 Mounika U. L. Goruganthu
Mounika U. L. Goruganthu Anil Shanker
Anil Shanker Mikhail M. Dikov
Mikhail M. Dikov David P. Carbone1
David P. Carbone1- 1Department of Internal Medicine, James Comprehensive Cancer Center, The Ohio State University Wexner Medical Center, Columbus, OH, United States
- 2Department of Biochemistry, Cancer Biology, Neuroscience and Pharmacology, Meharry Medical College School of Medicine, Nashville, TN, United States
- 3Vanderbilt-Ingram Cancer Center, Nashville, TN, United States
Understanding and targeting Notch signaling effectively has long been valued in the field of cancer and other immune disorders. Here, we discuss key discoveries at the intersection of Notch signaling, cancer and immunology. While there is a plethora of Notch targeting agents tested in vitro, in vivo and in clinic, undesirable off-target effects and therapy-related toxicities have been significant obstacles. We make a case for the clinical application of ligand-derived and affinity modifying compounds as novel therapeutic agents and discuss major research findings with an emphasis on Notch ligand-specific modulation of immune responses.
Introduction
Notch signaling plays a variety of physiological roles including, but not limited to, cell proliferation, cell fate decisions, cellular differentiation and angiogenesis (1). The role and importance of Notch signaling in hematopoietic compartment now stands undisputed. Despite the improved clinical response compared to standard chemotherapy, the efficacy of immune checkpoint inhibitors (ICI) across a variety of solid tumors is limited to a fraction of patients (2, 3). It is therefore essential to develop therapeutic agents that show promise as single agent immunomodulators or can be used in combination with ICIs to elicit antitumor immune responses.
Developing and utilizing agents that could support the induction of antitumor T cell functions while also precluding effector immune cells from immunosuppression offers great promise. Findings from murine models of solid tumors, allergic responses and autoimmune disorders indicate great potential for the clinical application of Notch ligands and their derivatives as immunomodulatory agents for the management of malignant cancers (4, 5). Engineered Notch ligand-derived moieties could be used to induce desired immune responses and boost antitumor immunity (6, 7).
Activating mutations in Notch1 have been described in lung, breast, colorectal and pancreatic cancers to name a few. On the other hand, loss of function mutations in Notch in hepatocellular carcinomas and melanomas have established its role as a tumor suppressor (8). Notch can play a highly contextual role in tumoral, stromal and immune compartments, which adds to the signaling complexity and warrants the need to pursue its therapeutic targeting with great prudence.
In the following sections, we report findings that revealed the varied effects of Notch signaling in immune compartments driving T cell development, activation, differentiation, and regulation of effector immune responses. Non-canonical Notch signaling and its crosstalk with other signaling pathways, impact of Notch post-translational modifications on T cell differentiation, consensus and controversies and open questions in the field are discussed. We highlight how knowledge obtained by structural studies and studying the mechanisms of various steps involved in Notch activation and signal transduction offer therapeutic opportunities that enable its targeting with high specificity.
Brief Overview of Notch Signaling
Canonical Notch signaling is unique in being driven by juxtracrine cell membrane bound receptor-ligand interactions (9). The mammalian Notch system is comprised of four Type I transmembrane receptors (Notch1-4) and two classes of ligands – Delta-like (DLL 1,3,4) and Jagged (JAG 1,2). Upon ligand binding, a mechanical force triggers sequential proteolytic cleavages in the intracellular portions of the receptor, ultimately releasing the Notch intracellular domain (NICD) into the cytoplasm. NICD then migrates into the nucleus where it acts along with a host of other transcriptional coactivators, including RBP-Jκ and MAML1-3 (10). To ensure tight regulation of Notch signaling, C-terminal PEST domain provides a proteolytic target for degradation of active Notch (11–13). Recent developments have indicated that Notch can also exert its functions non-canonically by interacting with members of other signaling pathways such as Wnt/β-catenin, NF-κB, TGFβ and many others (14–19).
Role of Notch Signaling in Helper and Effector T Cells
Select Delta-like ligands have been shown to induce differentiation of hematopoietic stem cells (HSCs) into T cells. These findings were obtained using OP9 bone marrow stromal cells expressing DLL1 or DLL4 and similar effects were also observed using purified plate-bound ligands (20, 21). T cell differentiation of HSCs is dependent on both ligand identity and level of expression where low-level expression of the Delta-like ligands attenuates but does not eliminate the myeloid potential of HSCs. Such fine tuning of dose responses is a recurring theme in Notch signaling, faithful artificial recapitulation of which has eluded us so far.
Antigen presenting cells (APCs) expressing Delta-like ligands activate and polarize naïve CD4+ T cells to a Th1 phenotype, while JAG1/2 expressing APCs lead to Treg/Th2/Th17 polarization (22–24). Convincing results in this direction showed that the intracellular domain of Notch1 is directly involved in interactions with and expression of Th1 master transcriptional factor T-bet and production of cytokine IFNγ in CD4 T cells. Notch signaling promoted the development of CD8+ terminal effector T cells and suppressed memory-precursor fate in effector-memory T cell (TEM) subsets (25). Activation of the Notch pathway in TEM cells also suppressed memory-precursor fate. Transcription factors such as eomesodermin (EOMES) and T-bet were found to be directly regulated by Notch, further supporting the importance of Notch signaling in driving effector T cell responses (26–28).
The tumor microenvironment (TME) also plays a major role in influencing T cell responses. Notch1 and Notch2 were found to be downregulated in tumor infiltrating T cells but not in splenic T cells of tumor-bearing mice (29). This attenuation of infiltrating T cell responses was driven by Jag1/2 expressed by immunosuppressive myeloid-derived suppressor cells (MDSCs) which could be overcome by ectopic expression of Notch1 intracellular domain (N1ICD) in antigen specific T cells, indicating that the TME is programmed with mechanisms to suppress Notch signaling and evade T cell-mediated tumor cell death. This reveals another interesting aspect of Notch: the spatio-temporal regulation of Notch ligand and receptor expression.
While several studies demonstrated the involvement of Notch signaling in driving effector and helper T cell responses (summarized in Figure 1), the precise regulatory mechanisms behind cell surface expression of Notch ligands and receptors are only partly known. TCR stimulation has been shown to induce expression of Notch1, Notch2 and Hes1 (Notch target) in T cells (30) but T cell activation using CD28 beads alone or low-dose CD3 and CD28 stimulation induces expression of Notch ligands on T cells (31). Notch ligands were not expressed by T cells during in vitro activation with mature bone-marrow derived dendritic cells. The induced ligands also co-localized with Notch receptors on the surface indicating cis-inhibition. Notch ligand expression was abrogated in the presence of NF-κB inhibitor, demonstrating the combined role of Notch and NF-κB pathways in driving T cell functions downstream of TCR stimulus. The observations suggest additional regulatory mechanisms, possibly to prevent erroneous T cell activity in the absence of both TCR and co-stimulatory CD28 signals.
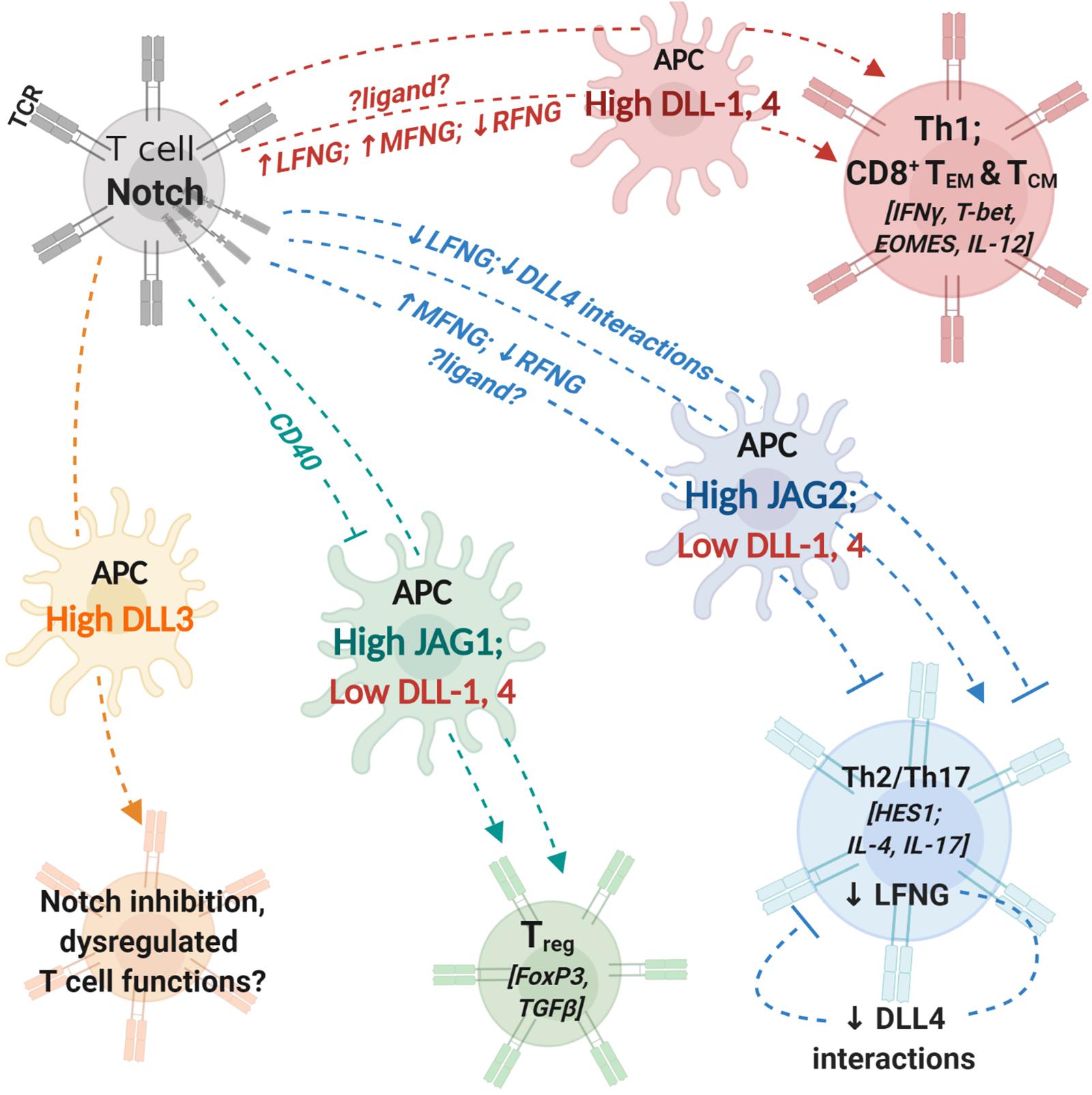
Figure 1. Notch interactions between antigen-presenting cells and T cells influence helper and effector T cell activity. T cells express T cell receptor (TCR) complex and Notch receptors. Antigen-presenting cells (APCs) express costimulatory molecules and Notch ligands. During T cell activation, the identity of Notch ligand present on the cell surface of APCs can influence T cell polarization and differentiation. Changes in expression levels of fringe glycosyl transferases can influence the process by modifying Notch receptor affinity to different ligands. Notch signaling in T cells regulates expression of transcription factors and cytokines (indicated within []) involved in helper and cytotoxic T cell functions. APCs with high expression levels of DLL1 or DLL4 can polarize CD4+ T cells into aTh1 phenotype and drive CD8+ T cell differentiation into memory cells. Increase (↑) in LFNG and MFNG expression and downregulation/loss (↓) of RFNG expression can enhance Th1 differentiation; identity of ligands involved in fringe-mediated Th1 differentiation are yet to be investigated (represented by ?ligand?). APCs with high JAG2 and low DLL1,4 expression drive helper T cell differentiation into Th2 or Th17 phenotypes. Expression of MFNG and downregulation of RFNG can block Th2 differentiation. Loss of LFNG in uncommitted T cells as well as Th2 polarized cells inhibits Notch interactions with DLL4 and attenuates Th2 responses. APCs with high JAG1 expression can induce T cell polarization into regulatory T cells (Treg). CD40 blockade together with JAG1 expression on APCs enhances immunosuppressive functions of Treg cells. APC, antigen presenting cell; DLL, Delta-like ligand; JAG, Jagged ligand; LFNG, lunatic fringe; MFNG, manic fringe; MHC, major-histocompatibility complex; TCR,; Th1, T helper type 1, Th2: T helper type 2; Th17, T helper type 17; Treg, T regulatory cell; TEM, effector-memory T cell; TCM, central-memory T cell; RFNG, radical fringe.
Notch extracellular domain (NECD) binding to cognate ligands is influenced by a variety of post-translational modifications, prominent among them being O-linked glycosylation by Fringe glycosyl transferases (32, 33). The three mammalian fringe proteins, Lunatic (Lfng), Manic (Mfng) and Radical (Rfng) extend O-Fucose moieties with GlcNAc at conserved serine or tyrosine residues in EGF repeats of NECD (34, 35). Glycosylation of Notch by Lfng and Mfng enhances interactions with Delta-like ligands while suppressing interactions with Jagged ligands. On the other hand, Notch glycosylation by Rfng enhances receptor interactions with both classes of ligands.
Tumor-mediated decrease in Lfng and Mfng expression levels have been shown to promote metastasis and poor survival (36, 37). Lfng interacts cooperatively with p53 to suppress tumors and Mfng suppresses tumorigenic activity of JAG1 and Notch3 (38–40). Lfng and Mfng thus appear to have a tumor-suppressive role in solid tumors and restoring their expression levels can be pursued as a therapeutic strategy to achieve tumor regression.
Fringe-mediated changes in Notch ligand-receptor interactions lead to dysregulations in thymic and ectopic T cell development resulting in altered T/B cell population ratios (41–44). Tumor burden and tumor-derived immunosuppressive cytokines also cause abnormalities in intrathymic T cell differentiation and development (45–47). Notch glycosylation by fringes influence the differentiation of mature T cell populations as well. It was found that Lfng and Mfng were downregulated and Rfng was upregulated in naïve CD4+ T cells in asthmatic rats (48). This was associated with more active Notch signaling in asthmatic naïve CD4+ T cells compared to control naïve T helper cells. Restoring Lfng and Mfng expression and silencing Rfng enhanced the number of Th1 cells while lowering Th2 cell differentiation. Lfng overexpression in naïve CD4+ T cells was able to drive Th1 and Th2 differentiation in a Notch-independent and dependent manner, respectively. These findings indicate that modulating Fringe expression levels can be potential therapeutic strategies for the management of allergic diseases. Moreover, the observation that fringe expression levels vary even in naïve CD4+ T cells under asthmatic conditions provide a basis for the hypothesis that there might be molecular factors that can alter T cell programs, which need to be elucidated. While Gu et al., were able to demonstrate the role of fringe glycosylation in influencing T helper cell differentiation, the source and identity of Notch ligands involved in this process were not identified.
Using a mouse airway allergic disease model, another study found that transcription of Lfng was driven by STAT5 in Th2 helper cells (49). Th2-mediated airway hyper-reactivity, mucus production and IL4 production was driven by DLL4-mediated Notch activation. Specifically, deletion of Lfng but not Mfng or Rfng in Th2 and CD4+ T cells resulted in reduced Th2 responses and inflammation. While STAT5 and GATA3 were previously known to drive Th2 differentiation independent of Notch signals (50, 51), the regulation of Lfng expression by STAT5 in Th2 subsets is a novel and interesting finding. It is likely that other inflammatory factors that can influence STAT5 signaling can potentially alter fringe expression levels. Notch activity in T cells thus can be profoundly influenced by complex intracellular networks of cytokines and signaling pathways involved in fine-tuning immune responses (52, 53).
Contrasting Observations and an Argument for Non-Canonical Notch Signaling in T Cells
Differentiation of complete T cell effector program has been observed to be dictated by the identity of Notch ligand expressed on APCs, which in turn is dictated by the type of antigenic stimulus encountered (22, 23). This is in stark contrast to observations from in vitro T cell differentiation by polarizing cytokines even in the absence of Notch ligands (54). In some in vitro experiments, Notch activity was shown to confer a proliferative effect in T cells but could not drive Th1/Th2 differentiation in the absence of polarizing cytokines (55). While some studies have demonstrated that DLL1/4 ligands can promote a Th1 polarization, others have argued that the Th1 phenotype is not acquired as a consequence of Notch signaling but by suppression of the alternative Th2/17 fate (56, 57). The disease model used, type of antigenic responses, stimuli involved in DC maturation and the relative expression levels of different Notch ligands are all factors that could potentially influence T cell polarization by APCs. Most studies, however, have produced convincing data in favor of Notch1-ICD binding directly to promoters of genes and transcription factors driving Th1 and cytotoxic responses. Non-canonical Notch signaling and crosstalk with NF-κB pathway is also observed in activated T cells (58). γ-secretase inhibitors reduced IFNγ production in in vitro activated CD8+ T cells but not in CD4+ cells, which can indicate that helper and cytotoxic T cells respond differently to Notch stimuli at least in vitro. It is likely that DC-borne ligands could orchestrate T cell survival and proliferation within an existing cytokine milieu instead of having an instructive role in naïve T cell differentiation (59–61).
These observations prompt a question: do Notch ligands play a deterministic/instructive role or do they simply enhance pre-existing T cell programs in an unbiased manner? It could be possible that Notch serves as a costimulatory signal that can set in motion any of the numerous downstream signaling pathways (62, 63). It might also be possible that Notch signaling might have different effects before, during and after T cell activation and differentiation. Majority of the studies on the role of Notch in immune cell functions have looked at Hes/Hey/Deltex family members, which are themselves transcriptional factors effecting expression of several genes. T cell functions might be ultimately dictated by a combinatorial framework in which terminal effector molecules are further regulated by Notch targets.
Unanswered Questions
Notch signaling does not always appear to operate as a simple ON/OFF switch. It has been shown to be regulated by a complex system of fine-tuning and crosstalk of input signals including relative expression levels of ligands and receptors, numerous post-translational modifications and a combination of cis- and trans- interactions (64–67). While attempts are being made to target Notch in various disease settings, a large number of therapies developed so far have led to undesirable side-effects and toxicities (7). To address these shortcomings, it is important to study the mechanistic and physical aspects of ligand-receptor interactions (68) and role of post-translational modifications such as ligand glycosylation and ubiquitination (32, 33, 35). It is also necessary to understand how the physiological consequences of ectopic Notch expression are similar to and differ from ligand-specific receptor activation and how different sources of ligands can influence differences in immunological outcomes. Redundancies in receptor and ligand paralogs also need to be resolved.
Therapeutic Strategies to Target Notch Signaling
Knowledge-based approaches on the activation mechanisms of Notch have led to the development of several Notch inhibitory agents. These include selective ligand/receptor-specific decoys, agents that block receptor cleavage, molecules that inhibit formation of Notch-CSL activator complex, antibodies, and post translational modifications influencing ligand-receptor interactions (Table 1 and Figure 2). In addition to being used as single agents in various clinical and preclinical studies, Notch inhibitors are also being studied in combination with current chemotherapeutic drugs. Despite being uncharacterized for the active component, some natural compounds show promising anti-proliferative effects on cancer cell lines and have traditionally been used as part of dietary modifications as chemo-preventative measures (69–71). Inhibition of the γ-secretase complex is the most widely employed method of blocking Notch signaling but has been fraught with toxicities (72, 73). There is a need to focus on Notch activators in the management of cancers like lung squamous cell carcinoma, where Notch acts as a tumor suppressor. The development of Notch modulators should be guided at every stage by the biological and physiological effects of the compounds being tested. Mechanism-based combinatorial regimens, biomarkers of response and contextual frameworks need to be developed and evaluated on a case by case basis.
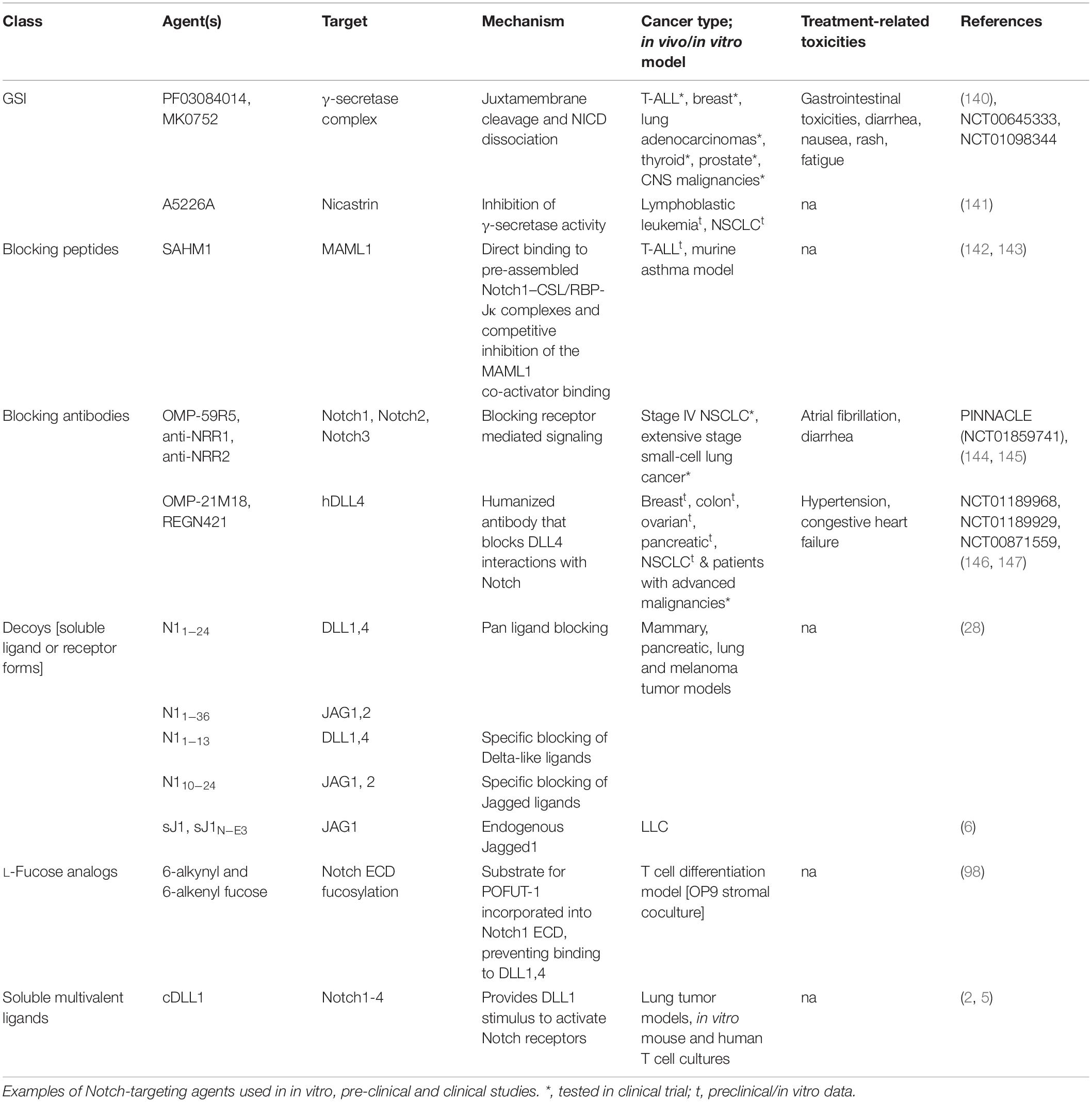
Table 1. Strategies to target Notch signaling.
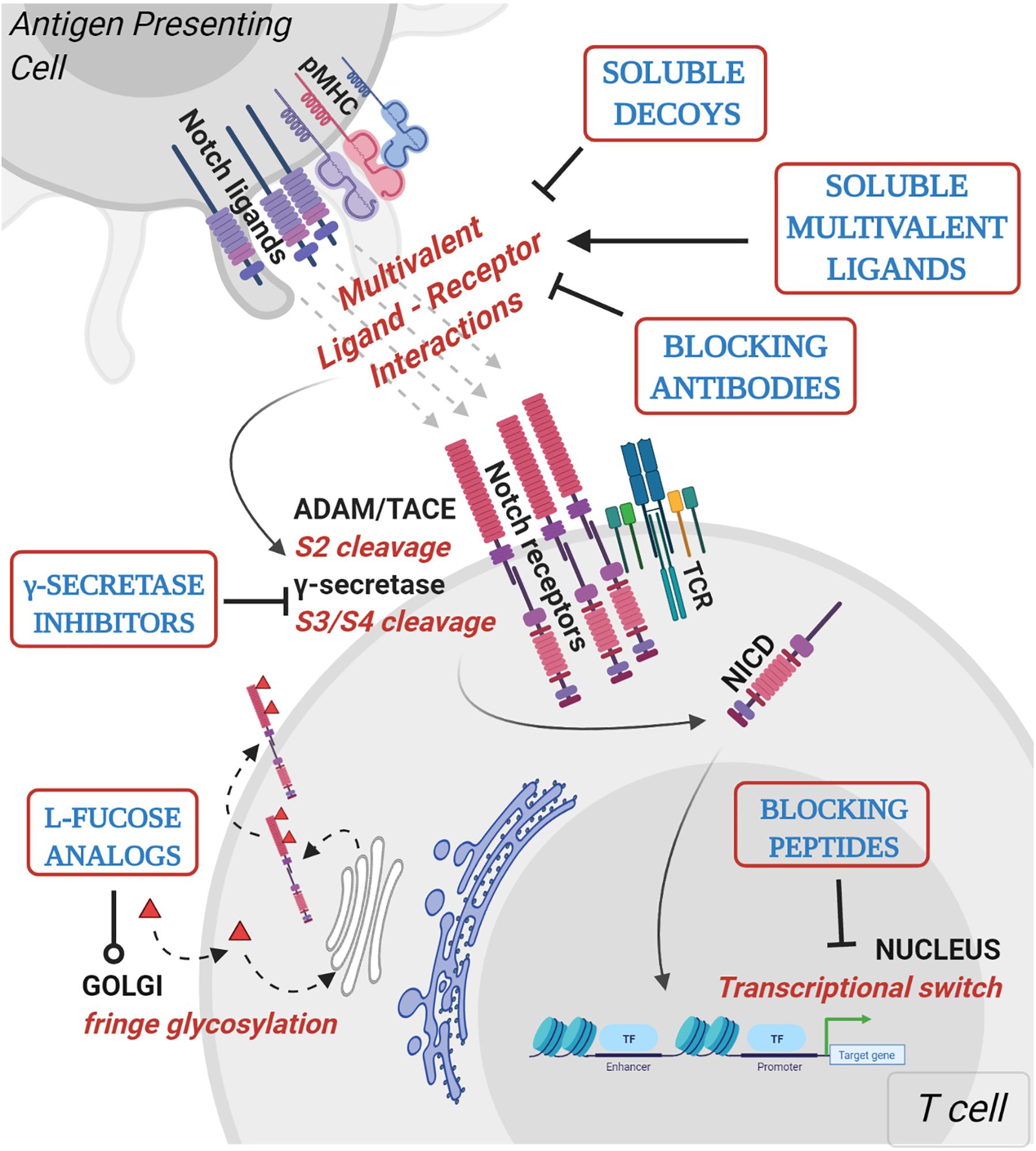
Figure 2. Mechanistic basis for therapeutic targeting of ligand-specific Notch signaling. Agents targeting Notch signaling can be grouped by the step or process in the Notch signaling pathway that is being affected. Soluble decoys comprise of extracellular portions of Notch ligands or receptors that can competitively inhibit multivalent receptor-ligand interactions. Soluble multivalent ligands comprise of clustered ligands that provide and/or augment ligand-specific Notch activation. Blocking antibodies block receptor interactions with ligands and are paralog-specific antagonists with high selectivity. γ-secretase inhibitors prevent NICD release by inhibiting S3 cleavage of Notch receptors at the juxtamembrane domain. L-fucose analogs (solid red triangles) are taken up by cells from media and incorporated into receptor extracellular domains. Fucose analogs on Notch receptors alter ligand-binding affinities and can be used to block selective ligand interactions. Blocking peptides target protein-protein interface in the nuclear Notch transcriptional complex and prevent transcription of Notch target genes. In vitro, pre-clinical and clinical studies demonstrating Notch-modulatory activities and anti-tumor efficacy of various classes of Notch therapeutics are presented in Table 1.
While most reagents presented in Table 1 were initially used to alter Notch signaling in stroma and the tumor microenvironment, recent focus has shifted to targeting Notch in tumor-infiltrating and circulating immune compartments. This has been done using agents directly targeting Notch receptors expressed by immune cells or in in vitro settings where Notch ligand-based agents are employed to activate, prime, and expand helper and effector T cell populations.
Notch-Based Reagents for Adoptive T Cell Therapy
As the biology of Notch signaling in driving T cell development began to be better understood, the system was applied to generate antigen-specific T cells in vitro. By coculturing with DLL1-expressing bone marrow stromal cells, embryonic and hematopoietic stem cells could be differentiated into immunocompetent T lymphocytes (74). NY-ESO-1–specific and human p53–specific, HLA-A2–restricted human TCR vectors were used to transduce human umbilical cord HSCs, which were then cultured on OP9-GFP or OP9-DL1 cells and expanded (75). The T cells thus generated displayed very little endogenous TCR and had a high expression of antigen-restricted tumor-reactive TCR. They were also less differentiated than in vitro expanded lymphocytes that are currently employed in clinic. The differentiated and expanded HSCs in this study expressed the NK cell markers CD56 and CD16 as well as the T cell markers CD3 and CD7 but did not express IFNγ and IL-4 as NK-T cells do. Both NY-ESO1 and p53 TCR-transduced and differentiated cells exhibited antigen-specific lysis of target cells indicating T cell properties. The p53-TCR transduced HSCs, however, lysed both specific and non-specific tumor cells, indicating an NK cell-like behavior. While these cells could be useful candidates for adoptive cell transfer in both HLA-restricted and HLA-independent settings, safety evaluations and detailed characterization of the observed dual T and NK cell behavior is needed. Activated and differentiated effector T cells possess enhanced tumor reactivity in vitro but they demonstrate reduced tumor attenuation compared to naïve and early effector cells in vivo (76). This was overcome by the generation of stem cell memory T cells [TSCM], a class of highly proliferative memory T cells, again using the OP9-DL1 coculture system, to generate ova-specific reactive T cells (77). Of note, the iTSCM cells displayed a loss of PD1 and CTLA4 expression, which contributed in part to enhanced cytolytic activity of the adoptively transferred naïve-like stem cell memory T cells. While Notch1 activity could upregulate PD-1 expression on CD8+ T cells activated with artificial APCs expressing both Delta-like and Jagged ligands (78), expression of inhibitory receptors was not seen with the OP9-DL1 coculture system. This indicates the advantage of employing ligand-specific T cell stimulation and expansion for therapeutic applications.
Notch as a Target of Other Therapeutic Agents and Pathways
Notch signaling has the unique feature of integrating signals from several pathways. This leads to an extensive “hyper-network” situation within a cell as well as at the multicellular level (18, 79). From a therapeutic standpoint, it becomes important to identify key regulatory nodes between different pathways. This will enable the development therapeutic agents with high specificity and prevent cross-pathway side effects. Some studies have identified how Notch in immune cells can be altered by therapeutic and experimental interventions targeting molecules in other signaling pathways.
While T cells can upregulate Notch expression a few hours after TCR stimulation, the exact molecular crosstalk between the two pathways is only partially known (30, 80). PKCθ has been linked to actin regulation as well as Notch induction, leading to the discovery of a spatio-temporal link between T cell stimulation by professional APCs and Notch activity (81). p38 MAPK was shown to induce Jagged1 as well as Notch1 during the maturation of macrophages (82).
Adenosine is an immunosuppressive ATP metabolite that is increased in the extracellular space in response to hypoxia and tissue injury, which can have profound effects on both lymphoid and myeloid cells that express adenosine receptors, predominantly, A2AR by T cells. A2AR agonists have been used in the treatment of inflammatory diseases while A2AR antagonists are being developed as novel cancer immunotherapeutics (83). Notch1 was identified as a target of A2AR-mediated immunosuppression. This is believed to be orchestrated by Cbl-mediated ubiquitination of Notch1 modulated by A2AR via cAMP. CD8+ T cells exposed to an A2AR agonist prior to TCR stimulation lowered Notch1 expression, heterodimer cleavage and reduced transcripts of Notch1 target genes Hes1 and Myc (84).
Thus, disparate pathways could potentially converge to drive Notch expression and function. This makes it all the more important to fully understand the fundamental cellular and molecular levels at which Notch signaling is regulated.
Novel Notch Modulatory Agents
Engineered Ligand-Specific Therapeutics
An important feature of Notch signaling is the stoichiometry of interactions: activation of Notch receptors requires polyvalent interactions between multiple receptors and ligands. On the other hand, interfering with even a few of the productive multivalent interactions can lead to disruption of Notch signaling. Therefore, soluble monovalent forms of Notch ligands or receptors can potentially act as efficient competitive inhibitors. In contrast, presenting Notch ligands in a multivalent form can provide or enhance ligand-specific stimulus. This mechanistic detail of Notch receptor-ligand interaction can be exploited to design ligand-based reagents that can uniquely stimulate or block Notch signaling.
Studies have demonstrated that endogenous Notch ligand-receptor interactions can be selectively blocked or enhanced to influence signaling in tumoral, stromal and immune compartments (5, 6, 85, 86). Although receptor-ligand interactions can be abrogated using blocking antibodies, this presents some limitations including high costs, low tissue penetration, unclear mode of action in vivo, cytotoxicity, and affinity for inhibitory Fc receptors which reduces their overall efficacy (87). Soluble decoys that can interfere with specific ligand-receptor interactions and are small enough to achieve good biodistribution in solid tumors present more attractive options. Extracellular domains of Notch1 that uniquely interact with Delta-like or Jagged classes of ligands have been used to develop Notch decoys achieving ligand-specific inhibition (28, 86, 88). A fragment of Notch1 ECD comprised of EGF repeats 10 to 24 (N110–24) could selectively inhibit Notch1-JAG interactions without interfering with Notch1-DLL interactions indicating competitive binding to Jagged ligands. N110–24 demonstrated potent antitumor effects in various murine tumor models by reducing angiogenic sprouting and disruption of tumor endothelium, both of which are phenotypes associated with JAG1-driven Notch signaling. Similarly, the Notch1 ECD fragment comprised of EGF repeats 1 to 13 (N11–13) could specifically inhibit DLL4-mediated Notch signaling effects leading to hyper-sprouting and poor perfusion. On the other hand, a larger Notch1 decoy, N11–24, recapitulated the effects of inhibiting either JAG1 or DLL4 or both, depending on the tumor microenvironment and in vitro angiogenesis model used.
Soluble inhibitory receptor-derived decoys have been reported for Notch3 as well. Distinct, short peptides derived from EGF repeats 7–10 and 21–22 of Notch3 bound directly to JAG1 (89). The ligand-binding domains of Notch3 were distinct from those of Notch1 despite the high sequence similarity in conserved EGF repeats. The peptide forms as well as recombinant immunoglobulin Fc chimeras (IgG-Fc) of Notch3-derived peptides were able to induce apoptosis in tumor cells, preferentially reduced Notch3 activation and the expression of Notch3-specific target Hey1. Peptide-IgGFc chimeras could also suppress tumor growth in a Notch3-driven human lung cancer xenograft model.
Thus, certain regions of Notch receptor extracellular domains that uniquely interact with different ligand classes and paralogs can be used to design soluble inhibitory decoys with high specificity.
Full-length or partial extracellular domains of Notch ligands could be also be used to modulate ligand-specific Notch signaling events. Soluble monomeric fragments comprised of the DSL and first two EGF repeats of DLL1 (sDLL1) and the first five N-terminal domains of JAG1 (sJAG1N–E3) could selectively inhibit Notch1-DLL1 and Notch1-JAG1 interactions, respectively (6). The sDLL1 fragment attenuated in vitro T cell proliferation in cocultures with DLL1-bearing dendritic cells, indicating its potential ability to impair T cell responses by blocking endogenous DLL1. A short synthetic peptide derived from DSL region of JAG1 spanning residues 188–204 demonstrated Notch activation driving keratinocyte differentiation in vitro in its soluble form (90). Portions of the DSL domain of human JAG1 as well as the complete extracellular region of human JAG1 ligands could function as activators affecting differentiation of myeloid progenitors (91). In contrast, soluble purified human Jagged1-immunoglobulin IgG1 Fc chimera protein inhibited growth of myeloid colonies and macrophage progenitors from human cord blood, indicating its inhibitory properties (92). Therefore, the exact sequence of Notch ligand extracellular domains used (partial fragment versus full length ECD, specific portions of extracellular domains) could determine whether the soluble ligand forms act as activators or inhibitors. This might be driven by the area and structural conformation of ligand-receptor interface being bound by the soluble ligands. Detailed structural and binding studies need to be done to evaluate the mechanistic aspects of soluble ligands as Notch modulators.
Ligand multivalency is commonly mimicked by immobilizing the ligands on culture plates prior to seeding cells or by pre-clustering of ligand immunoglobulin Fc fragment chimeras by anti-Fc antibodies (2, 93). While plate-bound ligands can provide multimeric ligand stimulus, their use is restricted to in vitro applications. For in vivo administration, pre-clustered ligands provide a more suitable format. Further complexing anti-Fc antibodies is possible by tagging them with biotin, FLAG or other non-immunogenic short peptides or affinity tags (94) and using anti-tag antibodies to in turn complex those. This would greatly increase the valency of ligand being provided and could be used for pharmacological stimulation of ligand-specific responses. A multiplexed reagent called clustered DLL1 (cDLL1) was developed which is comprised of three components: a chimera of full length murine or human DLL1 and Fc region of IgG2A, biotinylated anti-IgG2Fc antibody, and NeutrAvidin (a deglycosylated version of avidin with unaltered affinity to biotin) (1). This produces a tertiary complex with multiple ligand extracellular domains being available for Notch activation. cDLL1 administration to tumor-bearing mice improved antigen-specific CD8+ T cell responses and attenuated tumor growth in preclinical murine models of lung cancer. Multivalent DLL1 stimulus provided by cDLL1 could enhance CD8+ T effector-memory cells and reduce the number of regulatory T cells in spleen.
Apart from providing in vitro therapeutic agents, plate-bound, cell-expressing, and multimerized Notch ligands could thus be used in various multivalent formats for cancer treatment.
Affinity-Modifying Compounds
Carbohydrate moieties at the ligand-receptor interface in trans interactions can influence canonical ligand-mediated Notch receptor activation via steric effects (95–97). L-fucose analogs that could be directly incorporated into Notch EGF repeats can be exploited to manipulate Notch receptor binding to cognate ligands. Peracetylated forms of O-Fucose, 6-alkynyl and 6-alkenyl fucose, act as substrates by Pofut-1 and can differentially modulate ligand binding (98). Fucose analogs incorporated into Notch1 EGF repeats inhibit trans interactions with DLL1 and DLL4 whereas interactions with JAG1 remain unaffected. Mutational and structural analysis revealed that fucosylation at Notch EGF8 is the site contributing to steric clashes and subsequent ablation of interactions with Delta-like ligands. This can be explained by a higher sensitivity of Delta-like ligands to Notch post-translational modifications compared to the Jagged class of ligands.
Notch ligand-based and affinity-modifying reagents thus offer the benefit of specific targeting along with fine-tuning and versatility to activate or inhibit Notch activity in a ligand-specific manner.
The following sections summarize evidence from preclinical and clinical studies that provided substantial evidence in favor of Notch ligand-based moieties as immunomodulatory agents driving anti-tumor T cell functions.
Restoring DLL1-Specific Notch Signaling Can Reverse Impaired T Cell Development in Tumor-Bearing Mice
Tumor presence alters a number of cytokine-mediated intracellular signaling pathways, expression of chemotactic ligands and receptors by thymic populations (99–101). This results increased apoptosis of TECs, dysregulated lineage-commitment checkpoints, diminished TCR repertoire and low thymic output, all of which ultimately dampen immunosurveillance and promote tumor escape. In immature DN2 T cell subsets, CCR7 is a target of Notch1 and is important for the migration of developing T cell precursors through the thymic cortex to medulla (102–104). Reduction in expression levels of Notch1 and its targets in thymic pre-T cells of tumor-bearing mice is mediated by IL-10 produced by thymic epithelial cells (TECs) (45). This is associated with an upregulation in of Ikaros and IRF8 signaling which shunts the developing pre-T cell toward differentiating into dendritic cells. A network of interactions between Notch, Wnt, Ikaros, and IL10 (among several others) is involved in determining the balance between T and myeloid lineage commitment under normal physiologic conditions (105–108). Given the indispensable role of Notch in ensuring normal thymic T cell development, therapeutic interventions to restore Notch activity in thymic and peripheral T cells can promote antitumor immunity (109, 110).
Advanced stage cancer patients have high mean serum levels of vascular endothelial growth factor (VEGF) compared to healthy humans. Mice infused with VEGF to mimic this pathophysiology showed thymic atrophy and decreased percentage of peripheral T cells in their spleen and lymph nodes (46, 111). This effect was coupled to a significant decrease in the number of CD4+CD8+ thymic populations. The reduction in CD4+CD8+ numbers was not due to an induction of thymocyte apoptosis or inhibition of thymocyte development, as the VEGF-exposed thymic cells could develop normally in mice without tumors and in in vitro fetal thymic organ cultures. Administration of anti-VEGFR2 but not anti-VEGFR1 antibody restored normal hematopoiesis revealing a mechanistic link between tumor derived VEGF and impaired peripheral immunity (112). The thymic atrophy observed in tumor-bearing mice could be a consequence of a pre-thymic event such as a VEGF-mediated block in emigration of thymic progenitors from the bone marrow.
Compared to age-matched controls, tumor-bearing mice have low DLL1 and DLL4 expression levels in bone marrow cells as well as low splenic T:B cell ratios. When VEGF-infused mice were irradiated and received bone marrow progenitors overexpressing DLL1, the inhibitory effects on T cell development were reversed, indicating that DLL1 stimulus alone is sufficient to resuscitate VEGF-driven impaired antitumor immunity. In order to mimic the effects of BM transplantation with DLL1-overexpressing hematopoietic precursors, the more pharmacologically relevant multivalent DLL1 form (cDLL1 – described in the previous section) was employed (5). Administration of cDLL1 significantly lowered tumor burden in treated mice compared to untreated tumor-bearing controls. Tumor regression was T-cell mediated, as was seen with the loss of cDLL1 efficacy in tumor-bearing Rag1–/– mice and mice receiving anti-CD8 antibody to deplete CD8+ T cells. This was associated with increased number of antigen-specific memory T cells, improved IFNγ production, and higher intracellular pSTAT1&2 in differentiated T cells. Additionally, the transcript levels of T-bet were significantly higher in CD4+ T cells after cDLL1 administration, providing direct evidence of tumor attenuating Th1 responses being enhanced (112–115). Stimulation of Notch signaling in effector CD8+ T cells was also able to achieve tumor regression in mutant EGFRL858R oncogene-driven tumor models. Patients with EGFR-driven non-small cell lung cancers treated with tyrosine kinase inhibitors (TKI) such as erlotinib eventually acquire drug resistance (116–118). The TKI treatment also shapes the tumor microenvironment leading to an upregulation of PD-L1 expression (119–121). Given the low response rate of EGFR-mutant tumors to ICI treatment (122–124), DLL1-mediated enhancement of Type I immune responses might provide therapeutic benefit when used in combination with ICI.
Different Notch ligand-receptor interactions can result in distinct downstream outcomes hence while DLL4 stimulation promotes angiogenesis, DLL1 signaling does not (125–127). Concurrently, the administration of cDLL1 to tumor-bearing mice did not result in vascular defects and, in fact, significantly decreased tumor vascularization.
In this manner, Notch ligand-based therapeutics can selectively stimulate helper and effector immune functions with high ligand and contextual specificity. Multivalent DLL1-derived Notch activators could thus potentially provide clinically relevant immunotherapeutic agents to overcome thymic atrophy and impaired T cell functions.
Regulatory T Cell Functions Can Be Modulated in a JAG1-Specific Manner
Interaction of T cells with APCs is necessary to induce effector T cell function and differentiation by providing TCR stimulus from cognate peptide-bound MHC (signal 1) and costimulatory CD28 (signal 2) (128–130). Several findings have revealed the additional role of interactions between specific Notch ligands presented by DCs and Notch receptors on T-cells in providing critical activation, differentiation, and polarization signals.
Adoptive transfer of antigen-pulsed, Jagged1 (JAG1)-expressing DCs inhibited established immune responses in immunized mice. This inhibition was CD4+ T cell-specific and long lived (131). With JAG1-expressing DC administration, peripheral naive CD4+ T cells were found to differentiate into regulatory cells. These cells could induce antigen-specific tolerance when transferred into naïve hosts. Similar effects were seen in human peripheral naïve blood cells; stimulation of CD45RA+ naïve T cells by allogeneic antigen-presenting cells overexpressing JAG1 resulted in reduced production of IFNγ, IL-2 and IL-5. The activated cells upregulated TGFβ and inhibited proliferative and cytotoxic immune responses in freshly stimulated lymphocyte cultures (132). This reveals the molecular basis of regulatory T cell induction when naïve cells are activated by JAG1-borne APCs. Further investigation revealed that the immunosuppression was antigen-specific and affected both CD4+ and CD8+ T cells (133).
Mouse bone marrow cells could be differentiated into tolerogenic dendritic cells by culturing them in the presence of GM-CSF (GM-BMDCs). GM-BMDCs were found to express JAG1, essential for induction of regulatory T cell phenotype in CD4+ T cells (134). Abrogating JAG1-Notch interactions by using anti-JAG1 blocking antibodies suppressed Treg proliferation. Similar results were obtained by shRNA-mediated knockdown of JAG1 in murine bone marrow mesenchymal stromal cells (MSC) (135). CD4+ T cells cocultured with JAG1-expressing MSC differentiated into tolerogenic Treg cells capable of producing anti-inflammatory cytokine, IL-10. Tregs thus obtained could also protect against inflammation in vivo in a mouse model of allergen-induced airway pathology.
Since DCs expressing JAG1 exhibit a tolerogenic potential, they could be used in a transplantation setting to inhibit immune responses and prolong allograft survival. Indeed, when JAG1-overexpressing DCs were used in along with CD40 blocking antibody, murine allograft heart transplants were better tolerated in recipient mice (136). This was achieved by induction of alloantigen-specific T cell suppression and upregulation of TGFβ and FoxP3-expressing Treg numbers driven by JAG1-Notch interactions expressed by transferred DCs and host T cells, respectively. While JAG1-driven Notch activation of host T cells could attenuate Th1 responses, it did not effect Th2 differentiation. By employing JAG1-transduced DCs, this study could provide mechanistic insights into the specific source and functions of Notch ligands. It is worth noting that overexpression of JAG1 could lead to ligand being in far in excess of receptors available on T cells and can diminish Notch activation owing to reduced ligand trans-endocytosis (137, 138).
Further evidence that Treg -mediated suppression of effector T cell responses was mediated by Notch came from a systematic lineage-specific deletion of Notch pathway components in Tregs. Targeted deletion of Pofut1, Rbpj and Notch1 enhanced Treg cell frequency and decreased CD4+ and CD8+ T-cell immune responses (139). On the other hand, overexpression of a constitutionally active Notch1 intracellular domain in Treg cells resulted in autoimmunity, skewing to a Th1 phenotype and apoptosis of regulatory T cells. Notch inhibition appears to dictate the balance between inflammatory effector T cells and tolerant regulatory T cells.
Tissue tissue-specific genetic ablation of Jagged1 and systemic administration of soluble inhibitory JAG1 provided further proof of ligand-mediated Notch activation in Tregs (6). CD11c-specific ablation of JAG2 did not have any effects on IFN-γ production but significantly decreased IL-4 production by activated T cells. On the other hand, CD11c-specific deletion of DLL1 resulted in accelerated tumor growth in murine tumor models coupled to a reduction in CD8+ T cell activation and reduced differentiation of antigen-specific cytotoxic T cells and memory cells. Tumor-bearing mice treated with inhibitory monomeric soluble JAG1 (sJAG1) showed a significant reduction in tumor burden concurrent with a decrease in splenic Treg cell numbers. This was associated with low tumor infiltration of CD11c+Gr1+ cells, thereby providing further evidence of JAG1 as a factor mediating immunosuppressive tolerogenic responses. In vitro T:DC coculture experiments in the presence of sJAG1 could also downregulate the expression of PD-1 on CD8+ T effector memory cells.
Taken together, these studies provide a strong evidence in favor of specifically targeting JAG1 to modulate regulatory T cell functions. The use of well designed, soluble inhibitory JAG1 decoys could provide a therapeutic edge in the context of enhancing antitumor immune responses and attenuating immunosuppression. Distinct ligand-specific effects provide a great opportunity to avoid undesirable effects associated with pan-Notch inhibition.
Concluding Remarks and Future Perspectives
From the perspective of both basic and applied immunology, study of Notch signaling in immune subsets can provide valuable insights into the management and cure of metastatic solid tumors that are recalcitrant to conventional treatments. As the field of immunology progresses, so will our understanding of the role that Notch plays in immune cell function and regulation. This can be accomplished by interdisciplinary and complementary techniques such as tissue and lineage-specific genetic ablation, biochemical and molecular modulation of ligand-receptor interactions, evaluation of antigen specific immune responses and computational analysis of large patient datasets. More fundamental approaches such as investigating ligand/receptor redundancies, effector differentiation by cytokines in combination with ligand-specific Notch activation and non-canonical Notch signaling are also needed. Outcomes from current therapeutic regimens can be improved by using Notch-ligand based reagents in combination with or prior to checkpoint blockade to prime the immune system. Preclinical studies using Notch ligand-derived selective activators and inhibitors also provide mechanistic insights into how the immune system can be modulated in a ligand-specific manner in cancer and other immunopathological conditions. Such agents constitute a novel class of immunomodulatory drugs addressing unmet medical needs.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Institutes of Health grants R01CA175370 (MD and DC) and R01CA138923 (MD), Dallapezze Fund (MD and DC), Team Science Award, The James Comprehensive Cancer Center, Ohio State University (DC and MD). AS is in part supported by funds from the NIH grants U54 CA163069 and SC1 CA182843.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CSL, CBF-1, suppressor of hairless, lag-2; DLL, Delta-like ligand; ECD, extracellular domain; GSI, γ -secretase inhibitor; HSC, hematopoietic stem cell; ICI, immune checkpoint inhibitor; JAG, Jagged ligand; LLC, Lewis lung carcinoma; MAML1, mastermind-like protein 1; NECD, Notch extracellular domain; NICD, Notch intracellular domain; NRR, negative regulatory region; NSCLC, non-small cell lung cancer; PEST, peptide sequence rich in proline (P), glutamic acid (E), serine (S) and threonine (T); RBP-Jκ, recombination signal binding protein for immunoglobulin Kappa J region; SAHM1, Stapled α -helical peptides derived from mastermind-like protein 1; T-ALL, T-cell acute lymphoblastic leukemia; TME, tumor microenvironment.
References
1. Kovall RA, Gebelein B, Sprinzak D, Kopan R. The canonical notch signaling pathway: structural and biochemical insights into shape, sugar, and force. Dev Cell. (2017) 41:228–41. doi: 10.1016/j.devcel.2017.04.001
2. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or Recurrent non–small-cell lung cancer. New Engl J Med. (2017) 376:2415–26. doi: 10.1056/NEJMoa1613493
3. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. (2019) 39:147–64. doi: 10.1200/EDBK_240837
4. Huang Y, Lin L, Shanker A, Malhotra A, Yang L, Dikov MM, et al. Resuscitating cancer immunosurveillance: selective stimulation of DLL1-Notch signaling in T cells rescues T cell function and inhibits tumor growth. Cancer Res. (2011) 71:6122–31. doi: 10.1158/0008-5472.CAN-10-4366
5. Biktasova AK, Dudimah DF, Uzhachenko RV, Park K, Akhter A, Arasada RR, et al. Multivalent forms of the notch ligand DLL-1 enhance antitumor T-cell immunity in lung cancer and improve efficacy of EGFR-targeted therapy. Cancer Res. (2015) 75:4728–41. doi: 10.1158/0008-5472.CAN-14-1154
6. Tchekneva EE, Goruganthu MUL, Uzhachenko RV, Thomas PL, Antonucci A, Chekneva I, et al. Determinant roles of dendritic cell-expressed Notch Delta-like and Jagged ligands on anti-tumor T cell immunity. J Immun Ther Cancer. (2019) 7:95. doi: 10.1186/s40425-019-0592-2
7. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling — are we there yet? Nat Rev Drug Discov. (2014) 13:357–78. doi: 10.1038/nrd4252
8. Aster JC, Pear WS, Blacklow SC. The varied roles of notch in cancer. Annu Rev Pathol. (2017) 12:245–75. doi: 10.1146/annurev-pathol-052016-100127
9. Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. (2016) 17:722–35. doi: 10.1038/nrm.2016.94
10. Kopan R, Ilagan MXG. The canonical notch signaling pathway: unfolding the activation mechanism. Cell. (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
11. Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. (2002) 16:1397–411. doi: 10.1101/gad.991602
12. Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. (2004) 16:509–20. doi: 10.1016/j.molcel.2004.10.014
13. Kopan R. All good things must come to an end: how is notch signaling turned off? Sci. Signal. (1999) 1999:e1. doi: 10.1126/stke.1999.9.pe1
14. Heitzler P. Chapter fourteen–biodiversity and noncanonical notch signaling. In: Kopan R editor. Current Topics in Developmental Biology. Cambridge, MA: Academic Press (2010). p. 457–81. doi: 10.1016/S0070-2153(10)92014-0
15. Perumalsamy LR, Nagala M, Banerjee P, Sarin A. A hierarchical cascade activated by non-canonical Notch signaling and the mTOR–Rictor complex regulates neglect-induced death in mammalian cells. Cell Death Different. (2009) 16:879–89. doi: 10.1038/cdd.2009.20
16. Genz B, Coleman MA, Irvine KM, Kutasovic JR, Miranda M, Gratte FD, et al. Overexpression of miRNA-25-3p inhibits Notch1 signaling and TGF-β-induced collagen expression in hepatic stellate cells. Sci Rep. (2019) 9:8541. doi: 10.1038/s41598-019-44865-1
17. Poellinger L, Lendahl U. Modulating Notch signaling by pathway-intrinsic and pathway-extrinsic mechanisms. Curr Opin Genet Dev Different Gene Regul. (2008) 18:449–54. doi: 10.1016/j.gde.2008.07.013
18. Borggrefe T, Lauth M, Zwijsen A, Huylebroeck D, Oswald F, Giaimo BD. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim Biophys Acta (BBA) Mol Cell Res. (2016) 1863:303–13. doi: 10.1016/j.bbamcr.2015.11.020
19. Krejčí A, Bernard F, Housden BE, Collins S, Bray SJ. Direct response to notch activation: signaling crosstalk and incoherent logic. Sci Signal. (2009) 2:ra1. doi: 10.1126/scisignal.2000140
20. Schmitt TM, Zúñiga-Pflücker JC. Induction of T Cell Development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. (2002) 17:749–56. doi: 10.1016/S1074-7613(02)00474-0
21. Cheng P, Nefedova Y, Corzo CA, Gabrilovich DI. Regulation of dendritic-cell differentiation by bone marrow stroma via different Notch ligands. Blood. (2007) 109:507–15. doi: 10.1182/blood-2006-05-025601
22. Rutz S, Mordmüller B, Sakano S, Scheffold A. Notch ligands Delta-like1, Delta-like4 and Jagged1 differentially regulate activation of peripheral T helper cells. Eur J Immunol. (2005) 35:2443–51. doi: 10.1002/eji.200526294
23. Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. (2004) 117:515–26. doi: 10.1016/S0092-8674(04)00451-9
24. Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4+ T cells toward distinct T−helper cell subsets. Immunol Rev. (2013) 252:12–23. doi: 10.1111/imr.12032
25. Backer RA, Helbig C, Gentek R, Kent A, Laidlaw BJ, Dominguez CX, et al. A central role for Notch in effector CD8+ T cell differentiation. Nat Immunol. (2014) 15:1143–51. doi: 10.1038/ni.3027
26. Amsen D, Gisbergen KPJM, van Hombrink P, van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. (2018) 19:538. doi: 10.1038/s41590-018-0114-2
27. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Notch controls generation and function of human effector CD8+ T cells. Blood. (2013) 121:2638–46. doi: 10.1182/blood-2012-07-442962
28. Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, et al. notch decoys that selectively block DLL/Notch or JAG/Notch disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. (2015) 5:182–97. doi: 10.1158/2159-8290.CD-14-0650
29. Sierra RA, Thevenot P, Raber PL, Cui Y, Parsons C, Ochoa AC. Trillo-Tinoco, J., CD8+ T cells overcomes tumor-induced T-cell suppression and enhances immunotherapy in cancer. Cancer Immunol Res. (2014) 2:800–11. doi: 10.1158/2326-6066.CIR-14-0021
30. Palaga T, Miele L, Golde TE, Osborne BA. TCR-Mediated Notch signaling regulates proliferation and IFN-γ production in peripheral T cells. J Immunol. (2003) 171:3019–24. doi: 10.4049/jimmunol.171.6.3019
31. Mitra A, Shanthalingam S, Sherman HL, Singh K, Canakci M, Torres JA, et al. CD28 signaling drives notch ligand expression on CD4 T cells. Front Immunol. (2020) 11:735. doi: 10.3389/fimmu.2020.00735
32. Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. Roles of pofut1 and O-fucose in mammalian notch signaling. J Biol Chem. (2008) 283:13638–51. doi: 10.1074/jbc.M802027200
33. Haines N, Irvine KD. Glycosylation regulates notch signalling. Nat Rev Mol Cell Biol. (2003) 4:786–97. doi: 10.1038/nrm1228
34. Rampal R, Li ASY, Moloney DJ, Georgiou SA, Luther KB, Nita-Lazar A, et al. Lunatic fringe, manic fringe, and radical fringe recognize similar specificity determinants in o-fucosylated epidermal growth factor-like repeats. J Biol Chem. (2005) 280:42454–63. doi: 10.1074/jbc.M509552200
35. Kakuda S, Haltiwanger RS. Deciphering the fringe-mediated notch code: identification of activating and inhibiting sites allowing discrimination between ligands. Dev Cell. (2017) 40:193–201. doi: 10.1016/j.devcel.2016.12.013
36. Del Castillo V-HM, van der Weyden L, Nsengimana J, Speak AO, Sjöberg MK, Bishop DT, et al. Comparative genomics reveals that loss of lunatic fringe (LFNG) promotes melanoma metastasis. Mol Oncol. (2018) 12:239–55. doi: 10.1002/1878-0261.12161
37. Zhang S, Chung W, Wu G, Egan SE, Xu K. Tumor-suppressive activity of lunatic fringe in prostate through differential modulation of notch receptor activation. Neoplasia. (2014) 16:158–67. doi: 10.1593/neo.131870
38. Chung W-C, Zhang S, Challagundla L, Zhou Y, Xu K. Lunatic fringe and p53 cooperatively suppress mesenchymal stem-like breast cancer. Neoplasia. (2017) 19:885–95. doi: 10.1016/j.neo.2017.08.006
39. López-Arribillaga E, Rodilla V, Colomer C, Vert A, Shelton A, Cheng JH, et al. Manic Fringe deficiency imposes Jagged1 addiction to intestinal tumor cells. Nat Commun. (2018) 9:2992. doi: 10.1038/s41467-018-05385-0
40. Yi F, Amarasinghe B, Dang TP. Manic fringe inhibits tumor growth by suppressing Notch3 degradation in lung cancer. Am J Cancer Res. (2013) 3:490–9.
41. Tsukumo S, Hirose K, Maekawa Y, Kishihara K, Yasutomo K. Lunatic fringe controls T cell differentiation through modulating notch signaling. J Immunol. (2006) 177:8365–71. doi: 10.4049/jimmunol.177.12.8365
42. Yuan JS, Tan JB, Visan I, Matei IR, Urbanellis P, Xu K, et al. Lunatic fringe prolongs delta/notch-induced self-renewal of committed αβ T-cell progenitors. Blood. (2011) 117:1184–95. doi: 10.1182/blood-2010-07-296616
43. Song Y, Kumar V, Wei H-X, Qiu J, Stanley P. Lunatic, manic, and radical fringe each promote T and B cell development. J Immunol. (2016) 196:232–43. doi: 10.4049/jimmunol.1402421
44. Visan I, Tan JB, Yuan JS, Harper JA, Koch U, Guidos CJ. Regulation of T lymphopoiesis by Notch1 and Lunatic fringe–mediated competition for intrathymic niches. Nat Immunol. (2006) 7:634–43. doi: 10.1038/ni1345
45. Guha I, Bhuniya A, Shukla D, Patidar A, Nandi P, Saha A, et al. Tumor arrests DN2 to DN3 Pro T cell transition and promotes its conversion to thymic dendritic cells by reciprocally regulating notch1 and ikaros signaling. Front Immunol. (2020) 11:898. doi: 10.3389/fimmu.2020.00898
46. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. (2003) 101:4878–86. doi: 10.1182/blood-2002-07-1956
47. Lopez DM, Charyulu V, Adkins B. Influence of breast cancer on thymic function in mice. J Mamm Gland Biol Neopl. (2002) 9:191–9. doi: 10.1023/A:1020356020542
48. Gu W, Xu W, Ding T, Guo X. Fringe controls Naïve CD4+T cells differentiation through modulating notch signaling in asthmatic rat models. PLoS One. (2012) 7:e47288. doi: 10.1371/journal.pone.0047288
49. Mukherjee S, Rasky AJ, Lundy PA, Kittan NA, Kunkel SL, Maillard IP, et al. STAT5-induced lunatic fringe during Th2 development alters Delta-like 4-mediated Th2 cytokine production in respiratory syncytial virus-exacerbated airway allergic disease. J Immunol. (2014) 192:996–1003. doi: 10.4049/jimmunol.1301991
50. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. (2003) 19:739–48. doi: 10.1016/S1074-7613(03)00292-9
51. Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol. (2015) 194:21. doi: 10.4049/jimmunol.1401867
52. Delgoffe GM, Murray PJ, Vignali DA. Interpreting mixed signals: the cell’s cytokine conundrum. Curr Opin Immunol. (2011) 23:632–8. doi: 10.1016/j.coi.2011.07.013
53. Verdeil G, Chaix J, Schmitt−Verhulst A-M, Auphan−Anezin N. Temporal cross-talk between TCR and STAT signals for CD8 T cell effector differentiation. Eur J Immunol. (2006) 36:3090–100. doi: 10.1002/eji.200636347
54. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. (2019) 16:634–43. doi: 10.1038/s41423-019-0220-6
55. Ong C-T, Sedy JR, Murphy KM, Kopan R. Notch and presenilin regulate cellular expansion and cytokine secretion but cannot instruct Th1/Th2 fate acquisition. PLoS One. (2008) 3:e2823. doi: 10.1371/journal.pone.0002823
56. Maekawa Y, Tsukumo S, Chiba S, Hirai H, Hayashi Y, Okada H, et al. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. (2003) 19:549–59. doi: 10.1016/S1074-7613(03)00270-X
57. Minter LM, Turley DM, Das P, Shin HM, Joshi I, Lawlor RG, et al. Inhibitors of γ-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat Immunol. (2005) 6:680–8. doi: 10.1038/ni1209x
58. Shin HM, Tilahun ME, Cho OH, Chandiran K, Kuksin CA, Keerthivasan S, et al. NOTCH1 can initiate NF-κB activation via cytosolic interactions with components of the T cell signalosome. Front Immunol. (2014) 5:249. doi: 10.3389/fimmu.2014.00249
59. Moser M, Murphy KM. Dendritic cell regulation of TH1-TH2 development. Nat Immunol. (2000) 1:199–205. doi: 10.1038/79734
60. Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity. (2001) 14:205–15. doi: 10.1016/S1074-7613(01)00103-0
61. Lehar SM, Bevan MJ. Immunology: polarizing a T-cell response. Nature. (2004) 430:150–1. doi: 10.1038/430150a
62. Laky K, Evans S, Perez-Diez A, Fowlkes BJ. Notch signaling regulates antigen sensitivity of naive CD4+ T cells by tuning co-stimulation. Immunity. (2015) 42:80–94. doi: 10.1016/j.immuni.2014.12.027
63. Tindemans I, Peeters MJW, Hendriks RW. Notch signaling in T helper cell subsets: instructor or unbiased amplifier? Front Immunol. (2017) 8:419. doi: 10.3389/fimmu.2017.00419
64. Nandagopal N, Santat LA, LeBon L, Sprinzak D, Bronner ME, Elowitz MB. Dynamic ligand discrimination in the notch signaling pathway. Cell (2018) 172:869–80.e19. doi: 10.1016/j.cell.2018.01.002
65. Yaron A, Sprinzak D. The cis side of juxtacrine signaling: a new role in the development of the nervous system. Trends Neurosci. (2012) 35:230–9. doi: 10.1016/j.tins.2011.12.003
66. LeBon L, Lee TV, Sprinzak D, Jafar-Nejad H, Elowitz MB. Fringe proteins modulate Notch-ligand cis and trans interactions to specify signaling states. Elife. (2014) 3:e02950. doi: 10.7554/eLife.04998
67. Boareto M, Jolly MK, Lu M, Onuchic JN, Clementi C, Ben-Jacob E. Jagged–Delta asymmetry in Notch signaling can give rise to a Sender/Receiver hybrid phenotype. Proc Natl Acad Sci USA. (2015) 112:E402–9. doi: 10.1073/pnas.1416287112
68. Hozumi K. Distinctive properties of the interactions between Notch and Notch ligands. Dev Growth Different. (2020) 62:49–58. doi: 10.1111/dgd.12641
69. Nobili S, Lippi D, Witort E, Donnini M, Bausi L, Mini E, et al. Natural compounds for cancer treatment and prevention. Pharmacol Res. (2009) 59:365–78. doi: 10.1016/j.phrs.2009.01.017
70. Huang C-Y, Ju D-T, Chang C-F, Muralidhar RP, Velmurugan BK. A review on the effects of current chemotherapy drugs and natural agents in treating non–small cell lung cancer. Biomedicine (Taipei). (2017) 7:23. doi: 10.1051/bmdcn/2017070423
71. Blagodatski A, Yatsunskaya M, Mikhailova V, Tiasto V, Kagansky A, Katanaev VL. Medicinal mushrooms as an attractive new source of natural compounds for future cancer therapy. Oncotarget. (2018) 9:29259–74. doi: 10.18632/oncotarget.25660
72. Kopan R, Ilagan MXG. γ-Secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. (2004) 5:499–504. doi: 10.1038/nrm1406
73. Olsauskas-Kuprys R, Zlobin A, Osipo C. Gamma secretase inhibitors of Notch signaling. Onco Targets Ther. (2013) 6:943–55. doi: 10.2147/OTT.S33766
74. de Pooter RF, Zúñiga-Pflücker JC. Generation of immunocompetent T cells from embryonic stem cells. In: Fairchild PJ editor. Immunological Tolerance: Methods and Protocols. Totowa, NJ: Humana Press (2007). p. 73–81. doi: 10.1007/978-1-59745-395-0_5
75. Zhao Y, Parkhurst MR, Zheng Z, Cohen CJ, Riley JP, Gattinoni L, et al. Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoietic stem cells via notch signaling. Cancer Res. (2007) 67:2425–9. doi: 10.1158/0008-5472.CAN-06-3977
76. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. (2005) 115:1616–26. doi: 10.1172/JCI24480
77. Kondo T, Morita R, Okuzono Y, Nakatsukasa H, Sekiya T, Chikuma S, et al. Notch-mediated conversion of activated T cells into stem cell memory-like T cells for adoptive immunotherapy. Nat Commun. (2017) 8:1–14. doi: 10.1038/ncomms15338
78. Mathieu M, Cotta−Grand N, Daudelin J-F, Thébault P, Labrecque N. Notch signaling regulates PD-1 expression during CD8+ T-cell activation. Immunol Cell Biol. (2013) 91:82–8. doi: 10.1038/icb.2012.53
79. Hurlbut GD, Kankel MW, Lake RJ, Artavanis-Tsakonas S. Crossing paths with Notch in the hyper-network. Curr Opin Cell Biol Cell Regulat. (2007) 19:166–75. doi: 10.1016/j.ceb.2007.02.012
80. Steinbuck MP, Arakcheeva K, Winandy S. Novel TCR-Mediated mechanisms of notch activation and signaling. J Immunol. (2018) 200:997–1007. doi: 10.4049/jimmunol.1700070
81. Britton GJ, Ambler R, Clark DJ, Hill EV, Tunbridge HM, McNally KE, et al. PKCθ links proximal T cell and Notch signaling through localized regulation of the actin cytoskeleton. ELife. (2017) 6:e20003. doi: 10.7554/eLife.20003
82. Monsalve E, Pérez MA, Rubio A, Ruiz-Hidalgo MJ, Baladrón V, García-Ramírez JJ, et al. Notch-1 Up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. J Immunol. (2006) 176:5362–73. doi: 10.4049/jimmunol.176.9.5362
83. Leone RD, Lo Y-C, Powell JD. A2aR antagonists: next generation checkpoint blockade for cancer immunotherapy. Comput Struct Biotechnol J. (2015) 13:265–72. doi: 10.1016/j.csbj.2015.03.008
84. Sorrentino C, Hossain F, Rodriguez PC, Sierra RA, Pannuti A, Hatfield S, et al. Adenosine A2A receptor stimulation inhibits TCR-induced notch1 activation in CD8+T-cells. Front Immunol. (2019) 10:162. doi: 10.3389/fimmu.2019.00162
85. Vas V, Szilágyi L, Pálóczi K, Uher F. Soluble Jagged-1 is able to inhibit the function of its multivalent form to induce hematopoietic stem cell self-renewal in a surrogate in vitro assay. J Leuk Biol. (2004) 75:714–20. doi: 10.1189/jlb.1003462
86. Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, et al. A Notch1 ectodomain construct inhibits endothelial notch signaling tumor growth, and angiogenesis. Cancer Res. (2008) 68:4727–35. doi: 10.1158/0008-5472.CAN-07-6499
87. Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. (2009) 157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x
88. Klose R, Berger C, Moll I, Adam MG, Schwarz F, Mohr K, et al. Soluble Notch ligand and receptor peptides act antagonistically during angiogenesis. Cardiovasc Res. (2015) 107:153–63. doi: 10.1093/cvr/cvv151
89. Lin L, Mernaugh R, Yi F, Blum D, Carbone DP, Dang TP. Targeting specific regions of the notch3 ligand-binding domain induces apoptosis and inhibits tumor growth in lung cancer. Cancer Res. (2010) 70:632–8. doi: 10.1158/0008-5472.CAN-09-3293
90. Nickoloff B, Qin J-Z, Chaturvedi V, Denning M, Bonish B, Miele L. Jagged-1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF-kB and PPARg. Cell Death Different. (2020) 14:842–55. doi: 10.1038/sj.cdd.4401036
91. Li L, Milner LA, Deng Y, Iwata M, Banta A, Graf L, et al. The human homolog of Rat jagged1expressed by marrow stroma inhibits differentiation of 32D cells through interaction with Notch1. Immunity. (1998) 8:43–55. doi: 10.1016/S1074-7613(00)80457-4
92. Masuya M, Katayama N, Hoshino N, Nishikawa H, Sakano S, Araki H, et al. The soluble notch ligand, Jagged-1, inhibits proliferation of CD34+ macrophage progenitors. Int J Hematol. (2002) 75:269–76. doi: 10.1007/BF02982040
93. Varnum-Finney B, Wu L, Yu M, Brashem-Stein C, Staats S, Flowers D, et al. Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J Cell Sci. (2000) 113(Pt 23):4313–8.
94. Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. (2003) 60:523–33. doi: 10.1007/s00253-002-1158-6
95. Stanley P, Guidos CJ. Regulation of notch signaling during T- and B-cell development by O-fucose glycans. Immunol Rev. (2009) 230:201–15. doi: 10.1111/j.1600-065X.2009.00791.x
96. Luca VC, Jude KM, Pierce NW, Nachury MV, Fischer S, Garcia KC. Structural basis for Notch1 engagement of Delta-like 4. Science. (2015) 347:847–53. doi: 10.1126/science.1261093
97. Luca VC, Kim BC, Ge C, Kakuda S, Wu D, Roein-Peikar M, et al. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science. (2017) 355:1320–4. doi: 10.1126/science.aaf9739
98. Schneider M, Kumar V, Nordstrøm LU, Feng L, Takeuchi H, Hao H, et al. Inhibition of Delta-induced Notch signaling using fucose analogs. Nat Chem Biol. (2018) 14:65–71. doi: 10.1038/nchembio.2520
99. Driss V, Quesnel B, Brinster C. Monocyte chemoattractant protein 1 (MCP-1/CCL2) contributes to thymus atrophy in acute myeloid leukemia. Eur J Immunol. (2015) 45:396–406. doi: 10.1002/eji.201444736
100. Carrio R, Torroella-Kouri M, Iragavarapu-Charyulu V, Lopez DM. Tumor-induced thymic atrophy: alteration in interferons and Jak/Stats signaling pathways. Int J Oncol. (2011) 38:547–53. doi: 10.3892/ijo.2010.870
101. Wang W, Thomas R, Sizova O, Su D-M. Thymic function associated with cancer development, relapse, and antitumor immunity – a mini-review. Front Immunol. (2020) 11:773. doi: 10.3389/fimmu.2020.00773
102. Nitta T, Nitta S, Lei Y, Lipp M, Takahama Y. CCR7-mediated migration of developing thymocytes to the medulla is essential for negative selection to tissue-restricted antigens. Proc Natl Aacd Sci USA. (2009) 106:17129–33. doi: 10.1073/pnas.0906956106
103. Ueno T, Hara K, Willis MS, Malin MA, Höpken UE, Gray DHD, et al. Role for CCR7 ligands in the emigration of newly generated T lymphocytes from the neonatal thymus. Immunity. (2002) 16:205–18. doi: 10.1016/S1074-7613(02)00267-4
104. Chen ELY, Thompson PK, Zúñiga-Pflücker JC. RBPJ-dependent Notch signaling initiates the T cell program in a subset of thymus-seeding progenitors. Nat Immunol. (2019) 20:1456–68. doi: 10.1038/s41590-019-0518-7
105. Zhou J, Cheng P, Youn J-I, Cotter MJ, Gabrilovich DI. Notch and wnt signaling cooperate in regulation of dendritic cell differentiation. Immunity. (2009) 30:845–59. doi: 10.1016/j.immuni.2009.03.021
106. Mastio J, Simand C, Cova G, Kastner P, Chan S, Kirstetter P. Ikaros cooperates with Notch activation and antagonizes TGFβ signaling to promote pDC development. PLoS Genet. (2018) 14:e1007485. doi: 10.1371/journal.pgen.1007485
107. Dumortier A, Jeannet R, Kirstetter P, Kleinmann E, Sellars M, dos Santos NR, et al. Notch activation is an early and critical event during T-cell leukemogenesis in Ikaros-deficient mice. Mol Cell Biol. (2006) 26:209–20. doi: 10.1128/MCB.26.1.209-220.2006
108. Kleinmann E, Geimer Le Lay A-S, Sellars M, Kastner P, Chan S. Ikaros represses the transcriptional response to notch signaling in T-cell development. Mol Cell Biol. (2008) 28:7465–75. doi: 10.1128/MCB.00715-08
109. Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, et al. Deficient T cell fate specification in mice with an induced inactivation of notch1. Immunity. (1999) 10:547–58. doi: 10.1016/S1074-7613(00)80054-0
110. Fiorini E, Merck E, Wilson A, Ferrero I, Jiang W, Koch U, et al. Dynamic regulation of notch 1 and notch 2 surface expression during T cell development and activation revealed by novel monoclonal antibodies. J Immunol. (2009) 183:7212–22. doi: 10.4049/jimmunol.0902432
111. Kaiserlian D, Savino W, Hassid J, Dardenne M. Studies of the thymus in mice bearing the Lewis lung carcinoma. III. Possible mechanisms of tumor-induced thymic atrophy. Clin Immunol Immunopathol. (1984) 32:316–25. doi: 10.1016/0090-1229(84)90275-7
112. Huang Y, Chen X, Dikov MM, Novitskiy SV, Mosse CA, Yang L, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood. (2007) 110:624–31. doi: 10.1182/blood-2007-01-065714
113. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naïve CD4 + T cells. Nat Immunol. (2002) 3:549–57. doi: 10.1038/ni794
114. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science. (2002) 295:338–42. doi: 10.1126/science.1065543
115. Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4+ cell properties during viral infection. Immunity. (2011) 35:633–46. doi: 10.1016/j.immuni.2011.08.016
116. Kibirova A, Mattes MD, Smolkin M, Ma PC. The journey of an EGFR-mutant lung adenocarcinoma through erlotinib, osimertinib and ABCP immunotherapy regimens: sensitivity and resistance. Case Rep Oncol. (2019) 12:765–76. doi: 10.1159/000503417
117. Santoni-Rugiu E, Melchior LC, Urbanska EM, Jakobsen JN, de Stricker K, Grauslund M, et al. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: differences and similarities with acquired resistance. Cancers (Basel). (2019) 11:923. doi: 10.3390/cancers11070923
118. Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K, et al. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer. (2019) 18:165. doi: 10.1186/s12943-019-1073-4
119. Isomoto K, Haratani K, Hayashi H, Shimizu S, Tomida S, Niwa T, et al. Impact of EGFR-TKI treatment on the tumor immune microenvironment in EGFR mutation–positive non–small cell lung cancer. Clin Cancer Res. (2020) 26:2037–46. doi: 10.1158/1078-0432.CCR-19-2027
120. Ayeni D, Miller B, Kuhlmann A, Ho P-C, Robles-Oteiza C, Gaefele M, et al. Tumor regression mediated by oncogene withdrawal or erlotinib stimulates infiltration of inflammatory immune cells in EGFR mutant lung tumors. J. Immunother Cancer. (2019) 7:172. doi: 10.1186/s40425-019-0643-8
121. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discovery. (2013) 3:1355–63. doi: 10.1158/1535-7163.TARG-13-B290
122. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. New Engl J Med. (2015) 373:1627–39. doi: 10.1056/NEJMoa1507643
123. Hastings K, Yu HA, Wei W, Sanchez-Vega F, DeVeaux M, Choi J, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann Oncol. (2019) 30:1311–20. doi: 10.1093/annonc/mdz141
124. Lee CK, Man J, Lord S, Links M, Gebski V, Mok T, et al. Checkpoint inhibitors in metastatic EGFR-mutated non–small cell lung cancer—a meta-analysis. J Thorac Oncol. (2017) 12:403–7. doi: 10.1016/j.jtho.2016.10.007
125. Andersson ER, Sandberg R, Lendahl U. Notch signaling simplicity in design, versatility in function. Development. (2011) 138:3593–612. doi: 10.1242/dev.063610
126. De La Coste A, Freitas AA. Notch signaling: distinct ligands induce specific signals during lymphocyte development and maturation. Immunol Lett. (2006) 102:1–9. doi: 10.1016/j.imlet.2005.06.014
127. Phng L-K, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. (2009) 16:196–208. doi: 10.1016/j.devcel.2009.01.015
128. Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci USA. (1999) 96:185–90. doi: 10.1073/pnas.96.1.185
129. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
130. Deborah J, Lenschow TL, Walunas JAB. CD28/B7 System Of T Cell Costimulation. Ann Rev Immunol. (1996) 14:233–58. doi: 10.1146/annurev.immunol.14.1.233
131. Hoyne GF, Le Roux I, Corsin-Jimenez M, Tan K, Dunne J, Forsyth LMG, et al. Serrate1-induced Notch signalling regulates the decision between immunity and tolerance made by peripheral CD4+ T cells. Int Immunol. (2000) 12:177–85. doi: 10.1093/intimm/12.2.177
132. Yvon ES, Vigouroux S, Rousseau RF, Biagi E, Amrolia P, Dotti G, et al. Overexpression of the Notch ligand, Jagged-1, induces alloantigen-specific human regulatory T cells. Blood. (2003) 102:3815–21. doi: 10.1182/blood-2002-12-3826
133. Vigouroux S, Yvon E, Wagner H-J, Biagi E, Dotti G, Sili U, et al. Induction of antigen-specific regulatory T cells following overexpression of a notch ligand by human B lymphocytes. J Virol. (2003) 77:10872–80. doi: 10.1128/JVI.77.20.10872-10880.2003
134. Gopisetty A, Bhattacharya P, Haddad C, Bruno JC, Vasu C, Miele L, et al. OX40L/Jagged1 cosignaling by GM-CSF–induced bone marrow-derived dendritic cells is required for the expansion of functional regulatory T cells. J Immunol. (2013) 190:5516–25. doi: 10.4049/jimmunol.1202298
135. Cahill EF, Tobin LM, Carty F, Mahon BP, English K. Jagged-1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Therapy. (2015) 6:19. doi: 10.1186/s13287-015-0021-5
136. Lin Y, Chen W, Li J, Yan G, Li C, Jin N, et al. Overexpression of Jagged-1 combined with blockade of CD40 pathway prolongs allograft survival. Immunol Cell Biol. (2015) 93:213–7. doi: 10.1038/icb.2014.84
137. Hansson EM, Lanner F, Das D, Mutvei A, Marklund U, Ericson J, et al. Control of Notch-ligand endocytosis by ligand-receptor interaction. J Cell Sci. (2010) 123:2931–42. doi: 10.1242/jcs.073239
138. Parks AL, Klueg KM, Stout JR, Muskavitch MA. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. (2000) 127:1373–85.
139. Charbonnier L-M, Wang S, Georgiev P, Sefik E, Chatila TA. Control of peripheral tolerance by regulatory T cell–intrinsic Notch signaling. Nat Immunol. (2015) 16:1162–73. doi: 10.1038/ni.3288
140. Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, et al. Evaluation of selective γ-secretase inhibitor PF-03084014 for Its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. (2010) 9:1618–28. doi: 10.1158/1535-7163.MCT-10-0034
141. Hayashi I, Takatori S, Urano Y, Miyake Y, Takagi J, Sakata-Yanagimoto M, et al. Neutralization of the γ-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene. (2012) 31:787–98. doi: 10.1038/onc.2011.265
142. Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Direct inhibition of the Notch transcription factor complex. Nature. (2009) 462:182–8. doi: 10.1038/nature08543
143. KleinJan A, Tindemans I, Montgomery JE, Lukkes M, de Bruijn MJW, van Nimwegen M, et al. The Notch pathway inhibitor stapled α-helical peptide derived from mastermind-like 1 (SAHM1) abrogates the hallmarks of allergic asthma. J Aller Clin Immunol. (2018) 142:76–85.e8. doi: 10.1016/j.jaci.2017.08.042
144. Pietanza MC, Spira AI, Jotte RM, Gadgeel SM, Mita AC, Hart LL, et al. Final results of phase Ib of tarextumab (TRXT, OMP-59R5, anti-Notch2/3) in combination with etoposide and platinum (EP) in patients (pts) with untreated extensive-stage small-cell lung cancer (ED-SCLC). JCO. (2015) 33:7508–7508. doi: 10.1200/jco.2015.33.15_suppl.7508
145. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. (2010) 464:1052–7. doi: 10.1038/nature08878
146. Smith DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, et al. A Phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (Anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res. (2014) 20:6295–303. doi: 10.1158/1078-0432.CCR-14-1373
147. Chiorean EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA, et al. A Phase I first-in-human study of enoticumab (REGN421), a fully human delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res. (2015) 21:2695–703. doi: 10.1158/1078-0432.CCR-14-2797
Keywords: Notch signaling, Notch therapeutics, engineered Notch ligands, cancer immunotherapy, immunosurveillance, T cells, antigen presenting cells, tumor escape
Citation: Goruganthu MUL, Shanker A, Dikov MM and Carbone DP (2020) Specific Targeting of Notch Ligand-Receptor Interactions to Modulate Immune Responses: A Review of Clinical and Preclinical Findings. Front. Immunol. 11:1958. doi: 10.3389/fimmu.2020.01958
Received: 28 April 2020; Accepted: 20 July 2020;
Published: 14 August 2020.
Edited by:
Virginie Lafont, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Alessandro Poggi, San Martino Hospital (IRCCS), ItalyArya Biragyn, National Institutes of Health (NIH), United States
Copyright © 2020 Goruganthu, Shanker, Dikov and Carbone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mikhail M. Dikov, bWlraGFpbC5kaWtvdkBvc3VtYy5lZHU=