
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 19 October 2020
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.554301
 Ming Liu1,2
Ming Liu1,2 Jason Saredy3
Jason Saredy3 Ruijing Zhang1,4
Ruijing Zhang1,4 Ying Shao1Yu Sun1William Y. Yang3,5Jirong Wang1,6
Ying Shao1Yu Sun1William Y. Yang3,5Jirong Wang1,6 Lu Liu3Charles Drummer IV1Candice Johnson1
Lu Liu3Charles Drummer IV1Candice Johnson1 Fatma Saaoud1Yifan Lu1Keman Xu1Li Li2Xin Wang7Xiaohua Jiang1,3Hong Wang3,8
Fatma Saaoud1Yifan Lu1Keman Xu1Li Li2Xin Wang7Xiaohua Jiang1,3Hong Wang3,8 Xiaofeng Yang1,3,8*
Xiaofeng Yang1,3,8*The mechanisms that underlie various inflammation paradoxes, metabolically healthy obesity, and increased inflammations after inflammatory cytokine blockades and deficiencies remain poorly determined. We performed an extensive –omics database mining, determined the expressions of 1367 innate immune regulators in 18 microarrays after deficiencies of 15 proinflammatory cytokines/regulators and eight microarray datasets of patients receiving Mab therapies, and made a set of significant findings: 1) proinflammatory cytokines/regulators suppress the expressions of innate immune regulators; 2) upregulations of innate immune regulators in the deficiencies of IFNγ/IFNγR1, IL-17A, STAT3 and miR155 are more than that after deficiencies of TNFα, IL-1β, IL-6, IL-18, STAT1, NF-kB, and miR221; 3) IFNγ, IFNγR and IL-17RA inhibit 10, 59 and 39 proinflammatory cytokine/regulator pathways, respectively; in contrast, TNFα, IL-6 and IL-18 each inhibits only four to five pathways; 4) The IFNγ-promoted and -suppressed innate immune regulators have four shared pathways; the IFNγR1-promoted and -suppressed innate immune regulators have 11 shared pathways; and the miR155-promoted and -suppressed innate immune regulators have 13 shared pathways, suggesting negative-feedback mechanisms in their conserved regulatory pathways for innate immune regulators; 5) Deficiencies of proinflammatory cytokine/regulator-suppressed, promoted programs share signaling pathways and increase the likelihood of developing 11 diseases including cardiovascular disease; 6) There are the shared innate immune regulators and pathways between deficiency of TNFα in mice and anti-TNF therapy in clinical patients; 7) Mechanistically, up-regulated reactive oxygen species regulators such as myeloperoxidase caused by suppression of proinflammatory cytokines/regulators can drive the upregulation of suppressed innate immune regulators. Our findings have provided novel insights on various inflammation paradoxes and proinflammatory cytokines regulation of innate immune regulators; and may re-shape new therapeutic strategies for cardiovascular disease and other inflammatory diseases.
Cardiovascular diseases (CVDs), which include coronary heart disease, hypertension, stroke, and peripheral artery disease, collectively comprise the number one cause of death globally (1, 2). Our and others’ recent reports showed that CVD stressors and risk factors such as hyperlipidemia (3, 4), hyperglycemia (5), hyperhomocysteinemia (6, 7), and chronic kidney disease (8–10), promote atherosclerosis and vascular inflammation via several mechanisms. These mechanisms include endothelial cell activation (3, 11–14) and injury (15); caspase-1/inflammasome activation (8, 10), mitochondrial reactive oxygen species (ROS) (4); differentiation of Ly6Chigh mouse monocytes and CD40+ human monocytes (7, 16–18); decreased/transdifferentiated CD4+Foxp3+ regulatory T cells (Treg) (19–22); impaired vascular repairability of bone marrow-derived progenitor cells (23, 24); downregulated histone modification enzymes (25) and increased expressions of trained immunity pathway enzymes (26). These reports have clearly demonstrated that inflammation mechanisms play significant roles in the initiation and pathogenesis of vascular inflammation and atherosclerosis.
Proinflammatory cytokines (PCs) are key regulators of inflammation, participating in acute (27) and chronic inflammation via a complex and sometimes seemingly contradictory network of interactions (28). Numerous reports of gene deficiency within mouse models showed that while PCs promote vascular inflammation and atherosclerosis; deficiencies of these cytokine genes lead to decreased atherosclerosis. In contrast, several anti-inflammatory cytokines inhibit vascular inflammation and atherosclerosis, and the deficiencies of those anti-inflammatory cytokine genes result in increased inflammation and atherosclerosis (29). This recent progress led to the development of many cytokine blockage-based therapies for inflammatory diseases and CVDs. The CANTOS trial with the monoclonal antibody (Mab) Canakinumab to block proinflammatory cytokine interleukin-1β (IL-1β) was a recent success in treating coronary artery disease (30). However, recent reports from our and others’ teams suggest that inhibition of one or more proinflammatory regulators such as cytokines or microRNAs (miRs) can lead to new waves of inflammation. In our previous studies, deficiency of proinflammatory microRNA-155 (miR155) in atherogenic apolipoprotein E knock-out (ApoE KO, or ApoE-/-) mice results in the establishment of the first metabolically healthy obesity (MHO) mouse model with decreased aortic atherosclerosis, increased obesity, white adipose tissue hypertrophy and non-alcoholic fatty liver disease but without insulin resistance (31). In another report we showed that, analyzing 109 microRNAs (miRs) reported in four hyperlipidemia-related diseases (HRDs) such as atherosclerosis, non-alcoholic fatty liver disease (NAFLD), obesity, and type II diabetes (T2DM), we found that miR155 and miR221 are significantly modulated in all four HRDs. We hypothesized that miR155 is a proinflammatory, proatherogenic but obesity-suppressed master regulator. Deficiency of miR155 results in a “second wave of inflammation” in the high-fat feeding MHO model, which is our proposed new concept. Indeed, our results showed that high-fat feeding leads to a new “second wave of inflammation”, as we termed, in miR155-/-/ApoE-/- MHO mice with increased plasma proinflammatory adipokines leptin and resistin in plasma and white adipose tissue (32). In addition, biological disease-modifying antirheumatic drugs (bDMARDs) targeting inflammatory cytokines have expanded the treatment options for patients with rheumatoid arthritis (RA) (33), inflammatory bowel disease, psoriatic arthritis, severe psoriasis, autoinflammatory disease, Castleman disease, and plaque psoriasis (Table S1) (34). As shown in Table 1, the therapies of tumor necrosis factor-α (TNF-α) targeting monoclonal antibody (Mab) adalimumab and IL-6 receptor (IL-6R) targeting Mab sarilumab could lead to injection site reactions (sarilumab), worsening RA (adalimumab) and increased incidences of infections (sarilumab: 28.8%; adalimumab: 27.7%) in patients (35). Moreover, it was reported that paradoxical inflammations such as psoriasiform lesions, arthritis are induced by anti-TNF Mabs in some patients with Crohn’s disease and ulcerative colitis (36). Furthermore, it has been found that anti-IL-1β Mab Canakinumab is associated with a higher incidence of fatal infection than placebo (30). Finally, it was reported that unfavorable responses on anti-IL-17A Mab secukinumab are driven by patients with elevated inflammatory markers such as C-reactive protein (37).
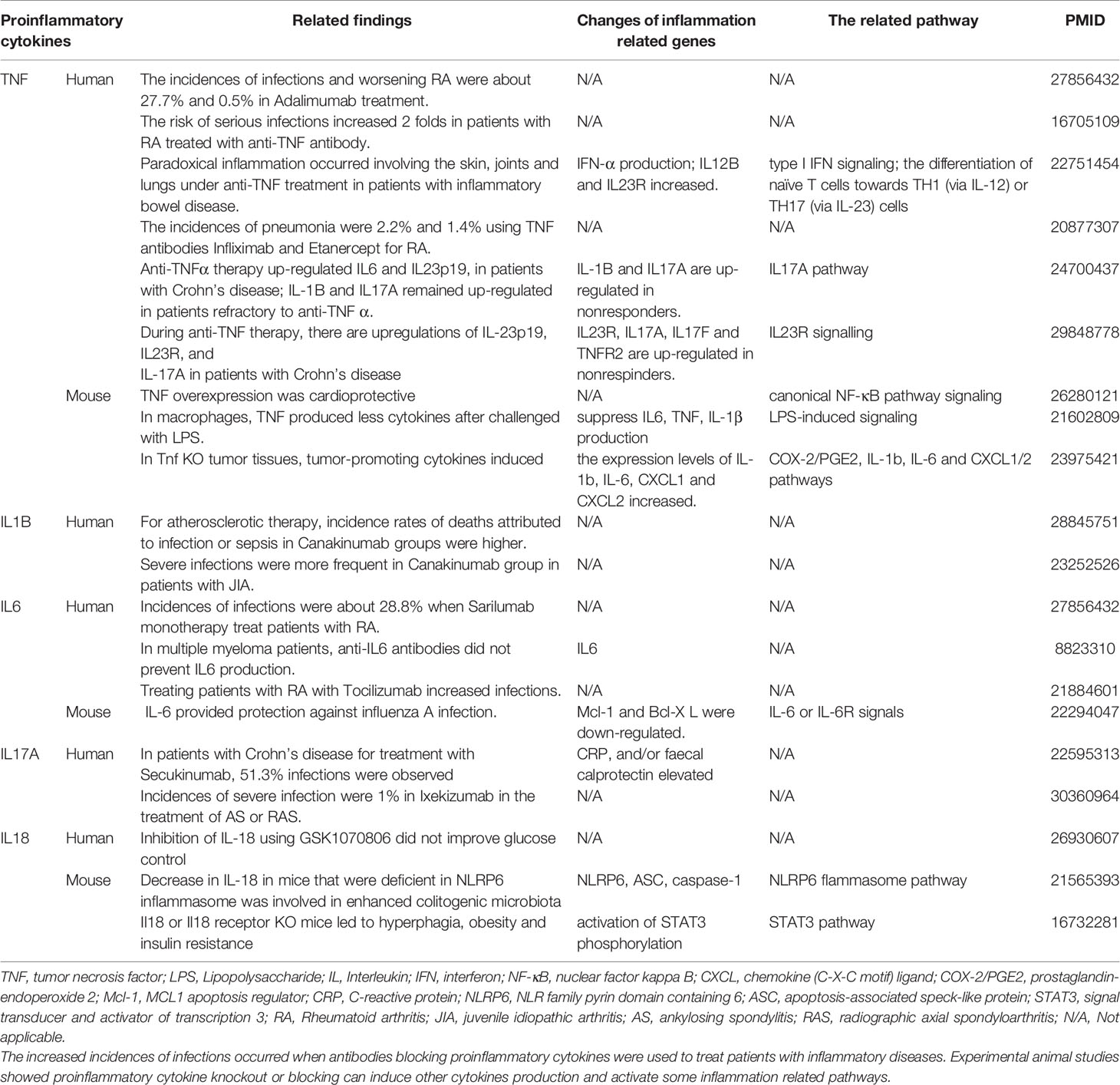
Table 1 Proinflammatory cytokine-blocking therapies paradoxically lead to increased inflammation.
In addition, various inflammation paradoxes have been reported including new inflammations occur when: i) particular cytokine genes and inflammatory regulators are mutated (38); ii) patients experience somatic mutations (39) and inflammageing (40); iii) PCs are weakened due to single nucleotide polymorphism (41); iv) cytokine blockage therapies are used; v) genes encoded PCs and other regulators are knocked-out in mice; vi) inflammation is resurged when MHO undergoes a transition to classical metabolically unhealthy obesity (31, 32) in response to the long term stimulation of metabolic disease risk factors such as hyperlipidemia, danger associated molecular patterns (DAMPs) and conditional DAMPs as we reported (42); vii) an obesity paradox exists, wherein obese individuals survive sepsis at higher rates than their normal-weight counterparts (43); viii) inflammation paradoxes are observed in the Amazon region showing that the indigenous Tsimane in Bolivia appears protected against non-communicable metabolic inflammatory diseases (NCDs) such as obesity, type 2 diabetes, and CVDs despite increased inflammatory markers (44); and ix) A widely discussed physiological puzzle of mammalian pregnancy is the immunological paradox, the semi-allogenic fetus is not attacked by the mother’s adaptive immune system (45). These inflammation paradoxes were summarized in Table S2. In an attempt to solve these paradoxes, we examined a significant issue that remains unknown: why proinflammatory regulator blockage therapies lead to a “secondary wave of inflammation” (32).
Similar to single cytokine targeting Mab therapies discussed above, one of the current research strategies is to use gene-deficient mouse models and transgenic mouse models to determine the dominant effects of these inflammatory regulators in disease models such as atherogenesis (29, 46). Numerous disease risk factors have been identified to induce metabolic CVDs and other inflammatory diseases, such as hyperlipidemia, hyperglycemia, hyperhomocysteinemia, obesity, hypertension, and cigarette smoke (29, 47). It has been documented that inflammation and metaflammation are evolutionally conserved; the underlying pathways are cross-talking (48). However, the issue remains unknown whether in proinflammatory “regulator A” deficiency conditions, disease risk factors promote the regulator A-suppressed secondary wave of inflammation as we termed in our recent report (32).
InnateDB (https://www.innatedb.com/) is a comprehensive database on innate immune regulatory genes, which has been developed to facilitate systems-level investigations of the mammalian (human, mouse and bovine) innate immune response (49). This list of innate immune regulators in the InnateDB database (innatome) is especially useful for us to analyze the expression changes of innate immune regulators in the presence and absence of certain immune regulators. In order to improve our understanding on the expression changes in the innatome in the presence and absence of proinflammatory regulators, we examined our novel hypothesis that proinflammatory cytokine blockages induce inflammatory regulators. To test this hypothesis, we performed an extensive –omics database mining, determined the expressions of 1367 innate immune regulators (innatomic genes, IGs) in 18 microarrays after deficiencies of 15 PCs and made a set of significant findings. Our findings will provide novel insights on various inflammation paradoxes and PC regulation of IGs; and may re-shape new therapeutic strategies for various inflammations.
The 18 murine microarray datasets of proinflammatory cytokine gene deficiencies and eight microarray datasets of patients receiving Mab therapies were collected from National Institutes of Health (NIH)-National Center for Biotechnology Information (NCBI)-Gene Expression Omnibus (GEO) databases (https://www.ncbi.nlm.nih.gov/gds/) and analyzed with an online software GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/). The detailed information of these GEO datasets was shown in Table 2A and 2B and related mechanism tables or figures. The original microarray experiments used different cells, which prevented us from comparing the effects of proinflammatory regulators in regulating IGs in the same cell types. Of note, our approach was well justified. For example, as a common practice, we (50) and others (51) often studied gene expression in non-ideal heterogenous peripheral blood mononuclear cell populations (PBMCs) in pathophysiological conditions, which are actually composed of many cell types (also see the Discussion section). The used IGs and ROS regulator gene -lists were listed in Table S3.
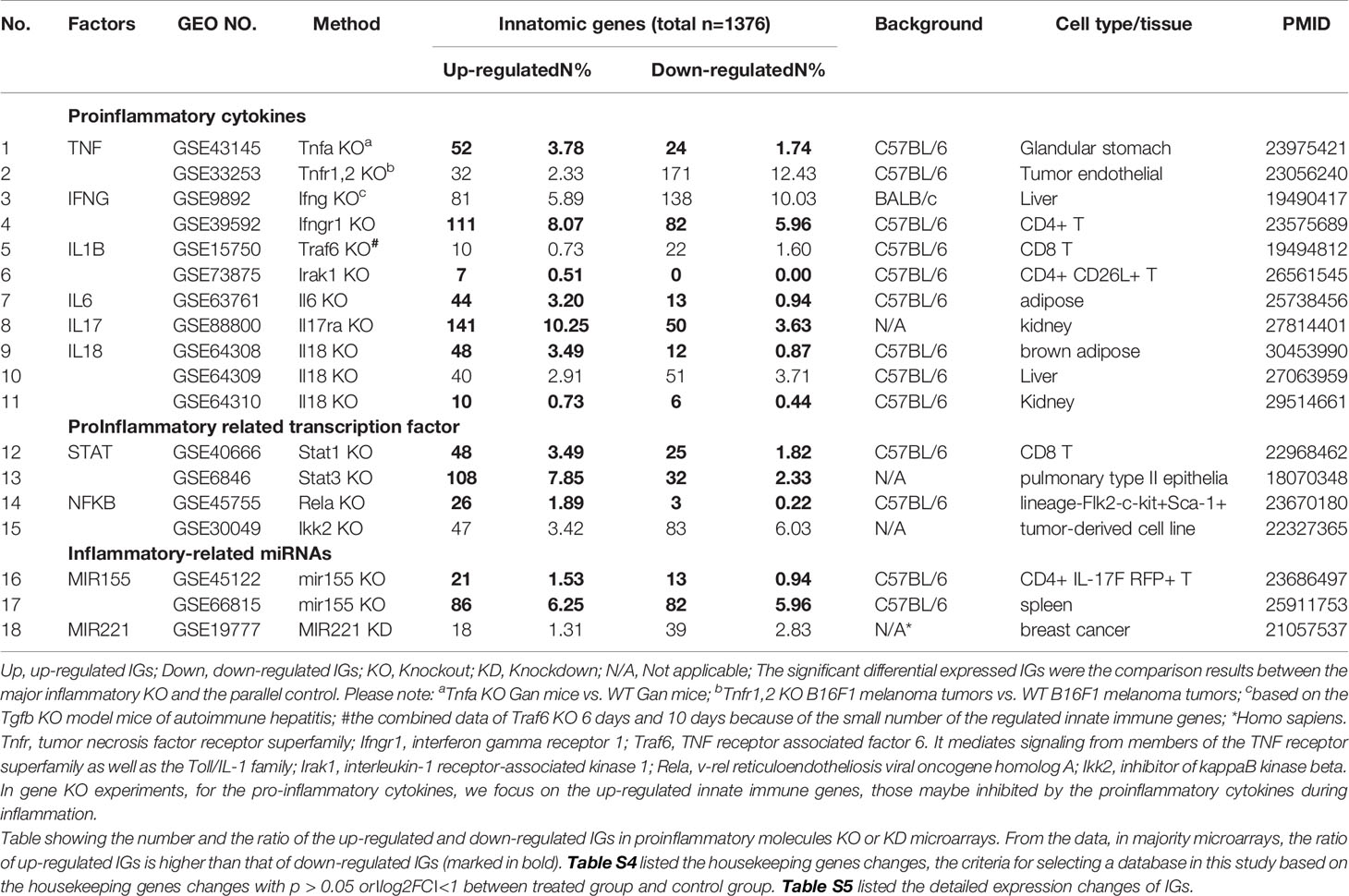
Table 2A 18 microarray datasets were collected to analyze the changes of innate immunity molecules (innatomic genes, IGs) in deficiencies of proinflammatory regulators (p < 0.05, ∣log2FC∣>1).
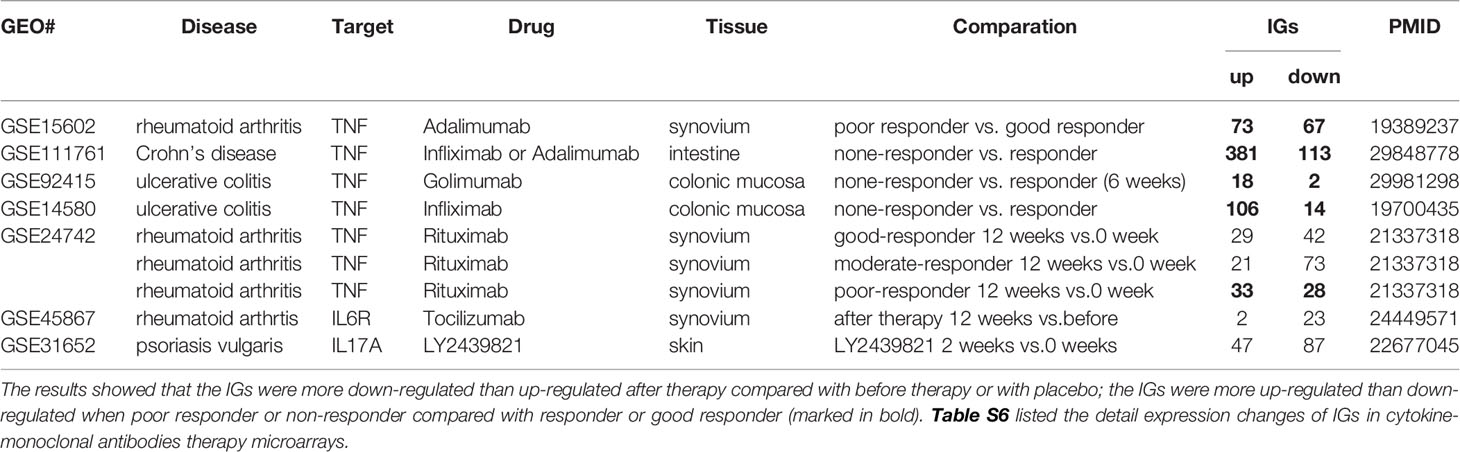
Table 2B The innatomic genes (IGs) were analyzed in cytokine-monoclonal antibodies therapy microarrays.
Six house-keeping genes including ACTB, CHMP2A, RPL27, SRP14, RPL22 and OAZ1 (Table S4) in all GEO datasets regardless of species that were chosen for this study. The house-keeping gene list was extracted from the list provided by Eisenberg and de Jonge (52, 53). Briefly, the mean fold change (FC) of house-keeping genes between treatment and control groups vary from 0.79 to 1.13 (53). As this variation was very narrow, we concluded that the datasets (Table 2A and 2B) were of high quality. Genes with expression changes more than 2-folds in microarrays were defined as the upregulated genes, while genes whose expressions decreased more than 2-fold in microarrays were defined as downregulated genes. Simply, genes with the expression changes at |log2FC|>1 and p<0.05 were defined as the differentially expressed genes.
We utilized Ingenuity Pathway Analysis (IPA, Qiagen, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/) to characterize clinical relevance and molecular and cellular functions related to the identified genes in our microarray analysis. Differentially expressed genes were identified and uploaded into IPA for analysis. The core and pathways analyses were used to identify molecular and cellular pathways, as we have previously reported (54, 55).
The studies using the compound gene-deficient mice established with specific cytokine deficiency crossing to two atherogenic mouse models such as apolipoprotein E deficient (ApoE-/-), and low-density lipoprotein receptor-deficient (LDLR-/-) background have significantly improved our understanding on the roles of these PCs on atherosclerotic progression. Deficiencies of TNFα, IL-1β, IL-18 and interferon-γ (IFNγ) lead to a significant reduction of atherosclerotic lesions. However, the parts of atherosclerotic lesions remain in those proinflammatory cytokine deficient and ApoE-/- double gene KO mice. The atherosclerotic lesions remain 66% for TNF-/-/ApoE-/-, 66% for IL-1β-/-/ApoE-/-, 65% for IL-18-/-/ApoE-/- and 25% for IFNγ-/-/LDLR-/- mice, respectively (Figure 1A). The remained atherosclerotic lesions may be contributed towards by the following factors (Figure 1B): 1) the specific cytokine “A”-independent proinflammatory, proatherogenic cytokines and factors, and 2) proinflammatory, proatherogenic cytokines and factors suppressed by the specific cytokine “A”. These two factors, especially the proinflammatory/proatherogenic cytokines/factors suppressed by the specific deficient cytokine “A”, remained unknown. In addition, we proposed a recently so-called “second wave of inflammatory responses” (32) for proinflammatory miR155 suppressed proinflammatory adipokines leptin and resistin, which may result from direct suppression of these proinflammatory adipokines by miR155 or indirect suppression of adipogenesis by miR155 (31). The second wave of inflammatory responses is required to protect the organism from infections or other pathologies at least when the following situations occur in the paradoxes introduced above when PCs are deficient and/or downregulated. Moreover, as shown in Table 1, proinflammatory cytokine-blocking therapies may paradoxically lead to increased inflammation, and increased incidences of infections. Finally, experimental animal studies (Table 1), with gene-deficient mouse models of PCs, proinflammatory cytokine knockout or blocking induced other cytokines production and activate some inflammation-related pathways was shown. In summary, our and others’ reports demonstrated that in some diseases and pathophysiological relevant conditions when proinflammatory regulators are deficient or inhibited, other PCs and IGs are upregulated.
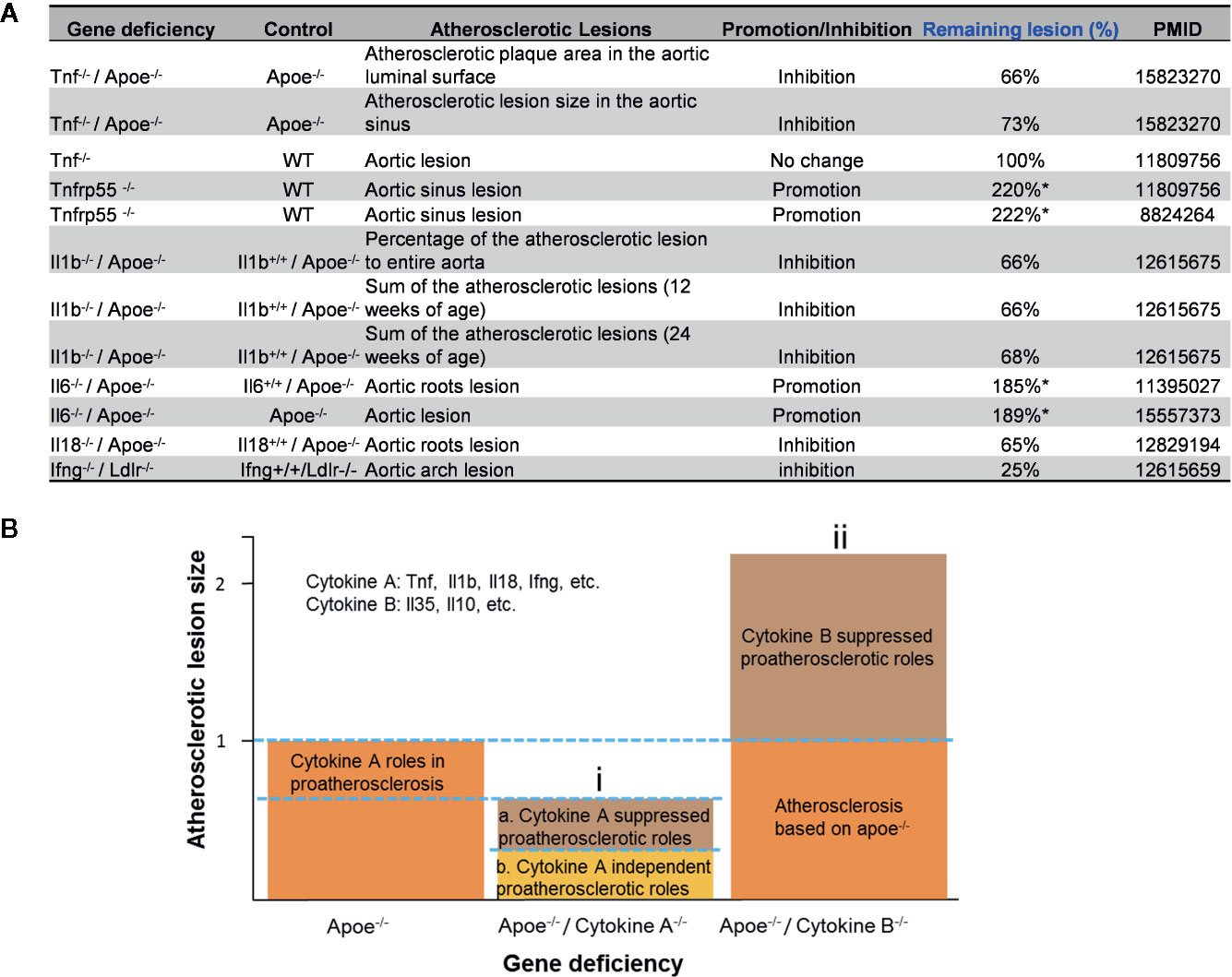
Figure 1 Proinflammatory cytokine-suppressed proinflammatory/proatherogenic mechanisms contribute to atherosclerotic lesions remained in mouse models with cytokine deficiencies. (A) Cytokine deficiency can only inhibit smaller atherosclerotic lesions even promote (Remaining lesion size > 100%) atherosclerotic lesions in murine models of atherosclerosis. (B) Cytokine roles were divided into two major groups: (i) Cytokines such as Tnf, Il1b, Il18 and Ifng were identified as cytokines with pro- atherosclerotic roles; and (ii) cytokines such as IL-35 and IL-10 (not focused in this study) were identified as cytokines with anti-atherosclerotic roles. In order to better compare the lesions size changes after cytokine deficiency, atherosclerotic lesions size is defined “100% (1)” in murine models of atherosclerosis (Apoe-/- mouse).
Since the pathways underlying inflammations and metaflammations are cross-talked (48), the interaction modes can be classified into three categories: i) agonism and synergy; ii) inhibition and antagonism; and iii) parallel and independence. To determine the mechanisms underlying cytokine targeting Mab therapies-induced inflammation and PC deficiencies-induced inflammation, we hypothesize that the deficiencies of PCs upregulate IGs in addition to downregulating IGs. We applied the -omics database mining methods and principles that we pioneered in 2004 (56, 57) to this study. The detailed features and justification of this data mining approach were highlighted in Table 1 of our recent paper (47). We examined a comprehensive list of IGs collected in the InnateDB database (https://www.innatedb.com/), which had a total of 1367 genes. Panoramic profiling of the expressions of IGs in PC deficiency microarray datasets would lead to high throughput characterization on transcriptomic regulation of the PCs on IGs. Table 2A and 2B are the results of the changes of IGs in deficiencies of PCs and in cytokine-monoclonal antibodies therapy microarrays, respectively. Table S5 and Table S6 listed the detailed expression changes of IGs. As shown in Table 2A, we found 11 cytokine gene knock-out (KO) microarray datasets, four proinflammatory transcription factor KO datasets, three proinflammatory microRNA KO datasets, including TNFα and TNFR1,2 KO, IFNγ and IFNγ receptor 1 (IFNγR1) KO, IL-1β pathway-related TNF receptor associated factor 6 (TRAF6) and interleukin-1 receptor-associated kinase 1 (IRAK1) KO, one IL-6 KO, one IL-17 receptor A (IL-17RA) KO, three IL-18 KO, one signal transducer and activator of transcription protein 1 (STAT1) KO, one STAT3 KO, one NF-kB subunit Rela KO, one Inhibitor of NFKB kinase subunit-β (IKK2) KO, two microRNA-155 (miR155) KO and one miR221 KD (knock-down). The results in Table 2A showed that: i) the deficiencies of all the 18 proinflammatory regulators lead to upregulation of IGs from 0.51% to 10.25% out of a total of 1367 IGs; ii) upregulated IGs in the deficiencies of IFNγ, IFNγR1, IL-17A, STAT3 and miR155 are more than that of the deficiencies of TNFα, IL-1β, IL-6, IL-18, STAT1, NF-kB, and miR221; and iii) the deficiencies of IFNγ, IFNγR1, IL-17RA, STAT3 and miR155 lead to high upregulation of IGs by 5.89%, 8.07%, 10.25%, 7.85% and 6.25%, respectively, in normal cell types and tissues including CD4+ T cells (IFNγ KO), kidney (IL-17 KO), pulmonary type II epithelial cells (STAT3 KO) and spleen (miR155 KO). Of note, the roles of IL-17 in promoting atherosclerosis development have been controversial as we (58), and others reported (46). However, the pro-atherogenic roles of TNFα and IFNγ have been well documented (46, 59). Therefore, our results have demonstrated for the first time that PCs suppress the expressions of some IGs; and upregulated IGs in the deficiencies of IFNγ, IFNγR1, IL-17RA, STAT3 and miR155 are more than that in the deficiencies of TNFα, IL-1β, IL-6, IL-18, STAT1, NF-kB, and miR221. These results have suggested that IFNγ, IFNγR1, IL-17RA, STAT3 and miR155 suppress more IGs expressions than other cytokines.
In microarray datasets of patients receiving Mab therapy (Table 2B), the results showed that more IGs were downregulated than upregulated when after therapy compared with the group before therapy or with placebo; more IGs were upregulated than downregulated when poor responder or non-responder compared with the group responders or good responders. On the one hand, the results confirm the efficacy of Mab in most of patients with autoimmune diseases, on the other hand, the results cannot deny the condition that in drug none-responders: “suppressed cytokines or innate immune regulator molecules” were upregulated.
We hypothesized that PCs are cross-talked and share their regulation on the expression of IGs. To test this hypothesis, we performed a Venn Diagram analysis on the upregulated IGs from proinflammatory cytokine KO datasets. Among 15 IGs lists shared by two proinflammatory cytokine pathways, the five shared cytokine pathways including TNFα/IFNγ, IFNγ/IL-18, IL-6/IL-17, IFNγ/IL-17, and IL-17/IL6 pathways have the higher numbers of shared IGs (Figures 2A, B). We further examined the cross-talking among all the five cytokine pathways using the Circos Plot (http://metascape.org/gp/index.html#/main/step1) (Figure 2C), Comparing with the data analyzed with the Venn Diagram, the Circos plot analysis has advantages in including the different genes fall into the same ontology term (Blue lines) among the IGs upregulated in the deficiencies of five PCs. The outer circle showed the upregulated IGs in each cytokine pathway. The dark orange section in the inner circle indicated the shared IGs, and the light orange bar indicated the cytokine pathway unique IGs. The results showed that: 1) each cytokine deficiency-upregulated IGs were shared with other cytokine pathways; 2) the dark orange section in the inner circle indicated the shared IGs (Purple lines link the same gene), which were the same as the cytokine-shared IGs in Figure 2A; and 3) IFNγ/IFNγR1 pathways have the highest numbers of upregulated IGs, which had minimal shared IGs with that of IL-6 pathway and had more shared IGs with that of IL-17, TNFα and IL18. Our findings were correlated well with that reported: IFNγ is an essential cytokine expressed highly in type 1 T helper cells (Th1); and IL-17/IFNγ double producing cells, the so-called Th17/Th1 plastic subset (21, 60), is a phenotype frequently observed in pathological conditions (61). These results have demonstrated that the deficiencies of PCs upregulate IGs, which can be in both cytokine-specific and shared manners.
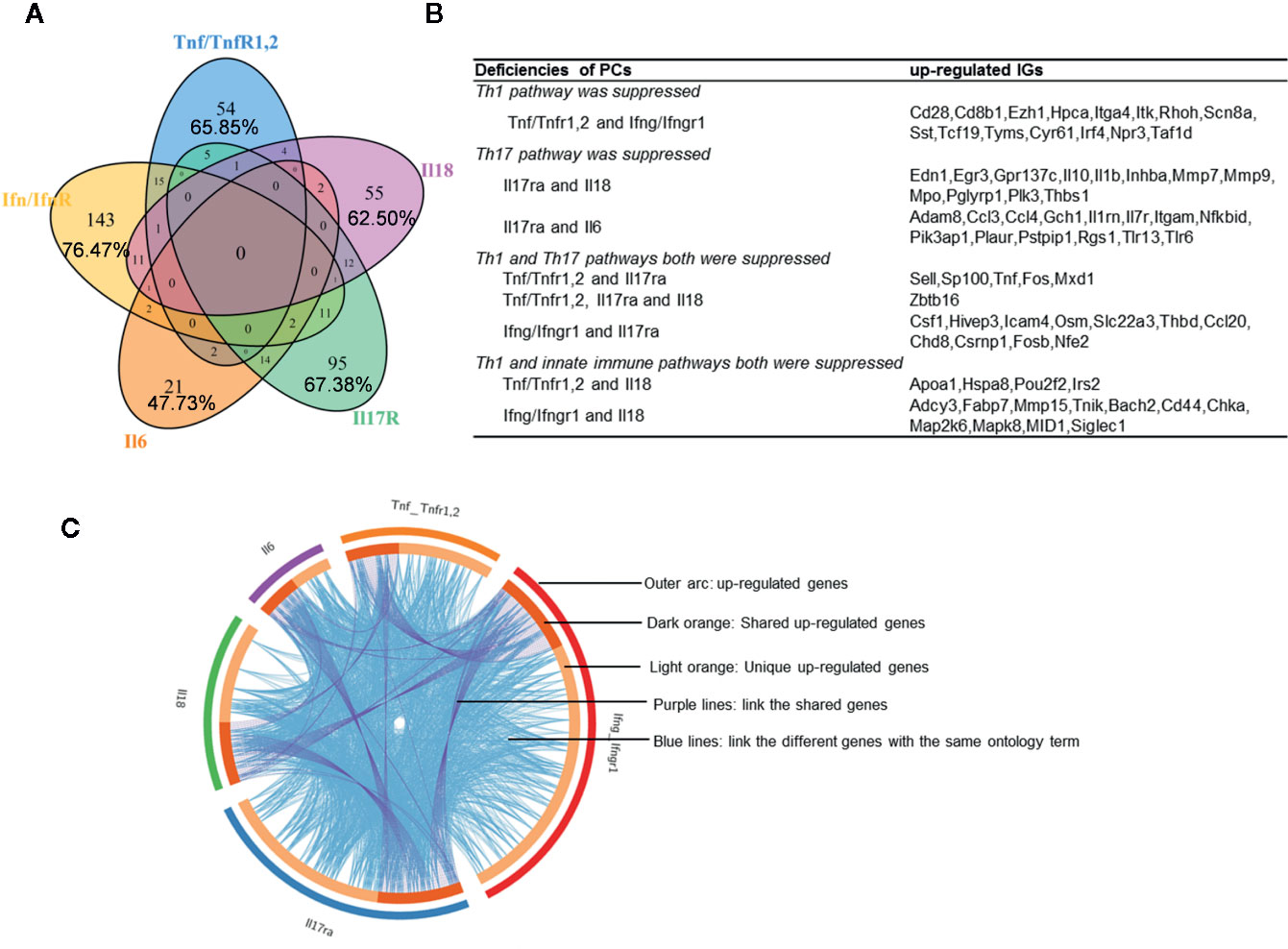
Figure 2 Deficiencies of proinflammatory cytokines shared the up-regulated innatomic genes. (A) 82, 187, 44, 141, and 88 up-regulated non-repeating innatomic genes were found in Tnf/Tnfr1,2 knock-out (KO), Ifn/Ifnr1 KO, Il6 KO, Il17ra KO and Il18 KO five groups according to the Table 2A, respectively. Venn Diagram (generated by using R) showed the majority of proinflammatory cytokine-suppressed innatomic genes were cytokine-specific and accounted for 65.85%, 76.47%, 47.73%, 67.38%, 62.50% in Tnf/Tnfr1,2 KO, Ifn/Ifnr1 KO, Il6 KO, Il17ra KO and Il18 KO groups, respectively. (B) There were several common genes between two or among three groups. In state of Th1 pathway was suppressed: in Tnf/Tnfr1,2 and Ifn/Ifnr1 KO groups, Cd28, isotype switching positive regulating gene, Cd8b1, activate T cell, Itga4, positively regulate leukocyte tethering or rolling, Itk, NK T cell differentiation and phospholipase C activity, and T-helper 17 cell lineage commitment and differentiation gene (Irf4) were commonly up-regulated. In state of Th17 pathway was suppressed: in Il17ra and Il18 KO groups, Egr3, has the role of regulating gamma-delta T cell differentiation and Il1b, positively regulate T-helper 1 cell cytokine production were up-regulated; in Il17ra and Il6 KO groups, Ccl3, which has the role of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum and Ccl4, positively regulate natural killer cell chemotaxis, Il7r, positively regulate T cell differentiation in thymus, Nfkbid, positively regulate T-helper 17 cell differentiation, and Pik3ap1, related to toll-like receptor 2,7,9 related signal pathways were up-regulated. In state of Th1 and Th17 pathways both were suppressed: in Tnf/Tnfr1,2, Il17ra KO groups, Sell, related to leukocyte tethering or rolling, neutrophil degranulation and leukocyte adhesion to vascular endothelial cell, was common up-regulated; in Tnf/TnfR1,2, Il17ra and Il18 KO groups, Zbtb16, which positively regulate NK T cell differentiation was up-regulated; in Ifn/Ifnr1 and Il17ra KO groups, Osm, positive regulation of interleukin-17 secretion, Thbd, negative regulation of platelet activation, and Ccl20, positive regulation of thymocyte, T cell and lymphocyte migration, were up-regulated. In state of Th1 and innate immune pathways both were suppressed: in Tnf/Tnfr1,2, and Il18 KO groups, Irs2, which has the role of negative regulation of plasma membrane long-chain fatty acid transport was up-regulated; in Ifn/Ifnr and Il18 KO groups, Cd44, which has the role of positive regulation of monocyte aggregation, was up-regulated. (C) The Circos plot (generated by using Metascape http://metascape.org/gp/index.html#/main/step1) showed how genes overlap from the up-regulated genes in these five groups, including the same genes (purple lines link the same genes that are shared by multiple proinflammatory cytokines KO) and genes with the same ontology term (blue lines link the different genes where they fall into the same ontology term) shared by the five groups.
We then examined the signaling pathways in upregulated IGs in the deficiencies of five PCs such as TNFα, IFNγ, IL-6, IL-17 and IL-18. We used IPA to perform this analysis, which is a web-based bioinformatics application that allows the uploading of microarray and RNA-Seq data for functional pathway analysis and integration. Table 3A and 3B are the IPA results of upregulated and downregulated IGs in deficiencies of PCs. The detailed IPA results were showed in Table S7 and Table S8. As shown in Table 3A, among 86 significant pathways identified, four pathways such as neuroinflammation, cardiac hypertrophy, leukocyte extravasation, and colorectal cancer metastasis were shared by four cytokine pathway deficiencies; 12 pathways including an additional eight pathways such as dendritic cell maturation, B cell receptor signaling, Tec kinase, nitric oxide and reactive oxygen species in macrophages, integrin, FGF signaling, ILK signaling and PI3 kinase in B cells, were shared by three cytokine pathway deficiencies, 28 pathways were shared by two cytokine pathways; the rest of the 58 pathways were induced by single cytokine deficiencies. In addition, the deficiencies by IFNγR1 KO and IL17R KO upregulate 59 and 39 pathways, respectively. The deficiencies of IFNγR1 and IL17RA resulted in the upregulation of many unique signaling pathways, suggesting that IFNγ and IL-17 inhibit many proinflammatory pathways. However, PD-1, PD-L1 cancer immunotherapy pathway was downregulated in IFNγ pathway deficiency, suggesting that IFNγ is required for PD-1, PD-L1 cancer immunotherapy presumably via expanding type 1 CD4+ T helper cell (Th1) pathways. Moreover, the deficiencies of TNFα, IL-6 and IL-18 resulted in fewer pathways upregulated, including four TNFα suppressed pathways (dendritic cell maturation, Tec kinase signaling, oxygen species in macrophages, and interferon signaling), five IL6 suppressed pathways (neuroinflammation signaling, leukocyte extravasation, colorectal cancer metastasis, B cell receptor signaling, and PI3K signaling in B cells), and five IL-18 suppressed pathways (neuroinflammation signaling, cardiac hypertrophy, NF-kB signaling, acute phase response signaling, endocannabinoid cancer initiation).

Table 3A Ingenuity Pathway Analysis (IPA) results showed the significant pathways (∣Z score∣> 2) of up-regulated innatomic genes (IGs) in proinflammatory cytokine KO microarray datasets.

Table 3B IPA results showed the significant pathways (∣Z score∣> 2) of down-regulated IGs in pro-inflammatory cytokine KO microarray datasets.
We also examined the signaling pathways in downregulated IGs in the deficiencies of four PCs such as TNFαR, IFNγ, IFNγR1, and IL-18. As shown in Table 3B, among 70 significant pathways identified, the dendritic cell maturation pathway was downregulated in all four cytokine deficiencies investigated. Sixteen pathways such as cardiac hypertrophy signaling, systemic Lupus erythematosus in B cell signaling, etc. were downregulated in three cytokine deficiencies. Thirty-seven pathways were downregulated in two cytokine deficiencies. In addition, TNFαR deficiency resulted in the downregulation of 51 pathways. IFNγ deficiency led to the downregulation of 38 pathways, and IFNγR1 deficiency resulted in the downregulation of 22 pathways. IL-18 deficiency led to the downregulation of 6 pathways. Of note, the IPA is an experimental database with the focus on pathway analysis, similar to the NIH-NCBI-PubMed database, rather than bioinformatic prediction. What the IPA found that IGs inhibited by IFNγR1 and IL-17 are functionally involved in 59 and 39 pathways in a statistically significant manner, suggesting that inhibitions of IGs by those two cytokines are multiple pathways-, and multiple function-based. In comparison, the IPA found that inhibitions of IGs by TNF-α, IL-6 and IL-18 are four to five pathways-based, much more focused and specific than that inhibited by IFNγR1 and IL-17. These results have demonstrated that IFNγ/IFNγR1 and IL-17 inhibit 59 and 39 pathways, respectively, whereas the deficiencies of TNFα, IL-6 and IL-17 inhibit four to five out of 86 pathways.
It has been well documented that IFNγ secreted from Th1 cells and natural killer cells (NK) play proinflammatory roles (62), and miR155 have strong proinflammatory/proatherogenic roles (63). However, our recent papers reported that IFNγ and TNFα stimulated endothelial cells upregulate co-stimulation receptors B7-H2, CD40, SEMA4A (a member of the semaphorin family of soluble and transmembrane proteins) and CD112 and immune checkpoint receptors Galectin 9, herpesvirus entry mediator (HVEM), B7-DC, and B7-H1 (PD-L1) (64); proinflammatory adipokines leptin and resistin are significantly upregulated in the plasma and white adipose tissue in miR155 KO mice (31, 32). These findings suggest that IFNγ may inhibit endothelial cells activation and inflammation via anti-inflammatory reverse signaling mediated by upregulated immune checkpoint receptors (65), and miR155 may suppress the second wave of inflammation as formulated in our recently proposed new concept (32). We hypothesized that IFNγ-promoted and -suppressed innate immune programs have shared pathways; and miR155 promoted and suppressed innate immune programs have shared pathways. The rationale for us to focus on these two inflammatory regulators is that among the proinflammatory regulators examined (Table 3A), both these two have long lists of promoted and suppressed pathways. IFNγ KO mice have 37 specific down-regulated innatome pathways, seven specific up-regulated innatome pathways, and four common pathways shared by both promoted and suppressed programs including cardiac hypertrophy, integrin-linked kinase (ILK), role of NFAT in regulation of the immune response and colorectal cancer metastasis signaling (Figure 3). IFNγR1 KO mice have 11 specific downregulated innatome pathways, 49 specific upregulated innatome pathways and 11 common pathways shared by both IFNγR1-promoted and suppressed programs including NF-kB, neuroinflammation, high mobility group box 1 (HMGB1), Toll-like receptors, production of reactive nitric oxide and ROS in macrophages, IL-1, cardiac hypertrophy, dendritic cell maturation, PI3K signaling in B cells, colorectal cancer metastasis, and cardiac hypertrophy (enhanced) (Figure 4). Of note, in addition to IFNγR1/2, IFNγ has also been shown to signal through alternative pathways, including signal transducer and activator of transcription 4 (STAT4), extracellular signal-regulated protein kinases 1 and 2 (Erk1/2), proline-rich tyrosine kinase 2 (Pyk2), and CRK like proto-oncogene (CrkL), among others (66, 67), which may explain the discrepancies between IFNγ signaling and IFNγR1 signaling. In addition, miR155 KO have 89 specific downregulated pathways, six specific upregulated innatome pathways and 13 common pathways shared by both miR155-promoted and suppressed programs including PI3K signaling in B cells, systemic lupus erythematosus in B cell signaling, CD28 signaling in T helper cells, B cell receptor, inducible T-cell co-stimulator (iCOS)-iCOSL signaling, protein kinase C theta (PKCθ) signaling, role of nuclear factor of activated T cells (NF-AT), dendritic cell maturation, Fcγ receptor (one antibody receptor)-mediated phagocytosis, Fc Epsilon RI, IL-6, Tec kinase, and adrenomedullin signaling (Figure 5).
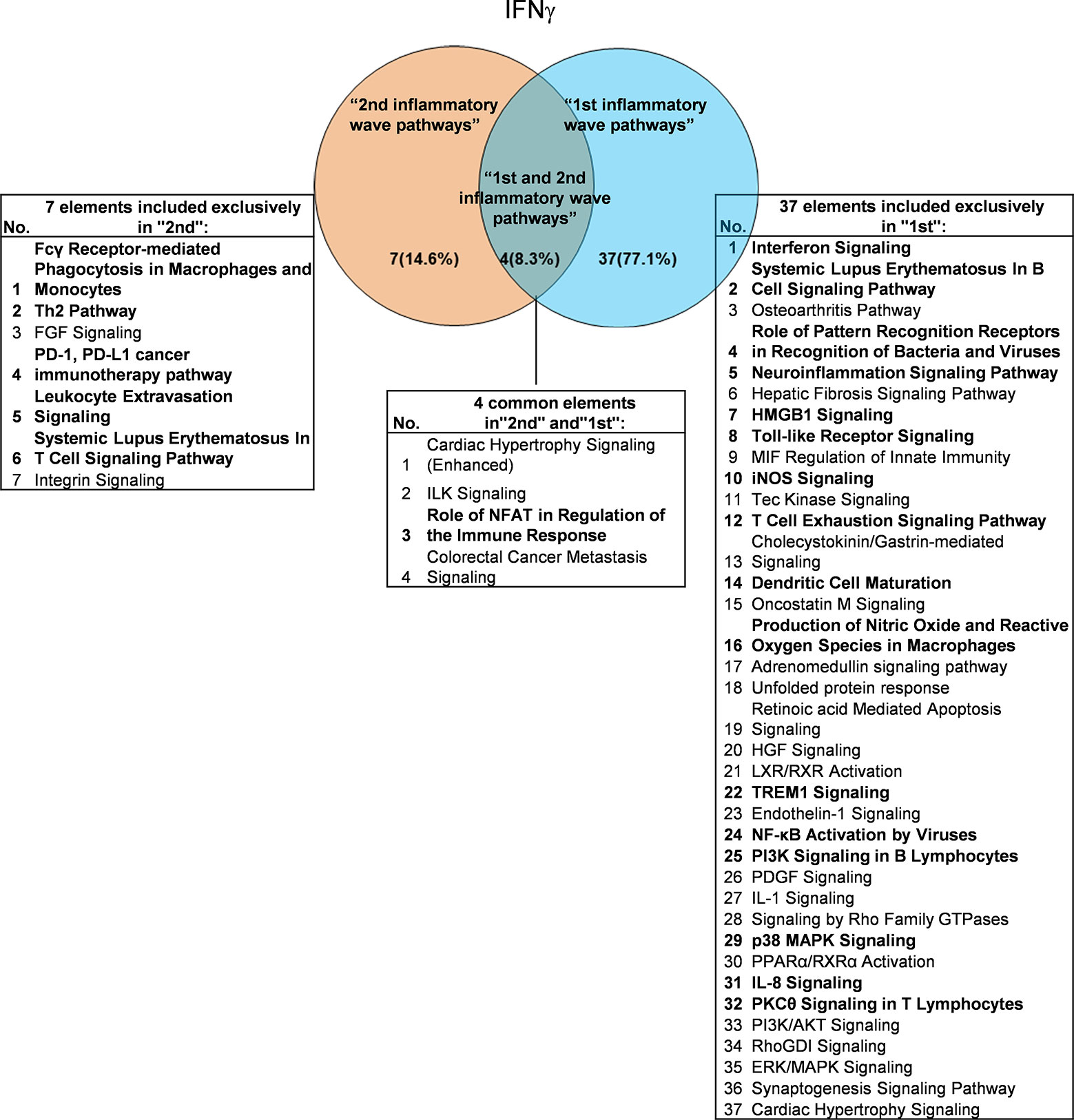
Figure 3 Venn diagram showing the overlapping significant pathways of up-regulated and down-regulated IGs in Ifng KO microarray dataset (GSE9892). The shared pathways by up-regulated and down-regulated IGs were called “2nd and 1st inflammatory wave pathways”. The data showed in 4 common significant “2nd and 1st inflammatory wave pathways”, Role of NFAT in Regulation of the Immune Response is immune response related pathway. The data suggested that the up-regulated IGs have the same immune function to the down-regulated IGs by Ifng KO. Fcγ Receptor-mediated Phagocytosis in Macrophages and Monocytes, Th2 Pathway, PD-1, PD-L1 cancer immunotherapy pathway, Leukocyte Extravasation Signaling and Systemic Lupus Erythematosus In T Cell Signaling Pathway were unique cellular immune response pathways of “2nd inflammatory wave pathways”. The suppression of Interferon Signaling, Systemic Lupus Erythematosus In B Cell Signaling Pathway, and Role of Pattern Recognition Receptors in Recognition of Bacteria and Viruses, etc. 16 pathways were unique cellular immune response pathways of “1st inflammatory molecules”. The cellular immune response pathways were marked in bold.
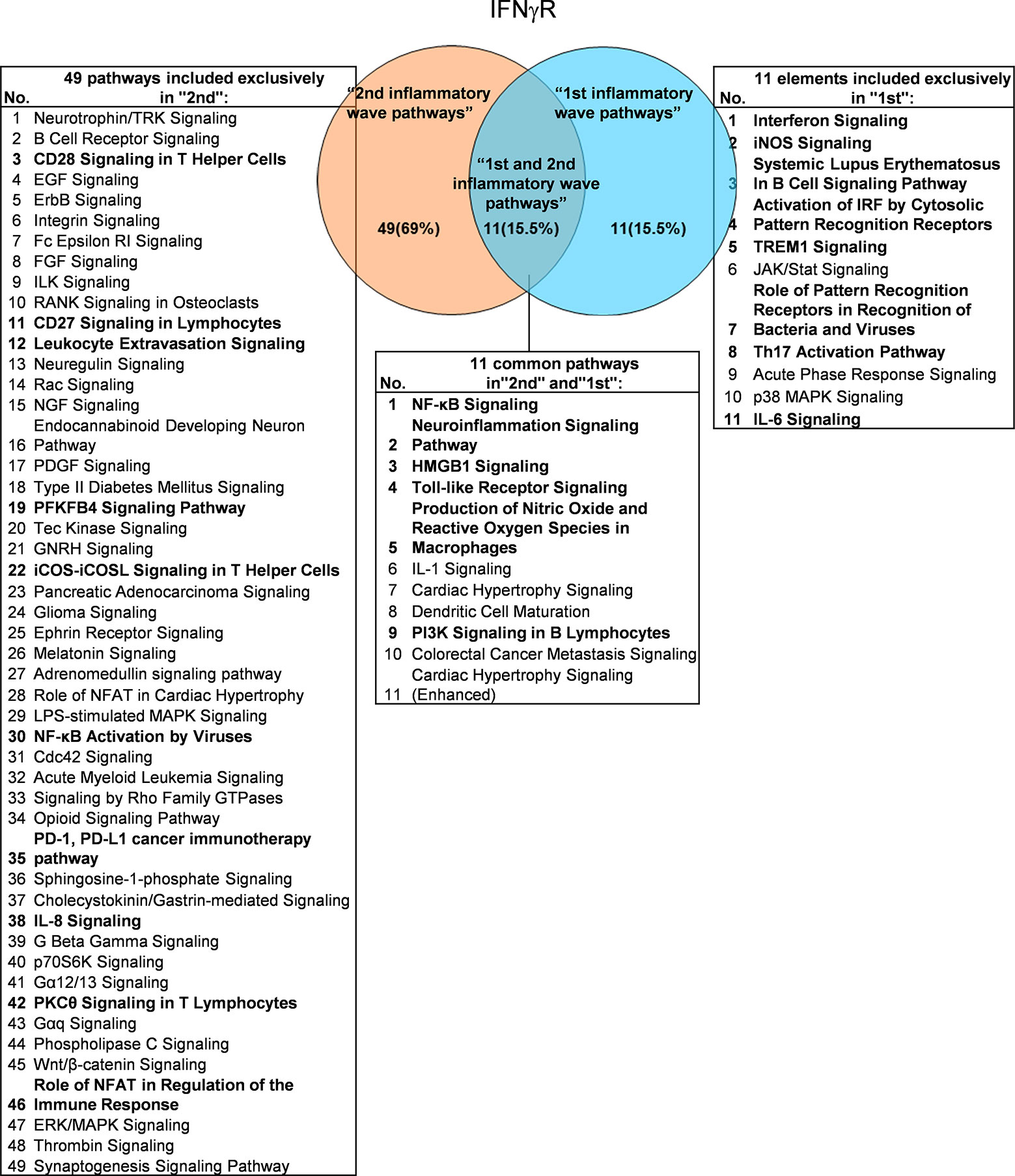
Figure 4 Venn diagram showing 11 overlapping significant pathways of up-regulated and down-regulated IGs in Ifngr KO microarray dataset (GSE39592). The data showed that in 11 common significant “2nd and 1st inflammatory wave pathways”, there were six cellular immune response related pathways. The data suggested that the up-regulated IGs have the same immune function to the down-regulated IGs by Ifngr KO. The suppression of PD-1, PD-L1 cancer immunotherapy pathway, and the activation of CD28 Signaling in T Helper Cells, CD27 Signaling in Lymphocytes, Leukocyte Extravasation Signaling, etc. ten pathways were unique cellular immune response pathways of “2nd inflammatory molecules”. Interferon Signaling, iNOS Signaling, Systemic Lupus Erythematosus etc. eight pathways were unique cellular immune response pathways of “1st inflammatory wave pathways”. The cellular immune response pathways were marked in bold.

Figure 5 Venn diagram showing 13 overlapping significant pathways of up-regulated and down-regulated genes in mir155 KO microarray dataset (GSE66815). The data showed in 13 common significant “2nd and 1st inflammatory wave pathways”, there were nine cellular immune response related pathways. The data suggested that the up-regulated IGs have the same immune function to the down-regulated IGs by inflammatory miRNA155 KO. Six pathways were unique pathways of “2nd inflammatory molecules” and three pathways were cellular immune response related pathways. 89 pathways were unique pathways of “1st inflammatory molecules” and 15 pathways were cellular immune response related pathways. That is, the main significant pathways of miR155 are “1st inflammatory wave pathways”. And in “1st inflammatory wave pathways”, Leptin Signaling in Obesity was inhibited, which can explain the pathogenesis of MHO partly. The cellular immune response pathways were marked in bold.
These results have demonstrated that: first, IFNγ promoted innate immune pathways (37 pathways) are more than that of IFNγ-suppressed pathways (7 pathways), suggesting that IFNγ plays roles in more upregulating IGs and less downregulating IGs; second, IFNγR1 promoted innate immune pathways (11 pathways) are less than that of IFNγR1-suppressed pathways (49 pathways), suggesting that IFNγR1 plays roles in less upregulating IGs and more downregulating IGs; third, miR155-promoted innate immune pathways (89 pathways) are more than that of miR155-suppressed pathways (6 pathways), suggesting that miR155 plays roles in more upregulating IGs and less downregulating IGs; fourth, IFNγ/IFNγR1 and miR155 both have their own promoted and suppressed pathways; and fifth, IFNγ/IFNγR1 has different shared pathways from that of miR155. These results have also suggested that proinflammatory molecules IFNγ and miR155 have negative-feedback mechanisms underlying downstream regulation.
We hypothesized that the common signaling pathways shared in PCs-promoted-, and suppressed programs play significant roles in the initiation and development of inflammation. To examine this hypothesis, we compiled all the significantly downregulated and upregulated pathways in the deficiencies of all the proinflammatory molecules examined (Figure 6). The results showed that proinflammatory molecules deficiencies downregulate 19 specific pathways such as activation of interferon regulatory factor (IRF) by cytosolic pattern recognition receptors, role of pattern recognition receptors in recognition of bacteria and viruses, Th17 activation, hepatocyte growth factor (HGF) signaling, Janus kinases (JAK)/STAT signaling, TNFR1 signaling, macrophage migration inhibitory factor (MIF)-mediated glucocorticoid regulation, p21-activated kinase (PAK) signaling, renin-angiotensin signaling, IL-7 signaling, RhoA signaling, phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT) signaling and cyclic adenosine monophosphate (cAMP)-mediated signaling, suggesting that these signaling pathways are essential for promoting inflammation and anti-infection innate immune responses. In addition, we found that 35 pathways were associated with upregulated IGs when proinflammatory molecules are deficient, suggesting that the 35 pathways are often suppressed by proinflammatory molecules; these 35 pathways serve as the key players in the second wave of inflammation when these PCs and molecules are deficient. Moreover, we identified 51 common pathways in IGs that are shared by proinflammatory molecules-promoted and suppressed programs, suggesting that these 51 common pathways allow the launching of effective inflammation, and innate and adaptive immune responses in the presence and absence of individual proinflammatory molecules. These common pathways are novel targets for future therapeutics to make proinflammatory cytokine blockade therapy more effective.

Figure 6 Venn diagram showing 51 overlapping significant pathways of up-regulated and down-regulated innatomic genes in proinflammatory cytokine KO microarray datasets. The significant pathways from down-regulated innate immune genes have the same expression pattern to proinflammatory cytokines. The data showed in 51 common significant pathways, there were about 22 cellular immune response related pathways (account for 43.14%). The data suggested that the up-regulated innate immune genes have the same immune function to the down-regulated innate immune genes by proinflammatory cytokines KO. Additionally, the suppression of PD-1, PD-L1 cancer immunotherapy pathway, Fcγ Receptor-mediated Phagocytosis in Macrophages and Monocytes, Th2 Pathway, Systemic Lupus Erythematosus In T Cell Signaling Pathway, CD27 Signaling in Lymphocytes, PFKFB4 Signaling Pathway and Inflammasome pathway were unique cellular immune response pathways of the up-regulated innate immunity genes after cytokines KO. Activation of IRF by Cytosolic Pattern Recognition Receptors, Th17 Activation Pathway, MIF-mediated Glucocorticoid Regulation, IL-7 Signaling Pathway were unique cellular immune response pathways of the down-regulated innate immunity genes after cytokines KO. The detailed IPA results were showed in Table S7, S8. The cellular immune response pathways were marked in bold.
We then hypothesized that the common signaling pathways shared in proinflammatory transcription factors (TFs) KO, including STAT1, STAT3, NF-KB Rela and IKK2-promoted-, and suppressed programs play significant roles on initiation and development of inflammation. To examine this hypothesis, we compiled all the significantly downregulated and upregulated pathways in the deficiencies of all the proinflammatory TFs (Figure 7). The results showed that proinflammatory TFs deficiencies-downregulate four pathways (systemic lupus erythematosus in B cell signaling, interferon signaling, activation of IRF by cytosolic pattern recognition receptors, and endothelin-1 signaling) suggesting that these four pathways are promoted by the four TFs. In addition, we found 67 pathways in the IGs upregulated in the four TF deficiencies, suggesting that these four TFs suppress a long list of innate immune pathways. Moreover, we found that eight pathways are shared in the IGs promoted and suppressed when the four TFs are deficient (Table S9 lists pathways of up-regulated IGs and down-regulated IGs in each microarray). Taken together, PCs-promoted and -suppressed innate immune programs have 51 shared pathways; and proinflammatory transcription factors suppress 67 innate immune pathways, which may become novel targets for the future therapeutics and also suggest strategical problems in targeting proinflammatory transcription factors for therapies.

Figure 7 Venn diagram showing eight overlapping significant pathways of up-regulated and down-regulated innatomic genes in pro-inflammatory related transcription factor KO microarray datasets. The significant pathways from down-regulated innate immune genes have the same expression pattern to proinflammatory related transcription factors. The data showed that in 8 common significant pathways, there were 3 cellular immune response related pathways. The data suggested that the up-regulated innate immune genes have the same immune function to the down-regulated innate immune genes by proinflammatory related transcription factors KO. There are 67 significant pathways in up-regulated innate immune genes including TREM1 Signaling, Leukocyte Extravasation Signaling, CD28 Signaling in T Helper Cells etc. cellular immune response pathways. The cellular immune response pathways were marked in bold. The detailed IPA results were showed in Tables S7–S9. The cellular immune response pathways were marked in bold.
As shown in Table 4 and Figure 8A, the results demonstrated a range from multiple cytokine deficiencies to single cytokine deficiency that: 1) deficiencies of all seven cytokines including IFNγ, IFNγR1, IL17RA, IL18, IL6, TNFα, and TNFR1,2 upregulated organismal injury and abnormalities; 2) deficiencies of five cytokines IFNγ, IFNγR1, IL6, TNFα, and TNFR1,2 upregulated immunological diseases; 3) deficiencies of four cytokines IFNγ, IL17ra, IL18, and IL6 upregulated inflammatory diseases; deficiencies of three cytokines IFNγ, IFNγR1, TNFR1,2 upregulated cancer; 4) deficiencies another three cytokines IL17RA, IL18 and IL6 upregulated inflammatory response, connective tissue disorders; 5) deficiencies of two cytokines IFNγR1 and TNFR1,2 upregulated hematological disease; 6) deficiencies of another two cytokines IL17ra and IL18 upregulated skeletal and muscular disorders; 7) deficiency of cytokine TNF upregulated gastrointestinal disease, endocrine system disorders and metabolic disease; 8) deficiency of cytokine TNFR1,2 upregulated tumor morphology; 9) deficiency of cytokine IFNγ upregulated connective tissue disorders; and 10) deficiency of cytokine receptor IFNγR1 increase cardiovascular disease. Similarly, we performed IPA for top disease and disorder associations of downregulated IGs in cytokine gene KO microarrays as shown in Table 4 and Figure 8B. The results are significant since some cytokine blockage therapies have not used for treatment of those top diseases yet. The results demonstrated that each cytokine KO decreased the likelihood to develop certain diseases, which provide novel valuable guidance for cytokine blockage therapies based on the cytokine modulating effects on the expression of IGs.
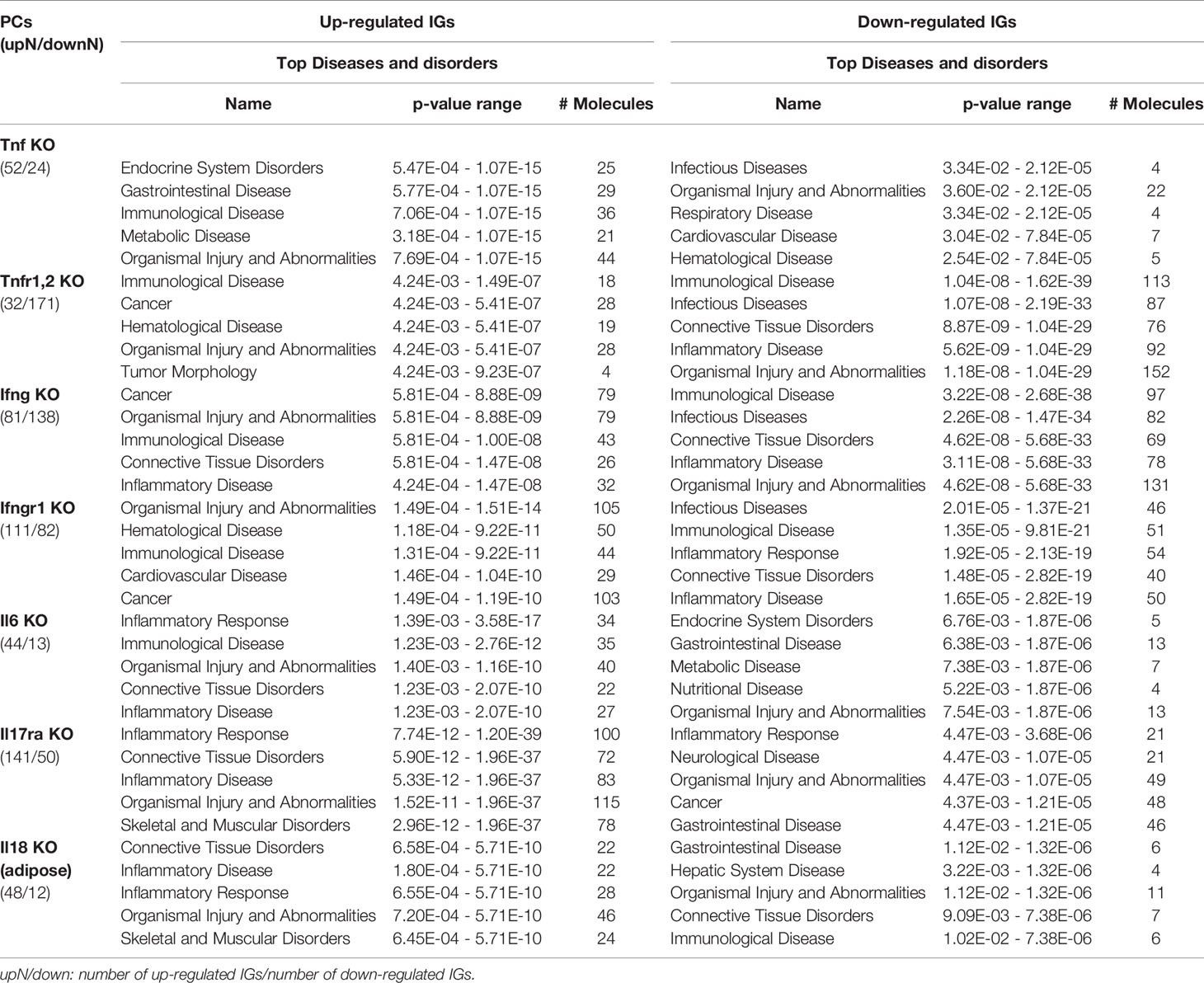
Table 4 The top 5 disease and disorders of the up-regulated and down-regulated IGs in cytokines KO microarrays.
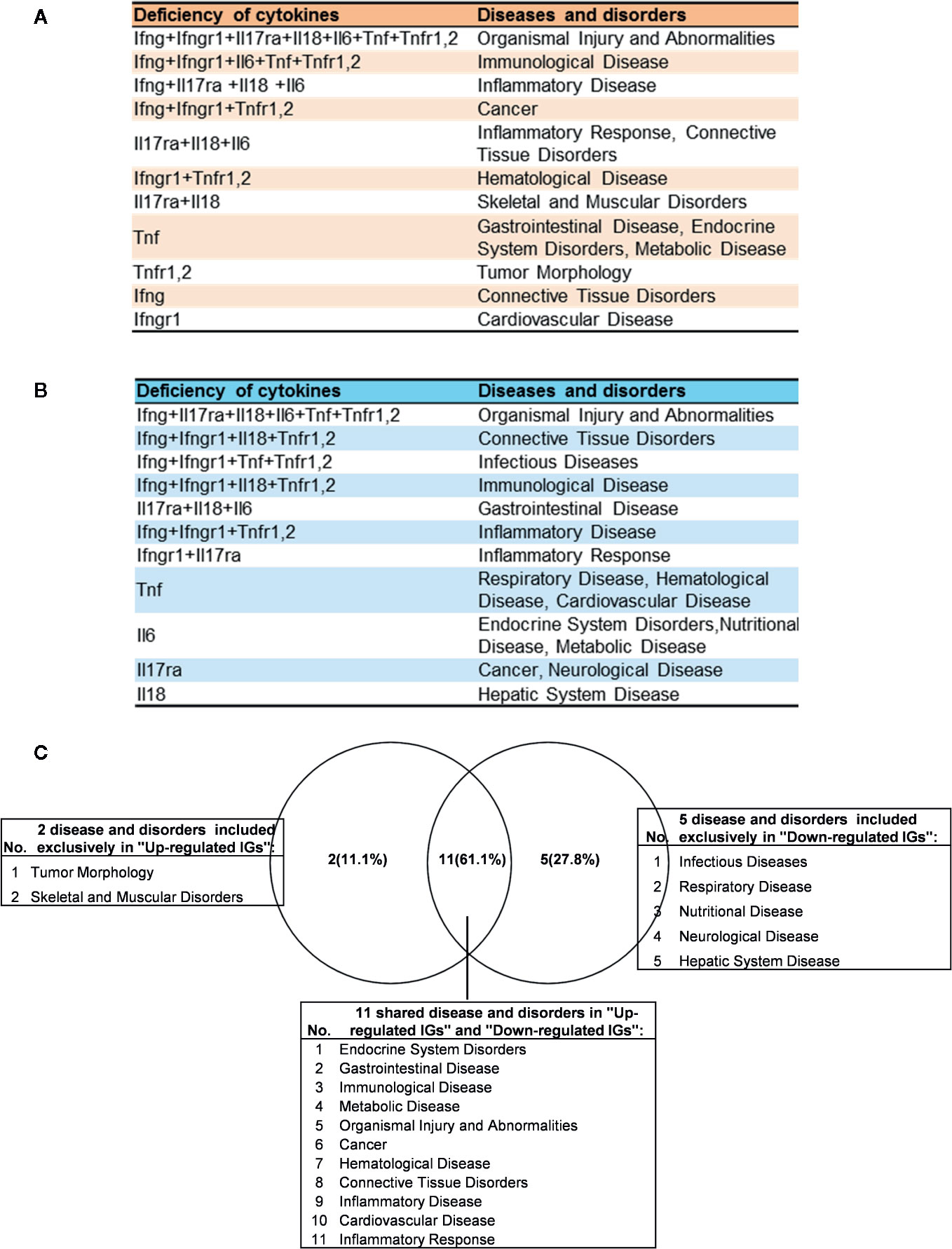
Figure 8 The up- and down-regulated innatomic genes (IGs) share inflammatory diseases in deficiencies of proinflammatory cytokines (PCs). (A) The upregulated IGs in PCs KO microarrays share 8 different diseases and disorders including inflammatory diseases. (B) The down-regulated IGs in PCs KO microarrays share 7 different diseases and disorders including infectious diseases (increased) and inflammatory diseases. (C) Venn diagram showed the upregulated IGs and the downregulated IGs share 11 diseases or disorders, two diseases or disorders included exclusively in “Up-regulated IGs” and five disease or disorders included exclusively in “Down-regulated IGs”. In them, cardiovascular disease is shared in up- and down-regulated IGs.
Of note, as shown in Figure 8C, comparing cytokine deficiencies-downregulated IGs related diseases to that of cytokine deficiencies-upregulated IGs related diseases, the results showed that first, the cytokine deficiencies-downregulated IGs related disease group has five diseases including infectious disease (increased, Figure S1), respiratory disease, nutritional disease, neurological disease, and hepatic system disease; second, cytokine deficiencies-upregulated IGs related disease group has two diseases such as tumor morphology and skeletal and muscular disorders; and third, the two groups share 11 diseases, suggesting that current therapeutic strategies have significant problems; and there are urgent needs to re-shape the strategies in designing cytokine blockage therapies. Taken together, our results have demonstrated that first, the deficiencies of PCs not only upregulate IGs and innate immune signaling pathways but also increase the likelihood to develop certain immune, inflammatory diseases and cancers; and second, the deficiencies of PCs not only downregulate IGs and innate immune signaling pathways but also decrease the likelihood to develop certain immune, inflammatory diseases and cancers and provide novel valuable guidance for cytokine blockage therapies.
We made an interesting finding that, the IGs were more upregulated than downregulated when poor responder or non-responder compared with the group responders or good responders in several microarray datasets of patients receiving monoclonal antibody (Mab) therapy (Table 2B). Then, we hypothesized that in drug none-responders, “suppressed cytokines or innate immune regulator molecules” were upregulated. To examine this hypothesis, Tnf KO (GSE43145, Tnf KO Gan mice vs. Gan mice) and anti-TNF therapy (GSE111761, non-responders vs. responders) microarrays were compared. The IPA results of the pathways and the involved IGs showed that four upregulated pathways in Tnf KO microarray also were all shared with that in patients receiving anti-TNF therapy; and several upregulated IGs involved in the pathways were shared (Figure 9A). IPA results of up-regulated innatomic genes in anti-TNF therapy microarray were listed in Table S10. All the significant differential expressed genes (p<0.05, ∣logFC∣>1) of these two microarrays were further analyzed by using GSEA for pathway enrichments (68, 69). The result showed that a total of 17 pathways were shared by Tnf KO and anti-TNF therapy microarrays including inflammatory response and cytokine signalings, which were activated in these two microarrays (Figure 9B). The Venn diagram showed that 97 significant differentially expressed IGs were shared by Tnf KO and anti-TNF therapy microarrays. The 97 genes were analyzed for additional enrichment analysis by using the Metascape database. The result showed that the five pathways such as type I interferon signaling pathway, lymphocyte differentiation, Adaptive Immune System, regulation of innate immune response, response to bacterium etc, were the significantly enriched GO or pathways (Figure 9C). Taken together, based on not only from the IPA results of significant differentially expressed IGs, but also the integrated analysis results from the GSEA and Metascape of all shared significant differentially expressed genes, our analyses have demonstrated that the new inflammatory responses will be activated when TNF is suppressed, thus supporting our novel hypothesis that the “2nd inflammatory wave” contributes to non-responder patients receiving anti-TNF Mab therapy partially. In fact, our findings were well correlated with the report about GSE111761 that in patients with Crohn’s disease receiving anti-TNF therapy, there were significant upregulations of mucosal Il-23p19, Il23R and Il17A in non-responders, but not in responders.
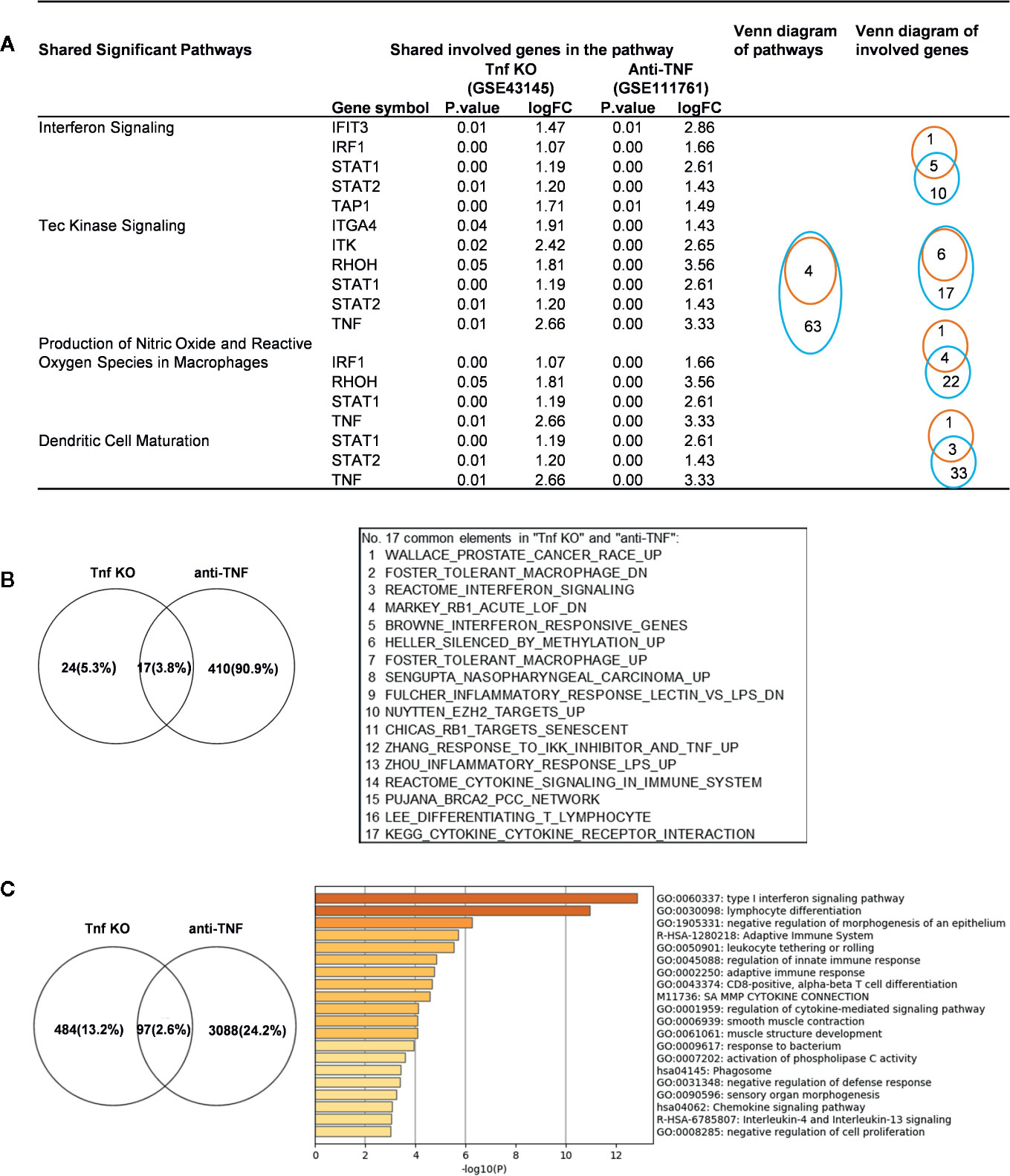
Figure 9 Up-regulated innatomic genes (IGs) in Tnf KO (GSE43145, Tnf KO Gan mice versus (vs.) Gan mice) and anti-TNF therapy (GSE111761, non-responder vs. responder) microarrays shared pathways. (A) IPA results of the pathways and the involved genes from innatome were compared. Some genes were involved more than two pathways. Orange line represents Tnf KO and blue line represents Anti-TNF therapy. (B) All the significant differential expressed genes (p<0.05, ∣logFC∣>1) of these two microarrays were enrichment analyzed by using GSEA. The result showed a total of 17 pathways were shared by Tnf KO and anti-TNF therapy microarrays. The result showed that except for interferon signaling, several cancer related, inflammatory response and cytokine signalings, were activated in these two microarrays. (C) Venn diagram showed 97 significantly differentially expressed genes were shared by Tnf KO and anti-TNF therapy microarrays. The 97 genes were carried out enrichment analysis by using metascape. The result showed the type I interferon signaling pathway, lymphocyte differentiation, Adaptive Immune System, regulation of innate immune response, response to bacterium etc. were the significantly enriched GO or pathways. Table S10 is IPA results of up- and down-regulated innatomic genes in anti-TNF therapy microarray.
Mitochondrial ROS (mtROS) are signaling molecules, which drive inflammatory cytokine production (70) and T cell activation (70, 71). In addition, CVDs, cancers, and autoimmune diseases all share a common feature of increased mtROS levels (72). Our recent study shed light on this important question and found that, during endothelial cell activation, mtROS could be upregulated in a proton leak-coupled, but ATP synthesis-uncoupled manner (72–75). As a result, endothelial cells could upregulate mtROS production for physiological endothelial cell activation without compromising mitochondrial membrane potential and ATP generation, and consequently without causing mitochondrial damage and endothelial cell death. Thus, a novel pathophysiological role of proton leak in driving mtROS production was uncovered for low-grade endothelial cell activation, patrolling immunosurveillance cell trans-endothelial migration and low-grade chronic inflammation without compromising cellular survival (72–76). One of the most evident features of the inflammatory response is the generation of a pro-oxidative environment due to the production of high fluxes of pro-oxidant species (77). We hypothesized that deficiencies of PCs and regulators upregulate some oxidative stress regulators. To test this hypothesis, we examined 165 ROS regulators collected in the Gene Set Enrichment Analysis (GSEA) database (https://www.gsea-msigdb.org/gsea/index.jsp). As shown in Table 5, 96 ROS regulators were modulated by deficiencies of PCs, transcription factors and miRs. Table S11 listed the detail expression changes of ROS regulator. Deficiencies of IL6, STAT1, NF-kB-Rela, miR155 resulted in no downregulation of ROS regulators. Venn Diagram analysis (Figure 10A) showed that: 1) deficiencies of proinflammatory regulators caused downregulation of 25 ROS regulators; 2) deficiencies of proinflammatory regulators caused upregulation of 32 ROS regulators; and 3) 39 ROS regulators were shared by upregulated and downregulated in deficiencies of proinflammatory regulators, suggesting that these 39 ROS regulators are required for the functions of modulated IGs in regardless of expressional levels of 9 proinflammatory regulators. Of note, the rest of the 69 ROS regulators were not significantly modulated in the deficiencies of major proinflammatory regulators.
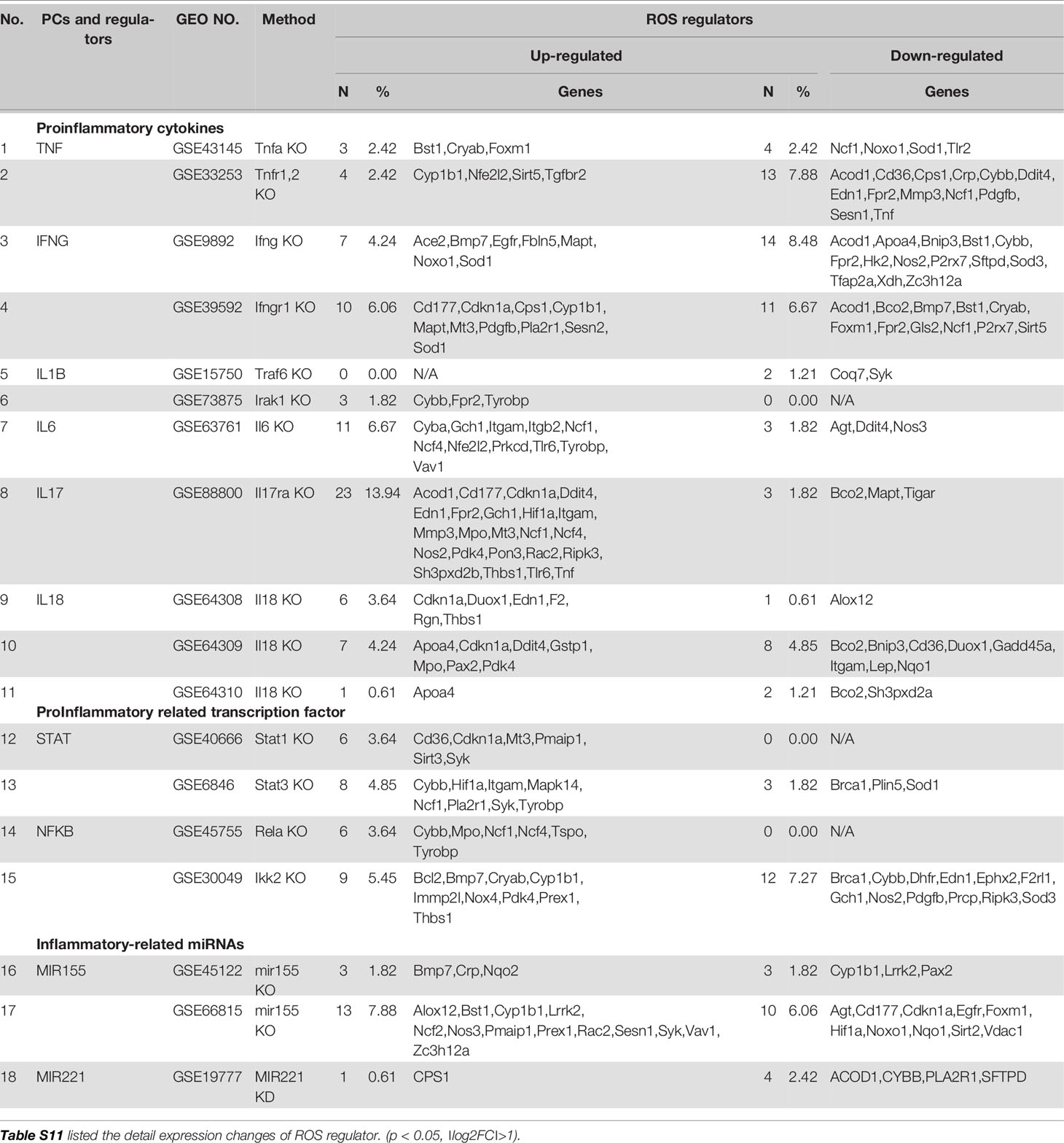
Table 5 165 ROS regulators were analyzed in inflammatory molecules KO microarrays.
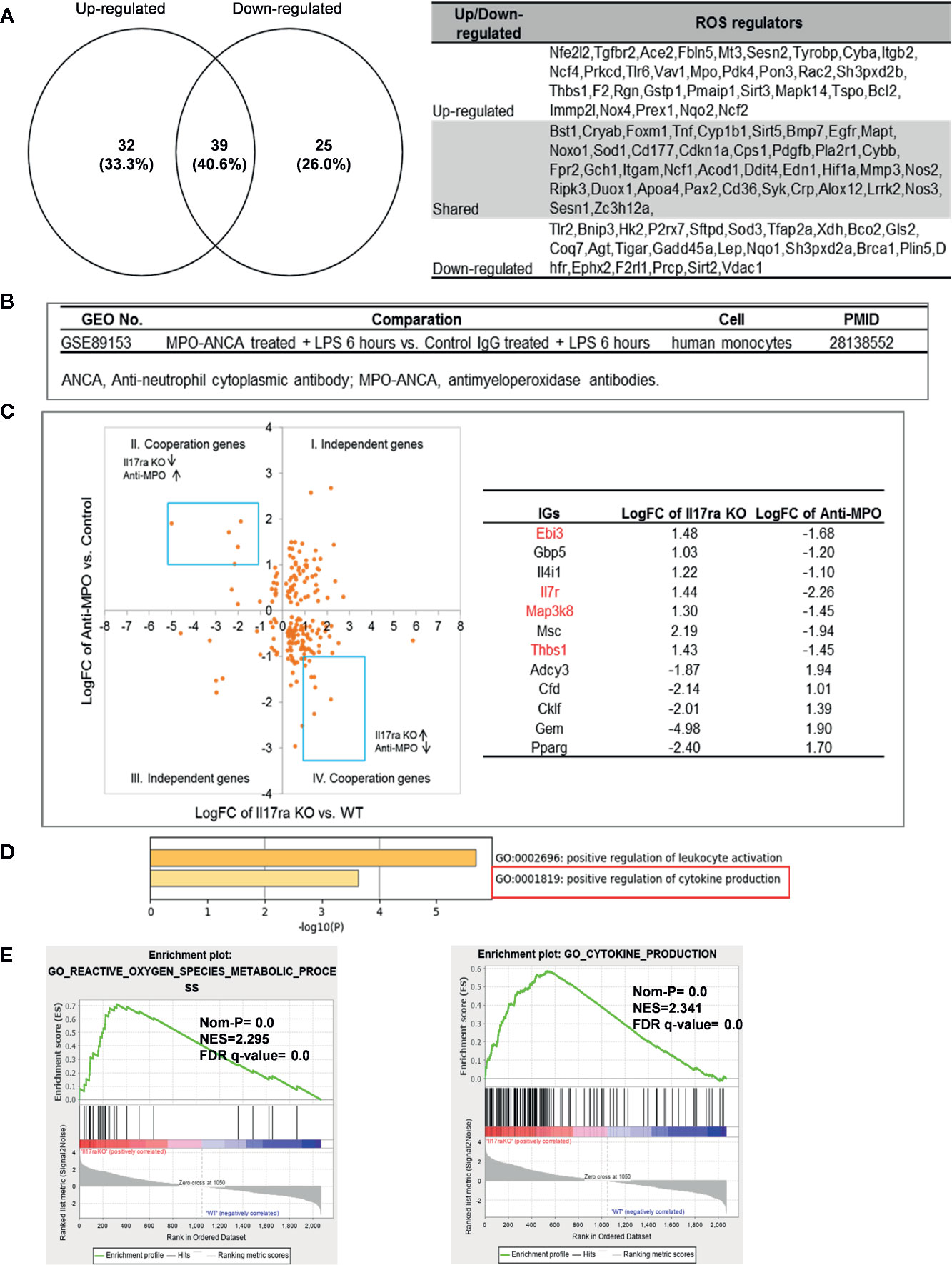
Figure 10 Mechanism: up-regulated reactive oxygen species (ROS) regulators can drive the upregulation of “suppressed innatomic genes”. (A) 32 ROS regulators were up-regulated, 25 ROS regulators were down-regulated and 39 ROS regulators were common in up-regulated and down-regulated. (p<0.05, ∣log2FC∣>1). Table S11 listed the detailed expression changes of ROS regulators. (B) Anti-myeloperoxidase (MPO) antibodies microarray GSE89153 was searched in GEO database to analyze the cooperation between ROS regulator MPO and proinflammatory cytokine Il17ra. (C) Cooperation analysis shows 12 genes were highly cooperatively regulated by MPO and Il17ra (Blue line box, p<0.05, ∣log2FC∣>1). (D) GO enrichment analyzed from Metascape software showed four in seven down-regulated genes (red in (b) were enriched in GO:0001819: positive regulation of cytokine production. (E) GSEA results of all the significant differential expression genes (p<0.05, ∣log2FC∣>1) in Il17ra KO showed the REACTIVE_OXYGEN_SPECIES_METABOLIC_PROCESS and POSITIVE_REGULATION_OF_CYTOKINE_PRODUCTION genes significantly enriched in the Il17ra KO groups. NES, Normalized Enrichment Score; FDR, false discovery rate.
Our detailed results showed that deficiencies of IL17RA, IL18 and NF-kB Rela result in upregulated heme peroxidase myeloperoxidase (MPO) expression (78). MPO and MPO-derived oxidants have been shown to contribute to the formation of foam cells, endothelial dysfunction and apoptosis, the activation of latent matrix metalloproteinases, and the expression of tissue factor that can promote the development of vulnerable plaque. Then, we hypothesized that IGs upregulated in deficiency of IL17RA are also upregulated by MPO. To examine this hypothesis, we found an anti-MPO antibodies microarray GSE89153 (Figure 10B) from the NIH-GEO database to analyze the cooperation between ROS generator MPO (Figure 10C) and proinflammatory cytokine receptor IL17RA. The cooperation analysis, as we reported (55), showed that among 197 common significantly differentially expressed IGs in IL17RA KO and Anti-MPO microarrays, 121 IGs (61.42%) were cooperatively regulated (p<0.05). 12 genes were highly cooperatively regulated (Blue line box, p<0.05, ∣log2FC∣>1). The GO enrichment (http://geneontology.org/) was analyzed by using Metascape software and GSEA (68, 69). The result showed that four in seven down-regulated genes (marked in red in Figure 10C) were enriched in GO:0001819: positive regulation of cytokine production (Figure 10D). The GSEA results of all the significant differential expression genes (p<0.05, ∣log2FC∣>1) in IL17RA KO showed the REACTIVE_OXYGEN_SPECIES_METABOLIC_PROCESS and POSITIVE_REGULATION_OF_CYTOKINE_PRODUCTION genes significantly enriched in the IL17RA KO groups with notable normalized enrichment score (NES; https://en.wikipedia.org/wiki/Gene_set_enrichment_analysis) and false discovery rate (FDR; https://en.wikipedia.org/wiki/False_discovery_rate) (Figure 10E). Our results have demonstrated a novel mechanism that deficiencies of IL-17RA and presumably IL-18 promote MPO upregulation, which drives a ROS-dependent inflammation.
These progresses lead to the development of many cytokine blockage-based therapies for inflammatory diseases and CVDs. The CANTOS trial with the Mab Canakinumab to block proinflammatory cytokine IL-1β was a recent success in treating coronary artery disease (30). However, recent reports from our and others’ teams suggest that inhibition of one proinflammatory regulators such as cytokines or microRNAs leads to new waves of inflammation. To approach these types of inflammation paradoxes, we performed an extensive -omics data mining analyses with the method that we pioneered in 2004 (56, 57, 79) and made a set of significant findings: 1) PCs suppress IGs; and upregulated IGs in the deficiencies of IFNγ, IFNγR1, IL-17A, STAT3 and miR155 are more than that after deficiencies of TNFα, IL-1β, IL-6, IL-18, STAT1, NF-kB, and miR221; 2) IFNγ/IFNγR1 and IL-17RA inhibit 10, 59 and 39 pathways, respectively; in contrast TNFα, IL-6 and IL-18 inhibits four to five pathways; 3) The IFNγ-promoted and -suppressed programs have 4 shared pathways, IFNγR1-promoted and -suppressed programs have 11 shared pathways; and miR155-promoted and -suppressed programs have 13 shared pathways, suggesting negative-feedback mechanisms in their regulatory pathways for IGs; 4) Deficiencies of PCs and transcription factors-suppressed, -promoted programs share the signaling pathways and likelihood to develop 11 diseases including cardiovascular disease; 5) There are shared IGs and pathways between deficiency of TNFα in mice and anti-TNF therapy in clinical patients; 6) Mechanistically, up-regulated ROS regulators such as MPO caused by suppression of major proinflammatory molecules can drive the upregulation of “suppressed IGs”.
The original microarray experiments used different cells, which prevented us from comparing the effects of proinflammatory regulators in regulating the expressions of IGs in the same cell types. Although our database mining approach was not ideal, however, as the first step to fill in the important knowledge gap this approach was justified. Actually, this was a common practice that we (50) and others (51) often used in studying gene expression in non-ideal, heterogenous peripheral blood mononuclear cell populations (PBMCs) in pathophysiological conditions versus healthy conditions, which are actually composed of many cell types, such as B cells (~15 %), T cells (~70 %), monocytes (~5 %), and natural killer (NK) cells (~10 %) among others (80).
Indeed, in addition to PC regulation of IGs transcription examined in our study, PCs could also regulate innate immune regulators in several other modes: 1) mRNA stability (81); 2) riboclustering (82); 3) alternative splicing (83); 4) microRNA regulation (84); 5) long non-coding RNA regulation (85); 6) circular RNAs regulation (86); 7) protein translation (87); 8) protein neddylation (88); 9) ubiquitination-proteasome regulation (89); 10) epigenetic regulation (90); and 11) immune metabolism and innate immune memory (91, 92). The proof of principles demonstrated in examination of inflammatory paradoxes performed in the current manuscript are far-reaching and can be extended to other fields of study such as cancers, infectious diseases such as COVID-19 and various pathologies.
To summarize our findings presented here, we propose a novel working model (Figure 11) to integrate these results as follows: first, since proinflammatory cytokines and regulators are interconnected through evolution, single cytokine blockade therapies result in significant upregulation of a long list of IGs and signaling pathways, presumably the “second wave of inflammation” as we proposed (32). The second wave of inflammation suppressed by PCs and regulators has never been identified in this comprehensive manner. The second wave of inflammation may be the underlying mechanisms for MHO, adverse effects observed in patients receiving Mab therapies blocking PCs. Novel therapeutic strategies need to be developed based on our findings to combine several related immune therapies as demonstrated by the synergy between immune checkpoint receptor CD279 blocking therapy and anti-TNFα therapy (93); second, the two groups of new IGs and their pathways have been identified as novel therapeutic targets including: i) IGs and their pathways shared by upregulated IGs and suppressed IGs after deficiencies of PCs and regulators; and ii) IGs and the pathways upregulated after deficiencies of PCs and regulators; third, deficiencies of PCs and regulators increase inflammatory diseases in up-regulated IGs. The new problems need to be seriously considered when designing blockade therapies for PCs and regulators; one new mechanism has been identified for modulating the expressions of IGs carrying out the second wave of inflammation: PCs blockade can regulate the expression of ROS regulators and up-regulated ROS regulators can drive the upregulation of “suppressed IGs”. Taken together, we propose that multiple pathway convergent points can be new therapeutic targets for the future development of novel inflammation modulation therapies. The proof of principles demonstrated in the examination of inflammatory paradoxes performed in the current manuscript are far-reaching and can be extended to other fields of study such as cancers, infectious diseases such as COVID-19 and various pathologies.
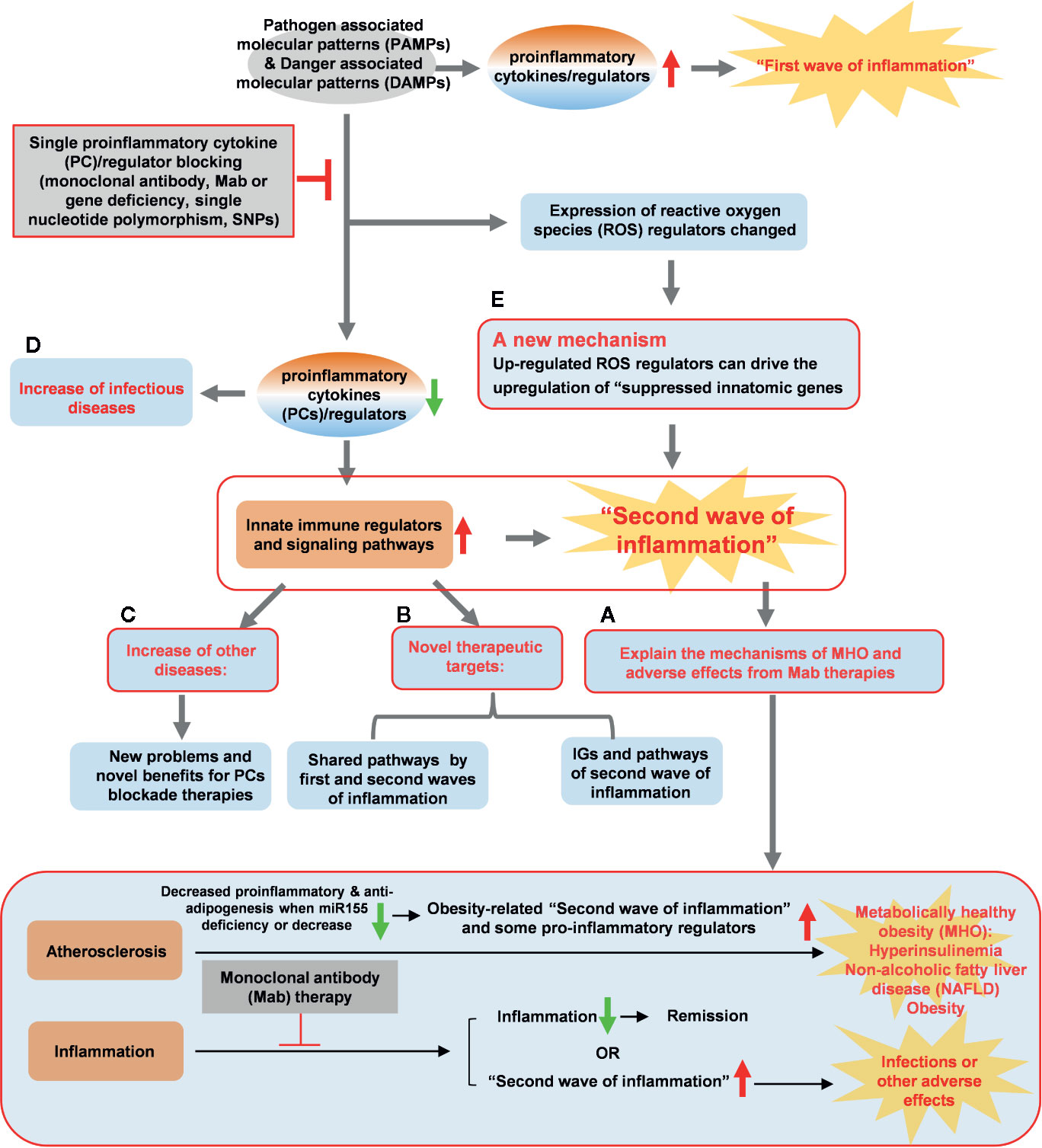
Figure 11 A new working model is proposed: Blocking proinflammatory regulators induces the proinflammatory regulators-suppressed “second waves of inflammation”. Single cytokine blockade therapies result in significant upregulation of innate immune regulators and signaling pathways, presumably “second wave of inflammation” as we proposed. (A) The second wave of inflammation may be the underlying mechanisms for metabolically healthy obesity (MHO), adverse effects observed in patients receiving monoclonal antibody (Mab) therapies in blocking proinflammatory cytokines. (B) The two groups of new innatome genes (IGs) and their pathways have been identified as novel therapeutic targets. (C) Deficiencies of proinflammatory cytokines and regulators upregulate innate immunomic genes reveal new problems and novel benefits for proinflammatory cytokine (PCs) blockade therapies. (D) Down-regulated IGs can increase infectious diseases. (E) PCs blockade can regulate the expression of ROS regulators and up-regulated ROS regulators can upregulate expression of IGs. A new mechanism, ROS regulators, has been identified for modulating the expressions of IGs carrying out the second wave of inflammation.
One limitation of the current study is that due to the low throughput nature of verification techniques, we could not verify every result we identified with the analyses of high throughput data. We acknowledge that carefully designed in vitro and in vivo experimental models will be needed to verify the PCs and regulators deficiencies-upregulated IGs further and underlying mechanisms we report here. In addition, when -omics data with well-controlled clinical samples become available, we need to further verify and consolidate some of our new findings identified in mice here. Our findings nevertheless provide novel insights on the roles of upregulated IGs in the pathogenesis of inflammatory diseases, novel pathways underlying the multi-pathway convergent point suppression therapeutics models as well as new targets for the future therapeutic interventions for various inflammations.
All the datasets used in this study are publicly available. The analyzed results in this study are included within the article and the Supplementary Materials.
ML carried out the data gathering, data analysis and prepared tables and figures. JS, RZ, YSh, YSu, WY, JW, LuL, CD, CJ, FS, YL, KX, LiL, XW, XJ, and HW aided with analysis of the data. XY supervised the experimental design, data analysis, and manuscript writing. All authors contributed to the article and approved the submitted version.
This work was partially supported by NIH grants to HW and XY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ML was supported by a fellowship from the School of Basic Medical Science, Shanxi Medical University.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.554301/full#supplementary-material
Supplementary Figure S1 | The heatmap of diseases related to down-regulated innatomic genes from IPA analysis comparison.
Supplementary Table S1 | Nine proinflammatory cytokine blocking monoclonal antibody therapies and clinical uses.
Supplementary Table S2 | Ten additional inflammation paradoxes related to this study which have been reported.
Supplementary Table S3 | The IGs and ROS regulators gene-lists used in this study.
Supplementary Table S4 | The expression changes of housekeeping genes selected in this study.
Supplementary Table S5 | The expression changes of up-regulated and down-regulated innatomic genes in deficiencies of proinflammatory cytokines and regulators. (p<0.05, ∣Log2FC∣>1)
Supplementary Table S6 | The expression changes of up-regulated and down-regulated innatomic genes in Mab therapy microarrays. (p<0.05, ∣Log2FC∣>1)
Supplementary Table S7 | IPA results of up-regulated innatomic genes in deficiencies of proinflammatory cytokines and regulators. (∣Z score∣> 2)
Supplementary Table S8 | IPA results of down-regulated innatomic genes in deficiencies of proinflammatory cytokines and regulators. (∣Z score∣> 2)
Supplementary Table S9 | Pathways from IPA results of up- and down-regulated innatomic genes in deficiencies of TFs. (∣Z score∣> 2)
Supplementary Table S10 | IPA results of up- and down-regulated innatomic genes in anti-TNF therapy microarray.
Supplementary Table S11 | The expression changes of ROS regulators in deficiencies of proinflammatory cytokines and regulators. (p<0.05, ∣Log2FC∣>1)
CVDs, cardiovascular diseases; IGs, innatomic genes (innate immune genes/regulators); PCs, proinflammatory cytokines/regulators; ROS, reactive oxygen species; Treg, regulatory T cells; IL-1β interleukin-1β; miRs: microRNAs; miR155, microRNA-155; KO, knock-out; MHO, metabolically healthy obesity; RA, rheumatoid arthritis; TNFα, tumor necrosis factor-α; Mab, monoclonal antibody; IL-6R, IL-6 receptor; DAMPs, danger associated molecular patterns; PAMPs, pathogen associated molecular patterns; NCDs, non-communicable diseases; NIH, National Institutes of Health; NCBI, National Center for Biotechnology Information; GEO, Gene Expression Omnibus; IPA: Ingenuity Pathway Analysis; GSEA, Gene Set Enrichment Analysis; IFNγ, interferon-γ; STAT1, signal transducer and activator of transcription protein 1; IKK2, Inhibitor of NFKB kinase subunit-β; TRAF6, TNF receptor associated factor 6; IRAK1, interleukin-1 receptor-associated kinase 1; PBMCs, peripheral blood mononuclear cell populations; Th1, type 1 T helper cells; HMGB1, high mobility group box 1; STAT4, signal transducer and activator of transcription 4; Erk1/2, extracellular signal-regulated protein kinases 1 and 2; Pyk2, proline-rich tyrosine kinase 2; CrkL, CRK like proto-oncogene; iCOS, inducible T-cell co-stimulator; NF-AT, nuclear factor of activated T cells; IRF, interferon regulatory factor; HGF, hepatocyte growth factor; JAK, Janus kinases; MIF: macrophage migration inhibitory factor; PAK, p21-activated kinase; PI3K, phosphatidylinositol-3-kinase; AKT, protein kinase B; cAMP, cyclic adenosine monophosphate; TFs, transcription factors; PD-1, programmed cell death protein-1; DC, dendritic cells; mtROS, mitochondrial ROS; MPO, myeloperoxidase; NES, normalized enrichment score; FDR, false discovery rate; NK, natural killer.
1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation (2017) 135:e146–603. doi: 10.1161/CIR.0000000000000491
2. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers (2019) 5:56. doi: 10.1038/s41572-019-0106-z
3. Yin Y, Li X, Sha X, Xi H, Li YF, Shao Y, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol (2015) 35:804–16. doi: 10.1161/ATVBAHA.115.305282
4. Li X, Fang P, Li Y, Kuo YM, Andrews AJ, Nanayakkara G, et al. Mitochondrial Reactive Oxygen Species Mediate Lysophosphatidylcholine-Induced Endothelial Cell Activation. Arterioscler Thromb Vasc Biol (2016) 36:1090–100. doi: 10.1161/ATVBAHA.115.306964
5. Fang P, Zhang D, Cheng Z, Yan C, Jiang X, Kruger WD, et al. Hyperhomocysteinemia potentiates hyperglycemia-induced inflammatory monocyte differentiation and atherosclerosis. Diabetes (2014) 63:4275–90. doi: 10.2337/db14-0809
6. Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X, et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ Res (2016) 118:1525–39. doi: 10.1161/CIRCRESAHA.116.308501
7. Yang J, Fang P, Yu D, Zhang L, Zhang D, Jiang X, et al. Chronic Kidney Disease Induces Inflammatory CD40+ Monocyte Differentiation via Homocysteine Elevation and DNA Hypomethylation. Circ Res (2016) 119:1226–41. doi: 10.1161/CIRCRESAHA.116.308750
8. Ferrer LM, Monroy AM, Lopez-Pastrana J, Nanayakkara G, Cueto R, Li YF, et al. Caspase-1 Plays a Critical Role in Accelerating Chronic Kidney Disease-Promoted Neointimal Hyperplasia in the Carotid Artery. J Cardiovasc Transl Res (2016) 9:135–44. doi: 10.1007/s12265-016-9683-3
9. Monroy MA, Fang J, Li S, Ferrer L, Birkenbach MP, Lee IJ, et al. Chronic kidney disease alters vascular smooth muscle cell phenotype. Front Biosci (Landmark Ed) (2015) 20:784–95. doi: 10.2741/4337
10. Sun Y, Johnson C, Zhou J, Wang L, Li YF, Lu Y, et al. Uremic toxins are conditional danger- or homeostasis-associated molecular patterns. Front Biosci (Landmark Ed) (2018) 23:348–87. doi: 10.2741/4595
11. Sha X, Meng S, Li X, Xi H, Maddaloni M, Pascual DW, et al. Interleukin-35 Inhibits Endothelial Cell Activation by Suppressing MAPK-AP-1 Pathway. J Biol Chem (2015) 290:19307–18. doi: 10.1074/jbc.M115.663286
12. Shao Y, Cheng Z, Li X, Chernaya V, Wang H, Yang XF. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction–a novel mechanism for maintaining vascular function. J Hematol Oncol (2014) 7:80. doi: 10.1186/s13045-014-0080-6
13. Li X, Wang L, Fang P, Sun Y, Jiang X, Wang H, et al. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J Biol Chem (2018) 293:11033–45. doi: 10.1074/jbc.RA118.002752
14. Li A, Sun Y, Drummer CT, Lu Y, Yu D, Zhou Y, et al. Increasing Upstream Chromatin Long-Range Interactions May Favor Induction of Circular RNAs in LysoPC-Activated Human Aortic Endothelial Cells. Front Physiol (2019) 10:433. doi: 10.3389/fphys.2019.00433
15. Lopez-Pastrana J, Ferrer LM, Li YF, Xiong X, Xi H, Cueto R, et al. Inhibition of Caspase-1 Activation in Endothelial Cells Improves Angiogenesis: A NOVEL THERAPEUTIC POTENTIAL FOR ISCHEMIA. J Biol Chem (2015) 290:17485–94. doi: 10.1074/jbc.M115.641191
16. Zhang CE, Wei W, Liu YH, Peng JH, Tian Q, Liu GP, et al. Hyperhomocysteinemia increases beta-amyloid by enhancing expression of gamma-secretase and phosphorylation of amyloid precursor protein in rat brain. Am J Pathol (2009) 174:1481–91. doi: 10.2353/ajpath.2009.081036
17. Zhang D, Fang P, Jiang X, Nelson J, Moore JK, Kruger WD, et al. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS-deficient mice. Circ Res (2012) 111:37–49. doi: 10.1161/CIRCRESAHA.112.269472
18. Fang P, Li X, Shan H, Saredy JJ, Cueto R, Xia J, et al. Ly6C(+) Inflammatory Monocyte Differentiation Partially Mediates Hyperhomocysteinemia-Induced Vascular Dysfunction in Type 2 Diabetic db/db Mice. Arterioscler Thromb Vasc Biol (2019) 39:2097–119. doi: 10.1161/ATVBAHA.119.313138
19. Xiong Z, Song J, Yan Y, Huang Y, Cowan A, Wang H, et al. Higher expression of Bax in regulatory T cells increases vascular inflammation. Front Biosci (2008) 13:7143–55. doi: 10.2741/3217
20. Xiong Z, Yan Y, Song J, Fang P, Yin Y, Yang Y, et al. Expression of TCTP antisense in CD25(high) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis (2009) 203:401–8. doi: 10.1016/j.atherosclerosis.2008.07.041
21. Yang WY, Shao Y, Lopez-Pastrana J, Mai J, Wang H, Yang XF. Pathological conditions re-shape physiological Tregs into pathological Tregs. Burns Trauma (2015) 3(1):1. doi: 10.1186/s41038-015-0001-0
22. Xu K, Yang WY, Nanayakkara GK, Shao Y, Yang F, Hu W, et al. gaTa3, hDac6, and Bcl6 regulate FOXP3+ Treg Plasticity and Determine Treg conversion into either novel antigen-Presenting cell-like Treg or Th1-Treg. Front Immunol (2018) 9:45. doi: 10.3389/fimmu.2018.00045
23. Nelson J, Wu Y, Jiang X, Berretta R, Houser S, Choi E, et al. Hyperhomocysteinemia suppresses bone marrow CD34+/VEGF receptor 2+ cells and inhibits progenitor cell mobilization and homing to injured vasculature-a role of beta1-integrin in progenitor cell migration and adhesion. FASEB J (2015) 29:3085–99. doi: 10.1096/fj.14-267989
24. Li YF, Huang X, Li X, Gong R, Yin Y, Nelson J, et al. Caspase-1 mediates hyperlipidemia-weakened progenitor cell vessel repair. Front Biosci (Landmark Ed) (2016) 21:178–91. doi: 10.2741/4383
25. Shao Y, Chernaya V, Johnson C, Yang WY, Cueto R, Sha X, et al. Metabolic Diseases Downregulate the Majority of Histone Modification Enzymes, Making a Few Upregulated Enzymes Novel Therapeutic Targets-”Sand Out and Gold Stays”. J Cardiovasc Trans Res (2016) 9(1):49–66. doi: 10.1007/s12265-015-9664-y
26. Lu Y, Sun Y, Drummer C IV, Nanayakkara GK, Shao Y, Saaoud F, et al. Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells - Novel qualification markers for chronic disease risk factors and conditional DAMPs. Redox Biol (2019) 24:101221. doi: 10.1016/j.redox.2019.101221
27. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol (2017) 39:517–28. doi: 10.1007/s00281-017-0639-8
28. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014
29. Yang XF, Yin Y, Wang H. Vascular Inflammation and Atherogenesis are Activated via Receptors for Pamps and Suppressed by Regulatory T Cells. Drug Discov Today Ther Strat (2008) 5:125–42. doi: 10.1016/j.ddstr.2008.11.003
30. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
31. Virtue A, Johnson C, Lopez-Pastrana J, Shao Y, Fu H, Li X, et al. MicroRNA-155 Deficiency Leads to Decreased Atherosclerosis, Increased White Adipose Tissue Obesity, and Non-alcoholic Fatty Liver Disease: A NOVEL MOUSE MODEL OF OBESITY PARADOX. J Biol Chem (2017) 292:1267–87. doi: 10.1074/jbc.M116.739839
32. Johnson C, Drummer CT, Virtue A, Gao T, Wu S, Hernandez M, et al. Increased Expression of Resistin in MicroRNA-155-Deficient White Adipose Tissues May Be a Possible Driver of Metabolically Healthy Obesity Transition to Classical Obesity. Front Physiol (2018) 9:1297. doi: 10.3389/fphys.2018.01297
33. Singh JA, Saag KG, Bridges SL Jr., Akl EA, Bannuru RR, Sullivan MC, et al. American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol (2015) 2016) 68:1–26. doi: 10.1002/art.39480
34. Monoclonal Antibodies. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases (2012–).
35. Burmester GR, Lin Y, Patel R, van Adelsberg J, Mangan EK, Graham NM, et al. Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): a randomised, double-blind, parallel-group phase III trial. Ann Rheum Dis (2017) 76:840–7. doi: 10.1136/annrheumdis-2016-210310
36. Cleynen I, Vermeire S. Paradoxical inflammation induced by anti-TNF agents in patients with IBD. Nat Rev Gastroenterol Hepatol (2012) 9:496–503. doi: 10.1038/nrgastro.2012.125
37. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
38. de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet (2017) 49:256–61. doi: 10.1038/ng.3760
39. Fuster JJ, Walsh K. Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circ Res (2018) 122:523–32. doi: 10.1161/CIRCRESAHA.117.312115
40. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol (2018) 15:505–22. doi: 10.1038/s41569-018-0064-2
41. Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front Med (Lausanne) (2018) 5:316. doi: 10.3389/fmed.2018.00316
42. Wang X, Li YF, Nanayakkara G, Shao Y, Liang B, Cole L, et al. Lysophospholipid Receptors, as Novel Conditional Danger Receptors and Homeostatic Receptors Modulate Inflammation-Novel Paradigm and Therapeutic Potential. J Cardiovasc Transl Res (2016) 9:343–59. doi: 10.1007/s12265-016-9700-6
43. Robinson J, Swift-Scanlan T, Salyer J, Jones T. The Obesity Paradox in Sepsis: A Theoretical Framework. Biol Res Nurs (2020) 22:287–94. doi: 10.1177/1099800420905889
44. Freese J, Klement RJ, Lotzerich H. The inflammation paradox: Why are Tsimane protected against Western diseases while Westerners are not? F1000Res (2018) 7:252. doi: 10.12688/f1000research.14052.2
45. Chavan AR, Griffith OW, Wagner GP. The inflammation paradox in the evolution of mammalian pregnancy: turning a foe into a friend. Curr Opin Genet Dev (2017) 47:24–32. doi: 10.1016/j.gde.2017.08.004
46. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
47. Lai B, Wang J, Fagenson A, Sun Y, Saredy J, Lu Y, et al. Twenty Novel Disease Group-Specific and 12 New Shared Macrophage Pathways in Eight Groups of 34 Diseases Including 24 Inflammatory Organ Diseases and 10 Types of Tumors. Front Immunol (2019) 10:2612. doi: 10.3389/fimmu.2019.02612
48. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature (2017) 542:177–85. doi: 10.1038/nature21363
49. Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res (2013) 41(Database issue):D1228–33. doi: 10.1093/nar/gks1147
50. Zhang R, Saredy J, Shao Y, Yao T, Liu L, Saaoud F, et al. End-stage renal disease is different from chronic kidney disease in upregulating ROS-modulated proinflammatory secretome in PBMCs - A novel multiple-hit model for disease progression. Redox Biol (2020) 20:101460. doi: 10.1016/j.redox.2020.101460
51. Li Y, Oosting M, Smeekens SP, Jaeger M, Aguirre-Gamboa R, Le KTT, et al. A Functional Genomics Approach to Understand Variation in Cytokine Production in Humans. Cell (2016) 167:1099–110 e14. doi: 10.1016/j.cell.2016.10.017
52. Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet (2013) 29:569–74. doi: 10.1016/j.tig.2013.05.010
53. de Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, et al. Evidence based selection of housekeeping genes. PLoS One (2007) 2:e898. doi: 10.1371/journal.pone.0000898
54. Wang L, Fu H, Nanayakkara G, Li Y, Shao Y, Johnson C, et al. Novel extracellular and nuclear caspase-1 and inflammasomes propagate inflammation and regulate gene expression: a comprehensive database mining study. J Hematol Oncol (2016) 9:122. doi: 10.1186/s13045-016-0351-5
55. Li YF, Nanayakkara G, Sun Y, Li X, Wang L, Cueto R, et al. Analyses of caspase-1-regulated transcriptomes in various tissues lead to identification of novel IL-1beta-, IL-18- and sirtuin-1-independent pathways. J Hematol Oncol (2017) 10:40. doi: 10.1186/s13045-017-0406-2
56. Ng B, Yang F, Huston DP, Yan Y, Yang Y, Xiong Z, et al. Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J Allergy Clin Immunol (2004) 114:1463–70. doi: 10.1016/j.jaci.2004.09.006
57. Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, et al. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol (2009) 22:311–22. doi: 10.1177/039463200902200208
58. Mai J, Nanayakkara G, Lopez-Pastrana J, Li X, Li YF, Wang X, et al. Interleukin-17A Promotes Aortic Endothelial Cell Activation via Transcriptionally and Post-translationally Activating p38 Mitogen-activated Protein Kinase (MAPK) Pathway. J Biol Chem (2016) 291:4939–54. doi: 10.1074/jbc.M115.690081
59. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol (2009) 27:165–97. doi: 10.1146/annurev.immunol.021908.132620
60. Cosmi L, Maggi L, Santarlasci V, Liotta F, Annunziato F. T helper cells plasticity in inflammation. Cytometry A (2014) 85:36–42. doi: 10.1002/cyto.a.22348
61. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature (2012) 484:514–8. doi: 10.1038/nature10957
62. Ivashkiv LB. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
63. Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene (2013) 532:1–12. doi: 10.1016/j.gene.2012.12.009
64. Shen H, Wu N, Nanayakkara G, Fu H, Yang Q, Yang WY, et al. Co-signaling receptors regulate T-cell plasticity and immune tolerance. Front Biosci (Landmark Ed) (2019) 24:96–132. doi: 10.2741/4710
65. Dai J, Fang P, Saredy J, Xi H, Ramon C, Yang W, et al. Metabolism-associated danger signal-induced immune response and reverse immune checkpoint-activated CD40(+) monocyte differentiation. J Hematol Oncol (2017) 10:141. doi: 10.1186/s13045-017-0504-1
66. Gotthardt D, Sexl V. STATs in NK-Cells: The Good, the Bad, and the Ugly. Front Immunol (2016) 7:694. doi: 10.3389/fimmu.2016.00694
67. Lee AJ, Ashkar AA. The Dual Nature of Type I and Type II Interferons. Front Immunol (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
68. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
69. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet (2003) 34:267–73. doi: 10.1038/ng1180
70. Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med (2011) 208:417–20. doi: 10.1084/jem.20110367
71. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18:488–98. doi: 10.1038/ni.3704
72. Li X, Fang P, Yang WY, Chan K, Lavallee M, Xu K, et al. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can J Physiol Pharmacol (2017) 95:247–52. doi: 10.1139/cjpp-2016-0515
73. Li X, Fang P, Li Y, Kuo Y-M, Andrews AJ, Nanayakkara G, et al. Mitochondrial Reactive Oxygen Species Mediate Lysophosphatidylcholine-Induced Endothelial Cell ActivationHighlights. Arteriosclerosis Thrombosis Vasc Biol (2016) 36:1090–100. doi: 10.1161/ATVBAHA.115.306964
74. Nanayakkara GK, Wang H, Yang X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation - A novel concept. Arch Biochem Biophys (2019) 662:68–74. doi: 10.1016/j.abb.2018.12.002
75. Cheng J, Nanayakkara G, Shao Y, Cueto R, Wang L, Yang WY, et al. Mitochondrial Proton Leak Plays a Critical Role in Pathogenesis of Cardiovascular Diseases. Adv Exp Med Biol (2017) 982:359–70. doi: 10.1007/978-3-319-55330-6_20
76. Li X, Shao Y, Sha X, Fang P, Kuo YM, Andrews AJ, et al. IL-35 (Interleukin-35) Suppresses Endothelial Cell Activation by Inhibiting Mitochondrial Reactive Oxygen Species-Mediated Site-Specific Acetylation of H3K14 (Histone 3 Lysine 14). Arterioscler Thromb Vasc Biol (2018) 38:599–609. doi: 10.1161/ATVBAHA.117.310626
77. Valacchi G, Virgili F, Cervellati C, Pecorelli A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front Physiol (2018) 9:858. doi: 10.3389/fphys.2018.00858
78. Teng N, Maghzal GJ, Talib J, Rashid I, Lau AK, Stocker R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep (2017) 22:51–73. doi: 10.1080/13510002.2016.1256119
79. Li X, Mai J, Virtue A, Yin Y, Gong R, Sha X, et al. IL-35 is a novel responsive anti-inflammatory cytokine–a new system of categorizing anti-inflammatory cytokines. PLoS One (2012) 7(3):e33628. doi: 10.1371/journal.pone.0033628
80. Corkum CP, Ings DP, Burgess C, Karwowska S, Kroll W, Michalak TI. Immune cell subsets and their gene expression profiles from human PBMC isolated by Vacutainer Cell Preparation Tube (CPT) and standard density gradient. BMC Immunol (2015) 16:48. doi: 10.1186/s12865-015-0113-0
81. Saaoud F, Wang J, Iwanowycz S, Wang Y, Altomare D, Shao Y, et al. Bone marrow deficiency of mRNA decaying protein Tristetraprolin increases inflammation and mitochondrial ROS but reduces hepatic lipoprotein production in LDLR knockout mice. Redox Biol (2020) 101609. doi: 10.1016/j.redox.2020.101609
82. Ganguly K, Giddaluru J, August A, Khan N. Post-transcriptional Regulation of Immunological Responses through Riboclustering. Front Immunol (2016) 7:161. doi: 10.3389/fimmu.2016.00161
83. Xiong Z, Shaibani A, Li YP, Yan Y, Zhang S, Yang Y, et al. Alternative splicing factor ASF/SF2 is down regulated in inflamed muscle. J Clin Pathol (2006) 59:855–61. doi: 10.1136/jcp.2005.032961
84. Wei Y, Schober A. MicroRNA regulation of macrophages in human pathologies. Cell Mol Life Sci (2016) 73:3473–95. doi: 10.1007/s00018-016-2254-6
85. Wang H, Liao S, Li H, Chen Y, Yu J. Long Non-coding RNA TUG1 Sponges Mir-145a-5p to Regulate Microglial Polarization After Oxygen-Glucose Deprivation. Front Mol Neurosci (2019) 12:215. doi: 10.3389/fnmol.2019.00215
86. Chen Y, Li C, Tan C, Liu X. Circular RNAs: a new frontier in the study of human diseases. J Med Genet (2016) 53:359–65. doi: 10.1136/jmedgenet-2016-103758
87. Mazumder B, Li X, Barik S. Translation control: a multifaceted regulator of inflammatory response. J Immunol (2010) 184:3311–9. doi: 10.4049/jimmunol.0903778
88. Chang FM, Reyna SM, Granados JC, Wei SJ, Innis-Whitehouse W, Maffi SK, et al. Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem (2012) 287:35756–67. doi: 10.1074/jbc.M112.397703
89. Zhang FF, Morioka N, Kitamura T, Hisaoka-Nakashima K, Nakata Y. Proinflammatory cytokines downregulate connexin 43-gap junctions via the ubiquitin-proteasome system in rat spinal astrocytes. Biochem Biophys Res Commun (2015) 464:1202–8. doi: 10.1016/j.bbrc.2015.07.105
90. Bam M, Yang X, Zhou J, Ginsberg JP, Leyden Q, Nagarkatti PS, et al. Evidence for Epigenetic Regulation of Pro-Inflammatory Cytokines, Interleukin-12 and Interferon Gamma, in Peripheral Blood Mononuclear Cells from PTSD Patients. J Neuroimmune Pharmacol (2016) 11:168–81. doi: 10.1007/s11481-015-9643-8
91. Lu Y, Sun Y, Drummer C4, Nanayakkara GK, Shao Y, Saaoud F, et al. Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells - Novel qualification markers for chronic disease risk factors and conditional DAMPs. Redox Biol (2019) 24:101221. doi: 10.1016/j.redox.2019.101221
92. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler Thromb Vasc Biol (2020) 40:e138–52. doi: 10.1161/ATVBAHA.120.314330
Keywords: proinflammatory cytokine blockage, proinflammatory cytokines, inflammation, innate immune regulators, reactive oxygen species
Citation: Liu M, Saredy J, Zhang R, Shao Y, Sun Y, Yang WY, Wang J, Liu L, Drummer C IV, Johnson C, Saaoud F, Lu Y, Xu K, Li L, Wang X, Jiang X, Wang H and Yang X (2020) Approaching Inflammation Paradoxes—Proinflammatory Cytokine Blockages Induce Inflammatory Regulators. Front. Immunol. 11:554301. doi: 10.3389/fimmu.2020.554301
Received: 21 April 2020; Accepted: 18 August 2020;
Published: 19 October 2020.
Edited by:
Francesca Granucci, University of Milano-Bicocca, ItalyReviewed by:
Min Wu, University of North Dakota, United StatesCopyright © 2020 Liu, Saredy, Zhang, Shao, Sun, Yang, Wang, Liu, Drummer, Johnson, Saaoud, Lu, Xu, Li, Wang, Jiang, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Yang, eGZ5YW5nQHRlbXBsZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.