 Martin A. Geiger
Martin A. Geiger Ana T. Guillaumon
Ana T. Guillaumon Francesco Paneni
Francesco Paneni Christian M. Matter
Christian M. Matter Sokrates Stein
Sokrates Stein
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 08 October 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.569358
This article is part of the Research TopicQuo Vadis Lipid Mediators – Lipid Mediators Implication in Inflammation and Chronic Inflammatory DiseasesView all 11 articles
Atherosclerotic cardiovascular disease is part of chronic immunometabolic disorders such as type 2 diabetes and nonalcoholic fatty liver disease. Their common risk factors comprise hypertension, insulin resistance, visceral obesity, and dyslipidemias, such as hypercholesterolemia and hypertriglyceridemia, which are part of the metabolic syndrome. Immunometabolic diseases include chronic pathologies that are affected by both metabolic and inflammatory triggers and mediators. Important and challenging questions in this context are to reveal how metabolic triggers and their downstream signaling affect inflammatory processes and vice-versa. Along these lines, specific nuclear receptors sense changes in lipid metabolism and in turn induce downstream inflammatory and metabolic processes. The transcriptional activity of these nuclear receptors is regulated by the nuclear receptor corepressors (NCORs), including NCOR1. In this review we describe the function of NCOR1 as a central immunometabolic regulator and focus on its role in atherosclerosis and associated immunometabolic diseases.
Atherosclerosis is characterized by the accumulation of immune cells, cholesterol species and other lipids in the intimal space of arteries. The disease primarily affects large, elastic, and high-pressure vessels, such as the coronary, renal, femoral, and carotid arteries. The complex pathophysiology is triggered by genetic and environmental risk factors, including lipid species and metabolites (e.g., cholesterol, specific fatty acids, carnitine), as well as by hypertension, diabetes and obesity (1–3). Molecular, genetic, dietary and pharmacological studies over the last decades suggest that hyperlipidemia, especially hypercholesterolemia, combined with the genetic predisposition is a major trigger of atherogenesis (4–9). Nevertheless, recent research showed that other metabolic and inflammatory processes are closely interconnected at the cellular level as well as via intra- and inter-organ communication (10–12) (Figure 1).
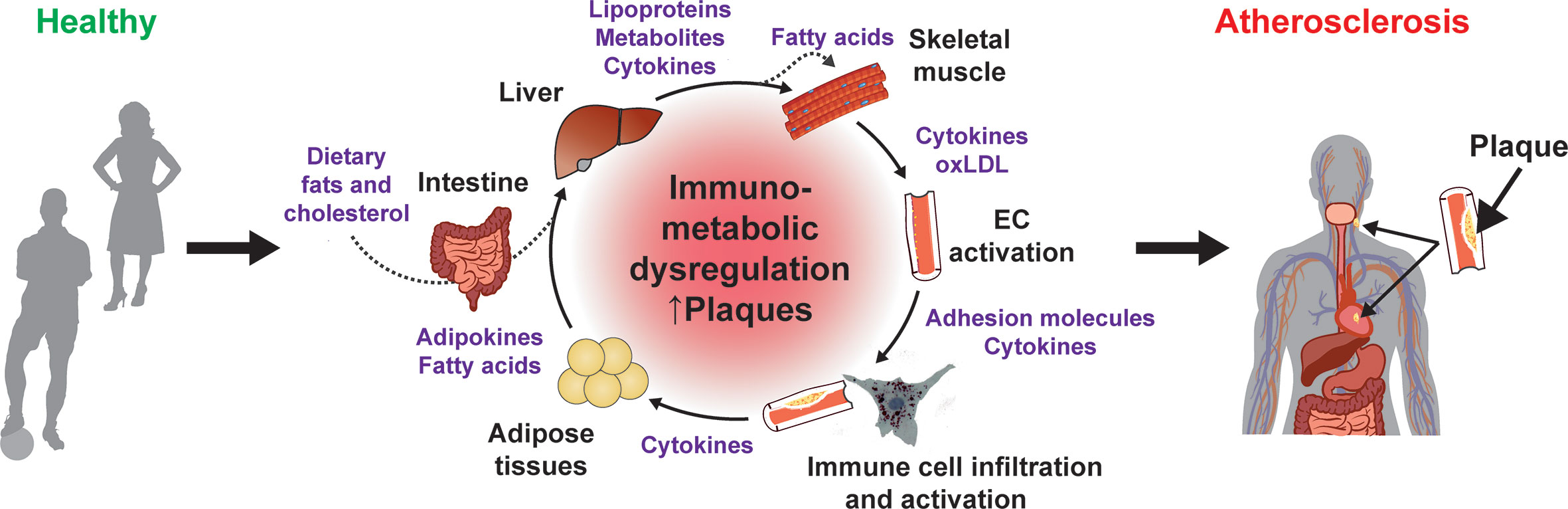
Figure 1 Immunometabolic dysregulation promotes atherogenesis. Contribution of different organs and immunometabolic mediators to the chronic dysregulation that promote atherogenesis.
The signaling cascades that are activated by inflammatory and metabolic triggers and/or mediators converge at key transcriptional regulators, which in turn coordinate the expression of specific target genes and atherogenic processes. Whether individual target genes are activated or repressed depends on several other factors, such as folding and compaction of the chromatin, posttranslational modifications of histones by chromatin-modifying enzymes, functional alterations by noncoding RNAs, and recruitment of the transcriptional machinery, including transcription factors and importantly also transcription cofactors, i.e. transcriptional corepressors and coactivators (13–16).
Nuclear receptors are a large family of druggable transcription factors that exert important functions in development, metabolism, and immune response (17, 18). Consequently, several nuclear receptors do affect the development of atherosclerosis and associated cardiometabolic diseases (reviewed in (19)). While the role of lipid-binding nuclear receptor in metabolism is well established, studies over the last decade demonstrated that several of these nuclear receptors, including PPARγ, LXRs, and LRH-1, mediate transrepression of pro-inflammatory molecules in the liver and/or immune cells, such as macrophages and T cells (20–25). Therefore, these nuclear receptors act as direct immunometabolic regulators and play an important role in atherogenesis.
Given their central role to integrate upstream information and regulate the expression of downstream target genes, specific transcriptional cofactors function as central immunometabolic regulators. About 300 transcription cofactors are known to exist in mice and human cells (26). However, only a fraction of those is expressed in a specific cell type or tissue, and their function is restricted to certain pathways and transcription factors (14, 15). Some of these factors are involved in inflammatory mechanisms, others known to exert metabolic functions.
Could research on (anti-)atherogenic corepressor complexes provide answers to the key question of causality between metabolic and inflammatory changes? One large nuclear receptor corepressor complex that has been studied extensively in vitro and in vivo in the last years is nuclear receptor corepressor 1 (NCOR1) (Figure 2).
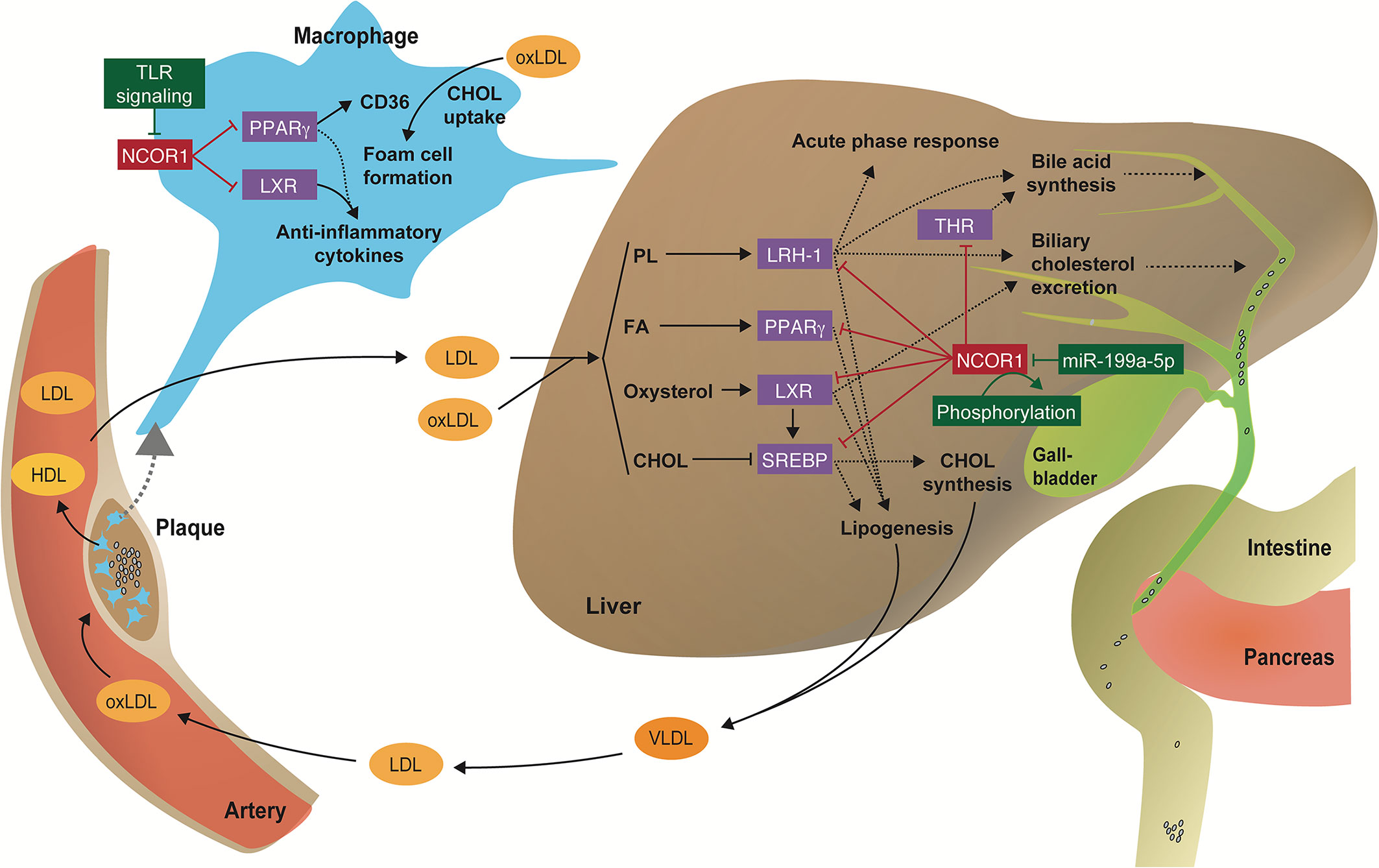
Figure 2 Regulation of immunometabolic processes by the NCOR1 complex. Scheme illustrating how transcriptional corepressor complexes repress the function of nuclear receptors and thus regulate key inflammatory and metabolic processes in the liver and macrophages that are involved in the pathophysiology of cardiometabolic diseases. CHOL, cholesterol; FA, fatty acid; oxLDL, oxidized LDL; PL, phospholipid.
NCOR1 serves as scaffolding protein that forms the basis for a large corepressor complex, including protein deacetylases [including class I (HDAC3), class II (HDAC4, 5, 7, and 9) and class III (SIRT1) KDACs], transducin beta-like 1 (TBL1) and TBLR1, two highly related F box/WD40-containing factors, and the G-protein-pathway suppressor 2 (GPS2) (27, 28). The molecular functions and (patho-)physiological role of HDAC3, class II HDACs, GPS2, and TBL1 has been extensively reviewed (29–31). Although germline Ncor1-/- and Ncor2-/- mice are embryonically lethal (32, 33), they enabled to establish important roles for NCOR1 in erythropoiesis, T-cell, and central nervous system development, whereas NCOR2 contributes to the morphological development of the heart (32–34). Targeted deletions of NCOR1 in immune cells, liver, adipose tissue, and muscle demonstrated that it affects pro- and anti-inflammatory gene signatures, mitochondrial function, lipid metabolism, insulin sensitivity, intestinal cholesterol absorption, thereby highlighting its immunometabolic functions, which will be discussed below (21, 35–41). To the best of our knowledge there are no studies addressing the tissue-specific deletion of Ncor2, which is essential to study the cell-specific function of a gene, especially for genes that are expressed in nearly all cells and organs and thus regulate multiple processes (e.g., a detailed expression pattern of Ncor1 and Ncor2 in mice can be found at Tabula muris (https://tabula-muris.ds.czbiohub.org). Therefore, this review will focus on tissue-specific functions of NCOR1.
Monocyte-derived macrophages are the most abundant immune cell population found in atherosclerotic lesions and plaques. They take up oxidized LDL (oxLDL) in an uncontrolled fashion, secrete pro-inflammatory cytokines that recruit T cells, and fall apart in a necrotic fashion, thus promoting plaque growth and destabilization (42, 43). Silencing of macrophage NCOR1 in vitro produces the same phenotype of activated macrophages: increased expression pro-inflammatory cytokines, chemokines and metalloproteases, and enhanced macrophage invasiveness (21). Mechanistically, it was proposed that the NCOR1 corepressor complex acts as a transcriptional checkpoint for these inflammatory genes: In the absence of a nuclear receptor ligand, NCOR1 is recruited to target genes and interacts with unphosphorylated c-Jun bound to target promoters, thus repressing pro-inflammatory gene expression. Upon stimulation with an innate inflammatory stimulus, such as LPS, a structural change in the ligand-binding domain of the nuclear receptor reduces its affinity to NCOR1, hence simultaneously increasing the affinity to coactivators (44). TBLR1 plays a crucial function as an E3-ligase that directs the ligand-dependent ubiquitination and proteasome-mediated clearance of NCOR1 and histone deacetylase 3 (HDAC3) from the promoter of target genes (45). Moreover, pro-atherosclerotic minimally oxidized LDL (oxLDL) promotes JNK-dependent derepression of AP-1 by releasing NCOR1 from the promoter of chemokine genes (40). Taken together, these in vitro studies suggest that absence of NCOR1 exacerbates the inflammatory response in macrophages.
Paradoxically, the in vivo myeloid cell-specific deletion of Ncor1 repressed inflammatory gene expression and improved insulin sensitivity in a diet-induced obesity mouse model (39). Li et al. suggest that deletion of Ncor1 in adipose tissue macrophages leads to a derepression of LXR target genes, increased expression of de novo lipogenesis and fatty acid desaturation genes, and subsequent production of anti-inflammatory ω3 fatty acids, which in turn suppress inflammatory activation of macrophages (39) (Figure 2).
Using a similar loss-of-function approach, Oppi et al. recently showed that the myeloid cell-specific deletion of Ncor1 increased atherosclerosis at the aortic sinus and the thoraco-abdominal aorta in Ldlr knockout mice (41). At the molecular level NCOR1 was found to bind to the Cd36 promoter thus repressing PPARγ-driven Cd36 expression. Consequently, peritoneal macrophages from Ncor1-deficient mice had increased CD36-mediated oxLDL accumulation and foam cell formation (Figure 2). Interestingly, Ncor1-deficient macrophages displayed an increased expression of both anti- and pro-inflammatory genes. Moreover, analyses of omics-datasets obtained from human plaque specimens suggested that NCOR1-driven PPARγ suppression is also protective in human plaque development and vulnerability (41).
The data from Oppi et al. and Li et al. are partially controversial (39, 41). Two major reasons could explain the differences. First, the genetic background of the mice: Myeloid cell-specific Ncor1 knockout mice on a ‘wildtype’ versus Ldlr-/- background. The Ldlr deficiency shifts the lipoprotein metabolism towards a humanized profile with very high LDL-cholesterol levels, which makes this mouse model prone to develop atherosclerosis and display a pro-inflammatory signature (9, 46). Second, the use of the experimental diets: High-fat diet to study the impact of NCOR1 on diet-induced obesity versus high-cholesterol diet to assess atherogenesis. It is known from numerous studies that diets differently affect major immune and metabolic processes and hence the development of chronic immunometabolic diseases, especially when investigating lipid-sensitive nuclear receptor signaling such as LRH-1 (47–51).
The phenotype of myeloid cell-specific Hdac3 knockout mice is partially resembling the data from Oppi et al. (41, 52). Hoeksema et al. showed that Hdac3-deficient mice develop more atherosclerotic plaques in the aortic sinus compared to control mice (52). Furthermore, Hdac3-deficient macrophages displayed an anti-inflammatory wound-healing like phenotype. On the other side, atherosclerotic plaques from myeloid cell-specific Hdac3 knockout mice displayed increased collagen disposition and enlarged protective fibrous caps, while the myeloid cell-specific Ncor1 knockout plaques had increased necrotic cores (41).
In line with the data from Oppi et al. (41), Fan et al. showed that myeloid cell-specific Gps2-knockout mice display an increased expression of pro-inflammatory cytokines and chemokines, such as Ccl2 and Ccl7, which is characteristic of pro-inflammatory M1-type macrophage activation (53). Moreover, the authors used palmitate as obesity-linked metabolic trigger of inflammation and observed that Gps2-deficient macrophages had an elevated pro-inflammatory gene signature (53). Vice-versa, transplantation of Gps2-overexpressing bone marrow into two mouse models of obesity reduced inflammation and improved insulin sensitivity in recipient mice (53). Moreover, docking of GPS2 and LXR stimulate H3K9 demethylation on the Abcg1 promoter, thus promoting its expression and mediating cholesterol efflux to HDL particles in monocytic THP-1 cells (54). Contrary to the induction of Abcg1 via LXR, GPS2 interacts with NF-κB to promote the expression of Abca1 upon LPS stimulation (55). These elegant studies demonstrate how GPS2 promotes macrophage cholesterol efflux upon different stimuli, i.e. via oxysterol-triggered LXR activation or via LPS-driven NF-κB activation. Interestingly, the activation of both processes by GPS2 is independent of NCOR1-HDAC3 recruitment (54, 55).
Aside from its effects on atherosclerotic plaques and metabolism, macrophage NCOR1 seems to play a pivotal role in the heart. Genetic deletion of Ncor1 in macrophages led to reduced infarct size and improved cardiac function in mice with experimental myocardial infarction (56). These findings were explained by suppression of inflammatory transcriptional programs (interleukin‐1β, interleukin‐6, AP-1 and nuclear factor‐κB) and reduced macrophage proliferation (due to inhibition of cell cycle progression). Hence, macrophage NCOR1 may act as an upstream regulator of myocardial inflammation thus participating to left ventricular hypertrophy, diastolic dysfunction and microvascular disease, key hallmarks of heart failure (57). In the same study, macrophage NCOR1 deficiency markedly inhibited neointimal hyperplasia and vascular remodeling in a mouse model of arterial wire injury (56). Taken together, these results suggest that selective modulation of NCOR1 in macrophages could have important implications for the prevention of myocardial ischemic damage, heart failure, coronary heart disease and intracoronary stent restenosis. Further molecular work and preclinical studies are needed to better explore the potential of NCOR targeting approaches in cardiovascular disease.
The liver plays a crucial role in the development of atherosclerosis by regulating metabolic and inflammatory processes, such as the expression of pro-inflammatory cytokines and acute phase response proteins, the secretion of VLDL particles, the uptake of cholesterol from the circulation, and the biliary cholesterol excretion. An immunometabolic dysregulation in the liver can promote nonalcoholic fatty liver disease and the development of atherosclerosis. Importantly, nonalcoholic fatty liver disease leads to adverse cardiovascular functions, such as increased oxidative stress and endothelial dysfunction, hypercoagulability, and accelerated development of atherosclerosis (58–60).
Several studies addressed the physiological functions of NCOR1, NCOR2, and HDAC3 in the liver, which target several lipid-responsive nuclear receptors (Figure 2) (24, 28, 29, 61–67). The role of NCOR1 in liver energy metabolism is particularly interesting during the fasting-feeding transition: both HDAC3 and NCOR1 are known to repress lipogenic genes, but paradoxically, NCOR1 was also reported to be critical for inhibition of PPARα, hepatic fatty oxidation and ketogenesis (63, 64). This is due to the ability of NCOR1 to select its repressor targets in a context-dependent manner to orchestrate liver energy metabolism depending on the energy status of the cell (37). Upon feeding, high levels of glucose and insulin activate the target of rapamycin complex 1 (mTORC1)-AKT signaling pathway, thus phosphorylating serine 1460 of NCOR1 (pS1460 NCOR1) (Figure 2). pS1460 decreases the ability of NCOR1 to interact with LXR, thus promoting the transcription of lipogenic LXRs target genes, and conversely, by fostering the interaction with PPARα and ERRα, with subsequent repression of downstream ketogenic and Oxphos genes (37).
Besides being regulated by phosphorylation in the liver, the translation of NCOR1 can also be blocked by the microRNA miR-199a-5p (68) (Figure 2). The authors used a bioinformatic approach to identify miRNAs affected in a non-alcoholic steatohepatitis (NASH) model induced by a methionine-choline−deficient (MCD) diet. The MCD diet increased miR-199a-5p expression, which in turn blocks the translation of the Ncor1 mRNA by binding to a conserved 3’ untranslated region (68).
To assess the function of NCOR1 in thyroid hormone receptor (TR) regulation, the group of A. Hollenberg generated a truncated form of NCOR1 which lacks the two main nuclear receptor interacting domains (NCOR1ΔID) and thus, for example, cannot interact with TRs or LXRs (69, 70). Interestingly, they found that mice with a disrupted nuclear receptor binding domain show a reduced content of cholesterol in the liver and an increased synthesis of alternative bile acid. In turn, these less hydrophobic bile acids have a lower capacity to bind fats and sterols in the intestine and thus reduce their absorption (38). The increased expression of alternative bile acid synthesis genes is a consequence of TRβ1 de-repression caused by the mutant NCOR1 (38).
GPS2 exerts protective function in the liver by interacting with SUMOylated nuclear receptors, such as LRH-1 and LXRβ, to repress inflammatory cytokine expression during the hepatic acute phase response (71). On the other side, hepatic deletion of GPS2 reduces nonalcoholic steatohepatitis via induction of PPARα-driven lipid catabolism (67). This repression of PPARα happens in cooperation with NCOR1, but not NCOR2 (67). Conversely to the GPS2 knockouts, hepatic deletion of TBL1 promotes hypertriglyceridemia and hepatic steatosis on both a normal chow or high-fat diet by impairing PPARα-driven lipid catabolism (72). The induction of PPARα was mediated by increased recruitment of NCOR1-HDAC3 complexes in the absence of TBL1 (72). These data underline that the repressive function and target specificity of the NCOR1 complex is largely dependent on its co-regulators.
Previous studies demonstrated that NCOR1 deficiency increases the activity of MEF2d in skeletal muscle (35). In a recent publication, Li et al. affirmed the suppressive role of NCOR1 in regulating the size of cardiomyocytes, presenting strong evidence of interactions among NCOR1, MEF2, and class IIa HDACs, being MEF2a and MEF2d key transcription factors interceded the impact of NCOR1 on cardiomyocyte size (73). As Li et al. described, NCOR1 may be considered as a stress-responsive and cardioprotective regulator during cardiac hypertrophy, showing that its deficiency led to cardiac hypertrophy under physiological condition and aggravated hypertrophy induced by pressure overload (73).
NCOR1 usually cooperates with HDACs to execute its repressive activities (28). The regulatory paradigm in cardiac hypertrophy involves alterations in gene expression that is mediated by chromatin remodeling. HDACs remove the acetyl group from histones, resulting in its hypoacetylation, which diminishes chromatin accessibility for transcription factors, leading to repression of transcription (74). The Class I HDAC3 enzyme participates in the repressive activities of NCOR1, and its deficiency in cardiomyocytes causes severe cardiac hypertrophy at an early stage. Interestingly, Li et al. identified class IIa to be involved in the process of cardiomyocytes, demonstrating that NCOR1 works more likely through class IIa instead of class I to affect cardiac hypertrophy (73).
B cell lymphoma-6 (BCL6) belongs to a class of zinc-finger transcription factors and acts as a transcriptional repressor. It regulates the development of germinal centers, B and T cells, coordinates the activation of macrophages, and was described as a proto-oncogene (75). Importantly, the interaction of BCL6 with NCOR complexes is essential to mediate its transrepressive activity in several biological processes (75, 76).
The PPARδ agonist GW0742 protects against angiotensin II (AngII)-accelerated atherosclerosis by inducing the expression of the Bcl6, and the regulators of G protein-coupled signaling (RGS) proteins RGS4 and RGS5, which in turn inhibit the expression of pro-inflammatory and atherogenic genes (77). Interestingly, ChIP-seq data demonstrate that the BCL6 and NCORs cistrome overlap in about 50% of all DNA binding sites, and that binding sites that are synergistically bound by BCL6 and NCORs are highly enriched for inflammatory and atherogenic NF-κB target genes (78).
The deletion of Bcl6 in the liver leads to a derepression of PPARα-driven enzymes mediating fatty acid oxidation and thus protects against high-fat diet-induced hepatic steatosis (79). Interestingly, binding of the corepressors NCOR1, NCOR2 and HDAC3 to BCL6-binding sites was reduced in Bcl6-deficient livers, and these sites displayed increased enhancer/promoter activity as shown by enhanced histone 3 lysine 27 acetylation (H3K27ac) (79). These data suggest that hepatic BCL6 recruits a subset of NCOR/HADC3 complexes to the promoter of specific target genes regulating lipid metabolism.
So far, the role of NCOR1 using an endothelial cell-specific genetic model has not been explored. Therefore, this section is limited to describe the function of its tight cofactor HDAC3 (28). HDAC3 is essential for endothelial monolayer survival and integrity. Studies demonstrated that HDAC3 is engaged in the differentiation of embryonic stem cells into endothelial progenitors and determinant for endothelial cell survival (64). Zampetaki et al. demonstrated that disturbed flow induces transient stabilization of the HDAC3 protein in endothelial cells. It happens through the activation of the VEGFR2 and PI3 kinase signaling pathways (80). HDAC3 expression increases near branch openings compared with areas of high laminar flow, which was also confirmed by exposure of endothelial cells to disturbed flow in vitro (80). It is therefore likely that NCOR1 might be involved in these processes as well.
To obtain a good overview about the function of NCOR1 in T cells we refer to an excellent review from Muller et al. (81). Most studies assessed the role of NCOR1 during T cell development, while only a few focused on its function in mature T cells in pathophysiological conditions. The lethal phenotype of the complete Ncor1 knockout mice developed by Jepsen et al. demonstrated that constitutive Ncor1-deficiency conferred impaired thymocyte development (33). Further studies using T cell-specific deletions of Ncor1 in mice revealed that the number of double negative thymocytes and peripheral T cells are decreased in the absence of NCOR1 (82, 83). Recently, it was shown that NCOR1 regulates the function of CD4+ and Th1 cells by regulating the expression of IFN-γ (84). Consistently, Zhang et al. showed that deletion of Ncor1 in male SJL T cells disrupted the fenofibrate-driven repression of IFN-γ synthesis, possibly also explaining differences in Th1 responses between male and female mice (85). Another interesting report demonstrated that NCOR1 represses the tolerogenic program in dendritic cells. As a consequence, dendritic cell-specific Ncor1 knockout mice display an increased number of FoxP3+ regulatory T cells (86). In terms of atherogenesis, this could turn into a protective effect.
Research over the past decades identified various molecular factors that affect atherogenesis, including inflammatory and metabolic regulators (2, 87, 88). In the last years, we have learned that immunometabolic pathways are often interconnected, and we are now starting to understand the major factors involved in these immunometabolic mechanisms (10, 11). Targeting immunometabolic pathways, e.g., those regulated by NCOR1, might become an attractive approach to prevent the development of atherosclerosis since they regulate various processes that promote the development of the disease. Moreover, since these pathways are also implicated in the development of other chronic immunometabolic diseases, such as obesity, type 2 diabetes, and nonalcoholic fatty liver disease, their mechanistic dissection will be relevant to understand development and progression of immunometabolic diseases on a broader scale.
Which could be suitable strategies to target NCOR1? One option would be to develop small molecule agonists or antagonists that interfere or facilitate the interaction of NCOR1 to a specific target nuclear receptor. Although this would be a specific approach, it would be very laborious and time-consuming. Another option is offered by current developments in gene therapy (including CRISPR therapeutics) and small interference RNA (siRNA) medicine, which offer excellent future targeting opportunities. The liver is an easily accessible target organ and new emerging drugs using siRNAs demonstrate that transcribed RNA can be targeted in a liver-specific fashion by conjugating the synthetic siRNA to triantennary N-acetylgalactosamine carbohydrates (e.g., as it is used to target PCSK9 in the liver (89, 90)). However, specifically targeting other organs or cells, such as cardiomyocytes and macrophages, continues to be challenging. Moreover, the identification of atherogenic targets, such as CD36 in macrophages (41), might lead to the identification of druggable targets and hence to the development of new therapeutic strategies to treat atherosclerotic disease patients that are at a very high risk to suffer a transient ischemic attack, stroke or acute coronary syndrome.
All authors analyzed the literature and wrote the manuscript. All authors contributed to the article and approved the submitted version.
SS was supported by the Swiss National Science Foundation (PZOOP3_161521), the SwissLife Jubiläumsstiftung, the Swiss Heart Foundation (FF19025), and the Foundation for Research in Science and the Humanities at the University of Zurich (STWF-20-008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med (2011) 17(11):1410–22. doi: 10.1038/nm.2538
2. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature (2011) 473(7347):317–25. doi: 10.1038/nature10146
3. Jonsson AL, Backhed F. Role of gut microbiota in atherosclerosis. Nat Rev Cardiol (2017) 14(2):79–87. doi: 10.1038/nrcardio.2016.183
4. Kessler T, Vilne B, Schunkert H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol Med (2016) 8(7):688–701. doi: 10.15252/emmm.201506174
5. Roberts R. Genetics of coronary artery disease. Circ Res (2014) 114(12):1890–903. doi: 10.1161/CIRCRESAHA.114.302692
6. Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. New Engl J Med (2015) 372(25):2387–97. doi: 10.1056/NEJMoa1410489
7. Nissen SE, Tuzcu EM, Schoenhagen P, Crowe T, Sasiela WJ, Tsai J, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. New Engl J Med (2005) 352(1):29–38. doi: 10.1056/NEJMoa042000
8. Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al. C-reactive protein levels and outcomes after statin therapy. New Engl J Med (2005) 352(1):20–8. doi: 10.1056/NEJMoa042378
9. Oppi S, Luscher TF, Stein S. Mouse Models for Atherosclerosis Research-Which Is My Line? Front Cardiovasc Med (2019) 6:46(46):46. doi: 10.3389/fcvm.2019.00046
10. Hotamisligil GS. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity (2017) 47(3):406–20. doi: 10.1016/j.immuni.2017.08.009
11. Lee YS, Wollam J, Olefsky JM. An Integrated View of Immunometabolism. Cell (2018) 172(1–2):22–40. doi: 10.1016/j.cell.2017.12.025
12. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest (2017) 127(1):1–4. doi: 10.1172/JCI92035. treatment of metabolic disease. J.M. Olefsky owns stock in Catabasis Pharmaceuticals and receives consulting income from Cymabay Inc., Second Genome, and AntriaBio.
13. Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet (2010) 11(4):285–96. doi: 10.1038/nrg2752
14. Smith E, Shilatifard A. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol Cell (2010) 40(5):689–701. doi: 10.1016/j.molcel.2010.11.031
15. Mouchiroud L, Eichner LJ, Shaw RJ, Auwerx J. Transcriptional coregulators: fine-tuning metabolism. Cell Metab (2014) 20(1):26–40. doi: 10.1016/j.cmet.2014.03.027
16. Costantino S, Mohammed SA, Ambrosini S, Paneni F. Epigenetic processing in cardiometabolic disease. Atherosclerosis (2019) 281:150–8. doi: 10.1016/j.atherosclerosis.2018.09.029
17. Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol (2006) 6(1):44–55. doi: 10.1038/nri1748
18. Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol (2003) 65:261–311. doi: 10.1146/annurev.physiol.65.092101.142528
19. Kurakula K, Hamers AA, de Waard V, de Vries CJ. Nuclear Receptors in atherosclerosis: a superfamily with many ‘Goodfellas’. Mol Cell Endocrinol (2013) 368(1–2):71–84. doi: 10.1016/j.mce.2012.05.014
20. Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell (2007) 25(1):57–70. doi: 10.1016/j.molcel.2006.11.022
21. Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, et al. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev (2009) 23(6):681–93. doi: 10.1101/gad.1773109
22. Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature (2005) 437(7059):759–63. doi: 10.1038/nature03988
23. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol (2010) 10(5):365–76. doi: 10.1038/nri2748
24. Treuter E, Venteclef N. Transcriptional control of metabolic and inflammatory pathways by nuclear receptor SUMOylation. Biochim Biophys Acta (2011) 1812(8):909–18. doi: 10.1016/j.bbadis.2010.12.008
25. Venteclef N, Jakobsson T, Steffensen KR, Treuter E. Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrinol Metabol: TEM (2011) 22(8):333–43. doi: 10.1016/j.tem.2011.04.004
26. Zhang HM, Chen H, Liu W, Liu H, Gong J, Wang H, et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res (2012) 40(Database issue):D144–9. doi: 10.1093/nar/gkr965
27. Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet (2010) 11(2):109–23. doi: 10.1038/nrg2736
28. Mottis A, Mouchiroud L, Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev (2013) 27(8):819–35. doi: 10.1101/gad.214023.113
29. Emmett MJ, Lazar MA. Integrative regulation of physiology by histone deacetylase 3. Nat Rev Mol Cell Biol (2019) 20(2):102–15. doi: 10.1038/s41580-018-0076-0
30. Liang N, Jakobsson T, Fan R, Treuter E. The Nuclear Receptor-Co-repressor Complex in Control of Liver Metabolism and Disease. Front Endocrinol (2019) 10:411. doi: 10.3389/fendo.2019.00411
31. Neele AE, Van den Bossche J, Hoeksema MA, de Winther MP. Epigenetic pathways in macrophages emerge as novel targets in atherosclerosis. Eur J Pharmacol (2015) 763(Pt A):79–89. doi: 10.1016/j.ejphar.2015.03.101
32. Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim HJ, Glass CK, et al. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature (2007) 450:415–9. doi: 10.1038/nature06270
33. Jepsen K, Hermanson O, Onami TM, Gleiberman AS, Lunyak V, McEvilly RJ, et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell (2000) 102(6):753–63. doi: 10.1016/S0092-8674(00)00064-7
34. Jepsen K, Gleiberman AS, Shi C, Simon DI, Rosenfeld MG. Cooperative regulation in development by SMRT and FOXP1. Genes Dev (2008) 22(6):740–5. doi: 10.1101/gad.1637108. doi: 22/6/740 [pii].
35. Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan W, Downes M, et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell (2011) 147(4):827–39. doi: 10.1016/j.cell.2011.10.017
36. Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell (2011) 147(4):815–26. doi: 10.1016/j.cell.2011.09.050
37. Jo YS, Ryu D, Maida A, Wang X, Evans RM, Schoonjans K, et al. Phosphorylation of the nuclear receptor corepressor 1 by protein kinase B switches its corepressor targets in the liver in mice. Hepatology (2015) 62(5):1606–18. doi: 10.1002/hep.27907
38. Astapova I, Ramadoss P, Costa-e-Sousa RH, Ye F, Holtz KA, Li Y, et al. Hepatic nuclear corepressor 1 regulates cholesterol absorption through a TRbeta1-governed pathway. J Clin Invest (2014) 124(5):1976–86. doi: 10.1172/JCI73419
39. Li P, Spann NJ, Kaikkonen MU, Lu M, Oh da Y, Fox JN, et al. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell (2013) 155(1):200–14. doi: 10.1016/j.cell.2013.08.054
40. Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res (2010) 107(1):56–65. doi: 10.1161/CIRCRESAHA.110.218420
41. Oppi S, Nusser-Stein S, Blyszczuk P, Wang X, Jomard A, Marzolla V, et al. Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARgamma signature. Eur Heart J (2020) 41(9):995–1005. doi: 10.1093/eurheartj/ehz667
42. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell (2011) 145(3):341–55. doi: 10.1016/j.cell.2011.04.005
43. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science (2013) 339(6116):161–6. doi: 10.1126/science.1230719
44. Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, et al. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc Natl Acad Sci U.S.A. (2004) 101(40):14461–6. doi: 10.1073/pnas.0405786101
45. Perissi V, Aggarwal A, Glass CK, Rose DW. Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell (2004) 116(4):511–26. doi: 10.1016/S0092-8674(04)00133-3
46. Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, et al. Animal models of atherosclerosis. Eur J Pharmacol (2017) 816:3–13. doi: 10.1016/j.ejphar.2017.05.010
47. Lefevre L, Authier H, Stein S, Majorel C, Couderc B, Dardenne C, et al. LRH-1 mediates anti-inflammatory and antifungal phenotype of IL-13-activated macrophages through the PPARgamma ligand synthesis. Nat Commun (2015) 6:6801. doi: 10.1038/ncomms7801
48. Miranda DA, Krause WC, Cazenave-Gassiot A, Suzawa M, Escusa H, Foo JC, et al. LRH-1 regulates hepatic lipid homeostasis and maintains arachidonoyl phospholipid pools critical for phospholipid diversity. JCI Insight (2018) 3(5). doi: 10.1172/jci.insight.96151
49. Stein S, Lemos V, Xu P, Demagny H, Wang X, Ryu D, et al. Impaired SUMOylation of nuclear receptor LRH-1 promotes nonalcoholic fatty liver disease. J Clin Invest (2017) 127(2):583–92. doi: 10.1172/JCI85499
50. Stein S, Oosterveer MH, Mataki C, Xu P, Lemos V, Havinga R, et al. SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab (2014) 20(4):603–13. doi: 10.1016/j.cmet.2014.07.023
51. Xu P, Oosterveer MH, Stein S, Demagny H, Ryu D, Moullan N, et al. LRH-1-dependent programming of mitochondrial glutamine processing drives liver cancer. Genes Dev (2016) 30(11):1255–60. doi: 10.1101/gad.277483.116
52. Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med (2014) 6(9):1124–32. doi: 10.15252/emmm.201404170
53. Fan R, Toubal A, Goni S, Drareni K, Huang Z, Alzaid F, et al. Loss of the co-repressor GPS2 sensitizes macrophage activation upon metabolic stress induced by obesity and type 2 diabetes. Nat Med (2016) 22(7):780–91. doi: 10.1038/nm.4114
54. Jakobsson T, Venteclef N, Toresson G, Damdimopoulos AE, Ehrlund A, Lou X, et al. GPS2 is required for cholesterol efflux by triggering histone demethylation, LXR recruitment, and coregulator assembly at the ABCG1 locus. Mol Cell (2009) 34(4):510–8. doi: 10.1016/j.molcel.2009.05.006
55. Huang Z, Liang N, Damdimopoulos A, Fan R, Treuter E. G protein pathway suppressor 2 (GPS2) links inflammation and cholesterol efflux by controlling lipopolysaccharide-induced ATP-binding cassette transporter A1 expression in macrophages. FASEB J (2019) 33(2):1631–43. doi: 10.1096/fj.201801123R
56. Du LJ, Sun JY, Zhang WC, Wang YL, Zhu H, Liu T, et al. Macrophage NCOR1 Deficiency Ameliorates Myocardial Infarction and Neointimal Hyperplasia in Mice. J Am Heart Assoc (2020) 9(15):e015862. doi: 10.1161/JAHA.120.015862
57. Metra M, Teerlink JR. Heart failure. Lancet (2017) 390(10106):1981–95. doi: 10.1016/S0140-6736(17)31071-1
58. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol (2013) 10(6):330–44. doi: 10.1038/nrgastro.2013.41
59. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol (2015) 62(1 Suppl):S47–64. doi: 10.1016/j.jhep.2014.12.012
60. Bhatia LS, Curzen NP, Calder PC, Byrne CD. Non-alcoholic fatty liver disease: a new and important cardiovascular risk factor? Eur Heart J (2012) 33(10):1190–200. doi: 10.1093/eurheartj/ehr453
61. Shimizu H, Astapova I, Ye F, Bilban M, Cohen RN, Hollenberg AN. NCoR1 and SMRT play unique roles in thyroid hormone action in vivo. Mol Cell Biol (2015) 35(3):555–65. doi: 10.1128/MCB.01208-14
62. Jeyakumar M, Liu XF, Erdjument-Bromage H, Tempst P, Bagchi MK. Phosphorylation of thyroid hormone receptor-associated nuclear receptor corepressor holocomplex by the DNA-dependent protein kinase enhances its histone deacetylase activity. J Biol Chem (2007) 282(13):9312–22. doi: 10.1074/jbc.M609009200
63. Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell (2013) 52(6):769–82. doi: 10.1016/j.molcel.2013.10.022
64. Alenghat T, Meyers K, Mullican SE, Leitner K, Adeniji-Adele A, Avila J, et al. Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature (2008) 456(7224):997–1000. doi: 10.1038/nature07541
65. Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science (2011) 331(6022):1315–9. doi: 10.1126/science.1198125
66. Zhang J, Xu Z, Gu J, Jiang S, Liu Q, Zheng Y, et al. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am J Physiol Endocrinol Metab (2018) 315(2):E150–E62. doi: 10.1152/ajpendo.00465.2017
67. Liang N, Damdimopoulos A, Goni S, Huang Z, Vedin LL, Jakobsson T, et al. Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARalpha. Nat Commun (2019) 10(1):1684. doi: 10.1038/s41467-019-09524-z
68. Zhang B, Wang R, Du J, Niu J, Zhang R, Xu S, et al. Upregulated microRNA-199a-5p inhibits nuclear receptor corepressor 1 translation in mice with nonalcoholic steatohepatitis. Mol Med Rep (2014) 10(6):3080–6. doi: 10.3892/mmr.2014.2592
69. Astapova I, Lee LJ, Morales C, Tauber S, Bilban M, Hollenberg AN. The nuclear corepressor, NCoR, regulates thyroid hormone action in vivo. Proc Natl Acad Sci U.S.A. (2008) 105(49):19544–9. doi: 10.1073/pnas.0804604105
70. Vella KR, Hollenberg AN. The actions of thyroid hormone signaling in the nucleus. Mol Cell Endocrinol (2017) 458:127–35. doi: 10.1016/j.mce.2017.03.001
71. Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev (2010) 24(4):381–95. doi: 10.1101/gad.545110. doi: 24/4/381 [pii].
72. Kulozik P, Jones A, Mattijssen F, Rose AJ, Reimann A, Strzoda D, et al. Hepatic deficiency in transcriptional cofactor TBL1 promotes liver steatosis and hypertriglyceridemia. Cell Metab (2011) 13(4):389–400. doi: 10.1016/j.cmet.2011.02.011
73. Li C, Sun XN, Chen BY, Zeng MR, Du LJ, Liu T, et al. Nuclear receptor corepressor 1 represses cardiac hypertrophy. EMBO Mol Med (2019) 11(11):e9127. doi: 10.15252/emmm.201809127
74. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J (2003) 370(Pt 3):737–49. doi: 10.1042/BJ20021321
75. Cardenas MG, Oswald E, Yu W, Xue F, MacKerell AD Jr., Melnick AM. The Expanding Role of the BCL6 Oncoprotein as a Cancer Therapeutic Target. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23(4):885–93. doi: 10.1158/1078-0432.CCR-16-2071
76. Huynh KD, Bardwell VJ. The BCL-6 POZ domain and other POZ domains interact with the co-repressors N-CoR and SMRT. Oncogene (1998) 17(19):2473–84. doi: 10.1038/sj.onc.1202197
77. Takata Y, Liu J, Yin F, Collins AR, Lyon CJ, Lee CH, et al. PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci U.S.A. (2008) 105(11):4277–82. doi: 10.1073/pnas.0708647105
78. Barish GD, Yu RT, Karunasiri MS, Becerra D, Kim J, Tseng TW, et al. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab (2012) 15(4):554–62. doi: 10.1016/j.cmet.2012.02.012
79. Sommars MA, Ramachandran K, Senagolage MD, Futtner CR, Germain DM, Allred AL, et al. Dynamic repression by BCL6 controls the genome-wide liver response to fasting and steatosis. eLife (2019) 8. doi: 10.7554/eLife.43922
80. Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation (2010) 121(1):132–42. doi: 10.1161/CIRCULATIONAHA.109.890491
81. Muller L, Hainberger D, Stolz V, Ellmeier W. NCOR1-a new player on the field of T cell development. J Leukoc Biol (2018) 104(6):1061–8. doi: 10.1002/JLB.1RI0418-168R
82. Muller L, Hainberger D, Stolz V, Hamminger P, Hassan H, Preglej T, et al. The corepressor NCOR1 regulates the survival of single-positive thymocytes. Sci Rep (2017) 7(1):15928. doi: 10.1038/s41598-017-15918-0
83. Wang J, He N, Zhang N, Quan D, Zhang S, Zhang C, et al. NCoR1 restrains thymic negative selection by repressing Bim expression to spare thymocytes undergoing positive selection. Nat Commun (2017) 8(1):959. doi: 10.1038/s41467-017-00931-8
84. Hainberger D, Stolz V, Zhu C, Schuster M, Muller L, Hamminger P, et al. NCOR1 Orchestrates Transcriptional Landscapes and Effector Functions of CD4(+) T Cells. Front Immunol (2020) 11:579:579. doi: 10.3389/fimmu.2020.00579
85. Zhang MA, Ahn JJ, Zhao FL, Selvanantham T, Mallevaey T, Stock N, et al. Antagonizing Peroxisome Proliferator-Activated Receptor alpha Activity Selectively Enhances Th1 Immunity in Male Mice. J Immunol (2015) 195(11):5189–202. doi: 10.4049/jimmunol.1500449
86. Ahad A, Stevanin M, Smita S, Mishra GP, Gupta D, Waszak S, et al. NCoR1: Putting the Brakes on the Dendritic Cell Immune Tolerance. iScience (2019) 19:996–1011. doi: 10.1016/j.isci.2019.08.024
87. Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev (2013) 93(3):1317–542. doi: 10.1152/physrev.00004.2012
88. Kolodgie FD, Yahagi K, Mori H, Romero ME, Trout HHR, Finn AV, et al. High-risk carotid plaque: lessons learned from histopathology. Semin Vasc Surg (2017) 30(1):31–43. doi: 10.1053/j.semvascsurg.2017.04.008
89. Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. New Engl J Med (2017) 376(15):1430–40. doi: 10.1056/NEJMoa1615758
Keywords: atherosclerosis, cardiometabolic, corepressor complex, NCoR1, nuclear receptor signaling, immunometabolism, mechanisms of disease, transcriptional regulation
Citation: Geiger MA, Guillaumon AT, Paneni F, Matter CM and Stein S (2020) Role of the Nuclear Receptor Corepressor 1 (NCOR1) in Atherosclerosis and Associated Immunometabolic Diseases. Front. Immunol. 11:569358. doi: 10.3389/fimmu.2020.569358
Received: 03 June 2020; Accepted: 24 September 2020;
Published: 08 October 2020.
Edited by:
Martin Giera, Leiden University Medical Center, NetherlandsReviewed by:
Bart Everts, Leiden University Medical Center, NetherlandsCopyright © 2020 Geiger, Guillaumon, Paneni, Matter and Stein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sokrates Stein, c29rcmF0ZXMuc3RlaW5AbWUuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.