
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 28 October 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.568630
 Pranav S. Renavikar
Pranav S. Renavikar Sushmita Sinha
Sushmita Sinha Ashley A. Brate
Ashley A. Brate Nicholas Borcherding
Nicholas Borcherding Michael P. Crawford
Michael P. Crawford Scott M. Steward-Tharp
Scott M. Steward-Tharp Nitin J. Karandikar*
Nitin J. Karandikar*Autoimmune diseases are characterized by regulatory deficit in both the CD4+ and CD8+ T-cell compartments. We have shown that CD8+ T-cells associated with acute relapse of multiple sclerosis are significantly deficient in their immune suppressive ability. We hypothesized that distinct CD8+ cytotoxic T-cell (Tc) lineages, determined by cytokine milieu during naïve T-cell differentiation, may harbor differential ability to suppress effector CD4+ T-cells. We differentiated purified human naïve CD8+ T-cells in vitro toward Tc0 (media control), Tc1 and Tc17 lineages. Using in vitro flow cytometric suppression assays, we observed that Tc0 and Tc17 cells had similar suppressive ability. In contrast, Tc1 cells showed significant loss of suppressive ability against ex vivo CD4+ T-cells and in vitro-differentiated Th0, Th1 and Th17 cells. Of note, Tc1 cells were also suboptimal in suppressing CD4-induced acute xenogeneic graft versus host disease (xGVHD) in vivo. Tc subtypes derived under various cytokine combinations revealed that IL-12-containing conditions resulted in less suppressive cells exhibiting dysregulated cytotoxic degranulation. RNA sequencing transcriptome analyses indicated differential regulation of inflammatory genes and enrichment in GM-CSF-associated pathways. These studies provide insights into the role of T-cell differentiation in CD8 suppressive biology and may reveal therapeutically targetable pathways to reverse suppressive deficit during immune-mediated disease.
As key regulators of the immune response, T-cells can serve both causative and protective roles during immune-mediated damage (1–8). Studies from our group and others have demonstrated an immunoregulatory and disease-suppressive function for CD8+ T-cells in both the autoimmune disease multiple sclerosis (MS) and its animal model (EAE) (9–20). Similar evidence has accumulated in the context of autoimmune diseases such as type 1 diabetes, colitis, SLE-like disease, Graves' disease, among others (21–25). These regulatory CD8+ T-cells function, in part, through suppression of autoreactive CD4+ responses (9, 14, 26–28).
Studies in MS point to a change in immune dynamics during disease relapse periods. We have demonstrated that acute MS relapses are characterized by a substantial deficit in the suppressive ability of patients’ CD8+ T-cells, as well as an increased resistance of patients’ CD4+ T-cells to suppression (29). Intriguingly, these functional deficits normalize during disease remission (30). This suggests that the relapse-associated inflammatory cytokine environment could influence CD8+ T-cells’ suppressive ability.
Similar to CD4+ T-helper (Th) cell subsets, lineages for CD8+ cytotoxic T-cells (Tc), such as Tc0 (media control), Tc1 (IL-2, IL-12, and anti-IL4), Tc17 (IL-6, IL-1β, TGF-β, anti-IL4, and anti-IFNγ), are predominantly determined by the cytokine milieu present during differentiation and defined by certain signature cytokines and transcription factors (31–34). We hypothesized that differentiation of naïve CD8+ T-cells along these pathways would result in variable immune-suppressive potential. Using both in vitro assay systems as well as an in vivo xenogeneic graft versus host disease (xGVHD) model, we discovered that CD8+ T-cells differentiated toward the Tc1 phenotype had significantly lower suppressive ability. Importantly, this inhibition was associated with IL-12-induced dysregulation of degranulation mechanisms and induction of multiple inflammatory pathways, revealing potential therapeutic targets for the reversal of the suppressive deficit.
Peripheral blood mononuclear cells (PBMC) from healthy subjects were isolated from de-identified leukocyte reduction system (LRS) cones containing leukocyte rich whole blood from platelet donors at the University of Iowa, DeGowin Blood Center. PBMC isolation was performed with Ficoll-Paque (GE Healthcare) density gradient centrifugation and frozen in DMSO-containing media for further use. Naïve CD8+ T-cells and/or naïve CD4+ T-cells were isolated from freshly thawed PBMC [RPMI 1640 (Corning, 10-040-CV) with DNase at 10KU/ml (Sigma D4513-1vl)] with manual LS column MACS sorting using human naïve CD8+ T-cell isolation kit (Miltenyi Biotech 130-093-244) and human naïve CD4+ T-cell isolation kit (Miltenyi Biotech 130-094-131) respectively according to manufacturer specifications. Sort purities were routinely above 95% by flow cytometric analysis (Supplementary Figure 1A). On the day of suppression assays, autologous CD4+ CD25- cells were sorted from thawed PBMC using human CD4+ T-cell isolation kit (Miltenyi Biotech, 130-096-533) and CD25+ microbeads (Miltenyi Biotech, 130-092-983). T-lymphocyte-depleted PBMC were used as antigen-presenting cells (APC).
Naïve CD8+ T-cells were sorted from PBMC and resuspended at 1 × 106 cells/ml in Stemline hematopoietic stem cell expansion serum-free media (Sigma, S0192), followed by stimulation in various differentiation conditions (Media Alone/Tc0, Tc1, and Tc17) (31, 35–37) as follows: (1) Media Alone/Tc0: no cytokines/antibodies added; (2) Tc1: anti-IL-4 (7 µg/ml, BD554481), IL-2 (10 ng/ml, BD554603), IL-12 (10 ng/ml, BD554613); (3) Tc17: anti-IL4 (7 µg/ml), anti-IFNγ (7 µg/ml, BD554698), TGFβ1 (10 ng/ml, eBioscience, 14-8348-62), IL-1β (10 ng/ml, BD554602), and IL-6 (50 ng/ml, BD550071). Cultures were activated with 0.5 µg/ml each of fixed anti-CD3 (eBioscience, 16-0037-85) and anti-CD28 (eBioscience, 16-0289-85) and incubated for 7 days at 37°C (Similar protocol was followed for experiments involving naïve CD4+ T-cell differentiation to Th0, Th1. and Th17 conditions). At day 7, cells were washed twice with PBS for suppression assay cultures. An aliquot of cells was washed, re-stimulated and supernatants were aliquoted 48 h later for ELISA assays. In some experiments, an aliquot of cultured cells was used for surface markers and intracellular cytokine staining to assess their state of differentiation.
ELISA was performed on supernatants per manufacturer protocol (eBioscience Human Platinum ELISA Kit for IL-17A (BMS2017). ELISA data were acquired on a BioTek Synergy H1 Hybrid Reader. Gen5 v2.09 was used for software analysis.
For surface and intracellular staining on day 7 of in vitro differentiation, cells were washed and cultured in media with 2 μL/ml of leukocyte activation cocktail with Golgi Plug (BD, 550583) for 5 h, followed by washing with 0.1% (w/v) sodium azide/phosphate-buffered saline and surface staining with anti-CD3 APC (BioLegend, 300458) and anti-CD8 BV786 (BD, 563823). In some experiments, anti-CD107a PE-Cy7 (BioLegend, 328617) was added during stimulation with cell activation cocktail (BioLegend, 423301) and Monensin (BD Golgi Stop, 554724) (38). Cells were fixed overnight at 4°C followed by permeabilization using fixation/permeabilization kit (eBioscience, 00-5523-00). Intracellular staining was performed using anti-IFNγ AlexaFluor700 (BD, 557995), anti-IL17A PE (eBioscience, 12-7179-42), and anti-Granzyme B APC (Miltenyi, 130-120-703). All cells were resuspended in staining buffer [0.1% (w/v) sodium azide/phosphate-buffered saline] for FACS analysis. Flow cytometric data were acquired on a 4-Laser, 17-color LSRII using BD FACSDiva Software v6.1.3 (Firmware v1.9). FlowJo version 9.1 was used for analysis.
CD8+ T-cells from the 7-day differentiation were placed in flow cytometric suppression assays, as described previously (29, 30). Briefly, responder ex vivo CD4+ CD25- T-cells were sorted or responder differentiated Th cells were obtained from 7-day cultures and stained with CFSE, followed by culture with irradiated APC and 1 µg/ml of soluble anti-CD3 (eBioscience, 16-0037-85) in the presence or absence of cultured CD8+ Tc cells. On day 7 of suppression culture, cells were stained for anti-CD3 AlexaFlour700 (BD, 557943), anti-CD8 BV786 (BD, 563823), anti-CD4 APC (BD, 555349), and anti-CD25 Pacific Blue (BioLegend, 356130) and flow cytometrically assessed for CD4+ proliferating fraction (CFSE dilution). % proliferation and % suppression were calculated as described previously (29).
Xenogeneic graft-vs-host-disease (xGVHD) was induced in female NSG mice (NOD-scid IL2RGnull, strain no. 005557, Jackson Laboratory), as described previously (39, 40). Briefly, ex vivo-purified human naïve CD8+ T-cells were first cultured for 7 days in Tc0 and Tc1 differentiation cultures, as described above. On day 7, Tc cells were washed, resuspended with PBS, and injected intravenously into 2 Gy irradiated 6–8-week- old female NSG mice admixed with ex vivo-sorted allogeneic bulk CD4+ CD25- T-cells at a 1:1 ratio (10 × 106 CD4 and 10 × 106 CD8 cells/mouse) in the indicated groups. 10 × 106 CD4+ CD25- T-cells/mouse without CD8+ T-cells (1:0 ratio) served as the control group. Some mice also received ex vivo-purified bulk CD8+ T-cells (Miltenyi Biotech, 130-045-201) or in vitro-differentiated Tc0 or Tc1 cells alone (10 × 106 cells/mouse, 0:1 ratio), representing CD8+ T-cell only groups. Mice were monitored for weight loss up to 15 days post injection. Number of mice per experimental group per experimental replicate is indicated in the figure legend.
Aliquot of cultured CD8+ Tc0 and Tc1A-E cells was flash frozen on day 7, and samples were submitted to the University of Chicago Genomics facility for RNA processing and sequencing. Single-end 50 bp sequencing was performed on the Illumina HiSeq 2000. Pseudoalignment was performed using kallisto (41) with the GRCh38 human genome build and processed using sleuth (v0.30) R package (41). Differential gene expression analysis was performed in the sleuth R package with the Wald test. Unless noted otherwise, differential genes were defined as log2-fold change >1 or <-1 and false discovery rate < 0.05. The significant genes were used for Ingenuity Pathway Analysis (Qiagen) using the same cut-points for significance as inputs.
Graphpad Prism v7.03 (La Jolla, CA) was used for statistical analyses (tests indicated in figure legends). P < 0.05 was considered significant. Data represent mean +/- SEM.
All experiments were performed on PBMC obtained from de-identified leukocyte reduction system (LRS) cones from healthy platelet donors at the University of Iowa DeGowin Blood Center, as approved by the University of Iowa IRB. All mice were kept at the University of Iowa Animal Care Facility under 12-h light/dark cycle, fed ad libitum, humanely cared for, and studied as approved by the University of Iowa’s Institutional Animal Care and Use Committee and in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications no. 8023, revised 1978).
We have shown that CD8+ T-cells isolated during acute relapse of MS are deficient in their ability to suppress myelin-specific CD4+ T-cell responses (29). Interestingly, the suppressive ability of patient CD8+ T-cells is regained during MS remission (30). We hypothesized that distinct categories of CD8+ cytotoxic T-cell (Tc) lineages may have variable suppressive function against effector CD4+ T-cell responses based on the cytokine milieu under which they were differentiated. To test this hypothesis, we obtained purified human naïve CD8+ T-cells from healthy donor peripheral blood mononuclear cells (PBMC) and differentiated them in vitro under the influence of published cytokine combinations (31, 35–37, 42–47) toward Tc0 (control), Tc1 and Tc17 lineages. At day 7 of culture, we confirmed via flow cytometry and cytokine ELISA that the differentiated populations of cells exhibited the expected functional phenotypes in terms of cytokine production (Supplementary Figure 1). In particular, we confirmed that the Tc0 control condition and the Tc1 condition showed predominantly IFNγ production but almost undetectable IL-17A, whereas the Tc17 conditions exhibited robust IL-17A production. As expected, Tc1 cells showed significantly higher proportion of cells with IFNγ expression (Supplementary Figure 1). Similar to previous studies from us and others involving human naïve CD4+ T-cell differentiation (48–50), a small proportion of the differentiated cells showed the presence of signature cytokine at the end of this short-term culture. However, after differentiation in these conditions, other cells within the culture are also known to show functional effects of this differentiation, regardless of production of the specific cytokine (such as IFNγ or IL-17A). Therefore, our studies included the entire population of “Tc1” or “Tc17”-differentiated cells for functional assessment of their suppressive ability, rather than focus on those producing specific cytokines. Thus, the entire Tc0 culture served as an appropriate control for the Tc1 and Tc17 cultures.
We therefore tested the ability of these differentiated Tc cells to suppress CD4+ T-cells using in vitro flow cytometric suppression assays, as described previously (29, 30). For these assays, ex vivo bulk CD4+ T-cells were stained with CFSE and served as responder T-cells. The cells were stimulated with autologous antigen-presenting cells (APC) and αCD3 and cultured in the presence (or absence) of differentiated autologous CD8+ T-cells. On day 7 of these suppression cultures, the proliferation of CD4+ T-cells was quantified (based on CFSE dilution) and then normalized to the 1:0 “no suppression” control condition (no CD8+ T-cells). As shown in Figures 1A, B, we observed that cells from Tc0 and Tc17 cultures had comparable suppressive ability. In contrast, cells from Tc1 cultures showed significantly less suppressive ability against bulk CD4+ T-cells.
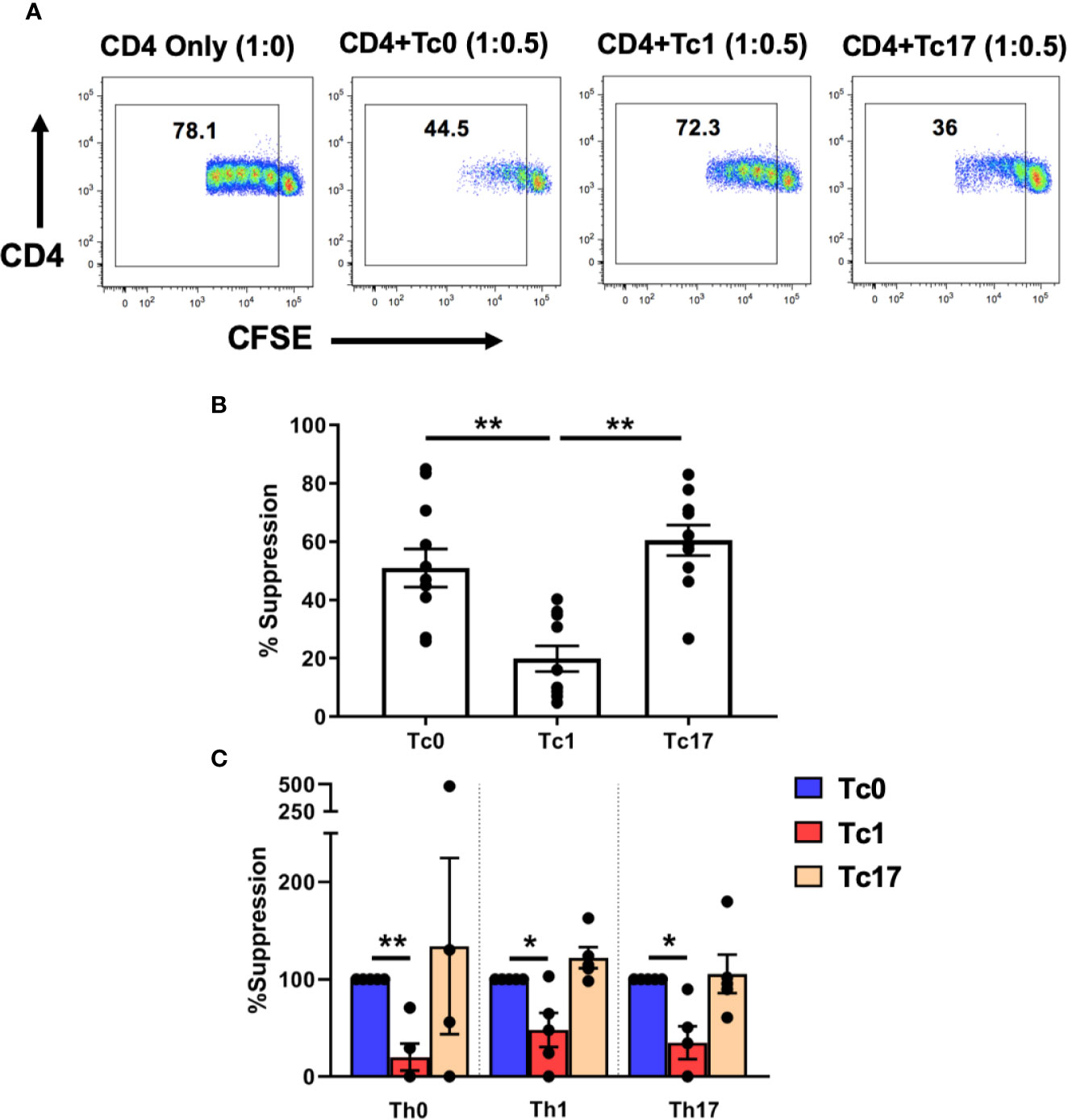
Figure 1 Tc lineages differ in their functional ability to suppress CD4+ immune responses. Cells obtained from Tc0, Tc1, and Tc17 cultures were assessed for suppressor ability against autologous CFSE-stained, ex vivo-sorted bulk CD4+ CD25- T-cells (A, B). Panel (A) shows representative flow cytometric plots demonstrating proliferation of CD4+ T-cells in the presence or absence of indicated Tc cells. Numbers indicate the proportion of cells within the proliferating fraction on day 7 of suppression assay. % suppression was calculated from the proliferation data. Panel (B) shows cumulative suppression data, indicating significantly decreased suppression by Tc1 cells (n = 10 independent samples). In Panel (C) ex vivo-sorted naïve CD4+ T-cells were first differentiated along Th0, Th1, and Th17 lineages for 7 days and then stained with CFSE and subjected to suppression assays using autologous differentiated Tc0, Tc1 and Tc17 cells (n = 5 independent samples). For all three Th types, suppression was normalized to the Tc0 control group and in every case, Tc1 suppression was significantly reduced compared to the Tc0 control. *p < 0.05 and **p < 0.01; paired t test.
We further tested this hypothesis by first generating various Th lineages from CD4+ T-cells and using these as responder cells in suppression assays. Thus, ex vivo-purified naïve CD4+ T-cells were cultured for 7 days in the presence of conditions similar to those used for CD8+ T-cells to obtain Th0, Th1, and Th17 cells. These cells were then stained with CFSE and subjected to suppression by autologous naïve-derived Tc0, Tc1, or Tc17 cultures. Similar to our findings with bulk ex vivo CD4+ T-cells, we found that cells from Tc0 and Tc17 cultures showed similar suppressive ability against all three Th lineages (Figure 1C). Again, Tc1-differentiated cells showed deficient suppressive ability against these Th cells. Therefore, based on these in vitro findings, we focused the rest of our studies on understanding the mechanisms responsible for reduced suppressive ability of cells from Tc1 cultures against bulk ex vivo CD4+ T-cells.
In order to validate our findings from the in vitro suppression assays, we utilized an acute xGVHD model using irradiated NOD-SCID-Gamma (NSG) immunodeficient mice, as described previously (40, 51). Thus, ex vivo-purified bulk human CD4+ T-cells were transferred into irradiated NSG mice, either with or without CD8+ T-cells differentiated in Tc0 or Tc1 conditions. Mice were then monitored for induction of acute xGVHD using weight loss as the primary parameter (40, 51–54).
We observed that transfer of ex vivo-purified bulk CD8+ T-cells or naïve-derived, in vitro-differentiated Tc0 or Tc1 cells did not result in disease (Figure 2A). In contrast to bulk or differentiated CD8+ T-cells, bulk CD4+ T-cells resulted in robust disease, as expected (Figure 2B). Of note, similar to our findings from in vitro suppression assays, the addition of cells from Tc0 control conditions resulted in robust suppression of xGVHD (Figure 2B). Interestingly, cells from Tc1 cultures were significantly deficient in their capacity to suppress CD4+ T-cell-driven disease (Figure 2B), matching with their deficit in in vitro suppression (Figure 1). We quantified % disease suppression on days 9, 12, and 15 by comparing % change in weight for the CD4+ T-cell only control group against CD4+Tc0 and CD4+Tc1 groups, normalizing to the Tc0 suppression as 100%. There was strong disease suppression in both the groups by day 9. However, the CD4+Tc1 group progressively lost disease suppressive capability (Figure 2C). The experiments were terminated by days 15–18 due to significant weight loss in the control group. These results showed that while cells from the Tc0 control cultures efficiently suppressed CD4+ T-cell driven pathology in vivo, cells from Tc1 cultures had an intrinsic suppressive defect.
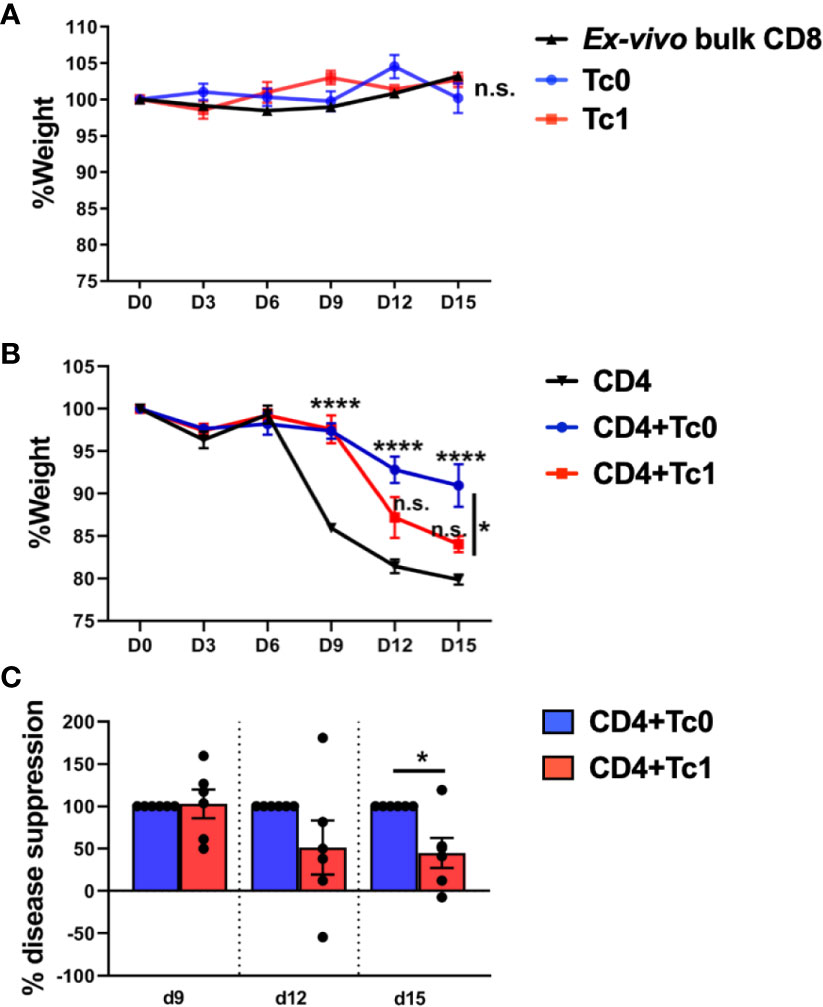
Figure 2 Naïve-derived Tc1 cells have deficient ability to suppress xGVHD in vivo. xGVHD was induced in 6–8-week-old female NSG mice by injecting bulk ex vivo-purified CD4+CD25- human T-cells with or without in vitro-generated, naïve-derived Tc0 (control), and Tc1 cells at a ratio of 1:1 (10 million cells each). Mice were monitored for weight loss over 15 days. Panels (A, B) show % weight (normalized to day 0) ± SEM. Panel (A) represents absence of xGVHD when only CD8+ T-cells were injected, either as ex vivo-purified bulk CD8+ T-cells (two independent replicates; n = 5 mice) or as in vitro-differentiated Tc0 or Tc1 cells (1 experiment, n = 3 mice each). Panel (B) demonstrates CD4-induced xGVHD with CD4+ T-cells alone or admixed with indicated Tc cells (two independent replicates; n = 6 mice per group). Panel (C) shows the same data represented as calculated % disease suppression on d9, d12, and d15 normalized to the Tc0 control group designated as “100% suppression”. *p < 0.05 and ****p < 0.0001; n.s., not significant. (A, B) Two-way ANOVA using Tukey’s multiple comparisons test and (C) unpaired t-test.
We next asked whether the functional differences in Tc1 CD8+ T-cells can be attributed to specific cytokines that they are exposed to during their differentiation. In the Tc1 differentiation condition, IL-12 was the dominant pro-inflammatory cytokine and seemed potentially accountable for mediating the loss of suppressive ability. We tested this hypothesis by setting up 6 different CD8+ T-cell culture conditions (Figure 3A). Thus, in addition to the media control (Tc0) and the standard Tc1 condition (now designated Tc1-A), we added 4 other variations that included different combinations of the same factors (IL-2, IL-12 and anti-IL-4). First, we utilized these differentiated CD8+ T-cells from all 6 conditions in suppression assays against autologous CD4+ T-cells. We observed that the 4 conditions that contained IL-12 (Tc1A-D) resulted in a significant loss of suppressive ability, when compared to the Tc0 condition (Figure 3B). When we grouped the conditions as containing or not containing IL-12, this confirmed that the populations exposed to exogenous IL-12 showed significantly lower suppressive ability (Figure 3C).
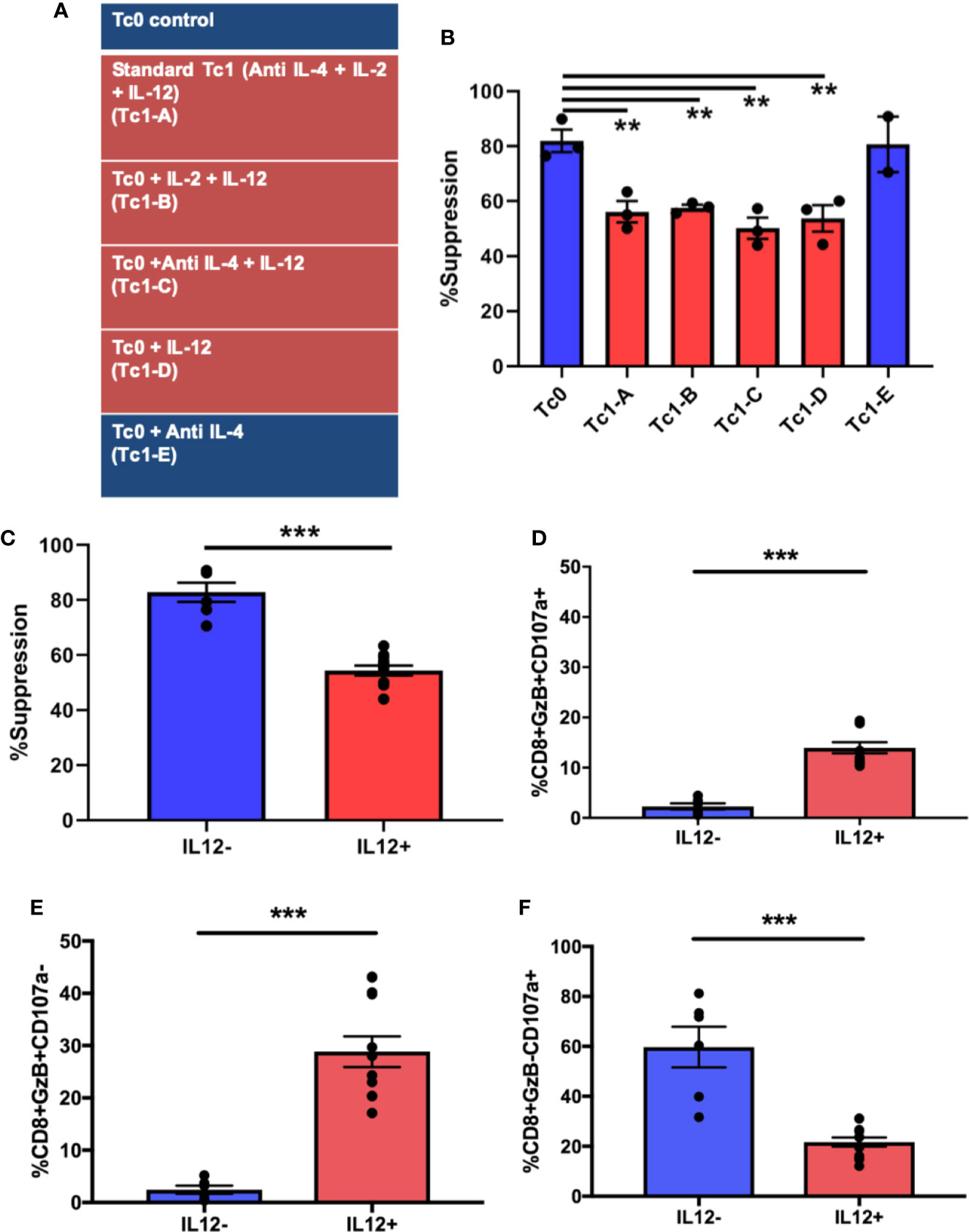
Figure 3 Exposure to IL-12 induces CD8+ T-cells characterized by suboptimal suppressive ability with dysregulated induction of cytotoxicity-related molecules. Six different culture conditions (A) were used to differentiate ex vivo-purified naïve CD8+ T-cells. On day 7, these Tc cells were assessed for suppressor ability against autologous CD4+CD25- T-cells. Panel (B) demonstrates %suppression for each of the conditions. Panel (C) shows the same data combined into two groups: cultures containing IL-12 (n = 12) and those without (n = 6). In parallel, cells from the 7-day cultures were stimulated with BioLegend Cell Activation cocktail with Monensin and anti-CD107a for 5h followed by intracellular staining for granzyme B. %GzB+CD107a+ dual positive CD8+ T-cells were quantified within the indicated groups (D), as were GzB+ (E) and CD107a+ (F) single-positive cells. **p < 0.01 and ***p < 0.001. (B) One-way ANOVA and (C–F) Mann-Whitney test.
In order to investigate the cytotoxic potential of these cells, they were surface-stained for the degranulation marker lysosomal-associated membrane protein 1 (LAMP1 or CD107a) (38, 55–57), followed by intracellular staining for the cytotoxic molecule, granzyme B (GzB). As shown in Figure 3D, the IL-12-exposed group exhibited a significantly greater proportion of dual-positive (granzyme B+CD107a+) cells compared to the group not exposed to IL-12 (including Tc0). In prior work, we have shown that suppressive ability of CD8+ T-cells is linked with their cytotoxic functions (9, 30). Therefore, it was somewhat unexpected that suboptimally suppressive Tc cells would show a greater proportion of cells expressing granzyme B. We therefore evaluated the two properties (degranulation vs. granzyme production) individually by quantifying single-positive and total-positive cells for each marker (representative dot plots shown in Supplementary Figure 2A). In this analysis, we saw that cells from IL-12-containing cultures showed significantly greater proportion of cells producing granzyme B, quantified either as single positive (Figure 3E) or as total positive (Supplementary Figure 2B), but demonstrated a significant reduction in the proportion of cells expressing surface CD107a (Figure 3F and Supplementary Figure 2B). Additionally, in linear regression analysis, whereas the cells from the IL-12(-) cultures demonstrated the expected positive correlation between suppression and degranulation, such correlation was lost in IL-12(+) cultures (Supplementary Figure 2C). Collectively, these findings suggest that exposure of naïve CD8+ T-cells to IL-12 may result in enhanced production of cytotoxic molecules but an inhibition in the granule transport and/or degranulation machinery, potentially translating to a reduced cytotoxic/suppressor capacity.
In an effort to dissect out the mechanisms responsible for these IL-12-induced functional changes, we differentiated naïve CD8+ T-cells for 7 days in various conditions depicted in Figure 3A, followed by RNAseq transcriptome analysis. We then compared differential gene expression between the groups that received or did not receive IL-12 during differentiation. First, we asked whether there was any evidence of greater apoptotic potential among the IL-12-exposed cells that could explain their inability to optimally suppress their targets. As shown in Supplementary Figure 3, we did not observe any significant differences in either pro-apoptotic (BCL2L11/Bim, BID, and BAD) or pro-survival factors (BCL2 and BCL2L1/Bcl-xl) (58).
Next, we asked which genes were differentially regulated in the IL-12(+) group, relative to the IL-12(-) group. Figure 4A shows a volcano plot of differentially expressed genes (451 significantly upregulated shown in red and 441 significantly downregulated shown in blue), with the top 10 significantly up- and downregulated genes shown in the bar graphs. In addition to evaluating the two broad groups, IL-12(+) and IL-12(-), we also compared differential expression between each of the IL-12-containing experimental conditions, relative to the Tc0 control (Supplementary Figure 4). In this manner, we were better able to tease out the specific effects of IL-12 within our in vitro system. Across all four comparisons, we saw a number of genes significantly up- and downregulated. The 31 common genes upregulated and 19 common genes downregulated between individual comparisons are represented in heatmaps in Supplementary Figure 4. Importantly, we performed Ingenuity Pathway Analysis (IPA) by utilizing the differential expression comparisons between IL-12-exposed (non-suppressive) group versus the IL-12-unexposed (suppressive) group. We saw 25 canonical pathways significantly upregulated in the IL-12(+) non-suppressive group (Figure 4B). IPA also performs estimated enrichments for upstream regulators of distinct genetic programs. We examined the upstream regulators with increased and decreased enrichment (Figure 4B and Supplementary Figure 5) across the IL-12(+) group. Interestingly, we found the highest increase in genetic expression patterns associated with the cytokine GM-CSF (Csf2), among other cytokines and transcription factors. These results suggested that IL-12 could act directly on naïve CD8+ T-cells to induce GM-CSF-related pathways.
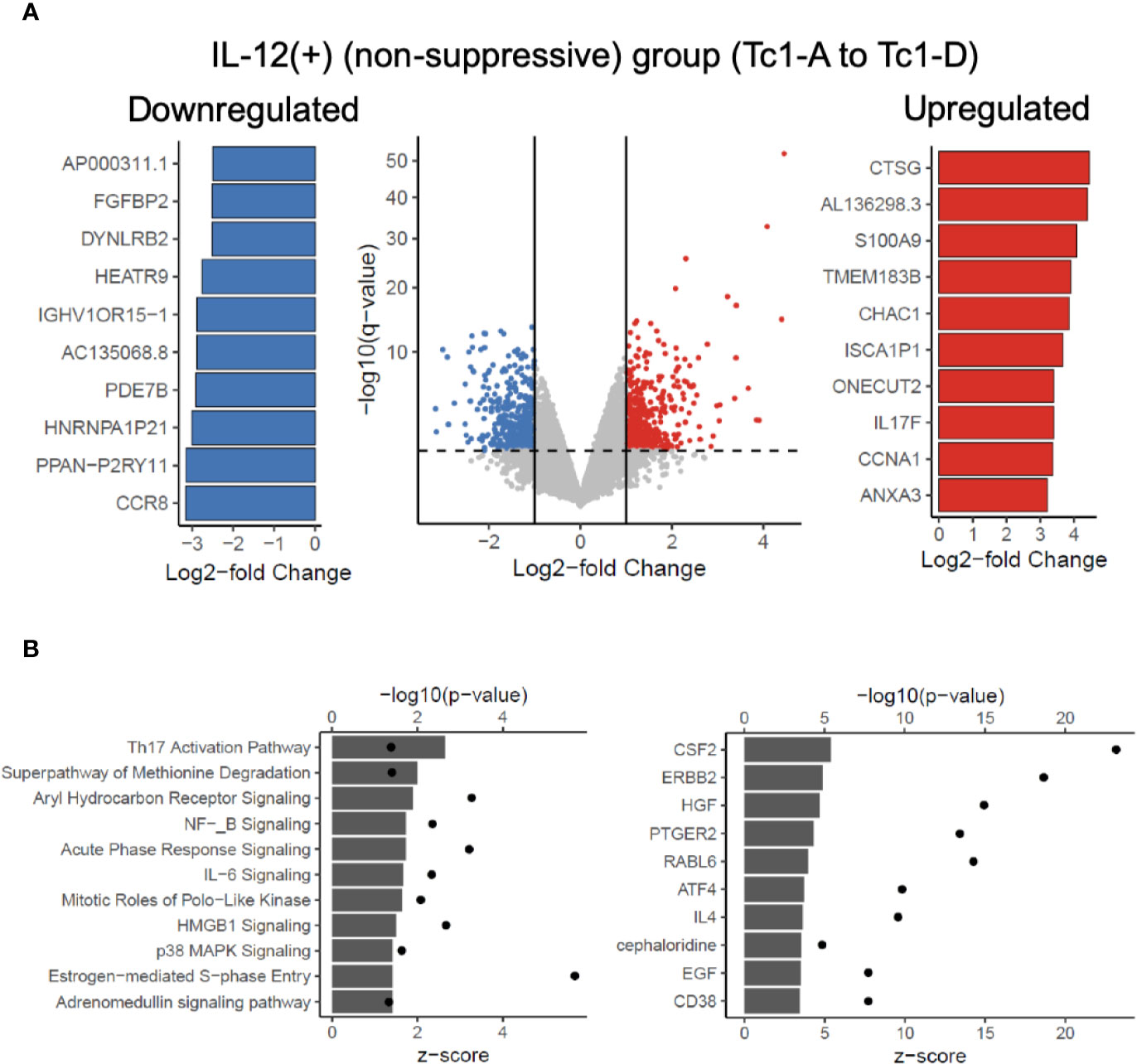
Figure 4 Transcriptome analysis reveals potentially targetable genes and pathways to reverse IL-12-induced suppressive deficit. RNAseq analysis was performed on 7-day differentiated Tc subtypes shown in Figure 3A. Panel (A) shows significant differential genes in the IL-12(+) (“non-suppressive”) group when compared against IL-12(-) (suppressive) group. Significant defined as log2-fold change > 1 or < -1 and adjusted p-value < 0.05. Top 10 significant differential genes upregulated (red) or downregulated (blue) as defined by log2-fold change are shown. Panel (B) depicts the IPA analysis of upregulated canonical pathways and upregulated predicted upstream regulators in the IL-12(+) (non-suppressive) group with z-score as bar charts and points as –log10(p-values).
Finally, in the context of our findings related to the disconnect between the production of granzyme B vs degranulation (Figure 3), we directly evaluated whether any of the genes associated with cytotoxicity and degranulation were different between the two groups. We observed significant differences in IFNG, GZMB, STX11, VAMP7, and LYST genes (Supplementary Figure 6). Corroborating our flow cytometry data, we found upregulation of IFNG and GZMB in the IL-12(+) group. There were no differences in message for CD107a or perforin. Interestingly, the LYST gene was significantly downregulated in IL-12-exposed cells (Supplementary Figure 6). The function of LYST (lysosomal trafficking regulator) is incompletely understood, but its mutation is known to cause Chediak-Higashi syndrome and some forms of hemophagocytic lymphohistiocytosis, characterized by defective lymphocyte degranulation due to changes in the morphology and function of secretory lysosomes (59–62). It is thought to be required for the maturation of cytotoxic granules into exocytosis-competent secretory granules (59), and therefore, the downregulation of this gene may offer a mechanistic explanation for the inhibited degranulation seen in Figure 3.
Several studies from our group and others have underscored the importance of CD8+ T-cells as regulators of destructive effector CD4+ T-cell responses during immune-mediated disease (8, 9, 14, 26). In these situations, deficient CD8-mediated regulation may be associated with greater disease severity, such as relapses of multiple sclerosis (29, 30). Therefore, it is critical to understand the genesis of such regulatory deficiency in order to devise intervention strategies.
In the current study, we addressed the hypothesis that cytokine-milieu-driven differentiation of CD8+ T-cells along various lineages results in differential ability to regulate effector CD4+ T-cell responses. While dissecting this biology, we have uncovered several fundamental concepts. First, we show that CD8+ T-cells differentiating under “Tc1 conditions” are significantly inhibited in their ability to suppress CD4+ effector T-cells, compared to either Tc0 controls or cells cultured under Tc17 conditions. Importantly, we show this using not only in vitro suppression assays but also in an in vivo model of xenogeneic GVHD in humanized NSG mice, validating the disease relevance of this finding. Further, we demonstrate that IL-12 exposure is the main driver of this phenotype and likely influences CD8+ suppressor function through induction of a dysregulated degranulation process affecting cytotoxicity. Finally, we conduct transcriptome analysis of these differentiated Tc cells, revealing the induction of multiple pro-inflammatory pathways, which could be potentially targetable for disease intervention. These hypothesis-generating data can help hone future investigation in the context of multiple clinical settings.
The inflammatory role of IL-12 revealed in the mid-1990s provided the rationale to develop the human IL-12-neutralizing antibodies, such as ustekinumab and briakinumab, which have been evaluated in a number of immune-mediated diseases like psoriasis, rheumatoid arthritis and multiple sclerosis, with psoriasis seeing the most advanced clinical development (63). High levels of IL-12 are detected in the aqueous humor, serum and synovial fluid of patients with different autoimmune conditions like autoimmune uveitis, multiple sclerosis and rheumatoid arthritis and are correlated with disease activity (64–67). This underscores the involvement of IL-12 in modulating immune-cell subsets in autoimmune diseases. Our findings suggest that IL-12 may exert its “pro-inflammatory” role, partly through inhibition of immune suppressive mechanisms. In particular, we observed that IL-12-induced Tc1 cells were suboptimal inhibitors of pathogenic CD4+ T-cells. This also speaks to a fundamental aspect of Tc1 differentiation wherein acquisition of certain effector functions is associated with inhibition of regulatory function.
In the current study, our in vitro differentiation cultures contained purified CD8+ T-cells, devoid of other immune cell types, such as APC or other lymphocytes. Therefore, our observations provide insights into a CD8+ T-cell-intrinsic processes downstream of IL-12 signaling in the context of global anti-CD3/anti-CD28-mediated stimulation. Some of our prior studies that have utilized PBMC cultures and specific autoantigens for the generation of autoregulatory CD8+ T-cells have shown the ability for IL-12 to enhance the ability of these cells to modulate autoantigen-specific effector responses (30). These differences suggest pleiotropic effects of this cytokine based on the overall immune environment, strength of antigenic stimulus and potentially the dose of cytokines in the inflammatory microenvironment. It will be important to dissect these features in future studies.
In a recent report, we have shown that naïve CD4+ T-cells attain distinct levels of resistance to CD8-mediated suppression (50). There we observed that “Th1” conditions rendered CD4+ T-cells more sensitive to suppression, whereas Th17 conditions resulted a cells that were significantly resistant to suppression. It is interesting that distinct cytokine milieus are responsible for inducing CD4 resistance (Th17 conditions) vs. CD8 suppressive deficit (Tc1 conditions). It will be critical to understand how various immune cell types respond within co-cultures that mimic a common set of in vivo inflammatory signals.
In the current study, we performed RNAseq and transcriptome analysis to gain insights into the mechanistic changes that may influence immune-suppressive behavior. First, we confirmed that pro-apoptotic (BCL2L11/Bim, BID and BAD) and pro-survival (BCL2 and BCL2L1/Bcl-xl) factors (58) were not altered in IL-12-induced Tc1 cultures (Supplementary Figure 3). Next, we looked at IL-12-mediated effects on maintaining cellular proliferation and activation. Prior studies have shown IL-12 to promote the expression of cell cycle associated transcripts, enhancing division of activated CD8+ T-cells by maintaining IL-2 signaling in vivo (58). IPA analysis of our in vitro data revealed cell-cycle related pathways like ‘estrogen mediated S phase entry’ significantly upregulated in IL-12(+) culture group (Figure 4B), and we also found Tc1 cultures to express the greatest proportion of CD25+ cells at the end of suppression assays (data not shown). We looked at modulation of markers associated with terminal differentiation and exhaustion and observed significant downregulation of CD160 and CTLA-4 whereas significant upregulation of LAG3 in IL-12-induced Tc1 cultures (Supplementary Figure 7).
In addition, we observed upregulation of several pro-inflammatory pathways in Tc1 cells, with an interesting involvement of GM-CSF and its associated pathways – NF-κB, MAPK, HMGB1 in the IL12-exposed non-suppressive groups. The role of CD4+ T-cell-derived GM-CSF has been implicated in autoimmune tissue inflammation in mouse models of neuroinflammation, arthritis, and myocarditis (68–71). Murine Th17 cells were identified as the chief source of GM-CSF, however multiple mouse and human studies have shown IL-12 to modulate Th1 cells and produce GM-CSF via an IL-23-independent pathway (72, 73). GM-CSF+ CD8+ T-cells have been shown to predominate in MS lesions (74). However, the role of CD8+ T-cell-derived GM-CSF and its regulation remains poorly understood. Our studies indicate activity of GM-CSF-related pathways in IL-12-differentiated human CD8+ T-cells, which will be an interesting aspect of future investigation, including whether this has a bearing on their disease regulatory role.
Finally, we evaluated the status of cytotoxic pathways in IL-12-induced Tc1 cells. We have shown in prior studies that CD8-mediated suppressor function relies on granzyme- and perforin-mediated cytotoxic killing of pathogenic CD4+ T-cells or pro-inflammatory APCs in autoimmune disease, both in mice and humans (9, 26, 29, 30). Therefore, we quantified granzyme B production as well as degranulation (as measured by surface CD107a) in Tc1 cells. While we observed higher proportion of granzyme B/CD107a double-positive cells within IL-12 exposed Tc1 populations, upon looking at these two properties individually, we saw that IL-12(+) cultures produced robust granzyme B, but demonstrated a significant reduction in surface CD107a expression (Figures 3E, F and Supplementary Figure 2B). Additionally, in a linear regression plot, the IL-12(-) cultures demonstrated a positive correlation between suppression and degranulation. Interestingly, this correlation was lost in IL-12(+) cultures with notable phenotypic clustering (Supplementary Figure 2C). Thus, it seems plausible that exogenous IL-12 within the microenvironment may render dysfunction in the granule transport and/or degranulation machinery leading to cytotoxic molecules being held up within the CD8+ Tc1-cell. In contrast, Tc0 cells may efficiently pump cytotoxic content outside the cells upon activation. We attempted to corroborate this functional observation within our transcriptome data. Of the various cytotoxicity-related genes (including perforin and CD107a), we found significant differences in IFNG, GZMB, STX11, VAMP7, and LYST genes. Importantly, granzyme B message was significantly higher in the IL-12-exposed group, with significant downregulation of LYST gene, which functions as a lysosomal trafficking regulator in lymphocytes (59). This discordant behavior in CD8+ T-cells has been shown in disorders associated with mutations in LYST gene like Chediak-Higashi syndrome and some forms of hemophagocytic lymphohistiocytosis (62, 75), characterized by defects in proteins regulating cytotoxic lymphocyte degranulation and shown to reduce surface CD107a expression (76–78). Surface CD107a has also been shown to protect NK cells from degranulation-associated damage (79). Overall, our studies indicate a novel angle of IL-12 acting directly on naïve CD8+ T-cells to induce a phenotype characterized by deficient immune-suppressive behavior, which may be the result of dysregulated cytotoxicity/granule transport. Prior studies have shown that this defect could be rescued by expression of effectors of lytic granule exocytosis, like Munc-13-4, Rab27a and Slp3 in cytotoxic T-lymphocytes (59). This may lead the way for potential translation of these types of interventions as a therapeutic strategy for autoimmune diseases.
To summarize, we have shown the novel finding that IL-12-mediated Tc1 differentiation results in reduced immune suppressive ability in CD8+ T-cells and have demonstrated plausible mechanistic explanations which will provide the basis for future investigation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/geo/, GSE151204.
The animal study was reviewed and approved by University of Iowa IACUC.
PR, SS, and NK contributed to conception and design of the study. PR, AB, NB, MC, SS, and SS-T performed the experiments and acquired and analyzed data. PR and NK organized the datasets and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported, in part, by grant awards from the NIH to NJK (R01 AI121567) and NB (F30 CA29655).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Drs. Alexander Boyden, Ashutosh Mangalam, Scott Lieberman, Ali Jabbari, and Chakrapani Vemulawada for the helpful discussions and advice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.568630/full#supplementary-material
1. Hoyer KK, Kuswanto WF, Gallo E, Abbas AK. Distinct roles of helper T-cell subsets in a systemic autoimmune disease. Blood J Am Soc Hematol (2009) 113(2):389–95. doi: 10.1182/blood-2008-04-153346
2. Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med (2008) 205(4):799–810. doi: 10.1084/jem.20071258
3. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med (2002) 8(5):500–8. doi: 10.1038/nm0502-500
4. Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest (2013) 123(1):247–60. doi: 10.1172/JCI63681
5. Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell (2000) 101(5):455–8. doi: 10.1016/S0092-8674(00)80856-9
6. Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, et al. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell (1995) 80(5):707–18. doi: 10.1016/0092-8674(95)90349-6
7. Chen Y, Kuchroo VK, Inobe J-I, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science (1994) 265(5176):1237–40. doi: 10.1126/science.7520605
8. von Herrath MG, Harrison LC. Antigen-induced regulatory T cells in autoimmunity. Nat Rev Immunol (2003) 3(3):223–32. doi: 10.1038/nri1029
9. Ortega SB, Kashi VP, Tyler AF, Cunnusamy K, Mendoza JP, Karandikar NJ. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J Immunol (2013) 191(1):117–26. doi: 10.4049/jimmunol.1300452
10. York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, et al. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun (2010) 35(1):33–44. doi: 10.1016/j.jaut.2010.01.003
11. Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol (2006) 176(11):7119–29. doi: 10.4049/jimmunol.176.11.7119
12. Karandikar NJ, Crawford MP, Yan X, Ratts RB, Brenchley JM, Ambrozak DR, et al. Glatiramer acetate (Copaxone) therapy induces CD8+ T cell responses in patients with multiple sclerosis. J Clin Invest (2002) 109(5):641–9. doi: 10.1172/JCI200214380
13. Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, et al. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood (2004) 103(11):4222–31. doi: 10.1182/blood-2003-11-4025
14. Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature (2019) 572(7770):481–7. doi: 10.1038/s41586-019-1467-x
15. Sarantopoulos S, Lu L, Cantor H. Qa-1 restriction of CD8+ suppressor T cells. J Clin Invest (2004) 114(9):1218–21. doi: 10.1172/JCI23152
16. Jiang H, Braunstein NS, Yu B, Winchester R, Chess L. CD8+ T cells control the TH phenotype of MBP-reactive CD4+ T cells in EAE mice. Proc Natl Acad Sci (2001) 98(11):6301–6. doi: 10.1073/pnas.101123098
17. Jiang H, Kashleva H, Xu LX, Forman J, Flaherty L, Pernis B, et al. T cell vaccination induces T cell receptor Vbeta-specific Qa-1-restricted regulatory CD8(+) T cells. Proc Natl Acad Sci U.S.A. (1998) 95(8):4533–7. doi: 10.1073/pnas.95.8.4533
18. Ben-Nun A, Wekerle H, Cohen IR. Vaccination against autoimmune encephalomyelitis with T-lymphocite line cells reactive against myelin basic protein. Nature (1981) 292(5818):60–1. doi: 10.1038/292060a0
19. Cohen IR, Weiner HL. T-cell vaccination. Immunol Today (1988) 9(11):332–5. doi: 10.1016/0167-5699(88)91330-8
20. Sinha S, Boyden AW, Itani FR, Crawford MP, Karandikar NJ. CD8+ T-Cells as Immune Regulators of Multiple Sclerosis. Front Immunol (2015) 6:619. doi: 10.3389/fimmu.2015.00619
21. Jiang H, Canfield SM, Gallagher MP, Jiang HH, Jiang Y, Zheng Z, et al. HLA-E–restricted regulatory CD8+ T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest (2010) 120(10):3641–50. doi: 10.1172/JCI43522
22. Wang R, Han G, Song L, Wang J, Chen G, Xu R, et al. CD8+ regulatory T cells are responsible for GAD-IgG gene-transferred tolerance induction in NOD mice. Immunology (2009) 126(1):123–31. doi: 10.1111/j.1365-2567.2008.02884.x
23. Endharti AT, Okuno Y, Shi Z, Misawa N, Toyokuni S, Ito M, et al. CD8+ CD122+ regulatory T cells (Tregs) and CD4+ Tregs cooperatively prevent and cure CD4+ cell-induced colitis. J Immunol (2011) 186(1):41–52. doi: 10.4049/jimmunol.1000800
24. Kim H-J, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8+ regulatory T cells is essential for self tolerance. Nature (2010) 467(7313):328–32. doi: 10.1038/nature09370
25. Saitoh O, Abiru N, Nakahara M, Nagayama Y. CD8+ CD122+ T cells, a newly identified regulatory T subset, negatively regulate Graves’ hyperthyroidism in a murine model. Endocrinology (2007) 148(12):6040–6. doi: 10.1210/en.2007-0300
26. Boyden AW, Brate AA, Karandikar NJ. Early IFNγ-mediated and late perforin-mediated suppression of pathogenic CD4 T cell responses are both required for inhibition of demyelinating disease by CNS-specific autoregulatory CD8 T cells. Front Immunol (2018) 9:2336. doi: 10.3389/fimmu.2018.02336
27. Balashov KE, Khoury SJ, Hafler DA, Weiner HL. Inhibition of T cell responses by activated human CD8+ T cells is mediated by interferon-gamma and is defective in chronic progressive multiple sclerosis. J Clin Invest (1995) 95(6):2711–9. doi: 10.1172/JCI117973
28. Yu P, Bamford RN, Waldmann TA. IL-15-dependent CD8+ CD122+ T cells ameliorate experimental autoimmune encephalomyelitis by modulating IL-17 production by CD4+ T cells. Eur J Immunol (2014) 44(11):3330–41. doi: 10.1002/eji.201444675
29. Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, et al. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J Autoimmun (2011) 36(2):115–24. doi: 10.1016/j.jaut.2010.12.003
30. Cunnusamy K, Baughman EJ, Franco J, Ortega SB, Sinha S, Chaudhary P, et al. Disease exacerbation of multiple sclerosis is characterized by loss of terminally differentiated autoregulatory CD8+ T cells. Clin Immunol (2014) 152(1):115–26. doi: 10.1016/j.clim.2014.03.005
31. Srenathan U, Steel K, Taams LS. IL-17+ CD8+ T cells: differentiation, phenotype and role in inflammatory disease. Immunol Lett (2016) 178:20–6. doi: 10.1016/j.imlet.2016.05.001
32. Yen H-R, Harris TJ, Wada S, Grosso JF, Getnet D, Goldberg MV, et al. Tc17 CD8 T cells: functional plasticity and subset diversity. J Immunol (2009) 183(11):7161–8. doi: 10.4049/jimmunol.0900368
33. Mosmann TR, Coffman R. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol (1989) 7(1):145–73. doi: 10.1146/annurev.iy.07.040189.001045
34. O’Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity (1998) 8(3):275–83. doi: 10.1016/S1074-7613(00)80533-6
35. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol (2008) 9(6):641–9. doi: 10.1038/ni.1610
36. Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal responsiveness to interleukin-12 and interferon-α specifies human CD8+ effector versus central memory T-cell fates. Blood J Am Soc Hematol (2009) 113(22):5516–25. doi: 10.1182/blood-2008-11-188458
37. Valenzuela J, Schmidt C, Mescher M. The Roles of IL-12 in Providing a Third Signal for Clonal Expansion of Naive CD8 T Cells. J Immunol (2002) 169(12):6842. doi: 10.4049/jimmunol.169.12.6842
38. Betts MR, Koup RA. Detection of T-cell degranulation: CD107a and b. Methods Cell Biol (2004) 75:497–512. doi: 10.1016/S0091-679X(04)75020-7
39. Ito R, Katano I, Kawai K, Yagoto M, Takahashi T, Ka Y, et al. A Novel Xenogeneic Graft-Versus-Host Disease Model for Investigating the Pathological Role of Human CD4+ or CD8+ T Cells Using Immunodeficient NOG Mice. Am J Transplant (2017) 17(5):1216–28. doi: 10.1111/ajt.14116
40. King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol (2009) 157(1):104–18. doi: 10.1111/j.1365-2249.2009.03933.x
41. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol (2016) 34(5):525–7. doi: 10.1038/nbt.3519
42. Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood (2011) 118(14):3890–900. doi: 10.1182/blood-2011-05-357111
43. Curtsinger JM, Lins DC, Mescher MF. Signal 3 Determines Tolerance versus Full Activation of Naive CD8 T Cells : Dissociating Proliferation and Development of Effector Function. J Exp Med (2003) 197(9):1141–51. doi: 10.1084/jem.20021910
44. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature (2008) 454(7202):350–2. doi: 10.1038/nature07021
45. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, et al. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nat Immunol (2008) 9(6):650–7. doi: 10.1038/ni.1613
46. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17–producing human T helper cells. Nat Immunol (2007) 8(9):942–9. doi: 10.1038/ni1496
47. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat Immunol (2007) 8(9):950–7. doi: 10.1038/ni1497
48. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol (2008) 9(6):641–9. doi: 10.1038/ni.1610
49. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol (2008) 9(6):650–7. doi: 10.1038/ni.1613
50. Crawford MP, Sinha S, Renavikar PS, Borcherding N, Karandikar NJ. CD4 T cell-intrinsic role for T helper 17 signature cytokine IL-17: effector resistance to immune suppression. Proc Natl Acad Sci U S A (2020) 117 (32):19408–14. doi: 10.1073/pnas.2005010117
51. Covassin L, Laning J, Abdi R, Langevin DL, Phillips NE, Shultz LD, et al. Human peripheral blood CD4 T cell-engrafted non-obese diabetic-scid IL2rγ(null) H2-Ab1 (tm1Gru) Tg (human leucocyte antigen D-related 4) mice: a mouse model of human allogeneic graft-versus-host disease. Clin Exp Immunol (2011) 166(2):269–80. doi: 10.1111/j.1365-2249.2011.04462.x
52. Bézie S, Meistermann D, Boucault L, Kilens S, Zoppi J, Autrusseau E, et al. Ex Vivo Expanded Human Non-Cytotoxic CD8(+)CD45RC(low/-) Tregs Efficiently Delay Skin Graft Rejection and GVHD in Humanized Mice. Front Immunol (2018) 8:2014–. doi: 10.3389/fimmu.2017.02014
53. Ali N, Flutter B, Rodriguez RS, Sharif-Paghaleh E, Barber LD, Lombardi G, et al. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rγnull mice display a T-effector memory phenotype. PloS One (2012) 7(8):e44219. doi: 10.1371/journal.pone.0044219
54. Hilger N, Glaser J, Müller C, Halbich C, Müller A, Schwertassek U, et al. Attenuation of graft-versus-host-disease in NOD scid IL-2Rγ–/–(NSG) mice by ex vivo modulation of human CD4+ T cells. Cytometry Part A (2016) 89(9):803–15. doi: 10.1002/cyto.a.22930
55. Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods (2003) 281(1-2):65–78. doi: 10.1016/S0022-1759(03)00265-5
56. Winchester BG. Lysosomal membrane proteins. Eur J Paediatric Neurol (2001) 5:11–9. doi: 10.1053/ejpn.2000.0428
57. Peters PJ, Borst J, Oorschot V, Fukuda M, Krähenbühl O, Tschopp J, et al. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med (1991) 173(5):1099–109. doi: 10.1084/jem.173.5.1099
58. Starbeck-Miller GR, Xue H-H, Harty JT. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med (2013) 211(1):105–20. doi: 10.1084/jem.20130901
59. Sepulveda FE, Burgess A, Heiligenstein X, Goudin N, Ménager MM, Romao M, et al. LYST controls the biogenesis of the endosomal compartment required for secretory lysosome function. Traffic (2015) 16(2):191–203. doi: 10.1111/tra.12244
60. Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, et al. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood J Am Soc Hematol (2014) 124(8):1331–4. doi: 10.1182/blood-2014-05-573105
61. Gil-Krzewska A, Saeed MB, Oszmiana A, Fischer ER, Lagrue K, Gahl WA, et al. An actin cytoskeletal barrier inhibits lytic granule release from natural killer cells in patients with Chediak-Higashi syndrome. J Allergy Clin Immunol (2018) 142(3):914–27. e6. doi: 10.1016/j.jaci.2017.10.040
62. Baetz K, Isaaz S, Griffiths GM. Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol (1995) 154(11):6122–31.
63. Teng MWL, Bowman EP, McElwee JJ, Smyth MJ, Casanova J-L, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med (2015) 21(7):719–29. doi: 10.1038/nm.3895
64. El-Shabrawi Y, Livir-Rallatos C, Christen W, Baltatzis S, Foster CS. High levels of interleukin-12 in the aqueous humor and vitreous of patients with uveitis. Ophthalmology (1998) 105(9):1659–63. doi: 10.1016/S0161-6420(98)99035-2
65. Balashov KE, Smith DR, Khoury SJ, Hafler DA, Weiner HL. Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci (1997) 94(2):599–603. doi: 10.1073/pnas.94.2.599
66. Comabella M, Balashov K, Issazadeh S, Smith D, Weiner HL, Khoury SJ. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Investigation (1998) 102(4):671–8. doi: 10.1172/JCI3125
67. Pope RM, Shahrara S. Possible roles of IL-12-family cytokines in rheumatoid arthritis. Nat Rev Rheumatol (2013) 9(4):252. doi: 10.1038/nrrheum.2012.170
68. McGeachy MJ. GM-CSF: the secret weapon in the T H 17 arsenal. Nat Immunol (2011) 12(6):521. doi: 10.1038/ni.2044
69. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med (2001) 194(7):873–82. doi: 10.1084/jem.194.7.873
70. Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol (1998) 161(7):3639–44.
71. Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6–dependent Th17 cell development and survival. J Exp Med (2008) 205(10):2281–94. doi: 10.1084/jem.20071119
72. Grifka-Walk HM, Giles DA, Segal BM. IL-12-polarized Th1 cells produce GM-CSF and induce EAE independent of IL-23. Eur J Immunol (2015) 45(10):2780–6. doi: 10.1002/eji.201545800
73. Noster R, Riedel R, Mashreghi M-F, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF Expression Are Antagonistically Regulated by Human T Helper Cells. Sci Trans Med (2014) 6(241):241ra80. doi: 10.1126/scitranslmed.3008706
74. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-β therapy. J Immunol (2015) 194(11):5085–93. doi: 10.4049/jimmunol.1403243
75. Chiang SC, Wood SM, Tesi B, Akar HH, Al-Herz W, Ammann S, et al. Differences in granule morphology yet equally impaired exocytosis among cytotoxic T cells and NK cells from Chediak–Higashi syndrome patients. Front Immunol (2017) 8:426. doi: 10.3389/fimmu.2017.00426
76. Rubin TS, Zhang K, Gifford C, Lane A, Choo S, Bleesing JJ, et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood J Am Soc Hematol (2017) 129(22):2993–9. doi: 10.1182/blood-2016-12-753830
77. Zhang J, Wang Z. Pedigree Gene Investigation and Parameters of NK Cell Activity, CD107a Degranulation Amd HLH Related Defective Protein Play Significant Role in the Diagnosis of Primary HLH. Washington, DC: American Society of Hematology (2016).
78. Hines MR, Nichols KE. Go with the flow: perforin and CD107a in HLH. Blood J Am Soc Hematol (2017) 129(22):2954–5. doi: 10.1182/blood-2017-04-773192
Keywords: immune regulation, CD8+ T cells, suppressor cells, autoimmunity, Tregs
Citation: Renavikar PS, Sinha S, Brate AA, Borcherding N, Crawford MP, Steward-Tharp SM and Karandikar NJ (2020) IL-12-Induced Immune Suppressive Deficit During CD8+ T-Cell Differentiation. Front. Immunol. 11:568630. doi: 10.3389/fimmu.2020.568630
Received: 01 June 2020; Accepted: 02 October 2020;
Published: 28 October 2020.
Edited by:
Dimitrios Petrou Bogdanos, University of Thessaly, GreeceReviewed by:
Maria Serena Longhi, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesCopyright © 2020 Renavikar, Sinha, Brate, Borcherding, Crawford, Steward-Tharp and Karandikar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nitin J. Karandikar, bml0aW4ta2FyYW5kaWthckB1aW93YS5lZHU=; orcid.org/0000-0002-6867-6950
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.