 Jui-Cheng Tseng1
Jui-Cheng Tseng1 James Cheng-Chung Wei2,3,4*
James Cheng-Chung Wei2,3,4* Atul Deodhar5Ruvie Martin6Brian Porter6
Atul Deodhar5Ruvie Martin6Brian Porter6 Suzanne McCreddin7
Suzanne McCreddin7 Zsolt Talloczy6
Zsolt Talloczy6- 1Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan
- 2Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan
- 3Department of Medicine, Chung Shan Medical University Hospital, Taichung, Taiwan
- 4Graduate Institute of Integrated Medicine, China Medical University, Taichung, Taiwan
- 5Oregon Health and Science University, Portland, OR, United States
- 6Novartis Pharmaceuticals Corporation, East Hanover, NJ, United States
- 7Clinical Development, Novartis Ireland Limited, Dublin, Ireland
Objectives: To present the long-term (4-year) efficacy and safety of secukinumab in Taiwanese patients with active AS in the MEASURE 1 extension study.
Methods: This post hoc analysis reports data from Taiwanese patients originally randomized to subcutaneous secukinumab 150 or 75mg or placebo every 4 weeks (following intravenous loading dose) who were invited to enter the 3-year extension study. Assessments at Week 208 included ASAS20/40 responses and other clinically relevant endpoints. Efficacy data are presented as observed. Safety analyses included all patients who received ≥1 dose of secukinumab.
Results: Of the 57 Taiwanese patients in the core trial, 48 entered the extension study and 87.5% patients (42/48) completed 4 years of treatment. Thirteen Taiwanese patients (including placebo-switchers) were escalated from 75 to 150mg (approved dose) at some point starting from Week 172. ASAS20/40 responses were sustained through 4 years in the Taiwanese patients who were originally randomized to secukinumab 150mg. Clinical responses were improved in those patients who received dose-escalation from 75 to 150mg during the study. No unexpected safety signals were reported.
Conclusion: Secukinumab 150mg demonstrated sustained efficacy over 4 years in Taiwanese patients with active ankylosing spondylitis. The safety profile of secukinumab was consistent with previous reports.
ClinicalTrials.gov identifier: NCT01863732.
Introduction
Ankylosing spondylitis (AS) is a chronic inflammatory arthritis that belongs to the family of spondyloarthritides and is clinically characterized by sacroiliitis, spinal ankylosis, extra-articular manifestations, and enthesitis (1, 2). AS causes progressive and irreversible structural damage of the sacroiliac and spinal joints, disability, and reduced quality of life (QoL) (3).
The prevalence of AS in the Chinese population is reported to be 0.3 to 0.5% (4, 5). A community-based epidemiologic study in Taiwan observed that the prevalence of AS in Taiwan Chinese was 0.38% (6).
The Assessment of SpondyloArthritis International Society (ASAS), the European League Against Rheumatism (EULAR), the American College of Rheumatology (ACR)/Spondylitis Association of America, and the Spondyloarthritis Research and Treatment Network (SPARTAN) treatment guidelines recommend non-steroidal anti-inflammatory drugs (NSAIDs) as the first-line treatment for active AS, followed by anti-tumor necrosis factor (TNF) agents or interleukin (IL)-17 inhibitors for patients who are nonresponsive to NSAIDs (2, 7). Currently, anti-TNF agents and IL-17 inhibitors are the only classes of biologics approved for the treatment of AS in Asia and worldwide (8). Up to 40% of AS patients experience an inadequate response or intolerance (IR) to anti-TNF therapies, and cases of relapse have also been reported, specifically in Asian patients, after completion of short-term treatment with anti-TNF therapies (9–12). Long-term studies with anti-TNF agents have shown a high incidence of tuberculosis and hepatitis B virus infections, as well as immunogenicity in Asian patients with AS (13–15).
Secukinumab, a fully human monoclonal immunoglobulin G (IgG)1κ antibody that directly inhibits IL-17A (expressed in a recombinant Chinese Hamster Ovary (CHO) cell line), has demonstrated sustained efficacy with a favorable and consistent safety profile through 5-years of treatment (16–19). Here we present the long-term (4-year) efficacy and safety data for secukinumab in a cohort of Taiwanese patients with active AS from the MEASURE 1 extension study.
Materials and Methods
Study Design and Patients
MEASURE 1 is a Phase 3, randomized, placebo-controlled 2-year study (NCT01358175) (16) with a 3-year extension trial (NCT1863732) (19). The study was double-blinded until Week 156 (3 years) followed by open-label treatment up to Week 260 (5 years). The Taiwanese subpopulation was assessed through Week 208 (4 years) as an interim analysis. The 52-week (1-year) results for the pooled Asian subpopulation from MEASURE 1 (NCT01358175) and MEASURE 2 (NCT01649375) phase 3 studies having 63 and 9 patients, respectively, have been reported previously (12).
In MEASURE 1, patients were initially randomized to receive intravenous (i.v.) secukinumab 10 mg/kg at baseline and Weeks 2 and 4, followed by either subcutaneous (s.c.) secukinumab 150 mg (i.v → 150 mg) or 75 mg (i.v. → 75 mg) every 4 weeks thereafter. Matched placebo was given on the same i.v. to s.c. dosing schedule, and placebo patients were re-randomized to secukinumab 150 mg s.c. or secukinumab 75 mg s.c. (hereafter referred to as placebo-switchers) by Week 16 (ASAS20 non-responders at Week 16) or Week 24 (ASAS20 responders at Week 16). After the 2-year core trial, patients receiving secukinumab 150 mg or 75 mg s.c. were invited to enter the 3-year extension trial. After protocol amendment, the study medication for patients on the secukinumab 75 mg treatment arm could be escalated to 150 mg every 4 weeks for patients whose overall therapeutic response was not fully achieved and could improve with a higher dose, as judged by the investigator. The dose escalation of the study medication could be determined at any site visit. For patients escalated to secukinumab 150 mg s.c. every 4 weeks, no dose reduction was performed and if the patient was unable to tolerate the 150 mg dose of secukinumab, alternative treatment options were considered only after discontinuation from the study. The first Taiwanese patient was dose escalated from secukinumab 75 mg to 150 mg at Week 172 whereas the first patient to receive escalated dose in the overall population was at Week 168 (as reported previously) (18). Patients were stratified according to previous anti-TNF therapy [patients who were naïve to anti-TNF therapy (anti-TNF-naïve) or those with a history of inadequate response or intolerance to no more than one of these agents (anti-TNF-IR)].
Detailed patient eligibility criteria have been previously reported (16). Briefly, patients aged ≥ 18 years with AS fulfilling the modified New York criteria with a score ≥ 4 (0–10) on the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and a spinal pain score ≥ 40 mm on a 0–100 mm visual analog scale, despite treatment with maximum tolerated doses of NSAIDs, were included in the core study. Concomitant sulfasalazine (≤ 3 g/day), methotrexate (≤ 25 mg/week), prednisone or equivalent (≤ 10 mg/day) and NSAIDs were permitted at stable doses. Patients were excluded if they had total spinal ankylosis, evidence of infection or malignancy on chest X-ray, or a history of human immunodeficiency virus or hepatitis B/C infection at screening. Patients with active systemic infection within 2 weeks prior to randomization or previous treatment with cell-depleting therapies or biologics other than anti-TNF therapy were also excluded. This study was conducted in accordance with the Declaration of Helsinki and was approved by the institutional review board or independent ethics committee at each participating center. Written informed consent was obtained from all enrolled patients.
Outcome and Assessments
Assessments through Week 208 (4 years) included ASAS20 response (defined as an improvement of ≥ 20% and absolute improvement of ≥ 1 unit on a scale of 10 in at least three of the four main ASAS domains and no worsening of ≥ 20% and ≥ 1 unit on a scale of 10 in the remaining domain), ASAS40 response (defined as an improvement of ≥ 40% and absolute improvement of ≥ 2 units on a scale of 10 in at least three of the four main ASAS domains and no worsening at all in the remaining domain), the proportion of patients achieving Ankylosing Spondylitis Disease Activity Score-C-reactive protein Inactive Disease (ASDAS-CRP ID), and mean change from baseline in high sensitivity CRP (hsCRP), BASDAI, Maastricht Ankylosing Spondylitis Enthesitis Score (MASES), Bath Ankylosing Spondylitis Functional Index (BASFI), Bath Ankylosing Spondylitis Metrology Index (BASMI), Functional Assessment of Chronic illnesses Therapy-Fatigue (FACIT-Fatigue), Ankylosing Spondylitis Quality of Life (ASQoL), and Short Form Survey-36 physical component summary (SF-36 PCS). ASAS5/6, BASDAI50 (50% improvement from baseline in BASDAI), and ASAS partial remission responses were also assessed.
Statistical Analysis
The sample size calculation and analysis of the primary and other efficacy endpoints have been reported previously for the overall study population (16). This post hoc analysis reports data from a Taiwanese patient subpopulation (N=57) who were initially randomized to the core trial and who continued in the extension trial (N=48) to Week 208 (4 years). Clinical outcomes are reported for Taiwanese patients originally randomized to secukinumab 150 or 75 mg, but not to placebo, to show the full 4-year efficacy of treatment, as well as separately for all Taiwanese patients who entered the extension study, i.e., including patients originally randomized to secukinumab and placebo switchers (hereafter, referred as the Any secukinumab 150 mg and Any secukinumab 75 mg groups). Efficacy data are presented as observed.
Safety analyses were pooled for both doses and included all Taiwanese patients who received ≥1 dose of secukinumab at any time throughout the core or extension trials. Descriptive statistics for observed safety data are provided.
Results
Patients
Of the 371 total randomized patients in the core study, 57 (~16%) were of Taiwanese origin. Overall 84% (48/57) Taiwanese patients completed the 2-year core trial and chose to enter the extension study with 21 (43.8%) and 27 (56.3%) patients in the Any secukinumab 150 mg and 75 mg groups, respectively. The overall retention rate at Week 208 was 87.5% (42/48) (Figure 1). A total of 5 patients discontinued in the Any secukinumab 150 mg group (3 due to patient decision, 1 due to lack of efficacy, and 1 was lost to follow-up); 1 patient discontinued in the Any secukinumab 75 mg group due to an adverse event (AE). A total of 13 Taiwanese patients (including placebo-switchers) dose-escalated from secukinumab 75 mg to 150 mg (approved dose) at various time points starting from Week 172.
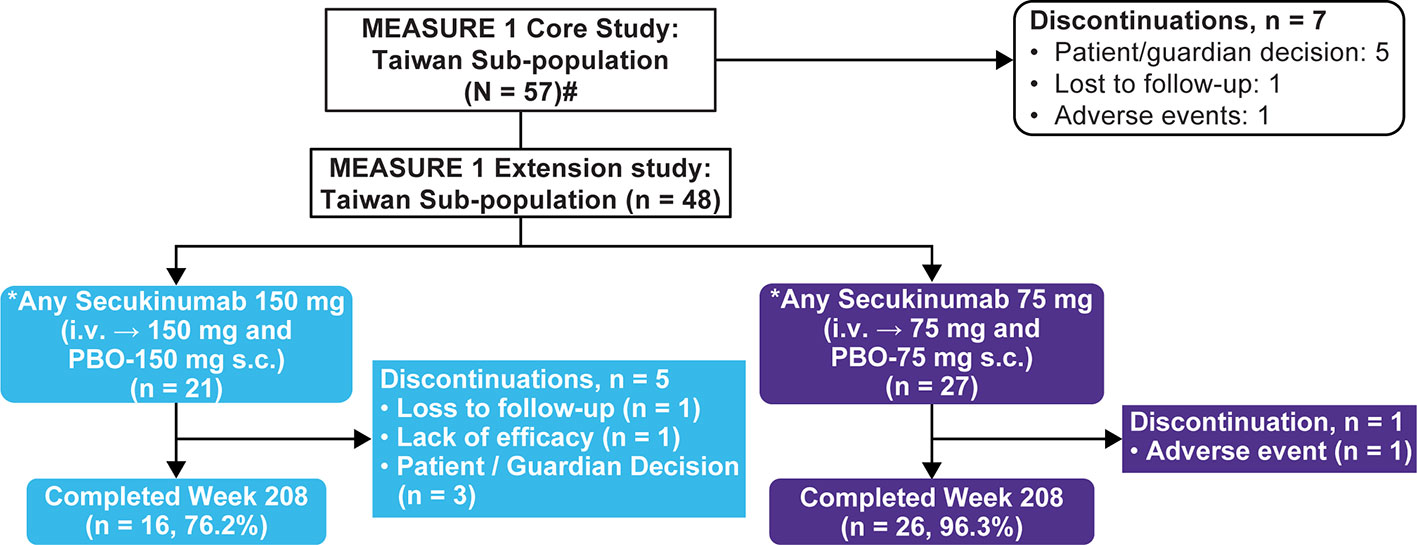
Figure 1 Patient disposition through 4 years. N, number of patients randomized; n, number of patients in a specific category i.v., Intravenous; s.c., Subcutaneous; PBO, Placebo. #Includes two Taiwanese patients who did not enter the extension phase. *Includes placebo-switchers.
Baseline demographic and disease characteristics were generally similar across the secukinumab and placebo groups in the Taiwanese subpopulation to that of the overall population, except for lower hsCRP levels and a higher percentage of HLA-B27 positive patients in the Taiwanese subpopulation (Table 1 and Supplementary Table S1). A total of 12.5% patients were inadequate responders to previous anti-TNF treatment.
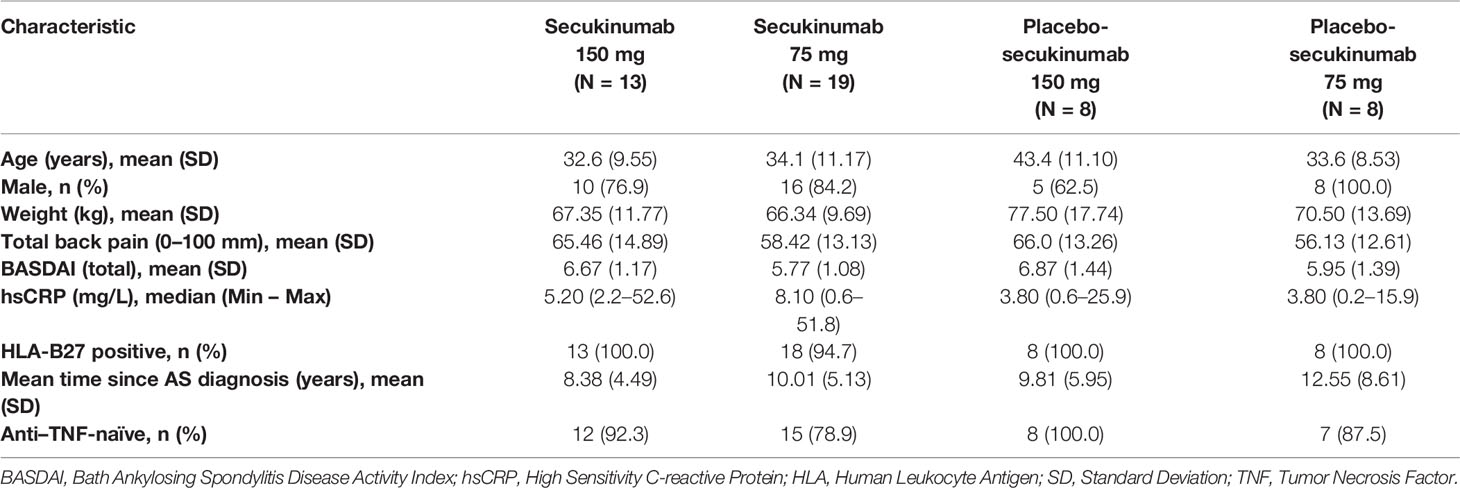
Table 1 Baseline demographic and clinical characteristics.
Efficacy
Efficacy results from the core study for the overall population have been published previously (primary endpoint of ASAS20 response at Week 16: 61% with secukinumab 150 mg vs. 29% with placebo, P < 0.001) (16).
Efficacy in Originally Randomized Patients to Secukinumab (Excluding Placebo-Switchers)
ASAS20/40 responses were sustained through 4 years in the Taiwanese patients who were originally randomized to secukinumab 150 mg or 75 mg (Figure 2A). ASAS20/40 responses were 83.3% each at 4 years in the secukinumab 150 mg group. In the secukinumab 75 mg group which included 9 patients who had dose-escalated to 150 mg, ASAS20/40 responses at 4 years for secukinumab 150 mg were 88.9% and 72.2%, respectively (Figure 2B). BASDAI scores and ASDAS-CRP ID clinical responses were sustained or further improved in the secukinumab 150 mg group through 4 years (Figure 2C and Table 2).
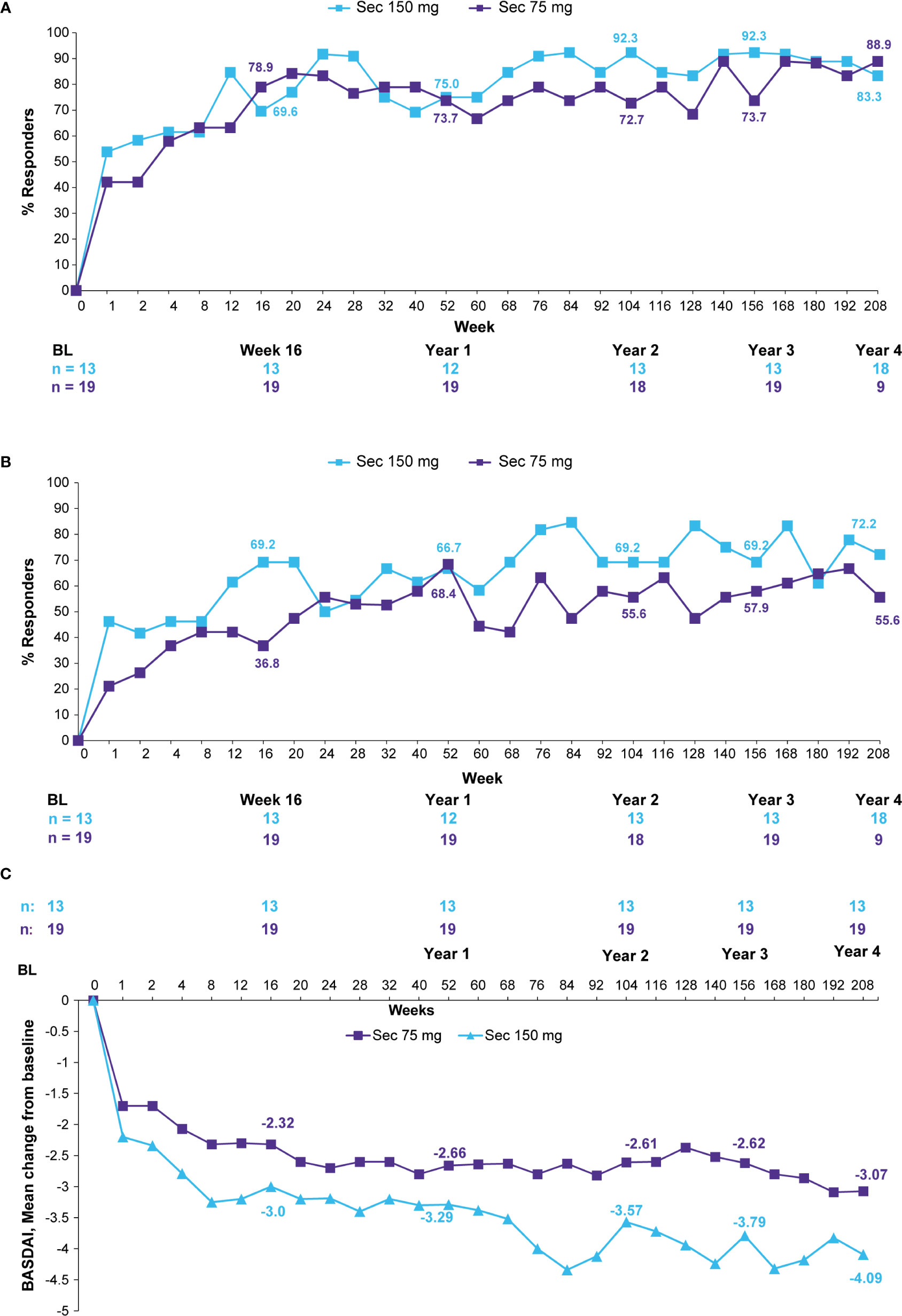
Figure 2 (A) ASAS20 responses, (B) ASAS40 responses, and (C) BASDAI mean change from baseline through 4 years (observed data for patients originally randomized to secukinumab). Observed data presented through Week 208. n, number of evaluable patients. The secukinumab 75 mg group included 9 patients who were dose-escalated from 75 to 150 mg at any time point starting from Week 172. Number of patients shown for Baseline, Weeks 16, 52 (Year 1), 104 (Year 2), 156 (Year 3), and 208 (Year 4). ASAS, Assessment of Spondyloarthritis International Society; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; Sec, Secukinumab.
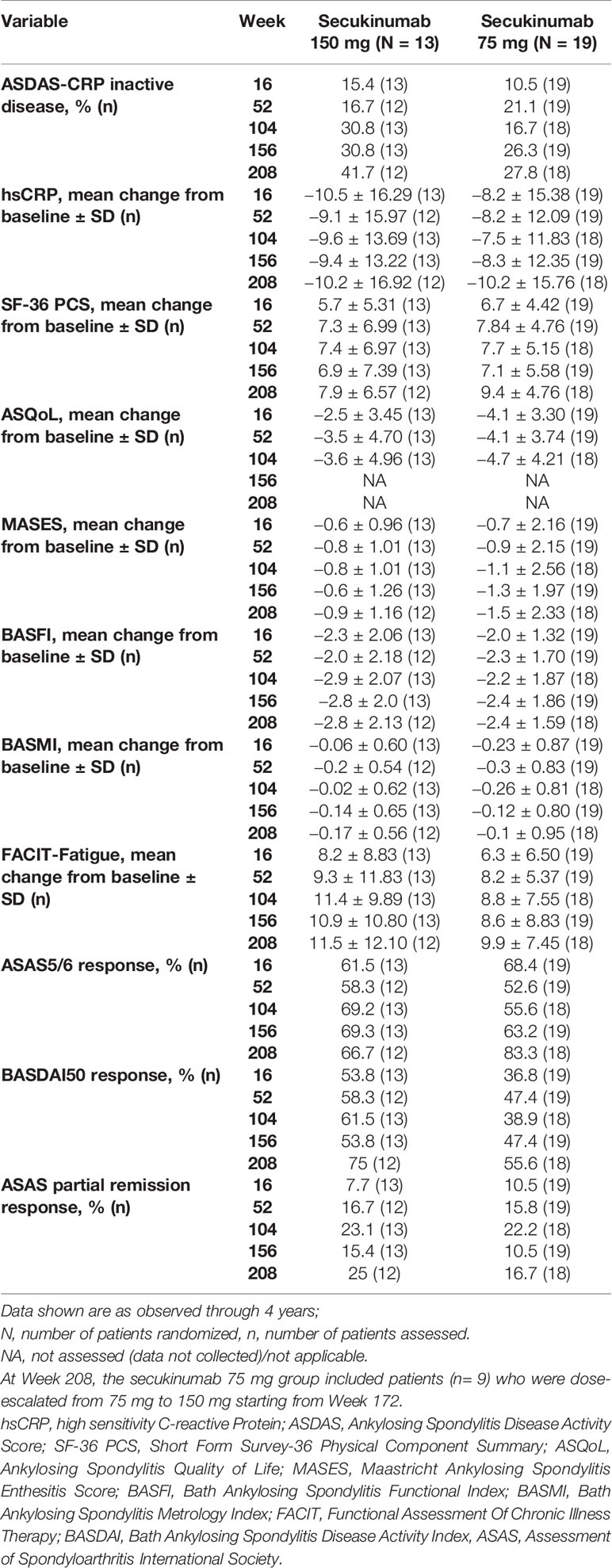
Table 2 Clinical improvements with secukinumab through Week 208 (observed data for patients originally randomized to secukinumab).
A comparison of efficacy outcomes in the overall population of patients originally randomized secukinumab 150 mg versus those of the Taiwanese subpopulation through 4 years are presented in Figure 3 and Supplementary Table S2.
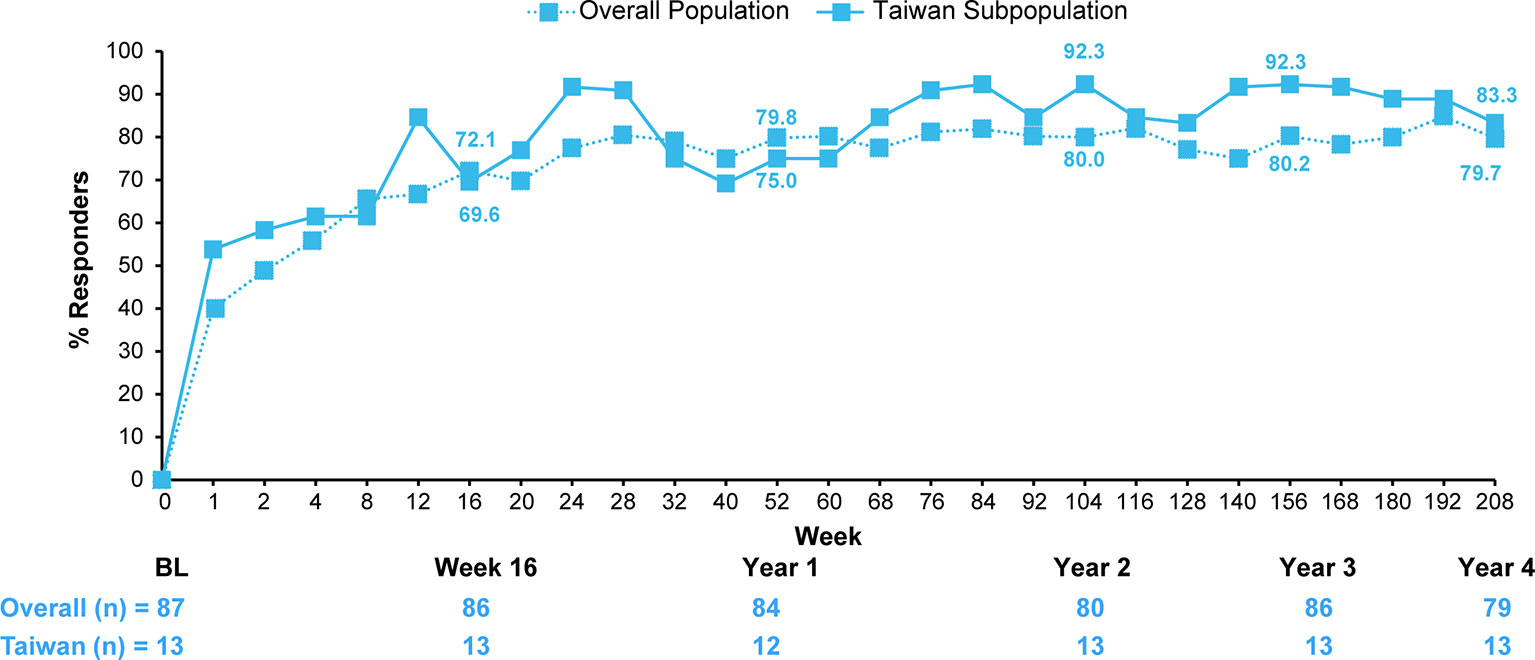
Figure 3 ASAS20 response of patients originally randomized to secukinumab 150 mg in the overall population versus the Taiwanese subpopulation. Data shown are as observed through 4 years; n, number of evaluable patients. Number of patients shown for Baseline, Weeks 16, 52 (Year 1), 104 (Year 2), 156 (Year 3), and 208 (Year 4). For overall population, the secukinumab 75 mg group included 25 patients who were dose-escalated from 75 mg to 150 mg starting from Week 168. For the Taiwanese subpopulation, the secukinumab 75 mg group included 9 patients who were dose-escalated from 75 mg to 150 mg starting from Week 172.
Efficacy in All Patients (Including Placebo-Switchers)
In the overall group of Taiwanese patients who received secukinumab 150 mg and 75 mg (i.e., including patients who switched from placebo to secukinumab at Week 16/24) efficacy was sustained through 4 years with similar responses to those observed in the originally randomized groups (Supplementary Table S3).
Efficacy Following Dose Escalation
A total of 13 out of the 27 (48.1%) patients who entered the extension study on 75 mg dose-escalated to 150 mg at some point starting from Week 172. Clinical responses were improved across most efficacy endpoints in patients who dose-escalated (Supplementary Table S3).
Safety
Rates of the most frequent treatment-emergent AEs (TEAEs) and selected AEs of interest in the Taiwanese subpopulation are shown in Table 3. Over the entire treatment period and combined for both secukinumab 150 mg and 75 mg doses, the exposure-adjusted incidence rates (EAIR) of TEAEs and serious adverse events (SAEs) were 251.9 and 3.4 events per 100 patient-years, respectively. Most of these AEs were mild to moderate. The most commonly reported TEAEs were dyslipidaemia (EAIR = 12.3) and viral upper respiratory tract infection (EAIR = 11.0). There were no cases of reactivation of hepatitis B or tuberculosis reported. Major adverse cardiovascular events including myocardial infarction (MI), stroke or cardiovascular death were reported in 1 patient (EAIR = 0.5). No cases of inflammatory bowel disease, including Crohn’s disease or ulcerative colitis, were reported. Uveitis was reported in 5 subjects (8.9%) (2 cases were de novo while 3 had flares out of the 14 patients who had pre-existing medical history of uveitis). The immunological assays were conducted for MEASURE 1 study and none of the Taiwanese patients had any treatment emergent anti-drug antibodies during the study. No cases of treatment-emergent suicidality-related AEs were reported during the entire treatment period.
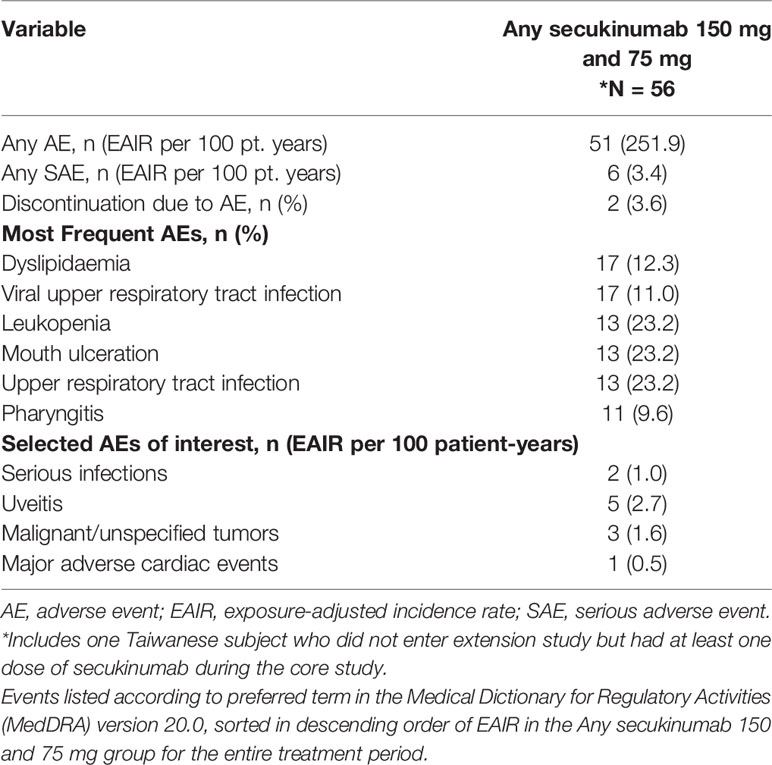
Table 3 Consolidated clinical safety for secukinumab doses during the entire treatment period.
Discussion
While the efficacy and safety of secukinumab in AS is well documented, (12, 16, 19–21) this is the first study demonstrating the long-term (4-year) efficacy, safety, and tolerability of secukinumab in Taiwanese patients with active AS.
These long-term results demonstrated that secukinumab 150 mg conferred sustained efficacy with a favorable and consistent safety profile over 4 years of treatment in Taiwanese patients with active AS. These findings are in line with previously reported 52-week pooled results in an Asian subpopulation from the MEASURE 1 and MEASURE 2 studies (12) and are consistent with previous long-term reports of the overall MEASURE 1 study population results (16–18, 22).
Overall, the study reported high retention rates. Clinical improvements were observed in ASAS20 response and in higher hurdle clinical endpoints, such as ASAS40, BASDAI50, and ASDAS-CRP ID with the approved secukinumab 150 mg dose that showed consistently better efficacy at most time points. These results were generally consistent with the overall population. Mean change from baseline in BASDAI score also showed higher improvement with secukinumab 150 mg than 75 mg at most time points. Improvements were also sustained in physical function (BASFI, SF-36 PCS), spinal mobility (BASMI), fatigue (FACIT-Fatigue) and QoL (ASQoL) over 4 years, further supporting the long-term efficacy of secukinumab in Taiwanese patients with AS. Following a protocol amendment, 48% of Taiwanese patients (13/27) receiving secukinumab 75 mg up-titrated to 150 mg during the study based on the clinical judgement of the treating physician, highlighting that 150 mg was a more efficacious dose for approximately half of the Taiwanese subpopulation.
The rates of SAEs and AEs leading to discontinuation were low with secukinumab. Secukinumab was well tolerated over 4 years with a safety profile consistent with previous reports in the overall MEASURE 1 population as well as pooled safety analysis of 21 secukinumab clinical trials across different indications (22, 23). Thus, the efficacy and safety of secukinumab in Taiwanese patients were observed to be similar to the non-Taiwanese population. The limitation of this post hoc analysis was the limited sample size and subsequent lack of statistical power to demonstrate the superiority of secukinumab versus placebo in Taiwanese patients. In addition, the MEASURE 1 study was not designed or powered to evaluate Taiwanese patients alone. Therefore, these results must be interpreted with caution.
In conclusion, secukinumab provided sustained long-term efficacy for the treatment of active AS in Taiwanese patients through 4 years. Secukinumab was well tolerated with a consistent safety profile through 4 years, with no new or unexpected safety risks identified. Overall, these long-term results support secukinumab 150 mg s.c. in the treatment of Taiwanese patients with active AS.
Data Availability Statement
The datasets generated and/or analyzed during the current study are not publicly available. Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved based on scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data may be requested from the corresponding author of the manuscript.
Ethics Statement
The studies involving human participants were reviewed and approved by institutional review board or independent ethics committee at each participating center. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this article, take responsibility for the integrity of the work as a whole, and were involved in the drafting and critical review of the manuscript. All authors agree to be accountable for all aspects of the work and attest to the accuracy and integrity of the work. All authors contributed to the article and approved the submitted version.
Funding
The clinical study was sponsored by Novartis Pharma AG, Basel, Switzerland, and designed by the scientific steering committee and Novartis personnel. Novartis funded the medical writing support.
Conflict of Interest
J-CT received honoraria for consulting or speaking for, or has received research grants from Pfizer, AbbVie, Jansen, UCB, Chugai, Roche, Bristol Myer Squibb (BMS), Eli Lilly, Novartis and Boehringer Ingelheim. JW received honoraria for consultation from TSH biopharm, Abbvie, BMS, Celgene, Chugai, Eisai, Janssen, Novartis, Pfizer, Sanofi-Aventis, and UCB pharma and research grant support from Abbvie, BMS, Celgene, Eli Lilly, Janssen, Novartis, Pfizer, and UCB. AD received honoraria for consulting or speaking for, or has received research grants from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myer Squibb (BMS), Eli Lilly, Glaxo Smith & Kline (GSK), Janssen, Novartis, Pfizer, and UCB. RM is an employee of Novartis with Novartis stock. BP is an employee of Novartis with Novartis stock. SM is an employee of Novartis with Novartis stock. ZT is an Employee of Novartis with Novartis stock.
Acknowledgments
The authors thank the patients who participated in this study, the study investigators, and John Gallagher (Novartis Pharmaceuticals UK Ltd.) for medical guidance and editorial support. Suchita Dubey (Novartis, India) provided medical writing support for this manuscript. The authors also thank Pin Su for coordination during the conduct of this study and Kok Yew Ngew for statistical support. This study was funded by Novartis Pharma AG, Basel, Switzerland in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.561748/full#supplementary-material
References
1. Braun J, Sieper J. Ankylosing spondylitis. Lancet (2007) 369(9570):1379–90. doi: 10.1016/S0140-6736(07)60635-7
2. Ward MM, Deodhar A, Akl EA, Lui A, Ermann J, Gensler LS, et al. American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network 2015 Recommendations for the Treatment of Ankylosing Spondylitis and Nonradiographic Axial Spondyloarthritis. Arthritis Rheumatol (Hoboken NJ) (2016) 68(2):282–98. doi: 10.1002/art.39298
3. Schett G, Coates LC, Ash ZR, Finzel S, Conaghan PG. Structural damage in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: traditional views, novel insights gained from TNF blockade, and concepts for the future. Arthritis Res Ther (2011) 13 Suppl 1(Suppl 1):S4–S. doi: 10.1186/1478-6354-13-S1-S4
4. Chou CT, Chen JM, Hsu CM, Chen SJ. HLA-B27 and its subtypes in 4 Taiwanese Aborigine tribes: a comparison to Han Chinese patients with ankylosing spondylitis. J Rheumatol (2003) 30(2):321–5.
5. Feltkamp TE, Mardjuadi A, Huang F, Chou CT. Spondyloarthropathies in eastern Asia. Curr Opin Rheumatol (2001) 13(4):285–90. doi: 10.1097/00002281-200107000-00007
6. Ho HH, Chen JY. Ankylosing spondylitis: Chinese perspective, clinical phenotypes, and associated extra-articular systemic features. Curr Rheumatol Rep (2013) 15(8):344. doi: 10.1007/s11926-013-0344-0
7. van der Heijde D, Ramiro S, Landewe R, Baraliakos X, Van den Bosch F, Sepriano A, et al. update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis (2017) 76(6):978–91. doi: 10.1136/annrheumdis-2016-210770
8. Cheung PP. Anti-IL17A in Axial Spondyloarthritis-Where Are We At? Front Med (Lausanne) (2017) 4:1. doi: 10.3389/fmed.2017.00001
9. Accortt NA, Bonafede MM, Collier DH, Iles J, Curtis JR. Risk of Subsequent Infection Among Patients Receiving Tumor Necrosis Factor Inhibitors and Other Disease-Modifying Antirheumatic Drugs. Arthritis Rheumatol (Hoboken NJ) (2016) 68(1):67–76. doi: 10.1002/art.39416
10. Sampaio-Barros PD, van der Horst-Bruinsma IE. Adverse effects of TNF inhibitors in SpA: are they different from RA? Best Pract Res Clin Rheumatol (2014) 28(5):747–63. doi: 10.1016/j.berh.2014.10.001
11. Zhang S, Li Y, Deng X, Huang F. Similarities and differences between spondyloarthritis in Asia and other parts of the world. Curr Opin Rheumatol (2011) 23(4):334–8. doi: 10.1097/BOR.0b013e32834640a9
12. Wei JC, Baeten D, Sieper J, Deodhar A, Bhosekar V, Martin R, et al. Efficacy and safety of secukinumab in Asian patients with active ankylosing spondylitis: 52-week pooled results from two phase 3 studies. Int J Rheum Dis (2017) 20(5):589–96. doi: 10.1111/1756-185x.13094
13. Jung SM, Kim HS, Kim HR, Kim NY, Lee JH, Kim J, et al. Immunogenicity of anti-tumour necrosis factor therapy in Korean patients with rheumatoid arthritis and ankylosing spondylitis. Int Immunopharmacol (2014) 21(1):20–5. doi: 10.1016/j.intimp.2014.04.006
14. Montiel PM, Solis JA, Chirinos JA, Casis B, Sanchez F, Rodriguez S. Hepatitis B virus reactivation during therapy with etanercept in an HBsAg-negative and anti-HBs-positive patient. Liver International: Off J Int Assoc Study Liver (2008) 28(5):718–20. doi: 10.1111/j.1478-3231.2007.01665.x
15. Navarra SV, Tang B, Lu L, Lin HY, Mok CC, Asavatanabodee P, et al. Risk of tuberculosis with anti-tumor necrosis factor-alpha therapy: substantially higher number of patients at risk in Asia. Int J Rheum Dis (2014) 17(3):291–8. doi: 10.1111/1756-185x.12188
16. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N Engl J Med (2015) 373(26):2534–48. doi: 10.1056/NEJMoa1505066
17. Braun J, Baraliakos X, Deodhar A, Baeten D, Sieper J, Emery P, et al. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann Rheum Dis (2017) 76(6):1070–7. doi: 10.1136/annrheumdis-2016-209730
18. Braun J, Baraliakos X, Deodhar A, Poddubnyy D, Emery P, Delicha EM, et al. Secukinumab shows sustained efficacy and low structural progression in ankylosing spondylitis: 4-year results from the MEASURE 1 study. Rheumatol (Oxford) (2019) 58(5):859–68. doi: 10.1093/rheumatology/key375
19. Baraliakos X, Kivitz AJ, Deodhar AA, Braun J, Wei JC, Delicha EM, et al. Long-term effects of interleukin-17A inhibition with secukinumab in active ankylosing spondylitis: 3-year efficacy and safety results from an extension of the Phase 3 MEASURE 1 trial. Clin Exp Rheumatol (2018) 36(1):50–5.
20. Marzo-Ortega H, Sieper J, Kivitz A, Blanco R, Cohen M, Delicha EM, et al. Secukinumab provides sustained improvements in the signs and symptoms of active ankylosing spondylitis with high retention rate: 3-year results from the phase III trial, MEASURE 2. RMD Open (2017) 3(2):e000592. doi: 10.1136/rmdopen-2017-000592
21. Pavelka K, Kivitz A, Dokoupilova E, Blanco R, Maradiaga M, Tahir H, et al. Efficacy, safety, and tolerability of secukinumab in patients with active ankylosing spondylitis: a randomized, double-blind phase 3 study, MEASURE 3. Arthritis Res Ther (2017) 19(1):285. doi: 10.1186/s13075-017-1490-y
22. Baraliakos X BJ, Deodhar A, Poddubnyy D, Kivitz AJ, Tahir H, van Den Bosch F, et al. Long-Term Evaluation of Secukinumab in Ankylosing Spondylitis: 5 Year Efficacy and Safety Results from a Phase 3 Trial [abstract]. Arthritis Rheumatol (2018) 70(suppl 10).
23. Deodhar A, Mease PJ, McInnes IB, Baraliakos X, Reich K, Blauvelt A, et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res Ther (2019) 21(1):111. doi: 10.1186/s13075-019-1882-2
Keywords: biologics, interleukin-17A inhibitor, Taiwan, secukinumab, ankylosing spondylitis
Citation: Tseng J-C, Wei JC-C, Deodhar A, Martin R, Porter B, McCreddin S and Talloczy Z (2020) Secukinumab Demonstrates Sustained Efficacy and Safety in a Taiwanese Subpopulation With Active Ankylosing Spondylitis: Four-Year Results From a Phase 3 Study, MEASURE 1. Front. Immunol. 11:561748. doi: 10.3389/fimmu.2020.561748
Received: 13 May 2020; Accepted: 27 October 2020;
Published: 26 November 2020.
Edited by:
Julia Kzhyshkowska, Heidelberg University, GermanyReviewed by:
Elizabeth Ann Repasky, University at Buffalo, United StatesSofia A. Casares, Naval Medical Research Center, United States
Copyright © 2020 Tseng, Wei, Deodhar, Martin, Porter, McCreddin and Talloczy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James Cheng-Chung Wei, amNjd2VpQGdtYWlsLmNvbQ==