 Oluwabukola T. Gbotosho
Oluwabukola T. Gbotosho Maria G. Kapetanaki2,3†
Maria G. Kapetanaki2,3† Gregory J. Kato
Gregory J. Kato
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 January 2021
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.561917
This article is part of the Research Topic Inflammatory Mechanisms of Hemolytic Diseases View all 21 articles
Hemolysis is a pathological feature of several diseases of diverse etiology such as hereditary anemias, malaria, and sepsis. A major complication of hemolysis involves the release of large quantities of hemoglobin into the blood circulation and the subsequent generation of harmful metabolites like labile heme. Protective mechanisms like haptoglobin-hemoglobin and hemopexin-heme binding, and heme oxygenase-1 enzymatic degradation of heme limit the toxicity of the hemolysis-related molecules. The capacity of these protective systems is exceeded in hemolytic diseases, resulting in high residual levels of hemolysis products in the circulation, which pose a great oxidative and proinflammatory risk. Sickle cell disease (SCD) features a prominent hemolytic anemia which impacts the phenotypic variability and disease severity. Not only is circulating heme a potent oxidative molecule, but it can act as an erythrocytic danger-associated molecular pattern (eDAMP) molecule which contributes to a proinflammatory state, promoting sickle complications such as vaso-occlusion and acute lung injury. Exposure to extracellular heme in SCD can also augment the expression of placental growth factor (PlGF) and interleukin-6 (IL-6), with important consequences to enthothelin-1 (ET-1) secretion and pulmonary hypertension, and potentially the development of renal and cardiac dysfunction. This review focuses on heme-induced mechanisms that are implicated in disease pathways, mainly in SCD. A special emphasis is given to heme-induced PlGF and IL-6 related mechanisms and their role in SCD disease progression.
Sickle Cell Disease (SCD) is an inherited hematological disorders, with a multi-organ complication affecting millions of people worldwide, especially in sub-Saharan Africa (1). In the United States, there are about 100,000 people with SCD. There are variability and often concurrent complications related to the disease, which may differ in frequency and severity. Accumulating evidence suggests that intravascular hemolysis and hemolysis byproducts including hemoglobin and heme instigate a series of events leading to vascular damage. While hemolysis is a prominent feature of SCD, it is certainly not unique to this disease. Red cell destruction may occur as a result of a hereditary hemolytic disorder, an infection, a medication, cancer, an autoimmune disorder, a cardiomyopathy, a hemorrhagic stroke, trauma or even a blood transfusion, to mention a few (2). The current review focuses on the heme-induced mechanisms that are implicated in disease pathways, mainly in SCD and downstream effects of non-bound (free) heme as a result of intravascular hemolysis caused by sickle cell anemia and other hemolytic disorders (Figure 1).
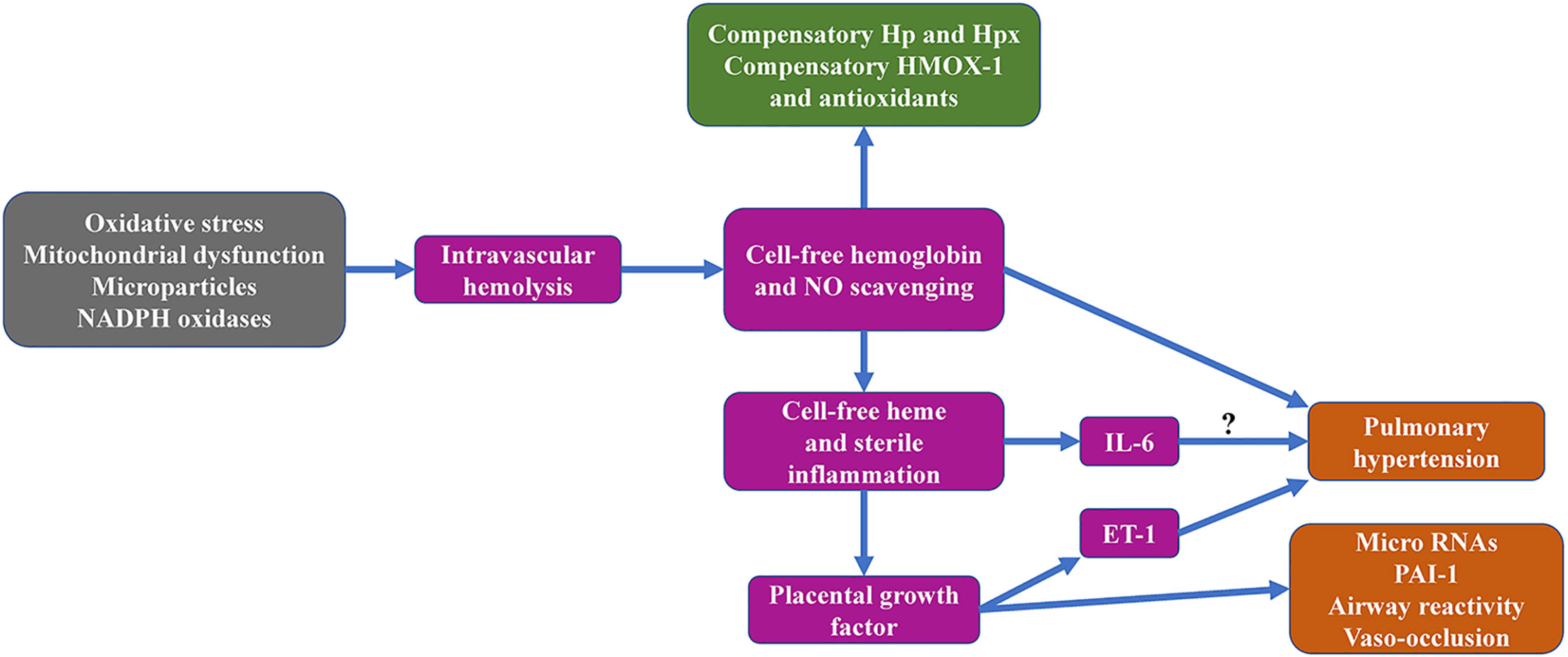
Figure 1 Graphical overview of sickle cell hemolysis-associated topics addressed in the current review manuscript.
Heme synthesis, transport and turnover occurs under normal physiological conditions, and it exerts a physiological signal that helps to control these pathways. For example, heme feeds back to the first committed step in porphyrin synthesis, α-levulinic acid synthase. Heme regulates the Ras-Mitogen Activated Protein Kinase (MAPK) pathway, and it regulates the BACH1 transcriptional repressor, impacting expression of HMOX-1 and β-globin. Heme-regulated inhibitor (HRI) is a eukaryotic initiation factor 2α kinase that coordinates protein synthesis with heme availability in reticulocytes (3). Heme is a crucial prosthetic group for activity of many hemoproteins, include oxygen transport, electron transport, oxygen reduction, and others (4). Heme modulates macrophage differentiation of monocytes to tissue-resident macrophages and stimulates macrophage inflammatory response (5). In sickle cell disease, heme from red cells is turned over via both intravascular and extravascular hemolysis pathways that leads to extensive pathology described in the remainder of this review.
Oxidative stress occurs due to dysregulation between production of reactive oxygen species (ROS) and antioxidants. ROS are vital for cell signaling and homeostasis and are produced as a natural by-product of the normal metabolism of oxygen or exogenously by ionizing radiation and xenobiotic compounds (6–8). Oxidative stress contributes to pathophysiological pathways that underlie inflammation in many hemolytic disorders including SCD (8), β-thalassemia (9, 10), paroxysmal nocturnal hemoglobinuria (11, 12), hereditary spherocytosis (13), and glucose-6-phosphate dehydrogenase deficiency (14–16). RBCs are constantly subjected to oxidative stress due to their role as an oxygen transporter and continuous exposure to both endogenous and exogenous sources of ROS that can damage the RBC and alter blood rheology in SCD patients (17, 18). ROS is generated in SCD through several pathways. Sickle hemoglobin (HbS) produces ROS such as superoxide anion (O2-), hydrogen peroxide (H2O2), peroxynitrite (OONO-) and hydroxyl radical (OH.) following auto-oxidation (19). Auto-oxidation is a normal physiological process that generates methemoglobin (metHb, Hb oxidized to Fe3+ state with no ability to bind O2) and in about 3% of the total Hb every day (19). A small rate of auto-oxidation can produce substantial levels of ROS due to the high concentration of oxygenated Hb (about 5 mM), which can cause enormous damage to the RBC itself, because RBCs make up 40% of the blood volume (20). Moreover, O2- is spontaneously converted to H2O2 by superoxide dismutase, thereby increasing ROS in the system (19). Excessive amounts of reactive oxygen metabolites is produced due to the unstable nature of HbS resulting in conformational change in the Hb in low O2 environment and the continuous auto-oxidation of iron in heme released from Hb (6–8). This heme can oxidize membrane lipids and proteins (21), as evidenced by elevated levels of products of lipid peroxidation including malondialdehyde (MDA) in the plasma of SCD patients (22). Other Hb oxidation products such as ferryl Hb which is also formed in RBCs under conditions of oxidative stress also occurs in HbS (23–25), causing actin remodeling, thereby compromising membrane integrity and transport (26, 27).
The major source of intracellular ROS is the mitochondria in most cells (28) but mature red blood cells (RBCs) from healthy individuals extrude their mitochondria and other organelles during the terminal process of erythropoiesis (29–32). In contrast, a higher percentage of mature RBCs from SCD patients and mice retain their mitochondria leading to excessive ROS accumulation and oxidative stress (25, 33, 34). It has been shown that treatment with products of hemolysis including ferric Hb, ferryl Hb or heme causes bioenergetics changes, abnormal membrane permeability and ROS-induced lipid peroxidation in endothelial and alveolar cells mitochondria (35, 36), which may contribute to inflammatory process and lung injury (37, 38). Additionally, platelets from SCD patients have abnormal mitochondrial activity resulting in oxidant generation and increased activation during vaso-occlusive crisis (VOC) (39). Exposure to cell-free hemoglobin exacerbates this aberrant platelet mitochondrial activity and correlates with markers of hemolysis, NO scavenging and severity of pulmonary arterial hypertension (40).
Another source of oxidative stress in SCD is erythrocyte-derived submicron membrane vesicles called microparticles (eMPs) (41–44). Plasma eMPs are elevated in sickle cell mice (25), in SCD patients at steady state (41, 44) and during vaso occlusive crisis (45, 46). These eMPs are generated during reoxygenation of sickled erythrocyte (42, 43) or during hemolysis (41, 47). Additionally, thrombospondin-1 (TSP1) may trigger shedding of phosphatidylserine positive eMPs and injection of these eMPs into SCD mice caused vaso occlusion in the kidney (48). These hemoglobin-laden eMPs can transfer heme to endothelial cells, adhere to vascular endothelium and scavenge NO thereby mediating oxidative stress (49–51). Staining of human renal biopsies has been shown to contain hemoglobin-laden eMPs adherent to the capillary endothelium in kidney tissue samples from hyperalbuminuric SCD patients, suggesting that eMPs may contribute to renal injury in SCD (51). Finally, other blood cells such as neutrophils and macrophages also release ROS into the plasma which are neutralized by anti-oxidants such as superoxide dismutase before they can be taken up by RBCs (52).
Vascular smooth muscle and phagocytic cells express nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, which can generate endogenous ROS (53). NADPH oxidase activity is mediated by activation of the small Ras-like GTPase Rac via protein kinase C (PKC) stimulation (53). Some plasma factors such as transforming growth factor β1 (TGFβ1) and endothelin-1 (ET-1) have also been shown to stimulate NADPH oxidase activity in neutrophils, monocytes and endothelial cells and many of these factors are present at higher levels in the plasma of SCD patients as a result of persistent inflammatory state associated with SCD (54). RBCs from SCD patients also contain NADPH oxidases, which can generate endogenous ROS, thereby contributing to RBC rigidity and fragility (55).
Accumulation of oxidative injury to the erythrocyte distorts membrane integrity, alters blood flow rheology, membrane transport abnormalities, exposure of phosphatidylserine, and cell death (56–58). Despite the numerous pathways by which ROS is generated in SCD, oxidative stress in patients appears to be compensated at steady state, and only becomes deleterious when the balance between ROS production and antioxidants is perturbed due to excessive ROS generation, low antioxidant levels or during crisis (59). Likewise, ROS production becomes markedly amplified in low antioxidant microenvironments, as found in SCD, resulting in damage of macromolecules including lipids (60, 61), DNA (62, 63), and proteins (64, 65).
However, studies of antioxidant levels in SCD patients have yielded variable results, with several studies reporting low (66–69) and others reporting high levels (70, 71) of activity of antioxidant enzymes including glutathione peroxidase (66, 67), superoxide dismutase (67, 70, 72), and catalase (68, 72). These differences may be due to variations in level of disease severity including hemolysis, lipid peroxidation, VOC, acute splenic sequestration and pulmonary hypertension reported in these patients (73–78). Irrespective of the levels detected, the total antioxidant capacity in SCD patients is insufficient to neutralize excess ROS, resulting in oxidative stress (79). Other non-enzymatic antioxidants such as vitamin C and E (80, 81), zinc (76), and selenium (69, 77, 80) are also decreased in SCD patients.
Several approaches to mitigate the harmful effects of oxidative stress in SCD have been proposed such as use of antioxidants (82), neutralization of products of hemolysis with haptoglobin (Hp) and hemopexin (Hpx) (83) and moderate strength and endurance exercise therapy (84). Recent studies showed that increase in physical activity improves blood rheology, increases NO bioavailability and reduction in oxidative stress and hemolysis in mice (85–87) and SCD patients (88).
Intravascular and extravascular hemolysis, due in large part to recurrent sickling and unsickling and oxidative stress discussed above, causes premature destruction of RBCs, and contributes to anemia in SCD (56, 89). Rapid production of RBCs ensues to compensate for anemia, resulting in an increased proportion of reticulocytes and younger RBCsin the circulation. Younger RBCs have a higher content of arginase, and with lysis of these younger cells, arginase is released into the plasma during hemolysis (90). This ectopic plasma arginase consumes plasma L-arginine (substrate needed for NO production), and together with consumption of endothelial NO by cell-free plasma Hb contributes to decreased NO bioavailability (91–93). Although consequences of hemolysis in SCD are multifactorial, induction of NO deficiency and oxidative stress by acute and chronic release of products of hemolysis into circulation are major sequelae of hemolysis (94). Depletion of NO promotes a chronic vasculopathy endophenotype that predisposes to pre-capillary pulmonary hypertension, leg ulceration, cerebrovascular arteriopathy, chronic kidney disease and priapism. Details of nitric oxide deficiency and pulmonary hypertension are beyond the scope of this review and have been reviewed in detail elsewhere (94–96).
Several distinct and overlapping mechanisms have evolved to mitigate the cytotoxic effect of products of hemolysis. Hb dimers are avidly bound by the serum glycoprotein haptoglobin (Hp), in the plasma to form Hb-Hp complex, which protects against oxidative damage (97–100). The Hb-Hp complex is recognized and internalized via its receptor, CD163, and subsequently cleared by the phagocytic cells in the reticuloendothelial system (97–99). Continuous formation of Hb-Hp complexes in diseases with severe intravascular hemolysis including SCD and paroxysmal nocturnal hemoglobinuria results in depletion of Hp to undetectable levels, leading to some accumulation in plasma of cell-free Hb (101, 102).
Cell-free Hb that becomes oxidized or denatured prior to clearance is prone to release free heme. Plasma free heme becomes elevated in SCD patients (103, 104). About 80% of total heme initially binds to plasma lipoproteins including low-density lipoproteins (LDLs) (105, 106) and high-density lipoproteins (HDLs) (107, 108), before being transferred to albumin and Hpx (107, 109). Low levels of these lipoproteins are reported in SCD patients which may be due to increased catabolism or decreased synthesis (110, 111), as low plasma levels also negatively correlated with markers of hemolysis in SCD patients (112–114). Free heme reversibly binds to albumin to form metalbumin (115–117), or with high affinity to hemopexin (Hpx) (118, 119), and α1-microglobulin (120–122).
Of all these plasma proteins, Hpx, a plasma glycoprotein produced in the liver has the highest affinity for binding free heme (118, 119, 123), resulting in the formation of Hpx-heme complexes that are removed by endocytosis via the Hpx receptor (CD91) in hepatocytes and macrophages (124, 125). After delivering heme to CD91-expressing cells for internalization and degradation by heme oxygenase 1 (HMOX-1), at least some of the Hpx molecules can be recycled back into plasma. Elevated eMPs also correlated with increase in hemolysis markers and low Hpx in SCD patients (126). In the same patients cohort, high eMPs positively correlated with elevated TRV, linking Hpx depletion to increased eMPs and hemolysis, which may predispose patients to pulmonary hypertension (126). In another study, low Hpx negatively correlated with lipid oxidation in human and mice with SCD, with postmortem analysis in SCD patients showing oxidized LDL deposits in the pulmonary artery (127). These reports showed that delayed clearance of heme in circulation due to low plasma Hpx may activate deleterious downstream pathological pathways that may contribute to morbidity and mortality in SCD patients.
HMOX-1 is an evolutionarily conserved and rate limiting enzyme that degrades heme into equimolar amount of iron, biliverdin and carbon monoxide (108, 128, 129). HMOX-1 is highly expressed in human and mice with SCD and further upregulated on exposure to heme (130, 131). Heme-induced oxidative stress exceeds the capacity of HMOX-1 to prevent cellular and organ injury in transgenic murine model of SCD. Augmentation of HMOX-1 level and activity via gene transfer approaches, or pharmacological activation through NRF2 (132), the transcription factor that regulates HMOX-1 expression, conferred protection from heme-induced lung injury (133), vaso-occlusion (134), liver injury (135), kidney injury (136), erythrocyte membrane damage (137), endothelium activation and adherence (135), activation of immune cells and production of inflammatory cytokines (138). Still, the effect of NRF2 activation on hemolysis, γ-globin levels or stress erythropoiesis in mouse model of SCD is controversial (136–138). Not all heme and Hb are bound to proteins or other macromolecules. Unbound heme or hemoglobin in circulation causes erythrocyte membrane damage and injury, activates proinflammatory signaling pathways in RBCs, immune and endothelial cells, hepatocytes, macrophages and neutrophils (105, 139).
Heme induces a program of antioxidant enzymes that compensate for its intrinsic oxidant stress. These include glutathione S-transferase pi (GSTpi) and NAD(P)H dehydrogenase [quinone] 1 (NQO1) (140).
Hemolysis is a major driver of sterile inflammation in pathological conditions including SCD (94, 103, 141), malaria (142, 143), sepsis (144, 145), and also a marker of severity and survival in these patients (146–149). Following hemolysis, Hb is oxidized to unstable methemoglobin resulting in release of free heme (139), which can intercalate into cell membrane and alter cellular structures or taken up by cells (150, 151).
Free heme accumulates in the plasma in both acute and chronic hemolysis when the rate of intravascular hemolysis exceeds the capacity of circulating heme-binding proteins (152), including Hp and Hpx, which are depleted in human and mice with SCD patients (59, 104, 114, 126, 127, 153–156). There is an emerging concept of small molecular weight scavenging protein such as α1-microglobulin, becoming the predominant heme scavenger when plasma Hpx is low (59). Binding of free heme to different scavenger impacts clinical manifestation of excess heme in circulation as heme-Hpx is trafficked to and recycled primarily in the liver while heme-bound α1-microglobulin are taken to the kidney (59). This phenomenon was demonstrated in a recent publication from Ofori-Acquah and colleagues. They showed that hemopexin deficiency correlates with a compensatory increase in α1-microglobulin in both human and mice with SCD (155). Elevated α1-microglobulin and low hemopexin was also associated with increase in acute kidney injury biomarkers urinary KIM-1 and serum NGAL in SCD patients. The authors showed that this heme-bound α1-microglobulin is directed to the kidney for clearance resulting in acute kidney injury in sickle cell mice (155). Also, acute kidney injury may occur via complement deposition in the kidney during intravascular hemolysis and in Hpx deficient condition in SCD mice (157). Patients with SCD with higher plasma levels of free heme also have greater frequency of VOC and acute chest syndrome (158). Accumulation of free heme in plasma is not only cytotoxic, but also mediates generation of free radicals via the Fenton pathway (159–161).
Assay of cell-free heme and Hb may be an important tool for diagnosis in disease conditions characterized by hemolysis (152, 162). Accurate quantification of heme species may result in early therapeutic intervention before irreversible damage to organs occurs. Currently, most commercially available assays measure total heme (free heme and heme bound to proteins) and are not specific for measuring cell-free heme or Hb. There is a possibility of overestimating or underestimating these heme species. Moreover, free heme is likely a more potent mediator of organ injury and signal transductions, its accurate quantification as a biomarker in disease conditions may be vital. Researchers have developed detection methods using the spectral deconvolution method, antibody capture ELISA or western blotting, reversed‐high‐performance liquid chromatography, and fluorescence-based assays to measure Hb and CFH (103, 152, 162–165). Although these are not commercially available currently, they present an opportunity to quantify different heme species in relation to pathogenesis and therapeutic efficacy in hemolytic conditions.
Free heme can induce inflammation via direct activation of RBCs (166, 167), macrophages (168–170), neutrophils (171), and endothelial cells (139, 172–174) to secret proinflammatory cytokines including toll-like receptors (TLRs), tumor necrosis factor (TNF), interleukin-6 (IL-6), placenta growth factor (PlGF), interleukin 1 beta (IL-1β) (105, 139, 169, 175, 176) and release of erythroid damage-associated molecular patterns (eDAMPs) that potentiates inflammation (177, 178). Heme has been shown to induce production of IL-1β by activated monocytes/macrophages, endothelial and smooth muscle cells through a nucleotide-binding domain and leucine-rich repeat-containing protein 3 (NLRP3) inflammasome dependent mechanism (139, 169, 172). High mobility group box 1 (HMGB1), a nuclear protein released during systemic inflammatory response, has also been shown to mediate ROS-dependent activation of endothelial cells to secrete IL-1β via NLRP3 activation (179, 180). Elevated circulating HMGB1 is associated with inflammation in hemolytic disorders including SCD and sepsis (181–184), suggesting a shared inflammatory signaling pathway through TLR4/Bruton tyrosine kinase for both heme and HMGB1 in SCD (185, 186). Heme can also directly affect the vasculature in mice, as recently shown with loss of heme exporter, feline leukemia virus subgroup C receptor 1a (FLVCR1a) in endothelial cells resulted in disruption of microvessel architecture (187).
Cell-free heme also contributes to inflammation by activating cell adhesion pathways. This includes activation of adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), selectins (L, P and E), all involved in mediating cell adhesion to the vascular endothelium via activation of integrin αMβ2 on neutrophils (188–192). Besides, several studies in the last decade have associated hemolysis and selectins expression with RBCs adhesion to endothelial cells (193–195), acute lung injury (196), vaso occlusion (197), pain (198, 199), liver injury (200–202), and kidney injury in SCD (83).
P-selectin is associated with platelet-neutrophil aggregate formation that contributes to inflammation, pulmonary dysfunction and lung vaso occlusion in SCD (200, 203). In addition, a recent study by Merle and colleagues, showed a direct link between heme-induced TLR4 and complement system activation on liver endothelium mediated by P-selectin, with genetic or pharmacological blockade of P-selectin or complement system ameliorating liver injury in mice (202). This expansive body of works culminated in clinical trial and eventual FDA approval of P-selectin blockade therapy for the prevention of pain crises in SCD (198, 199). Furthermore, persistent inducibility of endothelium-derived adhesion molecules by proinflammatory cytokines such as TNF-α and IL-6 coupled with chronic hemolysis in SCD patients ultimately results in VOC, organ dysfunction and early mortality (101, 204–208). There are several ongoing clinical trials in SCD looking at mediating the effect of inflammation-induced organ damage via some of the mechanisms discussed above.
Recent evidence supports a potential role of microRNAs (miRNAs) in complications of SCD (209, 210) and malaria (211, 212), both pathological conditions with hemolysis, suggesting a role for heme modulation of miRNAs. miRNAs are noncoding RNAs of 22 nucleotides in length that regulate the expression of their target genes post-transcriptionally (213). miRNAs are involved in important biological processes including apoptosis (214), hematopoietic differentiation (215) and cell proliferation (216). miRNAs are important regulatory molecules and activation of immune response during initiation and progression of many diseases inflammatory diseases such as cancer, Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus, and asthma, via expression of proinflammatory cytokines including TNF-α and TLRs (217–222). There are studies linking heme and miRNAs processing in mammalian cells. Heme binds directly to the RNA-binding protein DiGeorge critical region-8 (DGCR8), which is essential for the first miRNA processing step (213, 223–225). Hemolysis elevates the expression of several miRNAs found in RBCs including miR-16, miR-92a, miR-451, and miR-486 (226, 227). There is upregulation of some miRNAs including miR-16, miR-451 and miR-144 in reticulocytes from SCD patients (228, 229). Conversely, elevated levels of these miRNAs also correlated with severe anemia, increased sensitivity to oxidative stress, downregulation of NRF2 and decreased intracellular glutathione levels (230, 231). On the other hand, members of the miR-154, the miR-329 and miR-376 family, involved in TGF-β signaling pathway are downregulated in platelets of SCD patients (210). Although few numbers of studies have reported the involvement of miRNAs in complications of SCD (232), however, there is a gap in knowledge of how stress or heme regulation of these miRNAs and exposure of immune cells to proinflammatory cytokines that are elevated in SCD might play a role in organ dysfunction. Targeting these miRNAs in SCD might offer novel therapeutic strategy in preventing hemolysis-induced inflammation and end organ damage, especially in the heart, lung, liver, and kidney where miRNAs are abundant (222, 233–240).
SCD patients on average live longer today than 50 years ago. This is due to progress in understanding the mechanisms and risk factors of several complications of the disease, associated clinical findings and mouse models, approval of new treatment therapies, multi-disciplinary approach to care, penicillin prophylaxis and high-tech diagnostic tools (241). However, this reduction in childhood mortality gives rise to an older population of patients that develop age-related chronic organ damage, driven in part by hemolysis (94). Hemolysis-induced extensive and sometimes irreversible organ damage continues to be a major source of morbidity and mortality in SCD. Even transplanted organs are also at risk of failure in SCD patients due to hemolysis and sickling (242). Therefore, there is a need for research to understand the fundamental mechanisms involved in heme-mediated organ damage in SCD patients. Over the years, several studies in the general population as well as in SCD suggest that hemolysis causes injury to the kidney (243–245), lung (246), heart, and liver. We have summarized some of the impacts of hemolysis on different organs in Table 1.
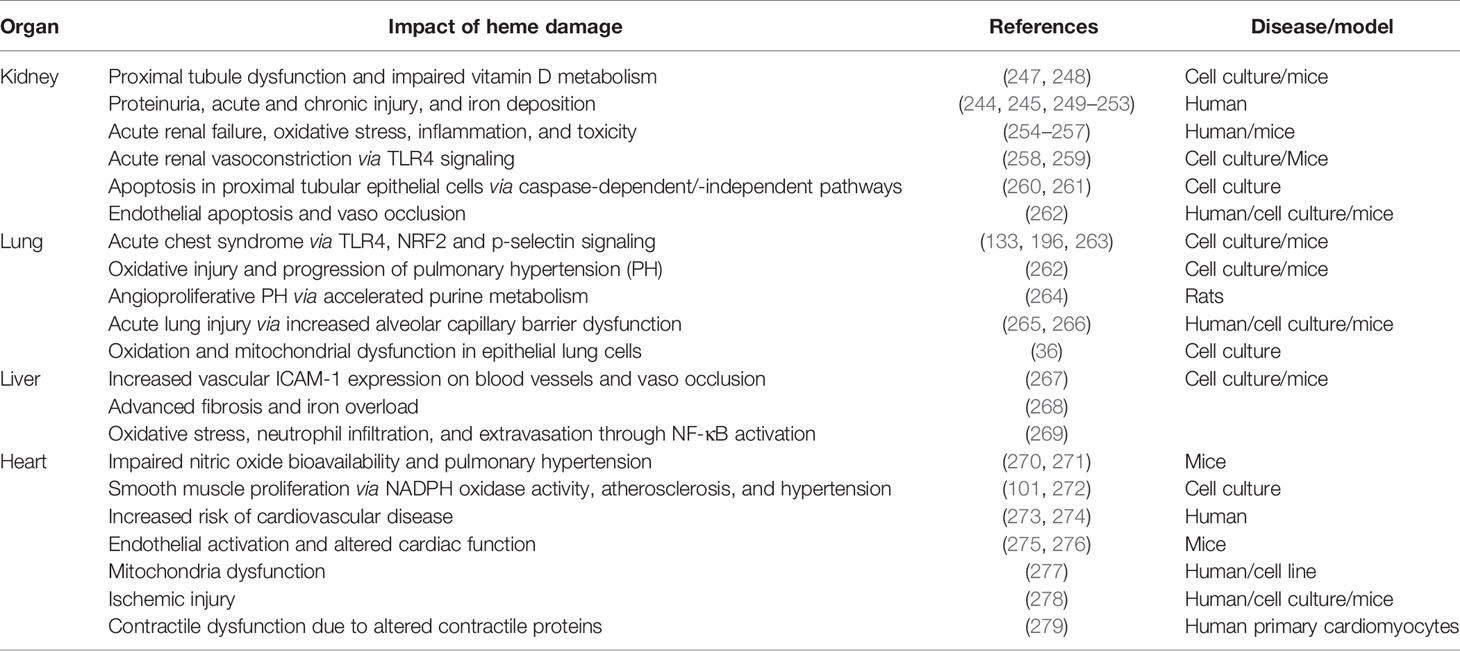
Table 1 Summary of current literature supporting a damaging role of hemolysis in different organs.
In addition to its role as a DAMP, heme promotes the expression and secretion of placenta growth factor (PlGF), a pleiotropic growth factor already known to influence multiple pathways contributing to the pathophysiology of SCD (167, 176, 280). PlGF is a member of the Vascular Endothelial Growth Factor (VEGF) family. It was originally cloned from a human placenta cDNA library in 1991 (281), hence the name, but since then it has been detected in a wide variety of tissues (282). PlGF has a partial sequence similarity to VEGF-A but the two molecules share a remarkable topological identity (283). There are four human isoforms (PlGF 1–4), which are generated by alternative splicing and are slightly different in size. PlGF-1 (131 aa) and PlGF-2 (152 aa) are the predominant isoforms in humans. On the contrary, mice carry a single isoform, PlGF-2 (140 aa).
PlGF exists as a homodimer or as a heterodimer with VEGF. PlGF is a ligand for the transmembrane and soluble form of the vascular endothelial growth factor receptor 1 (VEGFR-1, Flt-1) (284), which can also bind VEGF. Distinct from VEGF, PlGF does not bind vascular endothelial growth factor receptor 2 (VEGFR-2, Flk-1) but it can affect VEGFR-2 signaling in an indirect manner (285–287). PlGF-2 can also bind heparin and the transmembrane neuropilin receptors 1 and 2 (NRP1 and NRP2) (288, 289). In addition to its role as a receptor binding competitor of VEGF (284), PlGF can exert its own biological effect upon binding to VEGFR-1. Depending on the cell type, PlGF binding upregulates VEGF, fibroblast growth factor 2 (FGF2), platelet derived growth factor beta (PDGFB) and matrix metalloproteases (MMPs) (290, 291). Furthermore, PlGF receptor binding is shown to activate an intermolecular crosstalk regulator between VEGFR-1 and VEGFR-2, often resulting in enhancing VEGF/VEGFR-2 signaling (287). It is important to emphasize here that PlGF or VEGF binding to FLT1 results in discernible receptor phosphorylation patterns and induction of distinct signaling pathways (287, 292, 293). PlGF expression is induced by hypoxia, probably in a cell specific manner, but the exact mechanism remains elusive in the absence of hypoxia responsive elements (HRE) at the gene’s promoter region (294, 295). So far, the association of only a few transcription factors has been verified for the PlGF promoter: metal transcription factor 1 (MTF-1) (295), NF-kB (296), forkhead box D1 (FoxD) (297), erythroid Kruppel-like factor (EKLF) (167), nuclear factor erythroid 2 like 2 (NRF2) (176), glial cell missing 1 (GCM1) (298). Posttrascriptional regulation of PlGF has also been reported through the regulation of the protein kinase C (PKC), p38 mitogen activated protein kinases (p38 MAPK), c-jun N-terminal kinase (JNK) and Ras-dependent extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathways (299, 300).
Surprisingly, PlGF seems to have a redundant role under normal conditions (285) but becomes very important in disease situations, where fluctuations of its levels cause a variety of issues in multiple biological processes. Because of that reason, PlGF-based therapeutic approaches have been proposed as disease specific with minimal impact for healthy cells (301). The most well established role of PlGF is in angiogenesis and more specifically in neo-angiogenesis in pathological conditions such as ischemia or cancer (285, 302, 303). PlGF’s pleiotropic nature in evident in its angiogenic role where it exerts a paracrine or autocrine effect on endothelial cells, smooth-muscle cells, fibroblasts, bone marrow progenitor cells and monocytes, to orchestrate vessel growth and maturation (304). The description of the full spectrum of PlGF’s biological role is beyond the scope of this review but to mention a few, PlGF plays a role in inflammatory response (305, 306), promotes bone repair (307), sustains the proangiogenic M2 phenotype of tumor associated macrophages (308), affects dendritic cell differentiation and maturation (309), supports the generation of an inflammatory status driving adaptive cardiac remodeling (310). To summarize, all the evidence to date supports a role for PlGF in pathogenic angiogenesis and inflammation well outside the realm of pregnancy. Through mitogen and migratory effects on endothelial cells as well as macrophage activation and chemoattraction, PlGF emerges as a driver and marker of a plethora of seemingly diverse pathologies, especially angiogenesis and inflammation.
One of the least appreciated roles of PlGF is the one that it has in hematopoiesis (311, 312) and in hemoglobinopathies (313) (Figure 2). Plasma PlGF is elevated in SCD patients and the increase correlates with the severity of hemolysis, endothelin 1 (ET-1) expression, the occurrence of pulmonary hypertension (167, 280, 314, 315) and VOC (316, 317).
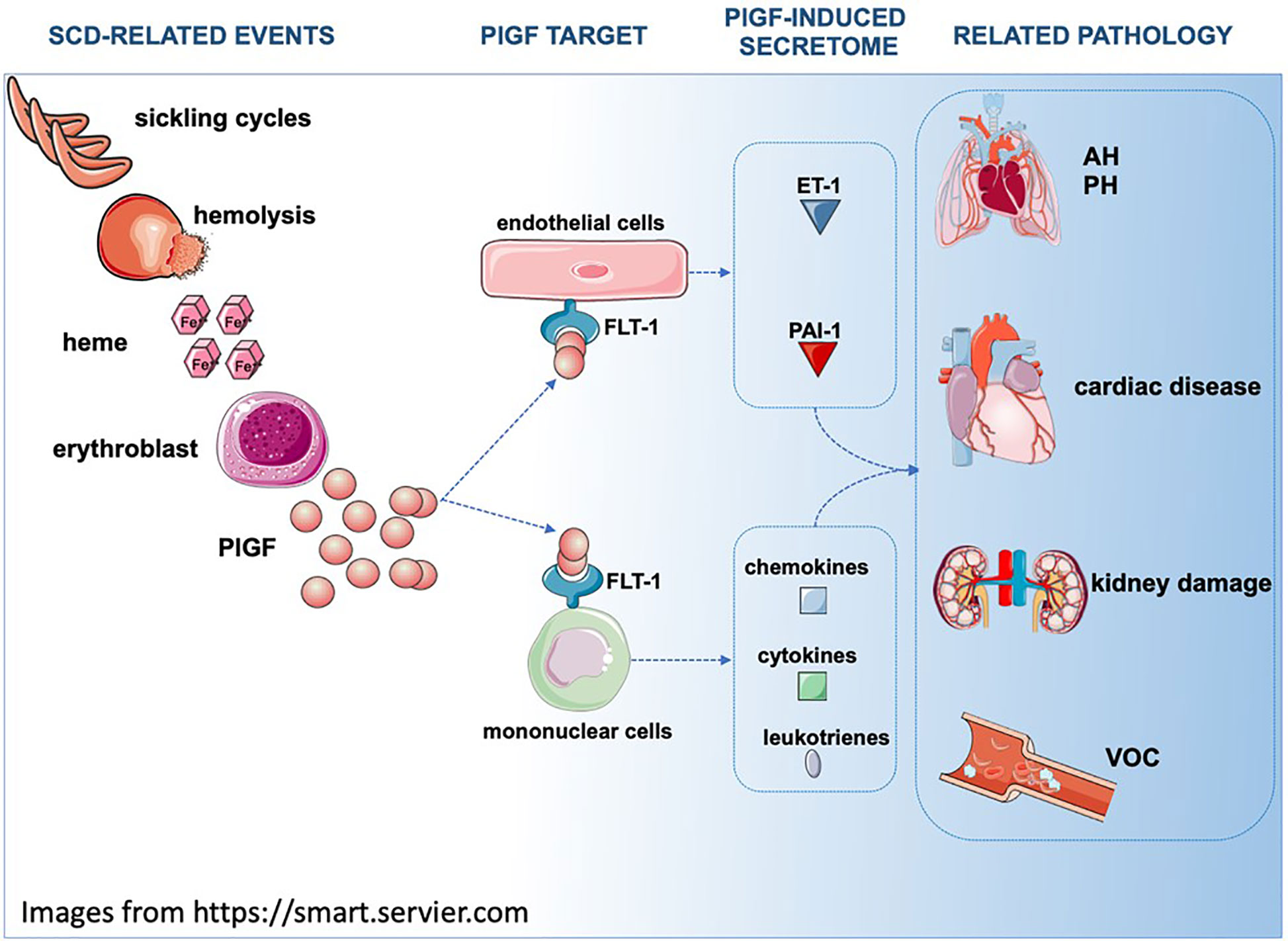
Figure 2 In SCD, repeating sickling cycles result in increased hemolysis. Hemolysis byproducts such as heme induce PlGF expression in multiple cell types (for simplicity purposes only erythroblasts are depicted). Secreted PlGF is a ligand for FLT-1 receptor and triggers the expression of ET-1, PAI-1, leukotrienes and cytochemokines, affecting the physiology of multiple organs. AH, Airway hyperreactivity; PH, Pulmonary hypertension; FLT1/VEGFR1, Fms related receptor tyrosine kinase 1; PlGF, placenta growth factor; ET-1, endothelin 1; PAI-1/Serpine1, plasminogen activator inhibitor 1.
PH is a serious complication in sickle cell patients, which is associated with high mortality (318). A variety of biological pathways and disease related pathologies contribute to the development of PH and many of them involve free heme and upregulation of PlGF. Along with PlGF, ET-1, a potent vasoconstrictor, is significantly higher in the blood of sickle patients (167, 316, 319, 320) suggesting a mechanistic link between the two factors. In support of this connection, the overexpression of PlGF in healthy mice using lentiviral gene transfer results in increased ET-1, increased right ventricle pressure and right ventricle hypertrophy as early as 8 weeks after PlGF gene transfer (280). In vitro PlGF stimulation of cultured human pulmonary microvascular endothelial cells (HPMVEC) revealed that ET-1 induction was mediated by PI-3 Kinase, NADPH-oxidase, and HIF-1a (314). Interestingly, HIF-1a stimulation of the ET-1 promoter is hypoxia independent and occurs upon the direct binding of HIF-1a on the HRE elements of the ET-1 promoter. In a similar manner, PlGF upregulates endothelin-B receptor (ET-BR) in monocytes, priming them to be over-stimulated by ET-1 and produce higher levels of chemokines MCP-1 and IL-8 (314). Both MCP-1 and IL-8 are elevated in SCD patients (321) supporting the PlGF-ET-1 synergy as another contributing factor to the development of PH in SCD.
On a post-transcriptional level, PlGF attenuates miR-648 and miR-454, which recognize and bind the 3’ UTR of ET-1 mRNA. The association of low miR-648/miR-454 with high ET-1 and PlGF levels is supported in both in vivo and in vitro studies (322, 323). Furthermore, PlGF attenuates miR-199-5p, which binds the 3’UTR of HIF-1a mRNA, creating another level of control over ET-1 expression (324). The molecular repression of miR-199-5p by PlGF is mediated by the upregulation of the activating transcription factor 3 (ATF3) which upon binding causes deacetylation and chromatin condensation at the miR-199-5p locus (325). Similar to miR-648, the association of low miR-199-5p levels with high PlGF and ET-1 levels is supported by in vivo and in vitro studies (324).
PlGF is also linked to the increase in PAI-1 levels in the plasma and lungs of sickle cell patients and humanized sickle mice respectively (326). PAI-1 is increased during steady state SCD but its expression is exacerbated during VOC. Elevation of PAI-1 levels is associated with decreased fibrinolytic capacity (327) and is believed to contribute to the SCD prothrombotic state and the development of PH (328). In vitro PlGF stimulation induced PAI-1 expression in pulmonary microvascular endothelial cells and monocytes through the activation of c-jun N-terminal kinase (JNK), hypoxia inducible factor 1a (HIF-1a) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (326). In addition, PlGF expression affects the stability of PAI-1 mRNA by downregulating microRNAs miR-454, miR-301a, and miR-30c which recognize and bind the PAI-1 3’-UTR. PlGF regulation of miR-454 and miR-301 is mediated by PPARa and HIF-1a (323). All of these microRNAs are detected in significantly lower levels in SCD patients compared to healthy controls (323, 329). In vivo experiments using PlGF null and SS sickle mice as well as adenoviral overexpression of PlGF, have confirmed that PlGF plays a significant role in PAI-1 regulation (326).
Airway hyper-reactivity is a common complication in SCD, especially in younger patients (330), and correlates with biomarkers of hemolysis (331). Patients show elevated levels of circulating leukotrienes (332) and their monocytes express higher levels of 5-lipoxygenase (5-LO) and 5-lipoxygenase activating protein (FLAP), both involved in leukotriene synthesis (333). Consistent to its proinflammatory nature, PlGF induces leukotriene production which in turn increases inflammation and airway hyper-reactivity, both key features of SCD. As in the case of PAI-1, the induction is mediated by HIF-1a and NADPH oxidase (333). Further studies have confirmed PlGF as an important regulator of leukotriene production and airway hyperactivity in SCD and asthma (332).
Activated leukocytes in sickle cell patients are considered a significant promoting factor for VOC (334). Activated mononuclear cells from SCD patients express high levels of the cytochemokines VEGF, IL-1β, monocyte chemotactic protein 1 (MCP-1), IL-8 and macrophage inflammatory protein-1 beta (MIP-1β). In vitro studies have shown that monocytes from healthy individuals can be activated by PlGF to increase the expression of proinflammatory cytokines and chemokines such as TNF-α, IL-1β, MCP-1, IL-8, and MIP-1β (316, 335). This activation is achieved by the PlGF-VEGFR-1 interaction and involves the PI-3 kinase/AKT and ERK-1/2 signaling pathways (335). Because VOC in SCD is promoted by inflammation and leukocyte adhesion stimulated by cytokines (197, 336, 337), antibody neutralization of PlGF was tried successfully for reduction of inflammation and vaso-occlusive complications in murine SCD models (317). Regulation of PlGF levels could also be achieved by manipulating factors that control its transcriptional or translational expression. Per instance, pharmacological upregulation of miR-214 which is known to bind PlGF 3’-UTR, could be engaged to reduce PlGF levels (338).
PlGF is significantly upregulated in the serum of patients with chronic kidney disease and decreased renal function, supporting a potential mechanistic link between PlGF and kidney function (339, 340). Sickle cell nephropathy (SCN) is an complex phenotype which encompasses almost every physiological process in the kidney, leading to complications that may range from common and relatively mild to rare and life-limiting (243). In SCD patients markers of renal dysfunction are associated with elevated ET-1 serum levels (341) and studies in sickle cell mice have shown that ET-1 can cause renal injury, likely mediated by ROS (342). Although it has not been shown experimentally, sickle cell-related elevated PlGF levels could possibly contribute to higher ET-1 levels (167, 314) driving renal dysfunction. However, administration of exogenous heme in control and sickle cell mice has been shown to result in the upregulation of PlGF in the murine kidneys in agreement with heme uptake from renal cells and HMOX-1induction (343). In addition to ET-1, PAI-1 has also been shown to play a role in nephropathies (344) but its role in SCD or its potential regulation by PlGF remains unexplored.
Cardiac complications are common in SCD patients and along with the pulmonary complications raise their morbidity and mortality risk (94, 345). There has been accumulating evidence that PlGF dysregulation is present in multiple heart conditions although it is often unclear if it is only a disease biomarker or it actively promotes disease pathogenesis. In patients with chronic kidney disease, PlGF levels are associated with higher incidence of cardiovascular events and mortality (340). In the same disease, PlGF is an independent risk predictor for left ventricular diastolic dysfunction (346). In human atherosclerotic plaques, the expression of PlGF is associated with plaque destabilization and disease manifestation (347). The pro-atherosclerotic role of PlGF is corroborated in rabbits where PlGF adenoviral expression promotes atherogenic intimal thickening and macrophage accumulation in the carotid artery (348). PlGF is also elevated in the plasma of patients with acute coronary syndromes where it can be used as a risk predicting biomarker (349). PlGF promotes cardiac hypertrophy via endothelial cell release of NO which induces cardiomyocyte growth (350) and by inducing the secretion of paracrine factors (IL-6, IL-1b, Cxcl1) from endothelia and fibroblasts that promote cardiac adaptation and hypertrophy (351–353). In the case of ischemic cardiomyopathy, PlGF has been reported both as promoting the disease (354) and as a potential therapeutic (355). The apparent controversy could be due to differences between a local and acute administration of an angiogenic factor (355) compared to a more systemic and chronic upregulation (354). Our research has shown that PlGF is elevated in the hearts of sickle mice and it is further induced after administering exogenous heme (343). Surprisingly, the level of PlGF induction is comparable to that of the liver which is considered the major heme detoxifying organ (343). An interesting finding of this study is that mouse hearts have high levels of HMOX-1, which are further increased by heme induction, and that they show no heme accumulation unless NRF2 is depleted. These data suggest that cardiac tissue has the ability to detoxify heme via the NRF2 antioxidant response pathway.
IL-6 is a ubiquitous and pleiotropic proinflammatory cytokine produced by many cells including macrophages (356, 357), neutrophils (358, 359), endothelial and smooth muscle cells (360, 361), cardiomyocytes (362) and fibroblasts (363), when stimulated by ligands for toll-like receptors or other pattern recognition receptors. IL-6 is a glycoprotein composed of 184 amino acids and of 26 kDa in molecular weight (364). Currently, there are ten cytokines belonging to the IL-6 family; IL-6, IL-11, ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), oncostatin M (OSM), cardiotropin-1 (CT-1), cardiotrophin-like cytokine (CLC), IL-27, neuropoietin (NP), and IL-31 (365). IL-6 regulates many biological functions including hematopoiesis (366), oncogenesis (367) and differentiation of B cells (368), induction of acute phase proteins and immune regulation (369). Additionally, IL-6 plays a vital role in chronic inflammatory processes in various cells and disease conditions (364). IL-6 signaling is through two pathways; classic/cis-mediated signaling via membrane-bound IL-6 receptor (mIL-6R) or trans-mediated signaling via the soluble form of IL-6R (sIL-6R) (364, 369). Classic/cis-signaling occurs in cells that express IL-6R such as hepatocytes, neutrophils and monocytes (365, 369). Conversely, trans-mediated signaling occurs after secretion of sIL-6R by RNA alternative splicing, ectodomain shedding or proteolytic cleavage of mIL-6R (370), which in turn stimulate cells (365, 369). Once IL-6 binds to mIL-6R or sIL-6R, the cytokine forms a complex with the ubiquitously expressed membrane protein gp130, a shared signal-transducing receptor of all IL-6 type cytokines (370). Dimerization of the receptor complex activates Janus kinases (JAKs) resulting in phosphorylation of the tyrosine residues in the cytoplasmic domain of gp130 (364, 371). Activation of JAKs triggers the extracellular-signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) signaling pathways (370, 371). However, IL-6 role in pathophysiology of chronic inflammation and diseases is driven via IL-6 trans-signaling because classic/cis-signaling via the mIL-6R is limited to few cells that express IL-6R (372). Blockade of IL-6 trans-signaling is effective in attenuating proinflammatory activities of IL-6 in several disease conditions (365).
Several studies in human and rodents found hemolysis and elevated IL-6 occurring concurrently. Hemolysis and elevated IL-6 are associated with disease severity in malaria (373, 374), sepsis (375) and pre-eclampsia (376), with cardiac dysfunction as an additional comorbidity in these diseases. Besides, elevated cardiac IL-6 is also associated with cardiac hypertrophy and fibrosis in the general population (362, 377) and in rodents (378, 379). In malaria, elevated IL-6 is found in patients with severe Plasmodium falciparum/vivax malaria and associated with development of cardiac complications (373, 374). Sepsis patients with elevated IL-6 are at a higher risk of developing cardiac dysfunction which may be due to direct negative inotropic effect of IL-6 mediated via altered production of myocardial nitric oxide (375), altered calcium homeostasis (380, 381) and impaired β-adrenergic signaling (382–384). Elevated IL-6 in pre-eclampsia patients result in reduced anti-inflammatory protection in the maternal vascular system (385) and stimulation of vasoactive substances including angiotensin II type 1 receptor and endothelin-1 (386). Although, elevated plasma IL-6 have been reported in human and mice with SCD (168, 387, 388), and hemolysis is a major comorbidity of SCD (94), however, there has been no direct link between these two processes. Conversely, left ventricular hypertrophy (LVH) is found in over 60% of children and 37% in adults with SCD (389, 390), with cardiopulmonary complications accounting for about 26% of deaths in adults with SCD (391). In this current issue and for the first time, our group investigated the expression of plasma and cardiac IL-6 and its inducibility by heme in Townes sickle cell (SS) mouse model (392). We observed significantly elevated cardiac IL-6 and direct heme induction of circulating and cardiac IL-6 transcripts and protein in SS mice compared to controls. We showed that this heme-induced IL-6 is NRF2-independent in the heart. Our results of heme-induced IL-6 is in agreement with elevated levels of IL-6 reported in cardiac cells treated with Hpx and in heart isolated from Hpx deficient mice (393). Because our data showed upregulation of cardiac hypertrophy genes following heme treatment in SS mice, there is a possibility that heme is inducing IL-6 in the heart and prolonged activation and exposure to IL-6 could contribute to LVH in SCD patients. We are currently investigating potential mechanism(s) and specific cell-types secreting IL-6 in the heart of SS mice. There are several pathways through which heme may induce IL-6 expression. It is possible that parallel heme-induced pathways are activating IL-6 indirectly and with continuous hemolysis forming a feedback loop. With elevated cardiac PlGF at baseline in SCD mice and further inducibility by heme (343), cardiac hypertrophy may develop via IL-6 signaling (350). Therefore, it can be envisaged that prolonged hemolysis induced PlGF and IL-6 in SCD feeds the vicious cycle of inflammation via an autocrine feedback system resulting in reactivation of genetic cardiac hypertrophy program.
The role of hemolysis and its attendant oxidant stress and inflammatory activation in SCD has been supported by the success of therapies that normalize these pathways. Hydroxyurea has pleiotropic effects that reduce hemolysis and offset its pathobiological consequences. The approval of hydroxyurea by the FDA in 1998 provided a watershed moment in the history of SCD (394, 395). Hydroxyurea treatment yielded an improved quality of life for SCD patients attributable to induction of fetal hemoglobin, slowing of chronic damage to several organs, including the brain (394–400). More than twenty years later, three new drugs; L-glutamine (Endari; reduction of pain-related hospital visit and length of stay) and crizanlizumab-tmca (Adakveo; reduction of frequency of VOC) and voxelotor (Oxbryta; inhibition of deoxygenated sickle hemoglobin polymerization), have been approved by the FDA for treatment of SCD (401). L-glutamine is thought to reverse the redox imbalance imposed by hemolysis and other sources of oxidative stress. Crizanlizumab blocks the inflammation-activated P-selection adhesive pathway. Voxelotor inhibits polymerization of sickle hemoglobin, with the most apparent effect of reduced hemolysis. Curative intent therapies have also shown evidence of reduced hemolysis. Although permanent cure afforded to patients through bone marrow transplant and gene therapy would be ideal, it would be quite expensive and the majority of patients with SCD live in areas lacking both economic and human resources needed to make these curative therapies broadly accessible (402). Importantly, the global majority of SCD patients live in resource-poor countries, with minimal access to these newer therapies and limited capacity for hematological monitoring requirements and other diagnostic equipment (1, 403). High childhood mortality rate ranging from 50–90% still prevail in these areas and acceptance of hydroxyurea as therapy is very low compared to developed countries (403–405).
Encouragingly, recent studies show the efficacy, safety and feasibility of using hydroxyurea treatment in children and adults with sickle cell anemia living in sub-Saharan Africa (406–408).
Clinical trials are underway to assess the potential of hemopexin intravenous infusion in the treatment of SCD (Clinicaltrials.gov identifier NCT04285827). In the Townes SCD mouse model, infusion of hemopexin reduced microvascular occlusion induced by hemoglobin infusion, hypoxia-reoxygenation, or lipopolysaccharide (83). Hemopexin mitigated induction of ICAM-1 and VCAM-1 via inhibition of NF-κB activation (83). In another study, treatment with Hpx attenuated free heme activation of complement pathways and kidney injury caused by complement deposition and inflammation in mice during hemolysis (157). Hemopexin also significantly decreased plasma heme concentration, pulmonary neutrophil extracellular trap (NET) formation, plasma DNA, neutrophil activation and NET-associated hypothermia in SCD mice (171).
Hemolysis is a feature of many diseases, and in most cases occurring with acute and chronic inflammation that contributes to organ injury. Products of hemolysis activate several inflammatory pathways in many cell types, including cells in the innate immune system. Hemolysis appears to serve as a priming stimulus that combines with TLR4 signaling to a cascade of production of inflammatory cytokines which activate downstream pathophysiology. Therapeutic intervention targeting the upstream effects of hemolysis has potential to mitigate downstream innate immune system response and inflammation in treating patients with intravascular hemolytic disease.
All authors drafted the review. The first two authors contributed equally. GK approved the final version of this review. All authors contributed to the article and approved the submitted version.
GK received support from NIH grants HL133864, MD009162 and from the Institute for Transfusion Medicine Hemostasis and Vascular Biology Research Institute at the University of Pittsburgh School of Medicine. OTG is supported by the American Society of Hematology Scholar Award.
GK is an employee of CSL Behring, LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PloS Med (2013) 10(7):e1001484. doi: 10.1371/journal.pmed.1001484
2. Phillips J, Henderson AC. Hemolytic Anemia: Evaluation and Differential Diagnosis. Am Fam Physician (2018) 98(6):354–61.
3. Mense SM, Zhang L. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res (2006) 16(8):681–92. doi: 10.1038/sj.cr.7310086
4. Shimizu T, Lengalova A, Martinek V, Martinkova M. Heme: emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem Soc Rev (2019) 48(24):5624–57. doi: 10.1039/C9CS00268E
5. Pradhan P, Vijayan V, Gueler F, Immenschuh S. Interplay of Heme with Macrophages in Homeostasis and Inflammation. Int J Mol Sci (2020) 21(3):1–14. doi: 10.3390/ijms21030740
6. Hebbel R, Morgan W, Eaton J, Hedlund B. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Natl Acad Sci USA (1988) 85(1):237–41. doi: 10.1073/pnas.85.1.237
7. Hebbel R. Beyond hemoglobin polymerization: The red blood cell membrane and sickle disease pathophysiology. Blood (1991) 77:214–37. doi: 10.1182/blood.V77.2.214.214
8. Hebbel R, Eaton J, Balasingam M, Steinberg M. Spontaneous oxygen radical generation by sickle erythrocytes. J Clin Investigation (1982) 70(6):1253–9. doi: 10.1172/JCI110724
9. Fibach E, Rachmilewitz E. The role of oxidative stress in hemolytic anemia. Curr Mol Med (2008) 8(7):609–19. doi: 10.2174/156652408786241384
10. Advani R, Rubin E, Mohandas N, Schrier SL. Oxidative red blood cell membrane injury in the pathophysiology of severe mouse beta-thalassemia. Blood (1992) 79(4):1064–7. doi: 10.1182/blood.V79.4.1064.1064
11. Amer J, Zelig O, Fibach E. Oxidative status of red blood cells, neutrophils, and platelets in paroxysmal nocturnal hemoglobinuria. Exp Hematol (2008) 36(4):369–77. doi: 10.1016/j.exphem.2007.12.003
12. Fibach E, Dana M. Oxidative stress in paroxysmal nocturnal hemoglobinuria and other conditions of complement-mediated hemolysis. Free Radical Biol Med (2015) 88(Pt A):63–9. doi: 10.1016/j.freeradbiomed.2015.04.027
13. Caprari P, Bozzi A, Ferroni L, Strom R, Salvati AM. Oxidative erythrocyte membrane damage in hereditary spherocytosis. Biochem Int (1992) 26(2):265–74.
14. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet (2008) 371(9606):64–74. doi: 10.1016/S0140-6736(08)60073-2
15. Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J (1995) 14(21):5209–15. doi: 10.1002/j.1460-2075.1995.tb00205.x
16. Schrier SL, Mohandas N. Globin-chain specificity of oxidation-induced changes in red blood cell membrane properties. Blood (1992) 79(6):1586–92. doi: 10.1182/blood.V79.6.1586.1586
17. Mohanty J, Nagababu E, Rifkind J. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front Physiol (2014) 5(84):1–6. doi: 10.3389/fphys.2014.00084
18. Caprari P, Massimi S, Diana L, Sorrentino F, Maffei L, Materazzi S, et al. Hemorheological Alterations and Oxidative Damage in Sickle Cell Anemia. Front Mol Biosciences (2019) 6:142. doi: 10.3389/fmolb.2019.00142
19. Nagababu E, Fabry M, Nagel R, Rifkind J. Heme degradation and oxidative stress in murine models for hemoglobinopathies: Thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells Molecules Diseases (2008) 41(1):60–6. doi: 10.1016/j.bcmd.2007.12.003
20. Johnson R, Goyette GJ, Ravindranath Y, Ho Y. Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radical Biol Med (2005) 39(11):1407–17. doi: 10.1016/j.freeradbiomed.2005.07.002
21. Rank B, Carlsson J, Hebbel R. Abnormal redox status of membrane-protein thiols in sickle erythrocytes. J Clin Investigation (1985) 75:1531–7. doi: 10.1172/JCI111857
22. Wood K, Hsu L, Gladwin M. Sickle cell disease vasculopathy: a state of nitric oxide resistance. Free Radical Biol Med (2008) 44(8):1506–28. doi: 10.1016/j.freeradbiomed.2008.01.008
23. Svistunenko D, Patel R, Voloshchenko S, Wilson M. The globin-based free radical of ferryl hemoglobin is detected in normal human blood. J Biol Chem (1997) 272(11):7114–21. doi: 10.1074/jbc.272.11.7114
24. Giulivi C, Davies KJ. A novel antioxidant role for hemoglobin. The comproportionation of ferrylhemoglobin with oxyhemoglobin. J Biol Chem (1990) 265:19453–60.
25. Jana S, Strader MB, Meng F, Hicks W, Kassa T, Tarandovskiy I, et al. Hemoglobin oxidation-dependent reactions promote interactions with band 3 and oxidative changes in sickle cell-derived microparticles. JCI Insight (2018) 3(21):1–20. doi: 10.1172/jci.insight.120451
26. Farah M, Sirotkin V, Haarer B, Kakhniashvili D, Amberg D. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton (2011) 68:340–54. doi: 10.1002/cm.20516
27. Cyrklaff M, Sanchez C, Kilian N, Bisseye C, Simpore J, Frischknecht F, et al. Hemoglobins S and C interfere with actin remodeling in Plasmodium falciparum-infected erythrocytes. Science (2011) 334(6060):1283–6. doi: 10.1126/science.1213775
28. Turrens J. Mitochondrial formation of reactive oxygen species. J Physiol (2003) 552(Pt 2):335–44. doi: 10.1113/jphysiol.2003.049478
29. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA (2007) 104(49):19500–5. doi: 10.1073/pnas.0708818104
30. Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy (2012) 8(9):1325–32. doi: 10.4161/auto.20764
31. Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood (2008) 112(4):1493–502. doi: 10.1182/blood-2008-02-137398
32. Gnanapragasam MN, McGrath KE, Catherman S, Xue L, Palis J, Bieker JJ. EKLF/KLF1-regulated cell cycle exit is essential for erythroblast enucleation. Blood (2016) 128(12):1631–41. doi: 10.1182/blood-2016-03-706671
33. Jagadeeswaran R, Vazquez BA, Thiruppathi M, Ganesh BB, Ibanez V, Cui S, et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp Hematol (2017) 50:46–52. doi: 10.1016/j.exphem.2017.02.003
34. Jagadeeswaran R, Rivers A. Evolving treatment paradigms in sickle cell disease. Hematol Am Soc Hematol Educ Program (2017) 2017(1):440–6. doi: 10.1182/asheducation-2017.1.440
35. Higdon AN, Benavides GA, Chacko BK, Ouyang X, Johnson MS, Landar A, et al. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am J Physiol Heart Circ Physiol (2012) 302(7):H1394–409. doi: 10.1152/ajpheart.00584.2011
36. Kassa T, Jana S, Strader MB, Meng F, Jia Y, Wilson MT, et al. Sickle Cell Hemoglobin in the Ferryl State Promotes betaCys-93 Oxidation and Mitochondrial Dysfunction in Epithelial Lung Cells (E10). J Biol Chem (2015) 290(46):27939–58. doi: 10.1074/jbc.M115.651257
37. Chintagari NR, Jana S, Alayash AI. Oxidized Ferric and Ferryl Forms of Hemoglobin Trigger Mitochondrial Dysfunction and Injury in Alveolar Type I Cells. Am J Respir Cell Mol Biol (2016) 55(2):288–98. doi: 10.1165/rcmb.2015-0197OC
38. Jana S, Meng F, Hirsch RE, Friedman JM, Alayash AI. Oxidized Mutant Human Hemoglobins S and E Induce Oxidative Stress and Bioenergetic Dysfunction in Human Pulmonary Endothelial Cells. Front Physiol (2017) 8:1082. doi: 10.3389/fphys.2017.01082
39. Cardenes N, Corey C, Geary L, Jain S, Zharikov S, Barge S, et al. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood (2014) 123(18):2864–72. doi: 10.1182/blood-2013-09-529420
40. Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood (2007) 110(6):2166–72. doi: 10.1182/blood-2006-12-061697
41. Westerman M, Pizzey A, Hirschman J, Cerino M, Weil-Weiner Y, Ramotar P, et al. Microvesicles in haemoglobinopathies offer insights into mechanisms of hypercoagulability, haemolysis and the effects of therapy. Br J Haematol (2008) 142(1):126–35. doi: 10.1111/j.1365-2141.2008.07155.x
42. Allan D, Limbrick AR, Thomas P, Westerman MP. Release of spectrin-free spicules on reoxygenation of sickled erythrocytes. Natur (1982) 295(5850):612–3. doi: 10.1038/295612a0
43. Lane PA, O’Connell JL, Marlar RA. Erythrocyte membrane vesicles and irreversibly sickled cells bind protein S. Am J Hematol (1994) 47(4):295–300. doi: 10.1002/ajh.2830470409
44. Mahfoudhi E, Lecluse Y, Driss F, Abbes S, Flaujac C, Garcon L. Red cells exchanges in sickle cells disease lead to a selective reduction of erythrocytes-derived blood microparticles. Br J Haematol (2012) 156(4):545–7. doi: 10.1111/j.1365-2141.2011.08897.x
45. van Tits LJ, van Heerde WL, Landburg PP, Boderie MJ, Muskiet FA, Jacobs N, et al. Plasma annexin A5 and microparticle phosphatidylserine levels are elevated in sickle cell disease and increase further during painful crisis. Biochem Biophys Res Commun (2009) 390(1):161–4. doi: 10.1016/j.bbrc.2009.09.102
46. van Beers EJ, Schaap MC, Berckmans RJ, Nieuwland R, Sturk A, van Doormaal FF, et al. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica (2009) 94(11):1513–9. doi: 10.3324/haematol.2009.008938
47. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. New Engl J Med (1994) 330(23):1639–44. doi: 10.1056/NEJM199406093302303
48. Camus SM, Gausseres B, Bonnin P, Loufrani L, Grimaud L, Charue D, et al. Erythrocyte microparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease. Blood (2012) 120(25):5050–8. doi: 10.1182/blood-2012-02-413138
49. Donadee C, Raat NJ, Kanias T, Tejero J, Lee JS, Kelley EE, et al. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation (2011) 124(4):465–76. doi: 10.1161/CIRCULATIONAHA.110.008698
50. Liu C, Zhao W, Christ GJ, Gladwin MT, Kim-Shapiro DB. Nitric oxide scavenging by red cell microparticles. Free Radical Biol Med (2013) 65:1164–73. doi: 10.1016/j.freeradbiomed.2013.09.002
51. Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, Lionnet F, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood (2015) 125(24):3805–14. doi: 10.1182/blood-2014-07-589283
52. Nagababu E, Rifkind J. Formation of fluorescent heme degradation products during the oxidation of hemoglobin by hydrogen peroxide. Biochem Biophys Res Commun (1998) 247(3):592–6. doi: 10.1006/bbrc.1998.8846
53. Bedard K, Krause K. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev (2007) 87(1):245–313. doi: 10.1152/physrev.00044.2005
54. Lanaro C, Franco-Penteado C, Albuqueque D, Saad S, Conran N, Costa F. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J Leukocyte Biol (2009) 85(2):235–42. doi: 10.1189/jlb.0708445
55. George A, Pushkaran S, Konstantinidis DG, Koochaki S, Malik P, Mohandas N, et al. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood (2013) 121(11):2099–107. doi: 10.1182/blood-2012-07-441188
56. Lew, Bookchin R. Ion transport pathology in the mechanism of sickle cell dehydration. Physiol Rev (2005) 85(1):179–200. doi: 10.1152/physrev.00052.2003
57. Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, Huber SM, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2005) 15(5):195–202. doi: 10.1159/000086406
58. Gbotosho OT, Cytlak UM, Hannemann A, Rees DC, Tewari S, Gibson JS. Inhibitors of second messenger pathways and Ca(2+)-induced exposure of phosphatidylserine in red blood cells of patients with sickle cell disease. Pflugers Archiv Eur J Physiol (2014) 466(7):1477–85. doi: 10.1007/s00424-013-1343-8
59. Detterich JA, Liu H, Suriany S, Kato RM, Chalacheva P, Tedla B, et al. Erythrocyte and plasma oxidative stress appears to be compensated in patients with sickle cell disease during a period of relative health, despite the presence of known oxidative agents. Free Radical Biol Med (2019) 141:408–15. doi: 10.1016/j.freeradbiomed.2019.07.004
60. Tappel AL. The mechanism of the oxidation of unsaturated fatty acids catalyzed by hematin compounds. Arch Biochem Biophys (1953) 44(2):378–95. doi: 10.1016/0003-9861(53)90056-3
61. Vincent SH, Grady RW, Shaklai N, Snider JM, Muller-Eberhard U. The influence of heme-binding proteins in heme-catalyzed oxidations. Arch Biochem Biophys (1988) 265(2):539–50. doi: 10.1016/0003-9861(88)90159-2
63. Gao JL, Lu Y, Browne G, Yap BC, Trewhella J, Hunter N, et al. The role of heme binding by DNA-protective protein from starved cells (Dps) in the Tolerance of Porphyromonas gingivalis to heme toxicity. J Biol Chem (2012) 287(50):42243–58. doi: 10.1074/jbc.M112.392787
64. Vasconcellos LR, Dutra FF, Siqueira MS, Paula-Neto HA, Dahan J, Kiarely E, et al. Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proc Natl Acad Sci USA (2016) 113(47):E7474–E82. doi: 10.1073/pnas.1608928113
65. Aft RL, Mueller GC. Hemin-mediated oxidative degradation of proteins. J Biol Chem (1984) 259(1):301–5.
66. Biswal S, Rizwan H, Pal S, Sabnam S, Parida P, Pal A. Oxidative stress, antioxidant capacity, biomolecule damage, and inflammation symptoms of sickle cell disease in children. Hematol (Amsterdam Netherlands) (2019) 24(1):1–9. doi: 10.1080/10245332.2018.1498441
67. Alsultan AI, Seif MA, Amin TT, Naboli M, Alsuliman AM. Relationship between oxidative stress, ferritin and insulin resistance in sickle cell disease. Eur Rev Med Pharmacol Sci (2010) 14(6):527–38.
68. Ama Moor VJ, Pieme CA, Chetcha Chemegne B, Manonji H, Njinkio Nono BL, Tchoula Mamiafo C, et al. Oxidative profile of sickle cell patients in a Cameroonian urban hospital. BMC Clin Pathol (2016) 16:15. doi: 10.1186/s12907-016-0037-5
69. Natta CL, Chen LC, Chow CK. Selenium and glutathione peroxidase levels in sickle cell anemia. Acta Haematol (1990) 83(3):130–2. doi: 10.1159/000205188
70. Renoux C, Joly P, Faes C, Mury P, Eglenen B, Turkay M, et al. Association between Oxidative Stress, Genetic Factors, and Clinical Severity in Children with Sickle Cell Anemia. J Pediatrics (2018) 195:228–35. doi: 10.1016/j.jpeds.2017.12.021
71. Mockesch B, Connes P, Charlot K, Skinner S, Hardy-Dessources MD, Romana M, et al. Association between oxidative stress and vascular reactivity in children with sickle cell anaemia and sickle haemoglobin C disease. Br J Haematol (2017) 178(3):468–75. doi: 10.1111/bjh.14693
72. Antwi-Boasiako C, Dankwah GB, Aryee R, Hayfron-Benjamin C, Donkor ES, Campbell AD. Oxidative Profile of Patients with Sickle Cell Disease. Med Sci (Basel Switzerland) (2019) 7(2):1–8. doi: 10.3390/medsci7020017
73. Schacter L, Warth JA, Gordon EM, Prasad A, Klein BL. Altered amount and activity of superoxide dismutase in sickle cell anemia. FASEB J (1988) 2(3):237–43. doi: 10.1096/fasebj.2.3.3350236
74. Farias ICC, Mendonca-Belmont TF, da Silva AS, do OK, Ferreira F, Medeiros FS, et al. Association of the SOD2 Polymorphism (Val16Ala) and SOD Activity with Vaso-occlusive Crisis and Acute Splenic Sequestration in Children with Sickle Cell Anemia. Mediterranean J Hematol Infect Diseases (2018) 10(1):e2018012. doi: 10.4084/mjhid.2018.012
75. Armenis I, Kalotychou V, Tzanetea R, Moyssakis I, Anastasopoulou D, Pantos C, et al. Reduced peripheral blood superoxide dismutase 2 expression in sickle cell disease. Ann Hematol (2019) 98(7):1561–72. doi: 10.1007/s00277-019-03709-8
76. Smith OS, Ajose OA, Adegoke SA, Adegoke OA, Adedeji TA, Oderinu KA. Plasma level of antioxidants is related to frequency of vaso-occlusive crises in children with sickle cell anaemia in steady state in Nigeria. Pediatr Hematol Oncol J (2019) 4(1):17–22. doi: 10.1016/j.phoj.2019.03.003
77. Delesderrier E, Cople-Rodrigues CS, Omena J, Kneip Fleury M, Barbosa Brito F, Costa Bacelo A, et al. Selenium Status and Hemolysis in Sickle Cell Disease Patients. Nutrients (2019) 11(9):1–11. doi: 10.3390/nu11092211
78. Manfredini V, Lazzaretti LL, Griebeler IH, Santin AP, Brandao VD, Wagner S, et al. Blood antioxidant parameters in sickle cell anemia patients in steady state. J Natl Med Assoc (2008) 100(8):897–902. doi: 10.1016/S0027-9684(15)31402-4
79. Morris C, Suh J, Hagar W, Larkin S, Bland D, Steinberg M, et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood (2008) 111(1):402–10. doi: 10.1182/blood-2007-04-081703
80. Hamdy MM, Mosallam DS, Jamal AM, Rabie WA. Selenium and Vitamin E as antioxidants in chronic hemolytic anemia: Are they deficient? A case-control study in a group of Egyptian children. J Adv Res (2015) 6(6):1071–7. doi: 10.1016/j.jare.2015.01.002
81. Arruda MM, Mecabo G, Rodrigues CA, Matsuda SS, Rabelo IB, Figueiredo MS, et al. and E supplementation increases markers of haemolysis in sickle cell anaemia patients: a randomized, double-blind, placebo-controlled trial. Br J Haematol (2013) 160(5):688–700. doi: 10.1111/bjh.12185
82. Muhammad A, Waziri AD, Forcados GE, Sanusi B, Sani H, Malami I, et al. Sickling-preventive effects of rutin is associated with modulation of deoxygenated haemoglobin, 2,3-bisphosphoglycerate mutase, redox status and alteration of functional chemistry in sickle erythrocytes. Heliyon (2019) 5(6):e01905. doi: 10.1016/j.heliyon.2019.e01905
83. Belcher JD, Chen C, Nguyen J, Abdulla F, Zhang P, Nguyen H, et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PloS One (2018) 13(4):e0196455. doi: 10.1371/journal.pone.0196455
84. Gellen B, Messonnier LA, Galacteros F, Audureau E, Merlet AN, Rupp T, et al. Moderate-intensity endurance-exercise training in patients with sickle-cell disease without severe chronic complications (EXDRE): an open-label randomised controlled trial. Lancet Haematol (2018) 5(11):e554–e62. doi: 10.1016/S2352-3026(18)30163-7
85. Chatel B, Messonnier LA, Barge Q, Vilmen C, Noirez P, Bernard M, et al. Endurance training reduces exercise-induced acidosis and improves muscle function in a mouse model of sickle cell disease. Mol Genet Metab (2018) 123(3):400–10. doi: 10.1016/j.ymgme.2017.11.010
86. Charrin E, Aufradet E, Douillard A, Romdhani A, Souza GD, Bessaad A, et al. Oxidative stress is decreased in physically active sickle cell SAD mice. Br J Haematol (2015) 168(5):747–56. doi: 10.1111/bjh.13207
87. Gouraud E, Charrin E, Dube JJ, Ofori-Acquah SF, Martin C, Skinner S, et al. Effects of Individualized Treadmill Endurance Training on Oxidative Stress in Skeletal Muscles of Transgenic Sickle Mice. Oxid Med Cell Longevity (2019) 2019:3765643. doi: 10.1155/2019/3765643
88. Grau M, Nader E, Jerke M, Schenk A, Renoux C, Dietz T, et al. Impact of A Six Week Training Program on Ventilatory Efficiency, Red Blood Cell Rheological Parameters and Red Blood Cell Nitric Oxide Signaling in Young Sickle Cell Anemia Patients: A Pilot Study. J Clin Med (2019) 8(12):1–16. doi: 10.3390/jcm8122155
89. Kato G, McGowan V, Machado R, Little J, Taylor J, Morris C, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood (2006) 107(6):2279–85. doi: 10.1182/blood-2005-06-2373
90. Morris C, Kato G, Poljakovic M, Wang X, Blackwelder W, Sachdev V, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. J Am Med Assoc (2005) 294(1):81–90. doi: 10.1001/jama.294.1.81
91. Reiter C, Wang X, Tanus-Santos J, Hogg N, Cannon RR, Schechter A, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med (2002) 8(12):1383–9. doi: 10.1038/nm1202-799
92. Palmer R, Ferrige A, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature (1987) 327:524 – 6. doi: 10.1038/327524a0
93. Arnold W, Mittal C, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA (1977) 74:3203–7. doi: 10.1073/pnas.74.8.3203
94. Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest (2017) 127(3):750–60. doi: 10.1172/JCI89741
95. Potoka KP, Gladwin MT. Vasculopathy and pulmonary hypertension in sickle cell disease. Am J Physiol Lung Cell Mol Physiol (2015) 308(4):L314–24. doi: 10.1152/ajplung.00252.2014
96. Gordeuk VR, Castro OL, Machado RF. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood (2016) 127(7):820–8. doi: 10.1182/blood-2015-08-618561
97. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature (2001) 409(6817):198–201. doi: 10.1038/35051594
98. Schaer DJ, Schaer CA, Buehler PW, Boykins RA, Schoedon G, Alayash AI, et al. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood (2006) 107(1):373–80. doi: 10.1182/blood-2005-03-1014
99. Nielsen MJ, Andersen CB, Moestrup SK. CD163 binding to haptoglobin-hemoglobin complexes involves a dual-point electrostatic receptor-ligand pairing. J Biol Chem (2013) 288(26):18834–41. doi: 10.1074/jbc.M113.471060
100. Smith A, McCulloh RJ. Hemopexin and haptoglobin: allies against heme toxicity from hemoglobin not contenders. Front Physiol (2015) 6:187. doi: 10.3389/fphys.2015.00187
101. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA (2005) 293(13):1653–62. doi: 10.1001/jama.293.13.1653
102. Immenschuh S, Vijayan V, Janciauskiene S, Gueler F. Heme as a Target for Therapeutic Interventions. Front Pharmacol (2017) 8:146. doi: 10.3389/fphar.2017.00146
103. Oh JY, Hamm J, Xu X, Genschmer K, Zhong M, Lebensburger J, et al. Absorbance and redox based approaches for measuring free heme and free hemoglobin in biological matrices. Redox Biol (2016) 9:167–77. doi: 10.1016/j.redox.2016.08.003
104. Thomas AM, Gerogianni A, McAdam MB, Floisand Y, Lau C, Espevik T, et al. Complement Component C5 and TLR Molecule CD14 Mediate Heme-Induced Thromboinflammation in Human Blood. J Immunol (2019) 203(6):1571–8. doi: 10.4049/jimmunol.1900047
105. Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Eaton JW, et al. Heme, heme oxygenase and ferritin in vascular endothelial cell injury. Mol Nutr Food Res (2005) 49(11):1030–43. doi: 10.1002/mnfr.200500076
106. Grinshtein N, Bamm VV, Tsemakhovich VA, Shaklai N. Mechanism of low-density lipoprotein oxidation by hemoglobin-derived iron. Biochemistry (2003) 42(23):6977–85. doi: 10.1021/bi020647r
107. Miller YI, Shaklai N. Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim Biophys Acta (1999) 1454(2):153–64. doi: 10.1016/S0925-4439(99)00027-7
108. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol (2010) 50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600
109. Fasano M, Mattu M, Coletta M, Ascenzi P. The heme-iron geometry of ferrous nitrosylated heme-serum lipoproteins, hemopexin, and albumin: a comparative EPR study. J Inorganic Biochem (2002) 91(3):487–90. doi: 10.1016/S0162-0134(02)00473-7
110. Sasaki J, Waterman MR, Buchanan GR, Cottam GL. Plasma and erythrocyte lipids in sickle cell anaemia. Clin Lab Haematol (1983) 5(1):35–44. doi: 10.1111/j.1365-2257.1983.tb00494.x
111. Akinlade KS, Adewale CO, Rahamon SK, Fasola FA, Olaniyi JA, Atere AD. Defective lipid metabolism in sickle cell anaemia subjects in vaso-occlusive crisis. Nigerian Med J J Nigeria Med Assoc (2014) 55(5):428–31. doi: 10.4103/0300-1652.140388
112. Zorca S, Freeman L, Hildesheim M, Allen D, Remaley AT, JGt T, et al. Lipid levels in sickle-cell disease associated with haemolytic severity, vascular dysfunction and pulmonary hypertension. Br J Haematol (2010) 149(3):436–45. doi: 10.1111/j.1365-2141.2010.08109.x
113. Yalcinkaya A, Unal S, Oztas Y. Altered HDL particle in sickle cell disease: decreased cholesterol content is associated with hemolysis, whereas decreased Apolipoprotein A1 is linked to inflammation. Lipids Health Disease (2019) 18(1):225. doi: 10.1186/s12944-019-1174-5
114. Vendrame F, Olops L, Saad STO, Costa FF, Fertrin KY. Differences in heme and hemopexin content in lipoproteins from patients with sickle cell disease. J Clin Lipidol (2018) 12(6):1532–8. doi: 10.1016/j.jacl.2018.08.002
115. Fasano M, Fanali G, Leboffe L, Ascenzi P. Heme binding to albuminoid proteins is the result of recent evolution. IUBMB Life (2007) 59(7):436–40. doi: 10.1080/15216540701474523
116. Ascenzi P, Fasano M. Serum heme-albumin: an allosteric protein. IUBMB Life (2009) 61(12):1118–22. doi: 10.1002/iub.263
117. Bunn HF, Jandl JH. Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem (1968) 243(3):465–75.
118. Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood (2005) 106(7):2572–9. doi: 10.1182/blood-2005-03-1185
119. Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Heme scavenging and the other facets of hemopexin. Antioxidants Redox Signaling (2010) 12(2):305–20. doi: 10.1089/ars.2009.2787
120. Allhorn M, Berggard T, Nordberg J, Olsson ML, Akerstrom B. Processing of the lipocalin alpha(1)-microglobulin by hemoglobin induces heme-binding and heme-degradation properties. Blood (2002) 99(6):1894–901. doi: 10.1182/blood.V99.6.1894
121. Meining W, Skerra A. The crystal structure of human alpha(1)-microglobulin reveals a potential haem-binding site. Biochem J (2012) 445(2):175–82. doi: 10.1042/BJ20120448
122. Allhorn M, Klapyta A, Akerstrom B. Redox properties of the lipocalin alpha1-microglobulin: reduction of cytochrome c, hemoglobin, and free iron. Free Radical Biol Med (2005) 38(5):557–67. doi: 10.1016/j.freeradbiomed.2004.12.013
123. Hahl P, Hunt R, Bjes ES, Skaff A, Keightley A, Smith A. Identification of oxidative modifications of hemopexin and their predicted physiological relevance. J Biol Chem (2017) 292(33):13658–71. doi: 10.1074/jbc.M117.783951
124. Paoli M, Anderson BF, Baker HM, Morgan WT, Smith A, Baker EN. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat Struct Biol (1999) 6(10):926–31. doi: 10.1038/13294
125. Gkouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochim Biophys Acta (2012) 1820(3):188–202. doi: 10.1016/j.bbagen.2011.10.013
126. Olatunya OS, Lanaro C, Longhini AL, Penteado CFF, Fertrin KY, Adekile A, et al. Red blood cells microparticles are associated with hemolysis markers and may contribute to clinical events among sickle cell disease patients. Ann Hematol (2019) 98(11):2507–21. doi: 10.1007/s00277-019-03792-x
127. Yalamanoglu A, Deuel JW, Hunt RC, Baek JH, Hassell K, Redinius K, et al. Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease. Am J Physiol Lung Cell Mol Physiol (2018) 315(5):L765–L74. doi: 10.1152/ajplung.00269.2018
128. Soares MP, Bach FH. Heme oxygenase-1: from biology to therapeutic potential. Trends Mol Med (2009) 15(2):50–8. doi: 10.1016/j.molmed.2008.12.004
129. Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, et al. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev (2003) 55(3):551–71. doi: 10.1124/pr.55.3.5
130. Alam J, Killeen E, Gong P, Naquin R, Hu B, Stewart D, et al. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol (2003) 284(4):F743–52. doi: 10.1152/ajprenal.00376.2002
131. Belcher JD, Beckman JD, Balla G, Balla J, Vercellotti G. Heme degradation and vascular injury. Antioxidants Redox Signaling (2010) 12(2):233–48. doi: 10.1089/ars.2009.2822
132. Boyle JJ, Johns M, Lo J, Chiodini A, Ambrose N, Evans PC, et al. Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arteriosclerosis thrombosis Vasc Biol (2011) 31(11):2685–91. doi: 10.1161/ATVBAHA.111.225813
133. Ghosh S, Hazra R, Ihunnah CA, Weidert F, Flage B, Ofori-Acquah SF. Augmented NRF2 activation protects adult sickle mice from lethal acute chest syndrome. Br J Haematol (2018) 182(2):271–5. doi: 10.1111/bjh.15401
134. Belcher JD, Vineyard JV, Bruzzone CM, Chen C, Beckman JD, Nguyen J, et al. Heme oxygenase-1 gene delivery by Sleeping Beauty inhibits vascular stasis in a murine model of sickle cell disease. J Mol Med (Berlin Germany) (2010) 88(7):665–75. doi: 10.1007/s00109-010-0613-6
135. Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Investig (2006) 116(3):808–16. doi: 10.1172/JCI26857
136. Belcher JD, Chen C, Nguyen J, Zhang P, Abdulla F, Nguyen P, et al. Control of Oxidative Stress and Inflammation in Sickle Cell Disease with the Nrf2 Activator Dimethyl Fumarate. Antioxidants Redox Signaling (2017) 26(14):748–62. doi: 10.1089/ars.2015.6571
137. Krishnamoorthy S, Pace B, Gupta D, Sturtevant S, Li B, Makala L, et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI Insight (2017) 2(20):1–16. doi: 10.1172/jci.insight.96409
138. Keleku-Lukwete N, Suzuki M, Otsuki A, Tsuchida K, Katayama S, Hayashi M, et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc Natl Acad Sci USA (2015) 112(39):12169–74. doi: 10.1073/pnas.1509158112
139. Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA (1993) 90(20):9285–9. doi: 10.1073/pnas.90.20.9285
140. Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol Cell Biol (2006) 26(7):2845–56. doi: 10.1128/MCB.26.7.2845-2856.2006
141. Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc (2018) 68(2-3):263–99. doi: 10.3233/CH-189012
142. Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med (2007) 13(6):703–10. doi: 10.1038/nm1586
143. Pereira MLM, Marinho CRF, Epiphanio S. Could Heme Oxygenase-1 Be a New Target for Therapeutic Intervention in Malaria-Associated Acute Lung Injury/Acute Respiratory Distress Syndrome? Front Cell Infection Microbiol (2018) 8:161. doi: 10.3389/fcimb.2018.00161
144. Ekregbesi P, Shankar-Hari M, Bottomley C, Riley EM, Mooney JP. Relationship between Anaemia, Haemolysis, Inflammation and Haem Oxygenase-1 at Admission with Sepsis: a pilot study. Sci Rep (2018) 8(1):11198. doi: 10.1038/s41598-018-29558-5
145. Adamzik M, Hamburger T, Petrat F, Peters J, de Groot H, Hartmann M. Free hemoglobin concentration in severe sepsis: methods of measurement and prediction of outcome. Crit Care (2012) 16(4):R125. doi: 10.1186/cc11425
146. Clark IA, Awburn MM, Harper CG, Liomba NG, Molyneux ME. Induction of HO-1 in tissue macrophages and monocytes in fatal falciparum malaria and sepsis. Malaria J (2003) 2(1):41. doi: 10.1186/1475-2875-2-41
147. Kato G, Gladwin M, Steinberg M. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev (2007) 21(1):37–47. doi: 10.1016/j.blre.2006.07.001
148. Takaki S, Takeyama N, Kajita Y, Yabuki T, Noguchi H, Miki Y, et al. Beneficial effects of the heme oxygenase-1/carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med (2010) 36(1):42–8. doi: 10.1007/s00134-009-1575-4
149. Janz DR, Bastarache JA, Sills G, Wickersham N, May AK, Bernard GR, et al. Association between haptoglobin, hemopexin and mortality in adults with sepsis. Crit Care (2013) 17(6):R272. doi: 10.1186/cc13108
150. Balla G, Jacob HS, Eaton JW, Belcher JD, Vercellotti GM. Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler Thromb J Vasc Biol (1991) 11(6):1700–11. doi: 10.1161/01.ATV.11.6.1700
151. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol (2014) 5:115. doi: 10.3389/fphar.2014.00115
152. Gouveia Z, Carlos AR, Yuan X, Aires-da-Silva F, Stocker R, Maghzal GJ, et al. Characterization of plasma labile heme in hemolytic conditions. FEBS J (2017) 284(19):3278–301. doi: 10.1111/febs.14192
153. Santiago RP, Guarda CC, Figueiredo CVB, Fiuza LM, Aleluia MM, Adanho CSA, et al. Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients. Blood Cells Mol Dis (2018) 72:34–6. doi: 10.1016/j.bcmd.2018.07.002
154. Vercellotti GM, Zhang P, Nguyen J, Abdulla F, Chen C, Nguyen P, et al. Hepatic Overexpression of Hemopexin Inhibits Inflammation and Vascular Stasis in Murine Models of Sickle Cell Disease. Mol Med (Cambridge Mass) (2016) 22:1–15. doi: 10.2119/molmed.2016.00063
155. Ofori-Acquah SF, Hazra R, Orikogbo OO, Crosby D, Flage B, Ackah EB, et al. Hemopexin deficiency promotes acute kidney injury in sickle cell disease. Blood (2020) 135(13):1044–8. doi: 10.1182/blood.2019002653
156. Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood (1968) 32(5):811–5. doi: 10.1182/blood.V32.5.811.811
157. Merle NS, Grunenwald A, Rajaratnam H, Gnemmi V, Frimat M, Figueres ML, et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight (2018) 3(12):1–17. doi: 10.1172/jci.insight.96910
158. Adisa OA, Hu Y, Ghosh S, Aryee D, Osunkwo I, Ofori-Acquah SF. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. Br J Haematol (2013) 162(5):702–5. doi: 10.1111/bjh.12445
159. Sadrzadeh SM, Graf E, Panter SS, Hallaway PE, Eaton JW. Hemoglobin. A biologic fenton reagent. J Biol Chem (1984) 259(23):14354–6.
160. Thomas DD, Espey MG, Vitek MP, Miranda KM, Wink DA. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: an alternative to the NO/O2- reaction. Proc Natl Acad Sci USA (2002) 99(20):12691–6. doi: 10.1073/pnas.202312699
161. Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett (1995) 82-83:969–74. doi: 10.1016/0378-4274(95)03532-x
162. Meng F, Alayash AI. Determination of extinction coefficients of human hemoglobin in various redox states. Anal Biochem (2017) 521:11–9. doi: 10.1016/j.ab.2017.01.002
163. Hanna DA, Harvey RM, Martinez-Guzman O, Yuan X, Chandrasekharan B, Raju G, et al. Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc Natl Acad Sci USA (2016) 113(27):7539–44. doi: 10.1073/pnas.1523802113
164. Newton LD, Pascu SI, Tyrrell RM, Eggleston IM. Development of a peptide-based fluorescent probe for biological heme monitoring. Org Biomol Chem (2019) 17(3):467–71. doi: 10.1039/C8OB02290A
165. Hargrove MS, Whitaker T, Olson JS, Vali RJ, Mathews AJ. Quaternary structure regulates hemin dissociation from human hemoglobin. J Biol Chem (1997) 272(28):17385–9. doi: 10.1074/jbc.272.28.17385
166. Anderson HL, Brodsky IE, Mangalmurti NS. The Evolving Erythrocyte: Red Blood Cells as Modulators of Innate Immunity. J Immunol (2018) 201(5):1343–51. doi: 10.4049/jimmunol.1800565
167. Wang X, Mendelsohn L, Rogers H, Leitman S, Raghavachari N, Yang Y, et al. Heme-bound iron activates placenta growth factor in erythroid cells via erythroid Krüppel-like factor. Blood (2014) 124(6):946–54. doi: 10.1182/blood-2013-11-539718
168. Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood (2016) 127(4):473–86. doi: 10.1182/blood-2015-08-663245
169. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA (2014) 111(39):E4110–8. doi: 10.1073/pnas.1405023111
170. Sparkenbaugh EM, Chantrathammachart P, Wang S, Jonas W, Kirchhofer D, Gailani D, et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica (2015) 100(3):308–14. doi: 10.3324/haematol.2014.114728
171. Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood (2014) 123(24):3818–27. doi: 10.1182/blood-2013-10-529982
172. Erdei J, Toth A, Balogh E, Nyakundi BB, Banyai E, Ryffel B, et al. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid Med Cell Longevity (2018) 2018:4310816. doi: 10.1155/2018/4310816
173. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood (2014) 123(3):377–90. doi: 10.1182/blood-2013-04-495887
174. Deuel JW, Vallelian F, Schaer CA, Puglia M, Buehler PW, Schaer DJ. Different target specificities of haptoglobin and hemopexin define a sequential protection system against vascular hemoglobin toxicity. Free Radical Biol Med (2015) 89:931–43. doi: 10.1016/j.freeradbiomed.2015.09.016
175. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem (2007) 282(28):20221–9. doi: 10.1074/jbc.M610737200
176. Kapetanaki MG, Gbotosho OT, Sharma D, Weidert F, Ofori-Acquah SF, Kato GJ. Free heme regulates placenta growth factor through NRF2-antioxidant response signaling. Free Radic Biol Med (2019) 143:300–8. doi: 10.1016/j.freeradbiomed.2019.08.009
177. Gladwin MT, Ofori-Acquah SF. Erythroid DAMPs drive inflammation in SCD. Blood (2014) 123(24):3689–90. doi: 10.1182/blood-2014-03-563874
178. Mendonca R, Silveira AA, Conran N. Red cell DAMPs and inflammation. Inflammation Res (2016) 65(9):665–78. doi: 10.1007/s00011-016-0955-9
179. Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, et al. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol (2011) 187(9):4809–17. doi: 10.4049/jimmunol.1102093
180. Vogel S, Thein SL. Platelets at the crossroads of thrombosis, inflammation and haemolysis. Br J Haematol (2018) 180(5):761–7. doi: 10.1111/bjh.15117
181. Maeda R, Kawasaki Y, Kume Y, Go H, Suyama K, Hosoya M. Involvement of high mobility group box 1 in the pathogenesis of severe hemolytic uremic syndrome in a murine model. Am J Physiol Renal Physiol (2019) 317(6):F1420–F9. doi: 10.1152/ajprenal.00263.2019
182. Ataga KI, Orringer EP. Hypercoagulability in sickle cell disease: a curious paradox. Am J Med (2003) 115(9):721–8. doi: 10.1016/j.amjmed.2003.07.011
183. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi: 10.1126/science.285.5425.248
184. Xu H, Wandersee NJ, Guo Y, Jones DW, Holzhauer SL, Hanson MS, et al. Sickle cell disease increases high mobility group box 1: a novel mechanism of inflammation. Blood (2014) 124(26):3978–81. doi: 10.1182/blood-2014-04-560813
185. Vogel S, Arora T, Wang X, Mendelsohn L, Nichols J, Allen D, et al. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv (2018) 2(20):2672–80. doi: 10.1182/bloodadvances.2018021709
186. Murthy P, Durco F, Miller-Ocuin JL, Takedai T, Shankar S, Liang X, et al. The NLRP3 inflammasome and bruton’s tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem Biophys Res Commun (2017) 483(1):230–6. doi: 10.1016/j.bbrc.2016.12.161
187. Petrillo S, Chiabrando D, Genova T, Fiorito V, Ingoglia G, Vinchi F, et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ (2018) 25(3):573–88. doi: 10.1038/s41418-017-0001-7
188. Wagener FA, Feldman E, de Witte T, Abraham NG. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc Soc Exp Biol Med Soc Exp Biol Med (1997) 216(3):456–63. doi: 10.3181/00379727-216-44197
189. Telen MJ. Cellular adhesion and the endothelium: E-selectin, L-selectin, and pan-selectin inhibitors. Hematology/Oncology Clinics North America (2014) 28(2):341–54. doi: 10.1016/j.hoc.2013.11.010
190. Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med (2009) 15(4):384–91. doi: 10.1038/nm.1939
191. Gee BE, Platt OS. Sickle reticulocytes adhere to VCAM-1. Blood (1995) 85(1):268–74. doi: 10.1182/blood.V85.1.268.bloodjournal851268
192. Kucukal E, Ilich A, Key NS, Little JA, Gurkan UA. Red Blood Cell Adhesion to Heme-Activated Endothelial Cells Reflects Clinical Phenotype in Sickle Cell Disease. Am J Hematol (2018) 93(8):1050–60. doi: 10.1002/ajh.25159
193. Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A. Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood (2001) 98(6):1955–62. doi: 10.1182/blood.V98.6.1955
194. Matsui NM, Varki A, Embury SH. Heparin inhibits the flow adhesion of sickle red blood cells to P-selectin. Blood (2002) 100(10):3790–6. doi: 10.1182/blood-2002-02-0626
195. Embury SH, Matsui NM, Ramanujam S, Mayadas TN, Noguchi CT, Diwan BA, et al. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood (2004) 104(10):3378–85. doi: 10.1182/blood-2004-02-0713
196. Ghosh S, Flage B, Weidert F, Ofori-Acquah SF. P-selectin plays a role in haem-induced acute lung injury in sickle mice. Br J Haematol (2019) 186(2):329–33. doi: 10.1111/bjh.15807
197. Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood (2010) 116(10):1779–86. doi: 10.1182/blood-2009-12-260513
198. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. New Engl J Med (2017) 376(5):429–39. doi: 10.1056/NEJMoa1611770
199. Kutlar A, Kanter J, Liles DK, Alvarez OA, Cancado RD, Friedrisch JR, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am J Hematol (2019) 94(1):55–61. doi: 10.1002/ajh.25308
200. Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, et al. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arteriosclerosis thrombosis Vasc Biol (2010) 30(12):2392–9. doi: 10.1161/ATVBAHA.110.211615
201. Keleku-Lukwete N, Suzuki M, Panda H, Otsuki A, Katsuoka F, Saito R, et al. Nrf2 activation in myeloid cells and endothelial cells differentially mitigates sickle cell disease pathology in mice. Blood Adv (2019) 3(8):1285–97. doi: 10.1182/bloodadvances.2018017574
202. Merle NS, Paule R, Leon J, Daugan M, Robe-Rybkine T, Poillerat V, et al. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc Natl Acad Sci USA (2019) 116(13):6280–5. doi: 10.1073/pnas.1814797116
203. Bennewitz MF, Jimenez MA, Vats R, Tutuncuoglu E, Jonassaint J, Kato GJ, et al. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight (2017) 2(1):e89761. doi: 10.1172/jci.insight.89761
204. Kato GJ, Martyr S, Blackwelder WC, Nichols JS, Coles WA, Hunter LA, et al. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br J Haematol (2005) 130(6):943–53. doi: 10.1111/j.1365-2141.2005.05701.x
205. Antwi-Boasiako C, Donkor ES, Sey F, Dzudzor B, Dankwah GB, Otu KH, et al. Levels of Soluble Endothelium Adhesion Molecules and Complications among Sickle Cell Disease Patients in Ghana. Diseases (2018) 6(2):1–7. doi: 10.3390/diseases6020029
206. Setty BN, Stuart MJ, Dampier C, Brodecki D, Allen JL. Hypoxaemia in sickle cell disease: biomarker modulation and relevance to pathophysiology. Lancet (2003) 362(9394):1450–5. doi: 10.1016/S0140-6736(03)14689-2
207. Elmariah H, Garrett ME, De Castro LM, Jonassaint JC, Ataga KI, Eckman JR, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol (2014) 89(5):530–5. doi: 10.1002/ajh.23683
208. Keikhaei B, Mohseni AR, Norouzirad R, Alinejadi M, Ghanbari S, Shiravi F, et al. Altered levels of pro-inflammatory cytokines in sickle cell disease patients during vaso-occlusive crises and the steady state condition. Eur Cytokine Netw (2013) 24(1):45–52. doi: 10.1684/ecn.2013.0328
209. Khalyfa A, Khalyfa AA, Akbarpour M, Connes P, Romana M, Lapping-Carr G, et al. Extracellular microvesicle microRNAs in children with sickle cell anaemia with divergent clinical phenotypes. Br J Haematol (2016) 174(5):786–98. doi: 10.1111/bjh.14104
210. Jain S, Kapetanaki MG, Raghavachari N, Woodhouse K, Yu G, Barge S, et al. Expression of regulatory platelet microRNAs in patients with sickle cell disease. PloS One (2013) 8(4):e60932. doi: 10.1371/journal.pone.0060932
211. Barker KR, Lu Z, Kim H, Zheng Y, Chen J, Conroy AL, et al. miR-155 Modifies Inflammation, Endothelial Activation and Blood-Brain Barrier Dysfunction in Cerebral Malaria. Mol Med (Cambridge Mass) (2017) 23:24–33. doi: 10.2119/molmed.2016.00139
212. Cohen A, Zinger A, Tiberti N, Grau GER, Combes V. Differential plasma microvesicle and brain profiles of microRNA in experimental cerebral malaria. Malaria J (2018) 17(1):192. doi: 10.1186/s12936-018-2330-5
213. Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol (2007) 14(1):23–9. doi: 10.1038/nsmb1182
214. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA (2005) 102(39):13944–9. doi: 10.1073/pnas.0506654102
215. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science (2004) 303(5654):83–6. doi: 10.1126/science.1091903
216. Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell (2003) 113(1):25–36. doi: 10.1016/S0092-8674(03)00231-9
217. Guo Z, Wu R, Gong J, Zhu W, Li Y, Wang Z, et al. Altered microRNA expression in inflamed and non-inflamed terminal ileal mucosa of adult patients with active Crohn’s disease. J Gastroenterol Hepatology (2015) 30(1):109–16. doi: 10.1111/jgh.12644
218. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature (2005) 435(7043):834–8. doi: 10.1038/nature03702
219. Alevizos I, Illei GG. MicroRNAs as biomarkers in rheumatic diseases. Nat Rev Rheumatol (2010) 6(7):391–8. doi: 10.1038/nrrheum.2010.81
220. Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheumatism (2008) 58(5):1284–92. doi: 10.1002/art.23429
221. Pekow JR, Kwon JH. MicroRNAs in inflammatory bowel disease. Inflammatory bowel Diseases (2012) 18(1):187–93. doi: 10.1002/ibd.21691
222. Tomankova T, Petrek M, Kriegova E. Involvement of microRNAs in physiological and pathological processes in the lung. Respiratory Res (2010) 11:159. doi: 10.1186/1465-9921-11-159
223. Weitz SH, Gong M, Barr I, Weiss S, Guo F. Processing of microRNA primary transcripts requires heme in mammalian cells. Proc Natl Acad Sci USA (2014) 111(5):1861–6. doi: 10.1073/pnas.1309915111
224. Nguyen TA, Park J, Dang TL, Choi YG, Kim VN. Microprocessor depends on hemin to recognize the apical loop of primary microRNA. Nucleic Acids Res (2018) 46(11):5726–36. doi: 10.1093/nar/gky248
225. Barr I, Smith AT, Chen Y, Senturia R, Burstyn JN, Guo F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc Natl Acad Sci USA (2012) 109(6):1919–24. doi: 10.1073/pnas.1114514109
226. Kirschner MB, Edelman JJ, Kao SC, Vallely MP, van Zandwijk N, Reid G. The Impact of Hemolysis on Cell-Free microRNA Biomarkers. Front Genet (2013) 4:94. doi: 10.3389/fgene.2013.00094
227. Pizzamiglio S, Zanutto S, Ciniselli CM, Belfiore A, Bottelli S, Gariboldi M, et al. A methodological procedure for evaluating the impact of hemolysis on circulating microRNAs. Oncol Lett (2017) 13(1):315–20. doi: 10.3892/ol.2016.5452
228. Chen SY, Wang Y, Telen MJ, Chi JT. The genomic analysis of erythrocyte microRNA expression in sickle cell diseases. PloS One (2008) 3(6):e2360. doi: 10.1371/journal.pone.0002360
229. Byon JC, Papayannopoulou T. MicroRNAs: Allies or foes in erythropoiesis? J Cell Physiol (2012) 227(1):7–13. doi: 10.1002/jcp.22729
230. Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood (2010) 116(20):4338–48. doi: 10.1182/blood-2009-04-214817
231. Li B, Zhu X, Ward CM, Starlard-Davenport A, Takezaki M, Berry A, et al. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Exp Hematol (2019) 70:85–96 e5. doi: 10.1016/j.exphem.2018.11.002
232. Desai AA, Zhou T, Ahmad H, Zhang W, Mu W, Trevino S, et al. A novel molecular signature for elevated tricuspid regurgitation velocity in sickle cell disease. Am J Respir Crit Care Med (2012) 186(4):359–68. doi: 10.1164/rccm.201201-0057OC
233. Ha TY. MicroRNAs in Human Diseases: From Lung, Liver and Kidney Diseases to Infectious Disease, Sickle Cell Disease and Endometrium Disease. Immune Netw (2011) 11(6):309–23. doi: 10.4110/in.2011.11.6.309
234. Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, et al. An analysis of human microRNA and disease associations. PloS One (2008) 3(10):e3420. doi: 10.1371/journal.pone.0003420
235. Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation (2010) 121(8):1022–32. doi: 10.1161/CIRCULATIONAHA.109.889048
236. Barringhaus KG, Zamore PD. MicroRNAs: regulating a change of heart. Circulation (2009) 119(16):2217–24. doi: 10.1161/CIRCULATIONAHA.107.715839
237. Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol (2009) 6(6):419–29. doi: 10.1038/nrcardio.2009.56
238. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science (2005) 309(5740):1577–81. doi: 10.1126/science.1113329
239. Wang K, Zhang S, Marzolf B, Troisch P, Brightman A, Hu Z, et al. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc Natl Acad Sci USA (2009) 106(11):4402–7. doi: 10.1073/pnas.0813371106
240. Pandey P, Brors B, Srivastava PK, Bott A, Boehn SN, Groene HJ, et al. Microarray-based approach identifies microRNAs and their target functional patterns in polycystic kidney disease. BMC Genomics (2008) 9:624. doi: 10.1186/1471-2164-9-624
241. Chaturvedi S, DeBaun MR. Evolution of sickle cell disease from a life-threatening disease of children to a chronic disease of adults: The last 40 years. Am J Hematol (2016) 91(1):5–14. doi: 10.1002/ajh.24235
242. Huang E, Parke C, Mehrnia A, Kamgar M, Pham PT, Danovitch G, et al. Improved survival among sickle cell kidney transplant recipients in the recent era. Nephrol Dial Transplant Off Publ Eur Dialysis Transplant Assoc - Eur Renal Assoc (2013) 28(4):1039–46. doi: 10.1093/ndt/gfs585
243. Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol (2015) 11(3):161–71. doi: 10.1038/nrneph.2015.8
244. Day TG, Drasar ER, Fulford T, Sharpe CC, Thein SL. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica (2012) 97(2):201–5. doi: 10.3324/haematol.2011.050336
245. Plewes K, Kingston HWF, Ghose A, Maude RJ, Herdman MT, Leopold SJ, et al. Cell-free hemoglobin mediated oxidative stress is associated with acute kidney injury and renal replacement therapy in severe falciparum malaria: an observational study. BMC Infect Dis (2017) 17(1):313. doi: 10.1186/s12879-017-2373-1
246. Gaggar A, Patel RP. There is blood in the water: hemolysis, hemoglobin, and heme in acute lung injury. Am J Physiol Lung Cell Mol Physiol (2016) 311(4):L714–L8. doi: 10.1152/ajplung.00312.2016
247. Gliozzi ML, Rbaibi Y, Long KR, Vitturi DA, Weisz OA. Hemoglobin alters vitamin carrier uptake and vitamin D metabolism in proximal tubule cells: implications for sickle cell disease. Am J Physiol Cell Physiol (2019) 317(5):C993–C1000. doi: 10.1152/ajpcell.00287.2019
248. van Swelm RP, Wetzels JF, Verweij VG, Laarakkers CM, Pertijs JC, van der Wijst J, et al. Renal Handling of Circulating and Renal-Synthesized Hepcidin and Its Protective Effects against Hemoglobin-Mediated Kidney Injury. J Am Soc Nephrol JASN (2016) 27(9):2720–32. doi: 10.1681/ASN.2015040461
249. Schein A, Enriquez C, Coates TD, Wood JC. Magnetic resonance detection of kidney iron deposition in sickle cell disease: a marker of chronic hemolysis. J Magn Reson Imaging (2008) 28(3):698–704. doi: 10.1002/jmri.21490
250. Vasavda N, Gutierrez L, House MJ, Drasar E, St Pierre TG, Thein SL. Renal iron load in sickle cell disease is influenced by severity of haemolysis. Br J Haematol (2012) 157(5):599–605. doi: 10.1111/j.1365-2141.2012.09093.x
251. Gurkan S, Scarponi KJ, Hotchkiss H, Savage B, Drachtman R. Lactate dehydrogenase as a predictor of kidney involvement in patients with sickle cell anemia. Pediatr Nephrol (2010) 25(10):2123–7. doi: 10.1007/s00467-010-1560-8
252. Saraf SL, Zhang X, Kanias T, Lash JP, Molokie RE, Oza B, et al. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. Br J Haematol (2014) 164(5):729–39. doi: 10.1111/bjh.12690
253. Barber BE, Grigg MJ, Piera KA, William T, Cooper DJ, Plewes K, et al. Intravascular haemolysis in severe Plasmodium knowlesi malaria: association with endothelial activation, microvascular dysfunction, and acute kidney injury. Emerging microbes Infect (2018) 7(1):106. doi: 10.1038/s41426-018-0105-2
254. Nath KA, Grande JP, Haggard JJ, Croatt AJ, Katusic ZS, Solovey A, et al. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol (2001) 158(3):893–903. doi: 10.1016/S0002-9440(10)64037-0
255. Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am J Pathol (2000) 156(5):1527–35. doi: 10.1016/S0002-9440(10)65024-9
256. Nath KA, Vercellotti GM, Grande JP, Miyoshi H, Paya CV, Manivel JC, et al. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int (2001) 59(1):106–17. doi: 10.1046/j.1523-1755.2001.00471.x
257. Rubio-Navarro A, Vazquez-Carballo C, Guerrero-Hue M, Garcia-Caballero C, Herencia C, Gutierrez E, et al. Nrf2 Plays a Protective Role Against Intravascular Hemolysis-Mediated Acute Kidney Injury. Front Pharmacol (2019) 10:740. doi: 10.3389/fphar.2019.00740
258. Nath KA, Belcher JD, Nath MC, Grande JP, Croatt AJ, Ackerman AW, et al. Role of TLR4 signaling in the nephrotoxicity of heme and heme proteins. Am J Physiol Renal Physiol (2018) 314(5):F906–F14. doi: 10.1152/ajprenal.00432.2017
259. Piazza M, Damore G, Costa B, Gioannini TL, Weiss JP, Peri F. Hemin and a metabolic derivative coprohemin modulate the TLR4 pathway differently through different molecular targets. Innate Immun (2011) 17(3):293–301. doi: 10.1177/1753425910369020
260. Wei Q, Hill WD, Su Y, Huang S, Dong Z. Heme oxygenase-1 induction contributes to renoprotection by G-CSF during rhabdomyolysis-associated acute kidney injury. Am J Physiol Renal Physiol (2011) 301(1):F162–70. doi: 10.1152/ajprenal.00438.2010
261. Gonzalez-Michaca L, Farrugia G, Croatt AJ, Alam J, Nath KA. Heme: a determinant of life and death in renal tubular epithelial cells. Am J Physiol Renal Physiol (2004) 286(2):F370–7. doi: 10.1152/ajprenal.00300.2003
262. Irwin DC, Baek JH, Hassell K, Nuss R, Eigenberger P, Lisk C, et al. Hemoglobin-induced lung vascular oxidation, inflammation, and remodeling contribute to the progression of hypoxic pulmonary hypertension and is attenuated in rats with repeated-dose haptoglobin administration. Free Radical Biol Med (2015) 82:50–62. doi: 10.1016/j.freeradbiomed.2015.01.012
263. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Investig (2013) 123(11):4809–20. doi: 10.1172/JCI64578
264. Bilan VP, Schneider F, Novelli EM, Kelley EE, Shiva S, Gladwin MT, et al. Experimental intravascular hemolysis induces hemodynamic and pathological pulmonary hypertension: association with accelerated purine metabolism. Pulmonary circulation (2018) 8(3):1–15. doi: 10.1177/2045894018791557
265. Shaver CM, Upchurch CP, Janz DR, Grove BS, Putz ND, Wickersham NE, et al. Cell-free hemoglobin: a novel mediator of acute lung injury. Am J Physiol Lung Cell Mol Physiol (2016) 310(6):L532–41. doi: 10.1152/ajplung.00155.2015
266. Singla S, Sysol JR, Dille B, Jones N, Chen J, Machado RF. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am J Respir Cell Mol Biol (2017) 57(3):307–14. doi: 10.1165/rcmb.2016-0287OC
267. Liu Y, Jing F, Yi W, Mendelson A, Shi P, Walsh R, et al. HO-1(hi) patrolling monocytes protect against vaso-occlusion in sickle cell disease. Blood (2018) 131(14):1600–10. doi: 10.1182/blood-2017-12-819870
268. Feld JJ, Kato GJ, Koh C, Shields T, Hildesheim M, Kleiner DE, et al. Liver injury is associated with mortality in sickle cell disease. Alimentary Pharmacol Ther (2015) 42(7):912–21. doi: 10.1111/apt.13347
269. Dey S, Bindu S, Goyal M, Pal C, Alam A, Iqbal MS, et al. Impact of intravascular hemolysis in malaria on liver dysfunction: involvement of hepatic free heme overload, NF-kappaB activation, and neutrophil infiltration. J Biol Chem (2012) 287(32):26630–46. doi: 10.1074/jbc.M112.341255
270. Hsu L, Champion H, Campbell-Lee S, Bivalacqua T, Manci E, Diwan B, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood (2007) 109(7):3088–98. doi: 10.1182/blood-2006-08-039438
271. Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell disease: role of nitric oxide and hemolytic anemia. Hematol Am Soc Hematol Educ Program (2005) 2005:51–7. doi: 10.1182/asheducation-2005.1.51
272. Moraes JA, Barcellos-de-Souza P, Rodrigues G, Nascimento-Silva V, Silva SV, Assreuy J, et al. Heme modulates smooth muscle cell proliferation and migration via NADPH oxidase: a counter-regulatory role for heme oxygenase system. Atherosclerosis (2012) 224(2):394–400. doi: 10.1016/j.atherosclerosis.2012.07.043
273. Qi L, van Dam RM, Rexrode K, Hu FB. Heme iron from diet as a risk factor for coronary heart disease in women with type 2 diabetes. Diabetes Care (2007) 30(1):101–6. doi: 10.2337/dc06-1686
274. Fang X, An P, Wang H, Wang X, Shen X, Li X, et al. Dietary intake of heme iron and risk of cardiovascular disease: a dose-response meta-analysis of prospective cohort studies. Nutrition metabolism Cardiovasc Dis NMCD (2015) 25(1):24–35. doi: 10.1016/j.numecd.2014.09.002
275. Ingoglia G, Sag CM, Rex N, De Franceschi L, Vinchi F, Cimino J, et al. Hemopexin counteracts systolic dysfunction induced by heme-driven oxidative stress. Free Radical Biol Med (2017) 108:452–64. doi: 10.1016/j.freeradbiomed.2017.04.003
276. Vinchi F, De Franceschi L, Ghigo A, Townes T, Cimino J, Silengo L, et al. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation (2013) 127(12):1317–29. doi: 10.1161/CIRCULATIONAHA.112.130179
277. Khechaduri A, Bayeva M, Chang HC, Ardehali H. Heme levels are increased in human failing hearts. J Am Coll Cardiol (2013) 61(18):1884–93. doi: 10.1016/j.jacc.2013.02.012
278. Sawicki KT, Shang M, Wu R, Chang HC, Khechaduri A, Sato T, et al. Increased Heme Levels in the Heart Lead to Exacerbated Ischemic Injury. J Am Heart Assoc (2015) 4(8):e002272. doi: 10.1161/JAHA.115.002272
279. Alvarado G, Jeney V, Toth A, Csosz E, Kallo G, Huynh AT, et al. Heme-induced contractile dysfunction in human cardiomyocytes caused by oxidant damage to thick filament proteins. Free Radical Biol Med (2015) 89:248–62. doi: 10.1016/j.freeradbiomed.2015.07.158
280. Sundaram N, Tailor A, Mendelsohn L, Wansapura J, Wang X, Higashimoto T, et al. High levels of placenta growth factor in sickle cell disease promote pulmonary hypertension. Blood (2010) 116(1):109–12. doi: 10.1182/blood-2009-09-244830
281. Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci U S A (1991) 88(20):9267–71. doi: 10.1073/pnas.88.20.9267
282. Persico MG, Vincenti V, DiPalma T. Structure, expression and receptor-binding properties of placenta growth factor (PlGF). Curr Top Microbiol Immunol (1999) 237:31–40. doi: 10.1007/978-3-642-59953-8_2
283. Iyer S, Leonidas DD, Swaminathan GJ, Maglione D, Battisti M, Tucci M, et al. The crystal structure of human placenta growth factor-1 (PlGF-1), an angiogenic protein, at 2.0 A resolution. J Biol Chem (2001) 276(15):12153–61. doi: 10.1074/jbc.M008055200
284. Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem (1994) 269(41):25646–54. doi: 10.1074/jbc.M008055200
285. Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med (2001) 7(5):575–83. doi: 10.1038/87904
286. Tarallo V, Vesci L, Capasso O, Esposito MT, Riccioni T, Pastore L, et al. A placental growth factor variant unable to recognize vascular endothelial growth factor (VEGF) receptor-1 inhibits VEGF-dependent tumor angiogenesis via heterodimerization. Cancer Res (2010) 70(5):1804–13. doi: 10.1158/0008-5472.CAN-09-2609
287. Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med (2003) 9(7):936–43. doi: 10.1038/nm884
288. Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher J, Klagsbrun M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J Biol Chem (2002) 277(27):24818–25. doi: 10.1074/jbc.M200730200
289. Gaur P, Bielenberg DR, Samuel S, Bose D, Zhou Y, Gray MJ, et al. Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin Cancer Res (2009) 15(22):6763–70. doi: 10.1158/1078-0432.CCR-09-1810
290. Roy H, Bhardwaj S, Babu M, Jauhiainen S, Herzig KH, Bellu AR, et al. Adenovirus-mediated gene transfer of placental growth factor to perivascular tissue induces angiogenesis via upregulation of the expression of endogenous vascular endothelial growth factor-A. Hum Gene Ther (2005) 16(12):1422–8. doi: 10.1089/hum.2005.16.1422
291. Marcellini M, De Luca N, Riccioni T, Ciucci A, Orecchia A, Lacal PM, et al. Increased melanoma growth and metastasis spreading in mice overexpressing placenta growth factor. Am J Pathol (2006) 169(2):643–54. doi: 10.2353/ajpath.2006.051041
292. Huang XX, McCaughan GW, Shackel NA, Gorrell MD. Up-regulation of proproliferative genes and the ligand/receptor pair placental growth factor and vascular endothelial growth factor receptor 1 in hepatitis C cirrhosis. Liver Int (2007) 27(7):960–8. doi: 10.1111/j.1478-3231.2007.01542.x
293. Clauss M, Weich H, Breier G, Knies U, Röckl W, Waltenberger J, et al. The Vascular Endothelial Growth Factor Receptor Flt-1 Mediates Biological Activities: Implications For A Functional Role Of Placenta Growth Factor In Monocyte Activation And Chemotaxis. J Biol Chem (1996) 271(30):17629–34. doi: 10.1074/jbc.271.30.17629
294. Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res (2003) 93(11):1074–81. doi: 10.1161/01.RES.0000102937.50486.1B
295. Green CJ, Lichtlen P, Huynh NT, Yanovsky M, Laderoute KR, Schaffner W, et al. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: a central role for metal transcription factor-1. Cancer Res (2001) 61(6):2696–703. doi: 10.1074/jbc.271.30.17629
296. Cramer M, Nagy I, Murphy BJ, Gassmann M, Hottiger MO, Georgiev O, et al. NF-kappaB contributes to transcription of placenta growth factor and interacts with metal responsive transcription factor-1 in hypoxic human cells. Biol Chem (2005) 386(9):865–72. doi: 10.1515/BC.2005.101
297. Zhang H, Palmer R, Gao X, Kreidberg J, Gerald W, Hsiao L, et al. Transcriptional activation of placental growth factor by the forkhead/winged helix transcription factor FoxD1. Curr Biol (2003) 13(18):1625–9. doi: 10.1016/j.cub.2003.08.054
298. Chiu YH, Yang MR, Wang LJ, Chen MH, Chang GD, Chen H. New insights into the regulation of placental growth factor gene expression by the transcription factors GCM1 and DLX3 in human placenta. J Biol Chem (2018) 293(25):9801–11. doi: 10.1074/jbc.RA117.001384
299. Yao YG, Yang HS, Cao Z, Danielsson J, Duh EJ. Upregulation of placental growth factor by vascular endothelial growth factor via a post-transcriptional mechanism. FEBS Lett (2005) 579(5):1227–34. doi: 10.1016/j.febslet.2005.01.017
300. Shaw JH, Lloyd PG. Post-transcriptional regulation of placenta growth factor mRNA by hydrogen peroxide. Microvasc Res (2012) 84(2):155–60. doi: 10.1016/j.mvr.2012.05.009
301. Dewerchin M, Carmeliet P. PlGF: A Multitasking Cytokine with Disease-Restricted Activity. Cold Spring Harbor Perspect Med (2012) 2(8):1227–34. doi: 10.1101/cshperspect.a011056
302. Rakic JM, Lambert V, Devy L, Luttun A, Carmeliet P, Claes C, et al. Placental growth factor, a member of the VEGF family, contributes to the development of choroidal neovascularization. Invest Ophthalmol Vis Sci (2003) 44(7):3186–93. doi: 10.1167/iovs.02-1092
303. Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med (2002) 8(8):831–40. doi: 10.1038/nm731
304. De Falco S. The discovery of placenta growth factor and its biological activity. Exp Mol Med (2012) 44(1):1–9. doi: 10.3858/emm.2012.44.1.025
305. Oura H, Bertoncini J, Velasco P, Brown LF, Carmeliet P, Detmar M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood (2003) 101(2):560–7. doi: 10.1182/blood-2002-05-1516
306. Yoo SA, Yoon HJ, Kim HS, Chae CB, De Falco S, Cho CS, et al. Role of placenta growth factor and its receptor flt-1 in rheumatoid inflammation: a link between angiogenesis and inflammation. Arthritis Rheumatol (2009) 60(2):345–54. doi: 10.1002/art.24289
307. Maes C, Coenegrachts L, Stockmans I, Daci E, Luttun A, Petryk A, et al. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest (2006) 116(5):1230–42. doi: 10.1172/JCI26772
308. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell (2011) 19(1):31–44. doi: 10.1016/j.ccr.2010.11.009
309. Lin YL, Liang YC, Chiang BL. Placental growth factor down-regulates type 1 T helper immune response by modulating the function of dendritic cells. J Leukoc Biol (2007) 82(6):1473–80. doi: 10.1189/jlb.0307164
310. Carnevale D, Cifelli G, Mascio G, Madonna M, Sbroggio M, Perrino C, et al. Placental growth factor regulates cardiac inflammation through the tissue inhibitor of metalloproteinases-3/tumor necrosis factor-alpha-converting enzyme axis: crucial role for adaptive cardiac remodeling during cardiac pressure overload. Circulation (2011) 124(12):1337–50. doi: 10.1161/CIRCULATIONAHA.111.050500
311. Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, et al. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med (2002) 8(8):841–9. doi: 10.1038/nm740
312. Carlo-Stella C, Di Nicola M, Longoni P, Cleris L, Lavazza C, Milani R, et al. Placental growth factor-1 potentiates hematopoietic progenitor cell mobilization induced by granulocyte colony-stimulating factor in mice and nonhuman primates. Stem Cells (2007) 25(1):252–61. doi: 10.1634/stemcells.2006-0020
313. Kalra VK, Zhang S, Malik P, Tahara SM. Placenta growth factor mediated gene regulation in sickle cell disease. Blood Rev (2018) 32(1):61–70. doi: 10.1016/j.blre.2017.08.008
314. Patel N, Gonsalves CS, Malik P, Kalra VK. Placenta growth factor augments endothelin-1 and endothelin-B receptor expression via hypoxia-inducible factor-1α. Blood (2008) 112(3):856–65. doi: 10.1182/blood-2007-12-130567
315. Brittain JE, Hulkower B, Jones SK, Strayhorn D, De Castro L, Telen MJ, et al. Placenta growth factor in sickle cell disease: association with hemolysis and inflammation. Blood (2010) 115(10):2014–20. doi: 10.1182/blood-2009-04-217950
316. Perelman N, Selvaraj SK, Batra S, Luck LR, Erdreich-Epstein A, Coates TD, et al. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood (2003) 102(4):1506–14. doi: 10.1182/blood-2002-11-3422
317. Gu J-M, Yuan S, Sim D, Abe K, Liu P, Rosenbruch M, et al. Blockade of placental growth factor reduces vaso-occlusive complications in murine models of sickle cell disease. Exp Hematol (2018) 60:73–82.e3. doi: 10.1016/j.exphem.2018.01.002
318. Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. New Engl J Med (2004) 350(9):886–95. doi: 10.1056/NEJMoa035477
319. Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M, McMillen MA. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood (1998) 92(7):2551–5. doi: 10.1182/blood.V92.7.2551
320. Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood (1998) 92(7):2594–6. doi: 10.1182/blood.V92.7.2594.2594_2594_2596
321. Qari MH, Dier U, Mousa SA. Biomarkers of inflammation, growth factor, and coagulation activation in patients with sickle cell disease. Clin Appl Thromb Hemost (2012) 18(2):195–200. doi: 10.1177/1076029611420992
322. Li C, Gonsalves CS, Eiymo Mwa Mpollo MS, Malik P, Tahara SM, Kalra VK. MicroRNA 648 Targets ET-1 mRNA and is cotranscriptionally regulated with MICAL3 by PAX5. Mol Cell Biol (2015) 35(3):514–28. doi: 10.1128/MCB.01199-14
323. Gonsalves CS, Li C, Malik P, Tahara SM, Kalra VK. Peroxisome proliferator-activated receptor-alpha-mediated transcription of miR-301a and miR-454 and their host gene SKA2 regulates endothelin-1 and PAI-1 expression in sickle cell disease. Biosci Rep (2015) 35(6):195–200. doi: 10.1042/BSR20150190
324. Li C, Mpollo MS, Gonsalves CS, Tahara SM, Malik P, Kalra VK. Peroxisome proliferator-activated receptor-alpha-mediated transcription of miR-199a2 attenuates endothelin-1 expression via hypoxia-inducible factor-1alpha. J Biol Chem (2014) 289(52):36031–47. doi: 10.1074/jbc.M114.600775
325. Li C, Zhou Y, Loberg A, Tahara SM, Malik P, Kalra VK. Activated Transcription Factor 3 in Association with Histone Deacetylase 6 Negatively Regulates MicroRNA 199a2 Transcription by Chromatin Remodeling and Reduces Endothelin-1 Expression. Mol Cell Biol (2016) 36(22):2838–54. doi: 10.1128/MCB.00345-16
326. Patel N, Sundaram N, Yang M, Madigan C, Kalra VK, Malik P. Placenta Growth Factor (PlGF), a Novel Inducer of Plasminogen Activator Inhibitor-1 (PAI-1) in Sickle Cell Disease (SCD). J Biol Chem (2010) 285(22):16713–22. doi: 10.1074/jbc.M110.101691
327. Nsiri B, Gritli N, Mazigh C, Ghazouani E, Fattoum S, Machghoul S. Fibrinolytic response to venous occlusion in patients with homozygous sickle cell disease. Hematol Cell Ther (1997) 39(5):229–32. doi: 10.1007/s00282-997-0229-7
328. Hillery CA, Panepinto JA. Pathophysiology of stroke in sickle cell disease. Microcirculation (2004) 11(2):195–208. doi: 10.1080/10739680490278600
329. Patel N, Tahara SM, Malik P, Kalra VK. Involvement of miR-30c and miR-301a in immediate induction of plasminogen activator inhibitor-1 by placental growth factor in human pulmonary endothelial cells. Biochem J (2011) 434(3):473–82. doi: 10.1042/BJ20101585
330. Leong MA, Dampier C, Varlotta L, Allen JL. Airway hyperreactivity in children with sickle cell disease. J Pediatr (1997) 131(2):278–83. doi: 10.1016/S0022-3476(97)70166-5
331. Field JJ, Stocks J, Kirkham FJ, Rosen CL, Dietzen DJ, Semon T, et al. Airway hyperresponsiveness in children with sickle cell anemia. Chest (2011) 139(3):563–8. doi: 10.1378/chest.10-1243
332. Eiymo Mwa Mpollo M-S, Brandt EB, Shanmukhappa SK, Arumugam PI, Tiwari S, Loberg A, et al. Placenta growth factor augments airway hyperresponsiveness via leukotrienes and IL-13. J Clin Invest (2016) 126(2):571–84. doi: 10.1172/JCI77250
333. Patel N, Gonsalves CS, Yang M, Malik P, Kalra VK. Placenta growth factor induces 5-lipoxygenase-activating protein to increase leukotriene formation in sickle cell disease. Blood (2009) 113(5):1129–38. doi: 10.1182/blood-2008-07-169821
334. Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci USA (2002) 99(5):3047–51. doi: 10.1073/pnas.052522799
335. Selvaraj SK, Giri RK, Perelman N, Johnson C, Malik P, Kalra VK. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood (2003) 102(4):1515–24. doi: 10.1182/blood-2002-11-3423
336. Kaul DK, Liu XD, Choong S, Belcher JD, Vercellotti GM, Hebbel RP. Anti-inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am J Physiol - Heart Circulatory Physiol (2004) 287(1 56-1):H293–301. doi: 10.1152/ajpheart.01150.2003
337. Mousavi Z, Yazdani Z, Moradabadi A, Hoseinpourkasgari F, Hassanshahi G. Role of some members of chemokine/cytokine network in the pathogenesis of thalassemia and sickle cell hemoglobinopathies: a mini review. Exp Hematol Oncol (2019) 8(1):21. doi: 10.1186/s40164-019-0145-x
338. Gonsalves CS, Li C, Mpollo MS, Pullarkat V, Malik P, Tahara SM, et al. Erythropoietin-mediated expression of placenta growth factor is regulated via activation of hypoxia-inducible factor-1alpha and post-transcriptionally by miR-214 in sickle cell disease. Biochem J (2015) 468(3):409–23. doi: 10.1042/BJ20141138
339. Zakiyanov O, Kalousová M, Zima T, Tesař V. Placental Growth Factor in Patients with Decreased Renal Function. Renal Failure (2011) 33(3):291–7. doi: 10.3109/0886022X.2011.560402
340. Matsui M, Uemura S, Takeda Y, Samejima K, Matsumoto T, Hasegawa A, et al. Placental Growth Factor as a Predictor of Cardiovascular Events in Patients with CKD from the NARA-CKD Study. J Am Soc Nephrol (2015) 26(11):2871–81. doi: 10.1681/ASN.2014080772
341. Ataga KI, Derebail VK, Caughey M, Elsherif L, Shen JH, Jones SK, et al. Albuminuria Is Associated with Endothelial Dysfunction and Elevated Plasma Endothelin-1 in Sickle Cell Anemia. PloS One (2016) 11(9):e0162652. doi: 10.1371/journal.pone.0162652
342. Heimlich JB, Speed JS, O’Connor PM, Pollock JS, Townes TM, Meiler SE, et al. Endothelin-1 contributes to the progression of renal injury in sickle cell disease via reactive oxygen species. Br J Pharmacol (2016) 173(2):386–95. doi: 10.1111/bph.13380
343. Gbotosho OT, Ghosh S, Kapetanaki MG, Lin Y, Weidert F, Bullock GC, et al. Cardiac expression of HMOX1 and PGF in sickle cell mice and haem-treated wild type mice dominates organ expression profiles via Nrf2 (Nfe2l2). Br J Haematol (2019) 187(5):666–75. doi: 10.1111/bjh.16129
344. Malgorzewicz S, Skrzypczak-Jankun E, Jankun J. Plasminogen activator inhibitor-1 in kidney pathology (Review). Int J Mol Med (2013) 31(3):503–10. doi: 10.3892/ijmm.2013.1234
345. Gladwin MT, Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol (2012) 59(13):1123–33. doi: 10.1016/j.jacc.2011.10.900
346. Peiskerová M, Kalousová M, Danzig V, Míková B, Hodková M, Němeček E, et al. Placental growth factor may predict increased left ventricular mass index in patients with mild to moderate chronic kidney disease–a prospective observational study. BMC Nephrol (2013) 14:142–. doi: 10.1186/1471-2369-14-142
347. Pilarczyk K, Sattler KJ, Galili O, Versari D, Olson ML, Meyer FB, et al. Placenta growth factor expression in human atherosclerotic carotid plaques is related to plaque destabilization. Atherosclerosis (2008) 196(1):333–40. doi: 10.1016/j.atherosclerosis.2006.10.038
348. Khurana R, Moons L, Shafi S, Luttun A, Collen D, Martin JF, et al. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation (2005) 111(21):2828–36. doi: 10.1161/CIRCULATIONAHA.104.495887
349. Heeschen C, Dimmeler S, Fichtlscherer S, Hamm CW, Berger J, Simoons ML, et al. Prognostic Value of Placental Growth Factor in Patients With Acute Chest Pain. JAMA (2004) 291(4):435–41. doi: 10.1001/jama.291.4.435
350. Jaba IM, Zhuang ZW, Li N, Jiang Y, Martin KA, Sinusas AJ, et al. NO triggers RGS4 degradation to coordinate angiogenesis and cardiomyocyte growth. J Clin Invest (2013) 123(4):1718–31. doi: 10.1172/JCI65112
351. Accornero F, van Berlo JH, Benard MJ, Lorenz JN, Carmeliet P, Molkentin JD. Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ Res (2011) 109(3):272–80. doi: 10.1161/CIRCRESAHA.111.240820
352. Harada E, Nakagawa O, Yoshimura M, Harada M, Nakagawa M, Mizuno Y, et al. Effect of interleukin-1 beta on cardiac hypertrophy and production of natriuretic peptides in rat cardiocyte culture. J Mol Cell Cardiol (1999) 31(11):1997–2006. doi: 10.1006/jmcc.1999.1030
353. Wang L, Zhang YL, Lin QY, Liu Y, Guan XM, Ma XL, et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur Heart J (2018) 39(20):1818–31. doi: 10.1093/eurheartj/ehy085
354. Nakamura T, Funayama H, Kubo N, Yasu T, Kawakami M, Momomura S, et al. Elevation of plasma placental growth factor in the patients with ischemic cardiomyopathy. Int J Cardiol (2009) 131(2):186–91. doi: 10.1016/j.ijcard.2007.10.050
355. Kolakowski S Jr., Berry MF, Atluri P, Grand T, Fisher O, Moise MA, et al. Placental growth factor provides a novel local angiogenic therapy for ischemic cardiomyopathy. J Card Surg (2006) 21(6):559–64. doi: 10.1111/j.1540-8191.2006.00296.x
356. Rolla S, Ingoglia G, Bardina V, Silengo L, Altruda F, Novelli F, et al. Acute-phase protein hemopexin is a negative regulator of Th17 response and experimental autoimmune encephalomyelitis development. J Immunol (2013) 191(11):5451–9. doi: 10.4049/jimmunol.1203076
357. Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T, et al. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem (2003) 278(39):36993–8. doi: 10.1074/jbc.M302942200
358. Zimmermann M, Aguilera FB, Castellucci M, Rossato M, Costa S, Lunardi C, et al. Chromatin remodelling and autocrine TNFalpha are required for optimal interleukin-6 expression in activated human neutrophils. Nat Commun (2015) 6:6061. doi: 10.1038/ncomms7061
359. Zimmermann M, Arruda-Silva F, Bianchetto-Aguilera F, Finotti G, Calzetti F, Scapini P, et al. IFNalpha enhances the production of IL-6 by human neutrophils activated via TLR8. Sci Rep (2016) 6:19674. doi: 10.1038/srep19674
360. Chi L, Li Y, Stehno-Bittel L, Gao J, Morrison DC, Stechschulte DJ, et al. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J interferon cytokine Res Off J Int Soc Interferon Cytokine Res (2001) 21(4):231–40. doi: 10.1089/107999001750169871
361. Zampetaki A, Zhang Z, Hu Y, Xu Q. Biomechanical stress induces IL-6 expression in smooth muscle cells via Ras/Rac1-p38 MAPK-NF-kappaB signaling pathways. Am J Physiol Heart Circ Physiol (2005) 288(6):H2946–54. doi: 10.1152/ajpheart.00919.2004
362. Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol (2005) 204(2):428–36. doi: 10.1002/jcp.20307
363. Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, et al. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem (2000) 275(38):29717–23. doi: 10.1074/jbc.M003128200
364. Fontes JA, Rose NR, Cihakova D. The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine (2015) 74(1):62–8. doi: 10.1016/j.cyto.2014.12.024
365. Su H, Lei CT, Zhang C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front Immunol (2017) 8:405. doi: 10.3389/fimmu.2017.00405
366. Peters M, Muller AM, Rose-John S. Interleukin-6 and soluble interleukin-6 receptor: direct stimulation of gp130 and hematopoiesis. Blood (1998) 92(10):3495–504. doi: 10.1182/blood.V92.10.3495.422k47_3495_3504
367. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev (2012) 38(7):904–10. doi: 10.1016/j.ctrv.2012.04.007
368. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature (1986) 324(6092):73–6. doi: 10.1038/324073a0
369. Lacroix M, Rousseau F, Guilhot F, Malinge P, Magistrelli G, Herren S, et al. Novel Insights into Interleukin 6 (IL-6) Cis- and Trans-signaling Pathways by Differentially Manipulating the Assembly of the IL-6 Signaling Complex. J Biol Chem (2015) 290(45):26943–53. doi: 10.1074/jbc.M115.682138
370. Scheller J, Garbers C, Rose-John S. Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin Immunol (2014) 26(1):2–12. doi: 10.1016/j.smim.2013.11.002
371. Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) (2012) 122(4):143–59. doi: 10.1042/CS20110340
372. Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest (2011) 121(9):3375–83. doi: 10.1172/JCI57158
373. Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I, et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infection Immunity (2004) 72(10):5630–7. doi: 10.1128/IAI.72.10.5630-5637.2004
374. Nayak KC, Meena SL, Gupta BK, Kumar S, Pareek V. Cardiovascular involvement in severe vivax and falciparum malaria. J Vector Borne Dis (2013) 50(4):285–91. doi: 10.1074/jbc.M115.682138
375. Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science (1992) 257(5068):387–9. doi: 10.1126/science.1631560
376. Burwick RM, Rincon M, Beeraka SS, Gupta M, Feinberg BB. Evaluation of Hemolysis as a Severe Feature of Preeclampsia. Hypertension (Dallas Tex 1979) (2018) 72(2):460–5. doi: 10.1161/HYPERTENSIONAHA.118.11211
377. Kumar S, Wang G, Zheng N, Cheng W, Ouyang K, Lin H, et al. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension (Dallas Tex 1979) (2019) 73(5):1058–70. doi: 10.1161/HYPERTENSIONAHA.118.12267
378. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension (Dallas Tex 1979) (2010) 56(2):225–31. doi: 10.1161/HYPERTENSIONAHA.109.148635
379. Dinh W, Futh R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, et al. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol (2009) 8:58. doi: 10.1186/1475-2840-8-58
380. Sugishita K, Kinugawa K, Shimizu T, Harada K, Matsui H, Takahashi T, et al. Cellular basis for the acute inhibitory effects of IL-6 and TNF- alpha on excitation-contraction coupling. J Mol Cell Cardiol (1999) 31(8):1457–67. doi: 10.1006/jmcc.1999.0989
381. Hagiwara Y, Miyoshi S, Fukuda K, Nishiyama N, Ikegami Y, Tanimoto K, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol (2007) 43(6):710–6. doi: 10.1016/j.yjmcc.2007.09.004
382. Drosatos K, Lymperopoulos A, Kennel PJ, Pollak N, Schulze PC, Goldberg IJ. Pathophysiology of sepsis-related cardiac dysfunction: driven by inflammation, energy mismanagement, or both? Curr Heart failure Rep (2015) 12(2):130–40. doi: 10.1007/s11897-014-0247-z
383. Zhang W, Qu X, Chen B, Snyder M, Wang M, Li B, et al. Critical Roles of STAT3 in beta-Adrenergic Functions in the Heart. Circulation (2016) 133(1):48–61. doi: 10.1161/CIRCULATIONAHA.115.017472
384. de Montmollin E, Aboab J, Mansart A, Annane D. Bench-to-bedside review: Beta-adrenergic modulation in sepsis. Crit Care (2009) 13(5):230. doi: 10.1186/cc8026
385. Wang Y, Lewis DF, Gu Y, Zhao S, Groome LJ. Elevated maternal soluble Gp130 and IL-6 levels and reduced Gp130 and SOCS-3 expressions in women complicated with preeclampsia. Hypertension (Dallas Tex 1979) (2011) 57(2):336–42. doi: 10.1161/HYPERTENSIONAHA.110.163360
386. Lamarca B, Brewer J, Wallace K. IL-6-induced pathophysiology during pre-eclampsia: potential therapeutic role for magnesium sulfate? Int J interferon cytokine Mediator Res (2011) 2011(3):59–64. doi: 10.2147/IJICMR.S16320
387. Sarray S, Saleh LR, Lisa Saldanha F, Al-Habboubi HH, Mahdi N, Almawi WY. Serum IL-6, IL-10, and TNFalpha levels in pediatric sickle cell disease patients during vasoocclusive crisis and steady state condition. Cytokine (2015) 72(1):43–7. doi: 10.1016/j.cyto.2014.11.030
388. Taylor SC, Shacks SJ, Mitchell RA, Banks A. Serum interleukin-6 levels in the steady state of sickle cell disease. J interferon cytokine Res Off J Int Soc Interferon Cytokine Res (1995) 15(12):1061–4. doi: 10.1089/jir.1995.15.1061
389. Lester LA, Sodt PC, Hutcheon N, Arcilla RA. Cardiac abnormalities in children with sickle cell anemia. Chest (1990) 98(5):1169–74. doi: 10.1378/chest.98.5.1169
390. Faro GB, Menezes-Neto OA, Batista GS, Silva-Neto AP, Cipolotti R. Left ventricular hypertrophy in children, adolescents and young adults with sickle cell anemia. Rev Bras Hematol Hemoter (2015) 37(5):324–8. doi: 10.1016/j.bjhh.2015.07.001
391. Crocker P, Werb Z, Gordon S, Bainton D. Ultrastructural localization of a macrophage-restricted sialic acid binding hemagglutinin, SER, in macrophage-hematopoietic cell clusters. Blood (1990) 76(6):1131–8. doi: 10.1182/blood.V76.6.1131.bloodjournal7661131
392. Gbotosho OT, Kapetanaki MG, Ghosh S, Villanueva FS, Ofori-Acquah SF, Kato GJ. Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Front Immunol (2020) 72(1):43–7. doi: 10.3389/fimmu.2020.01910
393. Ingoglia G, Sag CM, Rex N, De Franceschi L, Vinchi F, Cimino J, et al. Data demonstrating the anti-oxidant role of hemopexin in the heart. Data Brief (2017) 13:69–76. doi: 10.1016/j.dib.2017.05.026
394. Strouse JJ, Heeney MM. Hydroxyurea for the treatment of sickle cell disease: efficacy, barriers, toxicity, and management in children. Pediatr Blood Cancer (2012) 59(2):365–71. doi: 10.1002/pbc.24178
395. Kato GJ. New insights into sickle cell disease: mechanisms and investigational therapies. Curr Opin Hematol (2016) 23(3):224–32. doi: 10.1097/MOH.0000000000000241
396. Rees D, Williams T, Gladwin M. Sickle-cell disease. Lancet (2010) 376(9757):2018–31. doi: 10.1016/S0140-6736(10)61029-X
397. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers (2018) 4:18010. doi: 10.1038/nrdp.2018.10
398. Zimmerman SA, Schultz WH, Burgett S, Mortier NA, Ware RE. Hydroxyurea therapy lowers transcranial Doppler flow velocities in children with sickle cell anemia. Blood (2007) 110(3):1043–7. doi: 10.1182/blood-2006-11-057893
399. Platt OS. Hydroxyurea for the treatment of sickle cell anemia. New Engl J Med (2008) 358(13):1362–9. doi: 10.1056/NEJMct0708272
400. Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet (2011) 377(9778):1663–72. doi: 10.1016/S0140-6736(11)60355-3
401. Voelker R. New Option for Sickle Cell Disease. JAMA (2020) 323(1):18. doi: 10.1001/jama.2019.20640
402. Gluckman E. Allogeneic transplantation strategies including haploidentical transplantation in sickle cell disease. Hematol Am Soc Hematol Educ Program (2013) 2013:370–6. doi: 10.1182/asheducation-2013.1.370
403. Makani J, Ofori-Acquah SF, Nnodu O, Wonkam A, Ohene-Frempong K. Sickle cell disease: new opportunities and challenges in Africa. TheScientificWorldJournal (2013) 2013:193252. doi: 10.1155/2013/193252
404. Rogers DW, Clarke JM, Cupidore L, Ramlal AM, Sparke BR, Serjeant GR. Early deaths in Jamaican children with sickle cell disease. Br Med J (1978) 1(6126):1515–6. doi: 10.1136/bmj.1.6126.1515
405. Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Preventive Med (2011) 41(6 Suppl 4):S398–405. doi: 10.1016/j.amepre.2011.09.013
406. Tshilolo L, Tomlinson G, Williams TN, Santos B, Olupot-Olupot P, Lane A, et al. Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa. New Engl J Med (2019) 380(2):121–31. doi: 10.1056/NEJMoa1813598
407. Tayo BO, Akingbola TS, Saraf SL, Shah BN, Ezekekwu CA, Sonubi O, et al. Fixed Low-Dose Hydroxyurea for the Treatment of Adults with Sickle Cell Anemia in Nigeria. Am J Hematol (2018) 377(9778):1663–72. doi: 10.1002/ajh.25412
Keywords: hemolysis, sickle cell disease, free heme, inflammation, oxidative stress, IL-6, placental growth factor, pulmonary hypertension
Citation: Gbotosho OT, Kapetanaki MG and Kato GJ (2021) The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease. Front. Immunol. 11:561917. doi: 10.3389/fimmu.2020.561917
Received: 14 May 2020; Accepted: 04 December 2020;
Published: 27 January 2021.
Edited by:
Wilma Barcellini, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, ItalyReviewed by:
Claudio Canetti, Federal University of Rio de Janeiro, BrazilCopyright © 2021 Gbotosho, Kapetanaki and Kato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory J. Kato, Z3JlZ29yeWthdG9tZEBnbWFpbC5jb20=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.