 Charita Furumaya
Charita Furumaya Paula Martinez-Sanz
Paula Martinez-Sanz Panagiota Bouti
Panagiota Bouti Taco W. Kuijpers1,2
Taco W. Kuijpers1,2 Hanke L. Matlung
Hanke L. Matlung- 1Department of Blood Cell Research, Sanquin Research, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Pediatric Immunology, Rheumatology and Infectious Diseases, Emma Children’s Hospital, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
Over the last decades, cancer immunotherapies such as checkpoint blockade and adoptive T cell transfer have been a game changer in many aspects and have improved the treatment for various malignancies considerably. Despite the clinical success of harnessing the adaptive immunity to combat the tumor, the benefits of immunotherapy are still limited to a subset of patients and cancer types. In recent years, neutrophils, the most abundant circulating leukocytes, have emerged as promising targets for anti-cancer therapies. Traditionally regarded as the first line of defense against infections, neutrophils are increasingly recognized as critical players during cancer progression. Evidence shows the functional plasticity of neutrophils in the tumor microenvironment, allowing neutrophils to exert either pro-tumor or anti-tumor effects. This review describes the tumor-promoting roles of neutrophils, focusing on their myeloid-derived suppressor cell activity, as well as their role in tumor elimination, exerted mainly via antibody-dependent cellular cytotoxicity. We will discuss potential approaches to therapeutically target neutrophils in cancer. These include strategies in humans to either silence the pro-tumor activity of neutrophils, or to activate or enhance their anti-tumor functions. Redirecting neutrophils seems a promising approach to harness innate immunity to improve treatment for cancer patients.
Introduction
The new immunotherapies aimed at targeting of immunosuppressive checkpoint receptors, using antibodies against the inhibitory programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) pathway or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) pathway, have vigorously changed the landscape of anti-cancer immunotherapy in the last decades (1). This way of harnessing capacities of the immune system to combat cancer has recently delivered the first unprecedented clinical successes in the treatment of difficult-to-treat cancers, such as advanced stage metastatic melanoma (2), and non-small cell lung carcinoma (3), as well as more recently in other cancer types (4–6). Despite the encouraging efficacy seen for some cancer patients, where durable responses are observed for several years, others either fail to respond to these therapies or acquire resistance over time (7). Another recent and successful approach of cancer immunotherapy consists of the use of modified T cells with chimeric antigen receptors, the so-called CAR T cells. These have shown promising results for patients with advanced B-cell cancers in the early days (8) and more recently also in the case of other previously incurable malignancies, due to their specificity for a cell-surface antigen (9). Yet, these novel therapies still raise certain general concerns to clinicians, as they have been associated with serious toxicities (10). Although the current focus of cancer immunotherapies is on targeting adaptive immunity to fight the tumor, these therapies are limited to specific cancer types or need more efficient therapeutic control to reduce adverse effects. Besides, the cytotoxic anti-tumor capacities, as well as CAR T cell expansion, can easily be hampered by the interaction with other immune cells present in the tumor microenvironment (TME).
A different approach to immunotherapy in cancer focuses on the innate immune system. Cells belonging to the innate compartment are endowed with the capacity of reacting fast to invading pathogens that enter the body by recognizing sets of repeated patterns, as well as damage signals released from tissue injuries. In this way, innate immune cells have the outstanding talent of distinguishing self from non-self and respond to the latter in an appropriate manner. In addition, such cells are also able to distinguish self from “altered-self,” and this is the case for cancer. Innate immune cells such as natural killer (NK) cells, dendritic cells (DC), neutrophils, and macrophages are first-line effectors for the elimination of cancer cells before an adaptive immune response is mounted. In this way, NK cell-based therapies (i.e., used in the form of adoptive cell transfer or as CAR NK cells) (11), as well as DC-based therapies (i.e., used for vaccination against cancer) (12, 13) are currently being exploited for the treatment of cancer. However, the clinical application and efficacy of these therapies can be hampered as these approaches are still in early stages of development. Alternatively, neutrophils or polymorphonuclear (PMN) leukocytes, as the most abundant cell type among the circulating white blood cells in humans, can also be considered as compelling cells to address therapeutically.
Neutrophils: From Innate Effector to Myeloid Suppressor Cells
Neutrophils are commonly short-lived cells with a half-life in the circulation of less than 24 h and one or more days in the tissues depending on the extravascular milieu. Thus these cells are renewed in the bone marrow at an estimated rate of 1011 cells per day. The percentage of neutrophils in blood in healthy adults ranges from 50 to 70% (14), although these numbers may differ under pathological conditions (15). The mobilization of neutrophils from the bone marrow into the circulation is defined by the secretion of various stimuli from the site of injury or disease, to which they respond, resulting in variations in neutrophil numbers in blood. For example, granulocyte-colony stimulating factor (G-CSF), which is frequently produced by certain tumors or by cell types surrounding the tumor (16, 17), is able to skew the neutrophil retention/release balance in the bone marrow and ultimately lead to an increased release of neutrophils into the circulation (18). Moreover, chemokines, such as IL-8, can also be produced by tumor cells and attract myeloid cells, including neutrophils, to the TME, thereby affecting the number of neutrophils in the tumor (19). To date, many studies have looked at the neutrophil-to-lymphocyte ratio (NLR) to determine the correlation between the number of circulating neutrophils and cancer prognosis. In all cases, a high NLR has been recognized as a bad prognostic marker for all cancer types and stages of cancer (20–22). Moreover, the extent of intra-tumoral neutrophils also seems to have an unfavorable prognostic value in several cancer types (23–25). On the other hand, low neutrophil counts in blood has been proven advantageous for survival (26). Nonetheless, the option of persistent neutrophil depletion cannot be regarded in the context of cancer since prolonged neutropenia is considered a life-threatening condition due to the indispensable role of neutrophils in the protection of the host in the natural (mucosal) barriers against incoming microbial pathogens.
In fact, neutrophils in cancer may represent a heterogeneous population of cells, which can display different phenotypes and perform opposing functions. In mice, neutrophils found in tumor tissue have been referred to as tumor-associated neutrophils (TANs), which were classified as either anti-tumor neutrophils (N1), or tumor-promoting neutrophils (N2) (27). In experimental models, several factors of the TME have been suggested to determine the polarization of the recruited neutrophils in the tumor toward one or the other phenotype in vivo. Supportive evidence of such neutrophil subsets in human cancer tissue is, however, lacking to date. Studies in cancer patients have reported that circulating neutrophils can be classified into different subpopulations according to their densities upon isolation and centrifugation. Normal or high density neutrophils (HDNs) are associated with anti-tumor activity, whereas neutrophils in the low-density fraction (LDNs) are believed to expand in malignancy and display myeloid-derived suppressor cell (MDSC) and other pro-tumor activity. Similar to TANs, HDNs are capable of switching to LDNs in response to factors in the surrounding environment (28). Unfortunately, the current understanding of neutrophil subpopulations is still limited and debated due to the lack of specific molecular markers, non-uniform study approaches and variable expertise. Nonetheless, it is clear that neutrophils are being increasingly recognized as important players in cancer, and that they can carry both pro- and anti-tumoral properties depending on cancer type, stage and location of the disease (29).
In the following chapters, we aim to provide a summary of the current knowledge and latest findings on pro- and anti-tumor activities of neutrophils by describing the main mechanisms of action and highlight some conceivable ways by which we could silence tumor-promoting activity or redirect the activity of pro-tumor neutrophils into cytotoxic effector cells to help combat cancer.
Role of Neutrophils in Tumor Progression
Promotion of Proliferation, Angiogenesis, Invasion and Metastasis by Neutrophils
Neutrophils are capable of promoting tumor growth and progression, and their presence is often associated with poor clinical outcome (20). Mechanisms by which neutrophils have been shown to mediate tumor progression include enhancing proliferation, angiogenesis, invasion, metastasis, and immune suppression (30). Neutrophils in the TME have the ability to directly induce the proliferation of cancer cells, for example via the serine protease neutrophil elastase (NE) (31, 32). NE also plays a role in the migration and invasion of cancer cells (33). Other neutrophil granule components such as matrix metalloproteinase-9 (MMP-9) have been described to mediate angiogenesis and tumor cell invasion via degradation of the basement membrane (34). Additionally, a large body of literature demonstrates a pro-metastatic function of neutrophils. In a mouse breast cancer model, neutrophil-derived factors were shown to drive cancer spread (35). Furthermore, neutrophils have been suggested to promote cancer cell adherence, which was shown to be dependent on neutrophil Mac-1 (αMβ2 or CD11b/CD18), and thereby mediate metastasis in a murine model of liver metastasis (36). Concordantly, human neutrophils were shown to induce tumor cell migration and to interact with melanoma cells via β2 integrin (37).
Also the involvement of neutrophil extracellular traps (NETs) in cancer cell migration and extravasation is being investigated. Upon activation, neutrophils form NETs composed of released chromatin and granular proteins which trap and kill microbes (38). The neutrophil chemoattractant IL-8, which is produced in the TME, has been suggested to induce NET formation (39). The presence of NETs in the TME of patients with metastatic disease has been demonstrated, and additional studies in murine models have further suggested their role in cancer progression (40). NETs promoted cancer cell migration, invasion, and angiogenesis in vitro (41). Multiple studies illustrated the trapping of circulating murine tumor cells in NETs, which facilitated their extravasation and metastasis (42–44). Increased levels of NETs were also observed in patients suffering from different types of locally infiltrating cancer (45, 46), which was associated with adverse patient outcomes in colorectal cancer (47).
Immunosuppression by Neutrophils
In mice, MDSCs represent a heterogeneous group of pathologically activated immature myeloid cells with immunosuppressive properties (48). MDSCs accumulate under inflammatory conditions, including experimental cancer, and are divided into two major subsets depending on their lineage, either granulocytic (PMN-MDSCs) or monocytic (M-MDSCs) (49). The presence of PMN-MDSCs in patients has been shown to be associated with poor prognosis in different types of cancer (50–52). In mice, PMN-MDSCs are characterized as CD11b+Ly6G+ cells, while in humans the surface marker definition is CD11b+CD15+CD14–CD33+CD66b+HLA-DR– (53). However, based on these cell surface markers, PMN-MDSCs overlap with all circulating neutrophils, making an accurate discrimination between PMN-MDSCs and neutrophils impossible. Also other markers proposed to be more specific in identifying PMN-MDSCs, such as LOX-1 or CD10 (54, 55), have not been confirmed to discriminate circulating PMN-MDSCs in cancer patients (56).
While PMN-MDSCs were originally described as a subpopulation of immature myeloid cells capable of suppressing immune responses, mature neutrophils also have the ability to limit T cell activity and promote immune evasion (28, 57), but only upon cellular activation (56, 58). Thus the functional similarities between PMN-MDSCs and neutrophils further complicate the differentiation between the two populations. Functional plasticity of neutrophils suggests that a shift in neutrophil phenotype occurs, depending on signals from the TME, which lead to the acquisition of immunosuppressive activity or other pro-tumorigenic functions. To avoid confusion, we will mostly refer to these cells as immunosuppressive neutrophils. Such mature neutrophils with a T cell suppressive phenotype have been identified in various human cancers and are also associated with accelerated tumor progression and worse clinical outcomes (49, 58), illustrating their clinical relevance as potential targets to improve cancer immunotherapy.
Activation of Neutrophil Immunosuppressive Activity
Tumor cells and other cell types in the TME produce a wide range of inflammatory mediators, many of which have been demonstrated to contribute to the generation and recruitment of neutrophils with pro-tumor activity. High levels of the colony stimulating factor G-CSF released by tumors corresponds with the expansion of immunosuppressive neutrophils in cancer patients (50). Likewise, mature neutrophils of G-CSF-treated donors have been reported to display an activated immunosuppressive phenotype (55). Other signals implicated in the pathological activation of neutrophils include GM-CSF, TNFα, IL-1β, VEGF, IL-6, and IL-8 (59). However, our latest experiments in human neutrophils demonstrated that only fMLF, TLR ligands such as LPS, and TNFα act as activators of T cell suppressive activity in neutrophils (56, 60). The presence of soluble factors in ascites and malignant effusions from cancer patients was shown to induce a suppressive phenotype of neutrophils in the TME, which was dependent on complement factor C3 (58).
Mechanisms of Neutrophil Immunosuppressive Activity
In order to limit T cell mediated anti-tumor immune responses, suppressive neutrophils rely on several effector functions originally linked to their role as killers of invading pathogens. Degranulation refers to the process by which neutrophils release various factors stored in intracellular granules into phagosomes or the extracellular environment (61). Immunosuppression by neutrophils has been linked to the metabolism of L-arginine, which is converted into L-ornithine by arginase-1, an enzyme present in gelatinase granules (62, 63). Elevated arginase-1 plasma levels were observed in cancer patients, and the modulation of T cell responses was shown to be dependent on arginase-1 (50, 64, 65) via the depletion of L-arginine, an amino acid crucial for the expression of the T cell receptor ζ chain, which is in turn needed for T cell activation (66–68). Additionally, L-arginine shortage prevents the successful formation of immunological synapses due to impaired dephosphorylation of cofilin, which is an important player in the modulation of the actin cytoskeleton and the formation of an immunological synapse (69, 70). The dependence on arginase-1 and its regulation of the T cell receptor ζ chain was demonstrated using PBMCs from healthy donors (71, 72) and in cancer patients (64, 73). Accordingly, suppression of T cell mediated responses by activated human neutrophils was shown to depend on degranulation, which was elegantly confirmed using neutrophils from rare familial hemophagocytic lymphohistiocytosis (FHL)-5 patients, which show defective granule mobilization due to mutations in the STXBP2 gene (56, 74–76).
The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex generates oxidative stress by the production of reactive oxygen species (ROS) upon cellular activation, a process which is up regulated in different types of cancer (77). Hydrogen peroxide (H2O2) produced by the conversion of superoxide (O2–) suppresses T cell activation and proliferation via several mechanisms (78, 79). H2O2 can induce T cell apoptosis, or inhibit T cell activation by blocking NF-κB activation or affecting the availability of the T cell receptor ζ chain, which is essential for T cell activation, as described above (80, 81). T cell activation is additionally impaired by the interference of ROS with the regulation of cofilin (82, 83), as was also described as an effect of L-arginine deficiency. While L-arginine deficiency leads to impaired dephosphorylation of cofilin, ROS induce a conformational change in cofilin through its oxidation, both resulting in an inhibition of T cell response (69, 82, 83). Furthermore, H2O2 could be involved in preventing the metabolic switch from mitochondrial respiration to aerobic glycolysis that is required for T cell clonal expansion and cytokine production (84). Our data also indicate that ROS production is required for the suppression of T cell activation by human neutrophils (56). This was substantiated using neutrophils from patients with chronic granulomatous disease (CGD), a rare genetic defect in which neutrophils are incapable of ROS production due to a mutation in a subunit of the NADPH oxidase complex (85, 86).
T cell function could additionally be impaired by suppressive neutrophils via the interaction between T cell PD-1 and PD-L1 on neutrophils, which is known to block T cell proliferation and cytokine production (87). Crosstalk between PD-1 and PD-L1 plays an important role in T cell suppression in cancer (88). Neutrophil expression of the immune checkpoint surface molecule PD-L1 can be induced by IFNγ and GM-CSF or hypoxic conditions (89–91). Increased PD-L1 expression as well as suppression of T cell proliferation by neutrophils were demonstrated in patients with hepatocellular carcinoma (92). Moreover, inhibition of T cell function by neutrophils was dependent on PD-1 expression by T cells and PD-L1 expression by neutrophils. PD-L1+ neutrophils suppress T cell function by interacting with PD-1 on T cells, thus limiting the anti-tumor activity of T cells. This immune tolerance via neutrophil PD-L1 appears to contribute to human gastric cancer growth and progression (93).
In order for neutrophils to suppress T cell responses, the formation of close cell-cell contact via the expression of integrin Mac-1 on neutrophils is required (94). Singel et al. recently demonstrated the requirement of cell-cell contact for T cell suppression by human neutrophils activated by ovarian cancer ascites (58). This is in line with our findings, which demonstrate that suppression of T cells by neutrophils is dependent on CD11b-mediated interactions (56). Consistently, neutrophils from a patient with leukocyte adhesion deficiency type 1 (LAD-1), which lack the expression of β2 integrins, including Mac-1, were not capable of suppressing T cell proliferation (56). Blocking ICAM-1 on T cells decreased, but did not fully abrogate T cell suppression by activated neutrophils, indicating that additional ligands of Mac-1 on T cells are important for the interaction with neutrophils (56). In tumor-bearing mice, neutrophils were shown to induce T cell apoptosis in a contact-dependent manner (95). Similarly, an immunosuppressive myeloid cell population induced apoptosis of activated T cells in breast cancer patients (96). In our studies, live-cell imaging revealed intercellular contacts between neutrophils and T cells resulting in neutrophils containing pieces of T cell membrane (56), a process known as trogocytosis (97). During co-culture of activated neutrophils and T cells, a population of smaller T cells appeared, which could no longer be activated to proliferate. In contrast to earlier findings with respect to apoptosis induction, these small T cells displayed neither of the two common cell death mechanisms, i.e., apoptosis or necroptosis. Thus, the exact steps leading to T cell death upon suppression by neutrophils remain to be further investigated.
In addition to their own direct and suppressive effect on T cells, neutrophils can also inhibit NK cell activation in tumor-bearing mice (98, 99). Neutrophils suppressed NK cell cytotoxicity, which resulted in defective antitumor responses and promoted metastasis in mice (100, 101). Finally, further contribution of neutrophils to the immunosuppressive microenvironment within tumor tissue is made via the development and induction of regulatory T cells (Tregs) (102). Treg enrichment occurs due to reduced sensitivity of Tregs to oxidative stress in the TME (103, 104), which ultimately accounts for their preferential outgrowth and selection. Neutrophils also recruit Tregs through the secretion of CCL17, as was shown in murine tumors (105). Additionally, a positive feedback loop between neutrophils and Tregs, mediated by the anti-inflammatory cytokine IL-10, could support the immune suppression (106, 107). Induction of Tregs by suppressive neutrophils was demonstrated in patients with bladder cancer (108).
In summary, neutrophils can directly and indirectly mediate tumor progression via the promotion of proliferation, angiogenesis, invasion, and metastasis, in which mechanisms such as the release of granule components and NET formation are important. Furthermore, neutrophils inhibit anti-tumor responses by limiting T cell activation, which is dependent on degranulation, ROS production and cell-cell contact. Alternatively, neutrophils contribute to an immunosuppressive TME by affecting other immune cells, such as NK cells and Tregs. Apart from the pro-tumorigenic mechanisms by which neutrophils are activated to “adopt” their tumor-enhancing, immunosuppressive role, neutrophils can also exert strong anti-tumor activity.
Role of Neutrophils in Tumor Elimination
The vast majority of studies support the positive correlation of neutrophils with cancer progression by the mechanisms described above. Nevertheless, recent data also point into the opposite direction in which neutrophils can act as effector cells and combat cancer leading to the eradication of tumor cells (30, 109, 110). In fact, there are sufficient reasons to think of neutrophils as promising effector cells against cancer. One of the main advantages of this cell type is that they are found in high levels in blood under normal conditions, and in addition, their number can be incremented further upon in vivo treatment with G-CSF and GM-CSF cytokines (111, 112). Another feature that makes them unique is that neutrophils do not need ex vivo culturing and expansion for a later infusion into the patient, compared to most other immune cells used therapeutically, such as T cells, NK cells or DCs (113). Siders et al. showed that increasing the number of circulating neutrophils by a mere injection of G-CSF turned them into excellent cytotoxic cells that were able to kill alemtuzumab-opsonized cells in a xenograft model of a CD52+ tumor (114). This, however, must be regarded with caution as such effect may only be valid in the context of antibody therapy, as was also shown in other studies (115, 116). In other circumstances, the therapeutic effect of G-CSF treatment may mainly mobilize neutrophils of an immunosuppressive phenotype which are beneficial to the tumor, as discussed earlier in this review (50, 55).
Direct Killing Properties of Neutrophils Against Cancer
The mechanisms by which anti-tumor neutrophils induce cytotoxicity of the tumor cells are, however, not yet clear. Few studies demonstrated that the secretion of ROS through the respiratory burst comes into play once neutrophils are in direct contact with the tumor cells (117–119). Specifically, neutrophil secretion of H2O2 induced a lethal influx of Ca2+, mediated by the TRMP2 channel which ultimately killed the tumor cell. Furthermore, a mechanism was identified by which neutrophils isolated from healthy donors induced apoptotic cell death of tumor cells upon physical contact mediated through Fas ligand/Fas interaction (120). Although these contact-dependent processes can, under specific circumstances, induce tumor cell killing, the best well-established anti-tumor mechanism by which neutrophils mediate tumor cell death is through Fc receptor-dependent cytotoxicity against antibody-opsonized cells.
Antibody-Mediated Killing Properties of Neutrophils Against Cancer
Therapeutic monoclonal antibodies (mAbs) have become available for the treatment of cancer in the last decades and have provided significant improvement in the treatment outcome for a number of cancer subtypes. These mAbs can initiate direct tumor cell killing, through the F(ab’)2 domains of the immunoglobulin (Ig) interfering with the intrinsic function of the target cell (121, 122), or upon binding of the Fc part to the C1q component of the complement system inducing complement-dependent cytotoxicity (CDC) (123). Besides the direct mechanisms, the Fc region of the mAb can also bind to activating Fc receptors on innate immune cells which will in turn elicit indirect-mediated killing via antibody-dependent cellular cytotoxicity/phagocytosis (ADCC/P) of the opsonized cells (124–128).
The contribution of neutrophils in ADCC to eradicate antibody-opsonized tumor cells in vitro was already demonstrated some decades ago by Gale and Zighelboim (129). A few years later, Barker and colleagues showed a prominent role of neutrophils isolated from neuroblastoma patients in mediating the ex vivo lysis of neuroblastoma cells opsonized with anti-GD2 antibody. Interestingly, the cytotoxicity induced by neutrophils was in all cases higher than was seen for NK cells from the same donor (130). More recently, a number of studies have demonstrated the cytotoxic capacity of neutrophils in the context of antibody therapy in an in vivo setting of tumor-bearing mice which showed once again the indispensable role of neutrophils in achieving efficient antibody therapy responses (114, 131, 132). Most importantly, a considerable amount of literature has suggested the relevance of the anti-tumor capacities of neutrophils in a clinical setting. More specifically, different polymorphic variants found on the IgG Fc receptor IIa (FcγRIIa) explained the variability seen in the clinical outcome of breast cancer (133, 134), colorectal cancer (135), and neuroblastoma patients (136) in response to antibody treatment.
IgG Isotypes and Fcγ Receptor Involvement
Despite the fact that several Ig isotypes exist, as of yet, all approved therapeutic antibodies for cancer treatment in the clinic are IgG-based, particularly IgG1, IgG2, and IgG4 isotypes. The relevance of the IgG backbone used for such antibodies has to do with the different binding affinities to the several Fcγ receptors that are in turn differentially expressed on a number of immune cells (137, 138). This results in the induction of extremely diverse and highly regulated antibody responses, as the distinct affinities can also convey stronger or weaker effector functions by the different Fcγ receptor-expressing cells (139).
Six classical Fcγ receptors can be expressed by the human innate immune cells: FcγRI (CD64), FcγRIIa (CD32a), FcγRIIb (CD32b), FcγRIIc (CD32c), FcγRIIIa (CD16a), and FcγRIIIb (CD16b). From these, neutrophils constitutively express FcγRIIa and FcγRIIIb, and the FcγRI only upon activation. Depending on ethnic background, 15% of the population also expresses low levels of neutrophil FcγRIIc (140). All these Fcγ receptors on neutrophils can bind to the IgG opsonizing cancer cells (138, 141) and have their own capacity to contribute to ADCC activity. FcγRI and FcγRIIa/c are activating receptors and their signals are transduced by an immunoreceptor tyrosine-based activation motif (ITAM) that is either associated with the common FcRγ chain in the case of FcγRI, or present in the cytoplasmic tail of the Fcγ receptor itself for FcγRIIa/c. Subsequently, Syk tyrosine kinases can bind to these activating motifs and activate downstream signaling pathways, initiating ADCC, phagocytosis, cell migration, and degranulation processes (141, 142). FcγRIIIb, instead, is anchored to the cell membrane through a glycosyl phosphatidylinositol (GPI) molecule and lacks both transmembrane and cytoplasmic domains, restricting it from having signaling capacities (143). As mentioned above, resting neutrophils constitutively express both low- and intermediate- affinity FcγRIIa and FcγRIIIb in high levels (141). However, the high-affinity activating FcγRI is not detectable on resting neutrophils but can be up regulated upon activation of cells with G-CSF and IFNγ as demonstrated by us (30, 144) and others (145, 146). In addition, FcγRIIIb is actively shed from the surface of activated neutrophils when stimulated by various stimuli, including G-CSF and IFNγ (144, 147).
The specific contribution of each Fcγ receptor of neutrophils to mediate ADCC, however, has been found to differ per cancer type. With the use of blocking antibodies for the different Fcγ receptors, several studies have reported FcγRIIa to be the dominant receptor triggering neutrophil ADCC in solid tumors. This was seen for EGFR+ cancer cells opsonized with cetuximab (148), as well as for trastuzumab-coated HER2/neu+ human breast cancer SKBR3 cells (144, 149). Of interest, although FcγRI is the high-affinity receptor for IgG1 antibodies, no effect was found when monovalent Fc fragments were used for blockade. This might be explained by the incapacity to fully block the receptor by use of these monovalent Fc fragments or due to the relatively low expression levels on the surface of neutrophils (150). Nevertheless, contradicting results were described regarding the potential of FcγRI on neutrophils in mediating ADCC. It was shown that such receptor can very well contribute to tumor killing in a number of tumor types (151–154). With regard to the involvement of FcγRIIIb in the killing of tumor cells in the context of antibody therapy, we and others have shown this receptor to be a negative regulator of neutrophil ADCC as it scavenges available therapeutic antibody due to its high expression on the neutrophil surface (144, 148). For hematological tumors the evidence is slightly different. An in vivo study testing the efficacy of a FcγRI bispecific antibody in a mouse model of a rituximab-treated B-cell lymphoma reported the clearance of the tumor cells by G-CSF stimulated neutrophils (155), indicating that FcγRI may be the main receptor mediating neutrophil ADCC in such context. Another level of complexity of this receptor family comes from the several Fcγ receptor polymorphisms described in humans that could affect the degree of ADCC responses [extensively reviewed by Bruhns and Jonsson (138)], which ultimately can influence clinical responses to antibody therapy. It is, however, relevant to acknowledge that Fcγ receptors are not the only feature that makes neutrophils capable of killing cancer cells. Although falling outside the scope of this review, integrins, Mac-1 in particular, have also been shown to be indispensable in mediating ADCC processes (149).
Neutrophil-Mediated Antibody-Dependent Killing Mechanisms
Until recently, the mechanism by which ADCC leads to cell death remained largely unclear. A large body of evidence has reported that neutrophils have the ability to trogocytose, mainly in the context of antibody therapy (156). In 2002, trogocytosis was identified as an active mechanism that involves the transfer of plasma membrane and their associated molecules from a donor cell to an acceptor cell during intercellular contact (157). Although the purpose of the process has remained unclear (97), more recently, new evidence has accumulated to determine the importance of trogocytosis. In the context of infection, neutrophils were able to kill serum-opsonized Trichomonas vaginalis parasite using trogocytosis (158). Additionally, the tumoricidal effect of trogocytosis was shown in the study of Velmurugan et al., where they demonstrated that macrophage-trogocytosis led to efficient tumor cell death of trastuzumab-opsonized breast cancer cells (159). Most recently, we reported a direct association between neutrophil-mediated trogocytosis and tumor cell killing in antibody-opsonized solid cancer cells (149), by which the neutrophil takes “bites” from the plasma membrane of the cancer cells. Hereby, the neutrophils cause membrane damage eventually leading to a necrotic type of cancer cell death. Furthermore, neutrophil-mediated antibody-dependent destruction of cancer cells was found not to depend on their classic antimicrobial effector mechanisms, such as granule exocytosis (degranulation) and NADPH oxidase activity. In particular, neutrophils from FHL-5 patients, as well as neutrophils from patients with CGD, showed unexpectedly intact killing of HER2/neu+ breast cancer cells in the presence of the therapeutic antibody trastuzumab (149). Together, these findings support the idea of trogocytosis as a most relevant process involved in tumor killing in the context of antibody therapy.
Conversely, trogocytosis has also been described as a mechanism to escape ADCC, ADCP, or CDC, mainly in hematological cancers, as it involves the shaving of the target antigen from tumor cells by the effector cells. This was shown not only for chronic lymphocytic leukemia (CLL) upon rituximab treatment, where a partial loss of CD20 from CLL B cells accompanied trogocytosis events by neutrophils (160), but also anti-CD38 therapy with daratumumab directed against multiple myeloma (MM) which led to monocyte- and neutrophil-mediated shaving of CD38 from MM cells (161). Moreover, neutrophil-mediated trogocytosis might be the reason behind the significant reduction in HER2/neu expression, seen in a cohort of breast cancer in women treated with trastuzumab (162, 163). Therefore, depending on the circumstances, or perhaps tumor type, trogocytosis can be regarded either as a process initiating tumor killing or as a way for the tumor cells to evade immune activity.
Role of Neutrophils Regulating Adaptive Immune Response
For cancer therapy, it has now become clear that initiating potent adaptive immune responses is fundamental to establish long-term anti-tumor immunity. Although neutrophils have historically been regarded as strict innate cells with end-stage effector functions, new evidence has emerged manifesting their involvement in modulating the adaptive immune compartment (31, 32). At sites of infection, neutrophils were found to act as danger sensors by communicating the presence of inflammation or damage to DCs, which induces DC maturation, triggering in turn strong proliferation and Th1 polarization of naive CD4+ T cells (164, 165). In addition, neutrophils can also act as APCs themselves. In vitro, activated neutrophils stimulated with GM-CSF and IFNγ were able to present antigens to memory CD4+ T cells due to the acquisition of MHC-II molecule expression, as well as costimulatory molecules such as CD86, OX40L and 4-1BBL at early stages of tumorigenesis (166–168). Moreover, both human and mouse neutrophils were found to cross-present exogenous antigens to naive CD8+ T cells thereby turning them into cytotoxic T cells (169). Lastly, recent evidence suggests that upon antigen capture at the periphery, neutrophils can migrate to the lymph nodes in a CC-chemokine receptor 7 (CCR7)-dependent manner, under certain circumstances, as their presence has been found in lymphoid organs in vivo (170–172).
In the context of cancer, however, neutrophils can mediate opposite adaptive immune responses depending on the TME or stage of the tumor in vivo. For some cancer mouse models, neutrophil depletion led to a decrease in CD4 and CD8 T cell activation, thereby enhancing tumor growth (173), while in others their presence was able to suppress CD8 T cells and promote metastasis (174). The evidence for the role of neutrophils in inducing adaptive immune responses after antibody therapy of cancer is, unfortunately, more scarce. Yet, neutrophils were able to boost T-cell activation when combining a Fc/IL-2+TA99 antibody with adoptive T-cell transfer in a B16F10 melanoma model achieving significant tumor control (175). In general, neutrophils can not only play a role in innate immunity but also guide and support adaptive immune responses through cellular crosstalk.
Targeting Neutrophil Activity in Cancer Therapy
The different functions that neutrophils acquire in the context of cancer highlight their plasticity and ability to respond toward various targets within and outside of the TME. In this review, we have focused on the role of neutrophils in tumor progression or tumor elimination and we have described the major mechanisms that neutrophils utilize to achieve an efficient response. Although mechanistically not yet completely understood, some reports (28, 176) suggest TANs in several mouse models of cancer to display a gradual change during tumor progression, shifting from anti-tumor properties at the early stages toward pro-tumorigenic properties during the course of the disease.
As reviewed above, both immunosuppressive as well as anti-tumor neutrophils share some common characteristics, i.e., their need for activation through specific stimuli to be able to optimally exert their function (27, 50, 59, 177). Another necessity for neutrophils to perform their MDSC and anti-tumor function is Mac-1-mediated close contact, either between neutrophil and T cell in case of MDSC activity, or between neutrophil and antibody-opsonized tumor cell in case of ADCC (56, 58, 94, 149). At the same time, the fact that classical anti-microbial killing mechanisms, i.e., degranulation and ROS production, are essential for MDSC function of neutrophils, yet dispensable for neutrophil ADCC, shows that at least some of the required cellular mechanisms for MDSC and ADCC neutrophil activity are directly opposing (56, 149).
Together, in the following part of this review we will elaborate on ways to modulate pro-tumorigenic function, further enhance the anti-tumor response, or even redirect tumor-promoting neutrophils toward anti-tumor neutrophils (Figure 1). Interestingly, the first in vitro study showing a way to polarize human neutrophils toward the distinct N1 or N2 phenotype has recently been published (178).
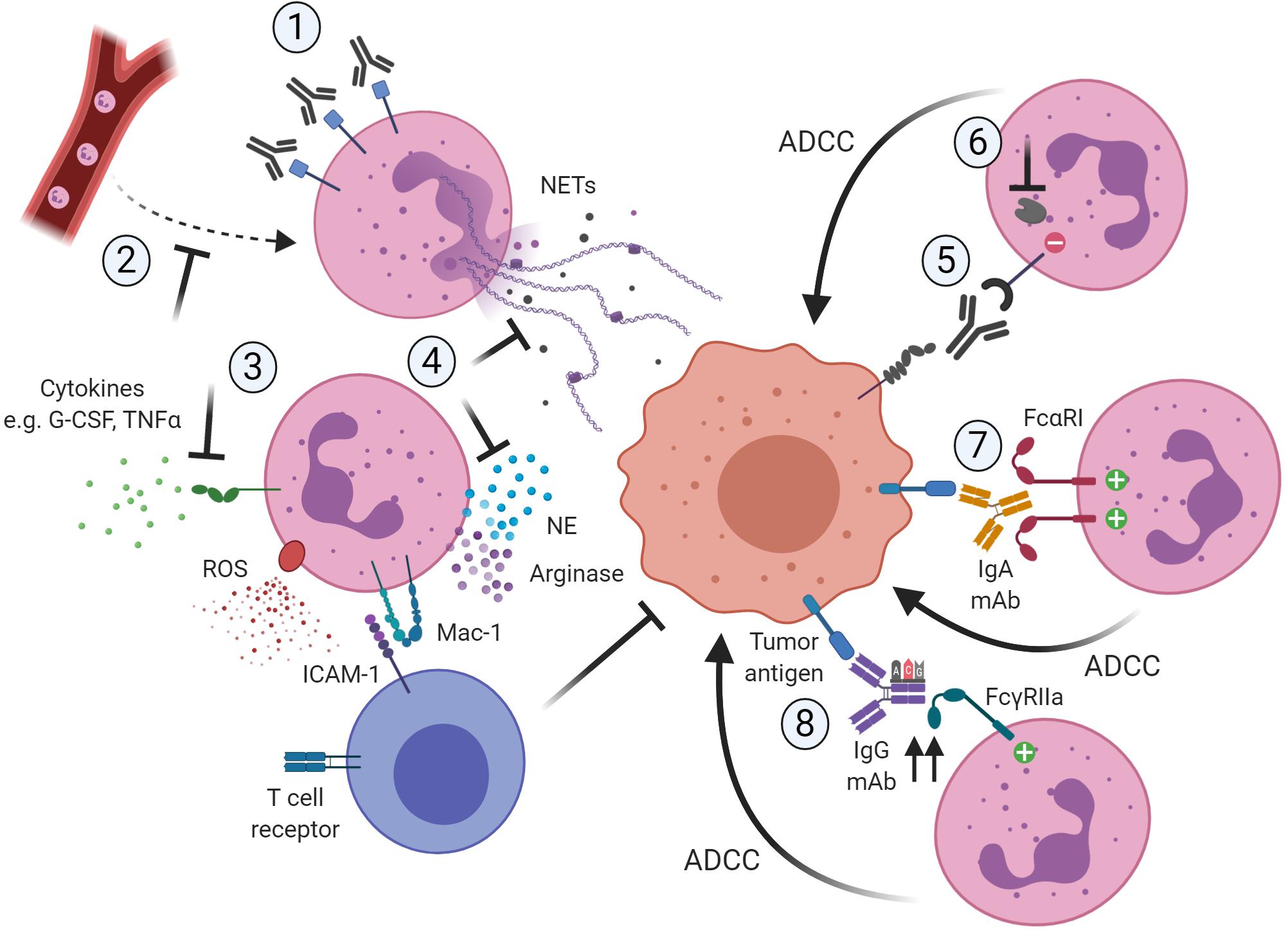
Figure 1. Potential ways to therapeutically target neutrophils in cancer by blocking their pro-tumor activity (1–4) or promoting their anti-tumor capacities (5–8). (1) Reduction of neutrophil numbers in the TME, for example by using antibodies targeting CD33 present on immunosuppressive neutrophils. (2) Blocking the recruitment of pro-tumor neutrophils to the TME, for example by inhibiting the chemokine receptor CXCR2. (3) Blocking of activation signals such as G-CSF or TNFα necessary for neutrophils to acquire a pro-tumor or immunosuppressive phenotype. (4) Neutrophils require ROS production and degranulation to exert their immunosuppressive role, which is dependent on close contact with T cells via Mac-1. Also, neutrophils rely on mechanisms such as NET formation and the release of granule components such as NE to exert their pro-tumor activity. Targeting these downstream mechanisms would limit the pro-tumorigenic activity of neutrophils. (5) Interfering with innate immune inhibitory checkpoints would restore antibody-mediated anti-tumor capabilities of neutrophils. (6) Targeting the recruitment and function of downstream regulators of the inhibitory receptor would further enhance antibody-mediated anti-tumorigenic capacities of neutrophils toward tumor cells. (7) The use of IgA-based therapeutic mAbs that can bivalently bind FcαRI on the neutrophil would induce stronger anti-tumor cytotoxic responses. (8) The use of protein engineering techniques to modify the Fc region of IgG therapeutic antibodies would increase the affinity to the activating FcγRIIa, resulting in more potent ADCC responses toward the opsonized tumor cells. Created with BioRender.com.
Limiting Pro-tumorigenic Capacity of Neutrophils
Reducing Neutrophil Numbers
The first possibility to interfere with the tumor-promoting role of neutrophils in cancer is the neutralization of these cells. Antibody-mediated depletion of neutrophils resulted in decreased metastasis in an intrasplenic model of liver metastasis and in a metastatic breast cancer mouse model (36, 174). Similarly, targeting of Ly6G+ cells in a murine model of pancreatic ductal adenocarcinoma increased intra-tumoral T cell accumulation and inhibited cancer progression (179).
The described promising results of antibody targeting in mice called for a similar approach to be undertaken in humans, in order to promote anti-cancer T cell responses. Based on immunophenotyping and functional assays to detect T cell suppression, the presence of suppressive neutrophils was determined in blood obtained from patients with different types of cancer, including prostate, lung, head and neck, and breast. The immunotoxin Gemtuzumab ozogamicin was used to deplete cells expressing CD33, identified as a surface marker on suppressive cells across cancer types. This depletion restored T cell proliferation, enhanced CAR T cell responses and tumor cell death (180).
Furthermore, a small-molecule receptor tyrosine kinase inhibitor has proven to successfully modulate the immunosuppressive TME. Sunitinib inhibits signaling through multiple receptor tyrosine kinases, including vascular endothelial growth factor receptors (VEGFRs), platelet-derived growth factor receptors (PDGFRα and PDGFRβ), stem cell factor receptor (c-Kit) and colony-stimulating factor-1 receptor (CSF-1R) (181). Treatment with sunitinib decreased the number of suppressive cells, enhanced CD8 and CD4 cell tumor infiltration and improved survival in tumor-bearing mice (182). In renal cell carcinoma patients, sunitinib also reversed T cell suppression and reduced Tregs at the tumor site (183). Moreover, treatment with sunitinib in human renal cell carcinoma improved the expansion of tumor-infiltrating lymphocytes (184).
Since neutrophils were shown to up regulate PD-L1, neutrophils may also be affected by therapies targeting immune checkpoints. For example ipilimumab treatment led to reduced PMN-MDSC numbers and less immunosuppressive activity in melanoma patients, which correlated with improved clinical outcome (185–187). Thus, increasing clinical evidence supports the notion that reducing neutrophil numbers in cancer could be beneficial to patients.
Targeting Neutrophil Recruitment and Activation of MDSC Activity
An alternative approach to targeting the pro-tumor activity of neutrophils is to inhibit their recruitment or activation. For instance, IL-8 secreted by tumor cells is responsible for the chemotactic recruitment of neutrophils to the TME via the receptors CXCR1 and CXCR2 (39). Already applied in patients suffering from other inflammatory diseases, inhibition of CXCR2 prevents the recruitment of immunosuppressive neutrophils (188, 189). Recent studies in mouse models of cancer have also shown promising effects of CXCR2 inhibition, which increased effector T cell accumulation in tumors and enhanced responses to immunotherapy, slowing tumorigenesis or preventing metastasis (190–192). The effect of the CXCR1 and CXCR2 inhibitors reparixin and SX-682 are currently being tested in clinical trials in metastatic breast cancer patients (NCT02370238, NCT03161431) (193).
Another example of the stimulation of anti-cancer responses achieved by limiting neutrophil recruitment is the inhibition of the receptor tyrosine kinase cMET (194). cMET is a receptor for hepatocyte growth factor (HGF), which shows increased levels and association with poor clinical outcome in human cancer (195). Limiting neutrophil recruitment by blocking cMET promotes the efficacy of adoptive T cell transfer and checkpoint therapy in murine melanoma (196). Antagonists of the HGF/cMET pathway have been developed and are being tested in multiple types of human cancer (197).
In addition, targeting pathways leading to the pathological activation of neutrophils could represent a strategy to limit their tumor-promoting effects. IL-17 production by γδ T cells was shown to induce G-CSF release in mice, resulting in the accumulation of neutrophils with a T cell-suppressive phenotype (174, 198). In human breast cancer, IL-17 and γδ T cells have also been described as poor prognostic factors (199, 200). Depletion of IL-17, G-CSF or γδ T cells resulted in decreased T cell suppression, and the absence of γδ T cells or neutrophils reduced metastases in a murine breast cancer model (174). Currently, IL-17 specific antibodies are being tested in psoriasis patients, but more preclinical studies are needed before this could also be applied in cancer patients (201).
In humans, G-CSF-mobilized neutrophils display immunosuppressive activity (55). Interestingly, we reported that neutrophil mobilization for granulocyte transfusion purposes silenced the ability of neutrophils to suppress T cell responses (202). The lack of MDSC activity was identified while our findings demonstrated unaltered anti-microbial effector activities, including motility, ROS formation, degranulation and killing of bacteria and fungi (202, 203). This important finding suggests that MDSC activity comprises common effector functions combined with a distinct and unique activity that is (only) involved upon MDSC induction. This finding raises the possibility of using a selective approach in patients to silence immunosuppressive neutrophils and to thereby enhance T cell activity in tumors. Alternatively, proteins of the complement system represent potential targets to block the immunomodulatory and pro-tumor activity of neutrophils, since the importance of C3 activation in T cell suppression by neutrophils from cancer patients was nicely illustrated (58).
Targeting Additional Pro-tumor Function of Neutrophils
As described in previous sections, neutrophils rely on several mechanisms to exert their pro-tumorigenic function. Interfering with these downstream tumor-enhancing effects could provide therapeutic benefit. For instance, increased uptake of fatty acids by murine PMN-MDSCs was demonstrated to support their immunosuppressive activity (204). Accordingly, PMN-MDSCs of patients with head and neck, lung, or breast cancer displayed lipid accumulation, along with increased expression of FATP2, a fatty acid transporting protein (205). FATP2 deletion in mice resulted in the loss of the ability to suppress T cell responses leading to a delay in tumor progression. However, most findings from these experimental studies were obtained in mouse models, which by definition cannot be easily extrapolated to human cancer (206).
Other studies have focused on targeting NE, which contributes to cancer progression through enhanced proliferation, invasion and metastasis (31–33, 35–37, 207, 208). Suppression of NE activity by the small molecule sivelestat resulted in reduced tumor growth in murine models of colorectal and prostate cancer (207, 208). The NE inhibitor sivelestat sodium hydrate has been successfully applied in esophageal cancer patients, although this study did not focus on tumor progression (209). Furthermore, NETs may represent a target for reducing the pro-tumor effects of neutrophils since their elimination by the DNA degrading enzyme DNase I is a method that has been established and is tested in clinical trials1, though not yet in cancer (210). More clinical studies are necessary to investigate the potential of targeting NE or NETs in human cancer patients. The potential ways to limit pro-tumorigenic activity of neutrophils are summarized in Figure 1; 1–4.
Promoting Anti-tumor Capacity of Neutrophils
Releasing the Off-Switch
Although the above-mentioned approaches to modulate MDSC activity or other pro-tumorigenic effector functions of neutrophils are very promising, redirecting the toxic MDSC activity against T cells toward an anti-tumorigenic role might contribute and steer the effector functions away from neutrophils as suppressor cells to effective tumor-killing in the TME. We have already described the efficacy of neutrophils attacking antibody-opsonized tumor cells, and there by we could speculate that the presence of a mAb could already shift MDSC activity to a certain extent by binding to the Fcγ receptor on their surface and subsequently inducing a cytotoxic response.
Recent research has been focusing on further augmenting the anti-tumor responses of neutrophils, i.e., by trying to switch off the brakes. Neutrophils express a variety of inhibitory receptors on their cell surface (211) providing potential therapeutic targets for checkpoint-blockade therapy. One well-established example of successful checkpoint-blockade on neutrophils is CD47-signal regulatory protein alpha (SIRPα) disruption (212). We and others have already shown the potency of blocking the interaction between CD47 and SIRPα on neutrophils, thereby enhancing the neutrophil’s ADCC capacity against both solid and hematological tumors in vitro and in vivo (213–215). Antibodies targeting either SIRPα or CD47 have been recently described, showing high efficacy and minimal to moderate toxicity effects (216–218) and a number of clinical trials with these CD47-SIRPα-interfering agents are ongoing1. In particular, Hu5F9-G4 against CD47 was tested in combination with rituximab for the treatment of patients with Non-Hodgkin’s lymphoma in a phase 1b clinical trial (219) inducing durable complete responses while no clinically significant toxicity events were observed. At the moment, Hu5F9-G4 is tested in combination with cetuximab for the treatment of colorectal cancers in a phase 1b/2 clinical trial (NCT02953782). Pre-clinical data further showed that blockade of the CD47-SIRPα checkpoint on innate cells eventually activates an anti-tumor response in T cells, bridging the innate with the adaptive immunity (220). Indeed, macrophages were shown to function as APCs and thereby activate the CD8+ T cell population while decreasing priming of CD4+ T cells after anti-CD47-induced phagocytosis of tumor cells (221). Also DCs are able to contribute to the therapeutic effect of anti-CD47 treatment through cross-priming of CD8+ T cells (222). Results from additional future clinical studies will demonstrate whether targeting the CD47-SIRPα axis in a clinical setting indeed will activate both innate as well as adaptive anti-tumor immunity.
Overall, most current studies considered macrophages or NK cells to be the major effector cells in innate checkpoint-blockade antibody therapy (223–225). Given that neutrophils are endowed with similar inhibitory receptors, they can potentially acquire a supporting anti-tumor response. Recent studies have elucidated that Sialic acid-binding immunoglobulin-like lectins or Siglec-blockade can augment the anti-tumor response both by reinvigorating the innate cells and by depleting MDSCs from the TME, thereby unleashing the T cells of the adaptive immune system (180, 226–228). In another perspective, targeting hypersialylation on tumor cells, a trait related to tumor progression and therapy resistance, could also inhibit the Siglec-sialoglycan axis and re-inforce the anti-tumor response (229). The role of Siglec checkpoint-blockade has been highlighted in neutrophils, where research on Siglec-9 or its murine equivalent Siglec-E showed up regulation of the protein receptor in the cytotoxic synapse formation between neutrophils and carcinoma cells, while incubation of neutrophils with anti-Siglec-9 mAbs resulted in significantly increased tumor cell killing by neutrophils (230–233).
Enhancement of the anti-tumor response of neutrophils can also be achieved by targeting signaling partners downstream of the inhibitory receptors. Even better, combination of ligand-receptor interaction disruption and simultaneous blockade of a protein functioning downstream, could have a substantial impact (234, 235). Protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) are two important regulators of immune cell responses. As they bind directly to the ITIM motif of the cytoplasmic tails of inhibitory receptors, drugs targeting their function could potentially amplify the neutrophil effector function against tumor cells (236). Despite the above-mentioned promising data on neutrophil effector function, evidence from in vivo experiments will shed more light on the complexity of neutrophil anti-tumor response.
Augmenting the On-Switch
A different perspective that could redirect pro-tumor and immunosuppressive activity of neutrophils to improve their cytotoxic responses toward cancer in the context of antibody therapy, could be the use of alternative antibody isotypes compared to the classical IgG1. Particularly, IgA antibodies, which specifically bind the IgA Fcα receptor I (FcαRI or CD89) present on cells of the myeloid lineage, including neutrophils (237), are currently considered as a promising approach in immunotherapy against cancer because of their superior ability to induce neutrophil-mediated ADCC. This was reported for a number of tumor-associated antigens such as Ep-CAM for colon carcinoma, HER2/neu for breast carcinoma, EGFR for epithelial, colorectal and renal cell carcinoma, HLA class II, CD20 and CD30 for B-cell lymphoma, and carcinoembryonic antigen as shown in in vitro studies (127, 128, 238–242). The induction of such stronger cytotoxic responses upon an IgA engagement of human neutrophils could be explained by the higher avidity of FcαRI which binds bivalently to IgA, and hence recruit more ITAMs to initiate a more robust signaling to activate effector functions (242). Noteworthy, immature neutrophils that were mobilized from the bone marrow after G-CSF treatment triggered a more efficient ex vivo tumor cell lysis in the presence of an IgA antibody compared to IgG (244, 245). Based on this we speculate that the use of IgA antibodies could unleash the cytotoxic potential of G-CSF mobilized immature neutrophils in antibody therapy of cancer that otherwise would be immunosuppressive, or even trigger re-polarization of PMN-MDSCs, due to FcαRI constitutive expression (128). In an in vivo setting, however, the use of IgA tumor-targeting antibodies is restricted due to the lack of an FcαRI homolog in mice. Nevertheless, the existing genetic engineering techniques have allowed the creation of FcαRI transgenic mouse models, which have been used to confirm the powerful capacity of IgA-mediated tumor killing by myeloid cells in a few studies for EGFR+ tumors (246, 247). An important limitation of IgA antibodies in vivo as well as in humans is the short half-life compared to that of IgG isotypes (15 h versus 4 days, respectively, in mice, and 5–6 days versus 21 days, respectively, in humans) (237, 246, 248). Attempts to extend the half-life of such promising antibodies or combine them with immune checkpoint blockade therapy are currently being studied (249, 250) and will bring new insights for human application in the near future.
Alternatively, several other approaches to increase ADCC activity of therapeutic antibodies are currently being studied. The majority of these approaches involve glyco- and protein-engineering of IgG1-Fc portions to improve the binding affinities to the activating Fcγ receptors on immune effector cells. On the one hand, it is now firmly established that the glycosylation patterns of the IgG-Fc region are essential for the activation of downstream biological mechanisms of the molecule. Consequently, interfering with such post-translational modifications can drastically influence the effector functions of the immune cells binding to it (137, 251). Specifically, core-fucosylation modifications of the IgG-Fc part are the ones showing a more significant effect (252), although Fc galactosylation and sialylation can have an influence as well (253, 254). In particular, the removal of the core fucose from Fc glycans of IgG1 was shown to increase the binding affinity to FcγRIIIa on NK cells, which resulted in a significant enhancement of ADCC activity for this particular effector cell type (255–257). In the case of neutrophils as effector cells, a reduction of the fucose content of the mAb actually abolished anti-tumor activity instead, indicating that antibody fucosylation differentially impacts cytotoxicity mediated by human NK cells and neutrophils (148). A similar finding was described upon deglycosylation of alemtuzumab (114). These observations may be explained by the fact that human neutrophils only express the decoy receptor FcγRIIIb, which was found to bind with high affinity to low-fucose antibodies, thereby impeding antibody efficacy. A better approach to specifically enhance neutrophil-mediated ADCC responses could be achieved by interfering with the amino acid sequences of the Fc region of the targeting antibody. Specific mutations in this region lead to a higher affinity of anti-EGFR mAbs to the activating FcγRIIa on neutrophils rather than to the decoy FcγRIIIb, which resulted in a restored ADCC activity by purified human neutrophils (257). This approach should be considered to enable the successful development of “next-generation” antibodies when targeting neutrophils as promising effector cells. The most encouraging ways to enhance the anti-tumorigenic activity of neutrophils are depicted in Figure 1; 5–8.
Concluding Remarks and Future Perspectives
Over time, neutrophils have been identified as important players in cancer, having the unique capacity to either promote or suppress tumor progression. In the present review, we have first provided an overview of such opposing functions of neutrophils that neutrophils can perform. Cancer progression is mediated by neutrophils via several mechanisms, such as the promotion of proliferation, angiogenesis, invasion and metastasis, as well as via suppression of anti-tumor T cell responses. Potential ways to limit the pro-tumor activity of neutrophils include the reduction of neutrophil numbers, or the inhibition of the recruitment or activation of immunosuppressive neutrophils. Conversely, neutrophils can efficiently act as effector cells toward cancer when triggered in the presence of a therapeutic antibody opsonizing the cancer cells, leading to tumor elimination. By releasing the brakes that suppress neutrophils (i.e., interfering with immune checkpoints) or by augmenting the affinity to the opsonizing antibody of interest, the anti-tumorigenic capacities of neutrophils can be significantly enhanced. As research focuses more and more on exploiting neutrophils against cancer, we anticipate that the aforementioned approaches will prove to be highly valuable to suppress the pro-tumor capacities of neutrophils and consequently fully unleash the anti-tumor potential of neutrophils. However, the similarity of these yet distinguished populations, may make the neutrophil-specific targeting difficult to accomplish in vivo. The coming years of neutrophil-related research will help understanding neutrophil behavior, while by using new developments we might witness a new era on harnessing neutrophil function against tumor progression.
Author Contributions
HM and TK are the principle investigators who designed and supervised the work. CF, PM-S, and PB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
CF is supported by the Louise Vehmeijer Foundation. HM is supported by the Dutch Cancer Society (grant #11537, awarded to HM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. R. van Bruggen for critically reading the manuscript.
Abbreviations
ADCC/P, antibody-dependent cellular cytotoxicity/phagocytosis; APC, antigen-presenting cell; CAR, chimeric antigen receptor; CCL, chemokine (C-C) ligand; CCR, C-C chemokine receptor; CDC, complement-dependent cytotoxicity; CGD, chronic granulomatous disease; CLL, chronic lymphocytic leukemia; CSF-1R, colony-stimulating factor-1 receptor; CTLA-4, cytotoxic T-lymphocyte -associated protein 4; CXCR, C-X -C chemokine receptor; DC, dendritic cell; EGFR, epidermal growth factor receptor; ERK, extracellular signal-related kinase; FATP2, fatty acid transport protein 2; Fc γ receptor, IgG Fc receptor; Fc α RI, IgA Fc receptor; FHL-5, familial hemophagocytic lymphohistiocytosis type 5; fMLF, N-formylmethionyl -leucyl-phenylalanine; G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; H2O2, hydrogen peroxide; HDN, high-density neutrophil; HGF, hepatocyte growth factor; HLA, human leukocyte antigen; ICAM, intercellular adhesion molecule; IFN, interferon; Ig, immunoglobulin; IL, interleukin; IRS-1, insulin receptor substrate 1; ITAM/ITIM, immunoreceptor tyrosine-based activation/inhibitory motif; LAD-1, leukocyte adhesion deficiency type 1; LDN, low-density neutrophil; LOX-1, lectin-type oxidized LDL receptor 1; LPS, lipopolysaccharide; mAb, monoclonal antibody; Mac-1, macrophage-1 antigen; MDSC, myeloid-derived suppressor cell; MHC, major histocompatibility complex; MM, multiple myeloma; MMP-9, matrix metalloproteinase-9; NADPH, nicotinamide adenine dinucleotide phosphate; NE, neutrophil elastase; NET, neutrophil extracellular trap; NF κ B, nuclear factor kappa B; NK, natural killer; NLR, neutrophil-to-lymphocyte ratio; PBMC, peripheral blood mononuclear cell; PD-1, programmed cell death protein 1; PDGFR, platelet-derived growth factor receptor; PD-L1, programmed death-ligand 1; PI3K, phosphoinositide 3-kinase; PMN, polymorphonuclear leukocyte; PTK, protein tyrosine kinase; PTP, protein tyrosine phosphatase; ROS, reactive oxygen species; Siglec, Sialic acid-binding immunoglobulin-like lectin; SIRP α, signal regulatory protein alpha; TAN, tumor-associated neutrophil; TGF- β, transforming growth factor- β; TLR, toll-like receptor; TME, tumor microenvironment; TNF α, tumor necrosis factor α; Treg, regulatory T cell; TRMP2, transient receptor potential cation channel, subfamily M, member 2; VEGF, vascular endothelial growth factor.
Footnotes
References
1. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (New York NY). (2015) 348:56–61. doi: 10.1126/science.aaa8172
2. Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. (2015) 16:522–30. doi: 10.1016/s1470-2045(15)70122-1
3. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. (2015) 373:1627–39.
4. Hu B, Jacobs R, Ghosh N. Checkpoint inhibitors hodgkin lymphoma and non-hodgkin lymphoma. Curr Hematol Malig Rep. (2018) 13:543–54. doi: 10.1007/s11899-018-0484-4
5. Teo MY, Rosenberg JE. Nivolumab for the treatment of urothelial cancers. Expert Rev Anticancer Ther. (2018) 18:215–21. doi: 10.1080/14737140.2018.1432357
6. Planes-Laine G, Rochigneux P, Bertucci F, Chretien AS, Viens P, Sabatier R, et al. PD-1/PD-L1 targeting in breast cancer: the first clinical evidences are emerging. A literature review. Cancers (Basel). (2019) 11:1033. doi: 10.3390/cancers11071033
7. O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
8. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33.
9. Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. (2020) 21:e168–78. doi: 10.1016/s1470-2045(19)30823-x
10. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy–assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62.
11. Shin MH, Kim J, Lim SA, Kim J, Kim SJ, Lee KM. NK Cell-Based Immunotherapies in Cancer. Immune Netw. (2020) 20:e14.
12. Kalinski P, Muthuswamy R, Urban J. Dendritic cells in cancer immunotherapy: vaccines and combination immunotherapies. Expert Rev Vaccines. (2013) 12:285–95. doi: 10.1586/erv.13.22
13. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. (2014) 11:509–24.
15. Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. (2006) 7:115.
16. Chen C, Duckworth CA, Zhao Q, Pritchard DM, Rhodes JM, Yu LG. Increased circulation of galectin-3 in cancer induces secretion of metastasis-promoting cytokines from blood vascular endothelium. Clin Cancer Res. (2013) 19:1693–704. doi: 10.1158/1078-0432.ccr-12-2940
17. Jablonska JLS, Sionov RV, Granot Z. The regulation of pre-metastatic niche formation by neutrophils. Oncotarget. (2007) 8:112132–44. doi: 10.18632/oncotarget.22792
18. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. (2016) 16:431–46. doi: 10.1038/nrc.2016.52
19. Gonzalez-Aparicio M, Alfaro C. Significance of the IL-8 pathway for immunotherapy. Hum Vaccin Immunother. (2019) 19:1–6. doi: 10.1080/21645515.2019.1696075
20. Guthrie GJ, Charles KA, Roxburgh CS, Horgan PG, McMillan DC, Clarke SJ. The systemic inflammation-based neutrophil-lymphocyte ratio: experience in patients with cancer. Crit Rev Oncol Hematol. (2013) 88:218–30. doi: 10.1016/j.critrevonc.2013.03.010
21. Templeton AJ, McNamara MG, Seruga B, Vera-Badillo FE, Aneja P, Ocana A, et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J Natl Cancer Inst. (2014) 106:dju124.
22. Grecian R, Whyte MKB, Walmsley SR. The role of neutrophils in cancer. Br Med Bull. (2018) 128:5–14. doi: 10.1093/bmb/ldy029
23. Caruso RA, Bellocco R, Pagano M, Bertoli G, Rigoli L, Inferrera C. Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in northern Italy. Mod Pathol. (2002) 15:831–7. doi: 10.1097/01.mp.0000020391.98998.6b
24. Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, et al. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol. (2011) 24:1612–9. doi: 10.1038/modpathol.2011.113
25. Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F, Von Der Maase H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J Clin Oncol. (2009) 27:4709–17. doi: 10.1200/jco.2008.18.9498
26. Shitara K, Matsuo K, Oze I, Mizota A, Kondo C, Nomura M, et al. Meta-analysis of neutropenia or leukopenia as a prognostic factor in patients with malignant disease undergoing chemotherapy. Cancer Chemother Pharmacol. (2011) 68:301–7. doi: 10.1007/s00280-010-1487-6
27. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
28. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
29. Shaul ME, Fridlender ZG. Cancer-related circulating and tumor-associated neutrophils - subtypes, sources and function. FEBS J. (2018) 285:4316–42. doi: 10.1111/febs.14524
30. Treffers LW, Hiemstra IH, Kuijpers TW, van den Berg TK, Matlung HL. Neutrophils in cancer. Immunol Rev. (2016) 273:312–28.
31. McGarry Houghton A, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. (2010) 16:219–23. doi: 10.1038/nm.2084
32. Nawa M, Osada S, Morimitsu K, Nonaka K, Futamura M, Kawaguchi Y, et al. Growth effect of neutrophil elastase on breast cancer: favorable action of sivelestat and application to anti-HER2 therapy. Anticancer Res. (2012) 32:13–9.
33. Gaida MM, Steffen TG, Gunther F, Tschaharganeh DF, Felix K, Bergmann F, et al. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur J Immunol. (2012) 42:3369–80. doi: 10.1002/eji.201242628
34. Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. (2011) 54:948–55. doi: 10.1016/j.jhep.2010.08.041
35. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. (2015) 528:413–7. doi: 10.1038/nature16140
36. Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, et al. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. (2012) 72:3919–27. doi: 10.1158/0008-5472.can-11-2393
37. Huh SJ, Liang S, Sharma A, Dong C, Robertson GP. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. (2010) 70:6071–82. doi: 10.1158/0008-5472.can-09-4442
38. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (New York NY). (2004) 303:1532–5. doi: 10.1126/science.1092385
39. Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. (2016) 22:3924–36. doi: 10.1158/1078-0432.ccr-15-2463
40. Berger-Achituv S, Brinkmann V, Abed UA, Kuhn LI, Ben-Ezra J, Elhasid R, et al. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. (2013) 4:48.
41. Jung HS, Gu J, Kim JE, Nam Y, Song JW, Kim HK. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS One. (2019) 14:e0216055. doi: 10.1371/journal.pone.0216055
42. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. (2013) 123:3446–58. doi: 10.1172/jci67484
43. Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. (2018) 216:176–94. doi: 10.1084/jem.20181170
44. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. (2016) 8:361ra138. doi: 10.1126/scitranslmed.aag1711
45. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. (2016) 76:1367–80. doi: 10.1158/0008-5472.can-15-1591
46. Oklu R, Sheth RA, Wong KHK, Jahromi AH, Albadawi H. Neutrophil extracellular traps are increased in cancer patients but does not associate with venous thrombosis. Cardiovasc Diagn Ther. (2017) 7(Suppl. 3):S140–9.
47. Richardson JJR, Hendrickse C, Gao-Smith F, Thickett DR. Neutrophil extracellular trap production in patients with colorectal cancer in vitro. Int J Inflam. (2017) 2017:4915062.
48. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
49. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci. (2014) 1319:47–65. doi: 10.1111/nyas.12469
50. Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. (2011) 13:591–9. doi: 10.1093/neuonc/nor042
51. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. (2011) 118:2254–65. doi: 10.1182/blood-2010-12-325753
52. Yamauchi Y, Safi S, Blattner C, Rathinasamy A, Umansky L, Juenger S, et al. Circulating and tumor myeloid-derived suppressor cells in resectable non-small cell lung cancer. Am J Respir Crit Care Med. (2018) 198:777–87.
53. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother. (2016) 65:161–9. doi: 10.1007/s00262-015-1782-5
54. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. (2016) 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
55. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10(+) and immature CD10(-) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood. (2017) 129:1343–56. doi: 10.1182/blood-2016-04-713206
56. Aarts CEM, Hiemstra IH, Beguin EP, Hoogendijk AJ, Bouchmal S, van Houdt M, et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv. (2019) 3:3562–74. doi: 10.1182/bloodadvances.2019031609
57. Hock BD, Taylor KG, Cross NB, Kettle AJ, Hampton MB, McKenzie JL. Effect of activated human polymorphonuclear leucocytes on T lymphocyte proliferation and viability. Immunology. (2012) 137:249–58. doi: 10.1111/imm.12004
58. Singel KL, Emmons TR, Khan ANH, Mayor PC, Shen S, Wong JT, et al. Mature neutrophils suppress T cell immunity in ovarian cancer microenvironment. JCI Insight. (2019) 4:e122311.
59. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. (2010) 185:2273–84. doi: 10.4049/jimmunol.1000901
60. Aarts CEM, Kuijpers TW. Neutrophils as myeloid-derived suppressor cells. Eur J Clin Invest. (2018) 48(Suppl. 2):e12989. doi: 10.1111/eci.12989
61. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
62. Jacobsen LC, Theilgaard-Monch K, Christensen EI, Borregaard N. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood. (2007) 109:3084–7. doi: 10.1182/blood-2006-06-032599
63. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. (2007) 13(2 Pt 2):721s–6s.
64. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. (2009) 69:1553–60. doi: 10.1158/0008-5472.can-08-1921
65. Sippel TR, White J, Nag K, Tsvankin V, Klaassen M, Kleinschmidt-DeMasters BK, et al. Neutrophil degranulation and immunosuppression in patients with GBM: restoration of cellular immune function by targeting arginase I. Clin Cancer Res. (2011) 17:6992–7002. doi: 10.1158/1078-0432.ccr-11-1107
66. Taheri F, Ochoa JB, Faghiri Z, Culotta K, Park HJ, Lan MS, et al. L-Arginine regulates the expression of the T-cell receptor ζ?chain (CD3ζ) in Jurkat cells. Clin Cancer Res. (2001) 7(Suppl. 3):958–65s.
67. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.can-04-0465
68. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3ζ chain expression by L-arginine. J Biol Chem. (2002) 277:21123–9.
69. Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. (2008) 26:233–59.
70. Feldmeyer N, Wabnitz G, Leicht S, Luckner-Minden C, Schiller M, Franz T, et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int Immunol. (2012) 24:303–13. doi: 10.1093/intimm/dxs004
71. Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, et al. L-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell Immunol. (2004) 232:21–31. doi: 10.1016/j.cellimm.2005.01.004
72. Munder M, Schneider H, Luckner C, Giese T, Langhans CD, Fuentes JM, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. (2006) 108:1627–34. doi: 10.1182/blood-2006-11-010389
73. Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH, et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14–/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. (2010) 136:35–45. doi: 10.1007/s00432-009-0634-0
74. de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. (2010) 10:568–79. doi: 10.1038/nri2803
75. Meeths M, Entesarian M, Al-Herz W, Chiang SC, Wood SM, Al-Ateeqi W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood. (2010) 116:2635–43. doi: 10.1182/blood-2010-05-282541
76. Zhao XW, Gazendam RP, Drewniak A, van Houdt M, Tool AT, van Hamme JL, et al. Defects in neutrophil granule mobilization and bactericidal activity in familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) syndrome caused by STXBP2/Munc18-2 mutations. Blood. (2013) 122:109–11. doi: 10.1182/blood-2013-03-494039
77. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. (2009) 182:5693–701. doi: 10.4049/jimmunol.0900092
78. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. (2001) 61:4756–60.
79. Szuster-Ciesielska A, Hryciuk-Umer E, Stepulak A, Kupisz K, Kandefer-Szerszen M. Reactive oxygen species production by blood neutrophils of patients with laryngeal carcinoma and antioxidative enzyme activity in their blood. Acta Oncol. (2004) 43:252–8. doi: 10.1080/02841860410029708
80. Malmberg KJ, Arulampalam V, Ichihara F, Petersson M, Seki K, Andersson T, et al. Inhibition of activated/memory (CD45RO+) T cells by oxidative stress associated with block of NF-κB activation. J Immunol. (2001) 167:2595–601. doi: 10.4049/jimmunol.167.5.2595
81. Whiteside TL. Down-regulation of ζ-chain expression in T cells: a biomarker of prognosis in cancer?Cancer Immunol Immunother. (2004) 53:865–78.
82. Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity. (2008) 29:404–13. doi: 10.1016/j.immuni.2008.06.016
83. Wabnitz GH, Goursot C, Jahraus B, Kirchgessner H, Hellwig A, Klemke M, et al. Mitochondrial translocation of oxidized cofilin induces caspase-independent necrotic-like programmed cell death of T cells. Cell Death Dis. (2010) 1:e58. doi: 10.1038/cddis.2010.36
84. Kramer PA, Prichard L, Chacko B, Ravi S, Overton ET, Heath SL, et al. Inhibition of the lymphocyte metabolic switch by the oxidative burst of human neutrophils. Clin Sci (Lond). (2015) 129:489–504. doi: 10.1042/cs20140852
85. Roos D. The genetic basis of chronic granulomatous disease. Immunol Rev. (1994) 138:121–57. doi: 10.1111/j.1600-065x.1994.tb00850.x
86. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
87. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
88. Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology. (2012) 1:1223–5. doi: 10.4161/onci.21335
89. Bankey PE, Banerjee S, Zucchiatti A, De M, Sleem RW, Lin CF, et al. Cytokine induced expression of programmed death ligands in human neutrophils. Immunol Lett. (2010) 129:100–7. doi: 10.1016/j.imlet.2010.01.006
90. de Kleijn S, Langereis JD, Leentjens J, Kox M, Netea MG, Koenderman L, et al. IFN-γ-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLoS One. (2013) 8:e72249. doi: 10.1371/journal.pone.0072249
91. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
92. He G, Zhang H, Zhou J, Wang B, Chen Y, Kong Y, et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. (2015) 34:141.
93. Wang TT, Zhao YL, Peng LS, Chen N, Chen W, Lv YP, et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut. (2017) 66:1900–11. doi: 10.1136/gutjnl-2016-313075
94. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. (2012) 122:327–36. doi: 10.1172/jci57990
95. Michaeli J, Shaul ME, Mishalian I, Hovav AH, Levy L, Zolotriov L, et al. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFalpha and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology. (2017) 6:e1356965. doi: 10.1080/2162402x.2017.1356965
96. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
97. Joly E, Hudrisier D. What is trogocytosis and what is its purpose?Nat Immunol. (2003) 4:815. doi: 10.1038/ni0903-815
98. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. (2005) 11:6713–21. doi: 10.1158/1078-0432.ccr-05-0883
99. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
100. Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. (2012) 72:3906–11. doi: 10.1158/0008-5472.can-11-3873
101. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. (2016) 6:630–49. doi: 10.1158/2159-8290.cd-15-1157
102. Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel). (2016) 4:28. doi: 10.3390/vaccines4030028
103. Mougiakakos D, Johansson CC, Jitschin R, Bottcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood. (2011) 117:857–61. doi: 10.1182/blood-2010-09-307041
104. Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood. (2009) 113:3542–5. doi: 10.1182/blood-2008-09-181040
105. Mishalian I, Bayuh R, Eruslanov E, Michaeli J, Levy L, Zolotarov L, et al. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17–A new mechanism of impaired antitumor immunity. Int J Cancer. (2014) 135:1178–86. doi: 10.1002/ijc.28770
106. Lewkowicz N, Klink M, Mycko MP, Lewkowicz P. Neutrophil-CD4+CD25+ T regulatory cell interactions: a possible new mechanism of infectious tolerance. Immunobiology. (2013) 218:455–64. doi: 10.1016/j.imbio.2012.05.029
107. Lewkowicz N, Mycko MP, Przygodzka P, Cwiklinska H, Cichalewska M, Matysiak M, et al. Induction of human IL-10-producing neutrophils by LPS-stimulated Treg cells and IL-10. Mucosal Immunol. (2016) 9:364–78. doi: 10.1038/mi.2015.66
108. Eruslanov E, Neuberger M, Daurkin I, Perrin GQ, Algood C, Dahm P, et al. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int J Cancer. (2012) 130:1109–19. doi: 10.1002/ijc.26123
109. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe?Carcinogenesis. (2012) 33:949–55. doi: 10.1093/carcin/bgs123
110. Rakic A, Beaudry P, Mahoney DJ. The complex interplay between neutrophils and cancer. Cell Tissue Res. (2018) 371:517–29. doi: 10.1007/s00441-017-2777-7
111. Lieschke GJ, Burgess AW. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor (1). N Engl J Med. (1992) 327:28–35.
112. Carulli G. Effects of recombinant human granulocyte colony-stimulating factor administration on neutrophil phenotype and functions. Haematologica. (1997) 82:606–16.
113. Gorabi AM, Hajighasemi S, Sathyapalan T, Sahebkar A. Cell transfer-based immunotherapies in cancer: a review. IUBMB Life. (2020) 72:790–800. doi: 10.1002/iub.2180
114. Siders WM, Shields J, Garron C, Hu Y, Boutin P, Shankara S, et al. Involvement of neutrophils and natural killer cells in the anti-tumor activity of alemtuzumab in xenograft tumor models. Leuk Lymphoma. (2010) 51:1293–304. doi: 10.3109/10428191003777963
115. Cornet S, Mathe D, Chettab K, Evesque A, Matera EL, Tredan O, et al. Pegfilgrastim enhances the antitumor effect of therapeutic monoclonal antibodies. Mol Cancer Ther. (2016) 15:1238–47. doi: 10.1158/1535-7163.mct-15-0759
116. Torka P, Patel P, Tan W, Wilding G, Bhat SA, Czuczman MS, et al. A phase II trial of rituximab combined with pegfilgrastim in patients with indolent B-cell non-hodgkin lymphoma. Clin Lymphoma Myeloma Leuk. (2018) 18:e51–60. doi: 10.1016/j.clml.2017.09.003
117. Zivkovic M, Poljak-Blazi M, Zarkovic K, Mihaljevic D, Schaur RJ, Zarkovic N. Oxidative burst of neutrophils against melanoma B16-F10. Cancer Lett. (2007) 246:100–8. doi: 10.1016/j.canlet.2006.02.002
118. Yan J, Kloecker G, Fleming C, Bousamra MII, Hansen R, Hu X, et al. Human polymorphonuclear neutrophils specifically recognize and kill cancerous cells. Oncoimmunology. (2014) 3:e950163. doi: 10.4161/15384101.2014.950163
119. Gershkovitz M, Fainsod-Levi T, Zelter T, Sionov RV, Granot Z. TRPM2 modulates neutrophil attraction to murine tumor cells by regulating CXCL2 expression. Cancer Immunol Immunother. (2019) 68:33–43. doi: 10.1007/s00262-018-2249-2
120. Sun B, Qin W, Song M, Liu L, Yu Y, Qi X, et al. Neutrophil suppresses tumor cell proliferation via fas/fas ligand pathway mediated cell cycle arrested. Int J Biol Sci. (2018) 14:2103–13. doi: 10.7150/ijbs.29297
121. Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. (2007) 44:3823–37. doi: 10.1016/j.molimm.2007.06.151
122. Hudis CA. Trastuzumab-mechanism of action and use in clinical practice. N Engl J Med. (2007) 357:39–51. doi: 10.1056/nejmra043186
123. Gelderman KA, Tomlinson S, Ross GD, Gorter A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. (2004) 25:158–64. doi: 10.1016/j.it.2004.01.008
124. Nimmerjahn F, Ravetch JV. Fc−receptors as regulators of immunity. Adv Immunol. (2007) 96:179–204. doi: 10.1016/s0065-2776(07)96005-8
125. Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. (2010) 10:301–16. doi: 10.1038/nri2761
126. van Egmond M, Bakema JE. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin Cancer Biol. (2013) 23:190–9. doi: 10.1016/j.semcancer.2012.12.002
127. Bakema JE, van Egmond M. Fc receptor-dependent mechanisms of monoclonal antibody therapy of cancer. Curr Top Microbiol Immunol. (2014) 382:373–92. doi: 10.1007/978-3-319-07911-0_17
128. Heemskerk N, van Egmond M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur J Clin Invest. (2018) 48(Suppl. 2:e12962. doi: 10.1111/eci.12962
129. Gale RP, Zighelboim J. Polymorphonuclear leukocytes in antibody-dependent cellular cytotoxicity. J Immunol. (1975) 114:1047–51.
130. Barker E, Mueller BM, Handgretinger R, Herter M, Yu AL, Reisfeld RA. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res. (1991) 51:144–9.
131. Hernandez-Ilizaliturri FJ, Jupudy V, Ostberg J, Oflazoglu E, Huberman A, Repasky E, et al. Neutrophils contribute to the biological antitumor activity of rituximab in a non-Hodgkin’s lymphoma severe combined immunodeficiency mouse model. Clin Cancer Res. (2003) 9(16 Pt 1):5866–73.
132. Albanesi M, Mancardi DA, Jonsson F, Iannascoli B, Fiette L, Di Santo JP, et al. Neutrophils mediate antibody-induced antitumor effects in mice. Blood. (2013) 122:3160–4. doi: 10.1182/blood-2013-04-497446
133. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. (2008) 26:1789–96. doi: 10.1200/jco.2007.14.8957
134. Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. FcgammaR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol. (2011) 22:1302–7. doi: 10.1093/annonc/mdq585
135. Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. (2007) 25:3712–8. doi: 10.1200/jco.2006.08.8021
136. Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol. (2006) 24:2885–90. doi: 10.1200/jco.2005.04.6011
137. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520.
138. Bruhns P, Jonsson F. Mouse and human FcR effector functions. Immunol Rev. (2015) 268:25–51. doi: 10.1111/imr.12350
139. Chen X, Song X, Li K, Zhang T. FcgammaR-binding is an important functional attribute for immune checkpoint antibodies in cancer immunotherapy. Front Immunol. (2019) 10:292.
140. Nagelkerke SQ, Tacke CE, Breunis WB, Tanck MWT, Geissler J, Png E, et al. Extensive ethnic variation and linkage disequilibrium at the FCGR2/3 locus: different genetic associations revealed in kawasaki disease. Front Immunol. (2019) 10:185.
141. Wang Y, Jonsson F. Expression, role, and regulation of neutrophil fcgamma receptors. Front Immunol. (2019) 10:1958.
142. Blank ULP, Benhamou M, Monteiro RC. Inhibitory ITAMs as novel regulators of immunity. Immunol Rev. (2009) 232:59–71. doi: 10.1111/j.1600-065x.2009.00832.x
143. Fleit HB, Wright SD, Unkeless JC. Human neutrophil Fc gamma receptor distribution and structure. Proc Natl Acad Sci USA. (1982) 79:3275–9.
144. Treffers LW, van Houdt M, Bruggeman CW, Heineke MH, Zhao XW, van der Heijden J, et al. FcgammaRIIIb restricts antibody-dependent destruction of cancer cells by human neutrophils. Front Immunol. (2018) 9:3124.
145. Repp R, Valerius T, Sendler A, Gramatzki M, Iro H, Kalden JR, et al. Neutrophils express the high affinity receptor for IgG (Fc gamma RI, CD64) after in vivo application of recombinant human granulocyte colony- stimulating factor. Blood. (1991) 78:885–9. doi: 10.1182/blood.v78.4.885.bloodjournal784885
146. van de Winkel JG, Capel PJ. Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications. Immunol Today. (1993) 14:215–21. doi: 10.1016/0167-5699(93)90166-i
147. Middelhoven PJ, van Buul JD, Kleijer M, Roos D, Hordijk PL. Actin polymerization induces shedding of FcgammaRIIIb (CD16) from human neutrophils. Biochem Biophys Res Commun.. (1999) 255:568–74. doi: 10.1006/bbrc.1999.0244
148. Peipp M, Lammerts van Bueren JJ. Schneider-Merck T, Bleeker WW, Dechant M, Beyer T, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. (2008) 112:2390–9. doi: 10.1182/blood-2008-03-144600
149. Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, et al. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep (2018) 23:3946–59.e6.
150. Treffers LW, Zhao XW, van der Heijden J, Nagelkerke SQ, van Rees DJ, Gonzalez P, et al. Genetic variation of human neutrophil Fcgamma receptors and SIRPalpha in antibody-dependent cellular cytotoxicity towards cancer cells. Eur J Immunol. (2018) 48:344–54. doi: 10.1002/eji.201747215
151. Valerius T, Repp R, de Wit TP, Berthold S, Platzer E, Kalden JR, et al. Involvement of the high-affinity receptor for IgG (Fc gamma RI; CD64) in enhanced tumor cell cytotoxicity of neutrophils during granulocyte colony-stimulating factor therapy. Blood. (1993) 82:931–9.
152. Michon J, Moutel S, Barbet J, Romet-Lemonne JL, Deo YM, Fridman WH, et al. In vitro killing of neuroblastoma cells by neutrophils derived from granulocyte colony-stimulating factor-treated cancer patients using an anti-disialoganglioside/anti-Fc gamma RI bispecific antibody. Blood. (1995) 86:1124–30. doi: 10.1182/blood.v82.3.931.bloodjournal823931
153. Keler TGR, Mandal A, Wallace PK, Fisher J, Guyre PM, Fanger MW, et al. BispecilIc antibody-dependent cellular cytotoxicity of HER2/neu-overexpressing tumor cells by fcy receptor type I-expressing effector cells. Cancer Res. (1997) 57:4008–14.
154. Heijnen IAGM, van de Winkel JG. Lysis of murine B lymphoma cells by transgenic phagocytes via a human FcgRI murine MHC class II bispecific antibody. Cancer Immunol Immunother. (1997) 45:166–70. doi: 10.1007/s002620050424
155. Honeychurch J, Tutt AL, Valerius T, Heijnen IA, Van De Winkel JG, Glennie MJ. Therapeutic efficacy of FcgammaRI/CD64-directed bispecific antibodies in B-cell lymphoma. Blood. (2000) 96:3544–52. doi: 10.1182/blood.v96.10.3544.h8003544_3544_3552
156. Taylor RP, Lindorfer MA. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: historical precedence and recent developments. Blood. (2015) 125:762–6. doi: 10.1182/blood-2014-10-569244
157. Tabiasco J, Espinosa E, Hudrisier D, Joly E, Fournie JJ, Vercellone A. Active trans-synaptic capture of membrane fragments by natural killer cells. Eur J Immunol. (2002) 32:1502–8. doi: 10.1002/1521-4141(200205)32:5<1502::aid-immu1502>3.0.co;2-y
158. Mercer F, Ng SH, Brown TM, Boatman G, Johnson PJ. Neutrophils kill the parasite Trichomonas vaginalis using trogocytosis. PLoS Biol. (2018) 16:e2003885. doi: 10.1371/journal.pbio.2003885
159. Velmurugan R, Challa DK, Ram S, Ober RJ, Ward ES. Macrophage-mediated trogocytosis leads to death of antibody-opsonized tumor cells. Mol Cancer Ther. (2016) 15:1879–89. doi: 10.1158/1535-7163.mct-15-0335
160. Valgardsdottir R, Cattaneo I, Klein C, Introna M, Figliuzzi M, Golay J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood. (2017) 129:2636–44. doi: 10.1182/blood-2016-08-735605
161. Krejcik J, Frerichs KA, Nijhof IS, van Kessel B, van Velzen JF, Bloem AC, et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin Cancer Res. (2017) 23:7498–511. doi: 10.1158/1078-0432.ccr-17-2027
162. Strizova Z, Vachtenheim JJr., Bartunkova J. The potential role of neutrophil trogocytosis and G-CSF in the loss of HER2 expression. Breast Cancer Res Treat. (2019) 178:247–8. doi: 10.1007/s10549-019-05386-7
163. Ignatov T, Gorbunow F, Eggemann H, Ortmann O, Ignatov A. Loss of HER2 after HER2-targeted treatment. Breast Cancer Res Treat. (2019) 175:401–8. doi: 10.1007/s10549-019-05173-4
164. van Gisbergen KP, Sanchez-Hernandez M, Geijtenbeek TB, van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med. (2005) 201:1281–92. doi: 10.1084/jem.20041276
165. Megiovanni AM, Sanchez F, Robledo-Sarmiento M, Morel C, Gluckman JC, Boudaly S. Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: a new link between leukocytes upstream of T lymphocytes. J Leukoc Biol. (2006) 79:977–88. doi: 10.1189/jlb.0905526
166. Sandilands GP, Ahmed Z, Perry N, Davison M, Lupton A, Young B. Cross-linking of neutrophil CD11b results in rapid cell surface expression of molecules required for antigen presentation and T-cell activation. Immunology. (2005) 114:354–68. doi: 10.1111/j.1365-2567.2004.02114.x
167. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. (2014) 124:5466–80. doi: 10.1172/jci77053
168. Eruslanov EB. Phenotype and function of tumor-associated neutrophils and their subsets in early-stage human lung cancer. Cancer Immunol Immunother. (2017) 66:997–1006. doi: 10.1007/s00262-017-1976-0
169. Beauvillain C, Delneste Y, Scotet M, Peres A, Gascan H, Guermonprez P, et al. Neutrophils efficiently cross-prime naive T cells in vivo. Blood. (2007) 110:2965–73. doi: 10.1182/blood-2006-12-063826
170. Maletto BA, Ropolo AS, Alignani DO, Liscovsky MV, Ranocchia RP, Moron VG, et al. Presence of neutrophil-bearing antigen in lymphoid organs of immune mice. Blood. (2006) 108:3094–102. doi: 10.1182/blood-2006-04-016659
171. Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. (2011) 117:1196–204. doi: 10.1182/blood-2009-11-254490
172. Voisin MB, Nourshargh S. Neutrophil trafficking to lymphoid tissues: physiological and pathological implications. J Pathol. (2019) 247:662–71. doi: 10.1002/path.5227
173. Suttmann H, Riemensberger J, Bentien G, Schmaltz D, Stockle M, Jocham D, et al. Neutrophil granulocytes are required for effective Bacillus Calmette-Guerin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res. (2006) 66:8250–7. doi: 10.1158/0008-5472.can-06-1416
174. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. (2015) 522:345–8. doi: 10.1038/nature14282
175. Zhu EF, Gai SA, Opel CF, Kwan BH, Surana R, Mihm MC, et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell. (2015) 27:489–501. doi: 10.1016/j.ccell.2015.03.004
176. Mishalian IBR, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol Immunother. (2013) 62:1745–56. doi: 10.1007/s00262-013-1476-9
177. Pylaeva E, Lang S, Jablonska J. The essential role of type I interferons in differentiation and activation of tumor-associated neutrophils. Front Immunol. (2016) 7:629.
178. Ohms M, Möller S, Laskay T. An attempt to polarize human neutrophils towards N1 and N2 phenotypes in vitro. Front Immunol. (2020) 11:532. doi: 10.3389/fimmu.2020.00532
179. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. (2014) 63:1769–81. doi: 10.1136/gutjnl-2013-306271
180. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine. (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
181. Roskoski RJr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. (2007) 356:323–8. doi: 10.1016/j.bbrc.2007.02.156
182. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. (2009) 69:2514–22. doi: 10.1158/0008-5472.can-08-4709
183. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. (2009) 15:2148–57. doi: 10.1158/1078-0432.ccr-08-1332
184. Guislain A, Gadiot J, Kaiser A, Jordanova ES, Broeks A, Sanders J, et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol Immunother. (2015) 64:1241–50. doi: 10.1007/s00262-015-1735-z
185. Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res. (2015) 21:5453–9. doi: 10.1158/1078-0432.ccr-15-0676
186. Pico de Coana Y, Poschke I, Gentilcore G, Mao Y, Nystrom M, Hansson J, et al. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunol Res. (2013) 1:158–62. doi: 10.1158/2326-6066.cir-13-0016
187. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, et al. Clinical Significance of circulating CD33+CD11b+HLA-DR– myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin Cancer Res. (2016) 22:5661–72.
188. De Soyza A, Pavord I, Elborn JS, Smith D, Wray H, Puu M, et al. A randomised, placebo-controlled study of the CXCR2 antagonist AZD5069 in bronchiectasis. Eur Respir J. (2015) 46:1021–32. doi: 10.1158/1078-0432.ccr-15-3104
189. Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, et al. CXCR2 antagonist MK-7123. a phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 191:1001–11. doi: 10.1164/rccm.201405-0992oc
190. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell. (2017) 32:654–68.e5.
191. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. (2016) 29:832–45. doi: 10.1016/j.ccell.2016.04.014
192. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. (2019) 4:e126853.
193. Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-negative metastatic breast cancer. Clin Cancer Res. (2017) 23:5358–65. doi: 10.1158/1078-0432.ccr-16-2748
194. Patnaik A, Swanson KD, Csizmadia E, Solanki A, Landon-Brace N, Gehring MP, et al. Cabozantinib eradicates advanced murine prostate cancer by activating antitumor innate immunity. Cancer Discov. (2017) 7:750–65. doi: 10.1158/2159-8290.cd-16-0778
195. Wislez M, Rabbe N, Marchal J, Milleron B, Crestani B, Mayaud C, et al. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res. (2003) 63:1405–12.
196. Glodde N, Bald T, van den Boorn-Konijnenberg D, Nakamura K, O’Donnell JS, Szczepanski S, et al. Reactive neutrophil responses dependent on the receptor tyrosine kinase c-MET limit cancer immunotherapy. Immunity (2017) 47:789–802.e9.
197. De Silva DM, Roy A, Kato T, Cecchi F, Lee YH, Matsumoto K, et al. Targeting the hepatocyte growth factor/Met pathway in cancer. Biochem Soc Trans. (2017) 45:855–70. doi: 10.1042/bst20160132
198. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. (2014) 40:785–800. doi: 10.1016/j.immuni.2014.03.013
199. Chen WC, Lai YH, Chen HY, Guo HR, Su IJ, Chen HH. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology. (2013) 63:225–33. doi: 10.1111/his.12156
200. Ma C, Zhang Q, Ye J, Wang F, Zhang Y, Wevers E, et al. Tumor-infiltrating γδ T lymphocytes predict clinical outcome in human breast cancer. J Immunol. (2012) 189:5029–36.
201. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
202. Aarts CEM, Hiemstra IH, Furumaya C, Van Bruggen R, Kuijpers TW. Different MDSC activity of G-CSF/dexamethasone mobilized neutrophils: benefits to the patient?Front Oncol. (2020) 10:1110. doi: 10.3389/fonc.2020.01110
203. Drewniak A, Boelens JJ, Vrielink H, Tool AT, Bruin MC, van den Heuvel-Eibrink M, et al. Granulocyte concentrates: prolonged functional capacity during storage in the presence of phenotypic changes. Haematologica. (2008) 93:1058–67. doi: 10.3324/haematol.12489
204. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. (2017) 6:e1344804. doi: 10.1080/2162402x.2017.1344804
205. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. (2019) 569:73–8. doi: 10.1038/s41586-019-1118-2
206. Eruslanov EB, Singhal S, Albelda SM. Mouse versus human neutrophils in cancer: a major knowledge gap. Trends Cancer. (2017) 3:149–60. doi: 10.1016/j.trecan.2016.12.006
207. Ho AS, Chen CH, Cheng CC, Wang CC, Lin HC, Luo TY, et al. Neutrophil elastase as a diagnostic marker and therapeutic target in colorectal cancers. Oncotarget. (2014) 5:473–80. doi: 10.18632/oncotarget.1631
208. Lerman I, Garcia-Hernandez ML, Rangel-Moreno J, Chiriboga L, Pan C, Nastiuk KL, et al. Infiltrating myeloid cells exert protumorigenic actions via neutrophil elastase. Mol Cancer Res. (2017) 15:1138–52. doi: 10.1158/1541-7786.mcr-17-0003
209. Suda K, Kitagawa Y, Ozawa S, Miyasho T, Okamoto M, Saikawa Y, et al. Neutrophil elastase inhibitor improves postoperative clinical courses after thoracic esophagectomy. Dis Esophagus. (2007) 20:478–86. doi: 10.1111/j.1442-2050.2007.00699.x
210. Hawes MC, Wen F, Elquza E. Extracellular DNA: A bridge to cancer. Cancer Res. (2015) 75:4260–4. doi: 10.1158/0008-5472.can-15-1546
211. van Rees DJ, Szilagyi K, Kuijpers TW, Matlung HL, van den Berg TK. Immunoreceptors on neutrophils. Semin Immunol. (2016) 28:94–108. doi: 10.1016/j.smim.2016.02.004
212. Zhang W, Huang Q, Xiao W, Zhao Y, Pi J, Xu H, et al. Advances in anti-tumor treatments targeting the CD47/SIRPalpha axis. Front Immunol. (2020) 11:18.
213. Zhao XW, van Beek EM, Schornagel K, Van der Maaden H, Van Houdt M, Otten MA, et al. CD47-signal regulatory protein-alpha (SIRPalpha) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci USA. (2011) 108:18342–7. doi: 10.1073/pnas.1106550108
214. Ring NG, Herndler-Brandstetter D, Weiskopf K, Shan L, Volkmer JP, George BM, et al. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci USA. (2017) 114:E10578–85.
215. Horrigan SK. Reproducibility project: cancer B. replication study: the CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Elife. (2017) 6:e18173.
216. Sim J, Sockolosky JT, Sangalang E, Izquierdo S, Pedersen D, Harriman W, et al. Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPalpha. MAbs. (2019) 11:1036–52. doi: 10.1080/19420862.2019.1624123
217. Voets E, Parade M, Lutje Hulsik D, Spijkers S, Janssen W, Rens J, et al. Functional characterization of the selective pan-allele anti-SIRPalpha antibody ADU-1805 that blocks the SIRPalpha-CD47 innate immune checkpoint. J Immunother Cancer. (2019) 7:340.
218. Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. (2019) 37:946–53. doi: 10.1200/jco.18.02018
219. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 Blockade by Hu5F9-G4 and rituximab in non-hodgkin’s lymphoma. N Engl J Med. (2018) 379:1711–21. doi: 10.1056/nejmoa1807315
220. McCracken MN, Cha AC, Weissman IL. Molecular pathways: activating T cells after cancer cell phagocytosis from blockade of CD47 “Don’t Eat Me” signals. Clin Cancer Res. (2015) 21:3597–601. doi: 10.1158/1078-0432.ccr-14-2520
221. Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci USA. (2013) 110:11103–8. doi: 10.1073/pnas.1305569110
222. Liu Q, Li Z, Gao JL, Wan W, Ganesan S, McDermott DH, et al. CXCR4 antagonist AMD3100 redistributes leukocytes from primary immune organs to secondary immune organs, lung, and blood in mice. Eur J Immunol. (2015) 45:1855–67. doi: 10.1002/eji.201445245
223. Meyaard L, Adema GJ, Chang C, Woollatt E, Sutherland GR, Lanier LL, et al. LAIR-1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity. (1997) 7:283–90. doi: 10.1016/s1074-7613(00)80530-0
224. Kubagawa H, Burrows PD, Cooper MD. A novel pair of immunoglobulin-like receptors expressed by B cells and myeloid cells. Proc Natl Acad Sci USA. (1997) 94:5261–6. doi: 10.1073/pnas.94.10.5261
225. Yi X, Zhang J, Zhuang R, Wang S, Cheng S, Zhang D, et al. Silencing LAIR-1 in human THP-1 macrophage increases foam cell formation by modulating PPARgamma and M2 polarization. Cytokine. (2018) 111:194–205. doi: 10.1016/j.cyto.2018.08.028
226. Chang YC, Nizet V. The interplay between Siglecs and sialylated pathogens. Glycobiology. (2014) 24:818–25. doi: 10.1093/glycob/cwu067
227. Hudak JE, Canham SM, Bertozzi CR. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. (2014) 10:69–75. doi: 10.1038/nchembio.1388
228. Haas Q, Boligan KF, Jandus C, Schneider C, Simillion C, Stanczak MA, et al. Siglec-9 regulates an effector memory CD8(+) T-cell subset that congregates in the melanoma tumor microenvironment. Cancer Immunol Res. (2019) 7:707–18. doi: 10.1158/2326-6066.cir-18-0505
229. Bull C, Stoel MA, den Brok MH, Adema GJ. Sialic acids sweeten a tumor’s life. Cancer Res. (2014) 74:3199–204. doi: 10.1158/0008-5472.can-14-0728
230. von Gunten S, Schaub A, Vogel M, Stadler BM, Miescher S, Simon HU. Immunologic and functional evidence for anti-Siglec-9 autoantibodies in intravenous immunoglobulin preparations. Blood. (2006) 108:4255–9. doi: 10.1182/blood-2006-05-021568
231. Laubli H, Alisson-Silva F, Stanczak MA, Siddiqui SS, Deng L, Verhagen A, et al. Lectin galactoside-binding soluble 3 binding protein (LGALS3BP) is a tumor-associated immunomodulatory ligand for CD33-related Siglecs. J Biol Chem. (2014) 289:33481–91. doi: 10.1074/jbc.m114.593129
232. McMillan SJ, Sharma RS, Richards HE, Hegde V, Crocker PR. Siglec-E promotes beta2-integrin-dependent NADPH oxidase activation to suppress neutrophil recruitment to the lung. J Biol Chem. (2014) 289:20370–6. doi: 10.1074/jbc.m114.574624
233. Adams OJ, Stanczak MA, von Gunten S, Laubli H. Targeting sialic acid-Siglec interactions to reverse immune suppression in cancer. Glycobiology. (2018) 28:640–7.
234. Chen S, Song Z, Zhang A. Small-molecule immuno-oncology therapy: advances, challenges and new directions. Curr Top Med Chem. (2019) 19:180–5. doi: 10.2174/1568026619666190308131805
235. Kerr WG, Chisholm JD. The next generation of immunotherapy for cancer: small molecules could make big waves. J Immunol. (2019) 202:11–9. doi: 10.4049/jimmunol.1800991
236. Dempke WCM, Uciechowski P, Fenchel K, Chevassut T. Targeting SHP-1, 2 and SHIP pathways: a novel strategy for cancer treatment?Oncology. (2018) 95:257–69. doi: 10.1159/000490106
238. Dechant MVT. IgA antibodies for cancer therapy. Crit Rev Oncol Hematol. (2001) 39:69–77. doi: 10.1016/s1040-8428(01)00105-6
239. Lohse S, Derer S, Beyer T, Klausz K, Peipp M, Leusen JH, et al. Recombinant dimeric IgA antibodies against the epidermal growth factor receptor mediate effective tumor cell killing. J Immunol. (2011) 186:3770–8. doi: 10.4049/jimmunol.1003082
240. Lohse S, Loew S, Kretschmer A, Jansen JHM, Meyer S, Ten Broeke T, et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Br J Haematol. (2018) 181:413–7. doi: 10.1111/bjh.14624
241. Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin A Fc receptor FcalphaRI; its function and potential as target in disease. Immunol Rev. (2015) 268:123–38.
242. Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, et al. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcalphaRI (CD89) binding. MAbs. (2015) 7:743–51. doi: 10.1080/19420862.2015.1047570
243. Brandsma AM, Bondza S, Evers M, Koutstaal R, Nederend M, Jansen JHM, et al. Potent Fc receptor signaling by IgA leads to superior killing of cancer cells by neutrophils compared to IgG. Front Immunol. (2019) 10:704.
244. Otten MA, Rudolph E, Dechant M, Tuk CW, Reijmers RM, Beelen RH, et al. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J Immunol. (2005) 174:5472–80. doi: 10.4049/jimmunol.174.9.5472
245. Otten MA, Leusen JH, Rudolph E, van der Linden JA, Beelen RH, van de Winkel JG, et al. FcR gamma-chain dependent signaling in immature neutrophils is mediated by FcalphaRI, but not by FcgammaRI. J Immunol. (2007) 179:2918–24. doi: 10.4049/jimmunol.179.5.2918
246. Boross P, Lohse S, Nederend M, Jansen JH, van Tetering G, Dechant M, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. (2013) 5:1213–26. doi: 10.1002/emmm.201201929
247. Lohse S, Meyer S, Meulenbroek LA, Jansen JH, Nederend M, Kretschmer A, et al. An anti-EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res. (2016) 76:403–17. doi: 10.1158/0008-5472.can-15-1232
248. Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn-mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin Immunol. (2007) 122:146–55. doi: 10.1016/j.clim.2006.09.001
249. Meyer S, Nederend M, Jansen JH, Reiding KR, Jacobino SR, Meeldijk J, et al. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. MAbs. (2016) 8:87–98. doi: 10.1080/19420862.2015.1106658
250. Treffers LW, Ten Broeke T, Rosner T, Jansen JHM, van Houdt M, Kahle S, et al. IgA-mediated killing of tumor cells by neutrophils is enhanced by CD47-SIRPalpha checkpoint inhibition. Cancer Immunol Res. (2020) 8:120–30.
251. Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci. (2009) 30:356–62. doi: 10.1158/2326-6066.cir-19-0144
252. Pereira NA, Chan KF, Lin PC, Song Z. The “less-is-more” in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs. (2018) 10:693–711. doi: 10.1080/19420862.2018.1466767
253. Scallon BJ, Tam SH, McCarthy SG, Cai AN, Raju TS. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol Immunol. (2007) 44:1524–34. doi: 10.1016/j.molimm.2006.09.005
254. Thomann M, Schlothauer T, Dashivets T, Malik S, Avenal C, Bulau P, et al. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS One. (2015) 10:e0134949. doi: 10.1371/journal.pone.0134949
255. Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. (2002) 277:26733–40. doi: 10.1074/jbc.m202069200
256. Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA. (2011) 108:12669–74. doi: 10.1073/pnas.1108455108
Keywords: neutrophils, cancer, tumor microenvironment, myeloid-derived suppressor cells, antibody-dependent cellular cytotoxicity, antibody therapy
Citation: Furumaya C, Martinez-Sanz P, Bouti P, Kuijpers TW and Matlung HL (2020) Plasticity in Pro- and Anti-tumor Activity of Neutrophils: Shifting the Balance. Front. Immunol. 11:2100. doi: 10.3389/fimmu.2020.02100
Received: 27 April 2020; Accepted: 03 August 2020;
Published: 02 September 2020.
Edited by:
Brahm Segal, Roswell Park Comprehensive Cancer Center, University at Buffalo, United StatesReviewed by:
Carlos Alfaro, Biotech Foods, SpainBipulendu Jena, Independent Researcher, San Diego, United States
Copyright © 2020 Furumaya, Martinez-Sanz, Bouti, Kuijpers and Matlung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanke L. Matlung, aC5tYXRsdW5nQHNhbnF1aW4ubmw=
†These authors have contributed equally to this work