
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 September 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.554725
 Gantsetseg Tumurkhuu1,2,3
Gantsetseg Tumurkhuu1,2,3 Shuang Chen2,4,5
Shuang Chen2,4,5 Erica N. Montano1,3
Erica N. Montano1,3 Duygu Ercan Laguna1,3Gabriela De Los Santos1,3Jeong Min Yu1Malcolm Lane2
Duygu Ercan Laguna1,3Gabriela De Los Santos1,3Jeong Min Yu1Malcolm Lane2 Michifumi Yamashita6Janet L. Markman2
Michifumi Yamashita6Janet L. Markman2 Luz P. Blanco7Mariana J. Kaplan7
Luz P. Blanco7Mariana J. Kaplan7 Kenichi Shimada2,4,5
Kenichi Shimada2,4,5 Timothy R. Crother2,4,5Mariko Ishimori1,5Daniel J. Wallace1,5
Timothy R. Crother2,4,5Mariko Ishimori1,5Daniel J. Wallace1,5 Caroline A. Jefferies1,3†
Caroline A. Jefferies1,3† Moshe Arditi2,4,5*†
Moshe Arditi2,4,5*†Systemic Lupus Erythematosus (SLE) is a chronic inflammatory autoimmune disease in which type I interferons (IFN) play a key role. The IFN response can be triggered when oxidized DNA engages the cytosolic DNA sensing platform cGAS-STING, but the repair mechanisms that modulate this process and govern disease progression are unclear. To gain insight into this biology, we interrogated the role of oxyguanine glycosylase 1 (OGG1), which repairs oxidized guanine 8-Oxo-2′-deoxyguanosine (8-OH-dG), in the pristane-induced mouse model of SLE. Ogg1−/− mice showed increased influx of Ly6Chi monocytes into the peritoneal cavity and enhanced IFN-driven gene expression in response to short-term exposure to pristane. Loss of Ogg1 was associated with increased auto-antibodies (anti-dsDNA and anti-RNP), higher total IgG, and expression of interferon stimulated genes (ISG) to longer exposure to pristane, accompanied by aggravated skin pathology such as hair loss, thicker epidermis, and increased deposition of IgG in skin lesions. Supporting a role for type I IFNs in this model, skin lesions of Ogg1−/− mice had significantly higher expression of type I IFN genes (Isg15, Irf9, and Ifnb). In keeping with loss of Ogg1 resulting in dysregulated IFN responses, enhanced basal and cGAMP-dependent Ifnb expression was observed in BMDMs from Ogg1−/− mice. Use of the STING inhibitor, H151, reduced both basal and cGAMP-driven increases, indicating that OGG1 regulates Ifnb expression through the cGAS-STING pathway. Finally, in support for a role for OGG1 in the pathology of cutaneous disease, reduced OGG1 expression in monocytes associated with skin involvement in SLE patients and the expression of OGG1 was significantly lower in lesional skin compared with non-lesional skin in patients with Discoid Lupus. Taken together, these data support an important role for OGG1 in protecting against IFN production and SLE skin disease.
SLE, a chronic systemic inflammatory autoimmune disease, occurs in about 1.8–7.6 people per 100,000 in the United States, with a 9:1 female to male ratio (1). Patients with SLE have a diverse range of immunological abnormalities that contribute to disease progression and pathology. Despite the heterogeneity of the disease, type I interferons (IFN) have emerged as key pathogenic cytokines in SLE and correlate with disease severity (2, 3). One source of extracellular DNA that initiates and drives type I IFN production is genomic DNA released from dead and dying cells, which is recognized by TLR9 in endosomes, driving IFNα expression. More recently, oxidized DNA from mitochondria (driven by ROS production) has also been recognized as a trigger for type I IFN production. In SLE, low density neutrophils/granulocytes are an important source of oxidized mitochondrial DNA (mtDNA), which is released from activated neutrophils as they undergo NETosis. Indeed, NETs are enriched in oxidized mtDNA, specifically 8-OH-dG motifs (4, 5). In addition, exposure to ultravliolet light can also drive oxidative stress, oxidized DNA damage, and accumulation of 8-OH-dG lesions in the skin. Oxidized DNA, including 8-OH-dG, is sensed by the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), which has recently emerged as an important contributor to elevated type I IFN in SLE (6, 7). Once activated, cGAS generates 2′3′-cyclic-dinucleotides which bind and activate the ER-resident adaptor protein stimulator of IFN genes (STING), and drive TBK1-IRF3 dependent induction of IFNβ (7–10).
Mechanisms regulating clearance of cytosolic DNA and RNA, including oxidized and damaged DNA, are critical to preventing triggering of IFN responses. OGG1 (oxoguanisine glycolase 1) is a DNA repair enzyme in the base excision repair (BER) pathway that excises and repairs 8-OH-dG DNA lesions (11). A single nucleotide polymorphism (SNP) in OGG1 is associated with development of lupus nephritis (12). This SNP encodes a serine-to-cysteine substitution at position 326 (S326C), which is thought to reduce function of OGG1 (13), thus conferring susceptibility to lupus nephritis by enabling accumulation of 8-OH-dG (4, 14–16).
Given that OGG1 plays an important role in protecting against accumulation of oxidized DNA lesions, we hypothesized that loss of Ogg1 may contribute to IFN-driven disease. We therefore investigated the role of OGG1 in the pristane-induced lupus (PIL) mouse model of IFN-driven SLE, which recapitulates numerous human SLE manifestations, including elevated type I IFNs, autoantibody generation, arthritis, and severe glomerulonephritis (17, 18). In this study, loss of Ogg1 resulted in increased oxidized DNA damage, enhanced recruitment of Ly6Chi monocytes and ISG expression in the peritoneal cavity, enhanced neutrophil activity, and systemic IFN-driven responses. Intriguingly, however, rather than increased kidney pathology, loss of Ogg1 resulted in cutaneous involvement in PIL, accompanied by enhanced IFN driven gene expression in the skin. Mechanistically, we observed enhanced signaling through the cGAS-STING pathway in Ogg1−/− bone marrow derived macrophages (BMDMs), an effect inhibited completely by blocking signaling through STING. In translating these results to human disease, we found decreased OGG1 expression in SLE patients associated with cutaneous involvement, and that lesional skin from patients with chronic cutaneous lupus erythematosus had reduced OGG1 expression compared to non-lesional areas. Taken together, our results indicate that OGG1 protects against IFN induction and cutaneous involvement in SLE by reducing 8-OH-dG driven IFN responses.
Wild-type C57BL/6 and Ogg1−/− mice (C57BL/6 background), female, 6–8 weeks old, received a single i.p. injection of 0.5 ml of pristane (2,6,10,14-Tetramethylpentadecane (TMPD), Sigma). Some mice were sacrificed at 1 week after pristane. Mice were bled at 7 months after pristane inoculation for complete blood cell count analysis. All mice were monitored for proteinuria by dipstick test once a month and sacrificed at 10 months after pristane treatment.
The following conjugated anti-mouse antibodies were used: anti-Ly-6G (1A8), anti-CD11b (M1/70), anti-Ly-6C (ER-MP20), anti-F4/80 (BM8), anti-CD4 (GK1.5), anti-CD8a (53–6.7), anti-CD11c (3.9), and anti-B220 (RA3-6B2) (eBiosciences). Cells were incubated in CD16/32 (Fc block; BD Biosciences) prior to staining. Cells were acquired on LSR II (BD Biosciences) and analyzed with the FlowJo software (Treestar).
Serum IgG subtypes (IgG1 and IgG2a) were measured by ELISA using the following coating antibodies: purified anti-mouse IgG1 and IgG2a and detection antibodies: biotin anti-mouse IgG1 and IgG2a (BD Pharmingen, San Jose, CA). Following Streptavidin-HRP (Sigma) incubation, the ELISA was developed with TMB substrate (Dako). The optical density (OD) for each well with a microplate reader set to 450 nm. MCP-1 and s100a8/9 levels were measured via ELISA according to the manufacturer's instruction (R&D). Total serum IgG and anti-dsDNA and anti-RNP Ab levels were quantified by ELISA using commercially available kits (Alpha Diagnostic International, San Antonio, TX), following manufacturer's protocols. An anti-mouse albumin ELISA kit was used to measure urine proteinuria (Bethyl labs).
Quantitative PCR (Q-PCR) was performed as previously described (19, 20). In brief, total RNA was extracted from 106 peritoneal cells using TRIzol reagent (Invitrogen, Carlsbad, CA), and cDNA was synthesized using the (Invitrogen) according to the manufacturer's protocol. qPCR was performed on a CFX96 Real-Time PCR Detection System (Biorad). A melting-curve analysis was performed to ensure specificity of the products. Gene expression was normalized to GAPDH, and expression relative to the PBS treatment was calculated using the ddCt method (18) Primer sequences are listed as follows:
mIsg15 Fw: GGTGTCCGTGACTAACTCCAT
mIsg15 Rv: CTGTACCACTAGCATCACTGTG
mMx1 Fw: GATCCGACTTCACTTCCAGATGG
mMx1 Rv: CATCTCAGTGGTAGTCAACCC
mIrf9 Fw: TGTCTGGAAGACTCGCCTAC
mIrf9 Rv: GCAACATCCATACGACCTCTCT
mIfnb Fw: CAGCTCCAAGAAAGGACGAAC
mIfnb Rv: GGCAGTGTAACTCTTCTGCAT
mOgg1 Fw: tgtgtaccgaggagacgaca
mOgg1 Rv: ctgtgccaggctgacatcta
hOGG1 Fw: CACACTGGAGTGGTGTACTAGC
hOGG1 Rv: CCAGGGTAACATCTAGCTGGAA
NETosis was measured with or without stimulation with the ionophore A23187 for 0, 60, and 120 min in bone marrow derived neutrophils (BMDN). Sytox (ST) measures external DNA and Pico Green (PG) measures total DNA and ratio ST/PG was calculated.
Skin samples from the shaved dorsal neck region were fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin. Frozen sections were prepared and stained with anti-mouse IgG-AF550 conjugated antibody. Kidneys were fixed in paraformaldehyde, embedded in paraffin, and sections were stained with H&E and periodic acid-Schiff (PAS). Kidney sections were scored by pathologist in a blinded fashion to determine a glomerular and interstitial inflammation score. Briefly, a mean glomerular score was calculated for each mouse by grading injury in fifty glomeruli. Glomeruli were scored as normal = 0, mesangial expansion =1, endocapillary proliferation = 2, capillaritis or necrotic changes = 3, or crescents = 4. The interstitial score was determined by examining 50 high power fields and scoring the interstitial inflammation on a scale from 0 to 4 as absent, involving <25%, 25–50%, or >50% of the interstitium. For immunofluorescence, sections were prepared from frozen kidneys in OCT (TissueTek). Glomerular immune complexes in kidney sections were detected by staining with Dylight 488 labeled goat anti-mouse IgG (minimal x-reactivity, Biolegend). Using the image analysis software, deposition of IC was scored by pathologist.
Cohorts of WT and Ogg1−/− mice were followed over time, and the onset of macroscopic skin disease was recorded along with the ages of the animals. Mice were considered affected when an area >0.5 cm of hair loss and ulceration was noted.
All animal experiments were performed according to the guidelines and approved protocols (IACUC #8299) of the Cedars-Sinai Medical Center Institutional Animal Care and Use Committee. Cedars-Sinai Medical Center is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC International) and abides by all applicable laws governing the use of laboratory animals. Laboratory animals are maintained in accordance with the applicable portions of the Animal Welfare Act and the guidelines prescribed in the DHHS publication, Guide for the Care, and Use of Laboratory Animals.
As described previously (21), all SLE patients (as per ACR diagnostic criteria) were recruited from Cedars-Sinai Medical Center, CA, USA. Age- and sex-matched healthy donors who had no history of autoimmune diseases or treatment with immunosuppressive agents were included. All participants provided informed written consent and the study received prior approval from the institutional ethics review board.
Peripheral blood mononuclear cells (PBMCs) were separated from whole blood by density-gradient centrifugation with Ficoll-Paque Plus (GE Healthcare). CD14+ monocytes were purified from fresh PBMCs by positive selection using magnetic CD14+ beads (Miltenyi Biotec) according to manufacturer's protocol.
All data are expressed as means ± SD. Statistical differences were measured using either an unpaired Student's t-test or 2-way analysis of variance (ANOVA) when appropriate with Bonferroni post-hoc test. Normality of data was assessed via a Shapiro–Wilk normality test. When the data analyzed was not distributed normally, we used the Mann–Whitney test or Kruskal–Wallis 1-way ANOVA with Dunn's post-hoc test. Data analysis was performed using Prism software version 7.0a (GraphPad, San Diego, CA). A P-value of < 0.05 was considered statistically significant. Asterisks in the figures represent the following: *P < 0.05; **P < 0.01; ***P < 0.001, and ****P < 0.0001.
To determine whether loss of Ogg1 exacerbated disease progression in PIL, we compared C57Bl/6 (WT) and Ogg1−/− mice 7 days after pristane treatment. Analysis of peritoneal lavage demonstrated that pristane treatment resulted in increased recruitment of inflammatory CD11b+Ly6Chi monocytes and expression of Siglec1, an IFN inducible gene, in Ogg1−/− mice compared to WT mice, whereas neutrophil (CD11b+Ly6G+) recruitment was similar across both genotypes (Figures 1A–D). In addition, significantly higher level of 8-OH-dG was determined in peritoneal lavage from Ogg1−/− mice compared to WT mice (Figure 1E). Taken together, lack of Ogg1 resulted in enhanced expression of IFN stimulated genes (ISGs)—Ifnb, Oas1, and Ip10 (Figure 1F).
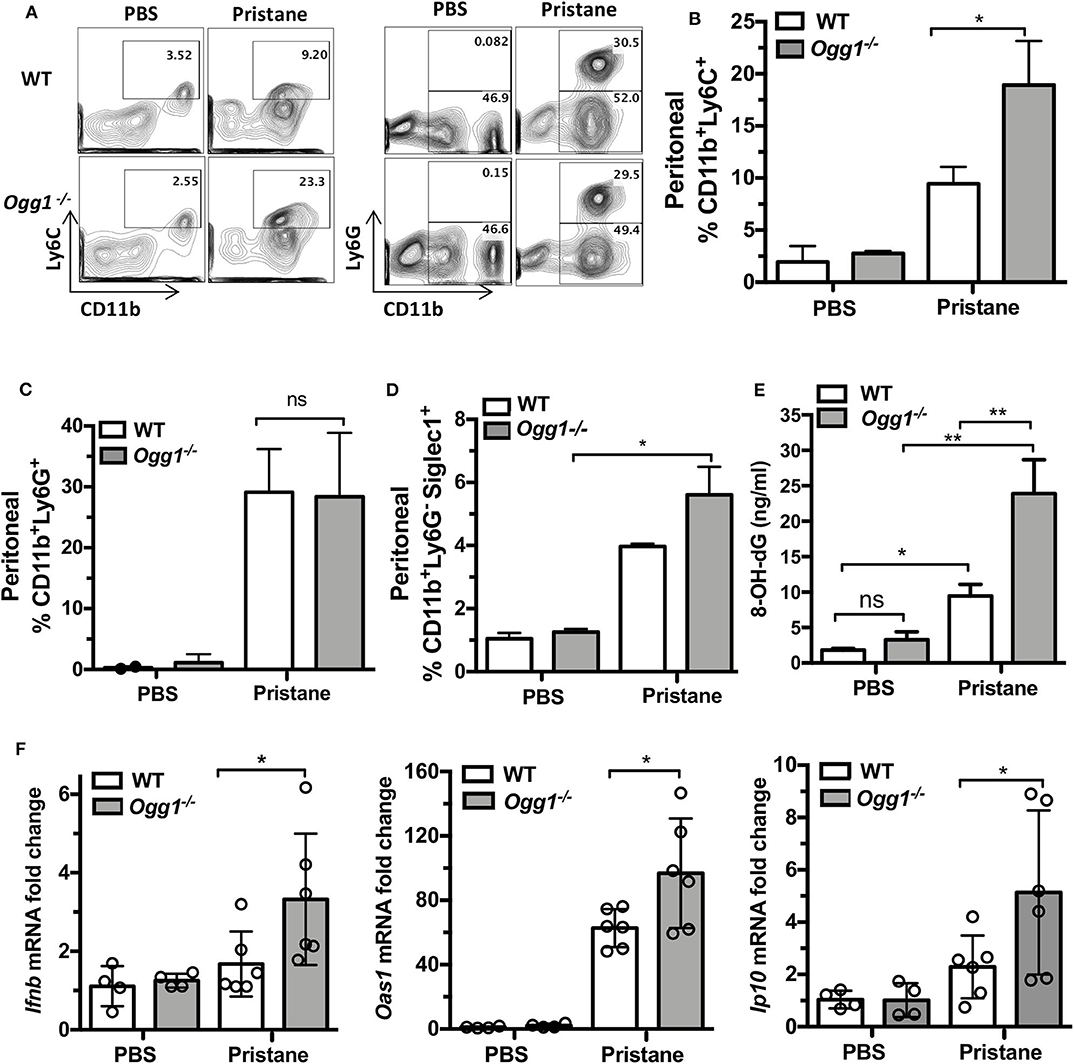
Figure 1. Early inflammatory response to pristane in WT and Ogg1−/−mice. (A) Representative pictures of FACS analysis of peritoneal cells day 7 after pristane. (B) % CD11bLy6C+ population, (C) % CD11bLy6G+ population, (D) Siglec1+ cells in CD11bLy6G− population. All data represent mean ± SD. (E) 8-OH-dG ELISA peritoneal lavage. (F) The expression of mRNA for IFN signature genes (Ifnb, Oas1, and Ip10) in peritoneal cells. Statistical significance was determined using Two-Way ANOVA with Bonferroni post-hoc test. *p < 0.05, **p < 0.01.
To determine the role of OGG1 in organ damage and SLE pathogenesis, we compared C57Bl/6 (WT) to Ogg1−/− mice 10 months after pristane injection. In female C57Bl/6 mice, pristane treatment significantly elevated the level of 8-OH-dG in urine 10 months after injection, indicating that pristane induced oxidative DNA damage (Figure 2A). Furthermore, Ogg1 expression was significantly lower in peritoneal cells from pristane-treated mice than untreated mice (Figure 2B), indicating that Ogg1 downregulation may permit the accumulation of oxidative DNA damage in PIL mice. In keeping with our analysis of the acute effects of pristane on IFN-inducible responses in Ogg1−/− mice we observed that expression of IFN stimulated genes (Irf9, Mx1, and Isg15) were significantly higher in the peritoneal cells of Ogg1−/−mice post pristane injection than in those of WT mice (Figure 2C), indicating a sustained systemic response to pristane treatment in the Ogg1−/− mice. In order to assess the level of lupus-like systemic inflammatory responses induced by pristane, we measured circulating levels of anti-dsDNA and anti-RNP autoantibodies, and found that all were substantially higher in Ogg1−/− mice than WT mice 10 months after pristane exposure (Figures 2D,E), whereas total IgG, IgG1, and IgG2a and other autoantibodies showed no difference between genotypes (Supplementary Figures 1A–D). Rather surprisingly whilst both pristane-treated WT and Ogg1−/− mice exhibited splenomegaly 10 months post-pristane treatment compared with untreated mice, with no statistically significant difference observed between treated WT or Ogg1−/− groups (Figure 3A). The splenomegaly observed was mirrored by a significant increase in the number of splenocytes (Figure 3B) and in circulating neutrophil numbers (as determined by complete blood count, Figure 3C and Supplementary Figure 2C) in pristane-injected WT and Ogg1−/− mice, again with no statistically significant difference observed between treated WT or Ogg1−/− groups. Similar changes were observed with respect to neutrophils (CD11b+Ly6G+) and inflammatory monocytes (CD11b+Ly6Chi) recruited to the peritoneal cavity following pristane treatment of both genotypes (Supplementary Figures 2A,B), whilst levels of S100 calcium binding protein A8/9 (s100a8/9, known as calprotectin), a potent chemotactic agent for neutrophils and monocytes both in vitro and in vivo (19), and MCP1 were similarly changed with no significant differences between treated groups (Supplementary Figures 2D,E). However, despite the lack of significant differences in absolute numbers between WT and Ogg1 KO treated groups, functionally lack of Ogg1 modified neutrophil function. Robust NET formation was observed following A23187 ionophore stimulation of bone-marrow derived neutrophils (BMDN) from both pristane-treated WT and Ogg1−/−mice. However, neutrophils from Ogg1−/− mice displayed altered kinetics, with NET formation starting earlier and being more pronounced (although not significantly) in Ogg1−/−BMDN compared with WT BMDN (Figure 3D). Overall, these results suggest that lack of Ogg1 leads to increased responses to 8-OH-dG, anti-dsDNA antibodies, and enhanced IFN responses in the pristane model.
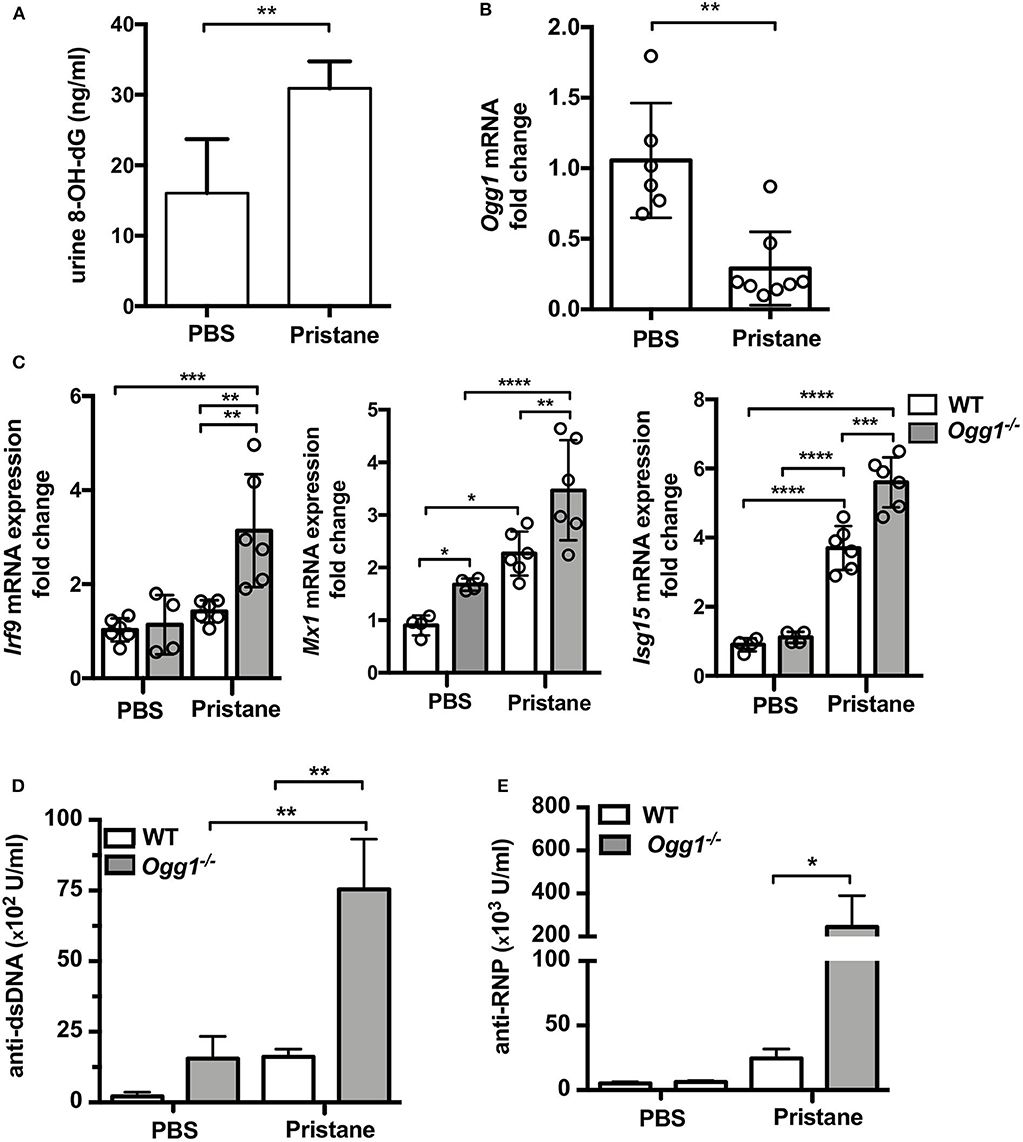
Figure 2. Increased inflammatory response to pristane in Ogg1−/− mice. (A) 8-OH-dG secretion in spot urine in WT mice with or without pristane. n = 3–4/group (B) Ogg1 mRNA expression in peritoneal cells in WT mice with or without pristane. n = 6–8/group (C) The expression of mRNA for IFN signature genes (Irf9, Mx1, and Isg15) in peritoneal cells. (D,E) The level of auto antibody, anti-dsDNA, and anti-RNP, in serum by ELISA. n = 10–15/group. All data represent mean ± SD. Statistical significance was determined using Two-way ANOVA with Bonferroni post-hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
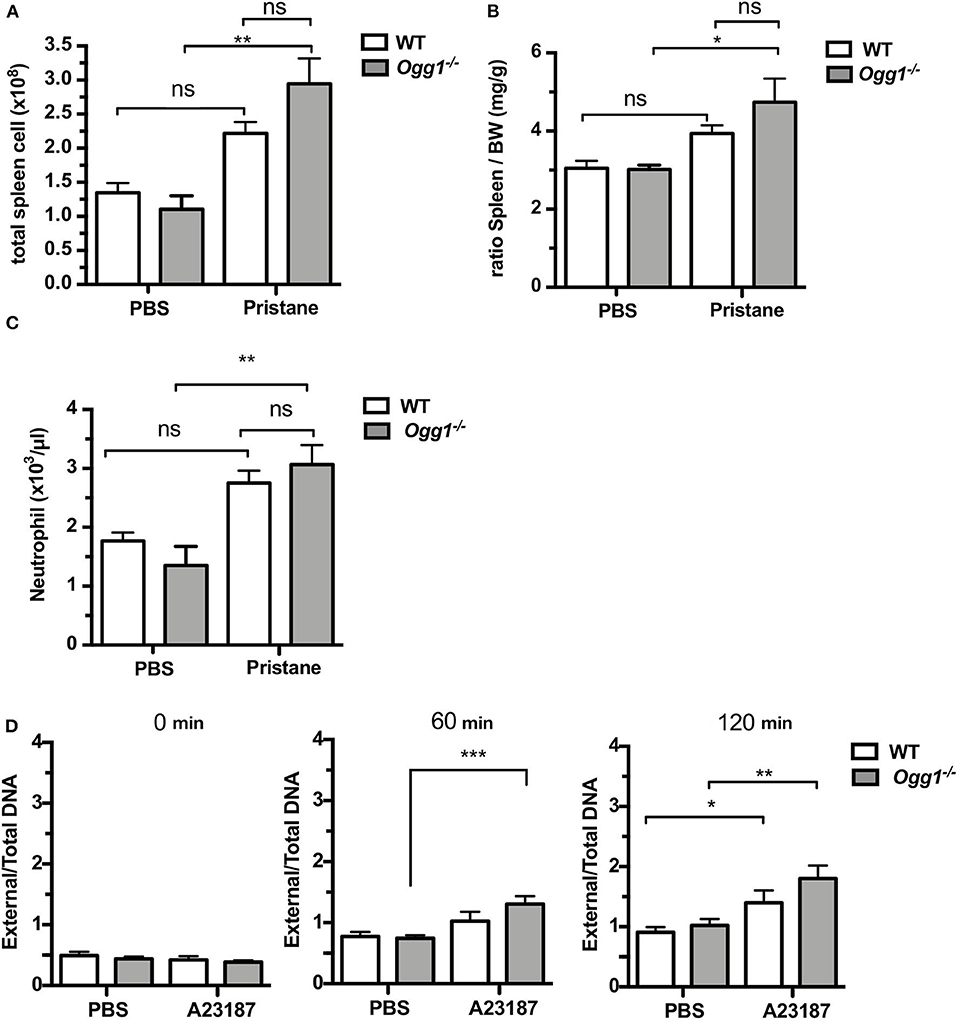
Figure 3. Increased inflammatory response to pristane in Ogg1−/−mice. (A,B) ratio of spleen to BW and Total cell number of spleen. (C) neutrophil counts by complete blood cell count (CBCC) analysis in peripheral blood. n = 4–6 per group (D) NETosis in A23187 stimulated BMDN obtained from pristane treated WT and Ogg1−/− mice. n = 4 in triplicates. All data represent mean ± SD. Statistical significance was determined using Two-Way ANOVA with Bonferroni post-hoc test. ns, not significant. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine the effect of Ogg1-deficiency on glomerulonephritis in this model of lupus, we measured proteinuria. None of the mice from untreated or treated groups scored 3+, although Ogg1−/−mice treated with pristane showed a non-significant trend toward increased pathological score (Supplementary Figure 3A). IgG deposition within the glomeruli was enhanced in both pristane-treated WT and Ogg1−/− mice compared with their untreated controls (Supplementary Figure 3B). We next scored kidney sections stained with PAS and Mesangial expansion/hypercellularity (M.E.) and Endocapillary Hypercellularity (H.E.) indices, which approximate the scoring system of human lupus nephritis. Both M.E and H.E were significantly increased in both pristane-treated WT and Ogg1−/− mice compared with their control groups, with a non-significant trend toward increased pathology in Ogg1−/− mice compared with WT (Supplementary Figure 3C). These results indicate that loss of Ogg1 may only play a minor role in the development of pristane induced lupus-like glomerulonephritis.
Surprisingly, we observed increased hair loss and differences in skin pathology in the Ogg1−/− compared to WT. Starting around 3–3.5 months of age, both untreated and treated Ogg1−/− mice lost hair in the dorsal neck area, whereas WT mice did not. In pristane-treated Ogg1−/− mice, hair loss spread to the frontal, and dorsal part of entire trunk, reaching the hind legs by 10 months of age (Figure 4A and Supplementary Figure 4A). Histopathological analysis of H&E stained sections revealed epidermal hyperplasia, consisting of acanthosis and hyperkeratosis, in pristane-treated Ogg1−/− mice (Figure 4B). Analysis of IgG deposition in the skin from the dorsal neck area revealed that skin of Ogg1−/− mice had visible IgG deposition, whereas WT had none (Figure 4C). These results indicate that Ogg1 deficiency promotes the development of pristane-induced skin disease.
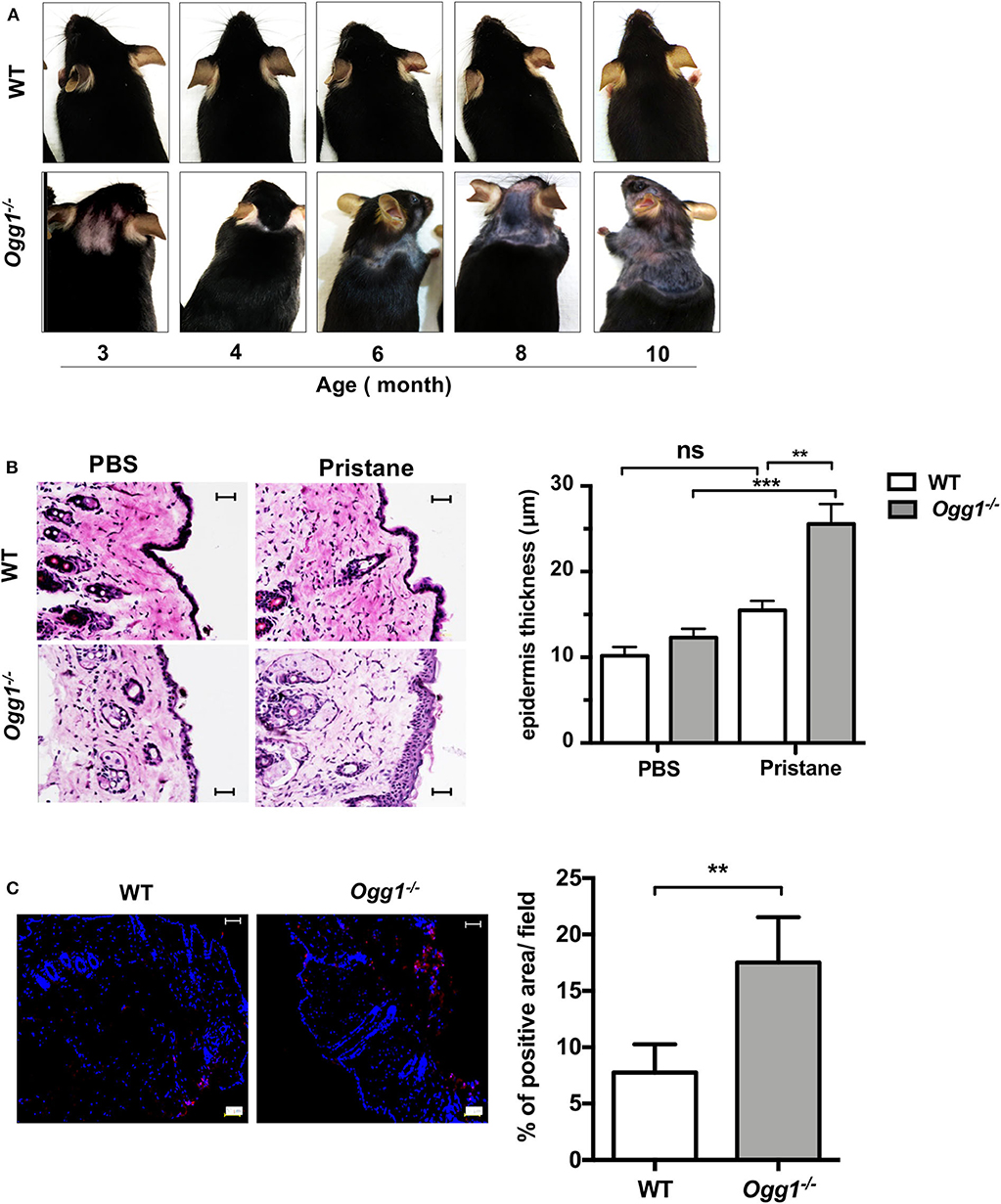
Figure 4. Ogg1 deficiency exacerbates skin changes in pristane treated mice. (A) Representative pictures of hair loss observed over the indicated time periods in pristane treated WT and Ogg1−/−mice. (B) Representative pictures of H&E staining of skin from dorsal neck area. Quantification of epidermal thickness. Scale bar = 20 μm. n = 10–12/group (C) Representative pictures and quantification of IgG deposition of dorsal neck area of skin of pristane treated WT and Ogg1−/−. n = 4/group. All data represent mean ± SD. Statistical significance was determined using Two-Way ANOVA with Bonferroni post-hoc test in panel (B) and two tails Student's t-test in panel (C). ns, not significant. **p < 0.01, ***p < 0.001.
To assess whether type I IFN may play a role in skin pathology in the pristane-treated Ogg1−/− mice, we measured IFN-stimulated gene expression in dorsal skin from pristane-treated mice. Pristane treatment induced significantly higher expression of Ifnb and type I IFN signature genes (Irf9 and Isg15) in dorsal neck skin of Ogg1−/−mice than WT mice (Figure 5A). Given the recently reported role of the cGAS-STING pathway in both recognition of 8-OH-dG DNA lesions and cutaneous LE (20), we interrogated the role of STING downstream of 8-OH-dG recognition in Ogg1−/− mice. Treatment of WT and Ogg1−/− BMDMs with 50 μM menodione to induce 8-OH-dG (22), resulted in enhanced Ifnb expression in Ogg1−/− BMDMs compared to controls (Figure 5B). Interestingly, menodione also amplified the response to cGAMP in Ogg1−/− BMDMs, indicating that loss of Ogg1 primed the cGAS-STING pathway. Confirming a role for STING in the enhanced Ifnb expression observed in Ogg1−/− BMDMs, the STING inhibitor H151 (1 μM), completely blocked menodione- and cGAMP-induced Ifnb expression in Ogg1−/− BMDM (Figure 5C). As a control, we did not observe any changes in polyIC induced Ifnb expression in H151-pretreated cells compared to non-treated cells (Supplementary Figure 5). Taken together, these data indicated that the cGAS-STING pathway may be responsible for increased type I IFN expression in Ogg1−/− cells.
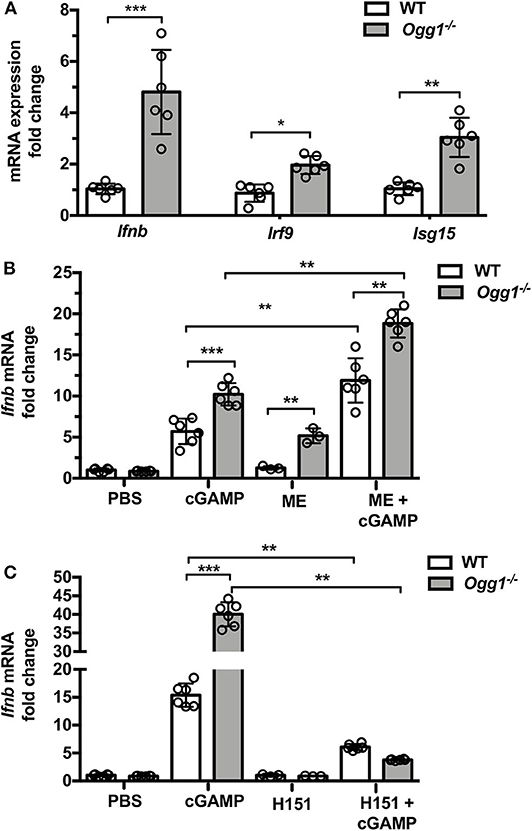
Figure 5. Loss of Ogg1 results in enhanced STING-driven type I IFN response. (A) The expression of mRNA for IFN signature genes (Ifnb, Irf9, and Isg15) affected area of skin, fold changes were Ogg1−/− WT. (B) BMDM from WT and Ogg1−/−mice, were stimulated with cGAMP with or without 50 μM menodione—ME (ROS inducer) pretreatment. Ifnb mRNA was measured by RT-PCR. (C) Ifnb mRNA in BMDM from WT and Ogg1−/−mice, were stimulated with cGAMP with or without H151 (STING inhibitor). n = 5–6 per group. All data represent mean ± SD. Statistical significance was determined using two tails Student's t-test (A) Two-way ANOVA with Bonferroni post-hoc test (B,C). *p < 0.05, **p < 0.01, ***p < 0.001.
To determine how our observations of cutaneous involvement in pristane-treated Ogg1−/− mice translated to human SLE, we looked at potential clinical associations of OGG1 expression in our SLE patient cohort (21). This cohort had been previously screened for increased expression of ISGs in peripheral blood mononuclear cells as shown in Smith et al. (21). Monocytes from these patients with skin and lung involvement expressed lower levels of OGG1 than monocytes from healthy controls, whereas no such decrease was observed in monocytes from patients with other organ involvement (Figure 6A). Analysis of gene expression profiling conducted by Werth et al. (23) on paired skin samples from lesional and non-lesion skin from patients with chronic cutaneous (Discoid) Lupus Erythematosus (DLE) showed that expression of OGG1 was significantly reduced in lesional skin compared with non-lesional skin (adj. p = 3.02 × 10−3; data accessible at NCBI GEO database (24, 25), accession GSE100093) (Figure 6B). Taken together, our data demonstrate that expression of OGG1 in SLE is negatively associated with skin damage observed in patients. This suggests that reduced expression or loss of OGG1 promotes inflammation and IFNβ production via recognition of oxidized DNA such as 8-OH-dG by the cGAS-STING pathway, contributing to both localized responses in the skin and systemic inflammation (Figure 6C).
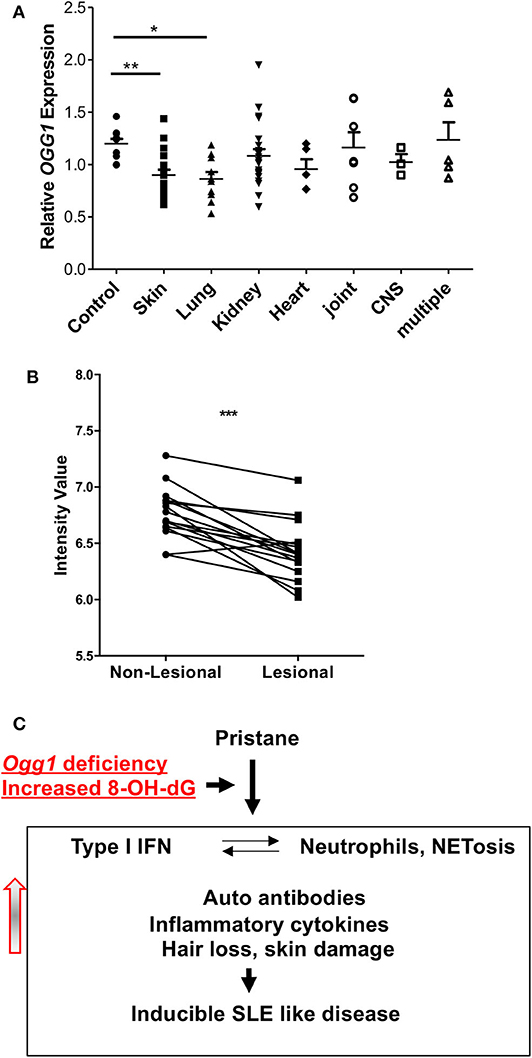
Figure 6. OGG1 expression in human patients. (A) OGG1 mRNA expression of PBMC of healthy control and SLE patients with different organ involvements. (B) OGG1 expression from paired lesional and non-lesional skin samples (n = 16) from patients with Discoid Lupus Erythematosus (DLE) (Data available at NCBI GEO database, accession GSE100093). Values are reported as mRNA expression levels, i.e., normalized fluorescent intensity. Lines indicate relationship between paired samples while *** indicates the difference is significant between the two groups at p < 0.00001 (Paired Student's t-test). (C) The proposed mechanism for increased inflammatory response to pristane in due to the loss of OGG1. *p < 0.05, **p < 0.01.
In this study, we explored the contribution of OGG1 to the pathogenesis of pristane-induced lupus. Deficiency of Ogg1 resulted in enhanced immune responses to pristane, namely increased recruitment of Ly6Chi inflammatory monocytes to the peritoneal cavity, enhanced NETosis, systemic expression of type I IFN signature genes, higher levels of autoantibodies and skin damage. Gene expression analysis showed decreased Ogg1 mRNA level in DLE lesions and in PBMCs from SLE patients with skin involvement. This demonstrates the importance of oxidized DNA repair enzyme OGG1 in protecting against oxidized DNA-induced IFN responses in lupus.
OGG1 has two isoforms: nuclear and mitochondrial (11). Multiple approaches have demonstrated that mitochondrial OGG1 is crucial for maintenance cell survival, mitochondrial function, and energetics (26, 27). Recognition and repair of 8-OH-dG lesions is carried out by the mitochondrial isoform, and we previously showed that Ogg1−/− BMDMs were more sensitive to menadione-induced mtDNA oxidative damage, and that inflammation caused by oxidized DNA damage and dyslipidemia-induced metabolic stress resulted in accelerated atherosclerosis in Ogg1−/−Ldlr−/− mice (22).
It is well-known that the catalytic activity of OGG1 is decreased in the setting of oxidative stress, resulting in an accumulation of 8-OH-dG (28). In turn, the accumulation of oxidized DNA drives both inflammation and induction of type I IFNs, which are thought to be the principal drivers of SLE. Our results show that Ogg1 deficiency results in increased proinflammatory monocytes and neutrophils, which are the main source of type I IFN in the pristane model. Abnormal accumulation of self DNA or oxidized DNA in the cytosol engages the DNA sensor cGAS, and promotes STING-IRF3-dependent signaling to elevate interferon-stimulated gene expression, potentiating type I IFN responses (29, 30). Indeed, recent evidence points to STING-mediated pathways as being overactivated in SLE patients (31). Here, the results of STING activator and inhibitor experiments (Figure 5) demonstrate that STING is essential for an increased expression of type I IFN in Ogg1−/− mice. During the past decade, significant progress has been made to elucidate a role for aberrant DNA repair in the development of lupus. Recent evidence indicates that patients with SLE have higher levels of DNA damage than normal subjects, and that polymorphisms in genes involved in the preservation of the genomic stability increase the risk for SLE (32–34). Furthermore, experience from animal models reinforces the importance of defective repair in the development of SLE-like disease (35), suggesting that therapeutic potential of targeting DNA damage and DNA repair responses in SLE pathogenesis (36, 37). Indeed, oxidized DNA has a greater immunostimulatory capacity to stimulate type I IFN than does normal DNA, possibly due to increased resistance to degradation by the 3′ repair exonuclease 1 TREX 1 (38). Our results show that the ROS inducer menodione more potently induces type 1 IFN expression from Ogg1−/− BMDMs, in keeping with the model that lack of Ogg1 results in accumulation of oxidized DNA, driving a more potent activation of the STING-dependent cytoplasmic DNA-sensing pathway.
There are 21 known polymorphisms in OGG1, 13 of which are associated with disease due to reduced capacity to repair DNA damage (13, 39, 40). In addition, OGG1 polymorphisms have been linked to SLE risk. For example, the OGG1 SNP rs1052133 is associated with the development of lupus nephritis and an observed increase of 8-OH-dG levels in plasma (12). Interestingly, however, we observed no exacerbated kidney pathology in the Ogg1−/− mice in response to pristane treatment. Instead, we find that loss of Ogg1 exacerbates pristane-induced skin pathology and increased expression of IFN stimulated genes in the skin of Ogg1−/− mice. Although the exact mechanism of skin involvement in SLE is unknown, ultraviolet light (UV), immune cells, cytokines, and deposition of immunoglobulins are all known to have a role in the development of skin inflammation in SLE (41–43). Recent studies also suggest a pathogenic role of endogenous nucleic acids in SLE with cutaneous involvement (44, 45). 8-OH-dG is abundant in skin lesions in SLE patients with cutaneous involvement, and colocalizes with an IFN gene signature (44, 46), suggesting that oxidized DNA damage is an important driver of pathology. In support of this finding, injection of oxidized DNA into the skin of lupus-prone MRL/Ipr mice induces lesions similar to those observed in patients (38). In keeping with these observations and consistent with our findings in the Ogg1−/− mice, we find that OGG1 expression is reduced in SLE patients with skin involvement and that OGG1 expression is decreased in skin lesions obtained from DLE patients compared to non-lesional matched samples. Taken together, our results indicate that OGG1 protects against IFN induction and cutaneous involvement in SLE by reducing 8-OH-dG-driven IFN reponses, via a mechanism that involves cGAS-STING mediated IFNβ production.
All datasets generated for this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Cedars Sinai ethics review board. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Cedars-Sinai Medical Center Institutional Animal Care and Use Committee.
GT, SC, MJK, KS, and TRC designed the experiments and conceived the overall research plan. GT, ENM, DEL, GD, JY, ML, LPB, JLM, and MY conducted the experiments and analyzed results. DJW and MI enrolled patients. GT and CAJ wrote the manuscript. All authors contributed to manuscript preparation. CAJ and MA contributed to supervision of the work.
This work was supported by funding from NIH (grant R21AI124211) to MA and CAJ, from Arthritis Foundation (AF2017-433570) to CAJ, AHA postdoctoral fellowship (16POST31010034) to GT and NIAMS/NIH (ZIAAR041199) to MJK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.554725/full#supplementary-material
1. Stojan G, Petri M. Epidemiology of systemic lupus erythematosus: an update. Curr Opin Rheumatol. (2018) 30:144–50. doi: 10.1097/BOR.0000000000000480
2. Kirou KA, Mavragani CP, Crow MK. Activation of type I interferon in systemic lupus erythematosus. Expert Rev Clin Immunol. (2007) 3:579–88. doi: 10.1586/1744666X.3.4.579
3. Rönnblom L. The type I interferon system in the etiopathogenesis of autoimmune diseases. UPSALA J Med Sci. (2011) 116:227–37. doi: 10.3109/03009734.2011.624649
4. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
5. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. doi: 10.1084/jem.20151876
6. An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:800–7. doi: 10.1002/art.40002
7. Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. (2015) 112:E5699–705. doi: 10.1073/pnas.1516465112
8. Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. (2013) 341:903–6. doi: 10.1126/science.1240933
9. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. (2013) 39:1019–31. doi: 10.1016/j.immuni.2013.10.019
10. Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. (2014) 35:88–93. doi: 10.1016/j.it.2013.10.010
11. Boiteux S, Radicella JP. The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch Biochem Biophys. (2000) 377:1–8. doi: 10.1006/abbi.2000.1773
12. Lee HT, Lin CS, Lee CS, Tsai CY, Wei YH. The role of hOGG1 C1245G polymorphism in the susceptibility to lupus nephritis and modulation of the plasma 8-OHdG in patients with systemic lupus erythematosus. Int J Mol Sci. (2015) 16:3757–68. doi: 10.3390/ijms16023757
13. Simonelli V, Camerini S, Mazzei F, Van Loon B, Allione A, D'Errico M, et al. Genotype-phenotype analysis of S326C OGG1 polymorphism: a risk factor for oxidative pathologies. Free Radic Biol Med. (2013) 63:401–9. doi: 10.1016/j.freeradbiomed.2013.05.031
14. Kohno T, Shinmura K, Tosaka M, Tani M, Kim SR, Sugimura H, et al. Genetic polymorphisms and alternative splicing of the hOGG1 gene, that is involved in the repair of 8-hydroxyguanine in damaged DNA. Oncogene. (1998) 16:3219–25. doi: 10.1038/sj.onc.1201872
15. Lunec J, Herbert K, Blount S, Griffiths HR, Emery P. 8-Hydroxydeoxyguanosine. A marker of oxidative DNA damage in systemic lupus erythematosus. FEBS Lett. (1994). 348:131–8. doi: 10.1016/0014-5793(94)00583-4
16. Lee HT, Lin CS, Lee CS, Tsai CY, Wei YH. Increased 8-hydroxy-2′-deoxyguanosine in plasma and decreased mRNA expression of human 8-oxoguanine DNA glycosylase 1, anti-oxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leucocytes in patients with systemic lupus erythematosus. Clin Exp Immunol. (2014) 176:66–77. doi: 10.1111/cei.12256
17. Leiss H, Niederreiter B, Bandur T, Schwarzecker B, Bluml S, Steiner G, et al. Pristane-induced lupus as a model of human lupus arthritis: evolvement of autoantibodies, internal organ and joint inflammation. Lupus. (2013) 22:778–92. doi: 10.1177/0961203313492869
18. Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. (2009) 30:455–64. doi: 10.1016/j.it.2009.06.003
19. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. (2018) 9:1298. doi: 10.3389/fimmu.2018.01298
20. Sontheimer C, Liggitt D, Elkon KB. Ultraviolet B irradiation causes stimulator of interferon genes-dependent production of protective type I interferon in mouse skin by recruited inflammatory monocytes. Arthritis Rheumatol. (2017) 69:826–36. doi: 10.1002/art.39987
21. Smith S, Fernando T, Wu PW, Seo J, Ni Gabhann J, Piskareva O, et al. MicroRNA-302d targets IRF9 to regulate the IFN-induced gene expression in SLE. J Autoimmun. (2017) 79:105–11. doi: 10.1016/j.jaut.2017.03.003
22. Tumurkhuu G, Shimada K, Dagvadorj J, Crother TR, Zhang W, Luthringer D, et al. Ogg1-dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ Res. (2016) 119:e76–90. doi: 10.1161/CIRCRESAHA.116.308362
23. Werth VP, Fiorentino D, Sullivan BA, Boedigheimer MJ, Chiu K, Wang C, et al. Brief report: pharmacodynamics, safety, and clinical efficacy of AMG 811, a human anti-interferon-gamma antibody, in patients with discoid lupus erythematosus. Arthritis Rheumatol. (2017) 69:1028–34. doi: 10.1002/art.40052
24. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. (2002) 30:207–10. doi: 10.1093/nar/30.1.207
25. Barrett T, Suzek TO, Troup DB, Wilhite SE, Ngau WC, Ledoux P, et al. NCBI GEO: mining millions of expression profiles–database and tools. Nucleic Acids Res. (2005) 33(Database issue):D562–6. doi: 10.1093/nar/gki022
26. Komakula SSB, Tumova J, Kumaraswamy D, Burchat N, Vartanian V, Ye H, et al. The DNA repair protein OGG1 protects against obesity by altering mitochondrial energetics in white adipose tissue. Sci Rep. (2018) 8:14886. doi: 10.1038/s41598-018-33151-1
27. Druzhyna NM, Hollensworth SB, Kelley MR, Wilson GL, Ledoux SP. Targeting human 8-oxoguanine glycosylase to mitochondria of oligodendrocytes protects against menadione-induced oxidative stress. Glia. (2003) 42:370–8. doi: 10.1002/glia.10230
28. Mohanty K, Dada R, Dada T. Oxidative DNA damage and reduced expression of DNA repair genes: Role in primary open angle glaucoma (POAG). Ophthalmic Genet. (2017) 38:446–50. doi: 10.1080/13816810.2016.1261904
29. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
30. Bhat N, Fitzgerald KA. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur J Immunol. (2014) 44:634–40. doi: 10.1002/eji.201344127
31. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:1507–15. doi: 10.1136/annrheumdis-2018-212988
32. Lopez-Lopez L, Nieves-Plaza M, Castro Mdel R, Font YM, Torres-Ramos CA, Vila LM, et al. Mitochondrial DNA damage is associated with damage accrual and disease duration in patients with systemic lupus erythematosus. Lupus. (2014) 23:1133–41. doi: 10.1177/0961203314537697
33. Meas R, Burak MJ, Sweasy JB. DNA repair and systemic lupus erythematosus. DNA Repair (Amst). (2017) 56:174–82. doi: 10.1016/j.dnarep.2017.06.020
34. Souliotis VL, Vougas K, Gorgoulis VG, Sfikakis PP. Defective DNA repair and chromatin organization in patients with quiescent systemic lupus erythematosus. Arthritis Res Ther. (2016) 18:182. doi: 10.1186/s13075-016-1081-3
35. Mireles-Canales MP, Gonzalez-Chavez SA, Quinonez-Flores CM, Leon-Lopez EA, Pacheco-Tena C. DNA damage and deficiencies in the mechanisms of its repair: implications in the pathogenesis of systemic lupus erythematosus. J Immunol Res. (2018) 2018:8214379. doi: 10.1155/2018/8214379
36. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
37. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. (2018) 215:1287–99. doi: 10.1084/jem.20180139
38. Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. (2013) 39:482–95. doi: 10.1016/j.immuni.2013.08.004
39. Corella D, Ramirez-Sabio JB, Coltell O, Ortega-Azorin C, Estruch R, Martinez-Gonzalez MA, et al. Effects of the Ser326Cys polymorphism in the DNA repair OGG1 gene on cancer, cardiovascular, and all-cause mortality in the PREDIMED study: modulation by diet. J Acad Nutr Diet. (2018) 118:589–605. doi: 10.1016/j.jand.2017.09.025
40. Leandro GS, Sykora P, Bohr VA. The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat Res. (2015) 776:31–9. doi: 10.1016/j.mrfmmm.2014.12.011
41. Deng GM, Tsokos GC. Pathogenesis and targeted treatment of skin injury in SLE. Nat Rev Rheumatol. (2015) 11:663–9. doi: 10.1038/nrrheum.2015.106
42. Pan Q, Chen J, Guo L, Lu X, Liao S, Zhao C, et al. Mechanistic insights into environmental and genetic risk factors for systemic lupus erythematosus. Am J Transl Res. (2019) 11:1241–54. doi: 10.2174/0929867326666190404140658
43. Callen JP. Cutaneous lupus erythematosus: reflecting on practice-changing observations over the past 50 years. Clin Dermatol. (2018) 36:442–9. doi: 10.1016/j.clindermatol.2018.04.002
44. Scholtissek B, Zahn S, Maier J, Klaeschen S, Braegelmann C, Hoelzel M, et al. Immunostimulatory endogenous nucleic acids drive the lesional inflammation in cutaneous lupus erythematosus. J Invest Dermatol. (2017) 137:1484–92. doi: 10.1016/j.jid.2017.03.018
45. Shrivastav M, Niewold TB. Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front Immunol. (2013) 4:319. doi: 10.3389/fimmu.2013.00319
Keywords: SLE, Ogg1, 8-OH-dG, cGAS-STING pathway, IFN-stimulated genes
Citation: Tumurkhuu G, Chen S, Montano EN, Ercan Laguna D, De Los Santos G, Yu JM, Lane M, Yamashita M, Markman JL, Blanco LP, Kaplan MJ, Shimada K, Crother TR, Ishimori M, Wallace DJ, Jefferies CA and Arditi M (2020) Oxidative DNA Damage Accelerates Skin Inflammation in Pristane-Induced Lupus Model. Front. Immunol. 11:554725. doi: 10.3389/fimmu.2020.554725
Received: 22 April 2020; Accepted: 12 August 2020;
Published: 24 September 2020.
Edited by:
Stefania Gallucci, Temple University, United StatesReviewed by:
Umesh S. Deshmukh, Oklahoma Medical Research Foundation, United StatesCopyright © 2020 Tumurkhuu, Chen, Montano, Ercan Laguna, De Los Santos, Yu, Lane, Yamashita, Markman, Blanco, Kaplan, Shimada, Crother, Ishimori, Wallace, Jefferies and Arditi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moshe Arditi, bW9zaGUuYXJkaXRpQGNzaHMub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.