 Katie Hudson
Katie Hudson Neil Cross
Neil Cross Nicola Jordan-Mahy
Nicola Jordan-Mahy Rebecca Leyland
Rebecca Leyland
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 21 October 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.568931
Programmed death-ligand 1 (PD-L1) is an immune checkpoint inhibitor that binds to its receptor PD-1 expressed by T cells and other immune cells to regulate immune responses; ultimately preventing exacerbated activation and autoimmunity. Many tumors exploit this mechanism by overexpressing PD-L1 which often correlates with poor prognosis. Some tumors have also recently been shown to express PD-1. On tumors, PD-L1 binding to PD-1 on immune cells promotes immune evasion and tumor progression, primarily by inhibition of cytotoxic T lymphocyte effector function. PD-1/PD-L1-targeted therapy has revolutionized the cancer therapy landscape and has become the first-line treatment for some cancers, due to their ability to promote durable anti-tumor immune responses in select patients with advanced cancers. Despite this clinical success, some patients have shown to be unresponsive, hyperprogressive or develop resistance to PD-1/PD-L1-targeted therapy. The exact mechanisms for this are still unclear. This review will discuss the current status of PD-1/PD-L1-targeted therapy, oncogenic expression of PD-L1, the new and emerging tumor-intrinisic roles of PD-L1 and its receptor PD-1 and how they may contribute to tumor progression and immunotherapy responses as shown in different oncology models.
Cancer immunotherapies work to re-establish immune-mediated tumor eradication (1). Despite the advancements in immunotherapy over the past decade, the interactions of cells within the tumor microenvironment continue to mediate immune evasion and tumor progression (2). The tumor consists of extracellular matrix components and diverse cell populations such as T cells, B cells, natural killer (NK) cells, macrophages, dendritic cells, fibroblasts and endothelial cells (3). Although immune cells such as NK, CD8+, and CD4+ T cells which migrate to the tumor display anti-tumor activity, over time the tumor microenvironment becomes immunosuppressive, favoring the emergence of tumor promoting cells such as regulatory T-cells (T-regs), myeloid derived suppressor cells (MDSCs) and M2 macrophages (1). This is known as the phenomenon cancer immuno-editing which involves three phases: elimination, equilibrium, and escape (Figure 1). Besides tumor cells acquiring the ability to escape immune recognition, they also employ immune-inhibitory mechanisms to evade the immune systems defenses (4). Immuno-editing occurs in patients with advanced cancers and this process can be influenced by therapies targeting immuno-inhibitory proteins (5, 6). One such mechanism is targeting of programmed death-ligand 1 (PD-L1) (7, 8).
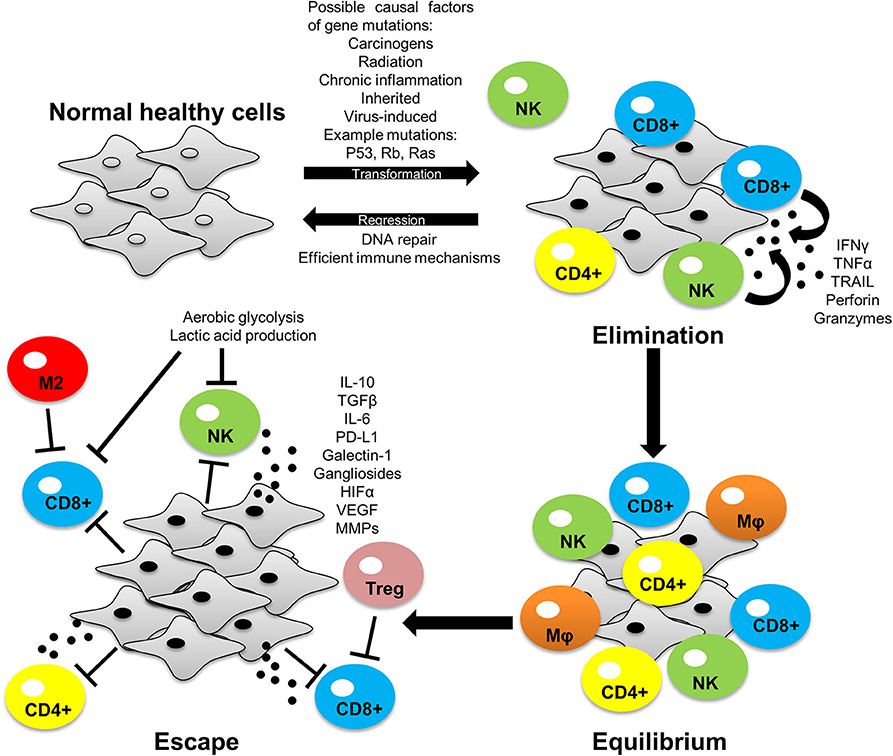
Figure 1. The process of cancer immuno-editing: elimination, equilibrium and escape. Normal healthy cells transform into tumor cells through acquiring mutations that allows for uncontrolled growth of the cell. In the elimination phase, immune cells can recognize and eliminate tumor cells by inducing apoptosis via granule and/or receptor-mediated mechanisms. Some tumor cells avoid immune destruction and enter dynamic equilibrium with immune cells whereby the immune system elicits a potent enough response to contain the tumor cells but not enough to eradicate them. During this phase tumor cells develop increased genetic instability and undergo immune selection, whereby the immune cells eliminate those tumor cells susceptible to immune-mediated killing, whilst selecting those tumor cells with mechanisms to evade the immune system. These selected tumor cells can now proliferate freely and expand leading to immune escape. PD-L1 expression is one of the many mechanisms employed by tumors to facilitate immune evasion and tumor development. Tumor-intrinsic mutations can induce PD-L1 expression and influence tumor cell-immune cell interactions within the tumor microenvironment to favor tumor growth (as discussed later in this review). Interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), interleukin 10 (IL-10), interleukin 6 (IL-6), transforming growth factor beta (TGF-β), hypoxic inducible factor 1/2 alpha (HIF-1/2α), vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs).
PD-L1, also known as B7-H1 and CD274, is an immune checkpoint inhibitor that binds to its receptor PD-1 expressed by T cells, B cells, dendritic cells, and monocytes (7). PD-L1 is expressed by T cells, B cells, NK cells, dendritic cells, macrophages, MDSCs, and many other cell types such as epithelial and endothelial cells (9, 10). The PD-1/PD-L1 signaling axis regulates immune responses to prevent exacerbated activation and autoimmunity (8, 10). Many tumors exploit this mechanism by overexpressing PD-L1 (11–13). Recently, tumors have also been shown to express PD-1 (14, 15). PD-L1 binding to PD-1 on immune cells induces inhibitory responses which in turn can promotes immune evasion and tumor progression.
Elevated expression of PD-L1 on tumors has been reported to strongly correlate with advanced disease state and unfavorable prognosis in melanoma (11), breast (13), gastric (16), ovarian (12), liver (17), kidney (18), pancreatic (19), and bladder (20) cancer. Immunotherapies targeting the PD-1/PD-L1 signaling axis have become the first-line treatment for some cancers due to their ability to promote durable anti-tumor immune responses in select patients with advanced cancers (21–23), leading to their approval by the USA Food and Drug Administration (FDA). Although PD-1/PD-L1-targeted therapies have demonstrated clinical benefits across a broad range of cancers, some patients are unresponsive, hyperprogressive, or develop resistance (24). The objective response rate of anti-PD-L1 monoclonal antibodies alone is ~15% in non-small-cell lung cancer (NSCLC) (21, 25), ~20% in urothelial carcinomas (23, 26) and ~30% in Merkel cell carcinomas (27, 28). Consequently, novel therapeutic strategies are required to enhance patient response rates through combining PD-1/PD-L1-targeted therapy with other immune approaches, small molecule inhibitors, chemotherapy, or other modalities. Patients considered eligible for PD-1/PD-L1-targeted therapy are those that present with PD-L1-positive tumors, circulating PD-1 positive/CD8+ T cells and/or tumors with high mutational burden (23, 29). However, patient tumors that have shown to lack PD-L1 have also responded positively to PD-1/PD-L1-targeted therapy (10, 30), suggesting that either blocking PD-L1 expression on tumors is not required for anti-tumor responses and inhibition of PD-L1 on immune cells alone may be sufficient or that more sensitive approaches to detecting PD-L1 expression on tumors is required. Conversely, some tumors with high PD-L1 expression have shown to be unresponsive to PD-1/PD-L1-targeted therapy (31), likely due to the lack of immune stimulatory cells present in the tumor microenvironment to elicit an effective anti-tumor immune response, but reasons for this remain to be fully elucidated. However, patients that are intrinsically unresponsive to PD-1/PD-L1-targeted therapy can also demonstrate “primary resistance” whereby their tumors display inadequate T cell infiltration, T cell exclusion, impaired IFNγR signaling, and/or local immune suppression (2, 31). Patients that initially respond to PD-1/PD-L1-targeted therapy also show “acquired resistance” whereby their tumors display loss of T cell function and/or disrupted antigen processing and presentation (31, 32). In recent years, approximately 10% of cancer patients have experienced pseudoprogression in response to PD-1/PD-L1-targeted therapy, whereby patients temporally exhibit rapid progression of their condition before responding successfully to treatment (33). On the other hand, some patients have experienced hyperprogression in response to PD-1/PD-L1-targeted therapy, which is characterized by rapid deterioration of their condition upon initialization of treatment without a successful response; giving patients <2 months to live from onset (33). The reasons for pseudoprogression and hyperprogression in response to PD-1/PD-L1-targeted therapy remain speculative and need to be explored. It is also important for clinicians to be able to distinguish between the two types of responses to inform patient selection for therapy. Further insight into the role of PD-L1 and PD-1 in the tumor microenvironment could allow the identification of more appropriate biomarkers predictive of clinical efficacy to PD-1/PD-L1-targeted therapy necessary to ensure patients receive the maximum clinical benefit whilst avoid immune-related adverse effects (24, 29), pseudoprogression and hyperprogression (33).
Immune checkpoint molecules expressed on T cells such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and PD-1 regulate immune responses by dampening T cell activation to prevent exacerbated activation and autoimmunity (4, 34). During cancer development anti-tumor immunity is suppressed and immunotherapies targeting CTLA-4 and PD-1 signaling axes have been developed to reactivate T cells to induce immune-mediated tumor eradication (35). Normally, T cell activation requires two signals. Signal one is the T cell receptor (TCR) recognizing and binding to an antigenic peptide presented on an MHC molecule on antigen presenting cells (APCs) or tumor cells. The second is a co-stimulatory signal through CD28 on T cells binding to CD80/CD86 on APCs. CTLA-4 prevents T cell activation by competing with the co-stimulatory molecule CD28 for the CD80/CD86 on APCs (36). Ipilimumab is a CTLA-4 inhibitor approved for the treatment of advanced or unresectable melanoma (36, 37). Unlike CTLA-4 expression restricted to T cells, PD-1 is expressed by activated T cells, B cells and monocytes. PD-1 binds to its two ligands, PD-L1 and PD-L2, expressed primarily by APCs and tumor cells (7). The function of PD-L2 however is not as widely known as PD-L1 (38). Activated PD-1 on T cells through PD-L1 binding counteracts the downstream signaling of the TCR and CD28 co-stimulatory signal by phosphorylating the cytoplasmic immunoreceptor tyrosine-based switch motif leading to the recruitment of Src homology region 2 domain containing phosphatases 1 and 2 (SHP1/2) and slam-associated protein (38, 39). SHP1/2 dephosphorylate the TCR and CD28 proximal signaling molecules including ZAP70 and PI3K, respectively, inhibiting T cell activation, cytokine production and promoting pro-apoptotic molecule expression, ultimately resulting in T cell anergy or apoptosis (2) (Figure 2). The overexpression of PD-L1 in many cancers causes functionally exhausted and unresponsive T cells, promoting immune evasion and tumor progression (7, 11–13) and abrogating PD-L1 expression on tumor cells can enhance sensitivity to T cell killing (40, 41). Signaling of PD-L1 intrinsically within immune cells has not been as well studied as PD-1, however macrophage treatment with anti-PD-L1 antibodies has been shown to upregulate mTOR pathway activity, and RNA-Seq analysis revealed upregulation of multiple macrophage inflammatory pathways (42).
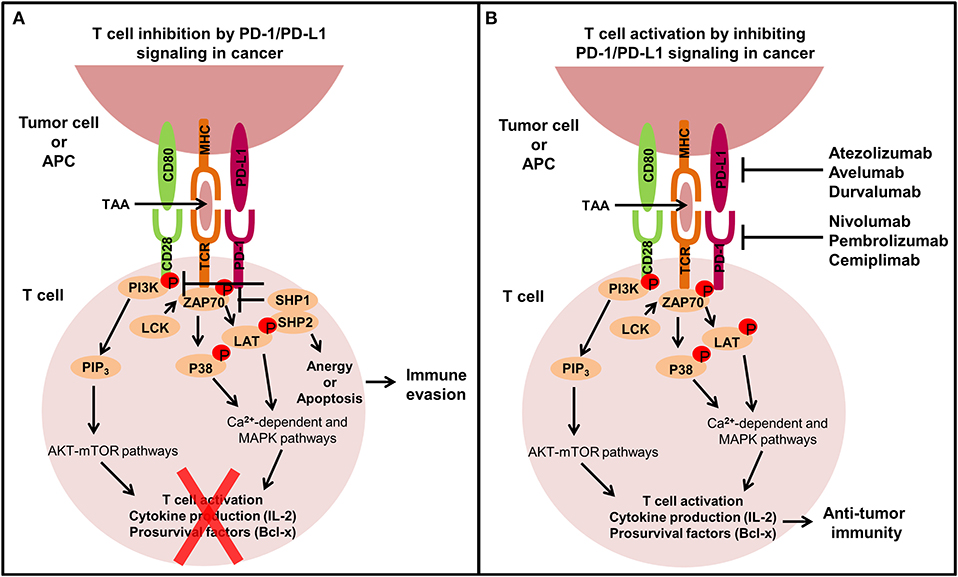
Figure 2. The extrinsic function of the PD-1/PD-L1 signaling axis in cancer. T cells play an important role in modulating immune responses against tumor cells, but tumors can exhibit immune inhibitory mechanisms like overexpressing PD-L1 to avoid T cell-mediated killing. (A) When PD-L1 binds to PD-1 expressed on the surface of T cells, T cells become inactivated through the recruitment of SHP1/2 which subsequently inhibits TCR and CD28 co-stimulatory signaling by preventing the phosphorylation of ZAP70 and PI3K, leading to T cell anergy or apoptosis and ultimately immune evasion. (B) Monoclonal antibodies targeting the PD-1/PD-L1 signaling axis have been developed to restore immune-mediated eradication of the tumor. PD-1/PD-L1 blockade allows co-stimulatory signal transduction from the TCR and CD28 on T cells upon interaction with APCs or tumor cells. TCR binding to the tumor-associated antigen (TAA) in the MHC complex leads to the phosphorylation of ZAP70, which then phosphorylates P38 and LAT resulting in activation of calcium-dependent and MAPK pathways. Simultaneously, CD80 binding to CD28 phosphorylates PI3K which activates PIP3 leading to AKT-mTOR pathway activation. These signaling pathways promote T cell activation, cytokine production and pro-survival factor expression stimulating anti-tumor immunity.
PD-1/PD-L1-targeted therapies yield remarkable anti-tumor immune responses with limited side effects in select patients with advanced cancers (24). They have shown to increase the proliferation of tumor-infiltrating lymphocytes and develop a more clonal TCR repertoire within the T cell population directed against the tumor (32). Currently there are six approved PD-1/PD-L1-targeted therapies for the treatment of multiple cancers as single agents (Table 1); some of which have gained accelerated approval and emerged as front-line treatments for some cancers (35). In 2014, the FDA approved the first anti-PD-1 monoclonal antibody, Nivolumab, for treatment of patients with unresectable or metastatic melanoma and disease progression following Ipilimumab (anti-CTLA-4), based on the CA209037 clinical trial data where Nivolumab alone achieved 31.7% objective response rate (43). Subsequently, Nivolumab was approved for the first-line treatment of metastatic melanoma (44) and second-line treatment for NSCLC (45) and renal cell carcinoma (46) following successful phase I-III clinical trials. Nivolumab has also been approved for classic Hodgkin lymphoma (47), head and neck squamous cell carcinoma (48), bladder cancer (49), and colorectal cancer with microsatellite instability or mismatch repair deficiency (50). Similarly, the anti-PD-1 monoclonal antibody Pembrolizumab is approved for the first-line treatment of metastatic melanoma (51) and NSCLC (25) and second-line treatment for metastatic head and neck squamous cell carcinoma (52) and refractory classical Hodgkin's lymphoma (53). In addition, it has also been approved for gastric/gastroesophageal junction adenocarcinomas (16), cervical cancer (54), and primary mediastinal large B-cell lymphoma (55). Pembrolizumab is also the first therapy to be approved for the treatment of all solid tumors with high mutational burden (56). More recently, Nivolumab and Pembrolizumab have gained accelerated approval for many more cancers (Table 1). Cemiplimab represents a newly approved anti-PD-1 monoclonal antibody for the treatment of metastatic cutaneous squamous cell carcinoma (57). Atezolizumab was the first anti-PD-L1 monoclonal antibody to be approved for treatment of advanced NSCLC and metastatic urothelial carcinoma. Atezolizumab promoted a tolerable and durable objective response rate of 23% and 15% in NSCLC (21, 58) and in metastatic urothelial carcinoma, respectively (22, 59), whilst anti-PD-L1 monoclonal antibodies Avelumab and Durvalumab are approved for the treatment of Merkel cell carcinoma (27, 28) and NSCLC (60), respectively and are both approved for treatment of metastatic urothelial carcinoma (26, 61). Furthermore, these PD-1/PD-L1-targeted therapies are also being investigated for treatment of colorectal, bladder, prostate and breast cancer as well as hematological malignancies and have shown promising results in the early clinical trials.
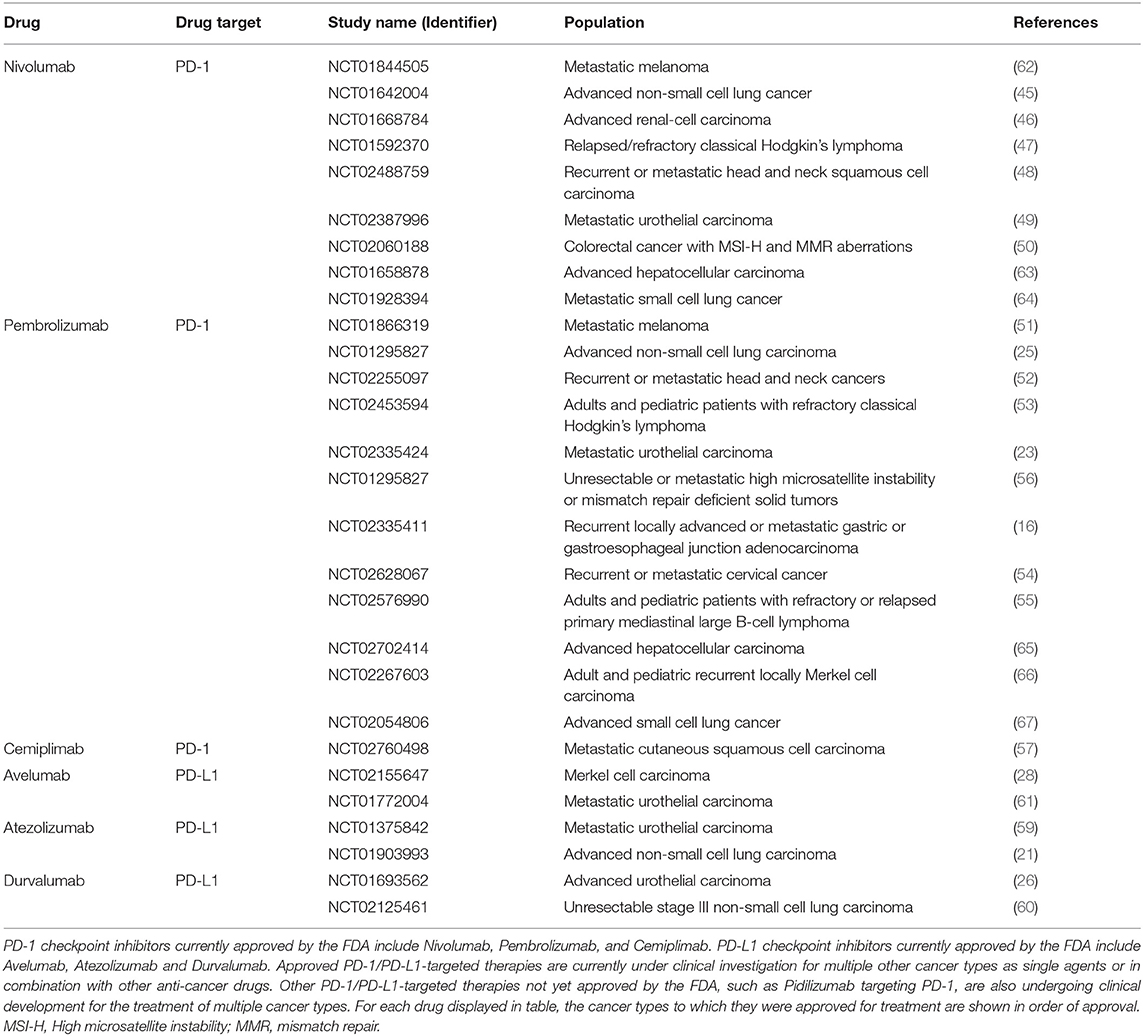
Table 1. FDA approved single agent use of PD-1/PD-L1-targeted therapy for a broad range of cancer types.
As single agents, immunotherapies targeting the PD-1/PD-L1 signaling axis have demonstrated unprecedented capabilities to elicit anti-tumor immune responses in some patients with advanced cancers (21–23), however the fact remains; there are a large percentage of non-responders or initial responders that acquire resistance (29, 68). Most research associated with PD-L1 and PD-1 has been focused on their extrinsic role to inhibit the immune system, but more recently a tumor-intrinsic role of PD-L1 and PD-1 is emerging in some cancer types; however these roles remain to be fully characterized in all cancers. Important questions to be addressed are the contribution of tumorigenic expression of PD-L1 and PD-1 to intrinsic signaling, whether monoclonal antibodies targeting the PD-1/PD-L1 signaling axis work sufficiently to block this new and emerging role of PD-L1 and PD-1 and whether the intrinsic roles of these proteins are contributing significantly to resistance, relapse to treatment, and hyperprogressive responses in patients.
The tumor-intrinsic PD-L1 pathway is aberrantly activated in many cancers (11–13, 69). There are several intrinsic and extrinsic mechanisms responsible for PD-L1 regulation in tumor cells, including genetic alterations, epigenetic modifications, oncogenic and tumor suppressor signals, inflammatory cytokines and other factors (Figure 3) (68–80).
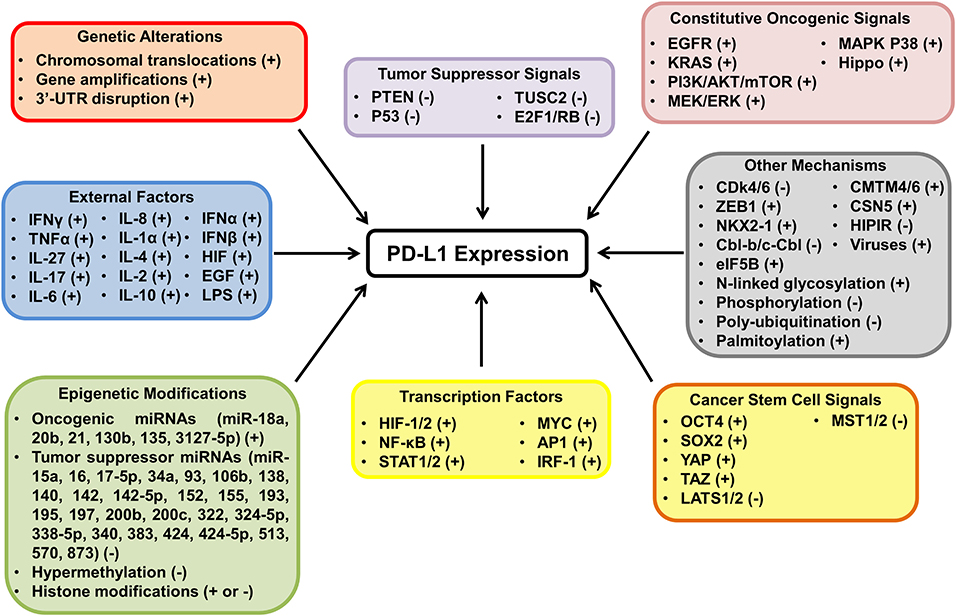
Figure 3. The mechanisms of PD-L1 activation and inactivation in cancer. The diagram highlights the many mechanisms behind PD-L1 regulation in tumor cells and whether the proposed mechanisms have been shown to upregulate (+) or downregulate (−) the expression of PD-L1. PD-L1 expression is regulated at the transcriptional, post transcriptional, translational, and post translational level in tumor cells. Many mechanisms have been shown to modulate PD-L1 expression including genetic aberrations, epigenetic modifications, oncogenic and tumor suppressor signals and extrinsic factors.
Several tumors harbor genetic aberrations of the chromosome 9p24.1 which CD274, the gene for PD-L1, resides ultimately affecting the expression of PD-L1 (72, 81, 82). Increased copy number alterations on chromosome 9p correlates directly with increased PD-L1 expression (72) and frequently occurs in primary mediastinal B-cell lymphoma (63%) (83), classical Hodgkin lymphoma (40%) (47), triple-negative breast cancer (29%) (84), and soft tissue carcinomas (21.1%) (85). A recent study analyzing 9,771 tumor samples from 22 cancer types revealed a high frequency of copy number gains in bladder, cervical, colorectal, ovarian, and head and neck cancer (more than 15% of tumors), but only a low frequency in pancreatic, renal cell, and papillary thyroid carcinoma (<5% of tumors) (72). In addition, copy number gains are also less frequently observed in gastric cancer (15%) (86), NSCLC (5.3%) (87), small cell lung cancer (1.9%) (88), and diffuse large B-cell lymphoma (3%) (89). PD-L1 copy number gains are associated with substantial therapeutic activity in some cancers due to the high levels of tumor PD-L1 and increased immune infiltrates that they have shown to promote (47, 90). There is some evidence to suggest that PD-L1 chromosomal translocations influence PD-L1 overexpression in certain diffuse large B cell lymphomas (81, 89). Disruption of the 3' untranslated region (UTR) of PD-L1 is another mechanism by which some tumors such as adult T-cell leukemia/lymphoma, diffuse large B-cell lymphoma, and gastric cancer display marked elevation of aberrant PD-L1 transcripts that have become stabilized by truncation of the 3'UTR (69, 82, 91–93). PD-L1 deletions however are more frequently observed in tumors than copy number gains (31 vs. 12%); particularly in melanoma and NSCLC where >50% of tumors harbor PD-L1 deletions (72). PD-L1 deletions, like PD-L1 copy number gains, are associated with a high tumor mutational load and poor prognosis, but the clinical significance of PD-L1 deletions is not yet clear (72, 84).
Epigenetic modifications including microRNAs (miRNAs), promoter DNA methylation, and histone modifications have been shown to modulate PD-L1 expression in different cancers (71, 79, 94–97). A number of miRNAs have been identified to directly or indirectly influence PD-L1 expression (93, 98); the majority of which inhibit PD-L1 expression by tumor cells (Figure 3). One miRNA identified across multiple cancers to inhibit PD-L1 expression is miR-200c which directly binds to the 3'UTR of PD-L1 in hepatocellular carcinoma (99), acute lymphoid leukemia (95), and NSCLC (100). Other miRNAs include: miR-140 (79, 101, 102), miR-142-5p (103, 104), miR-197 (105, 106), miR-34a (95, 107, 108), and miR-424/424-5p (109, 110). miRNAs that positively regulate PD-L1 expression include miR-135 (96) and miR-3127-5p (111) in NSCLC and miR-18a in cervical cancer (79). In colorectal cancer PTEN is directly targeted by miRNAs miR-130b, miR-20b, and miR-21 to indirectly induce PD-L1 expression via PI3K-AKT-mTOR pathway activation (71).
Recently, PD-L1 promoter methylation has been shown to negatively correlate with PD-L1 mRNA and/or protein expression in multiple cancer types including acute myeloid leukemia (78), glioblastoma (112), melanoma (113), head and neck cancer (114), colorectal cancer (115) and prostate cancer (116). The methylation status of the PD-L1 promoter has clinical significance for predicting the outcome of PD-1/PD-L1-targeted therapy (78, 97, 113–116). For example, in NSCLC patients, anti-PD-1 therapy enhanced PD-L1 promoter methylation and reduced PD-L1 expression which mediated resistance to anti-PD-1 immunotherapy Nivolumab in NSCLC patients (117). In addition, histone modifications including methylation and acetylation have been reported to modulate PD-L1 expression in some cancers (118–123). The histone methyltransferase, enhancer of zeste 2 polycomb repressive complex 2 subunit has been shown to suppress PD-L1 expression through mediating trimethylation of the PD-L1 promoter in hepatoma cells (120). Moreover, histone deacetylases have been reported to regulate PD-L1 expression in melanoma cells (122, 124, 125).
Oncogenic and tumor suppressor signaling pathways have been shown to regulate PD-L1 expression (126, 127). Oncogenic signals derived from aberrant receptors, effector molecules and transcription factors leads to the overexpression of PD-L1 by tumors and are associated with poor prognosis and patient response to PD-1/PD-L1-targeted therapy (69, 70, 127, 128). PI3K-AKT-mTOR and RAS-MAPK pathway activation is evidently linked to constitutive PD-L1 regulation in many cancers (69, 129–131). Loss of PTEN (a tumor suppressor that negatively regulates PI3K-AKT-mTOR signaling) or mutations in PIK3CA (a catalytic subunit of PI3K) leads to elevated PD-L1 expression via constitutive PI3K-ATK-mTOR pathway activation in squamous cell lung carcinoma (132, 133), NSCLC (130), gliomas (134), colorectal cancer (135), prostate cancer (136), and breast cancer (137). Some tumors harbor mutations in RAS, BRAF, and EGFR and exhibit constitutive RAS-MAPK pathway activation and consequently overexpress PD-L1 (70, 128, 129, 138). BRAF and EGFR mutations correlate with PD-L1 expression, poor prognosis and low patient response to PD-1/PD-L1-targeted therapy in melanoma (70, 138) and NSCLC (128), respectively. Moreover, oncogenic transcription factors including MYC (139), STAT (140), NFκB (141, 142), IRF-1 (143), AP-1 (144), and HIF (145, 146) have been reported to modulate PD-L1 expression at the transcriptional level. MYC expression is found elevated in 70% of cancers (147) and has recently been shown to bind to the PD-L1 promoter transcriptionally inducing PD-L1 expression (148). Similar to MYC, other oncogenic reprogramming factors have been implicated in PD-L1 regulation. OCT4 and SOX2 have both been shown to upregulate PD-L1 expression in cervical cancer (79) and hepatocellular carcinoma (149), respectively, highlighting the necessity of PD-L1 expression for tumor reprogramming functions.
Interferon gamma signaling in the tumor microenvironment is primarily responsible for PD-L1 upregulation by tumor cells in most cancer types (76, 150–154). This may be due in part to secretion of IFNγ from tumor specific T-cells within the tumor microenvironment. A study investigating IFNγ-mediated PD-L1 upregulation in multiple cancers including melanoma, renal cell carcinoma, head and neck cancer, and NSCLC, found that IFNγ was able to induce mRNA and protein PD-L1 expression by tumor cells regardless of constitutive PD-L1 expression (76). Although, IFNγ is a dominant driver of PD-L1 expression in various tumors, the mechanism by which IFNγ mediates PD-L1 upregulation appears to be distinct among different cancer types. For example, transcription factors JAK/STAT1, IRF-1 and NFκB are responsible for IFNγ-induced PD-L1 expression in hematopoietic tumors (155), lung cancer (143), and melanoma (141), respectively. IFNγ signaling is often associated with a positive patient response to PD-1/PD-L1-targeted therapy in metastatic melanoma, NSCLC, head and neck cancer, gastric cancer, and urothelial carcinoma (29, 156, 157). Moreover, loss of function mutations in molecules involved in the IFNγ signaling pathway such as JAK1, JAK2, and β2-microglobulin have been identified to render tumor cells unresponsive to IFNγ signaling and mediate intrinsic or acquired resistance to PD-1-targeted therapy (158–160).
Other inflammatory cytokines shown to promote PD-L1 expression by tumor cells include: TNFα in breast (161), prostate, colorectal cancer (162) and hepatocellular carcinoma (152); IL-27 in lung, prostate and ovarian cancer (163); and TGFβ in breast (164) and lung cancer (165). Additionally, some cytokines have been shown to work synergistically to upregulate PD-L1 expression in tumors such as TNFα with IFNγ (166) and with IL-17 (162). Besides inflammatory cytokines extrinsically modulating PD-L1 expression, hypoxia in the tumor microenvironment selectively elevates PD-L1 expression via HIF-1α activation in melanoma, breast, lung, thyroid and prostate cancer (9, 146, 167). In recent studies, HIF-2α has also been shown to correlate with PD-L1 expression in clear cell renal cell carcinoma (168, 169).
Despite the tremendous efforts of scientific researchers to provide insight into the mechanisms behind PD-L1 signal activation in cancer, the regulation of PD-L1 expression by tumors remains to be fully elucidated in all cancer types. Understanding the mechanisms of tumorigenic PD-L1 expression and signaling in different cancer types may provide therapeutic opportunities to alleviate PD-L1-induced intratumoural immunosuppression and overcome resistance to PD-1/PD-L1-targeted therapy. For greater improvement in the efficacy of PD-1/PD-L1-targeted therapy, it is necessary to identify and target tumor-intrinsic mechanisms that are both responsible for controlling PD-L1 expression and promoting tumor progression.
To date, there are less than twenty publications investigating the intrinsic role of PD-L1 in tumors; predominantly using RNA interference approaches in two dimensional (2D)-cultured mouse or human cancer cell lines and immunocompromised mouse models. There is an emerging role of PD-L1 to send pro-survival signals within tumor cells to promote cancer initiation, metastasis, development, and resistance to therapy (Figure 4). However, how these emerging pro-survival signals are conveyed intracellularly from cell surface PD-L1 is largely unknown. There is accumulating evidence that intracellular regions of PD-L1 are responsible for transducing survival signals in tumor cells (170–172). Three conserved amino acid sequences including RMLDVEKC, DTSSK, and QFEET motifs have been reported and shown to be located in the intracellular domain of PD-L1. RMLDVEKC and DTSSK motifs were reported to be associated with regulating PD-L1 stability and signal transduction due to the discovery of two specific ubiquitination sites located in the motifs (161, 171). Gato-Cañas et al., demonstrated that the RMLDVEKC motif was required to inhibit IFN-mediated cytotoxicity toward tumor cells via directly preventing STAT3 phosphorylation and caspase-mediated apoptosis. Another study also demonstrated that tumor cells expressing PD-L1 were refractory to Fas- and protein kinase inhibitor Staurosporine-mediated apoptosis (170), which could suggest that the intracellular motifs of PD-L1 may be involved in crosstalk with other signaling pathways; in particular signaling pathways that control tumor cell survival. Other studies have shown that PD-L1 agonists can induce crosslinking between PD-L1 and CD80/CD86 to transduce reverse signaling (173–175). Recently, PD-L1 has been shown to form a heterodimer with CD80, a shared ligand with CTLA-4 and CD28, in cis on APCs and tumor cells. This heterodimer was reported to weaken CD80:CTLA4 interaction, but not CD80:CD28 binding indicating that PD-L1 may prevent CTLA-4 inhibitory signals (174, 175). Furthermore, overexpression of CD80 on PD-L1 positive tumor cells was shown to blunt the pro-tumor role of PD-L1 (176). The above studies support the notation that PD-L1 reverse signaling exists in tumor cells. Research efforts should expand on this emerging concept of PD-L1 reverse signaling which has the potential to identify new mechanisms of PD-L1-targeted immunotherapy.
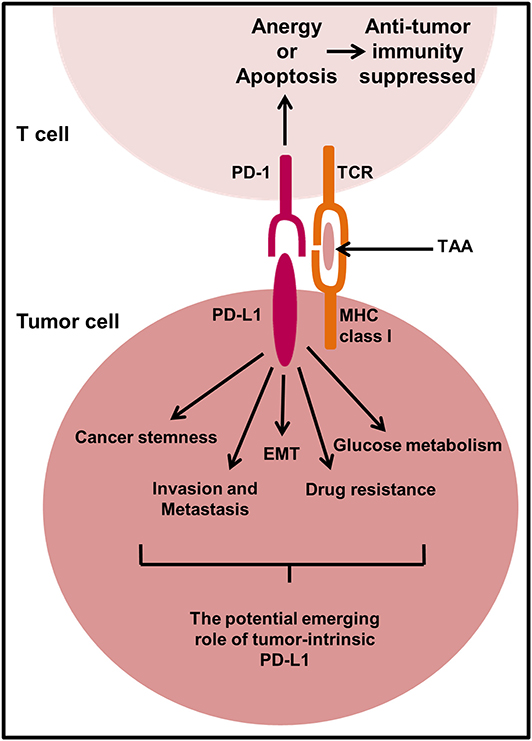
Figure 4. The proposed mechanism of action of PD-L1 in tumor cell signaling. In select cancer types, there is an emerging role of PD-L1 to send pro-survival signals in tumor cells. There is little known about the mechanisms behind PD-L1 signal transduction in tumor cells and more research is required to fully elucidate the potential mechanisms responsible. However, PD-L1 signaling in some tumor cells has been shown to promote cancer initiation, epithelial to mesenchymal transition (EMT), invasion and metastasis, regulate glucose metabolism, and contribute to drug resistance. TAA, Tumor-associated antigen.
PD-L1 expression has been shown to correlate with the cancer stem cell (CSC)-like characteristics including the expression of CD44 and/or CD133 at high levels on tumor cells. Human head and neck (177), lung (178), and colorectal (179) cancer cells that have CSC-like characteristics (CD44high/CD133high) were shown to preferentially express PD-L1 compared to CD44low/CD133low cancer cells in immunocompromised mouse models either inoculated with a patient-derived xenograft or human cancer cell lines mixed with Matrigel®, respectively. In breast and lung cancer cells CD44 was shown to be a key regulator of PD-L1 expression following shRNA-directed knockdown of CD44 in vitro and in vivo using a metastatic breast cancer xenograft mouse model (180). Additionally, primary tumor samples from breast and lung cancer patients expressed high levels of PD-L1 correlating with CD44 positivity (180), suggesting that CD44 regulation of PD-L1 expression observed in vitro could be similar to that of an in vivo human tumor.
OCT4 and Nanog are transcription factors critical for pluripotency and tumorigenesis (98). PD-L1 has been shown to promote OCT4 and Nanog expression via PI3K/AKT pathway in breast CSCs (131). PD-L1 knockdown compromised the capability of breast CSCs to self-renew themselves in vitro and in vivo using immune deficient nude mice. CSCs ability to self-renew and differentiate into heterogeneous lineages of cancer cells is thought to be responsible for drug resistance and relapse in cancer development and progression (98). A recent study showed that breast cancer stemness is regulated by miR-873 directly suppressing PD-L1 expression and thus PI3K/AKT and ERK1/2 signaling in breast cancer cells, which reduced CSC-like characteristics and enhanced chemosensitivity (181). Tumor PD-L1 has also been shown to promote the tumor-initiating cell generation in immunocompromised murine melanoma and ovarian cancer mouse models; a phenotype which was also verified in a human ovarian cancer cell xenograft mouse model (182, 183). This mechanism of intrinsic PD-L1 to drive tumor stemness was associated with increased mTORC1 signaling (182). However, CSC-like characteristics including high aldehyde dehydrogenase activity, reduced production of reactive oxygen species and a dormant state in the cell cycle were favored following knockdown of PD-L1 in cholangiocarcinoma cell tumors inoculated into mice compared to high PD-L1 expressing tumors (184), indicating that intrinsically PD-L1 may have different roles in different cancer types. Moreover, the CSC-like phenotype is shown to be associated with epithelial to mesenchymal transition (EMT) (98). Chen et al. (100) indirectly knocked down PD-L1 via the microRNA-200/ZEB1 axis in lung adenocarcinoma cells and found that PD-L1 expression correlated with EMT. Low miRNA-200 expressing cells transplanted into a syngeneic immunocompetent mouse model exhibited decreased intratumoural CD8+ T cells and increased metastatic potential due to lack of control over PD-L1 regulation.
Besides EMT playing a key role in invasion and metastasis, it has the ability to alter the tumor immune microenvironment to immunosuppressive and influence response to PD-1/PD-L1-targeted therapies (185). PD-L1 knockdown in cultured human gastric cancer cell lines SGC-7901 and AGS reduced cell proliferation, migration, invasion and induced cell cycle arrest in vitro and reduced tumor growth and EMT phenotypic marker expression in immunocompromised mice in vivo compared to gastric tumors expressing PD-L1 (186). Similarly, in cultured human Jurkat lymphoid leukemia cells and Raji lymphoma cells, PD-L1 knockdown by lentiviral transduction reduced their invasive ability via downregulation of extracellular matrix-degrading enzymes, matrix metalloproteinase 2 and 9 (187). PD-L1 silencing in murine B16 melanoma cells has also been shown to slow tumor growth and reduce metastases to the lungs of immunocompetent mice as well as immunodeficient mice via mechanisms that increase autophagy and reduce mTORC1 signaling (188). These findings may be linked to the intrinsic functions of PD-L1 to promote tumor stemness via mTORC1 signaling (182, 183). Tumor-initiating cells induced by intrinsic PD-L1 signaling are likely to show higher metastatic potential due to their self-renewal capabilities. Interestingly, the same therapeutic effect to reduce lung metastasis was absent in murine ovarian ID8agg cancer cells lacking PD-L1, in immunocompromised mice (188), suggesting the effects of intrinsic PD-L1 may be tumor specific and warrants further investigation. A recent study which knocked down PD-L1 in NCI-H1299 and Calu-1 cells showed enhanced proliferation in comparison to control cells, suggesting a tumor suppressor role of PD-L1 (15). Indeed, PD-L1 expression has been shown to correlate with EMT markers in many solid tumors including gastric, lung, breast, colon, and other common cancers (185, 189). With consideration co-targeting of EMT vulnerabilities and PD-1/PD-L1 signaling axis may have the potential to improve clinical efficacy of immunotherapy by limiting the shift of the tumor microenvironment from immunostimulatory to immunosuppressive during tumor development.
Within the tumor microenvironment, nutrient competition between tumor cells and immune cells may regulate tumor progression and PD-L1 has been reported to directly regulate the metabolism of several cancer cell lines (190, 191). Lactate derived from tumors can suppress the function of T cells by disrupting aerobic glycolysis, a process required for optimal T cell function (190). It has been reported that checkpoint blockade could induce an increase in the glucose concentration within a progressive tumor mouse model, which correlated with glycolytic capacity in tumor infiltrating lymphocytes and increased IFNγ production (191). Interestingly, treatment of B16 melanoma, MC38 colon cancer and sarcoma cancer cell lines in vitro with anti-PD-L1 antibodies was shown to reduce aerobic glycolysis mechanisms, including reduced glycolysis enzymes and Akt phosphorylation, indicating a tumor intrinsic role for PD-L1 in enhancing tumor glycolysis. The same results were achieved by shRNA mediated knockdown of PD-L1 (191), strongly suggesting that PD-L1 itself was the modulator of glycolysis in cancer cells. Hypoxic inducible factor, HIF-1α is a well-known modulator of glycolysis in cancer cells (192). The reduced glycolytic activity of cancer cells caused by PD-L1 blockade would subsequently induce an adaptive hypoxic response and stimulate the production of HIF-1α. HIF-1α also directly modulates immune cell activity in the tumor microenvironment to favor tumor growth and induces PD-L1 expression on tumor cells and immune cells; indirectly mediating immune escape and tumor progression (9). Under hypoxic conditions PD-L1 expression was directly induced by HIF-1α on MDSCs in B16-F10 tumor-bearing mice, and PD-L1 blockade increased MDSC-mediated T cell activation by downregulating IL-10 and IL-6 expression (193). Dual blockade of PD-L1 and HIF-1α could further reduce the glycolytic activity of cancer cells caused by PD-L1 blockade and enhance anti-tumor immunity, ultimately leading to cancer cell death.
PD-L1 exhibits an anti-apoptotic role in MDA-MB-231 breast cancer cells and silencing PD-L1 in these cells increased cancer cell apoptosis and enhanced cancer cell susceptibility to doxorubicin-induced apoptosis in vitro and in vivo (194), suggesting that PD-L1 not only prevents cancer cell apoptosis, but also promotes chemotherapy resistance. Likewise, CRISPR/Cas9 knockout of PD-L1 enhanced the sensitivity of human osteosarcoma KHOS and MNNG/HOS cells to doxorubicin and paclitaxel and compromised their ability to form three-dimensional (3D) spheroids in vitro (195). Further characterization of the role of PD-L1 in chemotherapy resistance in MDA-MB-231 breast cancer cells discovered that PD-L1 knockdown suppresses the expression of multidrug resistance 1/P-glycoprotein (MDR1/P-gp) via PI3K/AKT pathway in vitro (196); recognizing this has an additional therapeutic target. In fact, PD-1/PD-L1 interaction increased survival of breast cancer cells when exposed to doxorubicin (196), suggesting that PD-1/PD-L1-targeted therapy may increase chemotherapy efficacy by inhibiting MDR1/P-gp expression which usually confers resistance in breast cancer cells. Moreover, through culturing of breast (MDA-MB-231 and 4T1) and prostate (DU145) cancer cell lines with recombinant PD-1 or Jurkat T cells it has been shown how PD-1/PD-L1 interactions results in increased resistance to doxorubicin and docetaxel (197). Subsequent knockdown or blockade of PD-1 restored tumor cell chemo-sensitivity and reduced their metastatic potential in a synergistic breast cancer mouse model, suggesting blockade of intrinsic pathways is beneficial for therapy. Conversely, human colorectal cancer cells harboring a BRAFV600E mutation showed that the depletion of PD-L1 suppresses chemotherapy-induced apoptosis through the down regulation of BIM and BIK BH3-only proteins (198), even though depletion alone reduced tumor growth. The effect of PD-L1 on chemosensitivity was confirmed in BRAFV600E mutant MC38 murine tumor xenografts, where PD-L1 knockout cells were less sensitive to chemotherapy due to the suppression of pro-apoptotic molecules, BIM and BIK, compared to parental cells expressing PD-L1. This study highlights the importance of understanding the role of PD-L1 in each cancer type and its subtypes to design effective treatment regimens that will benefit cancer patients.
The tumor-intrinsic role of PD-L1 appears to be similar across all cancer types investigated in the literature to date, with the exceptions of cholangiocarcinoma and contradictory evidence in lung cancer, in that PD-L1 promotes tumor growth and development. However, the molecular mechanisms of PD-L1 exerting pro-tumor activity appear to be distinct amongst different cancer types. Notably, in the studies that have investigated the intrinsic role of PD-L1 in lung cancer the cells utilized were mesenchymal lung cancer cell lines which harbored KRAS and/or p53 mutations, suggesting that the tumor cells metastatic capacity and mutational status may not be determining factors as to whether PD-L1 exhibits a pro-tumor or anti-tumor role in lung cancer. Furthermore, Wang et al. (15) reported that PD-L1 expression reduced lung cancer cell proliferation, suggesting that although PD-L1 expression may limit tumor cell proliferation it is still affecting other tumor characteristics that influence tumor progression. The reasons behind this potential role of PD-L1 in lung cancer warrants further investigation.
Similar to PD-L1, the expression of PD-1 on T cells and its role to inhibit the immune system is well characterized, but recent studies have found intrinsic expression of PD-1 in tumor cells including melanoma (199), hepatic carcinoma cells (200), ovarian (201), bladder (201), lung (15, 202), and colorectal (15) cancer cells. In melanoma B16 tumors, a subpopulation of PD-1 expressing cancer cells were identified to modulate downstream mTOR signaling and promote tumorigenesis independent of adaptive immunity, in an in vivo mouse model lacking an adaptive immune system (Figure 5A) (199). This effect was abrogated with anti-PD-1 therapy, tumor-specific PD-1 knockdown and mutagenesis of intracellular signaling motifs downstream of PD-1, strongly suggesting an intrinsic function of PD-1 to promote tumorigenesis in melanoma. Similar to intrinsic PD-1 in melanoma cells, intrinsic PD-1 in liver cancer cells has been reported to mediate tumorigenesis in immunocompromised mice via regulating mTOR signaling (Figure 5A) (200) and thus combined inhibition of PD-1 and mTOR may be a potential therapeutic strategy for melanoma and liver cancer. Moreover, anti-PD-1 therapy has been reported to reduce the cell growth of bladder RT4 cancer cells cultured in 2D in the absence of adaptive immunity (201), implying that PD-1 expression is potentially oncogenic. Interestingly in murine NSCLC M109 cells, intrinsic PD-1 exhibited an anti-tumor role in immunocompromised mice and when NSCLC cells were treated with anti-PD-1 therapy they demonstrated increased proliferation and tumor growth (Figure 5B) (202). Consistent with this, silencing of PD-1 or therapeutic antibody blockade of PD-1 on the surface of NSCLC and colorectal cancer cells increased proliferation in vitro via activating PI3K and MAPK pathways (15), suggesting that PD-1 could be involved in development of resistance to immunotherapy blockade in NSCLC and could provide one explanation for why patients with NSCLC can display hyperprogressive disease following treatment with anti-PD-1 therapy (15, 203). The latter findings also suggest that the tumor suppressor role of PD-1 on cancer cells may not be limited to NSCLC. Although, Wang et al., demonstrated that PI3K and MAPK pathways were activated following anti-PD-1 therapy in NSCLC cells in vitro and in vivo, their study also showed that PD-1/PD-L1 dysfunction did not activate mTOR, illustrating that the mechanism behind tumor-intrinsic PD-1 to either induce or inhibit tumor growth may be different. However, mTOR activation has been shown to occur in the only two studies investigating tumor-intrinsic PD-1 where PD-1 has a pro-tumor role, which may suggest that mTOR signal activation is necessary for PD-1 to exhibit tumorigenic activity. Therefore, the molecular mechanism behind tumor-intrinsic PD-1 needs to be elucidated in other cancer types to confirm this potential role of mTOR in PD-1 signaling in tumor cells. Furthermore, studies investigating the role of tumor-intrinsic PD-1 in tumors have utilized tumor cell lines that exhibit invasive and metastatic potential. The metastatic potential of cells does not seem to be a factor in determining whether PD-1 is pro- or anti-tumorigenic and nor is it associated with enhanced tumor-intrinsic PD-1 activity (199). Additionally, studies have used both poorly- and well-differentiated tumor cells which have been shown to have the same PD-1-intrinsic function, implying that the differentiated state of the cell is also not a contributing factor to the role PD-1 in tumors. Yao et al., reanalyzed cancer transcriptomic and proteomic data from The Cancer Genomic Atlas Project and The Cancer Cell Line Encyclopedia Dataset to find that tumor-intrinsic PD-1 expression is widespread in many cancer types. This heterogeneity may explain the differential therapeutic effects of anti-PD-1 drugs and could provide crucial information required when selecting suitable patients for treatment dependent on the cancer cell type. However, further work in different cancers and tumor models could also shed more light into this area.
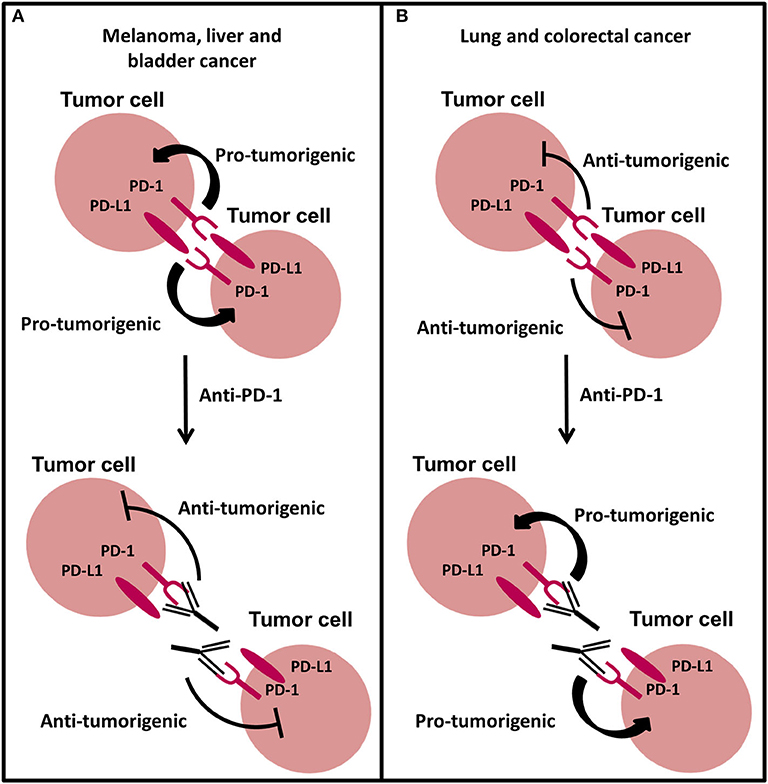
Figure 5. The new and emerging role of PD-1 signaling in cancer. (A) Intrinsic PD-1 signaling has been shown to promote tumorigenesis in melanoma, liver, and bladder cancer cells. Anti-PD-1 therapy abrogates this effect inhibiting tumor growth. (B) Intrinsic PD-1 signaling in NSCLC and colorectal cancer cells has been shown to inhibit tumorigenesis. Anti-PD-1 therapy preventing PD-1 signaling promotes tumor progression in NSCLC and colorectal cancer cells.
So far, most evidence for PD-L1 and PD-1 signaling in cancer cells is based on 2D cell culture models using murine and human cancer cells and immunodeficient mouse models that can fail to fully recapitulate the human in vivo tumor (24, 204). Therefore, more relevant models capable of recapitulating the heterogeneity of the tumor microenvironment during in vivo conditions could allow further predictive in vitro evaluation of the tumor-intrinsic role of PD-L1 and PD-1, and how these roles may be affected by immunotherapy treatment and influence immune cell function.
Recent reports discussed above suggest that the emerging intrinsic role of PD-L1 is largely pro-tumorigenic in a number of cancers, but that in lung cancer and cholangiocarcinoma, it may act as a tumor suppressor gene. Likewise, the new emerging tumor intrinsic role of PD-1 has also been reported to have differential roles in different cancer cell types and this remains to be further investigated. However, there are currently a limited number of reports investigating how immunotherapeutic drugs potentially modulate these intrinsic pathways. Theivanthiran et al. (205) demonstrated that PD-1 blockade on CTLs in a syngeneic mouse model was able to activate a PD-L1-NLRP3 inflammasome signaling pathway in tumor cells that promoted MDSC recruitment and infiltration into the tumor microenvironment. Intratumoural MDSCs can suppress T cell function (193) and thus may dampen the immune response and promote resistance to anti-PD-1 therapies. The effect of the immunotherapy drug Atezolizumab was measured on MDA-MB-231 breast cancer cells (206). In this study, RNA-Seq was utilized to assess the modulation of gene expression after treatment with Atezolizumab and it was reported that genes promoting cell migration, metastasis, EMT, cell growth, and hypoxia were downregulated whilst apoptosis genes were upregulated. This suggests that Atezolizumab may be able to modulate the signaling of PD-L1 in this cell line to some extent at the level of gene expression. In contrast, Wang et al. (15), investigated the effects of anti-PD-1 antibodies Nivolumab and Pembrolizumab or the anti-PD-L1 antibody Atezolizumab on Calu-1, SW480, HT-29, BxPC-3, SK-BR-3, and U-2 OS cells. All immunotherapy drugs were shown to increase cell proliferation compared to isotype control in vitro. To verify these findings in vivo, human lung cancer cells were inoculated into immunocompromised mice. Similar to in vitro studies, monoclonal antibody administration to block PD-1 or PD-L1 activated PI3K and MAPK pathways by phosphorylating AKT and ERK1/2, respectively, promoting tumor cell growth in vivo. These small numbers of studies suggests that immunotherapeutic antibodies may be able to modulate the intrinsic function of PD-L1 and PD-1 and potentially highlights another mechanism by which tumors may develop resistance to PD-1/PD-L1 targeting therapy through co-expressing PD-L1 and its receptor PD-1. The ability of immunotherapy drugs to modulate the intrinsic PD-L1 and PD-1 pathway in other cancers in more heterogeneous tumor models could also provide further important insight into the mechanism of immunotherapy treatment.
Tumor heterogeneity makes it challenging to identify novel therapeutic targets and potential biomarkers of immunotherapy response that could substantially enhance therapeutic efficacy. The scientific basis for numerous clinical trials has derived from 2D cell culture models and animal models, which can fail to fully replicate the human tumor microenvironment due to lack of heterogeneity and species-to-species variability, respectively, which could account for lack of transferability of PD-1/PD-L1-targeted antibodies into the clinic (24, 204). Furthermore, most evidence to date exploring the intrinsic role of PD-L1 and PD-1 has been based on 2D cell culture models using murine or human cancer cell lines or animal models, and thus limit the capacity to explore these roles in a relevant human tumor setting. Given the emerging intrinsic roles of PD-L1 and PD-1 and the differences between cancer types, utilizing models which closely mimic the heterogeneity of the human tumor microenvironment could allow a more predictive in vitro evaluation of the intrinsic role of PD-L1 and PD-1 in cancer and modulation by anti-cancer therapeutics. For example, human cancer cells implemented into different 3D cell culture models have shown to exhibit characteristics that more closely mimic in vivo human tumors, such as changes in morphology, proliferation, gene and protein expression, and response to treatment (204). Indeed the modulation of PD-L1 expression has been reported to be affected by the extracellular matrix stiffness of tumors in 3D culture (207) and a 3D model system utilizing patient-derived organoids that resembled the tumor immune microenvironment for the study of the PD-1/PD-L1 signaling axis has been developed (208). Furthermore, in a recent study a tumor-immune co-culture was utilized to assess the efficacy of immunotherapies Nivolumab and Durvalumab (209).
PD-1/PD-L1 checkpoint blockade is at the cutting edge of research offering cancer patients hope for new treatment regimens with potential to have substantial clinical benefit and prolong survival. PD-1/PD-L1-targeted therapies reactivate the immune system to induce immune-mediated tumor eradication, and although they have demonstrated success has single agents, they have also shown cooperation with conventional and targeted therapies in the clinic. Unfortunately, most patients are unresponsive or develop resistance to PD-1/PD-L1-targeted therapy. Further elucidating the tumor intrinsic role of PD-L1 and its receptor PD-1 in all cancer types will help understand the basis for or lack of response to immunotherapy and may allow the identification of novel therapeutic targets and biomarkers to enhance clinical efficacy.
KH and RL researched and wrote the manuscript. KH designed the figures. RL conceived the theme/direction. NC and NJ-M reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This project was funded by the Biomolecular Sciences Research Centre at Sheffield Hallam University. Open access funds are supported by Sheffield Hallam University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank the Biomolecular Sciences Research Centre and Sheffield Hallam University for funding this work.
NK, Natural killer cells; Tregs, Regulatory T cells; MDSCs, Myeloid-derived suppressor cells; PD-L1, Programmed death-ligand 1; PD-1, Programmed death 1; FDA, USA Food and Drug Administration; NSCLC, Non-small cell lung cancer; CTLA-4, Cytotoxic T lymphocyte antigen-4; TCR, T cell receptor; APCs, Antigen presenting cells; PD-L2, Programmed death-ligand 2; SHP1/2, Src-homology 2 domain-containing phosphatase 1/2; miRNA, microRNA; 3'UTR, 3’ untranslated region, ; GSK3β, Glycogen synthase kinase 3β; S/T, Serine/Threonine; ccRCC, Clear cell renal cell carcinoma; ER, Endoplasmic reticulum; 2D, Two-dimensional; CSC, Cancer stem cell; EMT, Epithelial to mesenchymal transition; 3D, Three-dimensional; MDR1/P-gp, Multidrug resistant 1/P-glycoprotein.
1. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
2. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. (2019) 18:10. doi: 10.1186/s12943-018-0928-4
3. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumour microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
4. Hanahan D, Weinberg RA. Hallmark of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. (2014) 65:185–202. doi: 10.1146/annurev-med-092012-112807
6. Teng MWL, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. (2015) 125:3338–46. doi: 10.1172/JCI80004
7. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
8. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
9. Johnson RMG, Dong H. Functional expression of programmed death-ligand 1 (B7-H1) by immune cells and tumor cells. Front Immunol. (2017) 8:961. doi: 10.3389/fimmu.2017.00961
10. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1 negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov. (2019) 9:1422–37. doi: 10.1158/2159-8290.CD-18-1259
11. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumour cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. (2010) 116:1757–66. doi: 10.1002/cncr.24899
12. Maine CJ, Aziz NH, Chatterjee J, Hayford C, Brewig N, Whilding L, et al. Programmed death ligand-1 over-expression correlates with malignancy and contributes to immune regulation in ovarian cancer. Cancer Immunol Immun. (2013) 63:215–24. doi: 10.1007/s00262-013-1503-x
13. Muenst S, Schaerli AR, Gao F, Däster S, Trella E, Droeser RA, et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat. (2014) 146:15–24. doi: 10.1007/s10549-014-2988-5
14. Yao H, Wang H, Li C, Fang JY, Xu J. Cancer cell-intrinsic PD-1 and implications in combinatorial immunotherapy. Front Immunol. (2018) 9:1774. doi: 10.3389/fimmu.2018.01774
15. Wang X, Yang X, Zhang C, Wang Y, Cheng T, Duan L, et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. PNAS. (2020) 117:6640–50. doi: 10.1073/pnas.1921445117
16. Fashoyin-Aje L, Donoghue M, Chen H, He K, Veeraraghavan J, Goldberg KB, et al. FDA Approval Summary: Pembrolizumab for Recurrent Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Adenocarcinoma Expressing PD-L1. Oncologist. (2019) 24:103–9. doi: 10.1634/theoncologist.2018-0221
17. Zeng Z, Shi F, Zhou L, Zhang MN, Chen Y, Chang XJ, et al. Upregulation of circulating PD-L1/PD-1 is associated with poor post-cryoablation prognosis in patients with HBV-related hepatocellular carcinoma. PLoS ONE. (2011) 6:e23621. doi: 10.1371/journal.pone.0023621
18. Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. (2006) 66:3381–5. doi: 10.1158/0008-5472
19. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. (2007) 13:2151–7. doi: 10.1158/1078-0432.CCR-06-2746
20. Inman BA, Sebo TJ, Frigola X, Dong H, Bergstralh EJ, Frank I, et al. PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer. (2007) 109:1499–505. doi: 10.1002/cncr.22588
21. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. The Lancet. (2016) 387:1837–46. doi: 10.1016/S0140-6736(16)00587-0
22. Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. (2016) 387:1909–20. doi: 10.1016/S0140-6736(16)00561-4
23. Balar AV, Castellano D, O'Donnell PH, Grivas P, Vuky J, Powels T, et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. (2017) 18:1483–92. doi: 10.1016/S1470-2045(17)30616-2
24. Wang D, Lin J, Yang X, Long J, Bai Y, Yang X, et al. Combination regimens with PD-1/PD-L1 immune checkpoint inhibitors for gastrointestinal malignancies. J Hematol Oncol. (2019) 12:42. doi: 10.1186/s13045-019-0730-9
25. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
26. Powles T, O'Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol. (2017) 3:e172411. doi: 10.1001/jamaoncol.2017.2411
27. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. (2016) 17:1374–85. doi: 10.1016/S1470-2045(16)30364-3
28. Kaufman HL, Russell JS, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Updated efficacy of avelumab in patients with previously treated metastatic merkel cell carcinoma after greater than or equal to 1 year of follow-up: JAVELIN merkel 200, a phase 2 clinical trial. J ImmunoTher Cancer. (2018) 6:7. doi: 10.1186/s40425-017-0310-x
29. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. (2018) 17:129. doi: 10.1186/s12943-018-0864-3
30. Chocarro de Erauso L, Zuazo M, Arasanz H, Bocanegra A, Hernandez C, Fernandez G, et al. Resistance to PD-L1/PD-1 blockade immunotherapy. A tumor-intrinsic or tumor-extrinsic phenomenon? Front Pharmacol. (2020) 11:441. doi: 10.3389/fphar.2020.00441
31. Bai J, Goa Z, Li X, Dong L, Han W, Nie J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget. (2017) 8:110693–707. doi: 10.18632/oncotarget.22690
32. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
33. Kocikowski M, Dziubek K, Parys M. Hyperprogression under immune checkpoint-based immunotherapy-current understanding, the role of PD-1/PD-L1 tumour-intrinsic signalling, future directions and a potential large animal model. Cancers (Basel). (2020) 12:e804. doi: 10.3390/cancers12040804
34. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways similarities, differences and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
35. Khair DO, Bax HJ, Mele S, Crescioli S, Pellizzari G, Khiabany A, et al. Combining immune checkpoint inhibitors: established and emerging targets and strategies to improve outcomes in melanoma. Front Immunol. (2019) 10:453. doi: 10.3389/fimmu.2019.00453
36. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
37. Pacheco JM, Camidge DR, Doebele RC, Schenk E. a changing of the guard: immune checkpoint inhibitors with and without chemotherapy as first line treatment for metastatic non-small cell lung cancer. Front Oncol. (2019) 9:195. doi: 10.3389/fonc.2019.00195
38. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
39. Peled M, Tocheva AS, Sandigursky S, Nayak S, Philips EA, Nichols KE, et al. Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc Natl Acad Sci USA. (2018) 115:E468–77. doi: 10.1073/pnas.1710437115
40. Teo PY, Yang C, Whilding LM, Parente-Pereira AC, Maher J, George AJT, et al. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: strategies to enhance T cell killing. Adv Healthc Mater. (2015) 4:1180–9. doi: 10.1002/adhm.201500089
41. Wu Y, Gu W, Li J, Chen C, Xu ZP. Silencing PD-1 and PD-L1 with nanoparticle-delivered small interfering RNA increases cytotoxicity of tumour-infiltrating lymphocytes. Nanomedicine. (2019) 14:955–67. doi: 10.2217/nnm-2018-0237
42. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. (2018) 6:1260–73. doi: 10.1158/2326-6066.CIR-17-0537
43. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. (2015) 16:375–84. doi: 10.1016/S1470-2045(15)70076-8
44. Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. (2017) 377:1824–35. doi: 10.1056/NEJMoa1709030
45. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhart WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. (2015) 373:1803–13. doi: 10.1056/NEJMoa1504627
46. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. (2015) 373:1803–13. doi: 10.1056/NEJMoa1510665
47. Ansell SM, Lesokhin AM, Borello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with Nivolumab in relapsed and refractory Hodgkin's lymphoma. N Engl J Med. (2015) 372:1277–90. doi: 10.1056/NEJMoa1411087
48. Ferris RL, Goncalves A, Baxi SS, Martens UM, Gauthier H, Langenburg M, et al. LBA46 - an open-label, multicohort, phase 1/2 study in patients with virus-associated cancers (CheckMate 358): safety and efficacy of neoadjuvant nivolumab in squamous cell carcinoma of the head and neck. Ann Oncol. (2017) 28:605–49. doi: 10.1093/annonc/mdx440.041
49. Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2017) 18:312–22. doi: 10.1016/S1470-2045(17)30065-7
50. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. (2017) 18:1182–91. doi: 10.1016/S1470-2045(17)30422-9
51. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
52. Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. J Clin Oncol. (2017) 35:1542–9. doi: 10.1200/JCO.2016.70.1524
53. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic hodgkin lymphoma. J Clin Oncol. (2017) 35:2125–32. doi: 10.1200/JCO.2016.72.1316
54. Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. (2019) 37:1470–8. doi: 10.1200/JCO.18.01265
55. Zinzani P, Thieblemont C, Melnichenko V, Osmanov D, Bouabdallah K, Walewski J, et al. Efficacy and safety of pembrolizumab in relapsed/refractory primary mediastinal large B-cell lymphoma (rrPMBCL): interim analysis of the KEYNOTE-170 phase 2 trial. Hematol Oncol. (2017) 35:62–3. doi: 10.1002/hon.2437_49
56. Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumours. Clin Cancer Res. (2015) 21:4286–93. doi: 10.1158/1078-0432.CCR-14-2607
57. Markham A, Duggan S. Cemiplimab: first global approval. Drugs. (2018) 78:1841–6. doi: 10.1007/s40265-018-1012-5
58. Horn L, Gettinger SN, Gordon MS, Herbst RS, Gandi L, Felip E, et al. Safety and clinical activity of atezolizumab monotherapy in metastatic non-small-cell lung cancer: final results from a phase I study. Eur J Cancer. (2018) 101:201–9. doi: 10.1016/j.ejca.2018.06.031
59. Petrylak DP, Powles T, Bellmunt J, Braiteh FY, Morales-Barrera R. Atezolizumab (MPDL3280A) monotherapy for patients with metastatic urothelial cancer long-term outcomes from a phase 1 study. JAMA Oncol. (2018) 4:537–44. doi: 10.1001/jamaoncol.2017.5440
60. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
61. Patel MR, Ellerton J, Infante JR, Agrawal M, Gordon M, Aljumaily R, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol. (2017) 19:51–64. doi: 10.1016/S1470-2045(17)30900-2
62. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey L, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:23–4. doi: 10.1056/NEJMoa1504030
63. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. (2017) 389:2492–502. doi: 10.1016/S0140-6736(17)31046-2
64. Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. (2016) 17:883–95. doi: 10.1016/S1470-2045(16)30098-5
65. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. KEYNOTE-224 investigators. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. (2018) 19:940–52. doi: 10.1016/S1470-2045(18)30351-6
66. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. (2016) 374:2542–52. doi: 10.1056/NEJMoa1603702
67. Ott PA, Elez E, Hiret S, Kim DW, Morosky A, Saraf S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the phase Ib KEYNOTE-028 study. J Clin Oncol. (2017) 5:3823–9. doi: 10.1200/JCO.2017.72.5069
68. Wang H, Yao H, Li C, Shi H, Lan J, Li Z, et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat Chem Biol. (2019) 15:42–50. doi: 10.1038/s41589-018-0161-x
69. Ju X, Zhang H, Zhou Z, Wang Q. Regulation of PD-L1 expression in cancer and clinical implications in immunotherapy. Am J Cancer Res. (2020) 10:1–11.
70. Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. (2013) 19:598–609. doi: 10.1158/1078-0432.CCR-12-2731
71. Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b,−21, and−130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. (2014) 75:348–53. doi: 10.1016/j.humimm.2014.01.006
72. Budczies J, Bockmayr M, Denkert C, Klauschen F, Gröschel S, Darb-Esfahani S, et al. Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274) – associations with gene expression, mutational load, and survival. Gene Chromosome Canc. (2016) 55:626–39. doi: 10.1002/gcc.22365
73. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. (2016) 7:12632. doi: 10.1038/ncomms12632
74. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. (2017) 549:106–10. doi: 10.1038/nature23669
75. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. (2018) 553:91–5. doi: 10.1038/nature25015
76. Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. JITC. (2019) 7:305. doi: 10.1186/s40425-019-0770-2
77. Suresh S, Chen B, Zhu J, Golden RJ, Lu C, Evers BM, et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nature Cancer. (2020) 1:533–45. doi: 10.1038/s43018-020-0056-0
78. Goltz D, Gevensleben H, Grünen S, Dietrich J, Kristiansen G, Landsberg J, et al. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia. (2017) 31:738–43. doi: 10.1038/leu.2016.328
79. Dong P, Xiong Y, Yu J, Chen L, Tao T, Yi S, et al. Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene. (2018) 37:5257–68. doi: 10.1038/s41388-018-0347-4
80. Wang S, Xu L, Che X, Li C, Xu L, Hou K, et al. E3 ubiquitin ligases Cbl-b and c-Cbl downregulate PD-L1 in EGFR wild-type non-small cell lung cancer. FEBS Lett. (2018) 592:621–30. doi: 10.1002/1873-3468.12985
81. Twa DDW, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. (2014) 123:2062–5. doi: 10.1182/blood-2013-10-535443
82. Kogure Y, Kataoka K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. (2017) 108:1719–25. doi: 10.1111/cas.13303
83. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. (2010) 116:3268–77. doi: 10.1182/blood-2010-05-282780
84. Barrett MT, Anderson KS, Lenkiewicz E, Andreozzi M, Cunliffe HE, Klassen CL, et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget. (2015) 6:26483–93. doi: 10.18632/oncotarget.4494
85. Budczies J, Mechtersheimer G, Denkert C, Klauschen F, Mughal SS, Chudasama P, et al. PD-L1 (CD274) copy number gain, expression, and immune cell infiltration as candidate predictors for response to immune checkpoint inhibitors in soft-tissue sarcoma. OncoImmunology. (2017) 6:e1279777. doi: 10.1080/2162402X.2017.1279777
86. Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. (2014) 513:202–9. doi: 10.1038/nature13480
87. Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J Thorac Oncol. (2016) 11:62–71. doi: 10.1016/j.jtho.2015.09.010
88. George J, Saito M, Tsuta K, Iwakawa R, Shiraishi K, Scheel AH, et al. Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin Cancer Res. (2017) 23:1220–6. doi: 10.1158/1078-0432.CCR-16-1069
89. Georgiou K, Chen L, Berglund M, Ren W, de Miranda NF, Lisboa S, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. (2016) 127:3026–34. doi: 10.1182/blood-2015-12-686550
90. Inoue Y, Yoshimura K, Mori K, Kurabe N, Kahyo T, Mori H, et al. Clinical significance of PD-L1 and PD-L2 copy number gains in non-small-cell lung cancer. Oncotarget. (2016) 7:32113–28. doi: 10.18632/oncotarget.8528
91. Wang W, Sun J, Li F, Li R, Gu Y, Liu C, et al. A frequent somatic mutation in CD274 3'-UTR leads to protein over-expression in gastric cancer by disrupting miR-570 binding. Hum Mutat. (2012) 33:480–4. doi: 10.1002/humu.22014
92. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature. (2016) 534:402–6. doi: 10.1038/nature18294
93. Shen X, Zhang L, Li J, Li Y, Wang Y, Xu ZX. Recent findings in the regulation of programmed death ligand 1 expression. Front Immunol. (2019) 10:1337. doi: 10.3389/fimmu.2019.01337
94. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC inhibitors enhance T cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2016) 22:4119–32. doi: 10.1158/1078-0432.CCR-15-2584
95. Pyzer AR, Stroopinsky D, Rosenblatt J, Anastasiadou E, Rajabi H, Washington A, et al. MUC1 inhibition leads to decrease in PD-L1 levels via upregulation of miRNAs. Leukemia. (2017) 31:2780–90. doi: 10.1038/leu.2017.163
96. Wang N, Zhang T. Down-regulation of microRNA-135 promotes sensitivity of non-small cell lung cancer to gefitinib by targeting TRIM16. Oncol Res. (2018) 26:1005–14. doi: 10.3727/096504017X15144755633680
97. Lv D, Xing C, Cao L, Zhuo Y, Wu T, Gao N. PD-L1 gene promoter methylation represents a potential diagnostic marker in advanced gastric cancer. Oncol Lett. (2020) 19:1223–34. doi: 10.3892/ol.2019.11221
98. Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front Oncol. (2018) 8:386. doi: 10.3389/fonc.2018.00386
99. Sun C, Lan P, Han Q, Huang M, Zhang Z, Xu G, et al. Oncofetal gene SALL4 reactivation by hepatitis B virus counteracts miR-200c in PD-L1-induced T cell exhaustion. Nat Commun. (2018) 9:1241. doi: 10.1038/s41467-018-03584-3
100. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. (2014) 5:5241. doi: 10.1038/ncomms6241
101. Xie WB, Liang LH, Wu KG, Wang LX, He X, Song C, et al. MiR-140 expression regulates cell proliferation and targets PD-L1 in NSCLC. Cell Physiol Biochem. (2018) 46:654–63. doi: 10.1159/000488634
102. Ji X, Wang E, Tian F. MicroRNA-140 suppresses osteosarcoma tumor growth by enhancing anti-tumor immune response and blocking mTOR signaling. Biochem Biophys Res Commun. (2018) 495:1342–8. doi: 10.1016/j.bbrc.2017.11.120
103. Jia L, Xi Q, Wang H, Zhang Z, Liu H, Cheng Y, et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem Biophys Res Commun. (2017) 488:425–31. doi: 10.1016/j.bbrc.2017.05.074
104. Kao SC, Cheng YY, Williams M, Kirschner MB, Madore J, Lum T, et al. Tumor suppressor microRNAs contribute to the regulation of PD-L1 expression in malignant pleural mesothelioma. J Thorac Oncol. (2017) 12:1421–33. doi: 10.1016/j.jtho.2017.05.024
105. Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther. (2015) 23:717–27. doi: 10.1038/mt.2015.10
106. Ahn H, Yang JM, Kim H, Chung JH, Ahn SH, Jeong WJ, et al. Clinicopathologic implications of the miR-197/PD-L1 axis in oral squamous cell carcinoma. Oncotarget. (2017) 8:66178–94. doi: 10.18632/oncotarget.19842
107. Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. (2015) 27:443–52. doi: 10.1016/j.cellsig.2014.12.003
108. Wang Y, Wang L. miR-34a attenuates glioma cells progression and chemoresistance via targeting PD-L1. Biotechnol Lett. (2017) 39:1485–92. doi: 10.1007/s10529-017-2397-z
109. Xu S, Tao Z, Hai B, Liang H, Shi Y, Wang T, et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun. (2016) 7:11406. doi: 10.1038/ncomms11406
110. Dastmalchi N, Hosseinpourfeizi MA, Khojasteh SMB, Baradaran B, Safaralizadeh R. Tumor suppressive activity of miR-424-5p in breast cancer cells through targeting PD-L1 and modulating PTEN/PI3K/AKT/mTOR signaling pathway. Life Sci. (2020) 2020:118239. doi: 10.1016/j.lfs.2020.118239
111. Tang D, Zhao D, Wu Y, Yao R, Zhou L, Lu L, et al. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J Cell Mol Med. (2018) 22:3847–56. doi: 10.1111/jcmm.13657
112. Heiland DH, Haaker G, Delev D, Mercas B, Masalha W, Heynckes S, et al. Comprehensive analysis of PD-L1 expression in glioblastoma multiforme. Oncotarget. (2017) 8:42214–25. doi: 10.18632/oncotarget.15031
113. Micevic G, Thakral D, McGeary M, Bosenberg MW. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. (2018) 32:435–40. doi: 10.1111/pcmr.12745
114. Franzen A, Vogt TJ, Müller T, Dietrich J, Schröck A, Golletz C, et al. PD-L1 (CD274) and PD-L2 (PDCD1LG2) promoter methylation is associated with HPV infection and transcriptional repression in head and neck squamous cell carcinomas. Oncotarget. (2017) 9:641–50. doi: 10.18632/oncotarget.23080
115. Goltz D, Gevensleben H, Dietrich J, Dietrich D. PD-L1 (CD274) promoter methylation predicts survival in colorectal cancer patients. Oncoimmunology. (2017) 6:e1257454. doi: 10.1080/2162402X.2016.1257454
116. Gevensleben H, Holmes EE, Goltz D, Dietrich J, Sailer V, Ellinger J, et al. PD-L1 promoter methylation is a prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients following radical prostatectomy. Oncotarget. (2016) 7:79943–55. doi: 10.18632/oncotarget.13161
117. Zhang Y, Xiang C, Wang Y, Duan Y, Liu C, Zhang Y. PD-L1 promoter methylation mediates the resistance response to anti-PD-1 therapy in NSCLC patients with EGFR-TKI resistance. Oncotarget. (2017) 8:101535–44. doi: 10.18632/oncotarget.21328
118. Lienlaf M, Perez-Villarroel P, Knox T, Pabon M, Sahakian E, Powers J, et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol. (2016) 10:735–50. doi: 10.1016/j.molonc.2015.12.012
119. Bae J, Hideshima T, Tai YT, Song Y, Richardson P, Raje N, et al. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia. (2018) 32:1932–47. doi: 10.1038/s41375-018-0062-8
120. Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. (2019) 7:300. doi: 10.1186/s40425-019-0784-9
121. Booth L, Roberts JL, Poklepovic A, Dent P. [pemetrexed + sildenafil], via autophagy-dependent HDAC downregulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol Ther. (2017) 18:705–14. doi: 10.1080/15384047.2017.1362511
122. Wang YF, Liu F, Sherwin S, Farrelly M, Yan XG, Croft A, et al. Cooperativity of HOXA5 and STAT3 Is critical for HDAC8 inhibition-mediated transcriptional activation of PD-L1 in human melanoma cells. J Invest Dermatol. (2018) 138:922–32. doi: 10.1016/j.jid.2017.11.009
123. Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, Aranda R, et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother. (2018) 67:381–92. doi: 10.1007/s00262-017-2091-y
124. Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. (2015) 3:1375–85. doi: 10.1158/2326-6066
125. Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget. (2017) 8:83155–70. doi: 10.18632/oncotarget.17950
126. Cao X, Zhao Y, Wang J, Dai B, Gentile E, Lin J, et al. TUSC2 downregulates PD-L1 expression in non-small cell lung cancer (NSCLC). Oncotarget. (2017) 8:107621–9. doi: 10.18632/oncotarget.22581
127. Janse van Rensburg HJ, Azad T, Ling M, Hao Y, Snetsinger B, Khanal P, et al. The hippo pathway component TAZ promotes immune evasion in human cancer through PD-L1. Cancer Res. (2018) 78:1457–70. doi: 10.1158/0008-5472.CAN-17-3139
128. Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. (2018) 33:843–52. doi: 10.1056/NEJMoa1801946
129. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. (2013) 3:1355–63. doi: 10.1158/2159-8290.CD-13-0310
130. Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. (2016) 76:227–38. doi: 10.1158/0008-5472.CAN-14-3362
131. Almozyan S, Colak D, Mansour F, Alaiya A, Al-Harazi O, Qattan A, et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J Cancer. (2017) 141:1402–12. doi: 10.1002/ijc.30834
132. Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. (2014) 259:590–604. doi: 10.1016/j.ccr.2014.03.033
133. McGowan M, Hoven AS, Lund-Iversen M, Solberg S, Helland Å, Hirsch FR, et al. PIK3CA mutations as prognostic factor in squamous cell lung carcinoma. Lung Cancer. (2017) 103:52–7. doi: 10.1016/j.lungcan.2016.11.018
134. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. (2007) 13:84–8. doi: 10.1038/nm1517
135. Song M, Chen D, Lu B, Wang C, Zhang J, Huang L, et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE. (2013) 8:e65821. doi: 10.1371/journal.pone.0065821
136. Crane CA, Panner A, Murray JC, Wilson SP, Xu H, Chen L, et al. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene. (2009) 28:306–12. doi: 10.1038/onc.2008.384
137. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. (2014) 2:361–70. doi: 10.1158/2326-6066.CIR-13-0127
138. Simeone E, Grimaldi AM, Festino L, Giannarelli D, Palla M, Caracò C, et al. Correlation between BRAF mutational status and clinical response to pembrolizumab in advanced melanoma patients. J Immunother Cancer. (2015) 3:P134. doi: 10.1186/2051-1426-3-S2-P134
139. Maeda T, Hiraki M, Jin C, Rajabi H, Tagde A, Alam M. MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res. (2017) 78:205–15. doi: 10.1158/0008-5472.CAN-17-1636
140. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1. B7-H1). PNAS. (2008) 105:20852–7. doi: 10.1073/pnas.0810958105
141. Gowrishhankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-κB. PLoS ONE. (2015) 10:e0123410. doi: 10.1371/journal.pone.0123410
142. Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, et al. Phosphorylated RB promotes cancer immunity by inhibiting NF-κB activation and PD-L1 expression. Mol Cell. (2018) 73:22–35. doi: 10.1016/j.molcel.2018.10.034
143. Lee SJ, Jang BC, Lee SW, Yang YI, Suhb SI, Park YM, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274). FEBS Lett. (2006) 580:755–62. doi: 10.1016/j.febslet.2005.12.093
144. Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O'Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. (2012) 18:1611–8. doi: 10.1158/1078-0432.CCR-11-1942
145. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol. (2015) 309:569–79. doi: 10.1152/ajpcell.00207.2015
146. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. (2014) 74:665–74. doi: 10.1158/0008-5472.CAN-13-0992
148. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. (2016) 352:227–31. doi: 10.1126/science.aac9935
149. Zhong F, Cheng X, Sun S, Zhou J. Transcriptional activation of PD-L1 by Sox2 contributes to the proliferation of hepatocellular carcinoma cells. Oncol Rep. (2017) 37:3061–7. doi: 10.3892/or.2017.5523
150. Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, et al. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep. (2017) 7:17810. doi: 10.1038/s41598-017-18132-0
151. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
152. Li N, Wang J, Zhang N, Zhuang M, Zong Z, Zou J, et al. Cross-talk between TNF-alpha and IFN-gamma signaling in induction of B7-H1 expression in hepatocellular carcinoma cells. Cancer Immunol Immunother. (2018) 67:271–83. doi: 10.1007/s00262-017-2086-8
153. Imai D, Yoshizumi T, Okano S, Itoh S, Ikegami T, Harada N, et al. IFN-γ promotes epithelial-mesenchymal transition and the expression of PD-L1 in pancreatic cancer. J Surg Res. (2019) 240:115–23. doi: 10.1016/j.jss.2019.02.038
154. Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, et al. PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. (2017) 22:1026–33. doi: 10.1007/s10147-017-1161-7
155. Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, et al. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunol. (2015) 4:e1008824. doi: 10.1080/2162402X.2015.1008824
156. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. (2017) 127:2930–40. doi: 10.1172/JCI91190
157. Karachaliou N, Gonzalez-Cao M, Crespo G, Drozdowskyj A, Aldeguer E, Gimenez-Capitan A, et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther Adv Med Oncol. (2018) doi: 10.1177/1758834017749748
158. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
159. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. (2017) 7:188–201. doi: 10.1158/2159-8290.CD-16-1223
160. Alavi S, Stewart AJ, Kefford RF, Lim SY, Shklovskaya E, Rizos H. Interferon signaling is frequently downregulated in melanoma. Front Immunol. (2018) 9:1414. doi: 10.3389/fimmu.2018.01414
161. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. (2016) 30:925–39. doi: 10.1016/j.ccell.2016.10.010
162. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett. (2017) 184:7–14. doi: 10.1016/j.imlet.2017.02.006
163. Carbotti G, Barisione G, Airoldi I, Mezzanzanica D, Bagnoli M, Ferrero S, et al. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget. (2015) 6:43267–80. doi: 10.18632/oncotarget.6530
164. Alsuliman A, Colak D, Al-Harazi O, Fitwi H, Tulbah A, Al-Tweigeri T, et al. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: significance in claudin-low breast cancer cells. Mol Cancer. (2015) 14:149. doi: 10.1186/s12943-015-0421-2
165. Kurimoto R, Iwasawa S, Ebata T, Ishiwata T, Sekine I, Tada Y, et al. Drug resistance originating from a TGF-beta/FGF-2-driven epithelial-to-mesenchymal transition and its reversion in human lung adenocarcinoma cell lines harboring an EGFR mutation. Int J Oncol. (2016) 48:1825–36. doi: 10.3892/ijo.2016.3419
166. Yee D, Shah KM, Coles MC, Sharp TV, Lago D. MicroRNA-155 induction via TNF-α and IFN-γ suppresses expression of programmed death ligand-1 (PD-L1) in human primary cells. J Biol Chem. (2017) 292:20683–93. doi: 10.1074/jbc.M117.809053
167. Zhou L, Cha G, Chen L, Yang C, Xu D, Ge M. HIF1α/PD-L1 axis mediates hypoxia-induced cell apoptosis and tumor progression in follicular thyroid carcinoma. Onco Targets Ther. (2019) 12:6461–70. doi: 10.2147/OTT.S203724
168. Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, et al. Renal cell carcinoma programmed death-ligand 1, a new direct target of hypoxia-inducible factor-2 alpha, is regulated by von hippel–lindau gene mutation status. Eur Urol. (2016) 70:633–4. doi: 10.1016/j.eururo.2015.11.029
169. Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer. (2016) 139:396–403. doi: 10.1002/ijc.30077
170. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. (2008) 111:3635–43. doi: 10.1182/blood-2007-11-123141
171. Gato-Cañas M, Zuazo M, Arasanz H, Ibañez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. (2017) 20:1818–29. doi: 10.1016/j.celrep.2017.07.075
172. Escors D, Gato-Cañas M, Zuazo M, Arasanz H, Gracía-Granda MJ, Vera R, et al. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct Target Ther. (2018) 3:26. doi: 10.1038/s41392-018-0022-9
173. Kim YS, Park GB, Lee HK, Song H, Choi IH, Lee WJ, et al. Cross-linking of B7-H1 on EBV-transformed B cells induces apoptosis through reactive oxygen species production, JNK signaling activation, and fasL expression. J Immunol. (2008) 181:6158–69. doi: 10.4049/jimmunol.181.9.6158
174. Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, Freeman GJ. PD-L1 binds to B7-1 only in cis on the same cell surface. Cancer Immunol Res. (2018) 6:921–9. doi: 10.1158/2326-6066.CIR-17-0316
175. Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity. (2019) 51:1059–73. doi: 10.1016/j.immuni.2019.11.003
176. Haile ST, Bosch JJ, Agu NI, Zeender AM, Somasundaram P, Srivastava MK, et al. Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J Immunol. (2011) 186:6822–9. doi: 10.4049/jimmunol.1003682
177. Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY, et al. CD44+ cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin Cancer Res. (2016) 22:3571–81. doi: 10.1158/1078-0432.CCR-15-2665
178. Nishino M, Ozaki M, Hegab AE, Hamamoto J, Kagawa S, Arai D, et al. Variant CD44 expression is enriching for a cell population with cancer stem cell-like characteristics in human lung adenocarcinoma. J Cancer. (2017) 8:1774–85. doi: 10.7150/jca.19732
179. Zhi Y, Mou Z, Chen J, He Y, Dong H, Fu X, et al. B7H1 expression and epithelial-to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PLoS ONE. (2015) 10:e0135528. doi: 10.1371/journal.pone.0135528
180. Kong T, Ahn R, Yang K, Zhu X, Fu Z, Morin G, et al. CD44 promotes PD-L1 expression and its tumor-intrinsic function in breast and lung cancers. Cancer Res. (2019) 80:444–57. doi: 10.1158/0008-5472.CAN-19-1108
181. Gao L, Guo Q, Li X, Yang X, Ni H, Wang T. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine. (2019) 41:395–407. doi: 10.1016/j.ebiom.2019.02.034
182. Gupta HB, Clark CA, Yuan B, Sareddy G, Pandeswara S, Padron AS, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct Tar. (2016) 1:16030. doi: 10.1038/sigtrans.2016.30
183. Kari SC, Kancharla A, Gupta HB, Risinger A, Curiel TJ. Tumor-intrinsic PD-L1 reduces actin cytoskeleton polymerization to promote mTORC1 signals driving tumor stemness. J Immunol. (2019) 202:137–8.
184. Tamai K, Nakamura M, Mizuma M, Mochizuki M, Yokoyama M, Endo H, et al. Suppressive expression of CD274 increases tumorigenesis and cancer stem cell like phenotypes in cholangiocarcinoma. Cancer Sci. (2014) 105:667–74. doi: 10.1111/cas.12406
185. Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, et al. A patient-derived, pan-cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial to mesenchymal transition. Clin Cancer Res. (2016) 22:609–20. doi: 10.1158/1078-0432.CCR-15-0876
186. Li J, Chen L, Xiong Y, Zheng X, Xie Q, Zhou Q, et al. Knockdown of PD-L1 in human gastric cancer cells inhibits tumor progression and improves the cytotoxic sensitivity to CIK therapy. Cell Physiol Biochem. (2017) 41:907–20. doi: 10.1159/000460504
187. Li Y, Wang J, Li C, Ke XY. Contribution of PD-L1 to on cogenesis of lymphoma and its RNAi-based targeting therapy. Leuk Lymphoma. (2012) 53:2015–23. doi: 10.3109/10428194.2012.673228
188. Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. (2016) 76:6964–74. doi: 10.1158/0008-5472.CAN-16-0258
189. Chen L, Xiong Y, Li J, Zheng X, Zhou Q, Turner A, et al. PD-L1 expression promotes epithelial to mesenchymal transition in human esophageal cancer. Cell Physiol Biochem. (2017) 42:2267–80. doi: 10.1159/000480000
190. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. (2008) 38:2438–50. doi: 10.1002/eji.200838289
191. Chang CH, Qui J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
192. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol. (2019) 7:4. doi: 10.3389/fcell.2019.00004
193. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
194. Ghebeh H, Lehe C, Barhoush E, Al-Romaih K, Tulbah A, Al-Alwan M, et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. (2010) 1:48. doi: 10.1186/bcr2605
195. Liao Y, Chen L, Feng Y, Shen J, Gao Y, Cote G, et al. Targeting programmed cell death ligand 1 by CRISPR/Cas9 in osteosarcoma cells. Oncotarget. (2017) 8:30276–87. doi: 10.18632/oncotarget.16326
196. Liu S, Chen S, Yuan W, Wang H, Chen K, Li D, et al. PD-1/PD-L1 interaction up-regulates MDR1/P-gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget. (2017) 8:99901–12. doi: 10.18632/oncotarget.21914
197. Black M, Barsoum IB, Truesdell P, Cotechini T, Macdonald-Goodfellow SK, Petroff M, et al. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget. (2016) 7:10557–15567. doi: 10.18632/oncotarget.7235
198. Feng D, Qin B, Pal K, Sun L, Dutta S, Dong H, et al. BRAF V600E-induced, tumour intrinsic PD-L1 can regulate chemotherapy-induced apoptosis in human colorectal cancer cells and in tumour xenografts. Oncogene. (2019) 38:6752–66. doi: 10.1038/s41388-019-0919-y
199. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. (2015) 162:1242–56. doi: 10.1016/j.cell.2015.08.052
200. Li H, Li X, Liu S, Guo L, Zhang B, Zhang J, et al. Programmed cell death-1 (PD-1) checkpoint blockade in combination with a mammalian target of rapamycin inhibitor restrains hepatocellular carcinoma growth induced by hepatoma cell-intrinsic PD-1. Hepatology. (2017) 66:1920–19933. doi: 10.1002/hep.29360
201. Osta E, Gupta HB, Zhang D, Kornepati A, Clark CA, Curiel TJ. Tumor cell-intrinsic programmed death protein 1 expression and induction in human cancer cell lines. J Immunol. (2018) 200:178.
202. Du S, McCall N, Park K, Guan Q, Fontina P, Ertel A, et al. Blockade of tumor-expressed PD-1 promotes lung cancer growth. Oncoimmunology. (2018) 7:e1408747. doi: 10.1080/2162402X.2017.1408747
203. Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. (2017) 23:1920–8. doi: 10.1158/1078-0432.CCR-16-1741
204. Hoarau-Véchot J, Rafii A, Touboul C, Pasquier J. Halfway between 2D and animal models: Are 3D cultures the ideal tool to study cancer-microenvironment interactions? Int J Mol Sci. (2018) 19:181. doi: 10.3390/ijms19010181
205. Theivanthiran B, Evans KS, DeVito NC, Plebanek M, Sturdivant M, Wachsmuth LP, et al. A tumor-intrinsic PD-L1-NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J Clin Invest. (2020) 130:2570–86. doi: 10.1172/JCI133055
206. Saleh R, Taha RZ, Sasidharan Nair V, Alajez NM, Elkord E. PD-L1 blockade by atezolizumab downregulates signaling pathways associated with tumor growth, metastasis, and hypoxia in human triple negative breast cancer. Cancers. (2019) 11:1050. doi: 10.3390/cancers11081050
207. Azadi S, Es HA, Bazaz SR, Thiery JP, Asadnia M, Warkiani ME. Upregulation of PD-L1 expression in breast cancer cells through the formation of 3D multicellular cancer aggregates under different chemical and mechanical conditions. Biochim Biophys Acta Mol Cell Res. (2019) 1866:118526. doi: 10.1016/j.bbamcr.2019.118526
208. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid modelling of the tumor immune microenvironment. Cell. (2018) 175:1972–88. doi: 10.1016/j.cell.2018.11.021
Keywords: programmed death-ligand 1, PD-1/PD-L1-targeted therapy, tumor-intrinsic role, oncology models, novel therapeutic strategies, biomarkers, immunotherapy, PD-1
Citation: Hudson K, Cross N, Jordan-Mahy N and Leyland R (2020) The Extrinsic and Intrinsic Roles of PD-L1 and Its Receptor PD-1: Implications for Immunotherapy Treatment. Front. Immunol. 11:568931. doi: 10.3389/fimmu.2020.568931
Received: 02 June 2020; Accepted: 27 August 2020;
Published: 21 October 2020.
Edited by:
Fang-Ping Huang, Shenzhen University, ChinaReviewed by:
Ekaterina Jordanova, Center for Gynaecologic Oncology Amsterdam, NetherlandsCopyright © 2020 Hudson, Cross, Jordan-Mahy and Leyland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebecca Leyland, ci5sZXlsYW5kQHNodS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.