 Florian Tran1,2*
Florian Tran1,2* Christine Klein3
Christine Klein3 Alexander Arlt2,4
Alexander Arlt2,4 Simon Imm1
Simon Imm1 Evelyn Knappe3
Evelyn Knappe3 Alison Simmons5,6
Alison Simmons5,6 Philip Rosenstiel1
Philip Rosenstiel1 Philip Seibler3
Philip Seibler3- 1Institute of Clinical Molecular Biology, University of Kiel, Kiel, Germany
- 2Klinik für Innere Medizin I, Universitätsklinikum Schleswig-Holstein, Kiel, Germany
- 3Institute of Neurogenetics, University of Lübeck, Lübeck, Germany
- 4University Department for Gastroenterology, Klinikum Oldenburg AöR, European Medical School (EMS), Oldenburg, Germany
- 5MRC Human Immunology Unit (MRC), University of Oxford, Oxford, United Kingdom
- 6Translational Gastroenterology Unit, University of Oxford, Oxford, United Kingdom
Individualised cellular models of disease are a key tool for precision medicine to recapitulate chronic inflammatory processes. Organoid models can be derived from induced pluripotent stem cells (iPSCs) or from primary stem cells ex vivo. These models have been emerging over the past decade and have been used to reconstruct the respective organ-specific physiology and pathology, at an unsurpassed depth. In cancer research, patient-derived cancer organoids opened new perspectives in predicting therapy response and provided novel insights into tumour biology. In precision medicine of chronic inflammatory disorders, stem-cell based organoid models are currently being evaluated in pre-clinical pharmacodynamic studies (clinical studies in a dish) and are employed in clinical studies, e.g., by re-transplanting autologous epithelial organoids to re-establish intestinal barrier integrity. A particularly exciting feature of iPSC systems is their ability to provide insights into organ systems and inflammatory disease processes, which cannot be monitored with clinical biopsies, such as immune reactions in neurodegenerative disorders. Refinement of differentiation protocols, and next-generation co-culturing methods, aimed at generating self-organised, complex tissues in vitro, will be the next logical steps. In this mini-review, we critically discuss the current state-of-the-art stem cell and organoid technologies, as well as their future impact, potential and promises in combating immune-mediated chronic diseases.
Stem Cells and Organoids – The Rise of Novel Model Systems
Chronic inflammatory diseases are characterised by either a persisting stimulus for inflammatory signals and/or an inadequate resolution of a response to tissue insults, leading to the development of cancer, neurodegenerative or autoinflammatory disorders. In this complex, disharmonised multi-cellular immune response, the epithelium, immune cells and the microbiome play central roles and thus the development of adequate in vitro models of disease, reflecting the complexity of immune interactions in chronic inflammation, is an urgent need in all biomedical fields. However, tissue biology is very challenging to study in mammals, and progress can be hindered by sample accessibility and ethical concerns in humans. One of the cardinal concepts underlying organoid technology is the idea that stem cells have the intrinsic ability to self-organise into 3D structures that resemble in vivo organs. A major breakthrough was achieved in 2009, when adult tissue-resident stem cells were found to proliferate and self-organise, in vitro, into organoids (1, 2) (Figure 1A). The method has since been adapted to generate murine and human organoids from epithelial tissues of major organs like skin, kidney, liver and intestine (3–6) (Figure 1B), and used for various physiological and disease-related studies, e.g. for complex disorders like inflammatory bowel disease (IBD). Using intestinal organoids, the functional link between the IBD risk gene ATG16L1, interleukin(IL)-22 signalling and STING-dependent type-I-interferon (IFN-I) response was discovered (7). Similarly, the role of the DNA damage repair gene RNASEH2B in intestinal inflammation and tissue regeneration was identified in organoid models (8). Purified intestinal epithelium from inflamed intestinal tissue obtained from IBD patients displayed distinct epigenetic and transcriptional alternations, which were retained in organoid cultures and correlated with disease outcome (9). Inflamed IBD tissue-derived organoids display increased activation of typical molecular hallmarks of IBD such as the ATF6 pathway as a branch of the unfolded protein response (UPR), which were used to identify novel anti-inflammatory ATF6 targeting compounds (10). As organoids can provide experimental manipulability and maintain at the same time biologic complexity, they function as a bridge between conventional 2D cell culture and animal models (2).
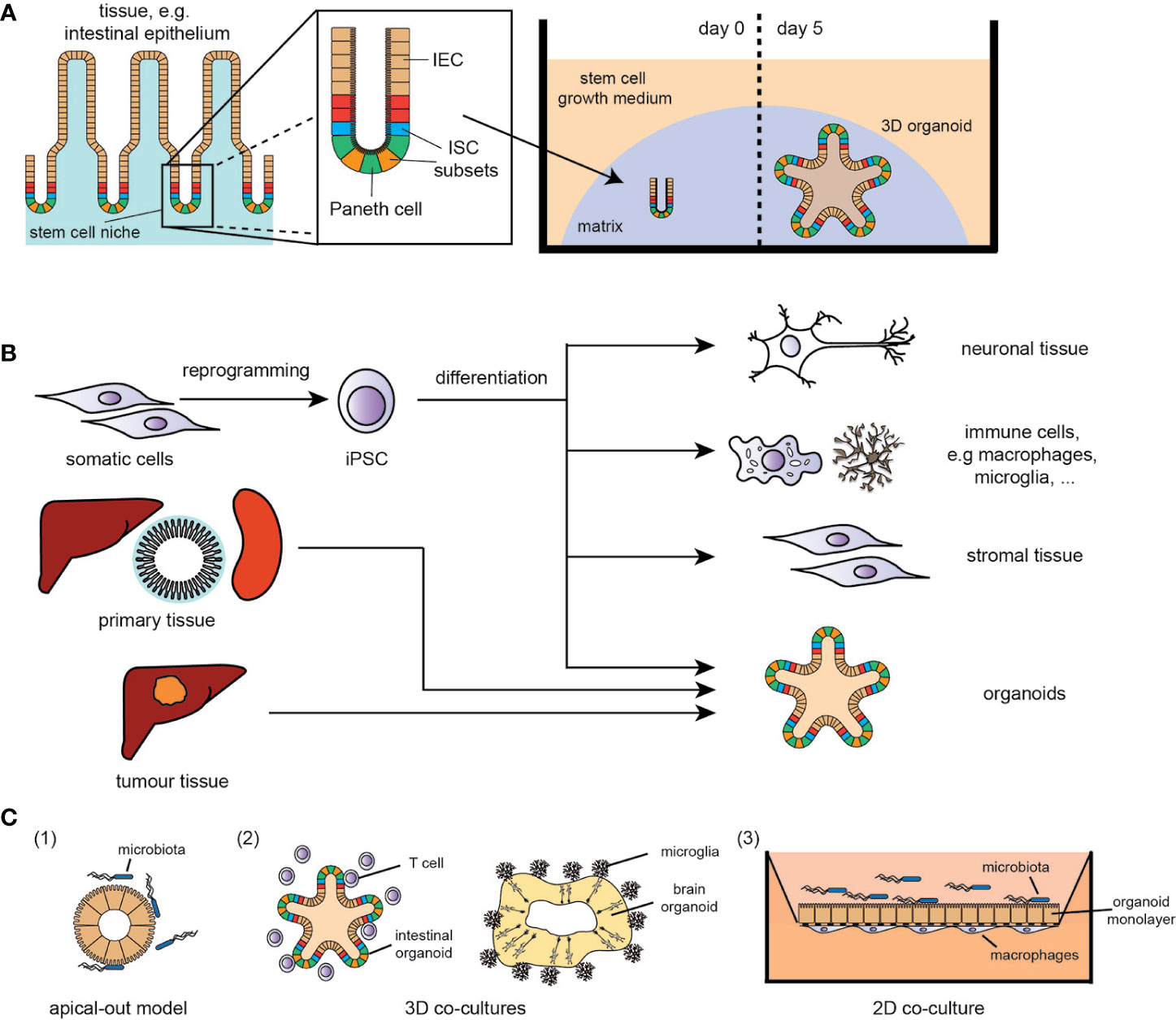
Figure 1 (A) Example of adult tissue-derived organoids (here: small intestine); IEC, intestinal epithelial cells; ISC, intestinal stem cell. (B) Overview of stem cell- and (tumour) tissue-derived in vitro models (selection); iPSC, induced pluripotent stem cells. (C) Overview of co-culture systems (simplified selection): (1) apical-out model of intestinal organoids for host-microbe-interaction studies, (2) 3D co-culture systems of immune cells with organoids, e.g., T cells/intestinal organoids and microglia/brain organoids, (3) 2D co-culture systems with a polarized organoid monolayer with either immune cell (basal interface) or microbe (apical interface).
A further breakthrough was the development of human induced pluripotent stem cell (iPSC)-derived organoid culture. Intestinal organoids were one of the first iPSC-derived tissues consisting of a polarised, columnar epithelium patterned into villus-like structures and crypt-like proliferative zones (11). The iPSC technology has the enormous potential to overcome the limitations of accessibility of specific tissues, such as brain or heart, providing improved patient-derived cellular models of human disease that can also be used for drug screens and personalised treatment strategies (Figure 1B). Cerebral brain organoids displayed various discrete though interdependent brain regions (12). This potential is further strengthened by combining iPSC technology with genome engineering - allowing the correction of mutations in patient-derived iPSCs (13) and the modification of reporter lines, thereby facilitating differentiation towards specific cell types (14).
In the following, we will discuss the technical progress and remaining limitations on using more complex cell culture models to understand inflammatory disorders, host–pathogen interactions or cancer.
Precise Disease Modelling: Co-Cultures and Tissue Engineering to Study Autoimmune Diseases and Infections
A wide range of co-culture and organoid-based disease models that reproduce genetic immune diseases (7, 8, 15) and host–pathogen interactions (16, 17) have already been developed and provide proof of principle that these complex cultures can show certain well-known pathological features. These model systems are the result of tremendous advances and bioengineering innovations of the past decade.
Impaired epithelial host-microbe interaction is one of the important features of IBD and thus, model systems focusing on this interplay are needed (18). Due to the “apical-in/basolateral-out” polarity of conventional organoid systems, microbes need to be microinjected into the lumen of the organoids to mimic physiological host-microbial cross-talks (19–21). This technique however is slow, even with standardised high-throughput approaches technically very challenging and the reproducibility of the results is low due to the heterogeneity of the organoids and their luminal content (22).
To combat this issue of “wrong” epithelial polarity, an “apical-out” system with reverse organoid polarity was developed (22) (Figure 1C), providing a more suitable and reliable model for processes and properties of the epithelium, e.g. examining nutrient absorption and host-pathogen interactions, such as inflammatory cytokines/chemokines, antimicrobial peptides and ROS production.
Another approach to assess host-pathogen interactions is the co-culturing of an epithelial monolayer of preserved apical-basolateral polarity with selected pathogens (Figure 1C). Using such models, the role of K. pneumoniae in priming TH17 response in primary sclerosing cholangitis (15) or the pathophysiology of chronic Helicobacter pylori infections (20, 23) were identified. Recently, microfluidic-based systems, i.e. the human microbial crosstalk (HuMiX) system (24, 25), were established, enabling co-culturing of human intestinal epithelial monolayers with anaerobic pathogens and the analysis of diet-microbiome-human interactions. Besides studies on antimicrobial response, the development of two-dimensional organoid cultures with an air-liquid-interface increased the complexity and maturation of the epithelial layer, providing a better approximation to the in vivo tissue, similar to colonic monolayers (26), and led to the discovery of a novel injury-related cell type, associated with IBD-related tissue regeneration.
The interaction of the epithelium with the stroma, including immune cells, mesenchymal cells and neuronal cells, is vital for organ development and homeostasis and needs to be taken into account when modelling complex immunological diseases like IBD (27). Stromal cells provide the niche for stem cell growth by production of ECM and secretion of essential growth factors, such as WNT proteins (27) and R-spondin (28). Various in vitro models (both 2D and 3D) have been co-cultured with different types of immune cells to assess immune-related orchestration of the epithelial barrier and vice versa the communication back to the specialised immune cells (29). E.g., co-culturing of intestinal organoids and macrophages led to enhanced barrier integrity (30), and the epithelial-macrophage communication enabled a more coordinated immune response to infection.
The enteric nervous system (ENS), controlling several functions, such as motility and permeability, has been linked to enteric neuropathies and gut disorders (31). To that end, a model of co-culturing of intestinal epithelial cells with ENS neurons and glia or subepithelial myofibroblasts was developed (32), showing that the ENS influences intestinal stem cell (ISC) fate by increasing differentiation towards the enteroendocrine lineage and thereby modulates intestinal barrier function. Vice versa, immune cells like tissue resident macrophages upon gastrointestinal infections modulate cellular fate of enteric neurons (33). Going one step further, human iPSC-derived intestinal tissue with a functional ENS was generated by combining intestinal organoids with neural crest cells, in order to investigate the pathophysiology of Hirschsprung’s disease (31). This model could also provide a platform for analysing the epithelium/ENS-axis in IBD.
Vice versa, environmental factors influencing the microbiome (e.g. intestinal infections) are suggested to play a key role in the initiation and progression of neurological disorders, such as Parkinson’s disease (PD) (34–36). PD is characterised by the loss of dopaminergic neurons and intracellular inclusions composed mainly of alpha synuclein, which can be identified in intestinal biopsies years prior to PD diagnosis (37). Notably, it was shown that inflammatory processes, e.g. mitochondrial stress-associated STING/IFN-I response in the absence of PD-linked Parkin or Pink1 function, are drivers of PD pathology (36, 38). Therefore, it is of interest not only to examine disease mechanisms of the gut-brain axis but also direct inflammatory processes in the brain triggered by microglia activation (39). In this regard, brain organoids in co-culture with microglia represent a major breakthrough in neuronal disease modelling techniques (40) and will serve to facilitate the development of more precise human brain models for basic mechanistic studies in neural-glial interactions and drug discovery.
There remains one crucial factor none of these models accounts for, that is blood vessels. Aside from transporting the vital contents of our blood to and from the demanding organs, vascularization is a critical component of physiological (and pathological) development. Recently, human pluripotent stem cells were engineered to induce endothelium development by expressing ETS variant 2, which contributed to forming a complex vascular-like network in human cortical organoids (41). Vascularized organoids displayed enhanced functional maturation and acquired several blood-brain barrier characteristics, including an increase in the expression of tight junctions, nutrient transporters and trans-endothelial electrical resistance. These cultures formed vasculature-like structures that resemble the status in early prenatal brain and present a robust model to study disease in vitro.
The key challenge of the next years will be the establishment of multi-compartment in vitro models for disease and beyond the two-compartment organoid co-culturing systems [e.g. organ/body-on-a-chip, including tissue cultures from multiple organs (42, 43)], to really represent complex, multi-organ immunological disorders.
Patient in a Dish: Excursus on Predicting Therapy Response in Inflammation-Driven Cancer
Inflammation is a driving aspect of oncogenesis and a necessary component of the established tumour’s mechanism of resistance. This aspect of the immune-oncological crosstalk has recently gained attention due to the use of immunotherapy in nearly all type of cancer (44). Previous oversimplified cell culturing methods lack the tumour’s micro-environment, which have only been partially resolved by co-culturing models of tumour cell lines with other somatic cells (45–47) (Figure 2A). Animal models, including genetic tumour models (GEM), murine-derived organoids (MDO) and xenotransplants, display higher degrees of tumour complexity, but are limited by inter-species differences in immunological mechanisms and chemokine signalling, compared to human cancer (48).
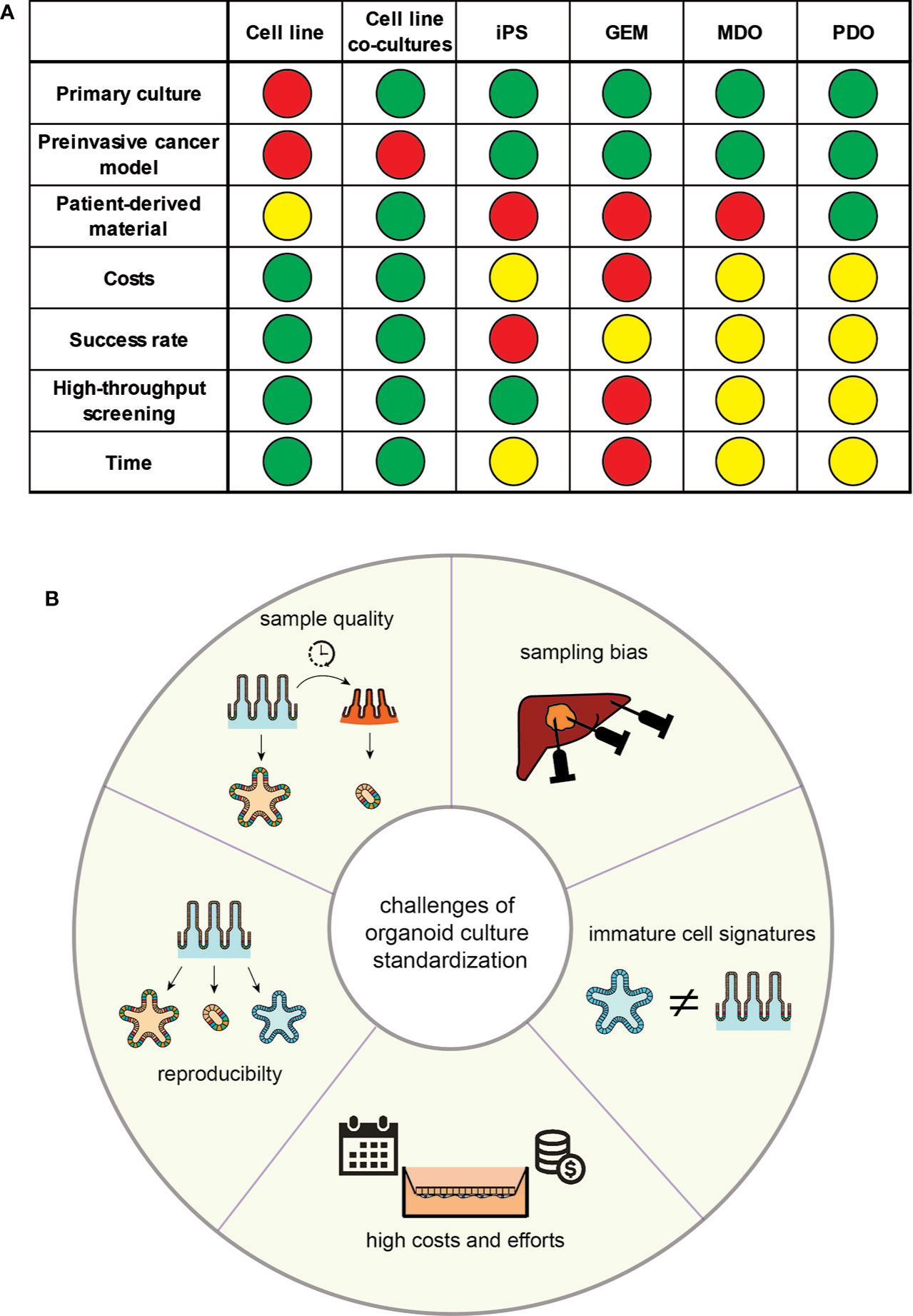
Figure 2 (A) Properties of the discussed models for translational research; GEM, genetically engineered mouse model; MDO, murine-derived organoid; iPS, induced pluripotential stem cell; PDO, patient-derived organoid; definition of colours: Green - possible/cheap (costs)/easy (success rate)/1–2 weeks (time); Yellow: medium (costs)/medium (success rate)/1–2 months (time); orange – 1–6 months (time); red - difficult/expansive (costs)/low (success rate)/over 6 months (time). (B) Overview over the proposed challenges in the standardisation of organoid culturing systems.
The possibility to easily generate primary tissue-derived organoids provides the basis for personalised ex vivo models, particularly in cancer research. Cancer-derived organoids display several traits that are similar to the original tumour and therefore allow the understanding of cancer biology by large-scale tumour bio-banking and high-throughput drug screenings, which gave rise to the discovery of novel anti-tumour compounds (49).
The availability of CRISPR/Cas9 systems enabled genetic engineering on (cancer) organoids with, however, highly variable efficiency of genome editing, depending on the route of CRISPR-Cas9 installation (50). As a proof of concept, sequential introduction of hallmark colorectal cancer mutations into human colonic organoids licenced growth factor independent proliferation of the organoids and mimicked aspects of the classical adenoma-carcinoma sequence (51). In the same study, re-implantation of these transformed organoids in a murine metastasis model demonstrated that, beside canonical cancer mutations, additional molecular lesions as chromosomal instability are necessary for an invasive cancer behaviour. Another example from the same group is the generation and usage of CRISPR/Cas9-guided knock-in of LGR5- and KRT20-reporters into cancer organoids to trace tumour stem cell behaviour in vivo (52).
On an individualised level, patient cancer-derived organoids (PDO) are suitable in the prediction of therapy response of an individual tumour or metastases by high-throughput screenings of therapies (e.g. compound screens, chemotherapy, irradiation) on a small specimen in neo-adjuvant settings (53, 54).
To depict the complexity of tumour microenvironments, including immuno-oncological cross-talks, distribution of drugs and metabolites in the tumour etc., the need for cancer organoid models exceeding the pure epithelial tissue was recently defined (55). The following two strategies are pursued and include the tumour epithelium and tumour-associated cell types such as, immune cells or stroma:
i. The cultivation of organotypic tumour spheroids or tumour cells on air-liquid interfaces is a more holistic approach and mimics the complete tumour, however it requires a more substantial (mostly surgical) specimen, which excludes the use for neo-adjuvant strategies.
ii. Co-culture systems of PDO with separately sampled cells of the microenvironment benefit from pre-established PDO culture protocols. These models have the advantage that specific cellular components of the microenvironment can be addressed, and only small tumour specimens are necessary for PDO generation.
Since the need for these complex PDO models and the strategies to address the microenvironment are rather new, there are some promising reports indicating that establishing complex PDO models harbouring different cellular compartments are feasible and representative for the original tumour (53, 56–59)
The advances of organoid research in both cancer and inflammation (non-malignant) gave rise to potential model systems, which, for instance, could depict the transition from chronic inflammation to inflammatory-driven carcinogenesis.
Standardisation Remains the Major Challenge for In Vitro Inflammation Models
As outlined, enormous advances in the stem cell and organoid field have emerged and their potential in translational research, and even healthcare, is obvious; however, current limitations of organoids remain an important caveat (Figure 2B).
A general limitation of organoid derivation is the high variability of the phenotypes that they can produce. Organoid and stem cell culture systems and their molecular outcome are critically dependent on the quality and properties of the sampled tissue. Variations in the pre-processing phase heavily interfere with immune signatures of these samples. Moreover, inflamed tissue is more likely to undergo cell death, might have different nutrient and culturing needs and thus the recovery rate of vital organoids from inflamed tissue is significantly lower (60). Whilst inflammatory signatures ex vivo are similar to their in vivo origin (9), particularly complex diseases such as IBD are characterised by a diversity of inflammatory flavours. Furthermore, biopsies are reflecting the biology of the sampled region, and therefore stem cells/organoids should be harvested from different regions. When using iPSCs, variations exist in organoids depending on the genetic background of the individual and the culture protocol used by the lab. Isogenic lines generated via gene-editing approaches can limit this variability in regard to genetic differences.
Because organoids from iPSCs are formed through differentiation of a homogeneous population, tissue-specific cell types and their microenvironment must be newly created. This challenges the use of iPSCs and despite significant similarities in structure and function between organoid and adult tissue, organoids often retain immature characteristics, making them more similar to foetal tissue. In an example of iPSC-derived intestinal organoid, this limitation could be resolved by either co-culturing it with T cells providing a more realistic growth niche (61), engineering of more sophisticated 3D scaffolds to improve organoid architecture and thus increase the similarity to the original tissue in vivo (62, 63), or in vivo transplantation, which resulted in mature intestinal epithelium with preserved intestinal stem cell niches, crypt/villus architecture and a laminated human mesenchyme, both supported by mouse vascular ingrowth (64). Apart from immature characteristics, organoids often show a limited lifespan once a certain size is reached. Due to the lack of diffusion, cells are not supplied with sufficient nutrients to support continued development. The use of bioreactors could improve the nutrient supply. Ideally, organoids are engineered towards the induction of endothelium development resulting in vascularization as shown recently (41). Another strategy would be to integrate endothelial cells, or their progenitors, during organoid development, and to include bioprinting methods to design 3D-scaffolds for the endothelial cells (65, 66). However, the original material tissue of iPSC-derived organoids is usually not inflamed, e.g. skin fibroblasts, and have been reprogrammed and cultured for many weeks to months in the absence of an inflammatory milieu. For the study of complex, multifactorial inflammation, iPSC-derived systems are therefore not the first choice in terms of a model. Vice versa, iPSC-derived organoids might be excellent tools to study mainly genetically driven inflammatory disorders, such as monogenic IBD.
Another important challenge is the magnitude of effort, time and expenses spent on organoid cultures. Whilst rather simple protocols are available for conventional organoid cultures at manageable costs, differentiation protocols include either expensive factors or culturing media or require conditioned media, which need to be produced by feeder cell lines. Co-culturing systems often include 2D models, which require much higher culturing efforts and time. Adding to this challenge, the generation of iPSC and their differentiation into specific tissue often require several weeks of intense culturing effort, which increases the risk of adverse events, e.g. contamination or undesired differentiation.
To combat the aforementioned issues, close collaboration of academic research and high-tech industrial partners may be a promising strategy to overcome the infrastructural challenges. Whilst academia can provide problem-derived ideas and a hypothesis-driven vision of novel disease models, industry can deliver the necessary technology, capacities for large-scale production and standardisation. Setting up these collaborative infrastructures are necessary and can foster future advances of stem cell technology.
Conclusions and Future Perspectives
Organoids have been shown to keep key multicellular, anatomical and to some degree even functional hallmarks of real organs. In order to exploit these improved disease models, the application of high-throughput analysis techniques and large-scale perturbation tools to organoids is required. Single-cell multi-omics (19, 20) and imaging technologies can provide insight into underlying pathological mechanisms at different regulatory molecular layers. Physiological interactions of different organs could be modelled by using microfluidics and organ-on-a-chip technologies to study key systemic interactions in diseases. The combination of advanced organoid models and these techniques might even enable to understand the early molecular mechanisms that cause cells to deviate from a healthy to a disease trajectory (67). This might lead to the detection of biomarkers for the prodromal disease state and the identification of new drug targets to intercept diseases before manifestation of symptoms.
One of the visions of stem cell research is the development of stem-cell-derived regenerative tissue for engraftment and transplantation purposes. Recently, a group successfully engrafted human iPSC-derived kidney organoids into immunodeficient NOD/SCID mice, which reached a higher degree of maturation compared to in vitro kidney organoids but still markedly immature compared to the neighbouring mouse kidney tissue (68). Besides tissue repair, this technology can be a promising vehicle for targeted gene therapy, especially for monogenic disorders. The advances in CRISPR/Cas9 technology already allow selective genome editing with a reduced number of off-target effects by using engineered secondary RNA structures (69) or by implementing prime editing guide RNA that specifies the target site and encodes the desired edit (70). However, only complete and definite exclusion of any off-target DNA alterations are acceptable for transplantation purposes.
It will be of great importance to implement organoids as a platform for screening and testing personalised medicine treatments, as they are cultures of primary patient material. Whilst this is already possible for cancer (54), it will in the future also be possible for inflammatory disorders that affect epithelial tissues. We expect implementation of these systems in drug discovery, therapy guidance and tissue regenerative medicine. Therefore, joint efforts of academia and industrial partners are mandatory to surpass challenges in regard to safety and reliability.
Author Contributions
All authors contributed to the discussion at the symposium workshop. The manuscript was conceptualised by FT, CK, AA, SI, PR, and PS, whilst FT coordinated the manuscript writing. The figures and tables were designed by FT, AA, and PS. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the German Research Foundation (PR, PS, and CK: ExC 2167 “Precision Medicine in Inflammation”, AA: project no. 414216991, PS and CK: FOR 2488, PR: CRC1182) and the EU Innovative Medicine Initiative 2 Joint Undertaking (PR: “3TR”, grant agreement no. 831434).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with several of the authors, CK, EK, and PS.
Acknowledgments
We highly appreciate all discussing participants of the workshops at the International Symposium of the Excellence Cluster “Precision Medicine in Inflammation”.
References
1. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature (2009) 459:262–5. doi: 10.1038/nature07935
2. Li M, Izpisua Belmonte JC. Organoids - Preclinical Models of Human Disease. N Engl J Med (2019) 380:569–79. doi: 10.1056/NEJMra1806175
3. Lee J, Bscke R, Tang PC, Hartman BH, Heller S, Koehler KR. Hair Follicle Development in Mouse Pluripotent Stem Cell-Derived Skin Organoids. Cell Rep (2018) 22:242–54. doi: 10.1016/j.celrep.2017.12.007
4. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature (2013) 494:247–50. doi: 10.1038/nature11826
5. Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature (2015) 526:564–8. doi: 10.1038/nature15695
6. Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology (2011) 141:1762–72. doi: 10.1053/j.gastro.2011.07.050
7. Aden K, Tran F, Ito G, Sheibani-Tezerji R, Lipinski S, Kuiper JW, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med (2018) 215(11):2868–86. doi: 10.1084/jem.20171029
8. Aden K, Bartsch K, Dahl J, Reijns MAM, Esser D, Sheibani-Tezerji R, et al. Epithelial RNase H2 Maintains Genome Integrity and Prevents Intestinal Tumorigenesis in Mice. Gastroenterology (2019) 156:145–59 e19. doi: 10.1053/j.gastro.2018.09.047
9. Howell KJ, Kraiczy J, Nayak KM, Gasparetto M, Ross A, Lee C, et al. DNA Methylation and Transcription Patterns in Intestinal Epithelial Cells From Pediatric Patients With Inflammatory Bowel Diseases Differentiate Disease Subtypes and Associate With Outcome. Gastroenterology (2018) 154:585–98. doi: 10.1053/j.gastro.2017.10.007
10. Stengel ST, Fazio A, Lipinski S, Jahn MT, Aden K, Ito G, et al. Activating Transcription Factor 6 Mediates Inflammatory Signals in Intestinal Epithelial Cells Upon Endoplasmic Reticulum Stress. Gastroenterology (2020) 159(4):1357–74.e10. doi: 10.1053/j.gastro.2020.06.088
11. Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature (2011) 470:105–9. doi: 10.1038/nature09691
12. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature (2013) 501:373–9. doi: 10.1038/nature12517
13. Sanjurjo-Soriano C, Erkilic N, Baux D, Mamaeva D, Hamel CP, Meunier I, et al. Genome Editing in Patient iPSCs Corrects the Most Prevalent USH2A Mutations and Reveals Intriguing Mutant mRNA Expression Profiles. Mol Ther Methods Clin Dev (2020) 17:156–73. doi: 10.1016/j.omtm.2019.11.016
14. Mithal A, Capilla A, Heinze D, Berical A, Villacorta-Martin C, Vedaie M, et al. Generation of mesenchyme free intestinal organoids from human induced pluripotent stem cells. Nat Commun (2020) 11:215. doi: 10.1038/s41467-019-13916-6
15. Nakamoto N, Sasaki N, Aoki R, Miyamoto K, Suda W, Teratani T, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol (2019) 4:492–503. doi: 10.1038/s41564-018-0333-1
16. Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Bottinger L, Klay D, et al. Long-term expanding human airway organoids for disease modeling. EMBO J (2019) 38:1–20. doi: 10.15252/embj.2018100300
17. Holokai L, Chakrabarti J, Broda T, Chang J, Hawkins JA, Sundaram N, et al. Increased Programmed Death-Ligand 1 is an Early Epithelial Cell Response to Helicobacter pylori Infection. PloS Pathog (2019) 15:e1007468. doi: 10.1371/journal.ppat.1007468
18. Caruso R, Lo BC, Nunez G. Host-microbiota interactions in inflammatory bowel disease. Nat Rev Immunol (2020) 20:411–26. doi: 10.1038/s41577-019-0268-7
19. Williamson IA, Arnold JW, Samsa LA, Gaynor L, DiSalvo M, Cocchiaro JL, et al. A High-Throughput Organoid Microinjection Platform to Study Gastrointestinal Microbiota and Luminal Physiology. Cell Mol Gastroenterol Hepatol (2018) 6:301–19. doi: 10.1016/j.jcmgh.2018.05.004
20. Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology (2015) 148:126–36 e6. doi: 10.1053/j.gastro.2014.09.042
21. Min S, Kim S, Cho SW. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp Mol Med (2020) 52:227–37. doi: 10.1038/s12276-020-0386-0
22. Co JY, Margalef-Catala M, Li X, Mah AT, Kuo CJ, Monack DM, et al. Controlling Epithelial Polarity: A Human Enteroid Model for Host-Pathogen Interactions. Cell Rep (2019) 26:2509–20 e4. doi: 10.1016/j.celrep.2019.01.108
23. Boccellato F, Woelffling S, Imai-Matsushima A, Sanchez G, Goosmann C, Schmid M, et al. Polarised epithelial monolayers of the gastric mucosa reveal insights into mucosal homeostasis and defence against infection. Gut (2018) 68(3):400–13. doi: 10.1136/gutjnl-2017-314540
24. Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, et al. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat Commun (2016) 7:11535. doi: 10.1038/ncomms11535
25. Greenhalgh K, Ramiro-Garcia J, Heinken A, Ullmann P, Bintener T, Pacheco MP, et al. Integrated In Vitro and In Silico Modeling Delineates the Molecular Effects of a Synbiotic Regimen on Colorectal-Cancer-Derived Cells. Cell Rep (2019) 27:1621–1632 e9. doi: 10.1016/j.celrep.2019.04.001
26. Wang Y, Chiang IL, Ohara TE, Fujii S, Cheng J, Muegge BD, et al. Long-Term Culture Captures Injury-Repair Cycles of Colonic Stem Cells. Cell (2019) 179:1144–59 e15. doi: 10.1016/j.cell.2019.10.015
27. Kabiri Z, Greicius G, Madan B, Biechele S, Zhong Z, Zaribafzadeh H, et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development (2014) 141:2206–15. doi: 10.1242/dev.104976
28. Sigal M, Logan CY, Kapalczynska M, Mollenkopf HJ, Berger H, Wiedenmann B, et al. Stromal R-spondin orchestrates gastric epithelial stem cells and gland homeostasis. Nature (2017) 548:451–5. doi: 10.1038/nature23642
29. Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell (2018) 175:1307–20 e22. doi: 10.1016/j.cell.2018.10.008
30. Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep (2017) 7:45270. doi: 10.1038/srep46790
31. Workman MJ, Mahe MM, Trisno S, Poling HM, Watson CL, Sundaram N, et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat Med (2017) 23:49–59. doi: 10.1038/nm.4233
32. Puzan M, Hosic S, Ghio C, Koppes A. Enteric Nervous System Regulation of Intestinal Stem Cell Differentiation and Epithelial Monolayer Function. Sci Rep (2018) 8:6313. doi: 10.1038/s41598-018-24768-3
33. Matheis F, Muller PA, Graves CL, Gabanyi I, Kerner ZJ, Costa-Borges D, et al. Adrenergic Signaling in Muscularis Macrophages Limits Infection-Induced Neuronal Loss. Cell (2020) 180:64–78 e16. doi: 10.1016/j.cell.2019.12.002
34. Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature (2019) 571:565–9. doi: 10.1038/s41586-019-1405-y
35. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell (2016) 167:1469–80 e12. doi: 10.1016/j.cell.2016.11.018
36. Haikal C, Chen QQ, Li JY. Microbiome changes: an indicator of Parkinson’s disease? Transl Neurodegener (2019) 8:38. doi: 10.1186/s40035-019-0175-7
37. Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evid 3 cases Mov Disord (2012) 27:716–9. doi: 10.1002/mds.25020
38. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
39. Lecours C, Bordeleau M, Cantin L, Parent M, Paolo TD, Tremblay ME. Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions? Front Cell Neurosci (2018) 12:282. doi: 10.3389/fncel.2018.00282
40. Park J, Wetzel I, Marriott I, Dreau D, D’Avanzo C, Kim DY, et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat Neurosci (2018) 21:941–51. doi: 10.1038/s41593-018-0175-4
41. Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang YJ, et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods (2019) 16:1169–75. doi: 10.1038/s41592-019-0586-5
42. Coppeta JR, Mescher MJ, Isenberg BC, Spencer AJ, Kim ES, Lever AR, et al. A portable and reconfigurable multi-organ platform for drug development with onboard microfluidic flow control. Lab Chip (2016) 17:134–44. doi: 10.1039/C6LC01236A
43. Edington CD, Chen WLK, Geishecker E, Kassis T, Soenksen LR, Bhushan BM, et al. Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies. Sci Rep (2018) 8:4530. doi: 10.1038/s41598-018-22749-0
44. Christofi T, Baritaki S, Falzone L, Libra M, Zaravinos A. Current Perspectives in Cancer Immunotherapy. Cancers (Basel) (2019) 11(10):1472. doi: 10.3390/cancers11101472
45. Geismann C, Schafer H, Gundlach JP, Hauser C, Egberts JH, Schneider G, et al. NF-kappaB Dependent Chemokine Signaling in Pancreatic Cancer. Cancers (Basel) (2019) 11(10):1445. doi: 10.3390/cancers11101445
46. Geismann C, Erhart W, Grohmann F, Schreiber S, Schneider G, Schafer H, et al. TRAIL/NF-kappaB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines. Int J Mol Sci (2018) 19(6):1661. doi: 10.3390/ijms19061661
47. Geismann C, Grohmann F, Dreher A, Hasler R, Rosenstiel P, Legler K, et al. Role of CCL20 mediated immune cell recruitment in NF-kappaB mediated TRAIL resistance of pancreatic cancer. Biochim Biophys Acta Mol Cell Res (2017) 1864:782–96. doi: 10.1016/j.bbamcr.2017.02.005
48. Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol (2006) 7:243. doi: 10.1186/gb-2006-7-12-243
49. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell (2015) 161:933–45. doi: 10.1016/j.cell.2015.03.053
50. Fujii M, Clevers H, Sato T. Modeling Human Digestive Diseases With CRISPR-Cas9-Modified Organoids. Gastroenterology (2019) 156:562–76. doi: 10.1053/j.gastro.2018.11.048
51. Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med (2015) 21:256–62. doi: 10.1038/nm.3802
52. Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature (2017) 545:187–92. doi: 10.1038/nature22081
53. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science (2019) 364:952–5. doi: 10.1126/science.aaw6985
54. Yao Y, Xu X, Yang L, Zhu J, Wan J, Shen L, et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell (2020) 26:17–26 e6. doi: 10.1016/j.stem.2019.10.010
55. Bar-Ephraim YE, Kretzschmar K, Clevers H. Organoids in immunological research. Nat Rev Immunol (2019) 20:279–93. doi: 10.1038/s41577-019-0248-y
56. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell (2018) 175:1972–1988 e16. doi: 10.1016/j.cell.2018.11.021
57. Zhang Z, Karthaus WR, Lee YS, Gao VR, Wu C, Russo JW, et al. Tumor Microenvironment-Derived NRG1 Promotes Antiandrogen Resistance in Prostate Cancer. Cancer Cell (2020) 38:279–296 e9. doi: 10.1016/j.ccell.2020.06.005
59. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discovery (2019) 9:282–301. doi: 10.1158/2159-8290.CD-18-0710
60. Konnikova L, Boschetti G, Rahman A, Mitsialis V, Lord J, Richmond C, et al. High-dimensional immune phenotyping and transcriptional analyses reveal robust recovery of viable human immune and epithelial cells from frozen gastrointestinal tissue. Mucosal Immunol (2018) 11:1684–93. doi: 10.1038/s41385-018-0047-y
61. Jung KB, Lee H, Son YS, Lee MO, Kim YD, Oh SJ, et al. Interleukin-2 induces the in vitro maturation of human pluripotent stem cell-derived intestinal organoids. Nat Commun (2018) 9:3039. doi: 10.1038/s41467-018-05450-8
62. Wang Y, Kim R, Gunasekara DB, Reed MI, DiSalvo M, Nguyen DL, et al. Formation of Human Colonic Crypt Array by Application of Chemical Gradients Across a Shaped Epithelial Monolayer. Cell Mol Gastroenterol Hepatol (2018) 5:113–30. doi: 10.1016/j.jcmgh.2017.10.007
63. Nikolaev M, Mitrofanova O, Broguiere N, Geraldo S, Dutta D, Tabata Y, et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature (2020) 585:574–8. doi: 10.1038/s41586-020-2724-8
64. Watson CL, Mahe MM, Munera J, Howell JC, Sundaram N, Poling HM, et al. An in vivo model of human small intestine using pluripotent stem cells. Nat Med (2014) 20:1310–4. doi: 10.1038/nm.3737
65. Zhang YS, Arneri A, Bersini S, Shin SR, Zhu K, Goli-Malekabadi Z, et al. Bioprinting 3D microfibrous scaffolds for engineering endothelialized myocardium and heart-on-a-chip. Biomaterials (2016) 110:45–59. doi: 10.1016/j.biomaterials.2016.09.003
66. Zhu W, Qu X, Zhu J, Ma X, Patel S, Liu J, et al. Direct 3D bioprinting of prevascularized tissue constructs with complex microarchitecture. Biomaterials (2017) 124:106–15. doi: 10.1016/j.biomaterials.2017.01.042
67. Rajewsky N, Almouzni G, Gorski SA, Aerts S, Amit I, Bertero MG, et al. LifeTime and improving European healthcare through cell-based interceptive medicine. Nature (2020) 587:377–86. doi: 10.1038/s41586-020-2715-9
68. Nam SA, Seo E, Kim JW, Kim HW, Kim HL, Kim K, et al. Graft immaturity and safety concerns in transplanted human kidney organoids. Exp Mol Med (2019) 51:1–13. doi: 10.1038/s12276-019-0336-x
69. Kocak DD, Josephs EA, Bhandarkar V, Adkar SS, Kwon JB, Gersbach CA. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat Biotechnol (2019) 37:657–66. doi: 10.1038/s41587-019-0095-1
Keywords: stem cell, cancer, precision medicine, co-culture, host-microbe, immune-epithelial interactions, induced pluripotent stem cells, patient derived organoids
Citation: Tran F, Klein C, Arlt A, Imm S, Knappe E, Simmons A, Rosenstiel P and Seibler P (2020) Stem Cells and Organoid Technology in Precision Medicine in Inflammation: Are We There Yet? Front. Immunol. 11:573562. doi: 10.3389/fimmu.2020.573562
Received: 17 June 2020; Accepted: 19 November 2020;
Published: 21 December 2020.
Edited by:
Gabriela Riemekasten, University of Lübeck, GermanyReviewed by:
Ranran Zhang, The State University of New Jersey, United StatesGilda Cobellis, University of Campania Luigi Vanvitelli, Italy
Copyright © 2020 Tran, Klein, Arlt, Imm, Knappe, Simmons, Rosenstiel and Seibler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florian Tran, Zi50cmFuQGlrbWIudW5pLWtpZWwuZGU=