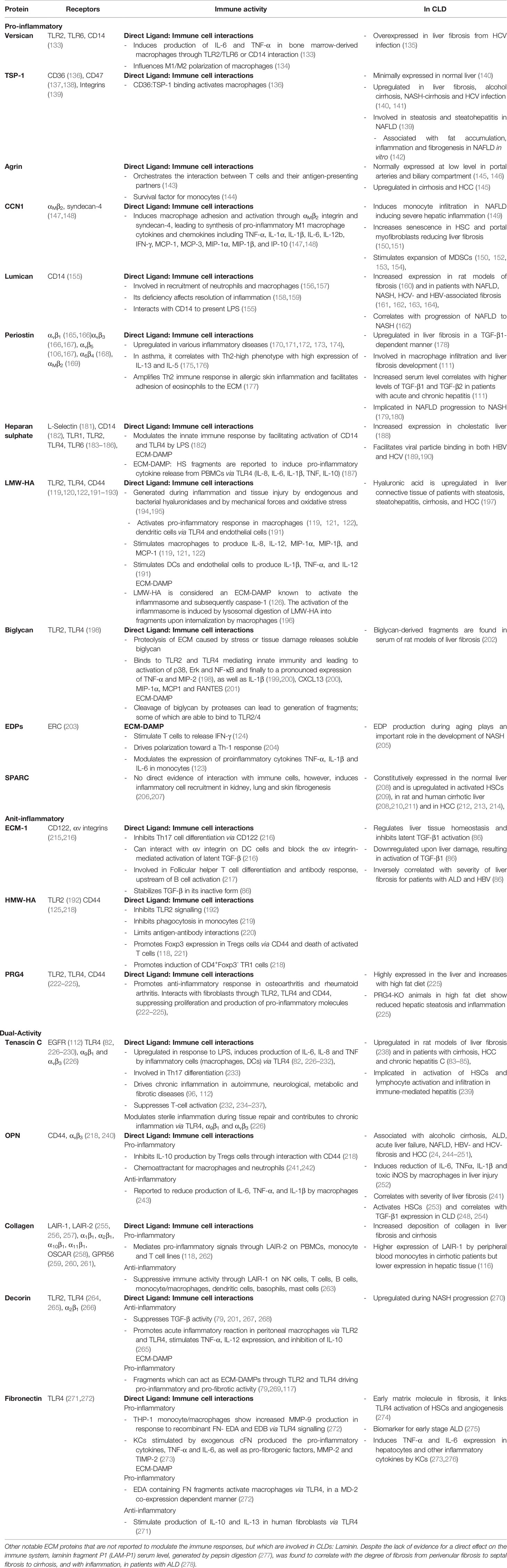
Table 3 Specific immune-modulatory properties of individual ECM proteins and known implications in CLDs.
ECM Proteins Which Drive Pro-Inflammatory Immune Responses
Versican
Versican is a chondroitin sulphate proteoglycan; the alternatively spliced GAG chain binding region gives rise to four versican isoforms: V0 (370 kDa), V1 (263 kDa), V2 (180 kDa), and V3 (74 kDa) (201, 279). V1 was shown to induce production of pro-inflammatory cytokines IL-6 and TNF-α in bone marrow-derived macrophages. This pro-inflammatory effect was mediated by interaction of V1 with TLR2 and CD14 on macrophages, but not TLR4. The response through TLR2 required TLR6 as co-receptor (133). Versican is considered to be involved in M1/M2 polarization of macrophages (134) and can also be synthesised and secreted by activated macrophages in inflammatory diseases, such as cardiovascular diseases and fibrosis, and in response to LPS (134, 280–282).
It has been reported that expression of versican increases in cirrhotic liver, where it localizes predominantly in the cytoplasm of hepatocytes, as well as in the perisinusoidal and stromal regions (135, 283). In a study by Ramnath and colleagues, the authors found V0 and V1 isoforms to be elevated in advanced liver fibrosis from either HCV or fatty liver disease, and these patients had elevated serum levels of versican (135). Versican is suggested to play a role in liver fibrosis by activating HSCs in in vitro experiments (283). It has been hypothesised that versican could have a pro-inflammatory effect in liver fibrosis as both the intact molecule and products of tissue remodeling by proteases are overexpressed in patients with advanced fibrosis (135).
Thrombospondin-1
Thrombospondin-1 (TSP-1) is a multi-functional glycoprotein which regulates cell-cell and cell-matrix interactions in tissue homeostasis and tissue repair and is upregulated under pathophysiological conditions (49). TSP-1 is recognized in the extracellular space by CD36 (expressed by endothelial cells, vascular smooth muscle cells, adipocytes and macrophages, among others (136, 284, 285), CD47 [expressed by endothelial cells, epithelial cells, smooth muscle cells, neutrophils, erythrocytes and chondrocytes (137, 138)] and integrins (139). TSP-1 regulates inflammation in a CD36- and CD47- dependent manner, for instance CD36:TSP-1 binding activates bone-marrow derived macrophage (BMDM)s (136).
The pro-inflammatory role of TSP-1 has been described in rheumatoid arthritis, inflammatory joint disease and in liver disease (73, 74, 139–142, 286–291). TSP-1 stimulates adipose tissue F4/80+ macrophage recruitment and activation in adipose tissue contributing to inflammation and insulin resistance resulting from high-fat diet-induced obesity (292). In NAFLD, TSP-1 is implicated in stimulating steatosis and steatohepatitis (139), seen by a reduction in lipid accumulation and TNF-α production in TSP-1 deficient mice (139). Similarly, in an in vitro model of NAFLD, TSP-1 expression by hepatocytes upon free fatty acid treatment, was associated with fat accumulation, inflammation and fibrogenesis (286). Also, in a partial hepatectomy mouse model, an immediate and transient TSP-1 expression was induced, primarily from endothelial cells and activated HSCs (293). TSP-1 expression was also increased alcohol-treated rats and in mouse models of liver fibrosis induced with CCl4 or 3,5- diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet (140).
Although TSP-1 is minimally expressed in the normal liver, hepatocytes and HSCs show deposition of large amounts of TSP-1 in patients with congenital liver fibrosis (291), and TSP-1 is upregulated in liver samples from patients with alcohol cirrhosis and NASH-related cirrhosis (140). In a HCV core transgenic mouse model, as well as in a co-culture model of hepatoma cells and HSCs, HBV core protein was able to induce expression of TSP-1 by hepatic cells and induce activation of TGF-β1 (294).
Agrin
Agrin is a large, heparin sulphate proteoglycan; its primary function is considered to be the establishment and maintenance of neuromuscular junctions (295), while its role in other tissues and organ microenvironments is less well understood. Agrin was found to orchestrate the formation of the “immunologic synapse” between T cells and their antigen-presenting partners (143), and was identified as a cell-autonomous survival factor for monocytes (144).
In the liver, agrin dramatically increases in the basal membrane of bile ductules and arterial walls in cirrhosis (145). Agrin can be secreted by HSCs activated by PDGF (296). PDGF receptor-inhibition by the multi-kinase inhibitor sorafenib, reduces the hepatocarcinogenesis mediated through agrin secretion and alleviates inflammation and fibrosis. A certain immunomodulatory activity by agrin is suggested by the interactions with T cells, monocytes and its ability to bind to growth factors in the ECM, such as TGF-β1 (87).
CCN1
The cysteine-rich protein 61, CCN1 or Cyr61, is an ECM-associated matricellular protein that plays a critical role in cell adhesion, migration, proliferation, apoptosis and survival, in both physiological and pathological tissue states (149, 297, 298). The CCN family of small cysteine-rich proteins are implicated in maintenance of normal liver function and pathogenesis of liver diseases (299). CCN1 is particularly expressed during inflammation or tissue repair. It induces macrophage adhesion and activation through integrin αMβ2 and syndecan-4, leading to expression of multiple pro-inflammatory cytokines and chemokines including TNF-α, IL-1α, IL-1β, IL-6, IL-12b, IFN-γ, MCP-1, MCP-3, macrophage inflammatory protein (MIP)-1α, MIP-1β, inflammatory protein 10 (IP-10), and others (147, 148), typical of M1 macrophage phenotype. These effects are a direct result of activation of NF-κB signaling in macrophages by CCN1, and does not involve TLR4 (147).
In a mouse model of NAFLD, CCN1 induced severe hepatic inflammation that was correlated with higher infiltration of monocytes in the liver via integrin αMβ2, confirmed by a decreased monocyte infiltration blocking integrin αMβ2 in presence of CCN1 (149). This study indicated that the pro-inflammatory process typical of NAFLD is initiated by CCN1-mediated induction of macrophage infiltration into the liver through direct chemotaxis and recruitment by chemokines. CCN1 is also able to induce expansion of myeloid-derived suppressor cells (MDSCs), a population of regulatory cells that accumulate in the liver (150, 152, 153, 154). CCN1 was secreted at high levels by injured cholangiocytes and hepatocytes in PBC, promoting expansion of MDSCs and contributing to suppression of T-cell proliferation (150).
Lumican
Lumican is a 40 kDa keratin sulphate proteoglycan involved in assembling collagen fibres and in regulating expression of TGF-β1 and α-SMA (300, 301).
Evidence of lumican participation in inflammatory signaling was seen upon analysing its interaction with the TNF superfamily member 6 that induces secretion of pro-inflammatory cytokines, which are involved in the recruitment of neutrophils and macrophages (156, 157). Furthermore, lumican deficiency reduces infiltration of neutrophils and negatively affects wound healing and resolution of inflammation (158, 159). Decreased sulforylation of lumican side chains stimulates macrophage adhesion and the cellular inflammatory response, suggesting that remodeling of the ECM could produce changes in the structure of lumican establishing the inflammatory microenvironment that promotes collagen deposition during fibrogenesis (302, 155). Interestingly, lumican does not seem to interact with TLR2 and TLR4, but with their adaptor molecule CD14 to present LPS and stimulate the relative immune response (155).
In the liver, lumican expression level increases during fibrosis in rat models of hepatic fibrosis (160) and in patients with NAFLD, NASH, HCV-, and HBV-associated fibrosis (161, 162, 163, 164), where it correlates with progression of fibrosis stage. In liver biopsies from patients with mild and severe NASH, lumican was found increased in sinusoids compared to tissue from obese patients with normal liver or with liver steatosis (162). This data suggests that lumican is expressed differentially across the progressive stages of NAFLD, indicating that this ECM component could be overexpressed at the beginning of the fibrogenic process in NAFLD patients with progressive disease.
Periostin
Periostin is a 93.3 kDa matricellular protein that interacts with other ECM structures (collagen I, fibronectin, tenascin-C and BMP-1) supporting processes like wound healing and fibrosis (170, 177, 303, 304). The main receptors reported to interact with periostin are integrin ανβ1 (165, 166), ανβ3 (166, 167), ανβ5 (166, 167), α6β4 (168), and αMβ2 (169).
Studies using animal models and patient samples have highlighted an active role of periostin in the pathobiology of various inflammatory diseases, including fibrosis, arthritis, chronic allergic skin inflammation, atherosclerosis and asthma (177, 171, 305, 306, 172, 173). In patients with asthma, periostin is upregulated and co-localizes with other proteins of the basement membrane such as tenascin-C, collagens I, III, and IV and fibronectin (174, 307), and correlates with “Th2-high” asthma, characterized by high expression of cytokines IL-13 and IL-5 (308, 175).
In allergic skin inflammation, activation of a Th2 immune reaction stimulates release of IL-4 and IL-3 that induce production of periostin by fibroblasts (177). Periostin is then able to activate keratinocytes through interaction with αν integrin to produce pro-inflammatory cytokines including thymic stromal lymphopoietin (TSLP), amplifying the type 2 immune response. Periostin also facilitates adhesion of eosinophils to the ECM (176). Thus, periostin is an important modulator in allergic inflammation by acting on epithelial cells, fibroblasts, eosinophils and possibly other immune cells.
Upregulation of periostin at both RNA and protein level was found in liver tissue of mouse models of liver fibrosis induced using CCl4 or BDL (178). Periostin is required for the activation of HSCs by TGF-β1 and promotes HSC migration. Periostin activity on HSC seems to be mediated by interaction with ανβ3 and ανβ5 integrins (178). These data were confirmed in studies with periostin-deficient mice, where CCl4- and BDL-induced acute and chronic liver fibrosis were drastically attenuated (111). In this experimental approach, periostin expression at liver tissue level and serum level was upregulated in mouse models of acute and chronic hepatic fibrosis, while this effect was abolished in the recovered livers after stopping CCl4 administration. Concomitantly, α-SMA and type I collagen upregulation upon liver injury was decreased in periostin-deficient animals. Periostin-deficient mice treated with CCl4 for 2 weeks also showed a significantly lower serum level of alanine transaminase (ALT) and aspartate amino transferase (AST), F4/80+ hepatic macrophage infiltration and fibrosis development compared to wild type treated mice. Expression of IL-6, IL-1β, and TNF-α was also decreased in the acute hepatic fibrosis model. In these models, TGF-β1 and TGF-β2 RNA expression and serum levels were correlated with presence of periostin: activated HSCs were able to produce periostin upon TGF-β1 stimulation, and periostin-deficient mice with acute and chronic hepatic fibrosis showed a significantly lower expression of TGF-β1 and TGF-β2 compared to wild-type mouse models of fibrosis. This evidence could indicate a potential reciprocal regulatory mechanism between TGF-β and periostin during liver fibrosis, where persistent liver injury and inflammation induces overexpression of periostin via TGF-β stimulation, and periostin, in turn, might induce macrophage infiltration and further production of TGF-β. This hypothesis was supported by analyses that indicate an increased serum level of TGF-β1, TGF-β2 and periostin in patients with acute and chronic hepatitis, compared to healthy controls (111). Other studies suggested that periostin expression could be induced by TNF-α and IL-17 (309, 310) contributing to hepatic fibrogenesis.
Periostin is implicated in the progression of NAFLD, where high expression of periostin was identified in high fat diet-fed mice and in obese mice (179, 311). Periostin-knockout mice fed with methionine-choline-deficient diet, which is known to induce NASH, exhibited a markedly lower level of steatosis, inflammation, and fibrosis in the liver compared to wild type animals (180). Similar evidence was reported in clinical research, where serum and liver tissue expression levels of periostin were higher in NAFLD patients in respect to controls (179, 312) and upregulation of periostin depended on the release of pro-inflammatory cytokines in NAFLD patient livers (313).
Heparan-Sulphate
Heparan sulphate (HS) chains are covalently bound to the core molecules of heparan sulphate proteoglycans (syndecans, perlecans, and glypicans). They are implicated in a variety of developmental and physiological processes as well as diseases including pulmonary, renal, and cystic fibrosis (314, 315, 181, 316), liver cholestasis (188), and viral hepatitis (189, 190).
HS can interact with a range of immune cells and modulate immune responses. It does this directly, by acting as a ligand for L-selectin, a transmembrane glycoprotein expressed on most leukocytes, as well as a binding platform for cytokines and chemokines (such as chemokine (C-X-C) ligand (CXCL) 8) (90) which modulate immune cell recruitment and activation, and is reported to play a role in immune cell infiltration during inflammation (181). HS is able to modulate the innate immune response by facilitating activation of CD14 and TLR4 by LPS (182). In addition to driving direct ligand-immune cell responses, HS fragments can also be recognized by immune cells as ECM-DAMPs. For example, HS fragments are reported to induce the expression of proinflammatory cytokines from human peripheral blood mononuclear cell (PBMC)s, including high levels of IL-8, IL-6, IL-1β, TNF, and IL-10 (187). HS fragments can also be released by heparanases (183) from perlecan (184), syndecans (185), and glypicans (186) and bind to TLR1, TLR2, TLR4, and TLR6. Nevertheless, the presence and activity of these ECM-derived fragments have not been investigated in CLDs.
Low Molecular Weight-Hyaluronic Acid
The GAG hyaluronic acid (HA) is composed of repeating disaccharides which form chains of variable lengths and molecular weights (118). Different molecular weight HA shows distinct effects on cellular phenotype and behaviour. HA breakdown products are generated during inflammation and tissue injury by endogenous and bacterial hyaluronidases (HYALs) and by mechanical forces and oxidative stress (194, 195). Low molecular weight HA (LMW-HA) (<15 saccharides; <3 kDa) produced during acute and persistent inflammation promotes angiogenesis (317), maturation of antigen-presenting cells (318, 319) and cell migration (320). LMW-HA acts as an endogenous activator of TLR2- and TLR4-promoted immune response and through CD44 (192, 193), which is highly expressed in patients with ALD (253). LMW-HA pro-inflammatory effects are mediated through activation of MMPs (321), plasminogen activator inhibitor (322), nitric oxide (323), and production of several pro-inflammatory cytokines including IL-8, IL-12, MIP-1α, MIP-1β, MCP-1 by macrophages (119, 120, 121, 122), and IL-1β, TNF-α, and IL-12 by DCs and endothelial cells (191). LMW-HA is considered an ECM-DAMP known to activate the NLRP3 inflammasome and subsequent cleavage of caspase- (126). The activation of the NLRP3 is induced by lysosomal digestion of LMW-HA into fragments upon internalization by macrophages in vivo and in vitro, a CD44-dependent process (196). Internalized LMW-HA interacts with the protein NLRP3 and induce release of IL-1β and CXCL2 chemokine.
In the liver, HA is synthesised mainly by HSCs (324), while degradation occurs principally by LSECs through HYAL1 and HYAL2 activity (324, 325). This produces LMW-HA that gathers at sites of active tissue catabolism and promotes inflammation. In a study by Mustonen and colleagues, the relationship between HA synthesis and degrading enzymes was demonstrated in histological stainings of liver sections from controls and patients with steatosis, steatohepatitis, cirrhosis and HCC (197), highlighting the importance of HA turnover in healthy liver and in CLD.
Biglycan
Biglycan is a secreted member of the small leucine-rich proteoglycans (326, 327, 198). ECM-bound biglycan does not induce any immune response as it is incapable of activating TLRs. Proteolysis of ECM caused by stress or tissue damage releases soluble biglycan, the first endogenous proteoglycan described to interact with TLRs (198). Soluble biglycan generates a rapid inflammatory response that can lead to de novo synthesis of new biglycan. In macrophages, biglycan binds to TLR2 and TLR4 mediating innate immunity and leading to activation of p38, Erk, and NF-κB and finally to a pronounced expression of TNF-α and MIP-2. This evidence was confirmed in experiments with TLR4-mutant, TLR2-/-, and myeloid differentiation factor 88 (MyD88)-/- macrophages, where the stimulatory effect of biglycan was significantly reduced, and in TLR2-/-/TLR4-mutant macrophages, that showed complete loss of activity (198).
Other downstream effector molecules involved in the signaling generated upon interaction of biglycan with TLR2/4 are IL-1β (199, 200), CXCL13 (200), MIP-1α, MCP-1, and RANTES (201). Biglycan is also considered an important link between the innate and adaptive immune response. In lupus nephritis, biglycan induces enhanced synthesis of CXCL13 which promotes recruitment of B1 cells, a subset of B cells involved in early and T-cell-independent antibody production (200).
Cleavage of biglycan by proteases can lead to generation of fragments; some of which are able to bind to TLR2/4. MMP-9 and MMP-12 are capable of degrading the ECM and generate specific biglycan-derived fragments that have been studied as biomarkers in liver fibrosis as they correlated with the severity of fibrosis, indicating that biglycan could mediate activation of sterile inflammatory responses (202).
Elastin-Derived Peptides
Elastin is composed of soluble tropoelastin monomers which aggregate and cross-link to form the insoluble protein core of elastin fibres (328). Tropoelastin and elastin degradation products can influence inflammation and tissue remodeling (329). Despite crosslinking, physiological, pathological and aging-related ECM remodeling can lead to elastin degradation and production of soluble elastin-derived peptides (EDPs) or elastokines (330). MMP-2, MMP-7, MMP-9, MMP-12, and neutrophil proteases are enzymes actively involved in elastin degradation (329). Most studies focus on a single enzyme family-induced production of EDPs, hindering the identification of EDPs with shared or distinct biological properties. For instance, some generated EDP harbour a GxxPG consensus motif (where x represents any amino acid) essential for their bioactivity as it allows the interaction with the elastin-binding protein, a component of the elastin receptor complex (ERC) (203). EDPs generated from MMP-7, MMP-9, and MMP-12 contain the bioactive peptide VGVAPG, but differences in the number of peptides produced have been identified among the three MMPs (331). EDPs generated by neutrophil-derived serine proteases also included the bioactive GxxPG motifs VGVAPG, but other new bioactive motifs were also identified, such as GVYPG, GFGPG, and GVLPG (332). Here as well, different enzymes (human leukocyte elastase, proteinase 3, and cathepsin G) showed differences in respect to the number of peptides produced, their sequence and the cleavage site specificity and preferences. Some peptides produced by proteinase 3 and cathepsin G contained multiple bioactive motifs that may enhance biological activity of EDPs due to an increased probability of interaction with EDP (333). Whether these distinct peptides possess distinct biological properties has yet to be investigated.
EDPs are potent stimulators of tissue repair, but continuous exposure of cells to the elastokines can induce a chronic inflammatory state (334). EDPs are able to attract peripheral blood monocytes and polymorphonuclear leucocytes (335, 336), and induce changes in gene expression of inflammatory cells at the site of injury (3). They are able to stimulate T cells to release IFN- γ, which correlates with emphysema severity (124), and can drive polarization toward a Th-1 response (204). In monocytes, EDPs can modulate the expression of proinflammatory cytokines TNF-α, IL-1β, and IL-6 (123).
In the normal liver, elastin is a minor component of the ECM, but tropoelastin is actively synthesised by hepatic myofibroblasts in liver fibrosis regardless of the aetiology (337, 338). In liver cirrhosis, elastin fibre accumulation is found around the main branches of the hepatic artery and the portal vein (329) and correlates with adverse liver-related outcomes in patients with advanced CLD (339). Elastin deposition in patients with chronic viral hepatitis occurs concomitantly with the formation of thick collagen bands, providing a correlation between age of the scars and elastin content (340, 341, 342). Nevertheless, elastin turnover is detected in liver fibrosis and cirrhosis, indicated by the presence of elastin fragments in the serum and markers of degradation of mature cross-linked elastin in the urine (343, 344). EDP production during aging plays an important role in the development of NASH. In the inflammatory-aging process, enzymatic activity of neutrophil elastase increases, inducing a high release of elastokines (205). These EDPs bind to the ERC and participate to the development and progression of diseases such as cancer, atherosclerosis (345), insulin resistance (346), and lipid accumulation and inflammation in the liver (330). Neutrophil elastase is known to increase its activity in mouse models of obesity and in obese patients. This results in the production of EDPs that stimulate accumulation of hepatic triglycerides, cytokine expression, inflammation, and hepatic ECM remodeling leading to strong fibrosis and NAFLD to NASH transition (330).
SPARC
Secreted protein, acidic and rich in cysteine (SPARC), or osteonectin, is a matricellular protein in the ECM that affects collagen fibre assembly and the activity of TGF-β1 (347) (4). SPARC is constitutively expressed in the normal liver (208) and is upregulated in activated HSCs (209), in rat and human cirrhotic liver (208, 210, 211) and in HCC (212–214). Its expression correlates with severity of liver fibrosis. Local production of SPARC was found to induce collagen deposition, inflammatory cell recruitment, TGF-β1 production, mesenchymal stem cell proliferation, and synthesis of ECM proteins in kidney, lung, and skin fibrogenesis (206, 207). The profibrogenic activity of SPARC has been connected to its ability to regulate TGF-β1 and collagen I synthesis (206). Decrease in TGF-β1 expression, obtained through siRNA silencing, decreased expression of SPARC; and decreased SPARC expression led to a decrease in TGF-β1 activity (348). In SPARC-/- mice, the amount of CD4+ T cells was greatly decreased compared to wild type animals, and BDL in SPARC-/- animals showed a strong decrease in collagen deposition when compared with control animals, possibly mediated by downregulation of TGF-β1 expression (349).
In a study by Mazzolini and colleagues, SPARC was detected in liver tissue samples of NAFLD patients, showing high levels of SPARC associated with higher mRNA levels of collagen Iα and TGF-β1 (350). They also showed that SPARC+/+ mice presented stronger inflammation than SPARC-/- animals, with higher expression of pro-inflammatory cytokines and chemokines IL-6, CXCL10, and FAS/CD95 and increased infiltration of F4/80+ hepatic macrophages in response to high fat diet. Fibrosis, inflammatory cells and chemokines were significantly reduced in SPARC-/- animals with hepatic injury induced by fatty acid accumulation, suggesting a role of SPARC in establishing a pro-inflammatory hepatic microenvironment.
Secreted modular calcium-binding protein 2 (SMOC2) belongs to the SPARC family of matricellular proteins and has also been suggested to be involved in fibrosis, inflammation, and cell differentiation and proliferation (351–353). SMOC2 has been identified as a key player in NAFLD progression. SMOC2 mRNA and protein levels were higher in liver samples of patients with NAFLD and in mice fed with high fat diet (354). SMOC2-knockdown mice fed with high fat diet showed lower collagen deposition compared to control animals, associated with a lower expression of TGF-β1 and α-SMA, and lower levels of pro-inflammatory cytokines IL-1β, IL-4, IL-6, MCP-1, and TNF-α in serum and liver samples. This study proved that SMOC2 ablation was protective against high fat diet-induced fibrosis and inflammation.
ECM Proteins Which Drive Anti-Inflammatory Immune Responses
Extracellular Matrix Protein-1
ECM-1 is a secreted glycoprotein that affects Th2 cell migration in asthma animal models and Th17 differentiation through interaction with CD122 receptor on T cells and αν integrins in experimental autoimmune encephalomyelitis (215, 216). It also regulates T follicular helper (TFH) cell differentiation (217). TFH cell differentiation in germinal centres is critical for their involvement in the production of high-affinity antibodies by activated B cells and plasma cells, and in mice it is positively regulated by IL-6 and IL-21 and suppressed by IL-2 (217, 355). IL-6 and IL-21 induce ECM-1 expression, which is a positive regulator of TFH cell differentiation in humoral immunity, germinal centre formation and antigen-specific antibody production by antagonising IL-2 signaling (217). Fan et al. described ECM-1 as a key mediator of liver tissue homeostasis (86). Depletion of ECM-1 severely affected liver architecture in mice, promoting liver fibrosis development, but without significant inflammation or hepatocyte damage (86). The authors found that ECM-1 was down-regulated in hepatocytes (and potentially other hepatic cell types) after liver damage, which activated TGF-β, probably via interacting with αν integrins, and releasing it from deposited latent TGF-β complexes to initiate HSC activation and fibrogenesis. Also, ECM-1 expression decreases in patients with liver fibrosis and inversely correlates with severity of fibrosis (86), but little is known about the regulation of the immune system by ECM-1 in CLD.
High Molecular Weight-Hyaluronic Acid
High-molecular weight (HMW-HA) (>2,000 saccharides; >400 kDa) is abundant in uninjured tissues and in healing tissues, and is considered inert or anti-inflammatory (131, 356). HMW-HA has been described as an inhibitor of TLR2 signaling (192) and phagocytosis by monocytes (219), and limits antigen-antibody interactions (220).
HA is known to bind to CD44 and TLR2 expressed on immune and non-immune cells (218) (12). T-cells and monocytes interact with HA only if CD44 is activated by pro-inflammatory cytokines, such as TNF-α and IFN-γ, or by T cell receptor (TCR) triggering (357). HMW-HA promotes the expression of Foxp3 by activated Tregs through interaction with CD44 (118, 221) and is able to provide a positive environment to support the persistence and suppressive function of Tregs pre-activated via their TCR complex (118). This property, together with the ability of HMW-HA to induce death of activated T cells (358), supports the role of this ECM component in regulating the inflammatory milieu. Bollyky and colleagues also described that HMW-HA modulates inflammation promoting induction of CD4+Foxp3- IL10-producing Treg (TR1) cells in peripheral tissues (218). These cells are important regulators of inflammation and maintain peripheral immunotolerance through the secretion of IL-10 (359, 360). HMW-HA was able to promote IL-10 production by murine CD4+Foxp3- cells mainly via cross-linking of CD44. In fact, CD44-/- mice showed a significantly lower up-regulation of IL-10 in response to HMW-HA. In human CD4+CD25- T cells, co-stimulation with HMW-HA significantly increased IL-10 production and generated TR1 cells that demonstrated active prevention of inflammation in a colitis model.
Other CD44 ligands impact the HA-mediated effect on TR1 induction. OPN, a known CD44 ligand which impacts IL-10 production (240, 361) is found in abundance in chronic inflammation and is known to aggravate autoimmunity (362, 363). OPN inhibits HA-mediated TR1 induction and production and mRNA expression of IL-10, irrespective of IL-2 supplementation. These effects are mediated through interaction with both CD44 and integrin ανβ3 receptor, suggesting that OPN and HA are part of an intricate network of ECM components that regulate TR1 pathways.
Proteoglycan 4
Proteoglycan 4 (PRG4), or lubrican, has been shown to promote an anti-inflammatory response in inflammatory osteoarthritis and rheumatoid arthritis (364). This proteoglycan interacts with TLR2, TLR4, and CD44 on synovial fibroblasts suppressing the production of a number of pro-inflammatory mediators and decreasing fibroblast proliferation (117, 222, 223, 224).
The liver presents high levels of PRG4 and the amount of PRG4 increases in response to high fat diet (225). PRG4 KO mice are protected from high fat diet-induced hepatic steatosis and exhibit an improved glucose tolerance and lower degree of white adipose tissue inflammation (225), suggesting a role for PRG4 in the regulation of nutritional homeostasis and inflammation in fatty liver disease.
ECM Proteins With Both Pro- and Anti-Inflammatory Effects on Immune Cells
Tenascins
The tenascin family includes four members: tenascin-C, tenascin-R, tenascin-X, and tenascin-W (365). Tenascin-C is a multifunctional and multimodular ECM glycoprotein that binds to a range of ECM proteins (95, 96) and binds receptors such as epidermal growth factor receptor (EGFR), TLR4 and integrins (96), as well as a wide range of growth factors [TGF-β, FGF, VEGF, PDGF, and insulin growth IGF-BPs (95, 96)]. The variety and number of interactions described for tenascin-C make it a multifunctional orchestrator of cell- and microenvironment-specific responses.
Tenascin-C is required for the inflammatory response to LPS in vivo. Piccinini et al. showed that, in a mouse model of experimental sepsis, tenascin-C-/- mice showed delayed and less severe symptoms of sepsis after LPS injection compared to tenascin-C+/+ mice (230). The authors suggested that upregulation of tenascin-C is an early response to LPS-induced TLR4 signaling, and this glycoprotein appears to specifically control the switch from anti- to pro-inflammatory cytokine response downstream to TLR4 activation (230). Tenascin-C activation of TLR4 induces soluble pro-inflammatory mediators, such as IL-6, IL-8, and TNF, important mediators of T-cell polarization, in a number of cell types, including macrophages (82, 226, 227, 228), DCs (229), fibroblasts (226, 366) and chondrocytes (231). Similarly, in a murine model of arthritic joint disease, Ruhmann and colleagues demonstrated that tenascin-C deficient mice show presence of DCs with diminished pro-inflammatory cytokine release and reduced capacity to drive naïve T cells toward Th17, but not Th1, Th2, or Treg cell differentiation, in response to LPS stimulation (233).
Tenascin-C is also involved in modulation of inflammation upon sterile tissue injury via activation of TLR4 and integrins, including α9β1 and ανβ3, promoting tissue repair but also contributing to chronic inflammation (230). Upon tissue injury, tenascin-C contributes to both early inflammatory response and subsequent tissue remodeling by activating cell-type specific responses via TLR4 (96). Persistent expression of tenascin-C has been implicated in driving chronic inflammation in autoimmune, neurological, metabolic, and fibrotic diseases, where it is likely to activate a positive feedback loop further driving disease (96, 112). It is associated with erosive joint disease and poor response to biological treatment in patients with rheumatoid arthritis (367), whereas mice lacking tenascin-C are protected from prolonged synovial inflammation and tissue destruction in joint disease (226, 233). This sterile inflammation seems to originate from distinct modes of TLR4 activation and downstream signaling compared to pathogenic TLR4 agonists (227).
In the liver, tenascin-C knockout mice with liver fibrosis induced by concanavalin A showed protection against collagen deposition and inflammatory cell infiltration, reduced IFN-γ, TNF, IL-4, and TGF-β1 production, and a lower number of activated HSCs in respect to wild type animals (239). This study suggested that tenascin-C may support the recruitment and activation of lymphocytes with cytokine upregulation in immune-mediated hepatitis conditions. Tenascin-C upregulation was detected in a rat model of liver fibrosis induced by CCl4 (238), probably produced by HSCs (368) and in the livers of patients with liver cirrhosis, HCC and chronic HCV, where it correlates with progressive disease activity (82, 83, 84, 85). Plasma levels of tenascin-C, together with AST, also provide an effective model to discriminate HCV cirrhotic patients with active infection from healthy controls and patients with undetectable levels of HCV RNA post HCV treatment regimen, highlighting tenascin-C as a potential indicator of ongoing hepatic injury and inflammation in these patients (369).
Interestingly, tenascin-C can also have immunosuppressive effects through regulating local concentration and activity of the immunosuppressive factor TGF-β (112, 125) and through suppression of T-cell activation induced by various stimuli (232, 234–237). Whether this local regulation of TGF-β by tenascin-C directly affects hepatocyte or cholangiocyte senescence in acute or chronic liver injury is unknown. This evidence supports the dual nature of tenascin-C, where from one side it mediates tissue repair, suppressing excessive inflammation and regulating tissue remodeling, while in other conditions it drives chronic inflammation and promotes fibrogenesis. Key aspects behind this dichotomy could be temporal and spatial control of tenascin-C expression.
The largest member of the tenascin family, tenascin-X, is involved in collagen deposition, collagen fibrillogenesis and development and maintenance of elastic fibers (370, 371, 372). In a mouse model of NASH induced by high-fat and high-cholesterol diet, absence of tenascin-X reduced inflammation and liver dysfunction in response to the diet compared with control same diet-fed wild-type animals. This study suggested that tenascin-X may promote high-fat and high-cholesterol diet-induced liver dysfunction through enhancement of the inflammatory response. Since tenascin-X has been reported to interact with the latent form of TGF-β regulating its activation into active form (373), it was hypothesized that the lack of tenascin-X in knockout animals could attenuate the activation of TGF-β upon feeding high-fat and high-cholesterol diet. This reduced TGF-β activation may then result in suppression of the inflammatory response. Similarly to tenascin-C, tenascin-X also binds to integrins, including β1-containing and α11β1 integrins (373, 374) on the cell surface, but the correlation between tenascin-X-integrin signaling and hepatic dysfunction and inflammation has yet to be investigated.
Osteopontin
OPN, also called secreted phosphoprotein-1 (SSP-1), is an extracellular structural glycoprotein that plays an important role in pathological conditions. Soluble and ECM-associated OPN are found in abundance in chronic inflammation, cancer and autoimmune diseases (218, 241). OPN binds to CD44 and integrin receptor αvβ3 (218, 240), the main mediators of OPN activity. The canonical CD44 ligand is HA. In a study by Bollyky and colleagues, OPN inhibited IL-10 production through interaction with CD44 on TR1 cells (218). OPN was able to block HA induction of TR1 binding to CD44 and integrin receptors, highlighting the role of OPN in controlling TR1 pathways.
In the liver, elevated expression of αvβ3 integrin is associated with high OPN levels in human alcoholic cirrhosis and mouse models of ALD (24). In addition, CD44 has been demonstrated to be involved in the regulation of inflammation in liver disease and is elevated in patients with ALD, including patients with alcoholic steatosis, alcoholic hepatitis and alcohol cirrhosis (253).
Initially, OPN was described in necrotic areas of the liver where it was contributing to local infiltration of KCs and resident macrophages (375). OPN has since been found to be involved in ALD, acute liver failure, NAFLD, liver fibrosis from HBV and HCV infection, and HCC (244–251), with increased circulating and ECM-associated OPN in patients with CLD. In animal models, OPN acts as a chemoattractant for hepatic resident macrophages and neutrophils (241, 242). OPN-deficient mice show impaired neutrophil infiltration, F4/80+ hepatic macrophage accumulation and release of pro-inflammatory cytokines (376).
Conversely, some studies suggest that OPN protects the liver from inflammatory injury, reducing production of IL-6, TNF-α, IL-1β, and toxic inducible nitric oxide synthase (iNOS) by F4/80+ hepatic macrophages, promoting hepatocyte survival (252). OPN deficiency in mice also protected against alcoholic hepatitis from chronic alcoholic steatohepatitis, showing increased induction of neutrophil infiltration in the alcoholic hepatitis model (377). In granuloma formation upon liver injury, OPN initially induces IL-12 and IFN-γ synthesis, while at late stages, OPN increases recruitment of CD4 T cells, F4/80+ macrophages and DCs into the liver and enhances TNFα production (243). In liver fibrosis, OPN correlates with disease severity and progression of disease (241). OPN can activate HSCs via CD44 (253) and via αvβ3 integrin resulting in upregulation of collagen I and transdifferentiation of HSCs (378). In patients with chronic liver fibrosis, hepatic OPN expression correlates with high TGF-β1 expression, portal space neutrophil-related inflammation and portal hypertension (248, 254). Portal inflammation is also predicted by high serum levels of OPN in patients with NAFLD (246). Mice treated with an OPN-neutralizing antibody show reduced obesity-driven hepatic inflammation and F4/80+ and Mac-2+ hepatic macrophage accumulation (379). In NASH, OPN produced by NKT cells induces activation of HSCs and progression of fibrogenesis (380).
The mechanism behind the dual role of OPN has not been specifically investigated, but it could be pathophysiological-dependent, spatiotemporal-dependent or cell/receptor-dependent. For example, OPN expression during early or late stages of inflammatory diseases seems to regulate the production of cytokines with distinct pro- or anti-inflammatory functions (381, 382, 383). Different isoforms, terminal fragments and post-transcriptional modifications of OPN may yield different impact as described in acute brain injury (384, 385, 386). Finally, OPN binds to multiple integrins and other receptors on multiple cell types, possibly mediating distinct phenotypic changes (387).
Collagen
Collagens are a large family of ECM proteins with 29 types, all characterized by a right-handed triple helix structure composed of three polypeptide chains. Receptors that bind directly to collagen have been identified on the surface of several cell types, e.g., α1β1, α2β1, α10β1, and α11β1 integrins. Hepatocytes and other cell types express the integrin receptor, α1β1 (255), which can bind to collagens I, III, IV, IX, XIII, XVI, and IV chain-derived peptide arresten (388, 389, 390). Other collagen-binding receptors include osteoclast-associated immunoglobulin-like receptor (OSCAR), known to have a role in the maturation of monocyte-derived DCs (258), and G-protein-coupled receptor (GPR) 56, present on fibroblasts, oligodendrocytes, melanoma cells, and cytotoxic NK and T lymphocytes (259, 260, 261). In addition, collagens I, II, III, and XVII are high-affinity ligands of leukocyte-associated immunoglobulin receptor-1 (LAIR-1 or CD305), expressed in both mice and humans on numerous immune cells, including NK cells, T cells, B cells, monocytes/macrophages, DCs, eosinophils, basophils, and mast cells (263). LAIR-1 is an inhibitory immune cell surface receptor which plays a key role in regulating inflammatory responses and LAIR-1:collagen interaction confers a general immunosuppressive activity (115, 256, 391). While intrahepatic immune cells are not exposed to collagens physiologically, collagens are ECM ligands that directly down-regulate immune responses through LAIR-1.
Surface-conjugated LAIR-1 significantly inhibits production of pro-inflammatory signaling molecules such as monokine induced by IFN-γ, MIP-1α, MIP-1β, MIP-2, and RANTES (256). Cross-linking of LAIR-1 with monoclonal antibodies or its ligands, inhibits the cytotoxic activity of NK and CD8+ T cells (392), differentiation and activation of B cells (393), the T cell receptor/CD3 complex signaling (394), the differentiation of peripheral blood precursors into DCs (395, 396) and the production of type-I interferon by CpG-activated plasmacytoid DCs and monocytes (397). Chronic autoimmune inflammation has also been linked to low expression of LAIR-1 (398).
Regulation of LAIR-1 is obtained by modulating its expression on the surface of immune cells during differentiation and activation (393, 394, 262, 399, 400). LAIR-1 can also be shed from immune cells upon cell activation (401) and can be regulated through competition with LAIR-2. LAIR-2 is a secreted protein that interacts with the same collagen sequences as LAIR-1, functioning as the decoy counterpart of LAIR-1. This soluble competitor to LAIR-1/collagen immunosuppressive activity (262) has been detected in primary PBMCs, monocytic, and T cell lines. PBMCs show increased LAIR-2 expression upon stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin, mainly produced by CD4+ T cells (262). The affinity for collagens I and III is comparable between the two receptors, suggesting that LAIR-2 may function as a pro-inflammatory mediator in vivo by decreasing the inhibitory potential of the immune inhibitor LAIR-1. In contrast, a reduced concentration of LAIR-2 may result in LAIR-1 binding to collagens, repressing immune cell activity at sites where it is not needed. Recently, the role of LAIR-1 expression on monocytes and macrophages in the development and progression of liver cirrhosis was studied in liver biopsies and peripheral blood samples of cirrhotic patients (116). LAIR-1 was expressed in hepatic resident CD68+ macrophages, and the number of LAIR-1-positive cells was reduced in cirrhotic livers. On the contrary, LAIR-1 was highly expressed in peripheral blood monocytes and their expression level of LAIR-1 [median fluorescence intensity (MFI)] was higher in cirrhotic patients than in healthy controls, in particular in the intermediate subset of monocytes (CD14++CD16+). These data suggest a role for LAIR-1/and LAIR-2/collagen interactions in the regulation of different immune cells involved in inflammatory responses in CLD.
Decorin
Decorin is a member of the small leucine-rich proteoglycans family. The protein core can interact with several receptor tyrosine kinases including EGFR (402), VEGF receptor (VEGFR) 2 (403), and with TLR2 and TLR4 receptors, evoking pro- and anti-inflammatory effects and recruiting macrophages (264, 265). Decorin is also a ligand for α2β1 integrin (266), widely expressed on epithelial and endothelial cells, fibroblasts, T-cells, myeloid cells, and others, with a role in inflammation during rheumatoid arthritis.
KO decorin mice show an increase in inflammation and fibrogenesis (79, 404, 405), while administration of recombinant decorin and decorin gene therapy showed anti-inflammatory effects (79, 406). Nevertheless, interaction between decorin and TLR2 and TLR4 on peritoneal macrophages triggers an acute inflammatory reaction with activation of p38, mitogen-activated protein kinase (MAPK), and NF-κB, increased synthesis of pro-inflammatory cytokines TNF-α and IL-12, and inhibition of anti-inflammatory IL-10 (265). This dual function is the result of ECM remodeling: cleavage of decorin by MMPs and other proteases can generate fragments of decorin which can act as ECM-DAMPs through TLR2 and TLR4, turning anti-fibrotic decorin into peptides with pro-inflammatory and pro-fibrotic activity (79, 117, 269).
In the liver, decorin accumulates along the sinusoidal walls (407), co-localizes with large amounts of TGF-β1 in fibrotic areas in chronic hepatitis and cirrhosis (408), and is upregulated during NASH progression (270).
Fibronectin
FN is the most abundant ECM glycoprotein in the liver and exists as several different forms. pFN, its soluble form, is synthesised by hepatocytes and assembled into the ECM where it can bind a plethora of growth factors. cFN, is found at low levels in the liver (24), but accumulates in response to injury and pathological stimuli (271). cFN contains an additional type III extra domain A and B (EDA+FN and EDB+FN). THP-1 monocyte/macrophages show increased MMP-9 production in response to recombinant EDA and EDB (409). EDA, in particular, was found to activate macrophages via TLR4, in a MD-2 (a TLR4 accessory protein) co-expression dependent manner. EDA-containing cFN activation of macrophages via TLR4 was confirmed in two studies by Doddapattar and colleagues, which showed that EDA+FN increased expression and activity of MMP-9 and potentiated dose-dependent NFκB-mediated inflammation (including secretion of TNF-α and IL-1β) in bone marrow–derived macrophages from atherosclerotic apolipoprotein E–deficient (Apoe−/−) mice (272). EDA-containing FN fragments are widely recognized as ECM-DAMPs, and are upregulated in several inflammatory diseases (myocardial infarction, cardiac fibrosis, stroke, cerebral ischemia, airway fibrosis, dermal fibrosis, liver fibrosis), where they activate both immune and non-immune cells prolonging inflammation and fibrosis (271, 274, 409–417). In dermal fibrosis, dermal fibroblasts recognize EDA via α4β1 integrin and TLR4, initiating a fibro-inflammatory response that includes synthesis of cytokines IL-10 and IL-13, and enrichment in the ratio of EDA+FN: total FN, indicating that EDA+FN further stimulates production of EDA+FN (271).
In the liver, FN was identified as an early matrix molecule that links TLR4 activation of HSCs with LSEC angiogenesis in a BDL mouse model of fibrosis (274). cFN was also identified as a biomarker of early stage ALD (275), appearing in the liver during the first 8 weeks of ethanol feeding in a rat model. Furthermore, in a rat model of ALD, Aziz-Seible et al (273). showed that KCs stimulated by exogenous cFN produced the pro-inflammatory cytokines, TNF-α and IL-6, as well as pro-fibrogenic factors, MMP-2 and TIMP-2.
Although FN is deposited during fibrosis and TLR4 is expressed on numerous liver cells including LSECs, KCs and HSCs, information regarding the potential role of EDA-containing FN fragments in regulating inflammatory microenvironments via TLR4 in CLD is limited.
Other ECM Proteins Involved in CLD
Laminin
Despite the lack of evidence for a direct effect on the immune system, laminin fragment P1 (LAM-P1) serum level, generated by pepsin digestion (277), was found to correlate with the degree of fibrosis from perivenular fibrosis to septal fibrosis to cirrhosis, and with inflammation, in patients with ALD (278). This evidence suggests that laminin and laminin fragments produced upon enzymatic digestion could be involved in regulation of the inflammatory progression in CLD. In the liver, only the β2 laminin chain is expressed in the sinusoids, portal biliary ducts, portal vascular structures and central veins, while other laminins are rare or absent (418). In liver fibrosis, deposition of laminin in the basement membrane increases together with that of type IV collagen and perlecan (418). Serum laminin level also correlates with degrees of fibrosis in alcoholic patients (419), it increases in cirrhosis in comparison to healthy controls (420), and in cirrhotic NAFLD patients compared to patients without cirrhosis (421).
Discussion
In this review, we summarize evidence that specific intact ECM components and enzymatically digested ECM fragments act as ECM-DAMPs and are able to modulate the immune system during inflammation in vitro and in vivo. Increasing understanding of the signaling pathways involved in sensing and responding to these ECM molecules highlights the key role of the ECM and its remodeling processes in the fibrogenic evolution of CLD. The paucity of curative therapies for inflammatory/autoimmune liver diseases necessitates further understanding of the mechanisms that exacerbate the disease condition, and focusing on the ECM-immune system crosstalk in CLDs may help identify new pathways and ultimately better targets for therapeutic interventions. Direct targeting of the ECM involved in the activation and actuation of pro- and anti-inflammatory/fibrogenic activities mediated by the ECM could represent fibrosis-specific therapy which remains a clinically unmet need. Although most of the proteins included in this review have been identified in liver fibrosis and correlate with different aetiologies and disease stage, direct protein-immune cell interactions remain poorly explored. Their immunomodulatory activity in other inflammatory diseases suggest that they could be the main protagonists in orchestrating localized tissue-specific inflammatory environments typical of CLDs.
ECM proteins have been studied in human samples and in different in vivo models of fibrosis and cirrhosis, particularly in NAFLD/NASH. Among them, matricellular proteins CCN1 and periostin appear to have a strong link with initiation of NAFLD and progression to NASH. CCN-1 is an important regulator of inflammation in NAFLD mouse models, but its overexpression also correlates with increased myofibroblast senescence, indicating that further studies should be directed to investigating CCN1 involvement in progression of NAFLD, particularly in the transition to NASH. Periostin and ECM-DAMPS EDPs are also involved in the progression of NAFLD to NASH in mouse models and have been linked to the inflammatory stage of NAFLD in patients. Since these proteins showed strong effects on T cells in other inflammatory diseases, and on macrophage infiltration in hepatic fibrosis, a deeper understanding of periostin and EDP involvement in regulating these immune cells in NAFLD livers could provide new insights on disease progression.
Other proteins such as TSP-1, OPN, proteoglycans lumican and agrin, and matricellular proteins SPARC and SMOC2 have a clear association with fibrosis in NAFLD patients and animal models. Their ability to drive inflammation and fibrosis is mainly indirect, modulating release and activation of TGF-β1. Other mechanisms of action such as recruitment of macrophages and regulation of T cell activity seem to be involved in the regulation of inflammatory processes by these proteins, but the direct protein-immune cell interaction remains unexplored in the context of CLDs and needs further investigation in specific liver disease models.
ECM-decorin also modulates several growth factors and inhibits TGF-β1 activity, but its role in orchestrating inflammation is controversial. Decorin has been described as capable of stimulating both pro- and anti-inflammatory responses. This duality could derive from differences in signaling pathways induced by the intact protein or upon ECM remodeling that produces decorin fragments, ECM-DAMPs with known pro-fibrotic and pro-inflammatory properties. Given the conflicting evidence regarding decorin activity, more studies need to focus on understanding its dynamic role in CLD. Similarly, evidence supports the dual nature of tenascin-C, where from one side it mediates tissue repair, suppressing excessive inflammation, and regulating tissue remodeling, while in other conditions it drives chronic inflammation and promotes fibrogenesis. In this case, key aspects behind this dichotomy could be temporal and spatial control of tenascin-C expression and investigation of tenascin-C presence and kinetics in different phases of CLD could provide important information regarding its role in regulating the microenvironment upon liver injury.
Only PRG4 and ECM-1 demonstrated protection from steatosis and inflammation in high fat diet mouse models, and an inversed correlation with severity of liver fibrosis, respectively. These proteins are able to both interact directly with immune cells through surface receptors (evidence from other inflammatory diseases) and indirectly by regulating local cytokine microenvironments and activation of TGF-β1. Given their protective role against liver disease, further studies are required to elucidate how these proteins interact with other components of the ECM, their dynamic turnover and whether they reveal immunomodulatory properties in CLDs.
Local TGF-β concentration at tissue level is a key component of the microenvironment and its tight regulation is a main participant in liver homeostasis, regeneration and response to injury. Although different ECM proteins are able to modulate or activate local TGF-β, it is not known whether a correlation between ECM remodeling, TGF-β activation, and induction/amplification of hepatic cell senescence could play a role in CLDs and this link should be further explored.
The study of specific ECM changes and relative effects on immune cells is significantly less frequent in autoimmune hepatitis or other autoimmune diseases and ALD, where inflammation has a central role in disease progression. Increased expression of tenascin-C, OPN and TSP-1 is associated with autoimmune diseases, while collagen immunosuppressive receptor LAIR-1 decreases in chronic autoimmune disease.
Very little is known regarding deposition and remodeling of specific ECM proteins in ALD; while cFN is a biomarker for early disease stages, high OPN levels in the liver, associated with elevated expression of receptors αvβ3 integrin and CD44, are detected in human alcoholic cirrhosis and mouse models of ALD. Some ECM-DAMPs with clear pro-inflammatory activities (biglycan fragments, EDPs, LAM-P1) were found to correlate with severity of fibrosis and inflammation in ALD and NAFLD/NASH, while others (LMW-HA, EDA-FN fragments, biglycan fragments, HS fragments) have not been identified or studied in CLDs. Since these ECM-DAMPs are able to bind to TLRs and derive from ECM proteins that are normally present in the liver and that change in proportion during hepatic fibrosis and ECM remodeling, their release and turnover should be specifically explored in CLDs to investigate whether they are actively involved in mediating inflammatory microenvironments.
ECM-DAMP involvement has been shown in a large number of liver inflammatory conditions. During the last 20 years, several mechanisms of ECM-DAMP generation, receptors and signaling pathways have been identified and they represent promising targets in the treatment of inflammatory conditions. However, there are still several gaps in knowledge that make translation of this field to effective therapies unsatisfactory. Our current knowledge regarding ECM-derived DAMPs, is still quite primitive, and it seems clear that there are many more types of DAMPs awaiting description. In our opinion, several key questions need to be addressed that concerns ECM-DAMPs, including: 1) what triggers ECM-DAMP release from the ECM and/or de novo synthesis upon tissue injury? 2) How do different ECM-DAMPs interact. 3) What ECM-DAMPs have additive effects or compete for binding to the same PRR (e.g., HA and OPN)? 4) The importance of timing and ECM-DAMPs local concentration on the interaction with PRRs and the downstream effects. 5) Do ECM-DAMPs and PAMPs interact during PAMP-dependent inflammation? The liver is exposed to microbial products from the gut and this translocation can impact the progression of CLD. ECM remodeling and release of ECM-DAMPs upon liver injury may directly stimulate sterile inflammation that in turn may have an effect on PAMP-induced activation of immune cells.
A potential avenue that should be further pursued is the use of ECM-DAMPs as biomarkers that might have diagnostic relevance in different aetiology and stages of CLD. ECM-DAMPs could represent non-invasive measures to facilitate the diagnosis of liver injuries and assess the fibrogenic evolution of CLD. Assessment of liver fibrosis is important to monitor the progression of CLD to end-stage cirrhosis, plan surveillance strategies and implement therapies. Quantifying serum biomarkers originated from ECM remodeling could be a valuable tool, in combination with other non-invasive methods, to overcome the disadvantages inherent in the use of liver biopsy approach, the recommended standard method for histological assessment of liver fibrosis. The potential use of ECM-DAMPs as biomarkers of liver disease stage and outcome prediction is supported by studies that highlight the detection of some products of ECM remodeling in the serum of patients. Examples were represented by elastin fragments in the serum of fibrotic and cirrhotic patients, the correlation of LAM-P1 serum level with the degree of fibrosis progression and inflammation in ALD and NAFLD patients, and the high levels of biglycan in the serum of rat models of liver fibrosis. Despite these advances, there is much that still could be addressed. Other ECM-DAMPs are likely to hold similar diagnostic or prognostic value if analysed in the appropriate animal model or cohort of CLD patients. For instance, EDA-containing and decorin fragments have demonstrated direct modulation of fibrosis; studies of their correlation with fibrosis stage or aetiology could reveal useful information for clinical application.
Moving forward, given the important role of the ECM in CLD, this central feature of the microenvironment needs to be taken into account when pathways of disease development and progression are studied. Conventional 2D in vitro models made of cell cultures on glass or plastic, are usually too simplified in terms of number and complexity of cell populations and microenvironment. This approach ignores the important interactions of hepatic and immune cells with the surrounding microenvironment (including the ECM) via integrins and other ECM-receptors, excluding related signaling pathways from the equation. On the other side, studies in animal models certainly present the advantage of using a physiologically relevant model that includes the ECM and cell-matrix interactions, but they are also associated with ethical concerns, limited reliability for predicting human responses to treatments, substantial differences in anatomy, immune responses, metabolic activity, and development in respect to humans, and low reproducibility (422). Most importantly, mechanistic studies on ECM components usually utilize specific -/- animals for a target protein, obtaining an “all or nothing” effect that does not translate into the complexity of human disease in terms of timing and overall effect when in presence of other compensatory mechanisms.
3D in vitro models may have the right features to provide more reliable platforms to understand health and disease biology in a more cost-effective, scalable and reproducible way than in vivo models. 3D cultures have demonstrated to be more effective in reproducing in vivo responses and cellular behaviours, as thoroughly reviewed by McCrary and colleagues (422). Given the critical role of the ECM in healthy tissue function and disease development and progression, incorporation of the ECM in these 3D culture models is essential to create the appropriate tissue microenvironment.
The lack of reliable and relevant 3D models of CLD that include the ECM and the immune system has hampered the study of the properties of the ECM. In addition, the tempo-spatial 3D signaling originated from the ECM and its remodeling should be considered when culture models are used to study inflammatory diseases such as CLD, e.g., generating and/or validating 3D models containing hepatic ECM from different stages of fibrosis. These models should include specific scaffolds with relative stiffness, combined with hepatic cells actively involved in the fibrotic process, and immune cells. To mimic the complex biochemical, physical and architectural features of the tissue microenvironment, one particularly promising approach is the use of native ECM as a biomaterial to develop 3D models, through a process called decellularization. Decellularization describes the treatment of a tissue or whole organ with a series of enzymes and detergents to effectively remove the native cellular content and DNA, while maintaining the ECM network. Decellularized scaffolds are tissue- and function-specific based on their origin and can be combined with different cell populations to create 3D models (423–425).
Decellularized ECM-scaffolds have demonstrated promising results in maintaining the native architecture and protein network for investigation of cell-to-matrix interaction. The advantages of using decellularized ECM-scaffolds in studying ECM composition and architecture and in disease modeling are vary; among the most important: i) decellularization eliminates the risk of negative immune responses due to removal of cellular content, making the scaffold “immuno-compatible” in the case of transplantation or co-culture with immune cells; ii) the ECM-scaffolds provide a natural template for the study of ECM composition using protemics profiling; iii) cell binding-sites are preserved in the ECM-scaffold, allowing for cell adhesion and studying of cell-ECM interactions; iv) disease and patient-specific phenotypes can be created using patient-derived tissue samples, replicating extremely specific microenvironments that may be difficult to be recreated by synthetic or more simple biomaterials.
The presence of a 3D ECM, as in the case of 3D models currently in use such as decellularized scaffold-based bioengineered constructs (423, 426, 427) and precision cut liver slices (428, 429), provides microenvironmental cues that could potentially participate in/modulate the signaling pathways relative to the scientific question studied. Most importantly, given the importance of the immune system in CLD and its interaction with the remodeling ECM, organ- and disease-specific immune cells should be integrated into 3D models for the study of ECM remodeling in CLD. Furthermore, bioreactors have been developed to support 3D cultures providing fluid-flow and active mechanical stimulation to recapitulate the physiological environment which cells are exposed to and that plays an important role in fibrosis pathogenesis. Integration of bioreactors and micro/macrofluidic devices can also improve longevity and function of 3D models and provide temporal control over cellular microenvironment (430) (Figure 3).
Figure 3 3D-dynamic perfusion-based immune competent systems to study extracellular matrix (ECM). Development of 3D-dynamic immune competent models to study ECM properties in vitro. (A) whole liver decellularization generates an acellular scaffold with maintained vasculature and ECM network. The resulting liver scaffold can be placed in a bioreactor which allows continuous circular perfusion of immune cells through the decellularized liver vasculature, facilitating immune cell:ECM interaction and monitoring of cellular phenotypes. (B) segments of liver tissue can be obtained from patient liver tissue and decellularized to generate acellular liver segments. These can be cultured in a multi-well bioreactor which supports perfusion of immune cells. The multi-well set up allows for direct comparison between eg healthy vs diseased tissue perfused with the same immune cells. Schematic was created using BioRender (https://app.biorender.com).
Although combinations of 3D microenvironment, immune cells, hepatic cells and a bioreactor-based culture are still rarely used in research around the study of ECM remodeling and CLD mechanisms, such complex multicellular dynamic 3D models are starting to emerge (431, 432). One example is a recent work published by Collin de L’Hortet et al. that described the transition from NAFLD to NASH in an in vitro model developed seeding decellularized rat livers with human mesenchymal cells, endothelial cells, fibroblasts, macrophages and induced pluripotent stem cell (iPSC)-derived hepatocytes (431). In this model, liver inflammation, lipid peroxidation and lipid accumulation in NAFLD were studied upon manipulation of deacetylase sirtuin-1 (SIRT1) to identify its involvement in NAFLD progression to NASH. The 3D model was maintained in culture by a perfusion bioreactor and included key disease features, showing the full potential of tissue engineered ECM-based models to study mechanisms of disease progression.
Despite the clear complexity in generating such models, development of physio-pathologically relevant multi-layered disease-specific culture systems is imperative to understand the role of the ECM in establishing inflammatory microenvironments.
The use of decellularized ECM-scaffolds in in vitro models is not exempt from limitations: limited mechanical instability and significant altered mechanical properties in comparison to the native counterpart; batch to batch variability due to variations in the decellularization process among studies, donor gender, sex, genetics, treatment, and anatomical tissue sampling; difficulties in assessing the exact protein content in different ECMs (422). Finally, most decellularization techniques, especially if not properly validated, inevitably removes some growth factors and ECM proteins from the matrix. To address this specific problem, highly informative advanced proteomic techniques have been recently employed to understand tissue-specific ECM protein and growth factor composition, which can be used to develop relevant in vitro models of disease and to gain insights to the ECM remodeling in disease. ECM composition analysis through proteomic profiling of decellularized tissues potentially removes interference from intracellular content. The type and number of ECM proteins that can be identified in a proteomic screen depend on several variables, including the reproducibility and reliability of the decellularization method, and parameters of the proteomic process such as the protocol to digest proteins into peptides, the extent of protein or peptide fractionation, and the mass spectrometry acquisition parameters (10). Furthermore, due to the destructive nature of the solubilization methods and the sampling of organs and tissues, proteomics does not provide information regarding specific regions and distribution of ECM proteins. Different preparation protocols for the creation of decellularized ECM-scaffolds diversely effect matrisome compositions, as described by a recent paper published by Daneshgar and colleagues (30). In this study, it was observed that the human liver matrisome constitutes of many relatively lower abundant matrisome-associated proteins, and that different preparation protocols influenced more the matrisome-associated fraction than the overall core matrisome.
In conclusion, ECM molecules and ECM-DAMPs and the signaling cascades that they activate represent new targets in potentially halting, reducing or reversing fibrogenesis and concomitant inflammatory state in CLD. ECM remodeling and its effects on the immune system should be studied in advanced dynamic 3D models that contain hepatic ECM. ECM proteins and fragments up- or down-regulated during CLD development and progression should be further investigated for their key role in modulating the immune system.
Author Contributions
CM and LU were involved in conceptualization, writing of the original draft, reviewing and editing. SC and RW were involved in reviewing and editing. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ford AJ, Rajagopalan P. Extracellular matrix remodeling in 3D: implications in tissue homeostasis and disease progression. Wiley Interdiscip Rev Nanomed Nanobiotechnol (2018) 10:e1503. doi: 10.1002/wnan.1503
2. Hastings JF, Skhinas JN, Fey D, Croucher DR, Cox TR. The extracellular matrix as a key regulator of intracellularsignalling networks. Br J Pharmacol (2018)176:82–92. doi: 10.1111/bph.14195
3. Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol (2008) 40:1101–10. doi: 10.1016/j.biocel.2007.12.005
4. Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol (2002) 14:608–16. doi: 10.1016/S0955-0674(02)00361-7
5. Boyd DF, Thomas PG. Towards integrating extracellular matrix and immunological pathways. Cytokine (2017) 98:79–86. doi: 10.1016/j.cyto.2017.03.004
6. Berger C, Bjørlykke Y, Hahn L, Mühlemann M, Kress S, Walles H, et al. Matrix decoded – A pancreatic extracellular matrix with organ specific cues guiding human iPSC differentiation. Biomaterials (2020) 244:119766. doi: 10.1016/j.biomaterials.2020.119766
7. Kanel GC. Liver: Anatomy, Microscopic Structure, and Cell Types. Textb Gastroenterol Fifth Ed (2009) 2:2057–72. doi: 10.1002/9781444303254.ch79
8. Bedossa P, Paradis V. Liver extracellular matrix in health and disease. J Pathol (2003) 200:504–15. doi: 10.1002/path.1397
9. Naba A, Clauser KR, Whittaker CA, Carr SA, Tanabe KK, Hynes RO, et al. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer (2014) 14:1–12. doi: 10.1186/1471-2407-14-518
10. Arteel GE, Naba A. The liver matrisome, looking beyond collagens. JHEP Rep (2020) 2:100115. doi: 10.1016/j.jhepr.2020.100115
11. Ueno T, Sata M, Tanikawa K. Cells Responsible for Extracellular Matrix Production in the Liver. Extracell Matrix Liver Approach Gene Ther (2003), 89–103. doi: 10.1016/B978-012525251-5/50007-5
12. Roderfeld M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol (2018) 68–69:452–62. doi: 10.1016/j.matbio.2017.11.011
13. Kim EJ, Cho HJ, Park D, Kim JY, Kim YB, Park TG, et al. Antifibrotic effect of MMP13-encoding plasmid DNA delivered using polyethylenimine shielded with hyaluronic acid. Mol Ther (2011) 19:355–61. doi: 10.1038/mt.2010.262
14. Siller-López F, Sandoval A, Salgado S, Salazar A, Bueno M, Garcia J, et al. Treatment with Human Metalloproteinase-8 Gene Delivery Ameliorates Experimental Rat Liver Cirrhosis. Gastroenterology (2004) 126:1122–33. doi: 10.1053/j.gastro.2003.12.045
15. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
16. Xuan J, Chen Y, Zhu L, Guo Y, Deng L, Zheng Y, et al. Ultrasound molecular imaging with cRGD-PLGA-PFOB nanoparticles for liver fibrosis staging in a rat model. Oncotarget (2017) 8:108676–91. doi: 10.18632/oncotarget.21358
17. Zhou X, Murphy FR, Gehdu N, Zhang J, Iredale JP, Benyon RC, et al. Engagement of αvβ3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem (2004) 279:23996–4006. doi: 10.1074/jbc.M311668200
18. Maher JJ. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis (2001) 21:417–26. doi: 10.1055/s-2001-17555
19. Seki E, Schwabe R. Hepatic Inflammation and Fibrosis: Functional Links and Key Pathways. Hepatology (2015) 61:1066–79. doi: 10.1002/hep.27332
20. Crawford JM, Burt AD. Anatomy, pathophysiology and basic mechanisms of disease. MacSween"sPathology of the Liver. Churchill Livingstone: Elsevier Ltd (2012). doi: 10.1016/B978-0-7020-3398-8.00001-5
21. Hernandez-Gea V, Friedman SL. Pathogenesis of Liver Fibrosis. Annu Rev Pathol Mech Dis (2011) 6:425–56. doi: 10.1146/annurev-pathol-011110-130246
22. Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver disease. Nat Rev DisPrim (2018) 4:1–22. doi: 10.1038/s41572-018-0014-7
23. Bellentani S. The epidemiology of non-alcoholic fatty liver disease. Liver Int (2017) 37:81–4. doi: 10.1111/liv.13299
24. Poole LG, Arteel GE. Transitional Remodeling of the Hepatic Extracellular Matrix in Alcohol-Induced Liver Injury. BioMed Res Int (2016) 2016:3162670. doi: 10.1155/2016/3162670
25. Morales-Ibanez O, Domínguez M, Ki SH, Marcos M, Chaves JF, Nguyen-Khac E, et al. Human and Experimental Evidence Supporting a Role for Osteopontin inAlcoholic Hepatitis. Hepatology (2014) 58:1742–56. doi: 10.1002/hep.26521
26. Aziz-Seible RS, Casey CA. Fibronectin: Functional character and role in alcoholic liver disease. World J Gastroenterol (2011) 17:2482–99. doi: 10.3748/wjg.v17.i20.2482
27. Parola M, Pinzani M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
28. Baiocchini A, Montaldo C, Conigliaro A, Grimaldi A, Correani V, Mura F, et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PloS One (2016) 11:1–14. doi: 10.1371/journal.pone.0151736
29. Novo E, Bocca C, Foglia B, Protopapa F, Maggiora M, Parola M, et al. Liver fibrogenesis: un update on established and emerging basic concepts. Arch Biochem Biophys (2020) 689:108445. doi: 10.1016/j.abb.2020.108445
30. Daneshgar A, Klein O, Nebrich G, Weinhart M, Tang P, Arnold A, et al. The human liver matrisome – Proteomic analysis of native and fibrotic human liver extracellular matrices for organ engineering approaches. Biomaterials (2020) 257:120247. doi: 10.1016/j.biomaterials.2020.120247
31. Mazza G, Telese A, Al-Akkad W, Frenguelli L, Levi A, Marrali M, et al. Cirrhotic Human Liver Extracellular Matrix 3D Scaffolds Promote Smad-Dependent TGF-β1 Epithelial Mesenchymal Transition. Cells (2019) 9:83. doi: 10.3390/cells9010083
32. Shao X, Taha IN, Clauser KR, Gao YT, Naba A. MatrisomeDB: the ECM-protein knowledge database. Nucleic Acids Res (2020) 48:D1136–44. doi: 10.1093/nar/gkz849
33. Ysno M, Mitsukamada H, Kage M, Ikeda K, Shimamatsu K, Inoue O, et al. The Long-term Pathological Evolution of Chronic HepatitisC. Hepatology (1996)23:1334–40. doi: 10.1002/hep.510230607
34. Wong F. Clinical Consequences of Infection in Cirrhosis: Organ Failures and Acute-on-Chronic Liver Failure. Clin Liver Dis (2019) 14:92–7. doi: 10.1002/cld.813
35. Koyama Y, Brenner DA, Koyama Y, Brenner DA. Liver inflammation and fibrosis Find the latest version: Liver inflammation and fibrosis. J Clin Invest (2017) 127:55–64. doi: 10.1172/JCI88881
36. Heymann F, Tacke F. Immunology in the liver-from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
37. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat Rev Immunol (2014) 14:181–94. doi: 10.1038/nri3623
38. Pierantonelli I, Svegliati-Baroni G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation (2019) 103:E1–E13. doi: 10.1097/TP.0000000000002480
39. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
40. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
41. Alsamman S, Christenson SA, Yu A, Ayad NME, Mooring MS, Segal JM, et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci Transl Med (2020) 12:1–15. doi: 10.1126/scitranslmed.aay8798
42. Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol (2010) 190:693–706. doi: 10.1083/jcb.201004082
43. Manmadhan S, Ehmer U. Hippo signaling in the liver - A long and ever-expandingstory. Front Cell Dev Biol (2019)7(43):1–9. doi: 10.3389/fcell.2019.00033
44. Chen G, Xia B, Fu Q, Huang X, Wang F, Chen Z, et al. Matrix mechanics as regulatory factors and therapeutic targets in hepatic fibrosis. Int J Biol Sci (2019) 15:2509–21. doi: 10.7150/ijbs.37500
45. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol (2017) 18:758–70. doi: 10.1038/nrm.2017.87
46. Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, et al. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator With PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology (2020) 71:1813–30. doi: 10.1002/hep.30928
47. Gruber E, Heyward C, Cameron J, Leifer C. Toll-like receptor signaling in macrophages is regulated by extracellular substrate stiffness and Rho-associated coiled-coil kinase (ROCK1/2). Int Immunol (2018) 30:267–78. doi: 10.1093/intimm/dxy027
48. Hinz B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol (2015) 47:54–65. doi: 10.1016/j.matbio.2015.05.006
49. Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X, et al. Transforming growth factor-beta in stem cells and tissue homeostasis. Bone Res (2018) 6:2. doi: 10.1038/s41413-017-0005-4
50. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell (1999) 96:319–28. doi: 10.1016/S0092-8674(00)80545-0
51. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-beta structure and activation. Nature (2011) 474:343–9. doi: 10.1038/nature10152
52. Murphy-Ullrich JE, Suto MJ. Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease. Matrix Biol (2018) 68–69:28–43. doi: 10.1016/j.matbio.2017.12.009
53. Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, et al. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci U.S.A. (1992) 89:5408–12. doi: 10.1073/pnas.89.12.5408
54. Chen X, Yang Y, Zhou Q, Weiss JM, Howard OZ, McPherson JM, et al. Effective chemoimmunotherapy with anti-TGFbeta antibody and cyclophosphamide in a mouse model of breast cancer. PloS One (2014) 9:e85398. doi: 10.1371/journal.pone.0085398
55. Esebanmen GE, Langridge WHR. The role of TGF-beta signaling in dendritic cell tolerance. Immunol Res (2017) 65:987–94. doi: 10.1007/s12026-017-8944-9
56. Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol (2010) 31:220–7. doi: 10.1016/j.it.2010.04.002
57. Huda N, Liu G, Hong H, Yan S, Khambu B, Yin X-M, et al. Hepatic senescence, the good and the bad. World J Gastroenterol (2019) 25:5069–81. doi: 10.3748/wjg.v25.i34.5069
58. Campisi J, D’Adda Di Fagagna F. Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8:729–40. doi: 10.1038/nrm2233
59. Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med (2010) 16:238–46. doi: 10.1016/j.molmed.2010.03.003
60. Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology (2013) 14:617–28. doi: 10.1007/s10522-013-9473-0
61. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasomecontrols paracrine senescence. Nat Cell Biol (2013)15:978–90. doi: 10.1038/ncb2784
62. Paradis V, Youssef N, Dargère D, Bâ N, Bonvoust F, Deschatrette J, et al. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum Pathol (2001) 32:327–32. doi: 10.1053/hupa.2001.22747
63. Wiemann S, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J (2002) 16:935–42. doi: 10.1096/fj.01-0977com
64. Ferreira-Gonzalez S, Lu WY, Raven A, Dwyer B, Man TY, O’Duibhir E, et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat Commun (2018) 9:1–15. doi: 10.1038/s41467-018-03299-5
65. Gutierrez-Reyes G, de Leon M del CG, Varela-Fascinetto G, Valencia P, Tamayo RP, Rosado CG, et al. Cellular senescence in livers from children with end stage liverdisease. PloS Onae (2010) 5:4–8. doi: 10.1371/annotation/6082f3f8-2b92-42a2-8d6f-b9210d2f25bf
66. Frey N, Venturelli S, Zender L, Bitzer M. Cellular senescence in gastrointestinal diseases: From pathogenesis to therapeutics. Nat Rev Gastroenterol Hepatol (2018) 15:81–95. doi: 10.1038/nrgastro.2017.146
67. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
68. Bird TG, M ller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, et al. TGFβ inhibition restores a regenerative response in acute liverinjury by suppressing paracrine senescence. Sci Transl Med(2018) 10:1–15. doi: 10.1126/scitranslmed.aan1230Setting
69. Ceci L, Francis H, Zhou T, Giang T, Yang Z, Meng F, et al. Knockout of the Tachykinin Receptor 1 in the Mdr2-/- (Abcb4-/-)Mouse Model of Primary Sclerosing Cholangitis Reduces Biliary Damage and LiverFibrosis. Am J Pathol (2020)190:2251–66. doi: 10.1016/j.ajpath.2020.07.007
70. Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol (1993) 122:923–32. doi: 10.1083/jcb.122.4.923
71. Schultz-Cherry S, Ribeiro S, Gentry L, Murphy-Ullrich JE. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J Biol Chem (1994) 269:26775–82.
72. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci (2003) 116:217–24. doi: 10.1242/jcs.00229
73. Bige N, Shweke N, Benhassine S, Jouanneau C, Vandermeersch S, Dussaule JC, et al. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int (2012) 81:1226–38. doi: 10.1038/ki.2012.21
74. Yang K, Vega JL, Hadzipasic M, Schatzmann Peron JP, Zhu B, Carrier Y, et al. Deficiency of thrombospondin-1 reduces Th17 differentiation and attenuates experimental autoimmune encephalomyelitis. J Autoimmun (2009) 32:94–103. doi: 10.1016/j.jaut.2008.12.004
75. Pierson BA, Gupta K, Hu WS, Miller JS. Human natural killer cell expansion is regulated by thrombospondin-mediated activation of transforming growth factor-beta 1 and independent accessory cell-derived contact and soluble factors. Blood (1996) 87:180–9. doi: 10.1182/blood.V87.1.180.bloodjournal871180
76. Mir FA, Contreras-Ruiz L, Masli S. Thrombospondin-1-dependent immune regulation by transforming growth factor-beta2-exposed antigen-presenting cells. Immunology (2015) 146:547–56. doi: 10.1111/imm.12517
77. Droguett R, Cabello-Verrugio C, Riquelme C, Brandan E. Extracellular proteoglycans modify TGF-beta bio-availability attenuating its signaling during skeletal muscle differentiation. Matrix Biol (2006) 25:332–41. doi: 10.1016/j.matbio.2006.04.004
78. Tang T, Thompson JC, Wilson PG, Nelson C, Williams KG, Tannock LR, et al. Decreased body fat, elevated plasma transforming growth factor-beta levels, and impaired BMP4-like signaling in biglycan-deficient mice. Connect Tissue Res (2013) 54:5–13. doi: 10.3109/03008207.2012.715700
79. Järvinen TAH, Prince S. Decorin: A Growth Factor Antagonist for Tumor Growth Inhibition. BioMed Res Int (2015) 79:1–11. doi: 10.1155/2015/654765
80. Iwasaki A, Sakai K, Moriya K, Sasaki T, Keene DR, Akhtar R, et al. Molecular Mechanism Responsible for Fibronectin-controlled Alterations in Matrix Stiffness in Advanced Chronic Liver Fibrogenesis. J Biol Chem (2016) 291:72–88. doi: 10.1074/jbc.M115.691519
81. Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, Schultze-Mosgau S, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta (2009) 1792:746–56. doi: 10.1016/j.bbadis.2009.06.004
82. Benbow JH, Thompson KJ, Cope HL, Brandon-Warner E, Culberson CR, Bossi KL, et al. Diet-Induced Obesity Enhances Progression of Hepatocellular Carcinoma through Tenascin-C/Toll-Like Receptor 4 Signaling. Am J Pathol (2016) 186:145–58. doi: 10.1016/j.ajpath.2015.09.015
83. El-Karef A, Kaito M, Tanaka H, Ikeda K, Nishioka T, Fujita N, et al. Expression of large tenascin-C splice variants by hepatic stellate cells/myofibroblasts in chronic hepatitis C. J Hepatol (2007) 46:664–73. doi: 10.1016/j.jhep.2006.10.011
84. Yamauchi M, Mizuhara Y, Maezawa Y, Toda G. Serum tenascin levels in chronic liver disease. Liver (1994) 14:148–53. doi: 10.1111/j.1600-0676.1994.tb00064.x
85. Tanaka H, El-Karef A, Kaito M, Kinoshita N, Fujita N, Horiike S, et al. Circulating level of large splice variants of tenascin-C is a marker of piecemeal necrosis activity in patients with chronic hepatitis C. Liver Int (2006) 26:311–8. doi: 10.1111/j.1478-3231.2005.01229.x
86. Fan W, Liu T, Chen W, Hammad S, Longerich T, Hausser I, et al. ECM1 Prevents Activation of Transforming Growth Factor beta, Hepatic Stellate Cells, and Fibrogenesis in Mice. Gastroenterology (2019) 157:1352–1367 e13. doi: 10.1053/j.gastro.2019.07.036
87. Banyai L, Sonderegger P, Patthy L. Agrin binds BMP2, BMP4 and TGFbeta1. PloS One (2010) 5:e10758. doi: 10.1371/journal.pone.0010758
88. McCaffrey TA, Falcone DJ, Du B. Transforming growth factor-beta 1 is a heparin-binding protein: identification of putative heparin-binding regions and isolation of heparins with varying affinity for TGF-beta 1. J Cell Physiol (1992) 152:430–40. doi: 10.1002/jcp.1041520226
89. Lyon M, Rushton G, Gallagher JT. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J Biol Chem (1997) 272:18000–6. doi: 10.1074/jbc.272.29.18000
90. Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol (2006) 6:633–43. doi: 10.1038/nri1918
91. Lortat-Jacob H, Garrone P, Banchereau J, Grimaud JA. Human interleukin 4 is a glycosaminoglycan-binding protein. Cytokine (1997) 9:101–5. doi: 10.1006/cyto.1996.0142
92. Lortat-Jacob H, Esterre P, Grimaud JA. Interferon-gamma, an Anti-fibrogenic Cytokine which Binds to Heparan Sulfate. Pathol Res Pract (1994) 190:920–2. doi: 10.1016/S0344-0338(11)80996-9
93. Gilat D, Hershkoviz R, Mekori YA, Vlodavsky I, Lider O. Regulation of adhesion of CD4+ T lymphocytes to intact or heparinase-treated subendothelial extracellular matrix by diffusible or anchored RANTES and MIP-1 beta. J Immunol (1994) 153:4899–4906.
94. Ariel A, Hershkoviz R, Cahalon L, Williams DE, Akiyama SK, Yamada KM, et al. Induction of T cell adhesion to extracellular matrix or endothelial cell ligands by soluble or matrix-bound interleukin-7. Eur J Immunol (1997) 27:2562–70. doi: 10.1002/eji.1830271015
95. De Laporte L, Rice JJ, Tortelli F, Hubbell JA. Tenascin C promiscuously binds growth factors via its fifth fibronectin type III-like domain. PloS One (2013) 8:e62076. doi: 10.1371/journal.pone.0062076
96. Marzeda AM, Midwood KS. Internal Affairs: Tenascin-C as a Clinically Relevant, Endogenous Driver of Innate Immunity. J Histochem Cytochem (2018) 66:289–304. doi: 10.1369/0022155418757443
97. Spenlé C, Loustau T, Murdamoothoo D, Erne W, Beghelli-de la Forest Divonne S, Veber R, et al. Tenascin-C Orchestrates an Immune-Suppressive Tumor Microenvironment in Oral Squamous Cell Carcinoma. Cancer Immunol Res (2020) 8:1122–38. doi: 10.1158/2326-6066.CIR-20-0074
98. Vaday GG, Lider O. Extracellular matrix moieties, cytokines, and enzymes: Dynamic effects on immune cell behavior and inflammation. J Leukoc Biol (2000) 67:149–59. doi: 10.1002/jlb.67.2.149
99. Hershkoviz R, Goldkorn I, Lider O. Tumour necrosis factor-α interacts with laminin and functions as a pro-adhesive cytokine. Immunology (1995) 85:125–30. doi: 10.1002/eji.1830271015
100. Gregory KE, Ono RN, Charbonneau NL, Kuo CL, Keene DR, Bachinger HP, et al. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J Biol Chem (2005) 280:27970–80. doi: 10.1074/jbc.M504270200
101. Sengle G, Charbonneau NL, Ono RN, Sasaki T, Alvarez J, Keene DR, et al. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J Biol Chem (2008) 283:13874–88. doi: 10.1074/jbc.M707820200
102. Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL, et al. Microenvironmental regulation by fibrillin-1. PloS Genet (2012) 8:e1002425. doi: 10.1371/journal.pgen.1002425
103. Tufvesson E, Westergren-Thorsson G. Tumour necrosis factor-alpha interacts with biglycan and decorin. FEBS Lett (2002) 530:124–8. doi: 10.1016/S0014-5793(02)03439-7
104. Chen XD, Fisher LW, Robey PG, Young MF. The small leucine-rich proteoglycan biglycan modulates BMP-4-induced osteoblast differentiation. FASEB J (2004) 18:948–58. doi: 10.1096/fj.03-0899com
105. Mochida Y, Parisuthiman D, Yamauchi M. Biglycan is a positive modulator of BMP-2 induced osteoblast differentiation. Adv Exp Med Biol (2006) 585:101–13. doi: 10.1007/978-0-387-34133-0_7
106. Desnoyers L, Arnott D, Pennica D. WISP-1 binds to decorin and biglycan. J Biol Chem (2001) 276:47599–607. doi: 10.1074/jbc.M108339200
107. Nili N, Cheema AN, Giordano FJ, Barolet AW, Babaei S, Hickey R, et al. Decorin inhibition of PDGF-stimulated vascular smooth muscle cell function: Potential mechanism for inhibition of intimal hyperplasia after balloon angioplasty. Am J Pathol (2003) 163:869–78. doi: 10.1016/S0002-9440(10)63447-5
108. Vial C, Gutiérrez J, Santander C, Cabrera D, Brandan E. Decorin interacts with connective tissue growth factor (CTGF)/CCN2 by LRR12 inhibiting its biological activity. J Biol Chem (2011) 286:24242–52. doi: 10.1074/jbc.M110.189365
109. Wohl AP, Troilo H, Collins RF, Baldock C, Sengle G. Extracellular Regulation of Bone Morphogenetic Protein Activity by the Microfibril Component Fibrillin-1. J Biol Chem (2016) 291:12732–46. doi: 10.1074/jbc.M115.704734
110. Alon R, Cahalon L, Hershkoviz R, Elbaz D, Reizis B, Wallach D, et al. TNF-alpha binds to the N-terminal domain of fibronectin and augments the beta 1-integrin-mediated adhesion of CD4+ T lymphocytes to the glycoprotein. J Immunol (1994) 152:1304–1313.
111. Huang Y, Liu W, Xiao H, Maitikabili A, Lin Q, Wu T, et al. Matricellular protein periostin contributes to hepatic inflammation and fibrosis. Am J Pathol (2015) 185:786–97. doi: 10.1016/j.ajpath.2014.11.002
112. Aungier SR, Cartwright AJ, Schwenzer A, Marshall JL, Dyson MR, Slavny P, et al. Targeting early changes in the synovial microenvironment: A new class of immunomodulatory therapy? Ann Rheumatol Dis (2019) 78:186–91. doi: 10.1136/annrheumdis-2018-214294
113. Lebbink RJ, De Ruiter T, Adelmeijer J, Brenkman AB, Van Helvoort JM, Koch M, et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med (2006) 203:1419–25. doi: 10.1084/jem.20052554
114. Lebbink RJ, Raynal N, de Ruiter T, Bihan DG, Farndale RW, Meyaard L, et al. Identification of multiple potent binding sites for human leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol (2009) 28:202–10. doi: 10.1016/j.matbio.2009.03.005
115. Meyaard L. LAIR and collagens in immune regulation. Immunol Lett (2010) 128:26–8. doi: 10.1016/j.imlet.2009.09.014
116. Martinez-Esparza M, Ruiz-Alcaraz AJ, Carmona-Martinez V, Fernandez-Fernandez MD, Anton G, Munoz-Tornero M, et al. Expression of LAIR-1 (CD305) on Human Blood Monocytes as a Marker of Hepatic Cirrhosis Progression. J Immunol Res (2019) 2019:2974753. doi: 10.1155/2019/2974753
117. Frevert CW, Felgenhauer J, Wygrecka M, Nastase MV, Schaefer L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J Histochem Cytochem (2018) 66:213–27. doi: 10.1369/0022155417740880
118. Bollyky PL, Falk BA, Wu RP, Buckner JH, Wight TN, Nepom GT, et al. Intact extracellular matrix and the maintenance of immune tolerance: high molecular weight hyaluronan promotes persistence of induced CD4+CD25+ regulatory T cells. J Leukoc Biol (2009) 86:567–72. doi: 10.1189/jlb.0109001
119. Hodge-Dufour J, Noble PW, Horton MR, Bao C, Wysoka M, Burdick MD, et al. Induction of IL-12 and chemokines by hyaluronan requires adhesion-dependent priming of resident but not elicited macrophages. J Immunol (1997) 159:2492–500.
120. Horton MR, Burdick MD, Strieter RM, Bao C, Noble PW. Regulation of hyaluronan-induced chemokine gene expression by IL-10 and IFN-gamma in mouse macrophages. J Immunol (1998) 160:3023–30.
121. Horton MR, McKee CM, Bao C, Liao F, Farber JM, Hodge-DuFour J, et al. Hyaluronan fragments synergize with interferon-gamma to induce the C-X-C chemokines mig and interferon-inducible protein-10 in mouse macrophages. J Biol Chem (1998) 273:35088–94. doi: 10.1074/jbc.273.52.35088
122. McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, et al. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J Clin Invest (1996) 98:2403–13. doi: 10.1172/JCI119054
123. Baranek T, Debret R, Antonicelli F, Lamkhioued B, Belaaouaj A, Hornebeck W, et al. Elastin receptor (spliced galactosidase) occupancy by elastin peptides counteracts proinflammatory cytokine expression in lipopolysaccharide-stimulated human monocytes through NF-kappaB down-regulation. J Immunol (2007) 179:6184–92. doi: 10.4049/jimmunol.179.9.6184
124. Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med (2007) 13:567–9. doi: 10.1038/nm1583
125. Kelly A, Houston SA, Sherwood E, Casulli J, Travis MA. Regulation of Innate and Adaptive Immunity by TGFβ. Adv Immunol (2017) 134:137–233. doi: 10.1016/bs.ai.2017.01.001
126. Schaefer L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J Biol Chem (2014) 289:35237–45. doi: 10.1074/jbc.R114.619304
127. Liu J, Cao X. Cellular and molecular regulation of innate inflammatory responses. Cell Mol Immunol (2016) 13:711–21. doi: 10.1038/cmi.2016.58
128. Midwood KS, Williams LV, Schwarzbauer JE. Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol (2004) 36:1031–7. doi: 10.1016/j.biocel.2003.12.003
129. Tu Z, Bozorgzadeh A, Pierce RH, Kurtis J, Crispe IN, Orloff MS, et al. TLR-dependent cross talk between human Kupffer cells and NK cells. J Exp Med (2008) 205:233–44. doi: 10.1084/jem.20072195
130. Asimakopoulos F, Hope C, Johnson MG, Pagenkopf A, Gromek K, Nagel B, et al. Extracellular matrix and the myeloid-in-myeloma compartment: balancing tolerogenic and immunogenic inflammation in the myeloma niche. J Leukoc Biol (2017) 102(2):265–75. doi: 10.1189/jlb.3mr1116-468r
131. Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: An information-rich system. Eur J Cell Biol (2006) 185:699–715. doi: 10.1016/j.ejcb.2006.05.009
132. Petrey AC, de la Motte CA. Hyaluronan, a crucial regulator of inflammation. Front Immunol (2014) 5:1–13. doi: 10.3389/fimmu.2014.00101
133. Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature (2009) 457:102–6. doi: 10.1038/nature07623
134. Asplund A, Friden V, Stillemark-Billton P, Camejo G, Bondjers G. Macrophages exposed to hypoxia secrete proteoglycans for which LDL has higher affinity. Atherosclerosis (2011) 215:77–81. doi: 10.1016/j.atherosclerosis.2010.12.017
135. Ramnath D, Irvine KM, Lukowski SW, Horsfall LU, Loh Z, Clouston AD, et al. Hepatic expression profiling identifies steatosis-independent and steatosis-driven advanced fibrosis genes. JCI Insight (2018) 3:1–17. doi: 10.1172/jci.insight.120274
136. Li Y, Qi X, Tong X, Wang S. Thrombospondin 1 activates the macrophage Toll-like receptor 4 pathway. Cell Mol Immunol (2013) 10:506–12. doi: 10.1038/cmi.2013.32
137. Oldenborg PA. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol (2013) 2013:614619. doi: 10.1155/2013/614619
138. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol (2001) 11:130–5. doi: 10.1016/S0962-8924(00)01906-1
139. Li Y, Turpin CP, Wang S. Role of thrombospondin 1 in liver diseases. Hepatol Res (2017) 47:186–93. doi: 10.1111/hepr.12787
140. Smalling RL, Delker DA, Zhang Y, Nieto N, McGuiness MS, Liu S, et al. Genome-wide transcriptome analysis identifies novel gene signatures implicated in human chronic liver disease. Am J Physiol Gastrointest Liver Physiol (2013) 305:G364–74. doi: 10.1152/ajpgi.00077.2013
141. Contreras-Ruiz L, Regenfuss B, Mir FA, Kearns J, Masli S. Conjunctival inflammation in thrombospondin-1 deficient mouse model of Sjogren’s syndrome. PloS One (2013) 8:e75937. doi: 10.1371/journal.pone.0075937
142. McMorrow JP, Crean D, Gogarty M, Smyth A, Connolly M, Cummins E, et al. Tumor necrosis factor inhibition modulates thrombospondin-1 expression in human inflammatory joint disease through altered NR4A2 activity. Am J Pathol (2013) 183:1243–57. doi: 10.1016/j.ajpath.2013.06.029
143. Khan AA, Bose C, Yam LS, Soloski MJ, Rupp F. Physiological regulation of the immunological synapse by agrin. Sci (80 ) (2001) 292:1681–6. doi: 10.1126/science.1056594
144. Mazzon C, Anselmo A, Soldani C, Cibella J, Ploia C, Moalli F, et al. Agrin is required for survival and function of monocytic cells. Blood (2012) 119:5502–11. doi: 10.1182/blood-2011-09-382812
145. Tatrai P, Dudas J, Batmunkh E, Mathe M, Zalatnai A, Schaff Z, et al. Agrin, a novel basement membrane component in human and rat liver, accumulates in cirrhosis and hepatocellular carcinoma. Lab Invest (2006) 86:1149–60. doi: 10.1038/labinvest.3700475
146. Batmunkh E, Tatrai P, Szabo E, Lodi C, Holczbauer A, Paska C, et al. Comparison of the expression of agrin, a basement membrane heparan sulfate proteoglycan, in cholangiocarcinoma and hepatocellular carcinoma. Hum Pathol (2007) 38:1508–15. doi: 10.1016/j.humpath.2007.02.017
147. Bai T, Chen CC, Lau LF. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J Immunol (2010) 184:3223–32. doi: 10.4049/jimmunol.0902792
148. Schober JM, Chen N, Grzeszkiewicz TM, Jovanovic I, Emeson EE, Ugarova TP, et al. Identification of integrin alpha(M)beta(2) as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate-early gene products expressed in atherosclerotic lesions. Blood (2002) 99:4457–65. doi: 10.1182/blood.V99.12.4457
149. Bian Z, Peng Y, You Z, Wang Q, Miao Q, Liu Y, et al. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res (2013) 54:44–54. doi: 10.1194/jlr.M026013
150. Zhang H, Lian M, Zhang J, Bian Z, Tang R, Miao Q, et al. A functional characteristic of cysteine-rich protein 61: Modulation of myeloid-derived suppressor cells in liver inflammation. Hepatology (2018) 67:232–46. doi: 10.1002/hep.29418
151. Kim KH, Chen CC, Monzon R II, Lau LF. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol (2013) 33:2078–90. doi: 10.1128/MCB.00049-13
152. Jiao Z, Hua S, Wang W, Wang H, Gao J, Wang X, et al. Increased circulating myeloid-derived suppressor cells correlated negatively with Th17 cells in patients with rheumatoid arthritis. Scand J Rheumatol (2013) 42:85–90. doi: 10.3109/03009742.2012.716450
153. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell (2016) 30:533–47. doi: 10.1016/j.ccell.2016.09.003
154. Ilkovitch D, Lopez DM. The liver is a site for tumor-induced myeloid-derived suppressor cell accumulation and immunosuppression. Cancer Res (2009) 69:5514–21. doi: 10.1158/0008-5472.CAN-08-4625
155. Wu F, Vij N, Roberts L, Lopez-Briones S, Joyce S, Chakravarti S, et al. A novel role of the lumican core protein in bacterial lipopolysaccharide-induced innate immune response. J Biol Chem (2007) 282:26409–17. doi: 10.1074/jbc.M702402200
156. Vij N, Roberts L, Joyce S, Chakravarti S. Lumican regulates corneal inflammatory responses by modulating Fas-Fas ligand signaling. Invest Ophthalmol Vis Sci (2005) 46:88–95. doi: 10.1167/iovs.04-0833
157. Carlson EC, Lin M, Liu CY, Kao WW, Perez VL, Pearlman E, et al. Keratocan and lumican regulate neutrophil infiltration and corneal clarity in lipopolysaccharide-induced keratitis by direct interaction with CXCL1. J Biol Chem (2007) 282:35502–9. doi: 10.1074/jbc.M705823200
158. Hayashi Y, Call MK, Chikama T, Liu H, Carlson EC, Sun Y, et al. Lumican is required for neutrophil extravasation following corneal injury and wound healing. J Cell Sci (2010) 123:2987–95. doi: 10.1242/jcs.068221
159. Lohr K, Sardana H, Lee S, Wu F, Huso DL, Hamad AR, et al. Extracellular matrix protein lumican regulates inflammation in a mouse model of colitis. Inflammation Bowel Dis (2012) 18:143–51. doi: 10.1002/ibd.21713
160. Krull NB, Gressner AM. Differential expression of keratan sulphate proteoglycans fibromodulin, lumican and aggrecan in normal and fibrotic rat liver. FEBS Lett (1992) 312:47–52. doi: 10.1016/0014-5793(92)81407-D
161. Bracht T, Schweinsberg V, Trippler M, Kohl M, Ahrens M, Padden J, et al. Analysis of disease-associated protein expression using quantitative proteomics-fibulin-5 is expressed in association with hepatic fibrosis. J Proteome Res (2015) 14:2278–86. doi: 10.1021/acs.jproteome.5b00053
162. Charlton M, Viker K, Krishnan A, Sanderson S, Veldt B, Kaalsbeek AJ, et al. Differential expression of lumican and fatty acid binding protein-1: new insights into the histologic spectrum of nonalcoholic fatty liver disease. Hepatology (2009) 49:1375–84. doi: 10.1002/hep.22927
163. Krishnan A, Li X, Kao WY, Viker K, Butters K, Masuoka H, et al. Lumican, an extracellular matrix proteoglycan, is a novel requisite for hepatic fibrosis. Lab Invest (2012) 92:1712–25. doi: 10.1038/labinvest.2012.121
164. Furuta K, Sato S, Yamauchi T, Ozawa T, Harada M, Kakumu S, et al. Intrahepatic gene expression profiles in chronic hepatitis B and autoimmune liver disease. J Gastroenterol (2008) 43:866–74. doi: 10.1007/s00535-008-2237-y
165. Butcher JT, Norris RA, Hoffman S, Mjaatvedt CH, Markwald RR. Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev Biol (2007) 302:256–66. doi: 10.1016/j.ydbio.2006.09.048
166. Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, et al. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med (2007) 13:962–9. doi: 10.1038/nm1619
167. Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD, et al. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res (2002) 62:5358–64.
168. Baril P, Gangeswaran R, Mahon PC, Caulee K, Kocher HM, Harada T, et al. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene (2007) 26:2082–94. doi: 10.1038/sj.onc.1210009
169. Johansson MW, Annis DS, Mosher DF. alpha(M)beta(2) integrin-mediated adhesion and motility of IL-5-stimulated eosinophils on periostin. Am J Respir Cell Mol Biol (2013) 48:503–10. doi: 10.1165/rcmb.2012-0150OC
170. Maruhashi T, Kii I, Saito M, Kudo A. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem (2010) 285:13294–303. doi: 10.1074/jbc.M109.088864
171. Kudo A. Periostin in fibrillogenesis for tissue regeneration: periostin actions inside and outside the cell. Cell Mol Life Sci (2011) 68:3201–7. doi: 10.1007/s00018-011-0784-5
172. Conway SJ, Izuhara K, Kudo Y, Litvin J, Markwald R, Ouyang G, et al. The role of periostin in tissue remodeling across health and disease. Cell Mol Life Sci (2014) 71:1279–88. doi: 10.1007/s00018-013-1494-y
173. Hakuno D, Kimura N, Yoshioka M, Mukai M, Kimura T, Okada Y, et al. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J Clin Invest (2010) 120:2292–306. doi: 10.1172/JCI40973
174. Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet (1989) 1:520–4. doi: 10.1016/S0140-6736(89)90067-6
175. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med (2009) 180:388–95. doi: 10.1164/rccm.200903-0392OC
176. Blanchard C, Mingler MK, McBride M, Putnam PE, Collins MH, Chang G, et al. Periostin facilitates eosinophil tissue infiltration in allergic lung and esophageal responses. Mucosal Immunol (2008) 1:289–96. doi: 10.1038/mi.2008.15
177. Izuhara K, Arima K, Ohta S, Suzuki S, Inamitsu M, Yamamoto K, et al. Periostin in allergic inflammation. Allergol Int (2014) 63:143–51. doi: 10.2332/allergolint.13-RAI-0663
178. Sugiyama A, Kanno K, Nishimichi N, Ohta S, Ono J, Conway SJ, et al. Periostin promotes hepatic fibrosis in mice by modulating hepatic stellate cell activation via alphav integrin interaction. J Gastroenterol (2016) 51:1161–74. doi: 10.1007/s00535-016-1206-0
179. Lu Y, Liu X, Jiao Y, Xiong X, Wang E, Wang X, et al. Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARalpha. J Clin Invest (2014) 124:3501–13. doi: 10.1172/JCI74438
180. Li Y, Wu S, Xiong S, Ouyang G. Deficiency of periostin protects mice against methionine-choline-deficient diet-induced non-alcoholic steatohepatitis. J Hepatol (2015) 62:495–7. doi: 10.1016/j.jhep.2014.10.005
181. Talsma DT, Katta K, Ettema MAB, Kel B, Kusche-Gullberg M, Daha MR, et al. Endothelial heparan sulfate deficiency reduces inflammation and fibrosis in murine diabetic nephropathy. Lab Investig (2018) 98:427–38. doi: 10.1038/s41374-017-0015-2
182. O’Callaghan P, Li JP, Lannfelt L, Lindahl U, Zhang X. Microglial Heparan Sulfate Proteoglycans Facilitate the Cluster-of-Differentiation 14 (CD14)/Toll-like Receptor 4 (TLR4)-Dependent Inflammatory Response. J Biol Chem (2015) 290:14904–14. doi: 10.1074/jbc.M114.634337
183. Goldberg R, Meirovitz A, Hirshoren N, Bulvik R, Binder A, Rubinstein AM, et al. Versatile role of heparanase in inflammation. Matrix Biol (2013) 32(5):234–40. doi: 10.1016/j.matbio.2013.02.008
184. Patel VN, Knox SM, Likar KM, Lathrop CA, Hossain R, Eftekhari S, et al. Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development (2007) 134(23):4177–86. doi: 10.1242/dev.011171
186. Ding K, Sandgren S, Mani K, Belting M, Fransson LA. Modulations of glypican-1 heparan sulfate structure by inhibition of endogenous polyamine synthesis. Mapping of spermine-binding sites and heparanase, heparin lyase, and nitric oxide/nitrite cleavage sites. J Biol Chem (2001) 276:46779–91. doi: 10.1074/jbc.M105419200
187. Goodall KJ, Poon IKH, Phipps S, Hulett MD. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PloS One (2014) 9:1–13. doi: 10.1371/journal.pone.0109596
188. Roskams T, Rosenbaum J, De Vos R, David G, Desmet V. Heparan sulfate proteoglycan expression in chronic cholestatic human liver diseases. Hepatology (1996) 24:524–32. doi: 10.1002/hep.510240310
189. Barth H, Schäfer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, et al. Cellular Binding of Hepatitis C Virus Envelope Glycoprotein E2 Requires Cell Surface Heparan Sulfate. J Biol Chem (2003) 278:41003–12. doi: 10.1074/jbc.M302267200
190. Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology (2007) 46:1759–68. doi: 10.1002/hep.21896
191. Termeer CC, Hennies J, Voith U, Ahrens T, Weiss JM, Prehm P, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med (2002) 195:99–111. doi: 10.1084/jem.20001858
192. Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR, et al. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol (2006) 177:1272–81. doi: 10.4049/jimmunol.177.2.1272
193. Noble PW, McKee CM, Cowman M, Shin HS. Hyaluronan fragments activate an NF-kappa B/I-kappa B alpha autoregulatory loop in murine macrophages. J Exp Med (1996) 183:2373–8. doi: 10.1084/jem.183.5.2373
194. Sironen RK, Tammi M, Tammi R, Auvinen PK, Anttila M, Kosma VM, et al. Hyaluronan in human malignancies. Exp Cell Res (2011) 317:383–91. doi: 10.1016/j.yexcr.2010.11.017
195. Agren UM, Tammi RH, Tammi M II. Reactive oxygen species contribute to epidermal hyaluronan catabolism in human skin organ culture. Free Radic Biol Med (1997) 23:996–1001. doi: 10.1016/S0891-5849(97)00098-1
196. Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem (2009) 284:12762–71. doi: 10.1074/jbc.M806084200
197. Mustonen AM, Salven A, Karja V, Rilla K, Matilainen J, Nieminen P, et al. Hyaluronan histochemistry-a potential new tool to assess the progress of liver disease from simple steatosis to hepatocellular carcinoma. Glycobiology (2019) 29:298–306. doi: 10.1093/glycob/cwz002
198. Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest (2005) 115:2223–33. doi: 10.1172/JCI23755
199. Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem (2009) 284:24035–48. doi: 10.1074/jbc.M109.014266
200. Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, et al. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest (2010) 120:4251–72. doi: 10.1172/JCI42213
201. Frey H, Schroeder N, Manon-Jensen T, Iozzo RV, Schaefer L. Biological interplay between proteoglycans and their innate immune receptors in inflammation. FEBS J (2013) 280:2165–79. doi: 10.1111/febs.12145
202. Genovese F, Barascuk N, Larsen L, Larsen MR, Nawrocki A, Li Y, et al. Biglycan fragmentation in pathologies associated with extracellular matrix remodeling by matrix metalloproteinases. Fibrogenes Tissue Repair (2013) 6:9. doi: 10.1186/1755-1536-6-9
203. Brassart B, Fuchs P, Huet E, Alix AJP, Wallach J, Tamburro AM, et al. Conformational Dependence of Collagenase (Matrix Metalloproteinase-1) Up-regulation by Elastin Peptides in Cultured Fibroblasts. J Biol Chem (2001) 276:5222–7. doi: 10.1074/jbc.M003642200
204. Debret R, Antonicelli F, Theill A, Hornebeck W, Bernard P, Guenounou M, et al. Elastin-derived peptides induce a T-helper type 1 polarization of human blood lymphocytes. Arter Thromb Vasc Biol (2005) 25:1353–8. doi: 10.1161/01.ATV.0000168412.50855.9f
205. Maurice P, Blaise S, Gayral S, Debelle L, Laffargue M, Hornebeck W, et al. Elastin fragmentation and atherosclerosis progression: the elastokine concept. Trends Cardiovasc Med (2013) 23:211–21. doi: 10.1016/j.tcm.2012.12.004
206. Francki A, Bradshaw AD, Bassuk JA, Howe CC, Couser WG, Sage EH, et al. SPARC regulates the expression of collagen type I and transforming growth factor-beta1 in mesangial cells. J Biol Chem (1999) 274:32145–52. doi: 10.1074/jbc.274.45.32145
207. Wang JC, Lai S, Guo X, Zhang X, de Crombrugghe B, Sonnylal S, et al. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res Ther (2010) 12:R60. doi: 10.1186/ar2973
208. Frizell E, Liu SL, Abraham A, Ozaki I, Eghbali M, Sage EH, et al. Expression of SPARC in normal and fibrotic livers. Hepatology (1995) 21:847–54. doi: 10.1002/hep.1840210335
209. Blazejewski S, Le Bail B, Boussarie L, Blanc JF, Malaval L, Okubo K, et al. Osteonectin (SPARC) expression in human liver and in cultured human liver myofibroblasts. Am J Pathol (1997) 151:651–7.
210. Kristensen DB, Kawada N, Imamura K, Miyamoto Y, Tateno C, Seki S, et al. Proteome analysis of rat hepatic stellate cells. Hepatology (2000) 32:268–77. doi: 10.1053/jhep.2000.9322
211. Lamireau T, Le Bail B, Boussarie L, Fabre M, Vergnes P, Bernard O, et al. Expression of collagens type I and IV, osteonectin and transforming growth factor beta-1 (TGFbeta1) in biliary atresia and paucity of intrahepatic bile ducts during infancy. J Hepatol (1999) 31:248–55. doi: 10.1016/S0168-8278(99)80221-9
212. Le Bail B, Faouzi S, Boussarie L, Guirouilh J, Blanc JF, Carles J, et al. Osteonectin/SPARC is overexpressed in human hepatocellular carcinoma. J Pathol (1999) 189:46–52. doi: 10.1002/(SICI)1096-9896(199909)189:1<46::AID-PATH392>3.0.CO;2-X
213. Goldenberg D, Ayesh S, Schneider T, Pappo O, Jurim O, Eid A, et al. Analysis of differentially expressed genes in hepatocellular carcinoma using cDNA arrays. Mol Carcinog (2002) 33:113–24. doi: 10.1002/mc.10027
214. Lau CP, Poon RT, Cheung ST, Yu WC, Fan ST. SPARC and Hevin expression correlate with tumour angiogenesis in hepatocellular carcinoma. J Pathol (2006) 210:459–68. doi: 10.1002/path.2068
215. Li Z, Zhang Y, Liu Z, Wu X, Zheng Y, Tao Z, et al. ECM1 controls T(H)2 cell egress from lymph nodes through re-expression of S1P(1). Nat Immunol (2011) 12:178–85. doi: 10.1038/ni.1983
216. Su P, Chen S, Zheng YH, Zhou HY, Yan CH, Yu F, et al. Novel Function of Extracellular Matrix Protein 1 in Suppressing Th17 Cell Development in Experimental Autoimmune Encephalomyelitis. J Immunol (2016) 197:1054–64. doi: 10.4049/jimmunol.1502457
217. He L, Gu W, Wang M, Chang X, Sun X, Zhang Y, et al. Extracellular matrix protein 1 promotes follicular helper T cell differentiation and antibody production. Proc Natl Acad Sci U.S.A. (2018) 115:8621–6. doi: 10.1073/pnas.1801196115
218. Bollyky PL, Wu RP, Falk BA, Lord JD, Long SA, Preisinger A, et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc Natl Acad Sci U.S.A. (2011) 108:7938–43. doi: 10.1073/pnas.1017360108
219. Forrester JV, Balazs EA. Inhibition of phagocytosis by high molecular weight hyaluronate. Immunology (1980) 40:435–46.
220. Delmage JM, Powars DR, Jaynes PK, Allerton SE. The selective suppression of immunogenicity by hyaluronic acid. Ann Clin Lab Sci (1986) 16:303–10.
221. Bollyky PL, Lord JD, Masewicz SA, Evanko SP, Buckner JH, Wight TN, et al. Cutting edge: high molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J Immunol (2007) 179:744–7. doi: 10.4049/jimmunol.179.2.744
222. Al-Sharif A, Jamal M, Zhang LX, Larson K, Schmidt TA, Jay GD, et al. Lubricin/Proteoglycan 4 Binding to CD44 Receptor: A Mechanism of the Suppression of Proinflammatory Cytokine-Induced Synoviocyte Proliferation by Lubricin. Arthritis Rheumatol (2015) 67:1503–13. doi: 10.1002/art.39087
223. Alquraini A, Garguilo S, D'Souza G, Zhang LX, Schmidt TA, Jay GD, et al. The interaction of lubricin/proteoglycan 4 (PRG4) with toll-like receptors 2 and 4: an anti-inflammatory role of PRG4 in synovial fluid. Arthritis Res Ther (2015) 17:353. doi: 10.1186/s13075-015-0877-x
224. Alquraini A, Jamal M, Zhang L, Schmidt T, Jay GD, Elsaid KA, et al. The autocrine role of proteoglycan-4 (PRG4) in modulating osteoarthritic synoviocyte proliferation and expression of matrix degrading enzymes. Arthritis Res Ther (2017) 19:89. doi: 10.1186/s13075-017-1301-5
225. Nahon JE, Hoekstra M, van Harmelen V, Rensen PCN, Willems van Dijk K, Kooijman S, et al. Proteoglycan 4 deficiency protects against glucose intolerance and fatty liver disease in diet-induced obese mice. Biochim Biophys Acta Mol Basis Dis (2019) 1865:494–501. doi: 10.1016/j.bbadis.2018.11.009
226. Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med (2009) 15:774–80. doi: 10.1038/nm.1987
227. Piccinini AM, Zuliani-Alvarez L, Lim JM, Midwood KS. Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Sci Signal (2016) 9:ra86. doi: 10.1126/scisignal.aaf3596
228. Zuliani-Alvarez L, Marzeda AM, Deligne C, Schwenzer A, McCann FE, Marsden BD, et al. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat Commun (2017) 8:1595. doi: 10.1038/s41467-017-01718-7
229. Machino-Ohtsuka T, Tajiri K, Kimura T, Sakai S, Sato A, Yoshida T, et al. Tenascin-C aggravates autoimmune myocarditis via dendritic cell activation and Th17 cell differentiation. J Am Hear Assoc (2014) 3:e001052. doi: 10.1161/JAHA.114.001052
230. Piccinini AM, Midwood KS. Endogenous Control of Immunity against Infection: Tenascin-C Regulates TLR4-Mediated Inflammation via MicroRNA-155. Cell Rep (2012) 2:914–26. doi: 10.1016/j.celrep.2012.09.005
231. Patel L, Sun W, Glasson SS, Morris EA, Flannery CR, Chockalingam PS, et al. Tenascin-C induces inflammatory mediators and matrix degradation in osteoarthritic cartilage. BMC Musculoskelet Disord (2011) 12:164. doi: 10.1186/1471-2474-12-164
232. Jachetti E, Caputo S, Mazzoleni S, Brambillasca CS, Parigi SM, Grioni M, et al. Tenascin-C Protects Cancer Stem-like Cells from Immune Surveillance by Arresting T-cell Activation. Cancer Res (2015) 75:2095–108. doi: 10.1158/0008-5472.CAN-14-2346
233. Ruhmann M, Piccinini AM, Kong PL, Midwood KS. Endogenous activation of adaptive immunity: tenascin-C drives interleukin-17 synthesis in murine arthritic joint disease. Arthritis Rheum (2012) 64:2179–90. doi: 10.1002/art.34401
234. Ruegg CR, Chiquet-Ehrismann R, Alkan SS. Tenascin, an extracellular matrix protein, exerts immunomodulatory activities. Proc Natl Acad Sci U.S.A. (1989) 86:7437–41. doi: 10.1073/pnas.86.19.7437
235. Hemesath TJ, Marton LS, Stefansson K. Inhibition of T cell activation by the extracellular matrix protein tenascin. J Immunol (1994) 152:5199–207.
236. Hibino S, Kato K, Kudoh S, Yagita H, Okumura K. Tenascin suppresses CD3-mediated T cell activation. Biochem Biophys Res Commun (1998) 250:119–24. doi: 10.1006/bbrc.1998.9258
237. Puente Navazo MD, Valmori D, Ruegg C. The alternatively spliced domain TnFnIII A1A2 of the extracellular matrix protein tenascin-C suppresses activation-induced T lymphocyte proliferation and cytokine production. J Immunol (2001) 167:6431–40. doi: 10.4049/jimmunol.167.11.6431
238. Chiquet-Ehrismann R, Chiquet M. Tenascins: regulation and putative functions during pathological stress. J Pathol (2003) 200:488–99. doi: 10.1002/path.1415
239. El-Karef A, Yoshida T, Gabazza EC, Nishioka T, Inada H, Sakakura T, et al. Deficiency of tenascin-C attenuates liver fibrosis in immune-mediated chronic hepatitis in mice. J Pathol (2007) 211:86–94. doi: 10.1002/path.2099
240. Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, et al. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Sci (80 ) (2000) 287:860–4. doi: 10.1126/science.287.5454.860
241. Wen Y, Jeong S, Xia Q, Kong X. Role of Osteopontin in Liver Diseases. Int J Biol Sci (2016) 12:1121–8. doi: 10.7150/ijbs.16445
242. Yang M, Ramachandran A, Yan HM, Woolbright BL, Copple BL, Fickert P, et al. Osteopontin is an initial mediator of inflammation and liver injury during obstructive cholestasis after bile duct ligation in mice. Toxicol Lett (2014) 224:186–95. doi: 10.1016/j.toxlet.2013.10.030
243. Morimoto J, Inobe M, Kimura C, Kon S, Diao H, Aoki M, et al. Osteopontin affects the persistence of beta-glucan-induced hepatic granuloma formation and tissue injury through two distinct mechanisms. Int Immunol (2004) 16:477–88. doi: 10.1093/intimm/dxh044
244. Arai M, Yokosuka O, Kanda T, Fukai K, Imazeki F, Muramatsu M, et al. Serum osteopontin levels in patients with acute liver dysfunction. Scand J Gastroenterol (2006) 41:102–10. doi: 10.1080/00365520510024061
245. Srungaram P, Rule JA, Yuan HJ, Reimold A, Dahl B, Sanders C, et al. Plasma osteopontin in acute liver failure. Cytokine (2015) 73:270–6. doi: 10.1016/j.cyto.2015.02.021
246. Yilmaz Y, Ozturk O, Alahdab YO, Senates E, Colak Y, Doganay HL, et al. Serum osteopontin levels as a predictor of portal inflammation in patients with nonalcoholic fatty liver disease. Dig Liver Dis (2013) 45:58–62. doi: 10.1016/j.dld.2012.08.017
247. Patouraux S, Bonnafous S, Voican CS, Anty R, Saint-Paul MC, Rosenthal-Allieri MA, et al. The osteopontin level in liver, adipose tissue and serum is correlated with fibrosis in patients with alcoholic liver disease. PloS One (2012) 7:e35612. doi: 10.1371/journal.pone.0035612
248. Zhao L, Li T, Wang Y, Pan Y, Ning H, Hui X, et al. Elevated plasma osteopontin level is predictive of cirrhosis in patients with hepatitis B infection. Int J Clin Pr (2008) 62:1056–62. doi: 10.1111/j.1742-1241.2007.01368.x
249. Huang W, Zhu G, Huang M, Lou G, Liu Y, Wang S, et al. Plasma osteopontin concentration correlates with the severity of hepatic fibrosis and inflammation in HCV-infected subjects. Clin Chim Acta (2010) 411:675–8. doi: 10.1016/j.cca.2010.01.029
250. Matsue Y, Tsutsumi M, Hayashi N, Saito T, Tsuchishima M, Toshikuni N, et al. Serum osteopontin predicts degree of hepatic fibrosis and serves as a biomarker in patients with hepatitis C virus infection. PloS One (2015) 10:e0118744. doi: 10.1371/journal.pone.0118744
251. Shang S, Plymoth A, Ge S, Feng Z, Rosen HR, Sangrajrang S, et al. Identification of osteopontin as a novel marker for early hepatocellular carcinoma. Hepatology (2012) 55:483–90. doi: 10.1002/hep.24703
252. Fan X, He C, Jing W, Zhou X, Chen R, Cao L, et al. Intracellular Osteopontin inhibits toll-like receptor signaling and impedes liver carcinogenesis. Cancer Res (2015) 75:86–97. doi: 10.1158/0008-5472.CAN-14-0615
253. Seth D, Duly A, Kuo PC, McCaughan GW, Haber PS. Osteopontin is an important mediator of alcoholic liver disease via hepatic stellate cell activation. World J Gastroenterol (2014) 20:13088–104. doi: 10.3748/wjg.v20.i36.13088
254. Pereira TA, Syn WK, Machado MV, Vidigal PV, Resende V, Voieta I, et al. Schistosome-induced cholangiocyte proliferation and osteopontin secretion correlate with fibrosis and portal hypertension in human and murine schistosomiasis mansoni. Clin Sci (2015) 129:875–83. doi: 10.1042/CS20150117
255. Gullberg D, Turner DC, Borg TK, Terracio L, Rubin K. Different beta 1-integrin collagen receptors on rat hepatocytes and cardiac fibroblasts. Exp Cell Res (1990) 190:254–64. doi: 10.1016/0014-4827(90)90194-F
256. Rowley AT, Nagalla RR, Wang SW, Liu WF. Extracellular Matrix-Based Strategies for Immunomodulatory Biomaterials Engineering. Adv Heal Mater (2019) 8:e1801578. doi: 10.1002/adhm.201801578
257. Chattopadhyay S, Raines RT. Review collagen-based biomaterials for wound healing. Biopolymers (2014) 101:821–33. doi: 10.1002/bip.22486
258. Schultz HS, Nitze LM, Zeuthen LH, Keller P, Gruhler A, Pass J, et al. Collagen induces maturation of human monocyte-derived dendritic cells by signaling through osteoclast-associated receptor. J Immunol (2015) 194:3169–79. doi: 10.4049/jimmunol.1402800
259. Chiang NY, Hsiao CC, Huang YS, Chen HY, Hsieh IJ, Chang GW, et al. Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. J Biol Chem (2011) 286:14215–25. doi: 10.1074/jbc.M110.183830
260. Peng YM, van de Garde MD, Cheng KF, Baars PA, Remmerswaal EB, van Lier RA, et al. Specific expression of GPR56 by human cytotoxic lymphocytes. J Leukoc Biol (2011) 90:735–40. doi: 10.1189/jlb.0211092
261. Yang L, Xu L. GPR56 in cancer progression: current status and future perspective. Futur Oncol (2012) 8:431–40. doi: 10.2217/fon.12.27
262. Lebbink RJ, van den Berg MC, de Ruiter T, Raynal N, van Roon JA, Lenting PJ, et al. The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J Immunol (2008) 180:1662–9. doi: 10.4049/jimmunol.180.3.1662
263. Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305). J Leukoc Biol (2008) 83:799–803. doi: 10.1189/jlb.0907609
264. Hsieh LT, Nastase MV, Zeng-Brouwers J, Iozzo RV, Schaefer L. Soluble biglycan as a biomarker of inflammatory renal diseases. Int J Biochem Cell Biol (2014) 54:223–35. doi: 10.1016/j.biocel.2014.07.020
265. Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhao JG, et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal (2011) 4:ra75. doi: 10.1126/scisignal.2001868
266. Fiedler LR, Schonherr E, Waddington R, Niland S, Seidler DG, Aeschlimann D, et al. Decorin regulates endothelial cell motility on collagen I through activation of insulin-like growth factor I receptor and modulation of alpha2beta1 integrin activity. J Biol Chem (2008) 283:17406–15. doi: 10.1074/jbc.M710025200
267. Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell (1991) 64:867–9. doi: 10.1016/0092-8674(91)90308-L
268. Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, et al. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature (1992) 360:361–4. doi: 10.1038/360361a0
269. Järvinen TAH, Ruoslahti E. Generation of a multi-functional, target organ-specific, anti-fibrotic molecule by molecular engineering of the extracellular matrix protein, decorin. Br J Pharmacol 176(1):16–25. doi: 10.1111/bph.14374
270. Hennig EE, Mikula M, Goryca K, Paziewska A, Ledwon J, Nesteruk M, et al. Extracellular matrix and cytochrome P450 gene expression can distinguish steatohepatitis from steatosis in mice. J Cell Mol Med (2014) 18:1762–72. doi: 10.1111/jcmm.12328
271. Kelsh-Lasher RM, Ambesi A, Bertram C, McKeown-Longo PJ. Integrin alpha4beta1 and TLR4 Cooperate to Induce Fibrotic Gene Expression in Response to Fibronectin’s EDA Domain. J Invest Dermatol (2017) 137:2505–12. doi: 10.1016/j.jid.2017.08.005
272. Doddapattar P, Jain M, Dhanesha N, Lentz SR, Chauhan AK. Fibronectin containing extra domain a induces plaque destabilization in the innominate artery of aged apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol (2018) 38:500–8. doi: 10.1161/ATVBAHA.117.310345
273. Aziz-Seible RS, Lee SM, Kharbanda KK, McVicker BL, Casey CA. Ethanol feeding potentiates the pro-inflammatory response of Kupffer cells to cellular fibronectin. Alcohol Clin Exp Res (2011) 35:717–25. doi: 10.1111/j.1530-0277.2010.01389.x
274. Zhu Q, Zou L, Jagavelu K, Simonetto DA, Huebert RC, Jiang ZD, et al. Intestinal decontamination inhibits TLR4 dependent fibronectin-mediated cross-talk between stellate cells and endothelial cells in liver fibrosis in mice. J Hepatol (2012) 56:893–9. doi: 10.1016/j.jhep.2011.11.013
275. Gillis SE, Nagy LE. Deposition of cellular fibronectin increases before stellate cell activation in rat liver during ethanol feeding. Alcohol Clin Exp Res (1997) 21:857–61. doi: 10.1111/j.1530-0277.1997.tb03849.x
276. Aziz-Seible RS, McVicker BL, Kharbanda KK, Casey CA. Cellular fibronectin stimulates hepatocytes to produce factors that promote alcohol-induced liver injury. World J Hepatol (2011) 3:45–55. doi: 10.4254/wjh.v3.i2.45
277. Risteli L, Timpl R. Isolation and characterization of pepsin fragments of laminin from human placental and renal basement membranes. Biochem J (1981) 193:749–55. doi: 10.1042/bj1930749
278. Nouchi T, Worner TM, Sato S, Lieber CS. Serum procollagen type III N-terminal peptides and laminin P1 peptide in alcoholic liver disease. Alcohol Clin Exp Res (1987) 11:287–91. doi: 10.1111/j.1530-0277.1987.tb01309.x
279. Tanaka Y, Tateishi R, Koike K. Proteoglycans are attractive biomarkers and therapeutic targets in hepatocellular carcinoma. Int J Mol Sci (2018) 19:1–17. doi: 10.3390/ijms19103070
280. Toeda K, Nakamura K, Hirohata S, Hatipoglu OF, Demircan K, Yamawaki H, et al. Versican is induced in infiltrating monocytes in myocardial infarction. Mol Cell Biochem (2005) 280:47–56. doi: 10.1007/s11010-005-8051-4
281. Wingrove JA, Daniels SE, Sehnert AJ, Tingley W, Elashoff MR, Rosenberg S, et al. Correlation of peripheral-blood gene expression with the extent of coronary artery stenosis. Circ Cardiovasc Genet (2008) 1:31–8. doi: 10.1161/CIRCGENETICS.108.782730
282. Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol (2002) 169:2253–63. doi: 10.4049/jimmunol.169.5.2253
283. Bukong TN, Maurice SB, Chahal B, Schaeffer DF, Winwood PJ. Versican: a novel modulator of hepatic fibrosis. Lab Invest (2016) 96:361–74. doi: 10.1038/labinvest.2015.152
284. Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal (2009) 2:re3. doi: 10.1126/scisignal.272re3
285. Maimaitiyiming H, Zhou Q, Wang S. Thrombospondin 1 Deficiency Ameliorates the Development of Adriamycin-Induced Proteinuric Kidney Disease. PloS One (2016) 11:e0156144. doi: 10.1371/journal.pone.0156144
286. Chavez-Tapia NC, Rosso N, Tiribelli C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol (2012) 12:20. doi: 10.1186/1471-230X-12-20
287. McMaken S, Exline MC, Mehta P, Piper M, Wang Y, Fischer SN, et al. Thrombospondin-1 contributes to mortality in murine sepsis through effects on innate immunity. PloS One (2011) 6:e19654. doi: 10.1371/journal.pone.0019654
288. Rico MC, Manns JM, Driban JB, Uknis AB, Kunapuli SP, Dela Cadena RA, et al. Thrombospondin-1 and transforming growth factor beta are pro-inflammatory molecules in rheumatoid arthritis. Transl Res (2008) 152:95–8. doi: 10.1016/j.trsl.2008.06.002
289. Lopez-Dee Z, Pidcock K, Gutierrez LS. Thrombospondin-1: multiple paths to inflammation. Mediat Inflammation (2011) 2011:296069. doi: 10.1155/2011/296069
290. Hiscott P, Armstrong D, Batterbury M, Kaye S. Repair in avascular tissues: fibrosis in the transparent structures of the eye and thrombospondin 1. Histol Histopathol (1999) 14:1309–20. doi: 10.14670/HH-14.1309
291. El-Youssef M, Mu Y, Huang L, Stellmach V, Crawford SE. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: possible role of the hepatic stellate cell. J Pediatr Gastroenterol Nutr (1999) 28:386–92. doi: 10.1097/00005176-199904000-00008
292. Li Y, Tong X, Rumala C, Clemons K, Wang S. Thrombospondin1 deficiency reduces obesity-associated inflammation and improves insulin sensitivity in a diet-induced obese mouse model. PloS One (2011) 6:e26656. doi: 10.1371/journal.pone.0026656
293. Hayashi H, Sakai K, Baba H, Sakai T. Thrombospondin-1 is a novel negative regulator of liver regeneration after partial hepatectomy through transforming growth factor-beta1 activation in mice. Hepatology (2012) 55:1562–73. doi: 10.1002/hep.24800
294. Benzoubir N, Lejamtel C, Battaglia S, Testoni B, Benassi B, Gondeau C, et al. HCV core-mediated activation of latent TGF-beta via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J Hepatol (2013) 59:1160–8. doi: 10.1016/j.jhep.2013.07.036
295. Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol (2003) 4:295–308. doi: 10.1038/nrm1074
296. Lv X, Fang C, Yin R, Qiao B, Shang R, Wang J, et al. Agrin para-secreted by PDGF-activated human hepatic stellate cells promotes hepatocarcinogenesis in vitro and in vivo. Oncotarget (2017) 8:105340–55. doi: 10.18632/oncotarget.22186
297. Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol (2009) 41:771–83. doi: 10.1016/j.biocel.2008.07.025
298. Perbal B. CCN proteins: multifunctional signalling regulators. Lancet (2004) 363:62–4. doi: 10.1016/S0140-6736(03)15172-0
299. Weiskirchen R. CCN proteins in normal and injured liver. Front Biosci (Landmark Ed) (2011) 16:1939–61. doi: 10.2741/3832
300. Chakravarti S, Magnuson T, Lass JH, Jepsen KJ, LaMantia C, Carroll H, et al. Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J Cell Biol (1998) 141:1277–86. doi: 10.1083/jcb.141.5.1277
301. Chakravarti S. Functions of lumican and fibromodulin: lessons from knockout mice. Glycoconj J (2002) 19:287–93. doi: 10.1023/A:1025348417078
302. Funderburgh JL, Mitschler RR, Funderburgh ML, Roth MR, Chapes SK, Conrad GW, et al. Macrophage receptors for lumican. A corneal keratan sulfate proteoglycan. Invest Ophthalmol Vis Sci (1997) 38:1159–67.
303. Kii I, Nishiyama T, Li M, Matsumoto K, Saito M, Amizuka N, et al. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem (2010) 285:2028–39. doi: 10.1074/jbc.M109.051961
304. Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, et al. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol (2006) 118:98–104. doi: 10.1016/j.jaci.2006.02.046
305. Masuoka M, Shiraishi H, Ohta S, Suzuki S, Arima K, Aoki S, et al. Periostin promotes chronic allergic inflammation in response to Th2 cytokines. J Clin Invest (2012) 122:2590–600. doi: 10.1172/JCI58978
306. Sen K, Lindenmeyer MT, Gaspert A, Eichinger F, Neusser MA, Kretzler M, et al. Periostin is induced in glomerular injury and expressed de novo in interstitial renal fibrosis. Am J Pathol (2011) 179:1756–67. doi: 10.1016/j.ajpath.2011.06.002
307. Laitinen A, Altraja A, Kampe M, Linden M, Virtanen I, Laitinen LA, et al. Tenascin is increased in airway basement membrane of asthmatics and decreased by an inhaled steroid. Am J Respir Crit Care Med (1997) 156:951–8. doi: 10.1164/ajrccm.156.3.9610084
308. Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U.S.A. (2007) 104:15858–63. doi:10.1073/pnas.0707413104
309. Amara S, Lopez K, Banan B, Brown SK, Whalen M, Myles E, et al. Synergistic effect of pro-inflammatory TNFalpha and IL-17 in periostin mediated collagen deposition: potential role in liver fibrosis. Mol Immunol (2015) 64:26–35. doi: 10.1016/j.molimm.2014.10.021
310. Chackelevicius CM, Gambaro SE, Tiribelli C, Rosso N. Th17 involvement in nonalcoholic fatty liver disease progression to non-alcoholic steatohepatitis. World J Gastroenterol (2016) 22:9096–103. doi: 10.3748/wjg.v22.i41.9096
311. Kobayashi T, Kanno K, Nguyen PT, Sugiyama A, Kawahara A, Otani Y, et al. Periostin antisense oligonucleotide prevents hepatic steatosis and fibrosis in a mouse model of non-alcoholic steatohepatitis. J Gastroenterol Hepatol (2020) May:0–3. doi: 10.1111/jgh.15088
312. Yang Z, Zhang H, Niu Y, Zhang W, Zhu L, Li X, et al. Circulating periostin in relation to insulin resistance and nonalcoholic fatty liver disease among overweight and obese subjects. Sci Rep (2016) 6:37886. doi: 10.1038/srep37886
313. Zhu JZ, Zhu HT, Dai YN, Li CX, Fang ZY, Zhao DJ, et al. Serum periostin is a potential biomarker for non-alcoholic fatty liver disease: a case-control study. Endocrine (2016) 51:91–100. doi: 10.1007/s12020-015-0735-2
314. Lu J, Auduong L, White ES, Yue X. Up-regulation of heparan sulfate 6-O-sulfation in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol (2014) 50:106–14. doi: 10.1165/rcmb.2013-0204OC
315. Westergren-Thorsson G, Bjermer L, Hallgren O, Zhou XH, Tykesson E, Åhrman E, et al. Increased deposition of glycosaminoglycans and altered structure of heparan sulfate in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol (2017) 83:27–38. doi: 10.1016/j.biocel.2016.12.005
316. Solic N, Wilson J, Wilson SJ, Shute JK. Endothelial activation and increased heparan sulfate expression in cystic fibrosis. Am J Respir Crit Care Med (2005) 172:892–8. doi: 10.1164/rccm.200409-1207OC
317. West DC, Hampson IN, Arnold F, Kumar S. Angiogenesis induced by degradation products of hyaluronic acid. Sci (80 ) (1985) 228:1324–6. doi: 10.1126/science.2408340
318. Takahashi Y, Li L, Kamiryo M, Asteriou T, Moustakas A, Yamashita H, et al. Hyaluronan fragments induce endothelial cell differentiation in a CD44- and CXCL1/GRO1-dependent manner. J Biol Chem (2005) 280:24195–204. doi: 10.1074/jbc.M411913200
319. Termeer CC, Hennies J, Voith U, Ahrens T, Weiss JM, Prehm P, et al. Oligosaccharides of hyaluronan are potent activators of dendritic cells. J Immunol (2000) 165:1863–70. doi: 10.4049/jimmunol.165.4.1863
320. Sugahara KN, Murai T, Nishinakamura H, Kawashima H, Saya H, Miyasaka M, et al. Hyaluronan oligosaccharides induce CD44 cleavage and promote cell migration in CD44-expressing tumor cells. J Biol Chem (2003) 278:32259–65. doi: 10.1074/jbc.M300347200
321. Horton MR, Shapiro S, Bao C, Lowenstein CJ, Noble PW. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J Immunol (1999) 162:4171–6.
322. Horton MR, Olman MA, Bao C, White KE, Choi AM, Chin BY, et al. Regulation of plasminogen activator inhibitor-1 and urokinase by hyaluronan fragments in mouse macrophages. Am J Physiol Lung Cell Mol Physiol (2000) 279:L707–15. doi: 10.1152/ajplung.2000.279.4.L707
323. McKee CM, Lowenstein CJ, Horton MR, Wu J, Bao C, Chin BY, et al. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor kappaB-dependent mechanism. J Biol Chem (1997) 272:8013–8. doi: 10.1074/jbc.272.12.8013
324. Vrochides D, Papanikolaou V, Pertoft H, Antoniades AA, Heldin P. Biosynthesis and degradation of hyaluronan by nonparenchymal liver cells during liver regeneration. Hepatology (1996) 23:1650–5. doi: 10.1002/hep.510230648
325. Bourguignon V, Flamion B. Respective roles of hyaluronidases 1 and 2 in endogenous hyaluronan turnover. FASEB J (2016) 30:2108–14. doi: 10.1096/fj.201500178R
326. Bianco P, Fisher LW, Young MF, Termine JD, Robey PG. Expression and localization of the two small proteoglycans biglycan and decorin in developing human skeletal and non-skeletal tissues. J Histochem Cytochem (1990) 38:1549–63. doi: 10.1177/38.11.2212616
327. Fisher LW, Termine JD, Young MF. Deduced protein sequence of bone small proteoglycan I (biglycan) shows homology with proteoglycan II (decorin) and several nonconnective tissue proteins in a variety of species. J Biol Chem (1989) 264:4571–6.
330. Romier B, Ivaldi C, Sartelet H, Heinz A, Schmelzer CEH, Garnotel R, et al. Production of Elastin-Derived Peptides Contributes to the Development of Nonalcoholic Steatohepatitis. Diabetes (2018) 67:1604–15. doi: 10.2337/db17-0490
331. Heinz A, Jung MC, Duca L, Sippl W, Taddese S, Ihling C, et al. Degradation of tropoelastin by matrix metalloproteinases - Cleavage site specificities and release of matrikines. FEBS J (2010) 277:1939–56. doi: 10.1111/j.1742-4658.2010.07616.x
332. Heinz A, Jung MC, Jahreis G, Rusciani A, Duca L, Debelle L, et al. The action of neutrophil serine proteases on elastin and its precursor. Biochimie (2012) 94:192–202. doi: 10.1016/j.biochi.2011.10.006
333. Duca L, Floquet N, Alix AJP, Haye B, Debelle L. Elastin as a matrikine. Crit Rev Oncol Hematol (2004) 49:235–44. doi: 10.1016/j.critrevonc.2003.09.007
334. Antonicelli F, Bellon G, Debelle L, Hornebeck W. Elastin-elastases and inflamm-aging. Curr Top Dev Biol (2007) 79:99–155. doi: 10.1016/S0070-2153(06)79005-6
335. Hauck M, Seres I, Kiss I, Saulnier J, Mohacsi A, Wallach J, et al. Effects of synthesized elastin peptides on human leukocytes. Biochem Mol Biol Int (1995) 37:45–55.
336. Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest (2006) 116:753–9. doi: 10.1172/JCI25617
337. Kanta J, Velebny V, Mergancova J, Ettlerova E, Chlumska A. Elastin content in human fibrotic and cirrhotic liver. Sb Ved Pr Lek Fak Karlov Univerzity Hradci Kral (1990) 33:489–94. doi: 10.3389/fphys.2016.00491
338. Liban E, Ungar H. Elastosis in fibrotic and cirrhotic processes of the liver. Arch Pathol (1959) 68:331–41.
339. Kendall TJ, Dolman GE, Duff CM, Paish EC, Zaitoun A, Irving W, et al. Hepatic elastin content is predictive of adverse outcome in advanced fibrotic liver disease. Histopathology (2018) 73:90–100. doi: 10.1111/his.13499
340. Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology (2004) 126:1795–808. doi: 10.1053/j.gastro.2004.03.009
341. Scheuer PJ, Maggi G. Hepatic fibrosis and collapse: histological distinction by orecin staining. Histopathology (1980) 4:487–90. doi: 10.1111/j.1365-2559.1980.tb02943.x
342. Thung SN, Gerber MA. The formation of elastic fibers in livers with massive hepatic necrosis. Arch Pathol Lab Med (1982) 106:468–9.
343. Leeming DJ, Karsdal MA, Byrjalsen I, Bendtsen F, Trebicka J, Nielsen MJ, et al. Novel serological neo-epitope markers of extracellular matrix proteins for the detection of portal hypertension. Aliment Pharmacol Ther (2013) 38:1086–96. doi: 10.1111/apt.12484
344. Afdhal NH, Keaveny AP, Cohen SB, Nunes DP, Maldonado N, O'Brien M, et al. Urinary assays for desmosine and hydroxylysylpyridinoline in the detection of cirrhosis. J Hepatol (1997) 27:993–1002. doi: 10.1016/S0168-8278(97)80142-0
345. Gayral S, Garnotel R, Castaing-Berthou A, Blaise S, Fougerat A, Berge E, et al. Elastin-derived peptides potentiate atherosclerosis through the immune Neu1-PI3Kgamma pathway. Cardiovasc Res (2014) 102:118–27. doi: 10.1093/cvr/cvt336
346. Blaise S, Romier B, Kawecki C, Ghirardi M, Rabenoelina F, Baud S, et al. Elastin-derived peptides are new regulators of insulin resistance development in mice. Diabetes (2013) 62:3807–16. doi: 10.2337/db13-0508
347. Trombetta-Esilva J, Bradshaw AD. The Function of SPARC as a Mediator of Fibrosis. Open Rheumatol J (2012) 6:146–55. doi: 10.2174/1874312901206010146
348. Zhou X, Tan FK, Guo X, Wallis D, Milewicz DM, Xue S, et al. Small interfering RNA inhibition of SPARC attenuates the profibrotic effect of transforming growth factor beta1 in cultured normal human fibroblasts. Arthritis Rheum (2005) 52:257–61. doi: 10.1002/art.20785
349. Atorrasagasti C, Peixoto E, Aquino JB, Kippes N, Malvicini M, Alaniz L, et al. Lack of the matricellular protein SPARC (secreted protein, acidic and rich in cysteine) attenuates liver fibrogenesis in mice. PloS One (2013) 8:e54962. doi: 10.1371/journal.pone.0054962
350. Mazzolini G, Atorrasagasti C, Onorato A, Peixoto E, Schlattjan M, Sowa JP, et al. SPARC expression is associated with hepatic injury in rodents and humans with non-alcoholic fatty liver disease. Sci Rep (2018) 8:725. doi: 10.1038/s41598-017-18981-9
351. Maier S, Paulsson M, Hartmann U. The widely expressed extracellular matrix protein SMOC-2 promotes keratinocyte attachment and migration. Exp Cell Res (2008) 314:2477–87. doi: 10.1016/j.yexcr.2008.05.020
352. Luo L, Wang CC, Song XP, Wang HM, Zhou H, Sun Y, et al. Suppression of SMOC2 reduces bleomycin (BLM)-induced pulmonary fibrosis by inhibition of TGF-beta1/SMADs pathway. BioMed Pharmacother (2018) 105:841–7. doi: 10.1016/j.biopha.2018.03.058
353. Shvab A, Haase G, Ben-Shmuel A, Gavert N, Brabletz T, Dedhar S, et al. Induction of the intestinal stem cell signature gene SMOC-2 is required for L1-mediated colon cancer progression. Oncogene (2016) 35:549–57. doi: 10.1038/onc.2015.127
354. Yuting Y, Lifeng F, Qiwei H. Secreted modular calcium-binding protein 2 promotes high fat diet (HFD)-induced hepatic steatosis through enhancing lipid deposition, fibrosis and inflammation via targeting TGF-beta1. Biochem Biophys Res Commun (2019) 509:48–55. doi: 10.1016/j.bbrc.2018.12.006
355. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi: 10.1016/j.immuni.2011.03.023
356. Ruppert SM, Hawn TR, Arrigoni A, Wight TN, Bollyky PL. Tissue integrity signals communicated by high-molecular weight hyaluronan and the resolution of inflammation. Immunol Res (2014) 58:186–92. doi: 10.1007/s12026-014-8495-2
357. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol (2005) 6:338–44. doi: 10.1038/ni1180
358. Ruffell B, Johnson P. Hyaluronan induces cell death in activated T cells through CD44. J Immunol (2008) 181:7044–54. doi: 10.4049/jimmunol.181.10.7044
359. Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK, et al. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev (2006) 212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x
360. Vieira PL, Christensen JR, Minaee S, O'Neill EJ, Barrat FJ, Boonstra A, et al. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol (2004) 172:5986–93. doi: 10.4049/jimmunol.172.10.5986
361. Heilmann K, Hoffmann U, Witte E, Loddenkemper C, Sina C, Schreiber S, et al. Osteopontin as two-sided mediator of intestinal inflammation. J Cell Mol Med (2009) 13:1162–74. doi: 10.1111/j.1582-4934.2008.00428.x
362. Cantor H, Shinohara ML. Regulation of T-helper-cell lineage development by osteopontin: the inside story. Nat Rev Immunol (2009) 9:137–41. doi: 10.1038/nri2460
363. Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L, et al. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol (2007) 8:74–83. doi: 10.1038/ni1415
364. Kosinska MK, Ludwig TE, Liebisch G, Zhang R, Siebert HC, Wilhelm J, et al. Articular Joint Lubricants during Osteoarthritis and Rheumatoid Arthritis Display Altered Levels and Molecular Species. PloS One (2015) 10:e0125192. doi: 10.1371/journal.pone.0125192
365. Yamaguchi S, Kawakami K, Satoh K, Fukunaga N, Akama K, Matsumoto K-I, et al. Suppression of hepatic dysfunction in tenascin−X−deficient mice fed a high−fat diet. Mol Med Rep (2017) 16:4061–7. doi: 10.3892/mmr.2017.7052
366. Maqbool A, Spary EJ, Manfield IW, Ruhmann M, Zuliani-Alvarez L, Gamboa-Esteves FO, et al. Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. World J Cardiol (2016) 8:340–50. doi: 10.4330/wjc.v8.i5.340
367. Page TH, Charles PJ, Piccinini AM, Nicolaidou V, Taylor PC, Midwood KS, et al. Raised circulating tenascin-C in rheumatoid arthritis. Arthritis Res Ther (2012) 14:R260. doi: 10.1186/ar4105
368. Yamada S, Ichida T, Matsuda Y, Miyazaki Y, Hatano T, Hata K, et al. Tenascin expression in human chronic liver disease and in hepatocellular carcinoma. Liver (1992) 12:10–6. doi: 10.1111/j.1600-0676.1992.tb00548.x
369. Benbow JH, Elam AD, Bossi KL, Massengill DL, Brandon-Warner E, Anderson WE, et al. Analysis of Plasma Tenascin-C in Post-HCV Cirrhosis: A Prospective Study. Dig Dis Sci (2018) 63:653–64. doi: 10.1007/s10620-017-4860-z
370. Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, et al. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat Genet (2002) 30:421–5. doi: 10.1038/ng850
371. Minamitani T, Ikuta T, Saito Y, Takebe G, Sato M, Sawa H, et al. Modulation of collagen fibrillogenesis by tenascin-X and type VI collagen. Exp Cell Res (2004) 298:305–15. doi: 10.1016/j.yexcr.2004.04.030
372. Zweers MC, Van Vlijmen-Willems IM, Van Kuppevelt TH, Mecham RP, Steijlen PM, Bristow J, et al. Deficiency of tenascin-X causes abnormalities in dermal elastic fiber morphology. J Invest Dermatol (2004) 122:885–91. doi: 10.1111/j.0022-202X.2004.22401.x
373. Alcaraz LB, Exposito JY, Chuvin N, Pommier RM, Cluzel C, Martel S, et al. Tenascin-x promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J Cell Biol (2014) 205:409–28. doi: 10.1083/jcb.201308031
374. Elefteriou F, Exposito JY, Garrone R, Lethias C. Cell adhesion to tenascin-X. Mapping of cell adhesion sites and identification of integrin receptors. Eur J Biochem (1999) 263:840–8. doi: 10.1046/j.1432-1327.1999.00563.x
375. Kawashima R, Mochida S, Matsui A, YouLuTu ZY, Ishikawa K, Toshima K, et al. Expression of osteopontin in Kupffer cells and hepatic macrophages and Stellate cells in rat liver after carbon tetrachloride intoxication: a possible factor for macrophage migration into hepatic necrotic areas. Biochem Biophys Res Commun (1999) 256:527–31. doi: 10.1006/bbrc.1999.0372
376. He CY, Liang BB, Fan XY, Cao L, Chen R, Guo YJ, et al. The dual role of osteopontin in acetaminophen hepatotoxicity. Acta Pharmacol Sin (2012) 33:1004–12. doi: 10.1038/aps.2012.47
377. Lazaro R, Wu R, Lee S, Zhu NL, Chen CL, French SW, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology (2015) 61:129–40. doi: 10.1002/hep.27383
378. Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, et al. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin alpha(V)beta(3) engagement and PI3K/pAkt/NFkappaB signaling. Hepatology (2012) 55:594–608. doi: 10.1002/hep.24701
379. Kiefer FW, Zeyda M, Gollinger K, Pfau B, Neuhofer A, Weichhart T, et al. Neutralization of osteopontin inhibits obesity-induced inflammation and insulin resistance. Diabetes (2010) 59:935–46. doi: 10.2337/db09-0404
380. Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology (2011) 53:106–15. doi: 10.1002/hep.23998
381. Xanthou G, Alissafi T, Semitekolou M, Simoes DCM, Economidou E, Gaga M, et al. Osteopontin has a crucial role in allergic airway disease through regulation of dendritic cell subsets. Nat Med (2007) 13:570–8. doi: 10.1038/nm1580
382. Weber GF, Cantor H. Differential roles of osteopontin/Eta-1 in early and late lpr disease. Clin Exp Immunol (2001) 126:578–83. doi: 10.1046/j.1365-2249.2001.01702.x
383. Weber GF, Ashkar S, Glimcher MJ, Cantor H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1). Sci (80 ) (1996) 271:509–12. doi: 10.1126/science.271.5248.509
384. Zhou Y, Yao Y, Sheng L, Zhang J, Zhang JH, Shao A, et al. Osteopontin as a candidate of therapeutic application for the acute brain injury. J Cell Mol Med (2020) 24:8918–29. doi: 10.1111/jcmm.15641
385. Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J Cell Commun Signal (2009) 3:311–22. doi: 10.1007/s12079-009-0068-0
386. Wolak T, Sion-Vardi N, Novack V, Greenberg G, Szendro G, Tarnovscki T, et al. N-terminal rather than full-length osteopontin or its C-terminal fragment is associated with carotid-plaque inflammation in hypertensive patients. Am J Hypertens (2013) 26:326–33. doi: 10.1093/ajh/hps043
387. Iida T, Wagatsuma K, Hirayama D, Nakase H. Is osteopontin a friend or foe of cell apoptosis in inflammatory gastrointestinal and liver diseases? Int J Mol Sci (2018) 19:(2018). doi: 10.3390/ijms19010007
388. Gardner H. Integrin alpha1beta1. Adv Exp Med Biol (2014) 819:21–39. doi: 10.1007/978-94-017-9153-3_2
389. Hamaia SW, Pugh N, Raynal N, Nemoz B, Stone R, Gullberg D, et al. Mapping of potent and specific binding motifs, GLOGEN and GVOGEA, for integrin alpha1beta1 using collagen toolkits II and III. J Biol Chem (2012) 287:26019–28. doi: 10.1074/jbc.M112.353144
390. Nykvist P, Tu H, Ivaska J, Kapyla J, Pihlajaniemi T, Heino J, et al. Distinct recognition of collagen subtypes by alpha(1)beta(1) and alpha(2)beta(1) integrins. Alpha(1)beta(1) mediates cell adhesion to type XIII collagen. J Biol Chem (2000) 275:8255–61. doi: 10.1074/jbc.275.11.8255
391. Zeltz C, Gullberg D. Post-translational modifications of integrin ligands as pathogenic mechanisms in disease. Matrix Biol (2014) 40:5–9. doi: 10.1016/j.matbio.2014.08.001
392. Meyaard L, Hurenkamp J, Clevers H, Lanier LL, Phillips JH. Leukocyte-associated Ig-like receptor-1 functions as an inhibitory receptor on cytotoxic T cells. J Immunol (1999) 162:5800–4. doi: 10.1189/jlb.0907609
393. van der Vuurst de Vries AR, Clevers H, Logtenberg T, Meyaard L. Leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1) is differentially expressed during human B cell differentiation and inhibits B cell receptor-mediated signaling. Eur J Immunol (1999) 29:3160–7. doi: 10.1002/(SICI)1521-4141(199910)29:10<3160::AID-IMMU3160>3.0.CO;2-S
394. Jansen CA, Cruijsen CW, de Ruiter T, Nanlohy N, Willems N, Janssens-Korpela PL, et al. Regulated expression of the inhibitory receptor LAIR-1 on human peripheral T cells during T cell activation and differentiation. Eur J Immunol (2007) 37:914–24. doi: 10.1002/eji.200636678
395. Son M, Santiago-Schwarz F, Al-Abed Y, Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci U.S.A. (2012) 109:E3160–7. doi: 10.1073/pnas.1212753109
396. Poggi A, Tomasello E, Ferrero E, Zocchi MR, Moretta L. p40/LAIR-1 regulates the differentiation of peripheral blood precursors to dendritic cells induced by granulocyte-monocyte colony-stimulating factor. Eur J Immunol (1998) 28:2086–91. doi: 10.1002/(SICI)1521-4141(199807)28:07<2086::AID-IMMU2086>3.0.CO;2-T
397. Bonaccorsi I, Cantoni C, Carrega P, Oliveri D, Lui G, Conte R, et al. The immune inhibitory receptor LAIR-1 is highly expressed by plasmacytoid dendritic cells and acts complementary with NKp44 to control IFNalpha production. PloS One (2010) 5:e15080. doi: 10.1371/journal.pone.0015080
398. Zhang Y, Lv K, Zhang CM, Jin BQ, Zhuang R, Ding Y, et al. The role of LAIR-1 (CD305) in T cells and monocytes/macrophages in patients with rheumatoid arthritis. Cell Immunol (2014) 287:46–52. doi: 10.1016/j.cellimm.2013.12.005
399. Verbrugge A, de Ruiter T, Geest C, Coffer PJ, Meyaard L. Differential expression of leukocyte-associated Ig-like receptor-1 during neutrophil differentiation and activation. J Leukoc Biol (2006) 79:828–36. doi: 10.1189/jlb.0705370
400. Maasho K, Masilamani M, Valas R, Basu S, Coligan JE, Borrego F, et al. The inhibitory leukocyte-associated Ig-like receptor-1 (LAIR-1) is expressed at high levels by human naive T cells and inhibits TCR mediated activation. Mol Immunol (2005) 42:1521–30. doi: 10.1016/j.molimm.2005.01.004
401. Ouyang W, Xue J, Liu J, Jia W, Li Z, Xie X, et al. Establishment of an ELISA system for determining soluble LAIR-1 levels in sera of patients with HFRS and kidney transplant. J Immunol Methods (2004) 292:109–17. doi: 10.1016/j.jim.2004.06.005
402. Iozzo RV, Moscatello DK, McQuillan DJ, Eichstetter I. Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem (1999) 274:4489–92. doi: 10.1074/jbc.274.8.4489
403. Khan GA, Girish GV, Lala N, Di Guglielmo GM, Lala PK. Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol Endocrinol (2011) 25:1431–43. doi: 10.1210/me.2010-0426
404. Jarvelainen H, Puolakkainen P, Pakkanen S, Brown EL, Hook M, Iozzo RV, et al. A role for decorin in cutaneous wound healing and angiogenesis. Wound Repair Regener (2006) 14:443–52. doi: 10.1111/j.1743-6109.2006.00150.x
405. Baghy K, Dezso K, Laszlo V, Fullar A, Peterfia B, Paku S, et al. Ablation of the decorin gene enhances experimental hepatic fibrosis and impairs hepatic healing in mice. Lab Invest (2011) 91:439–51. doi: 10.1038/labinvest.2010.172
406. Border WA, Ruoslahti E. Transforming growth factor-beta in disease: the dark side of tissue repair. J Clin Invest (1992) 90:1–7. doi: 10.1172/JCI115821
407. Meyer DH, Krull N, Dreher KL, Gressner AM. Biglycan and decorin gene expression in normal and fibrotic rat liver: cellular localization and regulatory factors. Hepatology (1992) 16:204–16. doi: 10.1002/hep.1840160131
408. Dudas J, Kovalszky I, Gallai M, Nagy JO, Schaff Z, Knittel T, et al. Expression of decorin, transforming growth factor-beta 1, tissue inhibitor metalloproteinase 1 and 2, and type IV collagenases in chronic hepatitis. Am J Clin Pathol (2001) 115:725–35. doi: 10.1309/J8CD-E9C8-X4NG-GTVG
409. Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem (2001) 276:10229–33. doi: 10.1074/jbc.M100099200
410. Gondokaryono SP, Ushio H, Niyonsaba F, Hara M, Takenaka H, Jayawardana ST, et al. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J Leukoc Biol (2007) 82:657–65. doi: 10.1189/jlb.1206730
411. Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: Synergism between the III-1 and EDA domains. PloS One (2014) 9:e102974. doi: 10.1371/journal.pone.0102974
412. McFadden JP, Basketter DA, Dearman RJ, Kimber IR. Extra domain A-positive fibronectin-positive feedback loops and their association with cutaneous inflammatory disease. Clin Dermatol (2011) 29:257–65. doi: 10.1016/j.clindermatol.2010.11.003
413. Booth AJ, Wood SC, Cornett AM, Dreffs AA, Lu G, Muro AF, et al. Recipient-derived EDA fibronectin promotes cardiac allograft fibrosis. J Pathol (2012) 226:609–18. doi: 10.1002/path.3010
414. Julier Z, Martino MM, de Titta A, Jeanbart L, Hubbell JA. The TLR4 agonist fibronectin extra domain A is cryptic, exposed by elastase-2; use in a fibrin matrix cancer vaccine. Sci Rep (2015) 5:8569. doi: 10.1038/srep08569
415. Khan MM, Gandhi C, Chauhan N, Stevens JW, Motto DG, Lentz SR, et al. Alternatively-spliced extra domain A of fibronectin promotes acute inflammation and brain injury after cerebral ischemia in mice. Stroke (2012) 43:1376–82. doi: 10.1161/STROKEAHA.111.635516
416. Dhanesha N, Ahmad A, Prakash P, Doddapattar P, Lentz SR, Chauhan AK, et al. Genetic Ablation of Extra Domain A of Fibronectin in Hypercholesterolemic Mice Improves Stroke Outcome by Reducing Thrombo-Inflammation. Circulation (2015) 132:2237–47. doi: 10.1161/CIRCULATIONAHA.115.016540
417. Kohan M, Muro AF, Bader R, Berkman N. The extra domain A of fibronectin is essential for allergen-induced airway fibrosis and hyperresponsiveness in mice. J Allergy Clin Immunol (2011) 127:435–9. doi: 10.1016/j.jaci.2010.10.021
418. Mak KM, Mei R. Basement Membrane Type IV Collagen and Laminin: An Overview of Their Biology and Value as Fibrosis Biomarkers of Liver Disease. Anat Rec (2017) 300:1371–90. doi: 10.1002/ar.23567
419. Lieber CS, Weiss DG, Paronetto F, Veterans Affairs Cooperative Study, G. Value of fibrosis markers for staging liver fibrosis in patients with precirrhotic alcoholic liver disease. Alcohol Clin Exp Res (2008) 32:1031–9. doi: 10.1111/j.1530-0277.2008.00664.x
420. Hirayama C, Suzuki H, Takada A, Fujisawa K, Tanikawa K, Igarashi S, et al. Serum type IV collagen in various liver diseases in comparison with serum 7S collagen, laminin, and type III procollagen peptide. J Gastroenterol (1996) 31:242–8. doi: 10.1007/BF02389524
421. Santos VN, Leite-Mor MM, Kondo M, Martins JR, Nader H, Lanzoni VP, et al. Serum laminin, type IV collagen and hyaluronan as fibrosis markers in non-alcoholic fatty liver disease. Braz J Med Biol Res (2005) 38:747–53. doi: 10.1590/S0100-879X2005000500012
422. McCrary MW, Bousalis D, Mobini S, Song YH, Schmidt CE. Decellularized tissues as platforms for in vitro modeling of healthy and diseased tissues. Acta Biomater (2020) 111:1–19. doi: 10.1016/j.actbio.2020.05.031
423. D’Angelo E, Natarajan D, Sensi F, Ajayi O, Fassan M, Mammano E, et al. Patient-Derived Sca ff olds of Colorectal Cancer Metastatic Microenvironment. Cancers (Basel) (2020) 12:1–20. doi: 10.3390/cancers12020364
424. Badylak SF, Taylor D, Uygun K. Whole-Organ Tissue Engineering: Decellularization and Recellularization of Three-Dimensional Matrix Scaffolds. Annu Rev Biomed Eng (2011) 13:27–53. doi: 10.1146/annurev-bioeng-071910-124743
425. Porzionato A, Stocco E, Barbon S, Grandi F, Macchi V, De Caro R, et al. Tissue-engineered grafts from human decellularized extracellular matrices: A systematic review and future perspectives. Int J Mol Sci (2018) 19:(2018). doi: 10.3390/ijms19124117
426. Mazza G, Rombouts K, Rennie Hall A, Urbani L, Vinh Luong T, Al-Akkad W, et al. Decellularized human liver as a natural 3D-scaffold for liver bioengineering and transplantation. Sci Rep (2015) 5:1–15. doi: 10.1038/srep13079
427. Urbani L, Camilli C, Phylactopoulos D, Crowley C, Natarajan D, Scottoni F, et al. Multi-stage bioengineering of a layered oesophagus with in vitro expanded muscle and epithelial adult progenitors. Nat Commun (2018) 9:1–16. doi: 10.1038/s41467-018-06385-w
428. Palma E, Doornebal EJ, Chokshi S. Precision-cut liver slices: a versatile tool to advance liver research. Hepatol Int (2019) 13:51–7. doi: 10.1007/s12072-018-9913-7
429. Palma E, Riva A, Moreno C, Odena G, Mudan S, Manyakin N, et al. Perturbations in Mitochondrial Dynamics Are Closely Involved in the Progression of Alcoholic Liver Disease. Alcohol Clin Exp Res (2020) 44:856–65. doi: 10.1111/acer.14299
430. Sacchi M, Bansal R, Rouwkema J. Bioengineered 3D Models to Recapitulate Tissue Fibrosis. Trends Biotechnol (2020) 38:623–36. doi: 10.1016/j.tibtech.2019.12.010
431. Collin de l’Hortet A, Takeishi K, Guzman-Lepe J, Morita K, Achreja A, Popovic B, et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab (2019) 30:385–401.e9. doi: 10.1016/j.cmet.2019.06.017
432. Wagner A, Röhrs V, Materne EM, Hiller T, Kedzierski R, Fechner H, et al. Use of a three-dimensional humanized liver model for the study of viral gene vectors. J Biotechnol (2015) 212:134–43. doi: 10.1016/j.jbiotec.2015.08.012
Glossary.

Keywords: extracellular matrix, extracellular matrix–damage-associated molecular patterns, fibrosis, chronic liver disease, microenvironment, inflammation, immunomodulation
Citation: McQuitty CE, Williams R, Chokshi S and Urbani L (2020) Immunomodulatory Role of the Extracellular Matrix Within the Liver Disease Microenvironment. Front. Immunol. 11:574276. doi: 10.3389/fimmu.2020.574276
Received: 19 June 2020; Accepted: 14 October 2020;
Published: 11 November 2020.
Edited by:
Walter Gottlieb Land, Université de Strasbourg, FranceReviewed by:
Gertraud Orend, INSERM Immuno Rhumatologie Moléculaire (IRM), FranceLara Campana, Resolution Therapeutics Ltd, United Kingdom
Copyright © 2020 McQuitty, Williams, Chokshi and Urbani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Urbani, bHVjYS51cmJhbmlAcmVzZWFyY2hpbmxpdmVyLm9yZy51aw==