 Zorica Stojić-Vukanić1†
Zorica Stojić-Vukanić1† Senka Hadžibegović2,3†
Senka Hadžibegović2,3† Olivier Nicole2,3
Olivier Nicole2,3 Mirjana Nacka-Aleksić4
Mirjana Nacka-Aleksić4 Sanja Leštarević4
Sanja Leštarević4 Gordana Leposavić4*
Gordana Leposavić4*- 1Department of Microbiology and Immunology, University of Belgrade-Faculty of Pharmacy, Belgrade, Serbia
- 2Institut des Maladies Neurodégénératives, CNRS, UMR5293, Bordeaux, France
- 3Institut des Maladies Neurodégénératives, Université de Bordeaux, UMR5293, Bordeaux, France
- 4Department of Pathobiology, University of Belgrade-Faculty of Pharmacy, Belgrade, Serbia
Neurocognitive impairment (NCI) is one of the most relevant clinical manifestations of multiple sclerosis (MS). The profile of NCI and the structural and functional changes in the brain structures relevant for cognition in MS share some similarities to those in Alzheimer's disease (AD), the most common cause of neurocognitive disorders. Additionally, despite clear etiopathological differences between MS and AD, an accumulation of effector/memory CD8+ T cells and CD8+ tissue-resident memory T (Trm) cells in cognitively relevant brain structures of MS/AD patients, and higher frequency of effector/memory CD8+ T cells re-expressing CD45RA (TEMRA) with high capacity to secrete cytotoxic molecules and proinflammatory cytokines in their blood, were found. Thus, an active pathogenetic role of CD8+ T cells in the progression of MS and AD may be assumed. In this mini-review, findings supporting the putative role of CD8+ T cells in the pathogenesis of MS and AD are displayed, and putative mechanisms underlying their pathogenetic action are discussed. A special effort was made to identify the gaps in the current knowledge about the role of CD8+ T cells in the development of NCI to “catalyze” translational research leading to new feasible therapeutic interventions.
Introduction
Neurocognitive impairment (NCI) is an important feature of multiple sclerosis (MS) and might be even more relevant to patients than mobility restrictions (1). More important, there is no efficient therapy for the NCI (2, 3). Hence, it is important to incorporate cognitive assessment into MS clinics and to stimulate research leading to effective interventions to moderate the NCI (4). To “catalyze” this research, we attempted to identify the major gaps in the current understanding of the immunopathogenesis of NCI. In this attempt, we considered that despite clear etiological differences, MS as Alzheimer's disease (AD) may be presented with dementia (major neurocognitive disorder in DSM-5 classification) (5–7), and that some similar immunohistopathological changes are found in cognitively relevant brain structures of MS and AD patients (8–12). Besides innate immunity cell-mediated neuroinflammation (13–15), accumulating evidence indicates involvement of the adaptive cellular immunity in the pathogenesis of not only MS but also AD. Although CD4+ T cells are shown to be the major driver of MS pathogenesis (16–19) and to be implicated in AD development (19–21), therapeutic strategies selectively altering/inhibiting the function of CD4+ T cells showed disappointing results in MS and AD alike (20, 22, 23). On the other hand, there are data indicating that (i) CD8+ T cells are the predominant type of T cells in MS brain lesions (8–10) and (ii) NCI development in AD patients and transgenic mouse AD models coincides with CD8+ T cells' infiltration into cognitively relevant brain structures (11, 12, 24–27). Thus, it may be hypothesized that CD8+ T cells contribute to the development of NCI in MS and AD. In this mini-review, the results from a side-by-side comparative analysis of literature data corroborating the role of CD8+ T cells are displayed as a starting point for translational research leading to feasible therapeutic interventions.
NCI And Histopathological Signature of Ms and Ad
NCI is seen in 43–70% of people diagnosed with MS (28); in the subclinical radiologically isolated syndrome, clinically isolated syndrome, and all phases of clinical MS (29). It is also detected in experimental autoimmune encephalomyelitis (EAE), the often used animal model of MS (30–36). The most commonly impaired cognitive domains in MS include memory, attention, executive functions, speed of information processing, and visual-spatial abilities (37). The neurocognitive profiles of MS patients substantially differ depending on (i) clinical subtype and duration of the disease (38–41); (ii) age, sex, and ethnicity (38–41); (iii) brain (reflects maximal lifetime brain volume determined by genetics) and cognitive (gains throughout life experience, e.g., education, intellectually enriching leisure activities) reserve (42); and (iv) differences in screening tools used for neurocognitive evaluation (41). A proportion of MS patients meets the criteria for dementia (5, 7). On the other hand, AD is shown to be the most common cause of dementia, accounting for 60–80% of dementia cases (www.alz.org/alzheimers-dementia). All cognitive domains may be affected in AD patients (43). The neurocognitive profile of AD patients depends on stage of the disease and brain and cognitive reserves (44). In the development of NCI in AD, changes corresponding to mild cognitive disorder in DSM-5 classification may also be detected (45). Of note, when cognitive functions in older MS patients were evaluated, similarities between their neurocognitive profiles and those of patients with mild cognitive disorder of the AD type (anamnestic mild cognitive impairment) were found (40).
In MS patients apart from characteristic inflammatory-demyelinating lesions of the white matter, inflammation and neurodegeneration in cortical and deep gray matter, which are associated with deficits in learning and memory in AD, was found (9, 46, 47). On the other hand, although AD is characterized by the gray matter damage, disruption of white matter integrity was also described (48).
The development of typical brain lesions in AD is linked with the neurotoxic β-amyloid peptide (Aβ) variants that form soluble oligomers and insoluble fibers, and Aβ-induced hyperphosphorylation of the microtubule-associated protein tau (49–52). Thus, in hippocampal and cortical regions of AD patients, extracellular aggregates of amyloid fibrils—senile plaques and intracellular aggregates of hyperphosphorylated tau—neurofibrillary tangles are typically present (49–52). Although aggregation of oligomeric Aβ (oAβ) and plaques formation is a major feature of AD (53), soluble oAβ, particularly those encompassing Aβ1−42, are neurotoxic (54). On the other hand, in MS despite the augmented expression of the amyloid precursor protein (APP), reflecting axonal damage (55, 56), and the increased levels of soluble α-APP and β-APP, intermediate products of APP proteolysis, in brain lesions (57), amyloid plaques have not been found (58–62). The latter could reflect an enhanced demyelinization and release of myelin basic protein, as this protein inhibits amyloid fibril formation (favoring the detrimental effect of their soluble precursors) (63–66) and/or their enhanced cleaning due to microglial activation (62). However, as TNF-α, a major proinflammatory cytokine, impairs autophagic flux of Aβ aggregates in microglia (67), the latter does not seem likely. Additionally, it is supposed that the generation/clearance of (o)Aβ in MS varies during the disease progression or depending on the phase of the disease (68, 69). This may explain the discrepancies between data on their concentration in cerebrospinal fluid (CSF) (70–78). Of note, although alterations in Aβ metabolism are implicated in the impairment of neural plasticity and the development of NCI in MS (79, 80), there are no data on concentration of neurotoxic (o)Aβ in brain tissue. Differently from amyloid plaques, characteristic insoluble hyperphosphorylated tau formation has been described in the brain in the neurodegenerative phase of EAE and MS (81–83).
Structural and Functional Alterations of Synapses
It appears that synaptic loss precedes neuronal loss in AD, and these effects are probably driven by amyloid and tau pathology (52, 84). Post-mortem analyses of synapses/synaptic markers (synapsin I, synaptophysin, and post-synaptic density protein 95) in hippocampal and frontotemporal tissue provide evidence for strong synapse loss in MS/EAE as well (32, 85–88). The synaptic loss is suggested to be the strongest correlate of NCI in AD (89–91). In EAE, a positive correlation between hippocampal-dependent memory impairment and synaptic loss was also found (32, 92).
While the density of synapses is a key determinant to control the complexity and diversity of neuronal networks, the ability of neurons to durably strengthen their connections, also called synaptic plasticity, is crucial to shape the neuronal networks necessary for learning and memory [reviewed in (93)]. Long-term potentiation (LTP) and long-term depression (LTD) are two forms of synaptic plasticity and leading candidates for mechanisms underlying learning and memory (94). Generally, in experimental models of AD soluble oAβ are shown to cause LTP impairment, as well as LTD depression and synaptic loss [reviewed in (95)]. Although LTP impairment is also found in EAE (96–101), there is no direct evidence for a role of soluble oAβ [reviewed in (95)].
It is likely that several post-synaptic receptors mediate soluble oAβ toxicity at the post-synaptic compartment (102). However, NMDA receptors (NMDARs) seem to be particularly important in both animal AD (103) and MS models (97). The alteration of synaptic plasticity in AD models has been linked with oAβ-induced increase in glutamate concentration at synapses (85, 104, 105) and its “spillover” out of the synaptic clefts (106). The latter causes recruitment of extrasynaptic NMDARs (107–109) and neuronal excitotoxicity (110, 111) resulting in progressive neuronal and memory loss (106). Additionally, specific activation of extrasynaptic NMDARs in animal AD models enhances the amyloidogenesis and Aβ release (112) and tau phosphorylation (106, 113–115), leading to a vicious circle and the disease progression (116). Thus, there is a positive correlation between oAβ release, tau pathology, and NCI in AD (117). On the other hand, the role of oAβ in the development of NCI in MS (79, 80) requires additional research.
Research Challenges
To further investigate the role of oAβ/phosphorylated tau in the development of NCI in MS/EAE.
CD8+ T Cells In Ms and Ad Lesions
CD8+ T Cells in MS Lesions
In MS lesions, including those relevant for NCI, the vast majority of CD3+ T cells were found to be clonally expanded CD8+ T cells (10, 118–124). Their number correlates with the severity of axonal damage (125). In NCI-related brain lesions of EAE-affected animals, T cells were also detected (36, 126), and the demyelination was shown to be more MHC class I- than MHC class II-dependent (127), suggesting an active role for CD8+ T cells in the destructive CNS immune response. To harmonize these findings with the long-standing view of MS as a CD4+ T-cell-driven disease (128), it was hypothesized that following the disease initiation CD4+ T cells (the key drivers of the disease initiation) in MS/EAE are eliminated by apoptosis (124, 129), so CD8+ T cells take on a leading role (130, 131). Furthermore, the genetics (HLA A*0301 and HLA A*0201 was associated with a higher risk for MS and a protective effect on MS, respectively) (132, 133) evinces CD8+ T cell involvement in MS (134). Consistently, in active MS lesions (125, 135) and CSF (136–138) classical cytotoxic CD8+ T lymphocytes (CTLs) were found. Of note, CTLs with polarized perforin/granzyme granules were observed in close proximity to oligodendrocytes/demyelinated nerve fibers (139, 140) and CD11b+ myeloid cells (135). In favor of CTL-mediated cytotoxicity, in relapsing-remitting MS the granzyme levels in CSF were higher at relapse compared with the remission and healthy controls (138). To additionally corroborate the pathogenetic role of CTLs, neuroantigen-specific CD8+ T cells from MS patients and EAE mice were shown to be capable of killing neuronal cells and releasing IFN-γ and TNF-α in vitro (141–146). Consistently, a role of IFN-γ- and TNF-α co-producing CD8+ T (Tc1) cells in MS pathogenesis is assumed (146). Additionally, in active acute and chronic MS lesions high frequency of IL-17-producing CD8+ T (Tc17) cells (147, 148) was observed. These cells co-produce GM-CSF (149, 150), which contributes to myeloid cell activation and inflammation (151, 152). Besides, a positive correlation between the frequency of Tc17 cells in CSF and MS-related disability was found (153). This, in conjunction with data showing that co-transfer of Tc17 cells with subpathogenic numbers of CD4+ T cells could induce the disease in mice resistant to EAE and deficient in both IL-17-producing CD4+ T cells and Tc17 cells (154), corroborates the important role of Tc17 cells in EAE/MS pathogenesis. Data from a rat EAE model are in the same vein (155). In EAE mice Tc17 cells do not exhibit cytotoxicity (156) but have a high plasticity to convert into IFN-γ-producing cells with strong cytotoxic activity (157). In relapsing-remitting MS the frequency of IFN-γ/TNF-α co-producing Tc17 cells is higher in peripheral blood (PB) at relapse compared with the remission (158). Moreover, the rise in the count of PB effector/memory CD8+ T cells re-expressing CD45RA (TEMRA) and secreting high levels of cytotoxic molecules and proinflammatory cytokines (IFN-γ and TNF-α) speaks in favor of an active CD8+ T-cell response in MS (159).
Although majority of CD8+ T cells in active MS lesion are recruited from the periphery (160), CD8+ cells with features of tissue-resident memory T (Trm) cells are also present in these lesions (123, 161) and suggested to have an important role (162–166).
Research Challenges
To investigate the contribution of CD8+ T cells in the development of NCI in MS considering the hypothesis that CD4+ T cells are involved in the initiation of brain lesions, whereas CD8+ T cells take on the leading role as the disease progresses.
CD8+ T Cells in AD Lesions
Similar to MS, in AD (11, 12, 24, 25, 167) and several mouse models of AD (26, 27, 167, 168), CD8+ T cells were found to be the predominant type of T cells in the brain structures related to cognition. Additionally, a strong correlation between CD8+ T cell infiltration and tau pathology in both humans and experimental animals has recently been described (11, 27). They are located in close proximity to neuronal processes and microglia (12, 25, 167). More important, granules loaded with granzyme A were detected in CD8+ T cells from AD-affected hippocampi (12). To additionally corroborate the active role of CD8+ T cells in the disease pathogenesis, greater count of IFN-γ/TNF-α-producing CD8+ TEMRA cells was found in PB from patients diagnosed with mild neurocognitive disorder of AD type/AD patients (12). The latter correlated with NCI development (12). Of note, in PB from AD patients, higher frequency of Tc17 cells was detected than in controls (169). Moreover, a substantial proportion of CD8+ T cells in their intrathecal immune compartment belonged to a clonally expanded TEMRA subset (12).
Finally, CD8+ T cells with characteristics of Trm cells were also detected in hippocampi and subcortical white matter in AD patients and animal AD models (11, 26, 161, 165). However, their role in AD requires further research.
Research Challenges
To elucidate the mechanistic immune signature of AD by exploring the role of CD8+ T cells in various experimental models of this disease.
Putative Mechanisms of CD8+ T Cells Pathogenetic Action in the Development of Ms and Ad
Considering the aforementioned data, CD8+ T cells most likely contribute to the propagation of the initial lesions in AD and MS. It may be supposed that myeloid cells, primarily microglia, sense initial tissue damage (inflammatory CD4+ T cell-mediated and neurotoxic Aβ/phosphorylated tau protein-mediated damage in MS and AD, respectively), activate, upregulate MHC-I expression, and start the production of inflammasome-related cytokines and chemokines to recruit CD8+ T cells (124, 167, 170). In brain tissue CD8+ T cells may communicate with cells of distinct types, including neurons, which are shown to upregulate MHC-I expression in MS and AD (134, 171). Conceptually, CD8+ T cells may directly affect neuronal integrity acting via perforin-dependent delivery of several granzymes or via Fas-ligand/Fas receptor interactions (172, 173) and/or causing their “collateral” injury (174–176). They cause the “collateral” injury through destruction of myelin sheath and/or oligodendrocytes (174–176). Of note, CTLs are capable of sequential and simultaneous killing of several target cells, which is followed by “spillover” of cytotoxic molecules from immunological synapses and consequent collateral death of neighboring cells [reviewed in (177)]. Apart from neuron apoptosis, CTLs may cause their electrical silencing by increasing intracellular Ca2+ levels through massive insertion of channel-forming perforin (174, 178). The intracellular Ca2+ load may be augmented also by enhanced glutamate release from activated CD8+ T-cells and/or the target neurons themselves (174, 178–180). Apart from cell-to-cell contacts, CD8+ T cells could contribute to neuronal damage and NCI by releasing IFN-γ, TNF-α, and IL-17. These cytokines increase the permeability of blood-brain barrier and promote T-cell immigration into the CNS parenchyma (181, 182). Additionally, they upregulate MHC-I and thereby sensitize neurons/non-neural cells to CD8+ T cell (183). They could also trigger cell apoptosis (184). Moreover, in MS/EAE and AD/animal AD models these cytokines contribute to alterations in synaptic plasticity, and consequently NCI (185–187), by increasing glutamate release and/or affecting expression/phosphorylation of glutamate receptors (188–191).
Contrary to in vitro studies, a recent immunocytochemical study failed to show direct communication between CD8+ T cells and neurons/oligodendrocytes in MS, but pointed to their direct communication with distinct subsets of CD11b+ myeloid cells, including microglia, with manifold functional consequences (135). Specifically, CD8+ T cells may be activated to kill the target cells, whereas the activation of CD11b+ myeloid cells is associated with secretion of a broad array of proinflammatory mediators, including reactive oxygen and nitrogen species, and proinflammatory cytokines that damage neighboring cells, including neurons (135, 170, 192–194). The influence of CD8+ T cells on microglial secretion of proinflammatory cytokines and iNOS expression in chronic infection corroborates this notion (195). A recent study showed that in EAE mice CD8+ T cells communicate with brain infiltrating monocytes/monocyte-derived cells in a Fas-ligand/Fas receptor dependent manner (131). Such an interaction may also trigger the activation of microglia and consequently the expression of multiple genes encoding proinflammatory mediators (196). Thus, it may be speculated that CD8+ T-cell to myeloid cell communication in the brain represents a hotspot in the immunopathogenesis of MS. Besides, considering the localization of CD8+ T cells in AD-affected brain (12, 167), it may be assumed that they also contribute to AD progression by enhancing microglial activation. Specifically, the presumption is that with AD progression, as with MS/EAE progression (98), initially phagocyting microglia become more activated and consequently dysfunctional/damaging, i.e., that along the disease trajectory microglia function as a “double-edged sword” (12, 167).
Finally, given that Trm “sessile” cells were found among CD8+ T cells in AD, and particularly in relapsing-remitting and progressive MS (123, 161, 164–166, 197, 198) and that drugs inhibiting T-cell recruitment into the brain in MS showed limited therapeutic efficacy (123), the role of CD8+ Trm cells in the progression of MS and AD should be examined. Generally, they are characterized by upregulated expression of inhibitory receptors (PD-1 and CTLA-4) and diminished production of cytotoxic enzymes (123, 161, 164, 198). However, as they have preserved capacity to secrete proinflammatory cytokines (IFN-γ, TNF-α, and GM-CSF), they may contribute to myeloid cell activation when stimulatory signals (e.g., increased generation of proinflammatory mediators and toxic metabolites) overcome the inhibitory signals (123, 161, 164, 198). Hence, the increased brain levels of toxic soluble (o)Aβ variants in AD and possibly in MS may contribute to this activational/functional “switch” (199–202). Namely, the engagement of Toll-like receptor 2 on CD8+ Trm cells by toxic (o)Aβ variants may convert their partial activation to the full activation and proinflammatory cytokine production (199–202). Thus, it seems that the role of CD8+ Trm cells in MS (166) and possibly AD pathogenesis could be dependent on the disease stage. Altogether, it may be hypothesized that not only effector/memory CD8+ T cells recruited from the periphery but also CD8+ Trm cells contribute to the deleterious changes in the activational status/functional properties of microglia, and thereby to MS/AD perpetuation and NCI worsening.
Research Challenges
To confirm the significance of CD8+ T cells and microglia communication in the progression of NCI in these diseases and particularly the contribution of CD8+ Trm cells to the detrimental output of this communication.
Conclusions
It may be speculated that CD8+ T cells recruited from the periphery together with CD8+ Trm cells contribute to MS and AD progression acting on neurons/neurites not only directly but also indirectly by affecting the functional properties of microglia (Figure 1). This concept may serve as a basis for further research to formulate therapeutic interventions targeting not only effector/memory CD8+ T cells but also CD8+ Trm cells to moderate NCI severity. This could be particularly important, as patients with MS are now more likely than ever to enter old age and develop AD, so a number of individuals with these complex comorbidities is expected to increase (203).
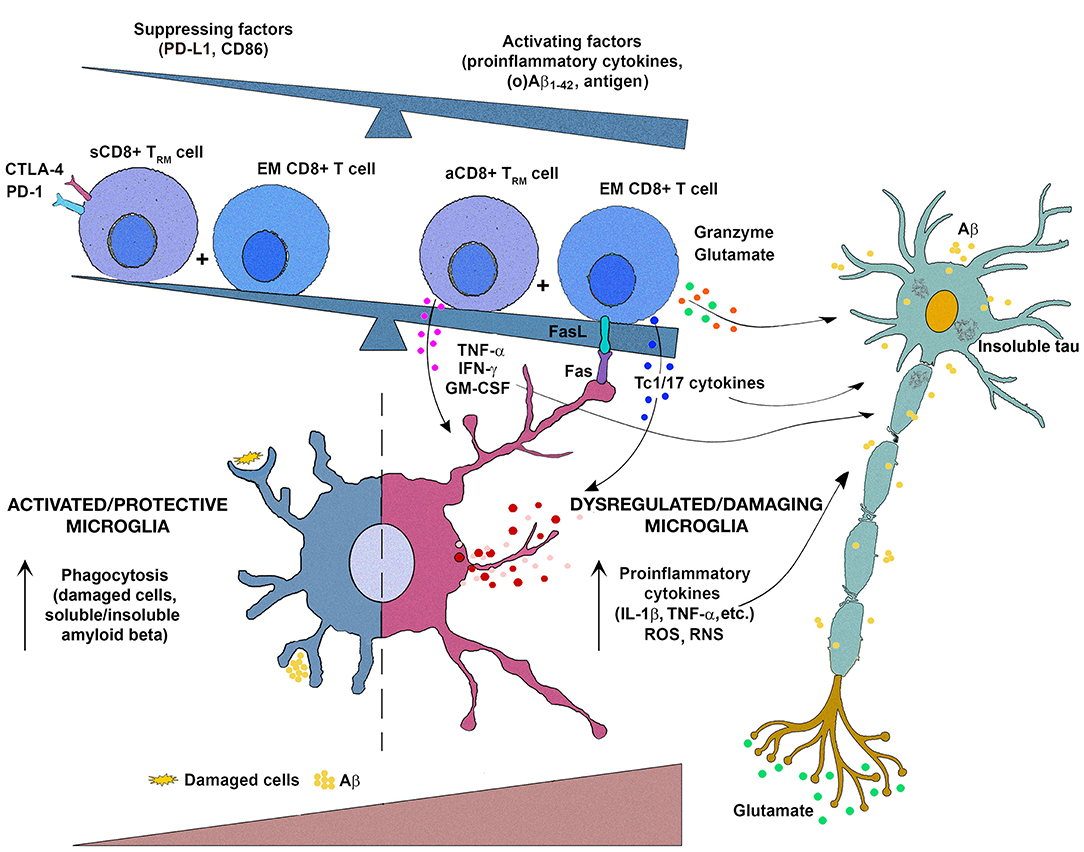
Figure 1. Schematic representation of the putative interactions between (re)activated CD8+ T cells and microglia in the progression of multiple sclerosis (MS) and Alzheimer's disease (AD). It may be hypothesized that upon entering in damaged (by CD4+ T cells and neurotoxic oligomeric amyloid β peptides [Aβ]/tau protein in MS and AD, respectively) brain tissue, effector/memory (EM) CD8+ T cells reactivate and additionally activate microglia through Fas ligand/Fas-mediated interactions and secretion of potentially damaging proinflammatory cytokines (IFN-γ, IL-17, TNF-α, GM-CSF). Consequently, microglia change their functional properties, viz. initially predominantly protective (phagocyting damaging cells, and Aβ variants and their soluble and insoluble assembly) microglia change to become dysfunctional/detrimental secreting damaging mediators, including proinflammatory cytokines (IL-1β, TNF-α), reactive oxygen species (ROS), and reactive nitrogen species (RNS) on the account of phagocyting ability. To this microglial transition also contribute CD8+ tissue-resident memory T (Trm) cells, as they in the response to alterations in the local microenvironment [mirrored in increasing accumulation of various activating mediators, e.g., proinflammatory cytokines, local metabolites, including Aβ1−42 and its soluble oligomers (o)Aβ1−42, which are shown to interact with TLR2 expressed on their cell surface] transit from a suppressed state (sCD8+ Trm cell) maintained by PD-L1- and CD86-mediated signaling to activated proinflammatory cytokine-secreting state (aCD8+ Trm cell). The damaging mediators derived from (re)activated EM CD8+ T cells (including glutamate, which is shown to contribute to activation of extrasynaptic NMDA receptors to promote cell death) and microglia, along with toxic metabolites/mediators from neurons themselves (Aβ, glutamate), contribute to neuron/neurite damage and further progression of the diseases.
Author Contributions
ZS-V, SH, ON, and GL wrote the manuscript. All authors participated in data collection and interpretation, critically revised the manuscript, and approved the final version for submission. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant No. 451-03-68/2020-14/200161).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Amato MP, Zipoli V, Portaccio E. Multiple sclerosis-related cognitive changes: a review of cross-sectional and longitudinal studies. J Neurol Sci. (2006) 245:41–6. doi: 10.1016/j.jns.2005.08.019
2. Amato MP, Langdon D, Montalban X, Benedict RH, DeLuca J, Krupp LB, et al. Treatment of cognitive impairment in multiple sclerosis: position paper. J Neurol. (2013) 260:1452–68. doi: 10.1007/s00415-012-6678-0
3. Macías Islas MA, Ciampi E. Assessment and impact of cognitive impairment in multiple sclerosis: an overview. Biomedicines. (2019) 7:22. doi: 10.3390/biomedicines7010022
4. Sumowski JF, Benedict R, Enzinger C, Filippi M, Geurts JJ, Hamalainen P, et al. Cognition in multiple sclerosis. Neurology. (2018) 90:278–88. doi: 10.1212/WNL.0000000000004977
5. Rahn K, Slusher B, Kaplin A. Cognitive impairment in multiple sclerosis: a forgotten disability remembered. Cerebrum. (2012) 2012:14.
6. Benedict RHB, Cookfair D, Gavett R, Gunther M, Munschauer F, Garg N, et al. Validity of the minimal assessment of cognitive function in multiple sclerosis (MACFIMS). J Int Neuropsychol Soc. (2006) 12:549–58. doi: 10.1017/S1355617706060723
7. Benedict RHB, Bobholz JH. Multiple sclerosis. Semin Neurol. (2007) 27:78–85. doi: 10.1055/s-2006-956758
8. Huseby ES, Huseby PG, Shah S, Smith R, Stadinsk BD. Pathogenic CD8 T cells in multiple sclerosis and its experimental models. Front Immunol. (2012) 3:64. doi: 10.3389/fimmu.2012.00064
9. Lucchinetti CF, Popescu BFG, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. (2011) 365:2188–97. doi: 10.1056/NEJMoa1100648
10. Salou M, Garcia A, Michel L, Gainche-Salmon A, Loussouarn D, Nicol B, et al. Expanded CD8 T-cell sharing between periphery and CNS in multiple sclerosis. Ann Clin Transl Neurol. (2015) 2:609–22. doi: 10.1002/acn3.199
11. Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT. Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an immunohistochemical study. Neurodegener Dis. (2018) 18:49–56. doi: 10.1159/000486200
12. Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer's disease. Nature. (2020) 577:399–404. doi: 10.1038/s41586-019-1895-7
13. Musella A, Gentile A, Rizzo FR, De Vito F, Fresegna D, Bullitta S, et al. Interplay between age and neuroinflammation in multiple sclerosis: effects on motor and cognitive functions. Front Aging Neurosci. (2018) 10:238. doi: 10.3389/fnagi.2018.00238
14. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. (2015) 16:229–36. doi: 10.1038/ni.3102
15. Wang MM, Miao D, Cao XP, Tan L, Tan L. Innate immune activation in Alzheimer's disease. Ann Transl Med. (2018) 6:177. doi: 10.21037/atm.2018.04.20
16. Chitnis T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol. (2007) 79:43–72. doi: 10.1016/S0074-7742(07)79003-7
17. Duffy SS, Lees JG, Moalem-Taylor G. The contribution of immune and glial cell types in experimental autoimmune encephalomyelitis and multiple sclerosis. Mult Scler Int. (2014) 2014:285245. doi: 10.1155/2014/285245
18. Segal BM. The diversity of encephalitogenic CD4+ T cells in multiple sclerosis and its animal models. J Clin Med. (2019) 8:120. doi: 10.3390/jcm8010120
19. Sommer A, Winner B, Prots I. The Trojan horse - neuroinflammatory impact of T cells in neurodegenerative diseases. Mol Neurodegener. (2017) 12:78. doi: 10.1186/s13024-017-0222-8
20. Monsonego A, Nemirovsky A, Harpaz I. CD4 T cells in immunity and immunotherapy of Alzheimer's disease. Immunology. (2013) 139:438–46. doi: 10.1111/imm.12103
21. Huang X, Reynolds AD, Mosley RL, Gendelman HE. CD 4+ T cells in the pathobiology of neurodegenerative disorders. J Neuroimmunol. (2009) 211:3–15. doi: 10.1016/j.jneuroim.2009.04.006
22. van Oosten BW, Lai M, Hodgkinson S, Barkhof F, Miller DH, Moseley IF, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, doubleblind, placebo-controlled, MR-monitored phase II trial. Neurology. (1997) 49:351–7. doi: 10.1212/WNL.49.2.351
23. Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. (2008) 7:796–804. doi: 10.1016/S1474-4422(08)70173-X
24. Itagaki S, McGeer PL, Akiyama H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer's disease brain tissue. Neurosci Lett. (1988) 91:259–64. doi: 10.1016/0304-3940(88)90690-8
25. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, et al. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. (2002) 124:83–92. doi: 10.1016/S0165-5728(01)00496-9
26. Ferretti MT, Merlini M, Späni C, Gericke C, Schweizer N, Enzmann G, et al. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer's disease-like cerebral amyloidosis. Brain Behav Immun. (2016) 54:211–25. doi: 10.1016/j.bbi.2016.02.009
27. Laurent C, Dorothée G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. (2017) 140:184–200. doi: 10.1093/brain/aww270
28. Chiaravalloti ND, DeLuca J. Cognitive impairment in multiple sclerosis. Lancet Neurol. (2008) 7:1139–51. doi: 10.1016/S1474-4422(08)70259-X
29. Jongen J, Ter Horst AT, Brands AM. Cognitive impairment in multiple sclerosis. Minerva Med. (2012) 103:73–96.
30. D'Intino G, Paradisi M, Fernandez M, Giuliani A, Aloe L, Giardino L, et al. Cognitive deficit associated with cholinergic and nerve growth factor down-regulation in experimental allergic encephalomyelitis in rats. Proc Natl Acad Sci USA. (2005) 102:3070–5. doi: 10.1073/pnas.0500073102
31. Tu JL, Zhao CB, Vollmer T, Coons S, Lin HJ, Marsh S, et al. APOE 4 polymorphism results in early cognitive deficits in an EAE model. Biochem Biophys Res Commun. (2009) 384:466–70. doi: 10.1016/j.bbrc.2009.04.153
32. Ziehn MO, Avedisian AA, Tiwari-Woodruff S, Voskuhl RR. Hippocampal CA1 atrophy and synaptic loss during experimental autoimmune encephalomyelitis, EAE. Lab Invest. (2010) 90:774–86. doi: 10.1038/labinvest.2010.6
33. Habbas S, Santello M, Becker D, Stubbe H, Zappia G, Liaudet N, et al. Neuroinflammatory TNFα impairs memory via astrocyte signaling. Cell. (2015) 163:1730–41. doi: 10.1016/j.cell.2015.11.023
34. LoPresti P. Glatiramer acetate guards against rapid memory decline during relapsing remitting experimental autoimmune encephalomyelitis. Neurochem Res. (2015) 40:473–9. doi: 10.1007/s11064-014-1491-z
35. Novkovic T, Shchyglo O, Gold R, Manahan-Vaughan D. Hippocampal function is compromised in an animal model of multiple sclerosis. Neuroscience. (2015) 309:100–12. doi: 10.1016/j.neuroscience.2015.03.008
36. Aharoni R, Schottlender N, Bar-Lev DD, Eilam R, Sela M, Tsoory M, et al. Cognitive impairment in an animal model of multiple sclerosis and its amelioration by glatiramer acetate. Sci Rep. (2019) 9:4140. doi: 10.1038/s41598-019-40713-4
37. Oreja-Guevara C, Ayuso Blanco T, Brieva Ruiz L, Hernández Pérez MA, Meca-Lallana V, Ramió-Torrentà L. Cognitive dysfunctions and assessments in multiple sclerosis. Front Neurol. (2019) 10:581. doi: 10.3389/fneur.2019.00581
38. Nabavi SM, Sangelaji B. Cognitive dysfunction in multiple sclerosis: Usually forgotten in the clinical assessment of MS patients. Res Med Sci. (2015) 20:533–4. doi: 10.4103/1735-1995.163984
39. Müller S, Saur R, Greve B, Melms A, Hautzinger M, Fallgatter AJ, et al. Recognition performance differentiates between elderly patients in the long term course of secondary progressive multiple sclerosis and amnestic mild cognitive impairment. Mult Scler. (2013) 19:799–805. doi: 10.1093/med/9780199732920.003.0001
40. Roy S, Drake A, Snyder S, Cline B, Khan A, Fuchs T, et al. Preliminary investigation of cognitive function in aged multiple sclerosis patients: challenges in detecting comorbid Alzheimer's disease. Mult Scler Relat Disord. (2018) 22:52–6. doi: 10.1016/j.msard.2018.03.008
41. Brochet B, Ruet A. Cognitive impairment in multiple sclerosis with regards to disease duration and clinical phenotypes. Front Neurol. (2019) 10:261. doi: 10.3389/fneur.2019.00261
42. Sumowski JF, Rocca MA, Leavitt VM, Riccitelli G, Comi G, DeLuca J, et al. Brain reserve and cognitive reserve in multiple sclerosis: what you've got and how you use it. Neurology. (2013) 80:2186–93. doi: 10.1212/WNL.0b013e318296e98b
43. Kelley BJ, Petersen RC. Alzheimer's disease and mild cognitive impairment. Neurol Clin. (2007) 25:577–v. doi: 10.1016/j.ncl.2007.03.008
44. Braskie MN, Thompson PM. Understanding cognitive deficits in Alzheimer's disease based on neuroimaging findings. Trends Cogn Sci. (2013) 17:510–6. doi: 10.1016/j.tics.2013.08.007
45. Petersen RC, Caracciolo B, Brayne C, Gauthier S, Jelic V, Fratiglioni L. Mild cognitive impairment: a concept in evolution. J Intern Med. (2014) 275:214–28. doi: 10.1111/joim.12190
46. Papadopoulou A, Müller-Lenke N, Naegelin Y, Kalt G, Bendfeldt K, Kuster P, et al. Contribution of cortical and white matter lesions to cognitive impairment in multiple sclerosis. Mult Scler. (2013) 19:1290–6. doi: 10.1177/1352458513475490
47. Jaroudi W, Garami J, Garrido S, Hornberger M, Keri S, Moustafa AA. Factors underlying cognitive decline in old age and Alzheimer's disease: the role of the hippocampus. Rev Neurosci. (2017) 28:705–14. doi: 10.1515/revneuro-2016-0086
48. Jang H, Kwon H, Yang JJ, Hong J, Kim Y, Kim KW, et al. Correlations between gray matter and white matter degeneration in pure Alzheimer's disease, pure subcortical vascular dementia, and mixed dementia. Sci Rep. (2017) 7:9541. doi: 10.1038/s41598-017-10074-x
49. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. (1992) 256:184–5. doi: 10.1126/science.1566067
50. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. (2002) 297:353–6. doi: 10.1126/science.1072994
51. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. (2011) 34:185–204. doi: 10.1146/annurev-neuro-061010-113613
52. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. (2019) 14:32. doi: 10.1186/s13024-019-0333-5
53. Reiss AB, Arai HA, Stecker MM, Siegart NM, Kasselman LJ. Amyloid toxicity in Alzheimer's disease. Rev Neurosci. (2018) 29:613–27. doi: 10.1515/revneuro-2017-0063
54. Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. (2008) 283:29639–43. doi: 10.1074/jbc.R800016200
55. Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. (1997) 120:393–9. doi: 10.1093/brain/120.3.393
56. Plummer S, Van den Heuvel C, Thornton E, Corrigan F, Cappai R. The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis. (2016) 7:163–79. doi: 10.14336/AD.2015.0907
57. Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. (2008) 451:1076–81. doi: 10.1038/nature06559
58. Dal Bianco A, Bradl M, Frischer J, Kutzelnigg A, Jellinger K, Lassmann H. Multiple sclerosis and Alzheimer's disease. Ann Neurol. (2008) 63:174–83. doi: 10.1002/ana.21240
59. Goodheart AE, Tamburo E, Minhas D, Aizenstein HJ, McDade E, Snitz BE, et al. Reduced binding of Pittsburgh compound-B in areas of white matter hyperintensities. Neuroimage Clin. (2015) 9:479–83. doi: 10.1016/j.nicl.2015.09.009
60. Zeydan B, Lowe VJ, Schwarz CG, Przybelski SA, Tosakulwong N, Zuk SM, et al. Pittsburgh compound-B PET white matter imaging and cognitive function in late multiple sclerosis. Mult Scler. (2018) 24:739–49. doi: 10.1177/1352458517707346
61. Pytel V, Matias-Guiu JA, Matías-Guiu J, Cortés-Martínez A, Montero P, Moreno-Ramos T, et al. Amyloid PET findings in multiple sclerosis are associated with cognitive decline at 18 months. Mult Scler Relat Disord. (2020) 39:101926. doi: 10.1016/j.msard.2020.101926
62. Zeydan B, Lowe VJ, Reichard RR, Przybelski SA, Lesnick TG, Schwarz CG, et al. Imaging biomarkers of alzheimer disease in multiple sclerosis. Ann Neurol. (2020) 87:556–67. doi: 10.1002/ana.25684
63. Hoos MD, Ahmed M, Smith SO, Van Nostrand WE. Inhibition of familial cerebral amyloid angiopathy mutant amyloid β-protein fibril assembly by myelin basic protein. J Biol Chem. (2007) 282:9952–61. doi: 10.1074/jbc.M603494200
64. Kotarba AME, Aucoin D, Hoos MD, Smith SO, Van Nostrand WE. Fine mapping of the amyloid β-protein binding site on myelin basic protein. Biochemistry. (2013) 52:2565–73. doi: 10.1021/bi4001936
65. Ou-Yang MH, Xu F, Liao MC, Davis J, Robinson JK, Van Nostrand WE. The N-terminal region of myelin basic protein reduces fibrillar amyloid-β deposition in Tg-5xFAD mice. Neurobiol Aging. (2015) 36:801–11. doi: 10.1016/j.neurobiolaging.2014.10.006
66. Matías-Guiu JA, Oreja-Guevara C, Cabrera-Martín MN, Moreno-Ramos T, Carreras JL, Matías-Guiu J. Amyloid proteins and their role in multiple sclerosis. Considerations in the use of amyloid-PET imaging. Front Neurol. (2016) 7:53. doi: 10.3389/fneur.2016.00053
67. Jin MM, Wang F, Qi D, Liu WW, Gu C, Mao CJ, et al. A critical role of autophagy in regulating microglia polarization in neurodegeneration. Front Aging Neurosci. (2018) 10:378. doi: 10.3389/fnagi.2018.00378
68. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. (2005) 128:2705–12. doi: 10.1093/brain/awh641
69. Felsky D, Patrick E, Schneider JA, Mostafavi S, Gaiteri C, Patsopoulos N, et al. Polygenic analysis of inflammatory disease variants and effects on microglia in the aging brain. Mol Neurodegener. (2018) 13:38. doi: 10.1186/s13024-018-0272-6
70. Valis M, Talab R, Stourac P, Andrys C, Masopust J. Tau protein, phosphorylated tau protein and beta-amyloid42 in the cerebrospinal fluid of multiple sclerosis patients. Neuro Endocrinol Lett. (2008) 29:971–6.
71. Hein Née Maier K, Köhler A, Diem R, Sättler MB, Demmer I, Lange P, et al. Biological markers for axonal degeneration in CSF and blood of patients with the first event indicative for multiple sclerosis. Neurosci Lett. (2008) 436:72–6. doi: 10.1016/j.neulet.2008.02.064
72. Sladkova V, Mareš J, Lubenova B, Zapletalova J, Stejskal D, Hlustik P, et al. Degenerative and inflammatory markers in the cerebrospinal fluid of multiple sclerosis patients with relapsing–remitting course of disease and after clinical isolated syndrome. Neurol Res. (2011) 33:415–20. doi: 10.1179/016164110X12816242542535
73. Szalardy L, Zadori D, Simu M, Bencsik K, Vecsei L, Klivenyi P. Evaluating biomarkers of neuronal degeneration and neuroinflammation in CSF of patients with multiple sclerosis-osteopontin as a potential marker of clinical severity. J Neurol Sci. (2013) 331:38–42. doi: 10.1016/j.jns.2013.04.024
74. Mattsson N, Axelsson M, Haghighi S, Malmeström C, Wu G, Anckarsäter R, et al. Reduced cerebrospinal fluid BACE1 activity in multiple sclerosis. Mult Scler. (2009) 15:448–54. doi: 10.1177/1352458508100031
75. Mori F, Rossi S, Sancesario G, Codecà C, Mataluni G, Monteleone F, et al. Cognitive and cortical plasticity deficits correlate with altered amyloid-β CSF levels in multiple sclerosis. Neuropsychopharmacology. (2011) 36:559–68. doi: 10.1038/npp.2010.187
76. Augutis K, Axelsson M, Portelius E, Brinkmalm G, Andreasson U, Gustavsson MK, et al. Cerebrospinal fluid biomarkers of β-amyloid metabolism in multiple sclerosis. Mult Scler. (2013) 19:543–52. doi: 10.1177/1352458512460603
77. Pietroboni AM, Schiano di Cola F, Scarioni M, Fenoglio C, Span B, Arighi A, et al. CSF β-amyloid as a putative biomarker of disease progression in multiple sclerosis. Mult Scler. (2017) 23:1085–9. doi: 10.1177/1352458516674566
78. Spitzer P, Lang R, Oberstein TJ, Lewczuk P, Ermann N, Huttner HB, et al. A specific reduction in Aβ1–42 vs. a universal loss of Aβ peptides in CSF differentiates Alzheimer's disease from meningitis and multiple sclerosis. Front Aging Neurosci. (2018) 10:152. doi: 10.3389/fnagi.2018.00152
79. Gentile A, Mori F, Bernardini S, Centonze D. Role of amyloid-β CSF levels in cognitive deficit in MS. Clin Chim Acta. (2015) 449:23–30. doi: 10.1016/j.cca.2015.01.035
80. Stampanoni Bassi M, Garofalo S, Marfia GA, Gilio L, Simonelli I, Finardi A, et al. Amyloid-β homeostasis bridges inflammation, synaptic plasticity deficits and cognitive dysfunction in multiple sclerosis. Front Mol Neurosci. (2017) 10:390. doi: 10.3389/fnmol.2017.00390
81. Anderson JM, Hampton DW, Patani R, Pryce G, Crowther RA, Reynolds R, et al. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain. (2008) 131:1736–48. doi: 10.1093/brain/awn119
82. Anderson JM, Patani R, Reynolds R, Nicholas R, Compston A, Spillantini MG, et al. Abnormal tau phosphorylation in primary progressive multiple sclerosis. Acta Neuropathol. (2010) 119:591–600. doi: 10.1007/s00401-010-0671-4
83. Tobin WO, Popescu BF, Lowe V, Pirko I, Parisi JE, Kantarci K, et al. Multiple sclerosis masquerading as Alzheimer-type dementia: clinical, radiological and pathological findings. Mult Scler. (2016) 22:698–704. doi: 10.1177/1352458515604382
84. Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochem Pharmacol. (2014) 88:508–16. doi: 10.1016/j.bcp.2014.01.015
85. Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA, et al. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol. (2011) 69:445–54. doi: 10.1002/ana.22337
86. Jürgens T, Jafari M, Kreutzfeldt M, Bahn E, Brück W, Kerschensteiner M, et al. Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain. (2016) 139:39–46. doi: 10.1093/brain/awv353
87. Papadopoulos D, Dukes S, Patel R, Nicholas R, Vora A, Reynolds R. Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol. (2009) 19:238–53. doi: 10.1111/j.1750-3639.2008.00177.x
88. Zhu B, Luo L, Moore GRW, Paty DW, Cynader MS. Dendritic and synaptic pathology in experimental autoimmune encephalomyelitis. Am J Pathol. (2003) 162:1639–50. doi: 10.1016/S0002-9440(10)64298-8
89. DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. (1990) 27:457–64. doi: 10.1002/ana.410270502
90. Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci Lett. (1994) 174:67–72. doi: 10.1016/0304-3940(94)90121-X
91. Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. (1991) 30:572–80. doi: 10.1002/ana.410300410
92. Mandolesi G, Grasselli G, Musumeci G, Centonze D. Cognitive deficits in experimental autoimmune encephalomyelitis: neuroinflammation and synaptic degeneration. Neurol Sci. (2010) 31:S255–9. doi: 10.1007/s10072-010-0369-3
93. Stampanoni Bassi M, Iezzi E, Gilio L, Centonze D, Buttari F. Synaptic plasticity shapes brain connectivity: implications for network topology. Int J Mol Sci. (2019) 20:E6193. doi: 10.3390/ijms20246193
94. Sweatt JD. Neural plasticity and behavior – sixty years of conceptual advances. J Neurochem. (2016) 139:179–99. doi: 10.1111/jnc.13580
95. Mango D, Saidi A, Cisale GY, Feligioni M, Corbo M, Nisticò R. Targeting synaptic plasticity in experimental models of Alzheimer's disease. Front Pharmacol. (2019) 10:778. doi: 10.3389/fphar.2019.00778
96. Di Filippo M, de Iure A, Giampà C, Chiasserini D, Tozzi A, Orvietani PL, et al. Persistent activation of microglia and NADPH drive hippocampal dysfunction in experimental multiple sclerosis. Sci Rep. (2016) 6:20926. doi: 10.1038/srep23855
97. Di Filippo M, Chiasserini D, Gardoni F, Viviani B, Tozzi A, Giampà C, et al. Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol Dis. (2013) 52:229–36. doi: 10.1016/j.nbd.2012.12.009
98. Dutra RC, Moreira ELG, Alberti TB, Marcon R, Prediger RD, Calixto JB. Spatial reference memory deficits precede motor dysfunction in an experimental autoimmune encephalomyelitis model: the role of kallikrein-kinin system. Brain Behav Immun. (2013) 33:90–101. doi: 10.1016/j.bbi.2013.06.002
99. Kim DY, Hao J, Liu R, Turner G, Shi FD, Rho JM. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS ONE. (2012) 7:e35476. doi: 10.1371/journal.pone.0035476
100. Mosayebi G, Soleyman MR, Khalili M, Mosleh M, Palizvan MR. Changes in synaptic transmission and long-term potentiation induction as a possible mechanism for learning disability in an animal model of multiple sclerosis. Int Neurourol J. (2016) 20:26–32. doi: 10.5213/inj.1632514.257
101. Planche V, Panatier A, Hiba B, Ducourneau EG, Raffard G, Dubourdieu N, et al. Selective dentate gyrus disruption causes memory impairment at the early stage of experimental multiple sclerosis. Brain Behav Immun. (2017) 60:240–54. doi: 10.1016/j.bbi.2016.11.010
102. Ittner LM, Götz J. Amyloid-β and tau–a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. (2011) 12:65–72. doi: 10.1038/nrn2967
103. Tan T, Xie J, Liu T, Chen X, Zheng X, Tong Z, et al. Low-frequency (1 Hz) repetitive transcranial magnetic stimulation (rTMS) reverses Aβ(1-42)-mediated memory deficits in rats. Exp Gerontol. (2013) 48:786–94. doi: 10.1016/j.exger.2013.05.001
104. Frigo M, Cogo MG, Fusco ML, Gardinetti M, Frigeni B. Glutamate and multiple sclerosis. Curr Med Chem. (2012) 19:1295–9. doi: 10.2174/092986712799462559
105. Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid ß peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci. (2013) 38:6–23. doi: 10.1503/jpn.110190
106. Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. (2013) 110:E2518–27. doi: 10.1073/pnas.1306832110
107. Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble A oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. (2011) 31:6627–38. doi: 10.1523/JNEUROSCI.0203-11.2011
108. Kervern M, Angeli A, Nicole O, Léveillé F, Parent B, Villette V, et al. Selective impairment of some forms of synaptic plasticity by oligomeric amyloid-β peptide in the mouse hippocampus: implication of extrasynaptic NMDA receptors. J Alzheimers Dis. (2012) 32:183–96. doi: 10.3233/JAD-2012-120394
109. Liu D, Yang Q, Li S. Activation of extrasynaptic NMDA receptors induces LTD in rat hippocampal CA1 neurons. Brain Res Bull. (2013) 93:10–6. doi: 10.1016/j.brainresbull.2012.12.003
110. Bading H. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J Exp Med. (2017) 214:569–78. doi: 10.1084/jem.20161673
111. Sun XY, Tuo QZ, Liuyang ZY, Xie AJ, Feng XL, Yan X, et al. Extrasynaptic NMDA receptor-induced tau overexpression mediates neuronal death through suppressing survival signaling ERK phosphorylation. Cell Death Dis. (2016) 7:e2449. doi: 10.1038/cddis.2016.329
112. Bordji K, Becerril-Ortega J, Buisson A. Synapses, NMDA receptor activity and neuronal Aβ production in Alzheimer's disease. Rev Neurosci. (2011) 22:285–94. doi: 10.1515/rns.2011.029
113. Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. (2014) 82:756–71. doi: 10.1016/j.neuron.2014.05.004
114. Shipton OA, Leitz JR, Dworzak J, Acton CEJ, Tunbridge EM, Denk F, et al. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J Neurosci. (2011) 31:1688–92. doi: 10.1523/JNEUROSCI.2610-10.2011
115. Pickett EK, Herrmann AG, McQueen J, Abt K, Dando O, Tulloch J, et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of alzheimer's disease. Cell Rep. (2019) 29:3592–604.e5. doi: 10.1016/j.celrep.2019.11.044
116. Rush T, Buisson A. Reciprocal disruption of neuronal signaling and Aβ production mediated by extrasynaptic NMDA receptors: a downward spiral. Cell Tissue Res. (2014) 356:279–86. doi: 10.1007/s00441-013-1789-1
117. Henstridge CM, Pickett E, Spires-Jones TL. Synaptic pathology: a shared mechanism in neurological disease. Ageing Res Rev. (2016) 28:72–84. doi: 10.1016/j.arr.2016.04.005
118. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. (2000) 192:393–404. doi: 10.1084/jem.192.3.393
119. Junker A, Ivanidze J, Malotka J, Eiglmeier I, Lassmann H, Wekerle H, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain. (2007) 130:2789–99. doi: 10.1093/brain/awm214
120. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. Neurol Sci. (1983) 62:219–32. doi: 10.1016/0022-510X(83)90201-0
121. Friese MA, Fugger L. Autoreactive CD8+ T cells in multiple sclerosis: a new target for therapy? Brain. (2005) 128:1747–63. doi: 10.1093/brain/awh578
122. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. (2009) 132:1175–89. doi: 10.1093/brain/awp070
123. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, Gabriely G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. (2018) 141:2066–82. doi: 10.1093/brain/awy151
124. Lassmann H. Multiple sclerosis pathology. Cold Spring Harb Perspect Med. (2018) 8:a028936. doi: 10.1101/cshperspect.a028936
125. Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. (2000) 123:1174–83. doi: 10.1093/brain/123.6.1174
126. Mangiardi M, Crawford DK, Xia X, Du S, Simon-Freeman R, Voskuhl RR, et al. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol. (2011) 21:263–78. doi: 10.1111/j.1750-3639.2010.00444.x
127. Storch MK, Bauer J, Linington C, Olsson T, Weissert R, Lassmann H. Cortical demyelination can be modeled in specific rat models of autoimmune encephalomyelitis and is major histocompatability complex (MHC) haplotype-related. J Neuropathol Exp Neurol. (2006) 65:1137–42. doi: 10.1097/01.jnen.0000248547.13176.9d
128. Legroux L, Arbour N. Multiple sclerosis and T lymphocytes: an entangled story. J Neuroimmune Pharmacol. (2015) 10:528–46. doi: 10.1007/s11481-015-9614-0
129. Schmied M, Breitschopf H, Gold R, Zischler H, Rothe G, Wekerle H, et al. Apoptosis of T lymphocytes in experimental autoimmune encephalomyelitis. Evidence for programmed cell death as a mechanism to control inflammation in the brain. Am J Pathol. (1993) 143:446–52.
130. Mars LT, Bauer J, Gross DA, Bucciarelli F, Firat H, Hudrisier D, et al. CD8 T cell responses to myelin oligodendrocyte glycoprotein-derived peptides in humanized HLA-A*0201-transgenic mice. J Immunol. (2007) 179:5090–8. doi: 10.4049/jimmunol.179.8.5090
131. Wagner CA, Roqué PJ, Mileur TR, Liggitt D, Goverman JM. Myelin-specific CD8+ T cells exacerbate brain inflammation in CNS autoimmunity. J Clin Invest. (2020) 130:203–13. doi: 10.1172/JCI132531
132. Fogdell-Hahn A, Ligers A, Grønning M, Hillert J, Olerup O. Multiple sclerosis: a modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens. (2000) 55:140–8. doi: 10.1034/j.1399-0039.2000.550205.x
133. Harbo HF, Lie BA, Sawcer S, Celius EG, Dai K-Z, Oturai A, et al. Genes in the HLA class I region may contribute to the HLA class II-associated genetic susceptibility to multiple sclerosis. Tissue Antigens. (2004) 63:237–47. doi: 10.1111/j.0001-2815.2004.00173.x
134. Hoftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, et al. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol. (2004) 14:43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x
135. Konjevic Sabolek M, Held K, Beltran E, Niedl AG, Meinl E, Hohlfeld R, et al. Communication of CD8 + T cells with mononuclear phagocytes in multiple sclerosis. Ann Clin Transl Neurol. (2019) 6:1151–64. doi: 10.1002/acn3.783
136. Jilek S, Schluep M, Rossetti AO, Guignard L, Le Goff Gr, Pantaleo G, et al. CSF enrichment of highly differentiated CD8+ T cells in early multiple sclerosis. Clin Immunol. (2007) 123:105–13. doi: 10.1016/j.clim.2006.11.004
137. Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonnière L, et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on α4 integrin. Brain. (2011) 134:3560–77. doi: 10.1093/brain/awr268
138. Malmestr?m C, Lycke J, Haghighi S, Andersen O, Carlsson L, Waden- vik H, et al. Relapses in multiple sclerosis are associated with increased CD8+ T-cell mediated cytotoxicity in CSF. J Neuroimmunol. (2008) 196:159–65. doi: 10.1016/j.jneuroim.2008.03.001
139. Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. (2002) 25:313–9. doi: 10.1016/S0166-2236(02)02154-9
140. Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. (2007) 17:210–8. doi: 10.1111/j.1750-3639.2007.00064.x
141. Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci USA. (1994) 91:10859–63. doi: 10.1073/pnas.91.23.10859
142. Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlén C, Goverman J. A pathogenic role for myelin-specific CD8+ T cells in a model for multiple sclerosis. J Exp Med. (2001) 194:669–76. doi: 10.1084/jem.194.5.669
143. Ford ML, Evavold BD. Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur J Immunol. (2005) 35:76–85. doi: 10.1002/eji.200425660
144. Denic A, Wootla B, Rodriguez M. CD8+ T cells in multiple sclerosis. Expert Opin Ther Targets. (2013) 17:1053–66. doi: 10.1517/14728222.2013.815726
145. Medana I, Martinic MA, Wekerle H, Neumann H. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol. (2001) 159:809–15. doi: 10.1016/S0002-9440(10)61755-5
146. Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol. (2004) 172:5120–7. doi: 10.4049/jimmunol.172.8.5120
147. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. (2008) 172:146–55. doi: 10.2353/ajpath.2008.070690
148. Lückel C, Picard F, Raifer H, Campos Carrascosa L, Guralnik A, Zhang Y, et al. IL-17+CD8+ T cell suppression by dimethyl fumarate associates with clinical response in multiple sclerosis. Nat Commun. (2019) 10:5722. doi: 10.1038/s41467-019-13731-z
149. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-β therapy. J Immunol. (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
150. Lotfi N, Thome R, Rezaei N, Zhang GX, Rezaei A, Rostami A, et al. Roles of GM-CSF in the pathogenesis of autoimmune diseases: an update. Front Immunol. (2019) 10:1265. doi: 10.3389/fimmu.2019.01265
151. Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res. (2006) 16:126–33. doi: 10.1038/sj.cr.7310017
152. Parajuli B, Sonobe Y, Kawanokuchi J, Doi Y, Noda M, Takeuchi H, et al. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflamm. (2012) 9:268. doi: 10.1186/1742-2094-9-268
153. Lolli F, Martini H, Citro A, Franceschini D, Portaccio E, Amato MP, et al. Increased CD8+ T cell responses to apoptotic T cell-associated antigens in multiple sclerosis. J Neuroinflammation. (2013) 10:94. doi: 10.1186/1742-2094-10-94
154. Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest. (2013) 123:247–60. doi: 10.1172/JCI63681
155. Stojić-Vukanić Z, Pilipović I, Djikić J, Vujnović I, Nacka-Aleksić M, Bufan B, et al. Strain specificities in age-related changes in mechanisms promoting and controlling rat spinal cord damage in experimental autoimmune encephalomyelitis. Exp Gerontol. (2018) 101:37–53. doi: 10.1016/j.exger.2017.11.002
156. Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, et al. A Th17-like developmental process leads to CD8+ Tc17 cells with reduced cytotoxic activity. Eur J Immunol. (2009) 39:1716–25. doi: 10.1002/eji.200939412
157. Liang Y, Pan HF, Ye DQ. Tc17 cells in immunity and systemic autoimmunity. Int Rev Immunol. (2015) 34:318–31. doi: 10.3109/08830185.2014.954698
158. Salehi Z, Doosti R, Beheshti M, Janzamin E, Sahraian MA, Izad M. Differential frequency of CD8+ T cell subsets in multiple sclerosis patients with various clinical patterns. PLoS ONE. (2016) 11:e0159565. doi: 10.1371/journal.pone.0159565
159. Tilly G, Yap M, Brouard S, Degauque N. TEMRA CD8 T cells are highly cytopathic cells that escape from costimulatory based-therapy. Transplantation. (2014) 98:318–9. doi: 10.1097/00007890-201407151-01012
160. Kivisäkk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. (2004) 55:627–38. doi: 10.1002/ana.20049
161. Smolders J, Heutinck KM, Fransen NL, Remmerswaal EBM, Hombrink P, ten Berge IJM, et al. Tissue-resident memory T cells populate the human brain. Nat Commun. (2018) 9:4593. doi: 10.1038/s41467-018-07053-9
162. Sasaki K, Bean A, Shah S, Schutten E, Huseby PG, Peters B, et al. Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J Immunol. (2014) 192:3029–42. doi: 10.4049/jimmunol.1302911
163. Boldison J, Chu CJ, Copland DA, Lait PJP, Khera TK, Dick AD, et al. Resident exhausted effector memory CD8+ T cells accumulate in the retina during chronic experimental autoimmune uveoretinitis. J Immunol. (2014) 192:4541–50. doi: 10.4049/jimmunol.1301390
164. Park C, Kupper T. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. (2015) 21:688–97. doi: 10.1038/nm.3883
165. Fransen NL, Hsiao CC, van der Poel M, Engelenburg HJ, Verdaasdonk K, Vincenten MCJ, et al. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain. (2020) 143:1714–30. doi: 10.1093/brain/awaa117
166. Smolders J, Fransen NL, Hsiao CC, Hamann J, Huitinga I. Perivascular tissue resident memory T cells as therapeutic target in multiple sclerosis. Expert Rev Neurother. (2020) 20:835–48. doi: 10.1080/14737175.2020.1776609
167. Unger MS, Marschallinger J, Kaindl J, Klein B, Johnson M, Khundakar AA, et al. Doublecortin expression in CD8+ T-cells and microglia at sites of amyloid-β plaques: A potential role in shaping plaque pathology? Alzheimers Dement. (2018) 14:1022–37. doi: 10.1016/j.jalz.2018.02.017
168. Monsonego A, Imitola J, Petrovic S, Zota V, Nemirovsky A, Baron R, et al. Aβ-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of Abeta in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. (2006) 103:5048–53. doi: 10.1073/pnas.0506209103
169. Pirker-Kees A, Schmied C, Dal-Bianco P. T-cells show increased production of cytokines and activation markers in Alzheimer's disease. Brain Disord Ther. (2013) 3:1. doi: 10.4172/2168-975X.1000112
170. Tröscher AR, Wimmer I, Quemada-Garrido L, Köck U, Gessl D, Verberk SGS, et al. Microglial nodules provide the environment for pathogenic T cells in human encephalitis. Acta Neuropathol. (2019) 137:619–35. doi: 10.1007/s00401-019-01958-5
171. Cebrián C, Loike JD, Sulzer D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson's and other brain diseases. Front Neuroanat. (2014) 8:114. doi: 10.3389/fnana.2014.00114
172. Esser MT, Haverstick DM, Fuller CL, Gullo CA, Braciale VL. Ca2 signaling modulates cytolytic T lymphocyte effector functions. J Exp Med. (1998) 187:1057–67. doi: 10.1084/jem.187.7.1057
173. Kessler B, Hudrisier D, Schroeter M, Tschopp J, Cerottini JC, Luescher IF. Peptide modification or blocking of CD8, resulting in weak TCR signaling, can activate CTL for Fas- but not perforin-dependent cytotoxicity or cytokine production. J Immunol. (1998) 161:6939–46.
174. Meuth SG, Herrmann AM, Simon OJ, Siffrin V, Melzer N, Bittner S, et al. Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J Neurosci. (2009) 29:15397–409. doi: 10.1523/JNEUROSCI.4339-09.2009
175. Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. (2001) 50:389–400. doi: 10.1002/ana.1123
176. Stadelmann C, Albert M, Wegner C, Bruck W. Cortical pathology in multiple sclerosis. Curr Opin Neurol. (2008) 21:229–34. doi: 10.1097/01.wco.0000318863.65635.9a
177. Melzer N, Meuth SG, Wiendl H. CD8+ T cells and neuronal damage: direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J. (2009) 23:3659–73. doi: 10.1096/fj.09-136200
178. Zhao D, Feng F, Zhao C, Wu F, Ma C, Bai Y, et al. Role of perforin secretion from CD8+ T-cells in neuronal cytotoxicity in multiple sclerosis. Neurol Res. (2018) 40:62–7. doi: 10.1080/01616412.2017.1398371
179. Nitsch R, Pohl EE, Smorodchenko A, Infante-Duarte C, Aktas O, Zipp F. Direct impact of T cells on neurons revealed by two-photon microscopy in living brain tissue. J Neurosci. (2004) 24:2458–64. doi: 10.1523/JNEUROSCI.4703-03.2004
180. Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol. (2008) 180:3866–73. doi: 10.4049/jimmunol.180.6.3866
181. Villegas-Mendez A, Greig R, Shaw TN, de Souza JB, Gwyer Findlay E, Stumhofer JS, et al. IFN-γ producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J Immunol. (2012) 189:968–79. doi: 10.4049/jimmunol.1200688
182. Cipollini V, Anrather J, Orzi F, Iadecola C. Th17 and cognitive Impairment: possible mechanisms of action. Front Immunol. (2019) 13:95. doi: 10.3389/fnana.2019.00095
183. Vass K, Lassmann H. Intrathecal application of interferon gamma. Progressive appearance of MHC antigens within the rat nervous system. Am J Pathol. (1990) 137:789–800.
184. Medana IM, Gallimore A, Oxenius A, Martinic MM, Wekerle H, Neumann H. MHC class I-restricted killing of neurons by virus-specific CD8+ T lymphocytes is effected through the Fas/FasL, but not the perforin pathway. Eur J Immunol. (2000) 30:3623–33. doi: 10.1002/1521-4141(200012)30:12<3623::AID-IMMU3623>3.0.CO;2-F
185. Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, Vanni V, et al. Necrosis factor and interleukin-1β modulate synaptic plasticity during neuroinflammation. Neural Plast. (2018) 2018:8430123. doi: 10.1155/2018/8430123
186. Cavanagh C, Wong TP. Preventing synaptic deficits in Alzheimer's disease by inhibiting tumor necrosis factor alpha signaling. IBRO Rep. (2018) 4:18–21. doi: 10.1016/j.ibror.2018.01.003
187. Monteiro S, Ferreira FM, Pinto V, Roque S, Morais M, de Sá-Calçada D, et al. Absence of IFNγ promotes hippocampal plasticity and enhances cognitive performance. Transl Psychiatry. (2016) 6:e707. doi: 10.1038/tp.2015.194
188. Mizuno T, Zhang G, Takeuchi H, Kawanokuchi J, Wang J, Sonobe Y, et al. Interferon-γ directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-γ receptor and AMPA GluRl receptor. FASEB J. (2008) 22:1797–806. doi: 10.1096/fj.07-099499
189. Kostic M, Dzopalic T, Zivanovic S, Zivkovic N, Cvetanovic A, Stojanovic I, et al. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol. (2014) 79:181–6. doi: 10.1111/sji.12147
190. Olmos G, Lladó J. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm. (2014) 2014:861231. doi: 10.1155/2014/861231
191. Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. (2005) 90:663–70. doi: 10.1113/expphysiol.2005.030734
192. Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. (2001) 76:846–54. doi: 10.1046/j.1471-4159.2001.00075.x
193. Sarlus H, Heneka MT. Microglia in Alzheimer's disease. J Clin Invest. (2017) 127:3240–9. doi: 10.1172/JCI90606
194. Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol. (2018) 8:1905. doi: 10.3389/fimmu.2017.01905
195. Schlüter D, Meyer T, Strack A, Reiter S, Kretschmar M, Wiestler OD, et al. Regulation of microglia by CD4+ and CD8+ T cells: selective analysis in CD45-congenic normal and Toxoplasma gondii-infected bone marrow chimeras. Brain Pathol. (2001) 11:44–55. doi: 10.1111/j.1750-3639.2001.tb00380.x
196. Krishnan A, Kocab AJ, Zacks DN, Marshak-Rothstein A, Gregory-Ksander M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J Neuroinflammation. (2019) 16:184. doi: 10.1186/s12974-019-1576-3
197. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. (2014) 41:886–97. doi: 10.1016/j.immuni.2014.12.007
198. Steinbach K, Vincenti I, Merkler D. Tissue-restricted immune resident-memory T cells in responses: for better or worse? Front Immunol. (2018) 9:2827. doi: 10.3389/fimmu.2018.02827
199. Mercier BC, Cottalorda A, Coupet CA, Marvel J, Bonnefoy-Berard N. TLR2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. J Immunol. (2009) 182:1860–7. doi: 10.4049/jimmunol.0801167
200. Cottalorda A, Mercier BC, Mbitikon-Kobo FM, Arpin C, Teoh DY, McMichael A, et al. TLR2 engagement on memory CD8+ T cells improves their cytokine-mediated proliferation and IFN-gamma secretion in the absence of Ag. Eur J Immunol. (2009) 39:2673–81. doi: 10.1002/eji.200939627
201. Salerno F, Freen-van Heeren JJ, Guislain A, Nicolet BP, Wolkers MC. Costimulation through TLR2 drives polyfunctional CD8+ T cell responses. J Immunol. (2019) 202:714–23. doi: 10.4049/jimmunol.1801026
202. Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol. (2012) 188:1098–107. doi: 10.4049/jimmunol.1101121
Keywords: multiple sclerosis, Alzheimer's disease, neurocognitive impairment, effector/memory CD8+ T cells, CD8+ tissue-resident memory T cells, microglia
Citation: Stojić-Vukanić Z, Hadžibegović S, Nicole O, Nacka-Aleksić M, Leštarević S and Leposavić G (2020) CD8+ T Cell-Mediated Mechanisms Contribute to the Progression of Neurocognitive Impairment in Both Multiple Sclerosis and Alzheimer's Disease? Front. Immunol. 11:566225. doi: 10.3389/fimmu.2020.566225
Received: 31 May 2020; Accepted: 17 August 2020;
Published: 19 November 2020.
Edited by:
Marcello Moccia, University of Naples Federico II, ItalyReviewed by:
Dejan Jakimovski, Buffalo Neuroimaging Analysis Center, United StatesTimo Jan Oberstein, University of Erlangen Nuremberg, Germany
Copyright © 2020 Stojić-Vukanić, Hadžibegović, Nicole, Nacka-Aleksić, Leštarević and Leposavić. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gordana Leposavić, Z29yZGFuYS5sZXBvc2F2aWNAcGhhcm1hY3kuYWMuYmcucnM=
†These authors have contributed equally to this work