 Malini Menon
Malini Menon Roli Budhwar
Roli Budhwar Rohit Nandan Shukla2
Rohit Nandan Shukla2 Udaykumar Ranga
Udaykumar Ranga
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 25 September 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.529614
The natural cysteine to serine variation at position 31 of Tat in HIV-1C disrupts the dicysteine motif attenuating the chemokine function of Tat. We ask if there exists a trade-off in terms of a gain of function for HIV-1C Tat due to this natural variation. We constructed two Tat-expression vectors encoding Tat proteins discordant for the serine 31 residue (CS-Tat vs. CC-Tat), expressed the proteins in Jurkat cells under doxycycline control, and performed the whole transcriptome analysis to compare the early events of Tat-induced host gene expression. Our analysis delineated a significant enrichment of pathways and gene ontologies associated with the angiogenic signaling events in CS-Tat stable cells. Subsequently, we validated and compared angiogenic signaling events induced by CS- vs. CC-Tat using human umbilical vein endothelial cells (HUVEC) and the human cerebral microvascular endothelial cell line (hCMEC/D3). CS-Tat significantly enhanced the production of CCL2 from HUVEC and induced an activated phenotype in endothelial cells conferring on them enhanced migration, invasion, and in vitro morphogenesis potential. The ability of CS-Tat to induce the activated phenotype in endothelial cells could be of significance, especially in the context of HIV-associated cardiovascular and neuronal disorders. The findings from the present study are likely to help appreciate the functional significance of the SAR (signature amino acid residues) influencing the unique biological properties.
HIV-1 genetic subtypes differ from one another in several molecular and biological properties, including genetic sequence variation, co-receptor usage, pathogenicity, replication fitness, transmission, and geographical distribution (1, 2). While factors such as the demographic variations, founder effect, host factor landscape differences, and socioeconomic parameters could significantly influence and modulate the observed differences among the HIV-1 genetic subtypes, natural genetic variations unique for individual viral subtypes play critical roles (3). The examination of the inherent genetic traits of viral subtypes could offer important clues to the survival advantage that the viral strains may have acquired in a specific population. Among several genetic subtypes of human immunodeficiency virus type I (HIV-1), subtype C (HIV-1C) is responsible for nearly half of HIV-1 infections worldwide (4). Signature sites are specific positions in an amino acid or nucleic acid alignment of variable sequences that are distinctly representative of a query set (viral genetic subtype), relative to a background set (constituted by the other genetic subtypes) (5). In other words, signature amino acid residues (SAR) are unique to one or a small number of viral genetic subtypes, present in a majority of the viral strains of the genetic subtypes, and either absent or present in only a small minority of other genetic subtypes. The study of the signature amino acid residues may offer valuable leads underpinning the biological differences among genetic subtypes of HIV-1 or other viruses and the survival advantage that the viral strains may have acquired in a specific population. In the present study, we used HIV-1 Tat as a model protein to understand the significance of S31, an important SAR of HIV-1C.
HIV-1 Tat regulates several functions critical for viral survival (6, 7) and modulates several host signaling pathways (8–10). Thus, the pleiotropic nature of Tat biology offers a variety of endpoints to examine the impact of SAR on the biological functions of Tat. The previous work from our laboratory identified seven SAR in HIV-1C Tat comprising H29, S31, L35, Q39, S57, P60, and E63 (11). Of these SARs, the biological significance of one of the SARs, S31, was experimentally evaluated in great detail (11–13). The substitution of a cysteine residue with a serine in HIV-1C, disrupts the dicysteine motif (C30C31), consequently attenuating the chemokine function of Tat (11–13). Our previous work, however, did not explore the evolutionary advantage of the natural variation of Tat, cysteine to serine substitution, unique to HIV-1C. In this backdrop, we investigated the functional significance of SAR S31 in HIV-1C Tat in the present work.
Several previous reports, including from our laboratory, suggested a neuroprotective function for the natural polymorphism of C31>S in HIV-1C Tat (11, 13–15). Li et al. examined if the interaction of Tat with the NMDA receptor, a key receptor involved in neurological processes such as learning, memory, and excitotoxicity, displayed subtype-specific differences. They found a significantly reduced neurotoxicity of C-Tat as compared to B-Tat, with the primary factor underlying this difference being the presence of C31 in B-Tat as against S31 in C-Tat (14). The residue, C31, forms a disulfide bond with the NR1 subunit of the NMDA receptor leading to activation. The presence of S31 in C-Tat attenuates this interaction and results in reduced neurotoxicity. These findings were corroborated by Mishra et al. when they demonstrated significantly reduced levels of apoptosis, chemokine secretion, and oxidative stress in the primary neurons and astrocytes exposed to C-Tat as compared to B-Tat (13). Further, Rao et al. demonstrated that the Tat dicysteine motif (C30C31) of C-Tat to function as the primary determinant of neurovirulence using a SCID mouse HIV-encephalitis model. The conclusions were drawn based on the monocyte migration, neurovirulence, and cognitive defects manifested following intracranial injection of MDM infected with Southern African HIV-1C isolate (from Zambia; 1084i) or HIV-1C isolate (Indie C1 from Southeast Asia) or an HIV-1B isolate (ADA from the US) (15). The most extensively examined impact of Tat-mediated EC activation and dysfunction in HIV-1 infection is in the context of HIV-associated neurocognitive disorders (HAND) and HIV-associated cardiovascular or angio-proliferative disorders (including Kaposi's sarcoma). In the context of the Tat dicysteine motif, reports from the South African population are of relevance (16). In South Africa, although the most prevalent viral strain is HIV-1C, a significant proportion of Tat in these viruses contains the intact C30C31 dicysteine motif. The results from the study suggested that the C31S substitution does not confer decreased impairment in cognitive performance. This study contradicted the earlier studies reporting substantial neurocognitive impairment in an HIV-1C-infected population despite the presence of the disrupted C30C31 motif in the Tat protein (17, 18).
Several studies previously attempted to compare the impact of the Tat proteins of HIV-1B and HIV-1C origin in modulating the functions of the host cell (1, 13, 19, 20). The HIV-1B Tat is seen to exhibit the superior potential to cause neuronal toxicity and synaptic plasticity, induce apoptosis in human primary neurons, and augment a pro-inflammatory cytokine response. The present study differs in important technical aspects from the previous reports by focusing on detecting the early intracellular events of Tat induction in certain respects, importantly, in exercising a tight control on Tat expression. Tat can modulate cell functions as an intracellular viral factor or when secreted into extracellular fluids as an extracellular protein (21–24). Tat, a powerful immunomodulator, can alter the expression of several cellular genes, including that of potent pro-inflammatory cytokines (21, 25, 26). The secondary amplification of cellular signaling events by pro-inflammatory cytokines and other immunomodulatory molecules could lead to a cascading effect, often masking of the primary impact of Tat. To detect only the early cellular events of Tat induction, and avoid the interference due to the secondary amplification of cellular signaling events, an efficient control on Tat expression was necessary.
To this end, we engineered Jurkat cells to express two variant forms of Tat—containing a serine (CS-Tat, wild type of HIV-1C) or a cysteine (CC-Tat, common in all the non-HIV-1C strains) residue at position 31, otherwise identical at all other positions. To detect only the early cellular events of Tat induction, we placed Tat expression under an inducible Tet-On promoter and generated stable Jurkat T-cells. Following induced expression of variant Tat in Jurkat cells, we evaluated the early cellular gene expression using RNA-Seq. The RNA-Seq analysis was suggestive of significant enrichment of the pathways and gene ontologies pertaining to angiogenic signaling events by CS-Tat, and the subsequent validation of these signaling events was executed using human umbilical vein endothelial cells (HUVEC) and the human cerebral microvascular endothelial cell line (hCMEC/D3).
The Jurkat cells (Clone E6-1) were procured from ATCC and cultured in RPMI 1640 (AL162S, HiMedia Laboratories, Mumbai, India), supplemented with 10% fetal bovine serum (10082147, Thermo Fisher Scientific, Waltham, USA), 2 mM L-glutamine (G8540, Sigma, St. Louis, USA), 100 units/ml penicillin G (P3032, Sigma, St. Louis, USA), and 100 μg/ml streptomycin (S9137, Sigma, St. Louis, USA). The HUVEC cells procured from ATCC, Virginia, USA, were used for the biological assays at passages 4 to 6. The cells were cultured in Ham's F-12K (Kaighn's) Medium (21127-022, Thermo Fischer Scientific), supplemented with 10% fetal bovine serum, 0.1 mg/ml heparin (H3149-100 KU, Sigma), and 0.03 mg/ml endothelial cell growth supplement, ECGS (E2759, Sigma). The HUVEC were seeded in tissue culture dishes coated with 0.1% gelatin (TCL059, HiMedia Laboratories, Mumbai, India). The immortalized HCMEC/D3 cells, procured from Cedarlane Cellutions Biosystems Inc. (CLU512), were cultured using EndoGRO MV complete media kit (SCME004, Millipore), on culture dishes coated with 150 μg/ml of type-I collagen from Rattail (C3867, Sigma). The Human Embryonic Kidney 293T (HEK293T) cells were procured from ATCC and cultured in Dulbecco's Modified Eagle's Medium (D1152, Sigma, St. Louis, USA), supplemented with 10% FBS, 2 mM L-glutamine, 100 units/ml penicillin G, and 100 μg/ml streptomycin.
To establish the doxycycline-inducible expression system, the Tet-inducible Tat-expression cassettes were cloned into the third-generation HIV-1-based lentivector expression vector, pCDH-CMV-MCS-EF1-copGFP (CD511B-1, System Biosciences, California, USA). The reverse tetracycline-controlled transactivator 3 gene (rtTA3) was amplified from pTRIPZ (GE Life Sciences, Chicago, USA) (FP: AGTTATGAATTCATGTCTAGGCTGGACAAGAGC and RP: ATACGAGGATCCTTACCCGGGGAGCATGTCAAGGTC). The second-generation Tet-On promoter or tetracycline response element (TRE), Ptight, was amplified from the commercial vector pTRE-Tight (631059, Clontech, California, USA) (FP: TAGGCGATTAATGAGGCCCTTTCGTCTTCACTC, RP: CCATGGGCTAGCCCGGTCCAGGCGATCTGACGG). The BL43 WT C-Tat (CS) and its SAR variant (CC) Tat were amplified from vectors p214.CS and p214.CC, respectively, generated previously in the laboratory and cloned into the MCS downstream of the TRE promoter using the enzyme sites, BglII and EcoRI for CS-Tat and SalI and BamHI for CC-Tat. The recombinant clones express the WT (CS) and variant (CC) Tat proteins, under the inducible Tet-responsive promoter, TRE.
To exercise tight control on the intracellular expression, we engineered the Tat expression under the control of a second-generation tetracycline/doxycycline-inducible promoter (Tet-On promoter). We established the Tet-On system for Tat expression by cloning two independent expression cassettes into the lentiviral expression vectors. The first lentiviral expression vector was designed to constitutively express rtTA3 and the puromycin resistance gene from a cytomegalovirus (CMV) promoter. The second vector was engineered for the inducible expression of the variant Tat proteins (Tet-On CS- or CC-Tat) from the Tet-On promoter. Additionally, an empty vector (Tet-On EV) that does not express Tat was also generated to serve as a control in all experiments. Of note, these vectors constitutively expressed the copepod green fluorescent protein (copGFP) as a reporter from the EF1α promoter. The transduced Jurkat cells were allowed to stabilize for 10 days following infection. The cells were subsequently analyzed using a flow cytometer for copGFP expression. The CS, CC, and EV Jurkat stable cells were established using fluorescence-assisted cell sorting (Supplementary Figure 1A).
The lentiviral vectors were used in combination with the packaging vectors (pVSV-G, pCMV-Rev, and psPAX2) to generate VSV-G pseudotyped lentiviruses in HEK293T for transduction. The Tat-stable cell lines were generated in two successive rounds of transduction. In the first round, the Jurkat cells were transduced with pseudotyped CMV-rtTA3-IRES-puromycin viruses (100 ng/ml p24 equivalent) for stable expression of rtTA3 and selected using puromycin. The rtTA3 expression from the transduced cells was confirmed using a quantitative PCR (FP: ACCCGCCCAACAGAGAAACAGTACGAAACC, and RP: CCTGTTCCTCCAATACGCAGCCCAGTGTAAAG) and stable cells were selected at a drug concentration of 800 ng/ml of puromycin. Several rtTA3 clonal cell lines were established by limit dilution and screened for the dox responsiveness in a separate experiment. The clone demonstrating the highest level of dox induction (~180-fold induction, following 12 h of induction) was selected for the subsequent Tat stable transduction. In the second round, the stable rtTA3-Jurkat cell line was transduced with the pseudotyped parental virus TRE-EF1α-copGFP (EV) or the Tat-expression variants (CS-Tat or CC-Tat). The rtTA3-Jurkat cells were infected with Tat viruses generated in HEK293T cells (the equivalent of 1.0, 0.1, and 0.01 ng/ml of p24 varying p24), and the expression of GFP was monitored using a flow cytometer. The viral infection at 0.1 ng/ml generated approximately 5% of GFP-positive cells, representing a single integration event per cell statistically (27). Using these conditions of viral infection, we established Tat-stable cell populations by GFP sorting. Optimal expression of Tat was induced from stable cell lines at a doxycycline concentration of 800 ng/ml. The expression of Tat transcript was quantitated employing a quantitative PCR (FP: GGAATCATCCAGGAAGTCAGCCCGAAAC, and RP: CTTCGTCGCTGTCTCCGCTTCTTCCTG).
Real-time PCR of the LTR was used to confirm a comparable number of integration events in the Tet-On Tat stable cells. Ten nanogram of genomic DNA isolated from the cells were amplified in a real-time PCR using primers targeting the strong-stop negative-strand DNA in the viral LTR. A serial 10-fold dilution of LTR copies from the genomic DNA extracted from TZM-bl cells (that contains two copies of stably integrated reporter provirus) was used in the LTR real-time PCR (FP: GATCTGAGCC(T/C)GGGAGCTCTCTG, and RP: TCTGAGGGATCTCTAGTTACCAGAGTC). A standard curve was constructed using serially diluted plasmid DNA containing HIV-1 LTR and to assess the sensitivity of the real-time PCR. Ct values of 24.07, 24.10, and 24.13 cycles were obtained for the CS, CC, and EV cell populations, respectively (Supplementary Figure 1B) suggesting comparable frequency of integration (28). We used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) reference gene PCR for normalization of the real-time PCR data (22.79, 22.56, and 22.99 Ct values, respectively).
We used the Illumina HiSeq 2000 platform to perform the RNA-Seq analysis and the TruSeq RNA Library preparation v2 kit (Illumina, California, USA) for library preparation. The reads of 100-bp length were obtained using a paired-end sequencing protocol. The tools used for resequencing analysis included- NGS QC Toolkit, TopHat, Cufflinks, DESeq, Microsoft Excel, and Cluster 3.0. The NGS QC Toolkit v2.3 was used for the quality control of the raw read data to obtain high-quality filtered reads. The high-quality filtered reads were aligned to the hg19 reference genome with TopHat (version 1.3.1). The gene model annotations and the known transcripts were deciphered using Hg19 assembly platform (UCSC genome browser). After the alignment to the reference genome, each RNAseq library was subjected to expression profiling using the tool package Cufflinks (version 1.1.0) (24). Differential gene expression profiling and differential analyses of the samples were performed for CS-Tat, CC-Tat, and EV (No Tat) using DESeq. Differentially expressed genes (DEGs) between the groups were calculated using the DESeq package in R version 3.2.5 (29). The threshold for DEGs was set as |log2 fold change| ≥1.0 and ≤ −1.0 and p ≤ 0.05. We computed both p-value and p-adjusted value (q-value) as a part of the standard analysis. To estimate the degree of biological variations between the replicates and to identify differentially expressed transcripts with acceptable false discovery rate, we proceeded with p-value as FDR to measure the total number of biologically meaningful differentially expressed genes (30). Identification of unique and overlapping genes within the DEG datasets was determined using Venny (bioinfogp.cnb.csic.es/tools/venny/index2.0.2.html). The differential gene lists were then subjected to perform significant biology wherein GO-Elite v1.2 (25) was used for Enrichment.
The conditioned media were harvested from the dox-induced Jurkat cells by centrifuging the cells at 1,500 × g for 5 min. One ml of conditioned media was incubated with one μg of E2.1 anti-Tat mouse monoclonal antibody (raised in-house) or IgG1 isotype control antibody (5415, Cell Signaling Technologies, Massachusetts, USA) for 30 min at room temperature with gentle agitation. The treated conditioned media were used in the assays.
The endothelial cell tubulogenesis was assayed using the growth factor reduced Matrigel-basement membrane matrix (354230, BD Biosciences). The Matrigel was spread evenly in wells of a 96-well plate; after 30 min of incubation at 37°C, HUVEC were suspended in the conditioned medium, seeded (0.12 million cells per ml) in the wells, and incubated for 24 h. The tube formation by HUVEC on Matrigel was assessed under an Olympus Inverted Microscope (100× magnification) and imaged at an interval of every 2 h. For the quantification of tube formation, the number of branch points of the formed tubes and other characteristics was evaluated using the Angiogenesis Analyzer plugin of the ImageJ software (31). Using this software, the original images (as captured at 100× magnification, three images per group) were transformed into the binary tree images and were treated with the “fill hole” method. The fill hole method minimizes the shadow effects in the image for quantification. Then, the images were skeletonized, and the binary trees were analyzed, and all the values were normalized with the analyzed area, making the area pixel number 19,20,000. Among several categories of quantification, the dimensional parameters such as a change in the size of the capillary-like network characterized by the number of nodes, number of junctions, number of segments, and number of branches were chosen for analysis. Since dimensional parameters do not fully characterize the architecture of the tubule patterns, the topological parameters, including total tubule length, total branching length, and total segment length, were also analyzed and compared among different groups (32, 33).
The polycarbonate membranes (8 μm pore size) in the transwells of a 24-well tissue culture plate (CLS3413, Sigma) were pre-coated with Matrigel (growth factor reduced basement membrane matrix; diluted to 300 μg/ml in a serum-free medium; 100 μl per transwell). The plates were incubated for 2 h at 37°C, and HUVEC were seeded at a density of 0.5 million cells per ml in the transwells (100 μl per transwell) in complete media. The Tat-conditioned media were added to the lower compartment of appropriate transwells. After 3–12 h of incubation at 37°C, the upper surface of the filter was scraped away with a cotton swab, the filters were fixed and stained with Giemsa stain (38723, Fischer Scientific), and five fields per well in duplicates were imaged at 200 × magnification (34). The images were quantified for the number of migrated cells using ImageJ image processing software suite from NIH. The transwell counter plugin of ImageJ was used for quantification, which allows for high-throughput automated counting of the number of cells on membranes in Transwell invasion and migration assays (35).
The trans-endothelial permeability of the hCMEC/D3 monolayer was assessed using 40 kDa FITC-Dextran (FD40, Sigma). The hCMEC/D3 cells were seeded on type-I collagen-coated 0.4-μm culture inserts and allowed to grow to confluence over 5 to 7 days. The confluent monolayers of cells were serum-starved for 12 h before the assay. The Jurkat-Tat conditioned media (12 h Dox-induced) were added to the confluent hCMEC/D3 monolayer on the inserts and wells. Following 24 h of incubation at 37°C, 0.1 μg/ml of FITC-dextran (40 kDa) was added apically to the inserts in the complete culture medium. The endothelial permeability was assessed by monitoring the flux of FITC-dextran across the endothelial monolayer by measuring the FITC-dextran fluorescence intensity in the spent media in the wells at regular intervals using a Fluorescence intensity spectral scanner (Varioskan Flash, Thermo Scientific).
The hCMEC/D3 endothelial cells were grown to confluence on collagen-coated coverslips in 24-well plates. Following 12 h of serum starvation, the monolayer was exposed for 24 h to Jurkat-Tat conditioned media (12 h Dox-induced). In parallel, the THP1 cells were treated with the Jurkat-Tat conditioned media for 12 h, washed with HBSS (14175-079, Thermo Fischer Scientific), and labeled with 5 μM CalceinAM (L3224, Thermo Fischer Scientific), for 30 min. The CalceinAM-labeled THP1 cell suspension was added at 0.5 million cells per treatment to the treated endothelial monolayer. Following 10 h of the addition of the labeled THP1 cells, the coverslips containing the endothelial monolayer were washed with HBSS. The adhered THP1 cells under each treatment were imaged using a fluorescence microscope.
All the statistical analyses were carried out using GraphPad Prism Version 5.0 for Windows, GraphPad Software, La Jolla, California, USA. The data sets were analyzed using one-way or two-way ANOVA with relevant post-tests for multiple comparisons. All the multiple comparison tests were based on the assumption that the values after each treatment were randomly drawn from populations with the same amount of scattering. One-way ANOVA was used where three or more groups were compared followed by the use Tukey–Kramer test for multiple comparison tests to compare all pairs of columns and find which groups are different from which other groups. The Tukey–Kramer test in GraphPad includes the extension by Kramer to allow for unequal sample size. Two-way ANOVA was used when a response was affected by two or more factors. GraphPad Prism suggests the use of the Bonferroni method of post-tests following two-way ANOVA. The Bonferroni correction lowers the P-value that is considered to be significant to 0.05 divided by the number of comparisons. This correction ensures that the 5% probability applies to the entire family of comparisons, and not separately to each comparison. For the differential gene expression analysis in NGS, both p-value and p-adjusted values (q-value) were computed as a part of the standard analysis. To estimate the degree of biological variation between replicates and to identify differentially expressed transcripts with acceptable false discovery rate (FDR), the p-value as FDR was used to measure the total number of biologically meaningful and differentially expressed genes (30). Transcripts with |log2 fold-change| ≥1.0 and ≤ −1.0 and p ≤ 0.05 were considered statistically significant.
Using lentiviral vectors, we established stable Jurkat cells expressing CS-Tat or CC-Tat under the control of the Dox-inducible system (Figure 1). To examine the primary events of Tat-induced host gene modulation, it was necessary to understand how soon the Tat transcripts are generated following Dox-induction. In a pilot analysis, we found 12 h of induction with 800 ng/ml of doxycycline to be optimal to detect Tat-induced early events (Supplementary Figure 2A). To examine the primary events of Tat-induced host gene modulation, it was necessary to understand how soon Tat transcripts are generated following Dox treatment of the cells. RNA extraction at the earliest time point of Tat induction is expected to be ideal for the whole transcriptome analysis (RNA-Seq). To this end, we induced rtTA3-CC-Tat Jurkat cells using 800 ng/ml of doxycycline (optimized using a Dox-dose response curve) and RNA was isolated from the cells at 6-h time intervals to monitor the modulation in the expression of Tat and a few cytokines comprising of TNF-α, IL-8, and IL-10 (Supplementary Figure 2A). Given that the gene expression of two important Tat-responsive genes, TNF-α and IL-8, peaked at 6–12 h, we selected the 12 h time point for the subsequent analyses. We confirmed similar levels of Tat expression at the RNA level in both CS and CC-Tat Jurkat cells using quantitative real-time PCR (p = 0.525, ns; Supplementary Figure 2B). Additionally, the modulation in the transcript level of cytokine/chemokine genes in CS and CC-Tat Jurkat cells was evaluated using gene-specific primers in a real-time PCR at a 12-h time point following Dox induction (Supplementary Figure 2C). These cytokines/chemokines are among many known to be modulated directly by Tat expression (20, 23, 36). Using the optimized experimental conditions, we performed the RNA-Seq analysis to gain a comprehensive overview of the global gene modulation following the induction of Tat variants in stable Jurkat cells (Supplementary Figure 3, and Supplementary Table 1). The RNA-Seq data of all the samples have been submitted to NCBI ((NCBI SRA accession number SUB7165990); http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89266). The differential expression analysis of CS-Tat vs. EV (empty vector) and CC-Tat vs. EV identified several genes to be significantly enriched (Supplementary Table 2 and Supplementary Figure 4). Of note, the CS and CC-Tat proteins being similar in sequence activated several common signaling pathways, but dysregulated specific pathways differentially. Some of the pathways commonly regulated by both the Tat proteins included metabolic processes of cellular macromolecules, regulation of gene expression, and nucleic acid metabolic processes, among others (Supplementary Table 3). Additionally, CS-Tat enriched for the regulation of the cellular membrane organization and regulation of I-kappaB kinase/NF-kappaB cascade and intracellular receptor-mediated signaling pathways (Supplementary Table 3). Some of the biological pathways significantly enriched by CS-Tat comprised VEGF, ERBB, EGF/EGFR, phosphatidylinositol, and angiopoietin receptor Tie2-mediated signaling. The data of CS-Tat induction collectively implied the involvement of pathways associated with growth factor receptors and angiogenic signaling (Supplementary Table 3). CC-Tat, in contrast, significantly enriched the pathways involved in apoptosis, TLR signaling, cell cycle, Jak-STAT, and p53 signaling (Supplementary Table 3).
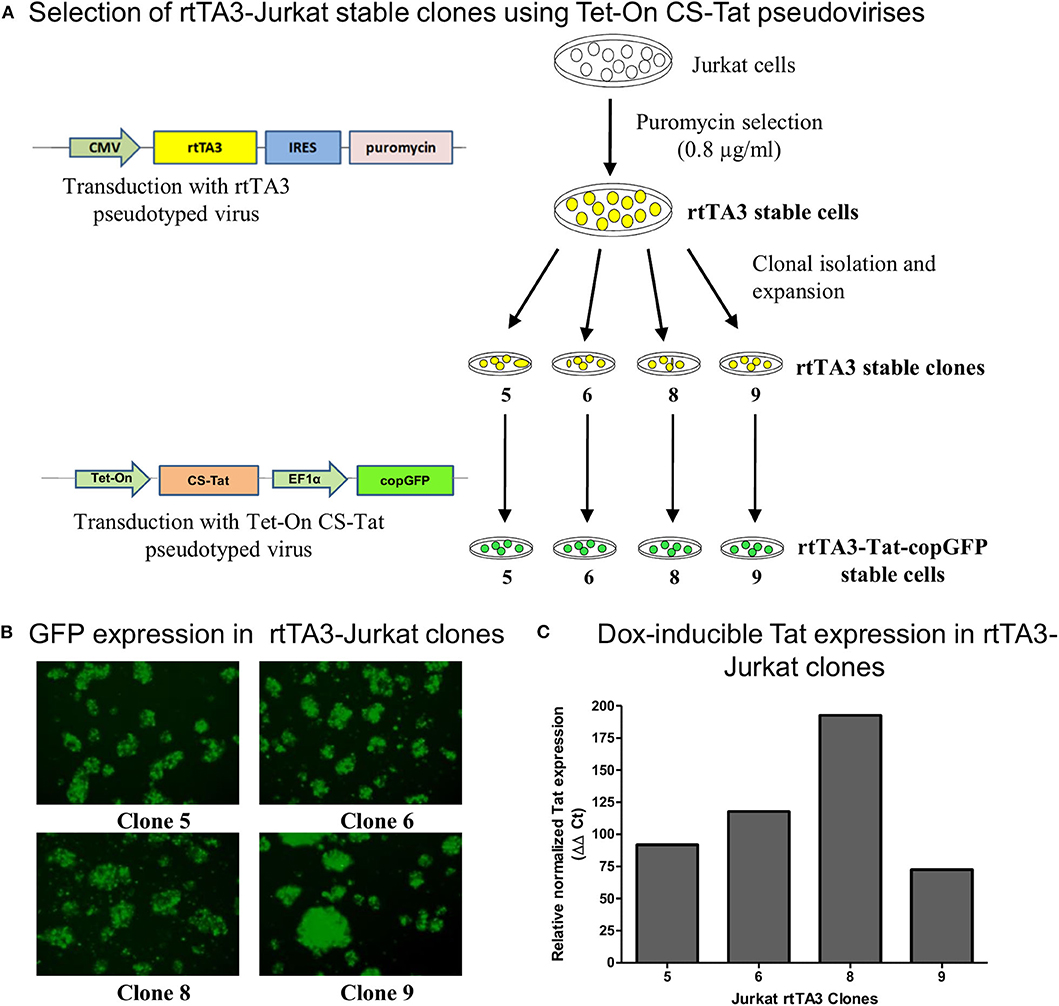
Figure 1. Generation and screening of rtTA3 stable cell lines. The Tat stable cells were obtained in two successive steps. (A) First, the Jurkat cells were transduced with the CMV-rtTA3-IRES-puromycin-pseudotyped lentivirus, and a pool of stable cells was obtained by selection with puromycin. The rtTA3-puro stable cells were subsequently subjected to limit dilution to obtain clonal cell lines, and each clone was infected with 30 ng/ml (p24 equivalent) of Tet-On CS-Tat EF1α-copGFP-pseudotyped virus. (B) The cells were imaged for copGFP expression at 72 h post-infection. (C) Each clonal population was subsequently treated with 800 ng/ml of Dox. A “No-Dox” control for each was used to evaluate the fold increase in Tat gene expression of each clone following doxycycline treatment. The cells were harvested at 24 h following Dox treatment, cDNA was synthesized from total cellular RNA, and a real-time PCR assay was performed to quantitate Tat transcripts. GAPDH was used as a reference gene control for normalization. The relative normalized Tat expression (using ΔΔCt method) was plotted.
Since a considerable amount of Tat and Tat-mediated cellular factors are secreted into the conditioned medium (37), we used the conditioned medium of Dox-induced Jurkat-Tat cells to examine the effects of secreted Tat and its secondary mediators on angiogenic responses (38). HUVEC are routinely used in studies investigating angiogenesis (31, 38–41). We used HUVEC cells to compare the angiogenic responses of CS-Tat vs. CC-Tat secreted into the t conditioned media of stable Jurkat cells. The interaction of Tat with the VEGFR-2/KDR or integrins on the cell surface causes the upregulation of chemokines such as CCL2 (monocyte chemoattractant protein, MCP-1) and cytokines such as IL-6 and IL-8 in endothelial cells (34–37).
The treatment of the HUVEC with conditioned medium from CS-Tat Jurkat cells induced a significantly higher level of CCL2 secretion from HUVEC (1137 ± 49.6 pg/ml, Figure 2) as compared to that of CC-Tat (782.5 ± 57.7 pg/ml, p < 0.0001) whereas no such differences were found between CC-Tat vs. EV. Since the HUVEC are primary cells, a considerable background level CCL2 secretion, SM (serum media control, 507.8 ± 12.77 pg/ml) was also observed; these levels, however, were significantly low to those of Tat treatment. The profiles of the CCL2 response remained mostly the same and significant regardless of the duration of the dox induction of Jurkat cells or the length of the treatment of the HUVEC cells with the conditioned medium. Since the two Tat proteins, CS and CC-Tat, differ from each other at only one signature amino acid residue (serine 31 vs. cysteine 31) in the cysteine-rich domain (CRD), the differential cytokine induction between the variant forms of Tat could be attributed to this single SAR difference. Of note, we confirmed that the conditioned medium did not contain any CCL2 by using the CS, CC, and EV conditioned media as control samples during the ELISA assay. The absorbance of these control samples was found to be equal to or lower than the assay blank (cell culture media), thus confirming that the CCL2 detected was induced from HUVEC.
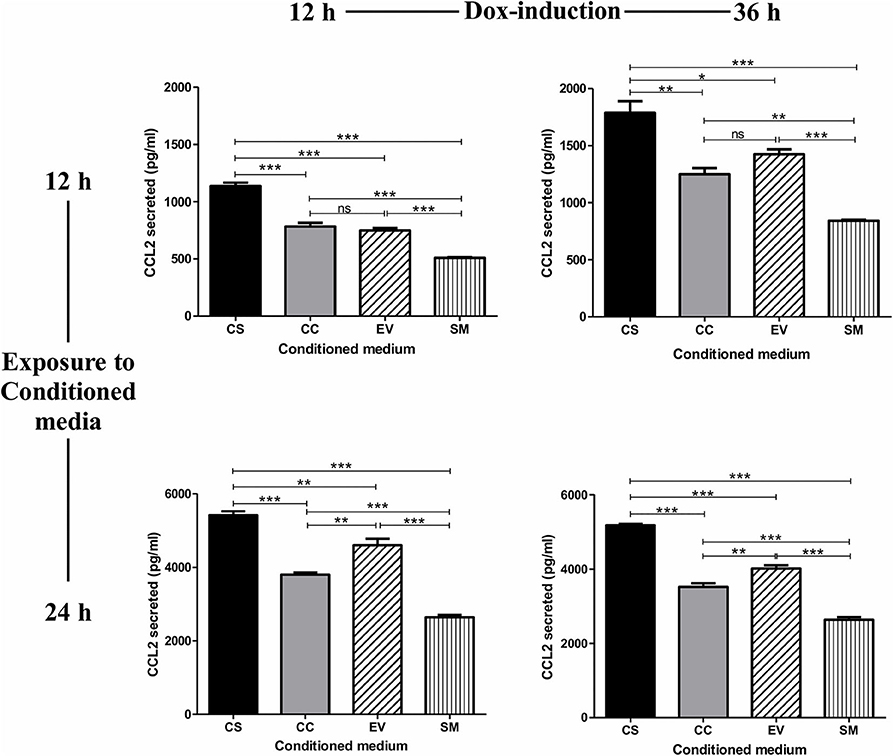
Figure 2. Conditioned medium of CS-Tat Jurkat cells augments CCL2 secretion from HUVEC. The cell-free conditioned media from the Jurkat cells (CS-Tat or CC-Tat or EV) were collected following 12 (left panel) or 36 h (right panel) of Dox induction. The conditioned media were added to HUVEC monolayers, and CCL2 secretion from the HUVEC was determined after 12 (top panel) or 24 h (bottom panel) of exposure to the conditioned media. The SM (HUVEC treated with RPMI supplemented with 10% FBS) served as the control for background CCL2 expression in the assay. The CCL2 levels were evaluated using an antigen-capture ELISA. EV, empty vector control; SM, serum medium control. The data are representative of three independent experiments. One-way ANOVA with Tukey–Kramer post-test (*p < 0.05, **p < 0.01, and ***p < 0.001).
To investigate if the differential induction of CCL2 in EC was Tat-specific, the conditioned media were subjected to Tat neutralization and, then, evaluated for CCL2 induction from the HUVEC (Figure 3). The treated conditioned media were added to HUVEC, and the CCL2 secreted into the medium at the end of 12 h was evaluated using an antigen-capture ELISA. Pretreatment of the CS-Tat-conditioned medium with a highly potent anti-Tat monoclonal antibody, but not an isotype control, reduced the secretion of CCL2 significantly (Figure 3). The mean CCL2 levels in the medium dropped from 1248 ± 74.64 pg/ml to 1018 ± 56.25 pg/ml following Tat neutralization, a level comparable to that of the CC-Tat medium (990.3 ± 24.33 pg/ml). In summary, although the influence of the other host factors in the conditional medium cannot be ruled out, the data of Tat neutralization confirms a significant role played by CS-Tat in inducing a significantly elevated concentration of CCL2.
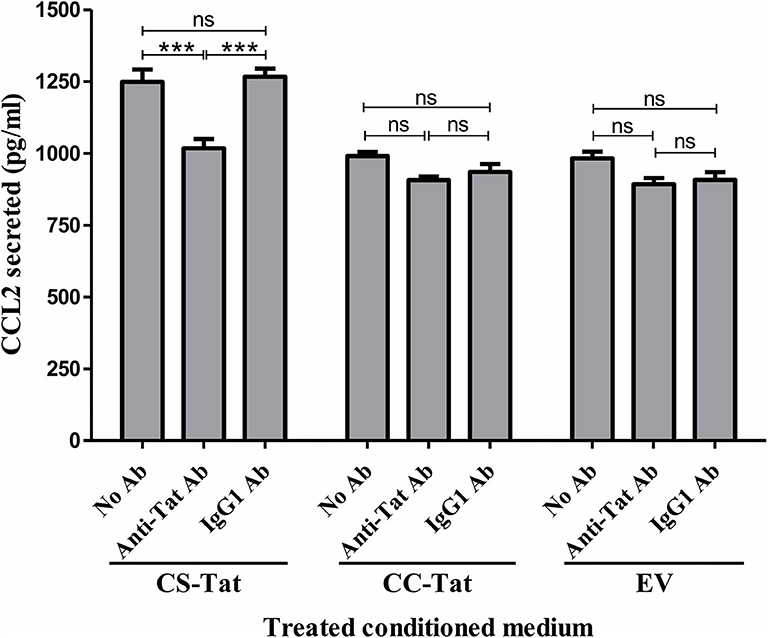
Figure 3. Tat neutralization in the conditioned media attenuates S31-specific differential CCL2 secretion. Conditioned media were collected from Jurkat-Tat cells at 12 h following Dox induction. The conditioned media were incubated with a highly potent anti-Tat monoclonal antibody (1 μg/ml of E2.1 antibody, raised in-house) or an IgG1 isotype control antibody or “no antibody” for 30 min at room temperature with gentle agitation. The HUVEC were exposed to the Tat-neutralized conditioned media for 12 h, and the CCL2 secretion was quantified using ELISA. EV: empty vector control. Two-way ANOVA with Bonferroni post-test (***p < 0.001).
The levels of CCL2 induced by the EV conditioned medium were significantly higher as compared to SM under most test conditions. Jurkat cells exhibit a basal level of an activated phenotype in culture, which can further be enhanced in response to several stimuli. The rtTA3Tet-On system engineered into EV Jurkat cells could also contribute to the background noise if activated by the cellular transcriptional noise. Of note, in our experiments, we observed comparable levels of CCL2 secretion by the conditioned media from parental EV cells as well as that of CC-Tat Jurkat cells, suggesting background noise.
We exposed HUVEC seeded on the Matrigel in a 96-well plate in triplicate wells to different conditioned media (CS-Tat, CC-Tat, and EV Jurkat conditioned media, 12 h dox induction) and measured the morphological features. Among the parameters evaluated, the number of junctions, segments, and branches did not show any significant difference between the treatment with the conditioned media of CS-Tat and CC-Tat. Importantly, however, we observed a significantly higher number of nodes in the CS-Tat treatment as compared to that of CC-Tat (545.33 ± 98.04 and 330 ± 32.7, respectively, p < 0.001; Figure 4A, and Supplementary Figure 5). We also found significantly longer tubes induced by CS-Tat as compared to CC-Tat (11,734.67 ± 2,017.9 and 7,546.66 ± 2,553.6, respectively, p < 0.05; Figure 4B). Additionally, the branching length of tubules for CS-Tat was significantly higher than that of CC-Tat (p < 0.05, Figure 4C). The response of HUVEC to rTat (purified recombinant Tat of HIV-1C origin) in different tubulogenesis parameters was comparable to that of the bFGF-positive control asserting the Tat-specific nature of the induction. Of note, the length of tubule segments induced by CS-Tat and CC-Tat did not differ significantly. Since tube formation assay serves as a robust measure of angiogenic potential, the above observations, when combined with the earlier finding of higher levels of expression of CCL2, allude to a superior angiogenic potential of CS-Tat that could be ascribed to the presence of the SAR S31 in C-Tat.
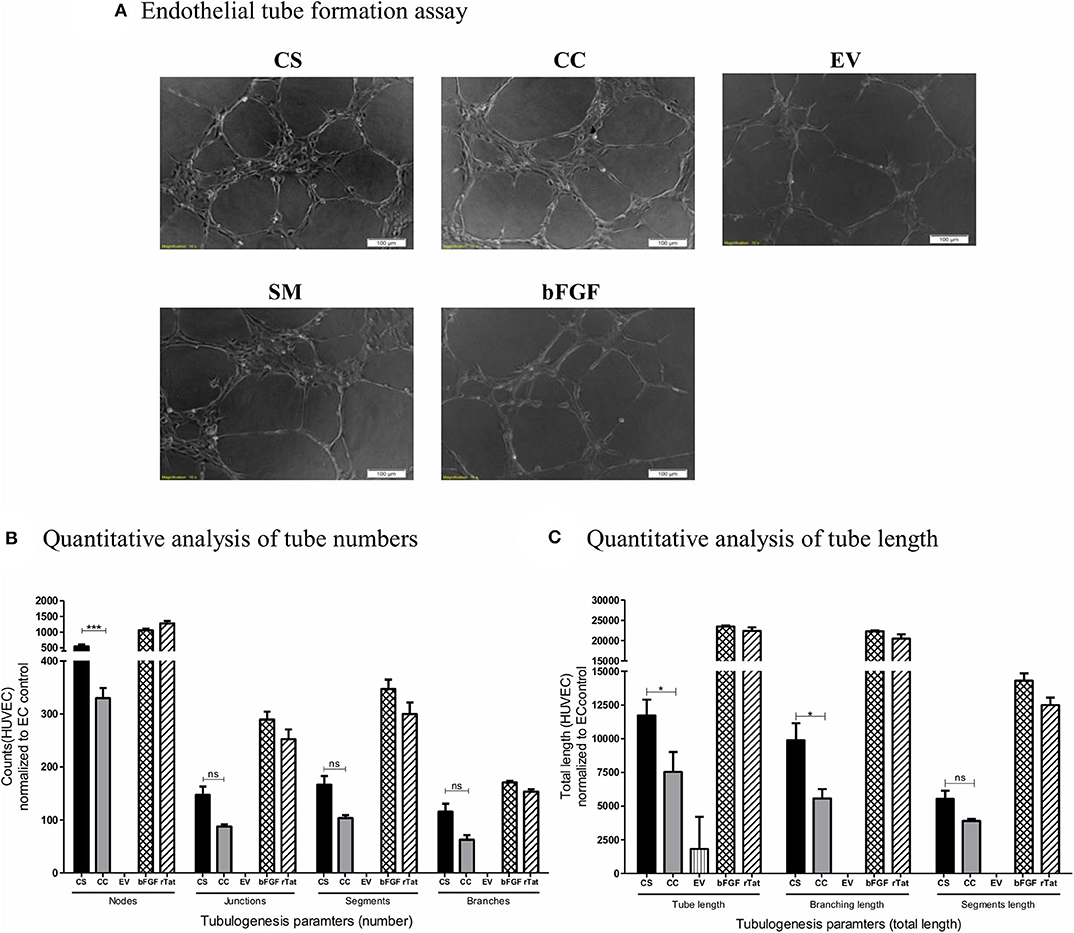
Figure 4. Tube formation of HUVEC following treatment with conditioned media. Conditioned media from Jurkat-Tat cells were harvested following 12 h of Dox-induction. 20,000 HUVEC in respective conditioned media were seeded over Matrigel in a 96-well plate. The HUVECs seeded in RPMI supplemented with 10% FBS served as the endothelial cell (SM) background control, and HUVECs treated with bFGF (2 ng/ml) and rTat (1 ng/ml) served as positive controls in the assay. (A) The cells were imaged at 2 h intervals for the tube formation, and the images captured at 4 h using a 10× objective are presented. The images were quantified using the Angiogenesis Analyzer plugin for ImageJ (NIH). The images were transformed to skeletonized binary tree form and analyzed for the tube parameters. The individual values were normalized to the SM control values for each parameter. (B) The normalized counts for the number of nodes, junctions, segments, and branches for CS, CC, EV, and bFGF treated HUVECs. (C) The total length, branching length, and segment length following different treatments. The counts and lengths ± SD are plotted, and the data are representative of three independent experiments. Two-way ANOVA with Bonferroni post-test (*p < 0.05, ***p < 0.001 and ns, non-significant).
CCL2, being a potent chemokine, can recruit monocytes and T-cells and augment their adhesion and transmigration across the endothelial monolayer (42, 43). We, therefore, explored if the augmented CCL2 secretion by CS-Tat can induce enhanced activation, migration, and invasion of endothelial cells and promote the migration of monocytes and T-cells across the endothelial monolayer.
To evaluate the migration and invasive behavior of HUVEC, the cells were suspended in serum-free medium and seeded on Matrigel-coated transwell inserts. The mean number of cells migrated per field under the influence of CS-Tat conditioned medium (95.8 ± 26.2) was significantly higher as compared to that of CC-Tat (51.4 ± 16.6, p < 0.01) or as compared to the EV and SM controls (Figures 5A,B, and Supplementary Figure 6). Since the migration of cells across the porous membrane necessarily requires the cells to migrate and invade through the matrigel, the assay takes into account both the invasive and migratory potential of cells.
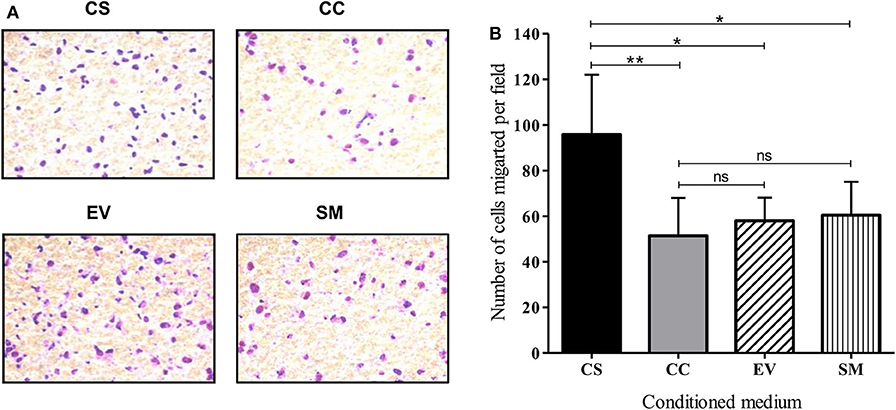
Figure 5. CS-Tat stimulates superior migration and invasion of HUVEC following an exposure to Jurkat-Tat-conditioned media. (A) HUVECs suspended in serum-free media were seeded in Matrigel (300 μg/ml) coated 8 μm cell culture inserts at a density of 0.5 × 106 cell/ml. The receiver wells contained conditioned media as labeled. HUVEC seeded in RPMI supplemented with 10% FBS served as the background (SM) control. The cells that did not migrate were scraped off using cotton swabs. The cells that migrated toward the lower chamber after 12 h of incubation were fixed, permeabilized, and stained with Giemsa for imaging using a 20× objective. (B) Five fields per treatment were imaged and the number of migrated cells were counted using ImageJ. The mean numbers of cells per field ± SD were plotted for each treatment. Conditioned media from Jurkat cells were harvested at 12 h post-dox induction. The data are representative of three independent experiments. One-way ANOVA with Tukey–Kramer post-test (*p < 0.05, **p < 0.01 and ns, non-significant).
In addition to serving as an inflammatory mediator, CCL2 serves as a potent chemokine modulating the expression levels of several cell surface adhesion molecules and junctional adhesion molecules, thus altering the trans-endothelial permeability properties of the blood–brain barrier (BBB) (44–46). To this end, we examined if the enhanced CCL2 response of CS-Tat treatment can translate into augmented trans-endothelial permeability. A confluent monolayer of hCMEC/D3 cells, extensively used as an in vitro model for BBB, seeded on collagen-coated 0.4-μm culture inserts served as a model for the BBB. We monitored the fluorescence intensity of FITC-dextran in culture media at regular intervals. CS-Tat conditioned media caused an increased FITC-dextran 40 flux across the monolayer (260.3 ± 6 and 237.9 ± 1.9 AU, respectively, for CS- and CC-Tat at the 6 h time point, p < 0.01; Figure 6). Of note, the data are consistent at different durations of treatment with the conditioned media. The disruption of the BBB was pronounced and highly significant as the duration of the treatment increased (380.9 ± 5.4 and 345.7 ± 5.1 AU, respectively, for CS and CC-Tat at the 10 h time point, p < 0.001).
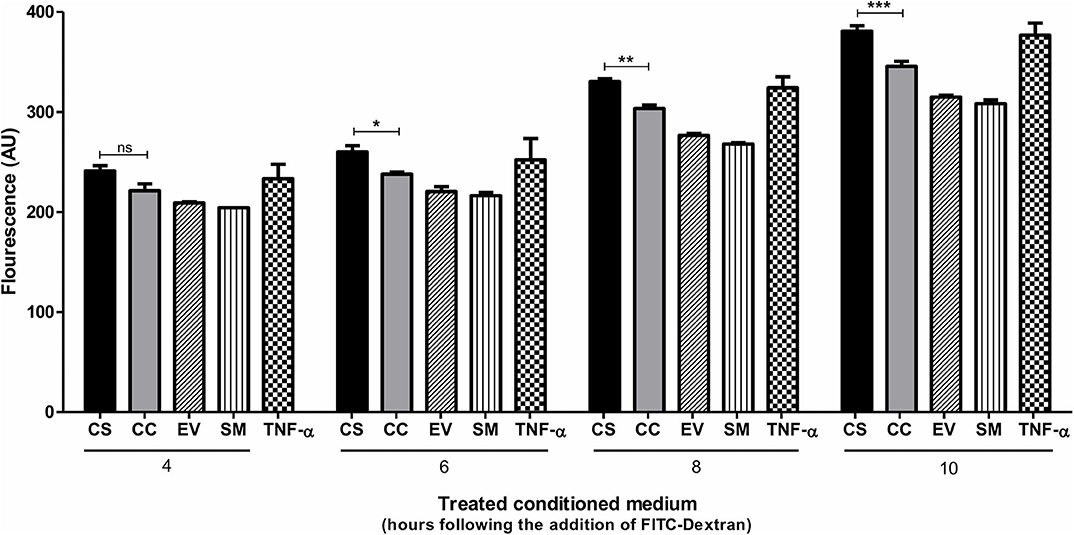
Figure 6. CS-Tat enhances trans-endothelial permeability of FITC-Dextran across the endothelial monolayer. hCMEC/D3 cells were seeded on collagen-coated 0.4-μm culture inserts and allowed to grow to confluence. The cells were serum-starved for 12 h before the assay. Jurkat-Tat-conditioned media collected at 12 h following dox induction were added to the confluent hCMEC/D3 monolayer on the inserts and the wells. Following 24 h of incubation at 37°C, a fresh medium supplemented with FITC dextran (0.1 μg/ml) was added apically to the inserts. Culture media were collected from the wells at regular intervals and monitored for fluorescence. hCMEC/D3 cells treated with 10 ng/ml of TNF-α served as the positive control in the assay. The data are representative of three independent experiments. Two-way ANOVA with Bonferroni post-test was used for the statistical evaluation (*p < 0.05, **p < 0.01, ***p < 0.001 and ns, non-significant).
HIV-1 Tat augments the expression of the integrin adhesion molecules (β2 integrins) on monocytes and T-cells and enhances their adhesion to endothelial cells (47, 48). To this end, we asked if exposing the hCMEC/D3 and THP1 cells to different Tat-conditioned media would lead to differential adhesion of monocytes to hCMEC/D3 cells. The adhesion of CalceinAM-labeled THP1 cells was monitored using a fluorescent microscope (Figure 7A). CS-Tat treatment resulted in a significantly higher level of cell adhesion as compared to that of CC-Tat, or the SM or the EV controls (64.33 ± 12.9, 40.67 ± 4.2, 20 ± 1.7, 14 ± 1.7, respectively, p < 0.05; Figure 7B).
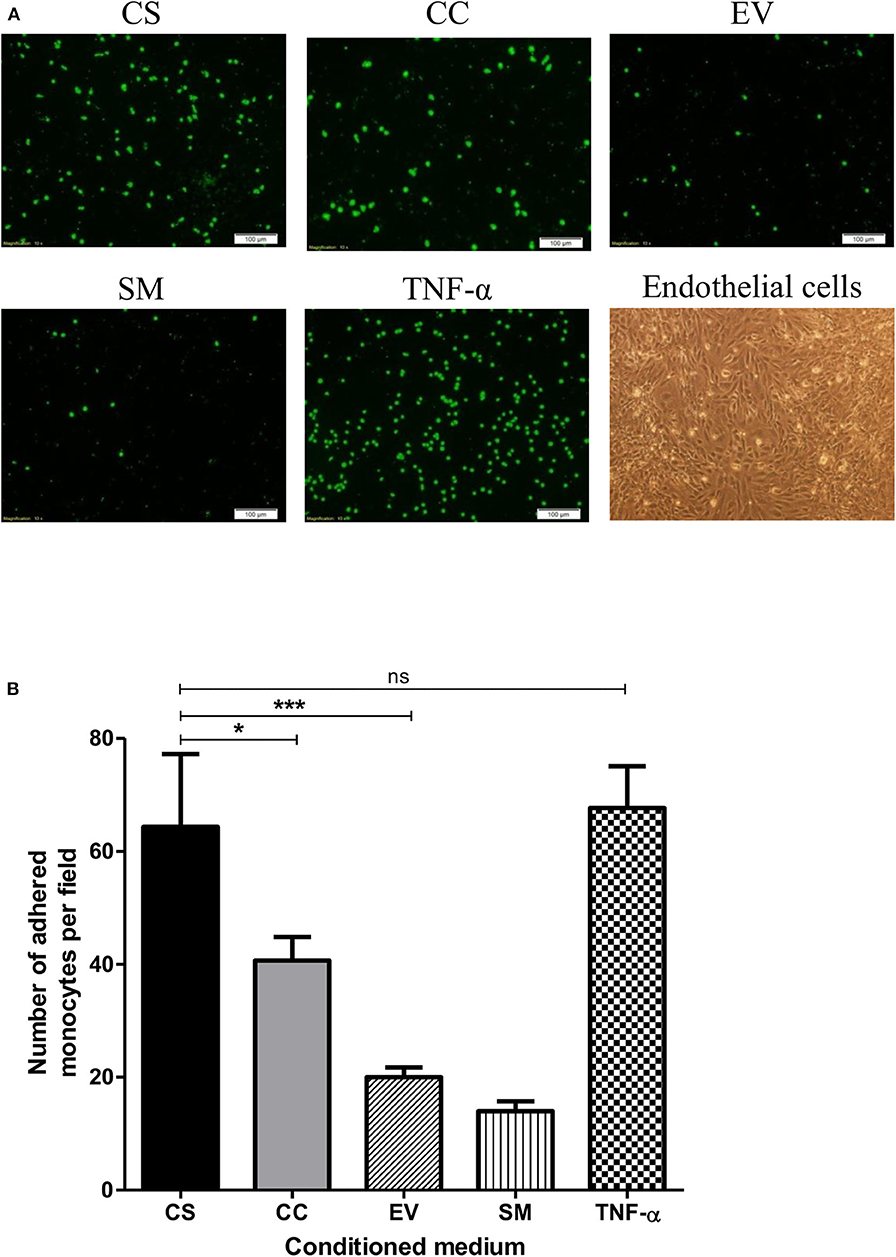
Figure 7. The treatment with CS-Tat Jurkat-conditioned medium augments monocyte-endothelial cell adhesion. hCMEC/D3 endothelial cells were grown to confluence on collagen-coated coverslips in 24-well plates. Following 12 h of serum starvation, the monolayer was exposed to Jurkat-Tat-conditioned media for 24 h. In parallel, THP1 cells were treated with the Jurkat-Tat conditioned media for 12 h, washed, and labeled with CalceinAM. The CalceinAM-labeled THP1 cell suspension was added to the endothelial monolayer, and following 10 h of the addition of THP1 cells, the coverslips containing the endothelial monolayer were washed with HBSS. (A) The adhered THP1 cells under each treatment were imaged using a fluorescent microscope. Endothelial cells treated with RPMI supplemented with 10% FBS served as the background control (SM) and cells treated with 10 ng/ml of TNF-α served as the positive control in the assay. (B) Five fields per treatment were used for the quantification, and the number of adhered monocytes per field were counted using ImageJ. The mean numbers of cells per field ± SD were plotted for each treatment. The assay was repeated twice, one-way ANOVA with Tukey–Kramer post-test (*p < 0.05, ***p < 0.001 and ns, non-significant).
Several groups, including ours, have previously demonstrated the crucial functions SAR may play in governing the biological properties of Tat (11, 37, 49, 50). Tat-mediated modulation of gene expression in cells of diverse lineage has been investigated extensively (10, 51–53). Using primary cultures and neuronal cell lines, Chang et al. examined the impact of HIV-1 Tat on the expression of miRNAs and demonstrated that Tat deregulates the levels of several miRNAs, including miR-34a, the most highly induced miRNAs in Tat-treated neurons (10). Evaluating gene expression modulation by Tat in H9-Tat cells using cDNA microarray technology, de la Fuente et al. reported Tat-mediated down-modulation of cell surface receptors, co-receptors including those associated with cellular receptor tyrosine kinase (RTK) activity and transcription coactivators such as p300/CBP and SRC-1 (51). Gibellini et al. performed a cDNA-membrane-array using CD4+ve Jurkat cells constitutively expressing Tat under serum-starved conditions and demonstrated 2- to 3-fold upregulation of several cellular genes including transcription factors, cellular receptors, adaptors, and mediators of signal transduction pathways (52). Given the highly diverse conditions of experimentation, including stable vs. transient expression of Tat, the lineage of the host cell, and the endpoint evaluation, the conclusions drawn are extremely variable making it difficult to draw unifying themes on Tat-mediated modulation of host cell transcription.
The present study is unique in three different qualities. First, unlike the previous studies, all of which employed Tat of HIV-1B origin, the present study used HIV-1C Tat. Given the subtype-specific molecular differences and SAR, an examination of viral factors of diverse genetic backgrounds is likely to throw more light on the biological differences among viral families. Second, in the present study, we used two different Tat proteins that are discordant for a single SAR at position 31—comparing serine vs. cysteine at this location. Our research, thus, is designed not only to examine the impact of Tat expression on the landscape of the host factors but also to compare the effect of Tat proteins discordant for a single amino acid residue. Lastly, unlike the previous studies, our work exercised control on Tat expression crucial to examining the earliest and direct targets of Tat, using RNA-Seq. The use of RNA-Seq gave the advantage of unbiased and easy identification of rare and low-abundant transcripts with increased sensitivity and specificity. One of the technical limitations of the present work is the absence of a primary cell model. Given that the study design required the establishment of double stable cells for controlled Tat expression, extending this work to primary CD4+ T-cells would be technically challenging. Of note, the role of SAR S31 in C-Tat has been examined in the absence of other viral proteins and regulatory elements. Whether the findings of the present analysis would be consistent in the context of full-length viral strains needs to be determined.
The present study stemmed from our earlier publication, which identified S31 to be a determinant of the defective chemokine function of HIV-1C Tat (11). It is intriguing, however, to find that C-Tat despite being a defective chemokine induces an enhanced CCL2 chemokine response from the endothelial cells. The exposure of endothelial cells to Tat causes a time- and dose-dependent release of CCL2 into the culture supernatant (42, 54). In the light of the salient functions of CCL2 as a chemokine, combined with its association with endothelial dysfunction and disruption of the BBB, it would be of interest to examine if the enhanced CCL2 response from EC following CS-Tat treatment would also translate into an altered expression of lymphocyte, monocyte, or endothelial cell surface adhesion molecules and increased expression of Matrix metalloproteinases (MMPs). Recent research has suggested an increase in virus production in the presence of CCL2 in HIV-infected macrophages, and this effect is seen to be Gag-mediated via the LYPX motif. This observation is seen to be subtype-specific, where in sharp contrast to HIV-1B, HIV-1C fails to show this response due to the absence of the LYPX motif (55). Thus, the CS-Tat-induced CCL2 production may influence biological functions other than the viral replication of HIV-1C.
The demonstration that S31 in CS-Tat predisposes Tat toward enhanced endothelial cell activation is a novel finding of the present study. The RNA-Seq analysis of CS-Tat revealed the activation of several signaling pathways, broadly suggesting a significantly modulated angiogenesis-related signaling process. In addition to the enhanced CCL2 induction in HUVEC, the CS-Tat-conditioned medium caused enhanced cell migration and invasion of endothelial cells. These observations, along with the in vitro tube formation results, provided convincing evidence to an angiogenic signaling program induced by the CS-Tat Jurkat-conditioned medium in HUVEC. The pro-angiogenic properties of extracellular HIV-1B Tat, especially in association with several proinflammatory cytokines, have been elegantly reviewed (56). The integrity of the arginine–glycine–aspartic acid (RGD) motif and the basic domain is critical for Tat to exert growth-promoting properties on the endothelial cells by binding to the α5β1 and αvβ3 integrins (38, 56–58). A vast majority of HIV-1C viral strains do not contain the RGD motif in exon 2 due to a natural variation. It would be necessary to understand if the natural variation of C31S in HIV-1C Tat, leading to significantly enhanced induction of CCL2 production, is a compensatory mechanism for the differences in the basic domain and the RGD motif.
Furthermore, HIV-associated cardiovascular diseases account for approximately 10% of the deaths in HIV-positive patients (59). Increased adhesion of leukocytes to the aortic epithelium and endothelial dysfunction is a pathogenic manifestation commonly observed in HIV-associated microvascular and angio-proliferative disorders (60). Since EC have not been reported to support HIV-1 replication, the observed vascular dysfunction is believed to be mediated by molecules released from HIV-infected cells, including viral proteins and induced cellular factors (61). Tat-mediated activation of EC associated with the enhanced monocyte adhesion to EC is mediated via the activation of MAP kinases and NF-κB (62). Additionally, Tat stimulates the upregulation of adhesion molecule ICAM-1 by downregulating miR-221 in an endothelial-specific manner. The HIV-1 Tat protein alongside inflammatory cytokines, synergistically enhances endothelial cell growth, resulting in the induction of angiogenic Kaposi's sarcoma like lesions (57, 58, 63, 64). Of note, the prevalence of Kaposi's sarcoma is rare in HIV-1C infection (65). The available data implicates a significant role for Tat in HIV-associated cardiomyopathies (62, 66). In the context of the present study, the evaluation of cardiomyopathy and KS cases in the HIV-1C-infected population becomes essential. Additionally, further research is required to evaluate the mechanistic details of the cellular pathways involved in the CS-Tat-mediated angiogenic response in EC.
In summary, the findings from the present study are likely to help appreciate the functional significance of the SAR residues influencing the unique biological properties of diverse genetic families of HIV-1. The ability of CS-Tat to induce an activated phenotype in endothelial cells conferring on them enhanced EC migration, invasion, and in vitro morphogenesis is significant, especially in the context of HIV-associated neuronal and cardiovascular disorders.
The datasets generated for this study can be found in the NCBI SRA accession number SUB7165990, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89266.
MM: conceptualization, data curation, investigation, validation, writing original draft, and reviewing and editing of the article. KB, MV, RB, and RS: resource provision, data curation, and validation. UR: conceptualization, fund acquisition, validation, writing, reviewing, and editing of the article. All authors contributed to the article and approved the submitted version.
This work was supported by grants to JNCASR from The Department of Biotechnology, Government of India (Grant No. BT/INF/22/SP27679/2018), the Council of Scientific and Industrial Research (Grant No. 27/320/16-EMR-II), and intramural funds from JNCASR. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
RB, RS, KB, and MV were employed by company Bionivid Pvt. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The TZM-bl cells were obtained through the National Institutes of Health AIDS Research and Reference Reagent Program, NIAID, NIH: TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc. MM was a recipient of the Council of Scientific and Industrial Research Fellowship from the Government of India.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.529614/full#supplementary-material
1. Li L, Dahiya S, Kortagere S, Aiamkitsumrit B, Cunningham D, Pirrone V, et al. Impact of tat genetic variation on HIV-1 disease. Adv Virol. (2012) 2012:123605. doi: 10.1155/2012/123605
2. Hu DJ, Buvé A, Baggs J, Van der Groen G, Dondero TJ. What role does HIV-1 subtype play in transmission and pathogenesis? An epidemiological perspective. AIDS. (1999) 13:873–81. doi: 10.1097/00002030-199905280-00002
4. Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global trends in molecular epidemiology of HIV-1 during 2000– 2007. AIDS. (2011) 25:679–89. doi: 10.1097/QAD.0b013e328342ff93
5. Korber B, Myers G. Signature pattern analysis: a method for assessing viral sequence relatedness. AIDS Res Hum Retroviruses. (1992) 8:1549–60. doi: 10.1089/aid.1992.8.1549
6. Li JC, Yim HC, Lau ASY. Role of HIV-1 Tat in AIDS pathogenesis: its effects on cytokine dysregulation and contributions to the pathogenesis of opportunistic infection. AIDS. (2010) 24:1609–23. doi: 10.1097/QAD.0b013e32833ac6a0
7. Huigen MCDG, Kamp W, Nottet HSLM. Multiple effects of HIV-1 trans-activator protein on the pathogenesis of HIV-1 infection. Eur J Clin Invest. (2004) 34:57–66. doi: 10.1111/j.1365-2362.2004.01282.x
8. Mischiati C, Pironi F, Milani D, Giacca M, Mirandola P, Capitani S, et al. Extracellular HIV-1 tat protein differentially activates the JNK and ERK/MAPK pathways in CD4 T cells. AIDS. (1999) 13:1637–45. doi: 10.1097/00002030-199909100-00006
9. Izmailova E, Bertley FMN, Huang Q, Makori N, Miller CJ, Young RA, et al. HIV-1 Tat reprograms immature dendritic cells to express chemoattractants for activated T cells and macrophages. Nat Med. (2003) 9:191–7. doi: 10.1038/nm822
10. Chang JR, Mukerjee R, Bagashev A, Del Valle L, Chabrashvili T, Hawkins BJ, et al. HIV-1 tat protein promotes neuronal dysfunction through disruption of microRNAs. J Biol Chem. (2011) 286:41125–34. doi: 10.1074/jbc.M111.268466
11. Ranga U, Shankarappa R, Siddappa NB, Ramakrishna L, Nagendran R, Mahalingam M, et al. Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J Virol. (2004) 78:2586–90. doi: 10.1128/JVI.78.5.2586-2590.2004
12. Rao VR, Neogi U, Talboom JS, Padilla L, Rahman M, Fritz-French C, et al. Clade C HIV-1 isolates circulating in Southern Africa exhibit a greater frequency of dicysteine motif-containing Tat variants than those in Southeast Asia and cause increased neurovirulence. Retrovirology. (2013) 10:61. doi: 10.1186/1742-4690-10-61
13. Mishra M, Vetrivel S, Siddappa NB, Ranga U, Seth P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: Significance of dicysteine C30C31 motif. Ann Neurol. (2008) 63:366–76. doi: 10.1002/ana.21292
14. Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, et al. NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci. (2008) 28:12190–98. doi: 10.1523/JNEUROSCI.3019-08.2008
15. Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, et al. HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci. (2008) 28:10010–16. doi: 10.1523/JNEUROSCI.2955-08.2008
16. Paul RH, Joska JA, Woods C, Seedat S, Engelbrecht S, Hoare J, et al. Impact of the HIV Tat C30C31S dicysteine substitution on neuropsychological function in patients with clade C disease. J Neurovirol. (2014) 20:627–35. doi: 10.1007/s13365-014-0293-z
17. Satishchandra P, Nalini A, Gourie-Devi M, Khanna N, Santosh V, Ravi V, et al. Profile of neurologic disorders associated with HIV/AIDS from Bangalore, South India (1989-96). Indian J Med Res. (2000) 111:14–23.
18. Wadia RS, Pujari SN, Kothari S, Udhar M, Kulkarni S, Bhagat SS, et al. Neurological manifestations of HIV disease. J Assoc Physicians India. (2001) 49:343–8.
19. Samikkannu T, Atluri VSR, Arias AY, Rao KVK, Mulet CT, Jayant RD, et al. HIV-1 subtypes B and C Tat differentially impact synaptic plasticity expression and implicates HIV-associated neurocognitive disorders. Curr HIV Res. (2014) 12:397–405. doi: 10.2174/1570162X13666150121104720
20. Gandhi N, Saiyed Z, Thangavel S, Rodriguez J, Rao KVK, Nair MPN. Differential effects of HIV type 1 clade B and clade C Tat protein on expression of proinflammatory and antiinflammatory cytokines by primary monocytes. AIDS Res Hum Retroviruses. (2009) 25:691–9. doi: 10.1089/aid.2008.0299
21. Pugliese A, Vidotto V, Beltramo T, Petrini S, Torre D. A review of HIV-1 Tat protein biological effects. Cell Biochem Funct. (2005) 23:223–27. doi: 10.1002/cbf.1147
22. Romani B, Engelbrecht S, Glashoff RH. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J Gen Virol. (2010) 91:1–12. doi: 10.1099/vir.0.016303-0
23. Johri MK, Mishra R, Chhatbar C, Unni SK, Singh SK. Tits and bits of HIV Tat protein. Expert Opin Biol Ther. (2011) 11:269–83. doi: 10.1517/14712598.2011.546339
24. Chiozzini C, Toschi E. Hiv-1 tat and immune dysregulation in aids pathogenesis: a therapeutic target. Curr Drug Targets. (2016) 17:33–45. doi: 10.2174/1389450116666150825110658
25. Scala G, Ruocco MR, Ambrosino C, Mallardo M, Giordano V, Baldassarre F, et al. The expression of the interleukin 6 gene is induced by the human immunodeficiency virus 1 TAT protein. J Exp Med. (1994) 179:961–71. doi: 10.1084/jem.179.3.961
26. Kedzierska K, Crowe SM. Cytokines and HIV-1: Interactions and clinical implications. Antivir Chem Chemother. (2001) 12:133–50. doi: 10.1177/095632020101200301
27. Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. (2005) 122:169–82. doi: 10.1016/j.cell.2005.06.006
28. Verma A, Rajagopalan P, Lotke R, Varghese R, Selvam D, Kundu TK, et al. Functional Incompatibility between the generic NF-κB motif and a subtype-specific Sp1III element drives the formation of the HIV-1 subtype C Viral Promoter. J Virol. (2016) 90:7046–65. doi: 10.1128/JVI.00308-16
29. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1101/002832
30. Kim J, Bang H. Three common misuses of P values. Dent Hypotheses. (2016) 7:73–80. doi: 10.4103/2155-8213.190481
31. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. (2010) 5:628–35. doi: 10.1038/nprot.2010.6
32. Leea E, Seung JL, Koskimakia JE, Hana Z, Niranjan B. Pandeya AS. Inhibition of breast cancer growth and metastasis by a biomimetic peptide. Sci Rep. (2014) 4:7139. doi: 10.1038/srep07139
33. Guidolin D, Albertin G, Ribatti D. Exploring in vitro angiogenesis by image analysis and mathematical modeling. Microsc Sci Technol Appl Educ. (2010) 2:876–84.
34. Albini A, Benelli R. The chemoinvasion assay: a method to assess tumor and endothelial cell invasion and its modulation. Nat Protoc. (2007) 2:504–11. doi: 10.1038/nprot.2006.466
35. O'brien J, Hayder H, Peng C. Automated quantification and analysis of cell counting procedures using ImageJ plugins. J Vis Exp. (2016) 2016:1–10. doi: 10.3791/54719
36. Wong JK, Campbell GR, Spector SA. Differential induction of interleukin-10 in monocytes by HIV-1 clade B and clade C Tat proteins. J Biol Chem. (2010) 285:18319–25. doi: 10.1074/jbc.M110.120840
37. Rayne F, Debaisieux S, Yezid H, Lin Y-L, Mettling C, Konate K, et al. Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J. (2010) 29:1348–62. doi: 10.1038/emboj.2010.32
38. Ismail M, Henklein P, Huang X, Braumann C, Rückert RI, Dubiel W. Identification of HIV-1 Tat peptides for future therapeutic angiogenesis. Eur J Haematol. (2006) 77:157–65. doi: 10.1111/j.1600-0609.2006.00682.x
39. Ma C, Wang XF. In vitro assays for the extracellular matrix protein-regulated extravasation process. Cold Spring Harb Protoc. (2008) 3:1–6. doi: 10.1101/pdb.prot5034
40. Cota-Gomez A, Flores NC, Cruz C, Casullo A, Aw TY, Ichikawa H, et al. the human immunodeficiency virus-1 tat protein activates human umbilical vein endothelial cell e-selectin expression via an nf-kappa b-dependent mechanism. J Biol Chem. (2002) 277:14390–9. doi: 10.1074/jbc.M108591200
41. Staton CA, Reed MWR, Brown NJ. A critical analysis of current in vitro and in vivo angiogenesis assays. Int J Exp Pathol. (2009) 90:195–221. doi: 10.1111/j.1365-2613.2008.00633.x
42. Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood. (2001) 97:352–8. doi: 10.1182/blood.V97.2.352
43. Zidovetzki R, Wang JL, Chen P, Jeyaseelan R, Hofman F. Human immunodeficiency virus Tat protein induces interleukin 6 mRNA expression in human brain endothelial cells via protein kinase C- and cAMP-dependent protein kinase pathways. AIDS Res Hum Retroviruses. (1998) 14:825–33. doi: 10.1089/aid.1998.14.825
44. Roberts TK, Eugenin EA, Lopez L, Romero IA, Weksler BB, Couraud P-O, et al. CCL2 disrupts the adherens junction: implications for neuroinflammation. Lab Investig. (2012) 92:1213–33. doi: 10.1038/labinvest.2012.80
45. Eugenin EA. CCL2/Monocyte chemoattractant protein-1 mediates enhanced transmigration of Human Immunodeficiency Virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and neuroAIDS. J Neurosci. (2006) 26:1098–106. doi: 10.1523/JNEUROSCI.3863-05.2006
46. Williams DW, Calderon TM, Lopez L, Carvallo-Torres L, Gaskill PJ, Eugenin EA, et al. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV Infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS ONE. (2013) 8:e69270. doi: 10.1371/journal.pone.0069270
47. Lafrenie RM, Wahl LM, Epstein JS, Hewlett IK, Yamada KM, Dhawan S. HIV-1-Tat modulates the function of monocytes and alters their interactions with microvessel endothelial cells. A mechanism of HIV pathogenesis. J Immunol. (1996) 156:1638–45.
48. Lafrenie RM, Wahl LM, Epstein JS, Yamada KM, Dhawan S. Activation of monocytes by HIV-Tat treatment is mediated by cytokine expression. J Immunol. (1997) 159:4077–83.
49. Ranjbar S, Rajsbaum R, Goldfeld AE. Transactivator of transcription from HIV type 1 subtype E selectively inhibits TNF gene expression via interference with chromatin remodeling of the TNF locus. J Immunol. (2006) 176:4182–90. doi: 10.4049/jimmunol.176.7.4182
50. Zhao X, Qian L, Qi D, Zhou D, Shen W, Liu Y, et al. The 57th amino acid conveys the differential subcellular localization of human immunodeficiency virus-1 Tat derived from subtype B and C. Virus Genes. (2016) 52:179–88. doi: 10.1007/s11262-015-1267-9
51. de la Fuente C, Santiago F, Deng L, Eadie C, Zilberman I, Kehn K, et al. Gene expression profile of HIV-1 Tat expressing cells: a close interplay between proliferative and differentiation signals. BMC Biochem. (2002) 3:14. doi: 10.1186/1471-2091-3-14
52. Gibellini D, Re MC, La Placa M, Zauli G. Differentially expressed genes in HIV-1 tat-expressing CD4+ T-cell line. Virus Res. (2002) 90:337–45. doi: 10.1016/S0168-1702(02)00253-8
53. Coiras M, Camafeita E, Ureña T, López JA, Caballero F, Fernández B, et al. Modifications in the human T cell proteome induced by intracellular HIV-1 Tat protein expression. Proteomics. (2006) 6(Suppl. 1):S63–73. doi: 10.1002/pmic.200500437
54. Arese M, Ferrandi C, Primo L, Camussi G, Bussolino F. HIV-1 Tat protein stimulates in vivo vascular permeability and lymphomononuclear cell recruitment. J Immunol. (2001) 166:1380–8. doi: 10.4049/jimmunol.166.2.1380
55. Ajasin DO, Rao VR, Wu X, Ramasamy S, Pujato M, Ruiz AP, et al. CCL2 mobilizes ALIX to facilitate Gag-p6 mediated HIV-1 virion release. Elife. (2019) 8:1–38. doi: 10.7554/eLife.35546
56. Barillari G, Ensoli B. angiogenic effects of extracellular human immunodeficiency virus type 1 tat protein and its role in the pathogenesis of AIDS-associated kaposi ' s sarcoma angiogenic effects of extracellular human immunodeficiency virus type 1 tat protein and its role in. Clin Microbiol Rev. (2002) 15:310–26. doi: 10.1128/CMR.15.2.310-326.2002
57. Ensoli B, Gendelman R, Markham P, Fiorelli V, Colombini S, Raffeld M, et al. Synergy between basic fibroblast growth factor and HIV-1 Tat protein in induction of Kaposi's sarcoma. Nature. (1994) 371:674–80. doi: 10.1038/371674a0
58. Cantaluppi V, Biancone L, Boccellino M, Doublier S, Benelli R, Carlone S, et al. HIV type 1 Tat protein is a survival factor for Kaposi's sarcoma and endothelial cells. AIDS Res Hum Retroviruses. (2001) 17:965–76. doi: 10.1089/088922201750290087
59. Bonnet F, Morlat P, Chene G, Mercie P, Neau D, Chossat I, et al. Causes of death among HIV-infected patients in the era of highly active antiretroviral therapy, Bordeaux, France, 1998-1999. HIV Med. (2002) 3:195–9. doi: 10.1046/j.1468-1293.2002.00117.x
60. Matzen K, Dirkx AEM, Oude Egbrink MGA, Speth C, Götte M, Ascherl G, et al. HIV-1 Tat increases the adhesion of monocytes and T-cells to the endothelium in vitro and in vivo: implications for AIDS-associated vasculopathy. Virus Res. (2004) 104:145–55. doi: 10.1016/j.virusres.2004.04.001
61. Mazzuca P, Caruso A, Caccuri F. HIV-1 infection, microenvironment and endothelial cell dysfunction. New Microbiol. (2016) 39:163–73.
62. Duan M, Yao H, Hu G, Chen XM, Lund AK, Buch S. HIV Tat induces expression of ICAM-1 in HUVECs: Implications for miR-221/-222 in HIV-associated cardiomyopathy. PLoS ONE. (2013) 8:e60170. doi: 10.1371/journal.pone.0060170
63. Ensoli B, Barillari G, Salahuddin SZ, Gallo RC, Wong-Staal F. Tat protein of HIV-1 stimulates growth of cells derived from Kaposi's sarcoma lesions of AIDS patients. Nature. (1990) 345:84–6. doi: 10.1038/345084a0
64. Zhou F, Xue M, Qin D, Zhu X, Wang C, Zhu J, et al. HIV-1 Tat Promotes Kaposi's Sarcoma-Associated Herpesvirus (KSHV) vIL-6-Induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3β signaling pathway. PLoS ONE. (2013) 8:e0053145. doi: 10.1371/journal.pone.0053145
65. Agarwala MK, George R, Sudarsanam TD, Chacko RT, Thomas M, Nair S. Clinical course of disseminated Kaposi sarcoma in a HIV and hepatitis B co-infected heterosexual male. Indian Dermatol Online J. (2015) 6:280–3. doi: 10.4103/2229-5178.160271
Keywords: HIV-1C, genetic subtypes, Tat, signature amino acid residue, CCL2, endothelial cells, angiogenesis
Citation: Menon M, Budhwar R, Shukla RN, Bankar K, Vasudevan M and Ranga U (2020) The Signature Amino Acid Residue Serine 31 of HIV-1C Tat Potentiates an Activated Phenotype in Endothelial Cells. Front. Immunol. 11:529614. doi: 10.3389/fimmu.2020.529614
Received: 27 January 2020; Accepted: 18 August 2020;
Published: 25 September 2020.
Edited by:
Zhiwei Wu, School of Medicine, Nanjing University, ChinaReviewed by:
Johan Van Weyenbergh, KU Leuven, BelgiumCopyright © 2020 Menon, Budhwar, Shukla, Bankar, Vasudevan and Ranga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Udaykumar Ranga, dWRheWt1bWFyQGpuY2Fzci5hYy5pbg==; cmFuZ2F1ZGF5a3VtYXJAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.