 Coen H. Wiegman
Coen H. Wiegman Feng Li
Feng Li Bernhard Ryffel
Bernhard Ryffel Dieudonnée Togbe
Dieudonnée Togbe Kian Fan Chung
Kian Fan Chung
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 September 2020
Sec. Inflammation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01957
This article is part of the Research Topic Ozone as a Driver of Lung Inflammation and Innate Immunity, and as a Model for Lung Disease View all 10 articles
Oxidative stress plays an important role in the pathogenesis of chronic obstructive pulmonary disease (COPD) caused by cigarette smoke and characterized by chronic inflammation, alveolar destruction (emphysema) and bronchiolar obstruction. Ozone is a gaseous constituent of urban air pollution resulting from photochemical interaction of air pollutants such as nitrogen oxide and organic compounds. While acute exposure to ozone induces airway hyperreactivity and neutrophilic inflammation, chronic ozone exposure in mice causes activation of oxidative pathways resulting in cell death and a chronic bronchial inflammation with emphysema, mimicking cigarette smoke-induced COPD. Therefore, the chronic exposure to ozone has become a model for studying COPD. We review recent data on mechanisms of ozone induced lung disease focusing on pathways causing chronic respiratory epithelial cell injury, cell death, alveolar destruction, and tissue remodeling associated with the development of chronic inflammation and AHR. The initial oxidant insult may result from direct effects on the integrity of membranes and organelles of exposed epithelial cells in the airways causing a stress response with the release of mitochondrial reactive oxygen species (ROS), DNA, and proteases. Mitochondrial ROS and mitochondrial DNA activate NLRP3 inflammasome and the DNA sensors cGAS and STING accelerating cell death pathways including caspases with inflammation enhancing alveolar septa destruction, remodeling, and fibrosis. Inhibitors of mitochondrial ROS, NLRP3 inflammasome, DNA sensor, cell death pathways, and IL-1 represent novel therapeutic targets for chronic airways diseases underlined by oxidative stress.
Ozone is a gaseous constituent of urban air pollution that is generated by interaction of rising constituents of air pollution such as nitrogen oxide and organic compounds, induced by sunlight. Early studies have documented the effects of a short acute exposure to ozone in inducing airway inflammation and bronchial hyperreactivity in humans and in various species including mice, rats, guinea pigs, and dogs, at levels of ozone that were much higher than those measured in a highly polluted traffic-dense environment during the summer months.
More than 92% of the world's population are regularly exposed to unhealthy levels of ozone and among the world's 11 most populous countries, population-weighted seasonal ozone concentrations range from about 45 ppb in Brazil to 68 ppb in China. In Europe, the average ozone levels are reported to be highest in Italy with an annual average of 65 ppb (https://www.eea.europa.eu/publications/air-quality-in-europe-2019). Ozone concentration EU targets for the protection of human health has been set not to exceed 60 ppb but these targets are very often exceeded in several EU countries (Table 1). The European Air Quality Report highlights that 17 EU member states and six other reporting countries registered concentrations of ozone above the target value more than 25 times.
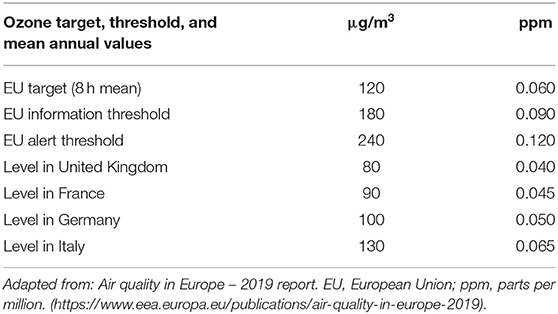
Table 1. Mean ozone concentration targets and thresholds in Europe.
Over the recent years, there has been growing evidence from epidemiological and human exposure studies of the detrimental effects of ozone on respiratory health. Thus, short-term changes in ozone levels have been associated with increased mortality (1, 2), and a positive association between ozone and hospital admissions for asthma and COPD in the elderly and between ozone levels and asthma emergency visits in children (3, 4). Indeed, more recent studies have linked long-term exposure to ozone with reduced lung function and an increasing risk of developing emphysema irrespective of being a cigarette smoker (5, 6).
The effects of ozone on inflammation have already been reviewed although these have usually looked at mainly acute exposures (7). One of the long-term interests of the effect of ozone has been to determine whether exposure to ozone can represent a good model for airways disease. Acute exposure to ozone in rats or mice and later in humans have been used as a model of bronchial hyperresponsiveness underlined by a neutrophilic inflammation, the mechanisms of this effect having been reviewed (7). However, it is the chronic exposure to ozone that led to a model of emphysema and chronic airway inflammation that has been of the most interest in recent years (8). This represented the most direct proof that oxidative stress could induce features similar to that seen in the condition labeled as chronic obstructive pulmonary disease (COPD).
In this review, we will consider by what mechanisms this oxidant stress can lead to chronic bronchial inflammation and lung destruction of emphysema, and how models of oxidative stress such as exposure to ozone particularly on a long term basis can inform us on the nature of COPD and can be used to examine potentially new treatments.
COPD is defined as a common, preventable, and treatable disease that is characterized by persistent respiratory symptoms and airflow obstruction that is due to airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases (9, 10). This is a disease with high degree of morbidity and mortality throughout the world. While initially, this was considered to be a disease mainly found or caused by cigarette smoking, it is now agreed that exposure to biomass fuel and increasingly to environmental pollution can also contribute to the pathogenesis of COPD (11).
Some of the pathological features of smoking-induced COPD consist of severe airflow obstruction which is associated partly to the inflammation in the small airways particularly by neutrophils, macrophages, and lymphocytes (12). In addition, there is airway wall remodeling of the small airways, which involves all the components of the airway wall including epithelium, lamina propria, and airway smooth muscle (13, 14). Finally, there is the presence of emphysema, with the destruction of alveolar airspaces, which has been attributed to an imbalance of protease and anti-protease activity with enhanced protease activity associated with neutrophil activation (15). Airway hyperreactivity (AHR) prevalence and pathology in COPD was found to be present in one out of two COPD patients with its presence influenced by decline in lung function and smoking status (16). As such, AHR can be seen as an independent predictor and a contributing factor to COPD development.
Various pathological pathways have been proposed including increased apoptosis mechanisms (17, 18) in addition to inflammageing and autoimmunity (19). There is growing evidence that COPD may represent a cellular senescence program associated with secretion of pro-inflammatory cytokines that cause chronic inflammation, that leads to the increased levels of reactive oxygen species that can induce oxidative stress (20). Oxidative stress mechanisms can also lead to activation of pro-inflammatory pathways in addition to causing DNA damage (21).
An increasing body of evidence has been accumulating linking COPD with the process of autoimmunity, with the presence of autoantibodies in the sera of COPD patients, and with some of the antibodies correlating with particular disease phenotypes (19). Thus, it has been proposed that autoimmunity may play an important role in the pathogenesis of COPD. Interestingly, ozone-exposed mice for 6 weeks exhibited increased antibody titres to carbonyl-modified protein, as well as activated antigen-presenting cells in lung tissue and splenocytes sensitized to activation of carbonyl-modified protein (22). This is evidence of oxidative stress induced antibodies supporting an auto-immune component, as has been described in COPD with the elevation of carbonyl-modified self-protein that correlated with the severity of disease. Cigarette smoke models have also supported this effect of ozone as has been shown in matrix metalloproteinase 12 (MMP12)-generated elastin fragments serving as a self-antigen and driving the cigarette smoke-induced autoimmune processes in mice that result in a bronchitis-like phenotype and airspace enlargement (23). These elastin autoantibodies and others have been described in advanced COPD (24). The fact that autoantibodies in animal models of COPD such as after ozone exposure are capable of inducing a COPD-like disease phenotype indicate an autoimmune mechanisms perhaps involving B cells, plasma cells, and B cell-rich lymphoid follicles that are present in COPD (25).
It is quite clear that ozone reacts with cellular membranes and with the epithelial lung lining fluid to generate bioactive mediators that cause oxidative stress, innate immune responses, and signaling (26, 27). When inhaled, there is direct contact between ozone and the first level of cells in the airway surface such as airway and alveolar epithelial cells, and airway macrophages. These cells release reactive oxygen species and various other inflammatory mediators including cytokines and lipids from oxidative damage to the airway epithelium (28, 29). In addition, ozone exposure has been shown to impair macrophage phagocytic and efferocytosis function (28, 30) which can cause prolonged injury and inflammation. The mechanisms underlying these changes have been previously reviewed in terms of the transcriptional effects of ozone in the lungs and airways (31, 32).
The increased oxidative stress as a direct effect of ozone is often investigated in relation to cellular dysfunction. Ozone affects several intracellular pathways in different cell types, but not always in the same way. Ozone exposure in cultured alveolar epithelial cells results in cytotoxicity but does not always affect the production of cytokines (33). However, when conditioned medium from ozone-exposed alveolar macrophages is added to alveolar epithelial cells, the cytokines chemokine (C-X-C) ligand 1 (CXCL1) and C-C motif chemokine ligand 2 (CCL2) were induced, mediated through interleukin-1α (IL-1α) (34). On the other hand, it has been reported that, although human alveolar macrophages are much more sensitive to ozone than epithelial cells, they do not produce increased amounts of IL-6, IL-8, or fibronectin following ozone exposure. This increased sensitivity of alveolar macrophages was observed in the form of increased cell death with a third of the cells lost after a low ozone exposure dose. However, whether macrophages are more sensitive to ozone in vivo is not known, but bronchial airway epithelial cells produce substantially more of all three proteins following ozone exposure, and both IL-6 and fibronectin are secreted vectorially toward the apical side at least in the first 4 and 24 h after ozone exposure (29). Ozone exposure has been shown to induce the expression of intracellular adhesion molecule-1 (ICAM-1) and neutrophil adhesion to human airway epithelial cells, which is mediated through the Canonical Transient Receptor Potential Channel 6 (TRPC6) (35). TRPC6 also impacts neutrophilic inflammation by nuclear factor-κB (NF-κB) activation and therefore promoting transcription of inflammatory mediators (35). Therefore, cellular interactions such as those between alveolar macrophages and, airway and alveolar epithelial cells which are the initial airway cells encountered by ozone may be important in the induction of its effects. The role of TRPC6 during oxidative stress conditions has been investigated in several cell types. Oxidative stress induces TRPC6 expression and function in podocytes (36), HEK293T cells (37), vascular myocytes (38), neutrophils (35), and macrophages (39). In a recent study, TRPC6 in bronchial epithelium cells, was shown to act as an oxidative stress sensor where the TRPC6-mediated calcium cascade leads to the activation of the extracellular signal-regulated kinase (ERK) pathway and inflammation (40). This could be a possible inflammatory response pathway in several cell types in response to ozone, but this has yet to be described in COPD.
Ozone exposure activates several types of innate immune cells which play a role in both the Th1 and Th2 inflammatory responses. While the role of neutrophils as part of the Th1 response is well-established, other innate cell such NK cells (41) and innate lymphoid type 2 (ILC2) cells (42) may also contribute. Surfactant protein D released by exposure to ozone has been shown to stimulate interferon (IFN)-γ by NK cells to initiate IFN-γ, IL-12 feedback circuit (41). Continuous exposure to ozone has been shown to result in the conversion of an initial neutrophilic inflammation to an eosinophilic inflammation in the nasal mucosa of mice (43). This was accompanied by an overexpression of type-2 cytokines in the nasal epithelium. The role of ILC2 cells was confirmed by showing that the epithelial-derived alarmins, IL-33, IL-25, and thymic stromal lymphopoietin (TSLP) that were increased in the nasal epithelium was dependent on ILC-2 cells, but not on adaptive T or B lymphoid cells (44). The involvement of ILC2 cells is also supported by the observation that IL-13, a product of such cells, can augment the effects of single ozone exposure, namely AHR and inflammation (45). In addition, ovalbumin sensitization and challenge can also augment ozone exposure effects (46).
Several intracellular pathways are involved in the effects of ozone exposure. Prolonged exposure to ozone induced gene expression levels of several hypoxia inducible factor-1α (HIF-1α) target genes including histone deacetylase 2 (HDAC2), vascular endothelial growth factor (VEGF), Kelch-like ECH-associated protein 1 (Keap1), and Macrophage migration inhibitory factor (MIF), in addition to a decrease in the antioxidative stress response as indicated by an increase in nuclear erythroid-related factor 2 (Nrf2) activity and protein level (47). The Toll like receptors (TLRs) and downstream adaptor proteins myeloid differentiation marker 88 (MyD88) and toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) appear to be involved in the inflammation response to ozone (48, 49). Ozone is able to induce TLR4 signaling through MyD88 in normal Tlr4 expressing C3H/HeOuJ mice. This in contrast to C3H/HeJ mice that express a defective dominant negative mutant Tlr4 gene (50). TLR4 can be activated by ligand binding and subsequently recruits adapter proteins including MyD88. The MyD88-dependant pathway signals through several signal transduction pathways including MAPK, NF-κB, and AP-1 to induce cytokine gene expression contributing to the inflammation response toward ozone. Heath shock protein 70 (HSP70) was identified as a downstream mediator to have a role the TLR4 mediated effects by ozone (50). The ozone-induced oxidative stress response might be involved in the activation of the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome complex (51). Both the acute and chronic exposure of mice to ozone induces lung inflammation through activation of ROS and of the NLRP3 inflammasome with caspase activation (52, 53) and release of the highly inflammatory mediator IL-1β, but also IL-1α (54), which are involved in the ozone injury response (55). Uric acid crystals cause NLRP3-dependent lung inflammation upon injury (56) and therefore it is not excluded that crystals formed during injury may contribute to ozone induced inflammation (57).
How this cytoplasmic multiprotein complex is activated upon ozone exposure remains to be determined. One of the mechanisms might involve reactive oxygen species (ROS) and oxidative stress induced danger signals as reviewed recently (58). The transcriptional effects of ozone and the subsequent impact on airway inflammation have recently been reviewed in an article in this series (31).
Ozone reacts with cellular membranes and with the epithelial lung lining fluid to generate bioactive mediators that cause oxidative stress, innate immune responses, and signaling (26, 27). Ozone-induced ROS at the cell membrane influences membrane integrity with the disruption of tight junction, cell stress, and death with leak of the respiratory barrier within 1–2 h. This first phase is independent of cellular ROS, inflammatory mediators, and inflammatory cells (58). These effects might be the result of ozone exposure induced changes in the expression of claudin (CLDN) proteins which are protein components of tight junctions. In mice exposed to ozone an increased expression levels of CLDN3 and CLDN4 while a reduction in CLDN14 was reported (59). These workers also showed that bronchial epithelial cell integrity was disrupted and that the trans-electrical resistance between cells was decreased leading to cell disintegration. These changes were associated with elevated ROS and increased expression of antioxidant defense system involving Nrf2 and Keap1 (59). Another potential mechanism is lipid peroxidation, which is induced by ozone in human alveolar epithelial cells (60) and in lung surfactant (61). Phospholipids and cholesterol in cell membranes and surfactant can react with ozone to form cytotoxic products which activate second messenger systems involving free arachidonic acid (62), platelet-activating factor (PAF) (63) and prostaglandin E2 (PGE2) (64). Ozone exposure induced changes to cell membrane integrity might involve several pathways, dependent on dose and duration of exposure.
The acute respiratory barrier disruption by ozone has been recently reviewed in an accompanying review in this series (65). This first phase is followed by a robust barrier injury with cell death, protein leak, and influx of ROS expressing myeloid cells including neutrophils, IL-1α and IL-33 production by epithelial and myeloid cells within 6–12 h. Thus, a biphasic response is observed, an immediate direct membrane damage by ROS and a second phase mediated by myeloid cells, which aggravates the damage of the epithelium and inflammation. In support of this hypothesis is the fact that neutrophil depletion attenuates the second phase (58). Oxidative stress peaks at ~18 h after a single ozone exposure during the second phase with increased ROS positive neutrophils and epithelial cells (66). This increase in ROS subsequently causes cell injury, mitochondrial dysfunction, formation, and release of toxic metabolites and even DNA damage. These events have been associated with and contribute to the development of tissue destruction leading to emphysema and lung remodeling.
The toxic effect of ozone was also observed in the distal part of the lung, the alveoli, with death of alveolar epithelial cells with defective repair resulting in enlarged air spaces and emphysema (8). Emphysema upon chronic ozone exposure is commonly found in mice and is dependent on ROS-dependent inflammation (8), but this does not involve IL-17 (67).
Understanding the initial physico-chemical and molecular events of ozone-induced cell membrane injury in vitro and in vivo is an area of intense research leading to cell death will be important. Inflammation and cell death are also associated with neutrophil extracellular trap (NET) formation with chromatin externalization (68) and release of extracellular DNA, which are inflammatory activating the DNA sensing pathways cGAS/STING as reviewed for lung inflammation (69). Aerosol particles such as silica and likely others including ozone cause extracellular release of nuclear and mitochondrial DNA from dying cells, which drive cGAS/STING-type I interferon dependent inflammation, which is inhibited by DNase administration or blockade of STING (70).
In ozone-exposed mice, ROS-induced cell-death was dependent on interactions of Keap1, the cellular ROS sensor and the phosphatase phosphoglycerate mutase family member 5 (PGAM5) with antioxidant function and apoptosis inducing factor mitochondria associated 1 (AIFM1), a pro-apoptotic factor. At high ROS concentration PGAM5 is released from the complex and activates the pro-apoptotic factor AIFM1 inducing apoptotic pathway resulting in a new form of cell death known as oxeiptosis (71–73). However, the contribution of the different cell death pathways such as necroptosis, apoptosis, and efferocytosis by ozone is yet resolved (74). These cell death pathways are likely to be responsible for the emphysematous process seen on oxidant-induced emphysema.
Ozone exposure in ex vivo and in vivo experimental models has focussed recently on the mitochondrial effects of ozone. Mitochondria are double membrane bound organelles that exist in most eukaryotic organisms. Mitochondria play an important role in production of adenosine triphosphate (ATP) and mitochondrial ROS production (75). Dysfunctional mitochondria influence airway contractility, gene expression, oxidative stress, cell proliferation, apoptosis and metabolism, and immune and inflammatory responses that are all implicated in airway diseases including COPD (76).
The toxic effects of ozone on mitochondria are closely dependent upon the dose and duration of ozone exposure and age of animals (Table 2). Short-term ozone exposure (2 ppm for 8 h or 4 ppm for 4 h) to adult rats caused decreases in mitochondrial O2 consumption, mitochondrial ATP synthesis and oxidation of thiol groups in mitochondria in lung, and an increase in lung mitochondrial permeability (77). In vitro mitochondrial respiration in lung homogenates and in lung mitochondria from adult rats (2–3 months) was decreased after acute ozone exposure (15 ppm, 20 min). Subacute ozone exposure (0.8 ppm, for 10–20 days) to adult rats led to increased mitochondrial O2 consumption in lung homogenates, increased mitochondrial number and mitochondrial respiratory activity in alveolar type II cells in the lung (81). Acute ozone exposure (3 ppm for 8 h) in both adult (4–6 months) and old rats (24–26 months) decreased mitochondrial respiration and mitochondrial O2 consumption in isolated lung mitochondria (78). In young (3 weeks) and adult rats (6 months), a more chronic ozone exposure (0.5 ppm, 12 h, 7 days) did not affect lung mitochondrial O2 consumption. However, in aged rats (20 months), ozone increased the mitochondrial O2 consumption and H2O2 release, which means increased mitochondrial ROS activity (80). This indicates that ozone may accelerate senescence process in the elderly.
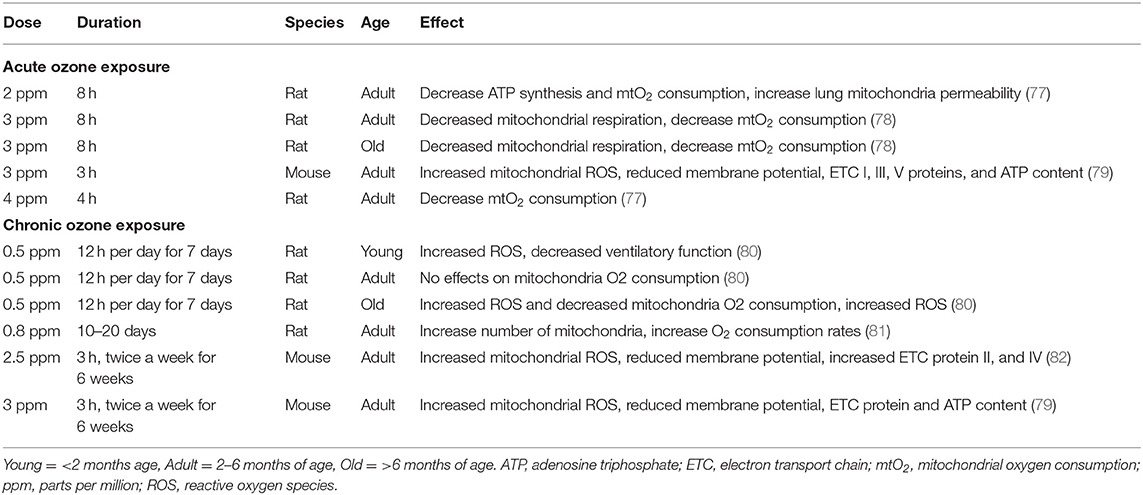
Table 2. Dose, duration, and effect of ozone exposures at different ages of rodents.
In mice, a single ozone exposure (3 ppm for 3 h) resulted in enhanced cellular and mitochondrial ROS levels and a reduced mitochondrial membrane potential (ΔΨm) in lung. (83). Upon acute and chronic ozone exposures, increased mitochondrial ROS levels, decreased ATP content, decreased electron transport chain (ETC) complex I enzyme activity, and reduced expression of ETC complex I, complex III, and complex V in the lungs of 1- and 6-week ozone-exposed mice (3 ppm, 3 h, twice a week) have been observed. Furthermore, ozone-induced inflammation, airway hyperreactivity, mitochondrial dysfunction, and ROS levels were reduced when mice were administered the mitochondrial antioxidant MitoQ (79). In addition, ozone induced inflammation, increased mitochondrial ROS, and expression of ETC complex II and IV in lung mitochondria in 6-week ozone exposed mice (2.5 ppm, 3 h, twice a week) was reduced by treatment with MitoTEMPO, another mitochondria-targeting antioxidant (82). Therefore, targeting the mitochondrial dysfunction to prevent or treat the ozone-induced associated inflammation, airway hyperresponsiveness, and oxidative stress could be a promising treatment strategy.
AHR is a hallmark of ozone effects on the airways (66, 67) and is exacerbated in allergen-sensitized mice (84–86). AHR is due to a direct effect of ROS on bronchial smooth muscle cells (79), but also neuronal cells (87). The effect of ROS on cell membrane integrity is likely due to a direct disruption of the cell membrane structure which needs further studies (88). This is further supported by the effect of N-acetylcysteine (NAC) in reversing established AHR after chronic exposure to ozone (89). Studies of the airway smooth muscle from mice exposed to ozone showed that the hyperresponsiveness to cholinergic contractile agents can be reproduced in vitro and that this oxidant stress-induced hyperresponsiveness was dependent on the activation of the p38 mitogen-activated kinase (MAPK) and inhibited by corticosteroids (52). The AHR induced by chronic ozone exposure was also dependent on IL-17 (67), as was also shown by Pichavant et al. who reported that NK cells producing IL-17 was important for the maintenance of ozone-induced AHR (86). The pulmonary inflammation induced by subacute ozone exposure has been reported to require γδ T cells and tumor necrosis factor-α (TNFα)-dependent recruitment of IL-17A + γδ T cells to the lung (90).
Several studies have investigated the effects of novel treatment strategies relating to the effects of ozone exposure in animal models (Table 3). Targeting inflammation, oxidative stress, and mitochondria could be part of future treatment strategies for COPD.
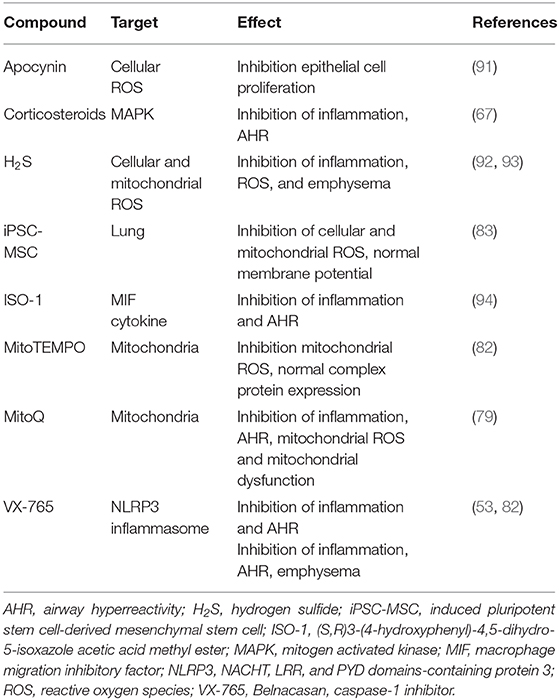
Table 3. Effect of different treatment strategies in ozone-exposed animal models.
Corticosteroids can attenuate the single exposure effects of ozone including AHR and lung inflammation (95–97). This includes the inhibition of expression of macrophage inflammatory protein 2 (MIP-2), inducible nitric oxidase synthase (iNOS) (98, 99) and NFκB (96), and the increased proliferation of the airway epithelium (91). However, chronic exposure to ozone itself can induce a state of corticosteroid insensitivity similar to what has been observed in COPD with reduced or little effect in preventing AHR, inflammation, and emphysema (94, 100). Corticosteroid insensitivity occurs in COPD through mechanisms induced by reactive oxygen species (101, 102). Several such mechanisms have been postulated, including a reduction in HDAC2 activity and expression, impaired corticosteroid activation of the glucocorticoid receptor (GR) and increased pro-inflammatory signaling pathways. Therefore, the chronic model of ozone exposure represents a good model of COPD to study the mechanisms of corticosteroid insensitivity.
Macrophage migration inhibitory factor (MIF) has been implicated as a driver of inflammation in COPD, and possibly as a driver of corticoidsteroid insensitivity in COPD. Using (S,R)3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1) which inhibits MIF tautomerase activity, the corticosteroid-insensitive lung inflammation and AHR after chronic ozone exposure was blocked (94). Thus, inhibition of MIF which is elevated in COPD may provide a novel anti-inflammatory approach in COPD. However, the contribution of mitogen-activated protein kinase phosphatase-1 (MKP-1) that has been proposed to underlie CS insensitivity in COPD was found to be negligible in the chronic ozone model (100). Single ozone exposure aggravated airway inflammation, airway remodeling, activation of p38 MAPK, and downregulation of MKP-1 in ovalbumin (OVA)-sensitized and -challenged mice, an effect that was ineffectively controlled by corticosteroids (46), this also supported the role of p38 MAPK activation as a likely pathway involved in corticosteroid insensitivity (103).
Hydrogen sulfide (H2S), a metabolic product of methionine, is synthesized from L-cysteine primarily by three key enzymes: cystathionine-c-lyase (CGL), cystathionine-b-synthetase (CBS) and 3-mercaptypyruvate sulfurtransferase (MPST). Identified as the third gasotransmitter, along with nitric oxide and carbon monoxide, H2S modulates a variety of physiological functions including anti-oxidative stress, anti-senescence/aging, and anti-apoptotic effects (104–106). H2S has been proposed as serving as a potent antioxidant through reactive oxygen species/reactive nitrogen species scavenging, or through post-translational modification of proteins by addition of a thiol (-SH) group onto reactive cysteine residues (107). In the lungs, H2S suppresses the airway smooth muscle proliferation and cytokine release, an effect that is less effective in muscle from COPD patients (108).
In addition, H2S content is reduced in the lungs of smokers and COPD patients (109), and H2S attenuates nicotine-induced endoplasmic reticulum stress and apoptosis in bronchial epithelial 16HBE cells (110). H2S is able to prevent and treat the development of inflammation, AHR, and oxidative stress in acute ozone-exposed mice (92). In addition, the ozone-induced increase in p38 MAK signaling was reduced in mice treated with H2S indicating that this intracellular signaling pathway might be involved (92). In chronic ozone-exposed mice, H2S is also able to prevent the inflammation, AHR, and remodeling of the lung but is not able to reverse these hallmarks of the model (93). Induction of p38 MAPK signaling is also reversed in both treatment strategies in the chronic ozone exposure model. In addition to this, the activation of the NLRP3 inflammasome and the ratio between cleaved caspase-1 to pro-caspase-1 were positively correlated with changes in lung function parameters and structural changes in the lung. H2S was able to prevent and treat the changes observed in NLRP3 activation (93). Taken together, the main difference between the acute and chronic ozone exposure models on the treatment effects of H2S on the ozone-induced changes suggests that H2S treatment only affects the damage inducing pathways (oxidative stress) and not the regenerative pathways of the lung (92, 93). It is of interest that H2S donor NaHS significantly inhibits cigarette smoke-induced mitochondrial dysfunction, oxidative damage, cell senescence, and apoptosis in alveolar epithelial A549 cells (111). These findings provide novel mechanisms underlying the protection of H2S against ozone and cigarette smoke-induced COPD and suggest that H2S donors targeted toward mitochondria may be beneficial in the treatment of COPD.
In addition to H2S, Belnacasan or VX-765 inhibits NLRP3 inflammasome activation effects by inhibiting caspase-1. In acute ozone exposed mice VX-765 is able to prevent bronchoalveolar lavage (BAL) inflammatory markers, and AHR. Mitochondrial oxidative stress was reduced and this was associated with lower expression levels of dynamin-related protein 1 (DRP1) and mitochondrial fission factor (MFF), and increased expression of Mitofusin 2 (MFN2) proteins involved in mitochondrial fission and fusion, respectively (53). Similar effects were observed in chronic ozone exposed mice were VX-765 is able to prevent inflammation, emphysema, airway remodeling, and oxidative stress while it decreased the expression of the fission protein DRP1 and MFF with affecting proteins involved in fusion dynamics. (82). Mitochondrial oxidative stress and NLRP3 inflammasome were driving ozone-induced inflammation processes and targeting these specifically might have therapeutic value in COPD.
Apocynin, reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor decreased the proliferation in bronchial epithelium after an acute exposure to ozone but not the inflammation (91). Ozone-induced inflammation, airway hyperreactivity, mitochondrial dysfunction, and ROS levels were reduced when chronically-exposed mice were pre-administered the mitochondrial directed antioxidant, MitoQ (79). Similarly, ozone-induced inflammation, increased mitochondrial ROS, and expression of ETC complex II and IV in lung mitochondria in 6-week ozone exposed mice was reduced by treatment with mitoTEMPO, another mitochondria-targeting antioxidant (82). However, airway remodeling and airflow obstruction were not (82). Similarly, in single exposure to ozone, mitoTEMPO inhibited mitochondrial ROS without affecting inflammation and bronchial hyperresponsiveness (53).
Mucolytic/antioxidant agents such as erdosteine, carbocysteine, and NAC reduced the risk of acute exacerbations in patients with COPD (112). In the chronic ozone exposure model, preventive NAC reduced the number of BAL macrophages and airway smooth muscle (ASM) mass while therapeutic NAC reversed AHR, and reduced ASM mass and apoptotic cells (89). Thus, NAC could represent a treatment for protecting against the oxidative effects of ozone and other pollutants, as well as an agent for reducing exacerbations of COPD.
As documented above, several intracellular pathways have been implicated in the effects of single or multiple exposures to ozone in the mouse. These pathways are related to the control of several key transcription regulatory factors including NF-κB, antioxidant factors such as Nrf2, the p38 MAPK, and priming of the immune system by up-regulating toll-like receptor expression. Thus, in the single ozone exposure model, AHR and inflammation was inhibited by a c-jun NH2 terminal kinase (JNK) inhibitor (SP600125) (113), p38 MAPK inhibitor (SD282) (46, 114) and NF-κB inhibitor (115). VX-765, an inhibitor of NLRP3 inflammasome, prevented lung inflammation and AHR caused by acute exposure to ozone (53), and also inhibited lung inflammation and emphysema from chronic exposure (82).
Mitochondrial transfer from induced pluripotent stem cell-derived mesenchymal stem cell (iPSC-MSC) offered protection against oxidative stress-induced mitochondrial dysfunction in human airway smooth muscle cells (ASMC) and in mouse lungs exposed to ozone while reducing airway inflammation and hyperresponsiveness (83). Direct co-culture of ASMCs with iPSC-MSCs protected the former from cigarette smoke-induced mitochondrial ROS production, mitochondrial depolarization, and apoptosis. When ASMCs were exposed to supernatants from iPSC-MSCs or transwell inserts with iPSC-MSCs, only cigarette smoke-induced mitochondrial ROS, but not mitochondrial depolarization and apoptosis in ASMCs, were improved, indicating that soluble factors from iPSC-MSCs reduced production of mitochondrial ROS. When there was direct contact between iPSC-MSCs and ASMCs, mitochondria were transferred from iPSC-MSCs to ASMCs, possibly through formation of tunneling nanotubes, an effect that was enhanced by cigarette smoke medium (CSM) treatment. iPSC-MSCs prevented, but did not reverse, ozone-induced mitochondrial dysfunction, AHR, and airway inflammation in the mouse model of single ozone exposure, an effect resulting from direct interaction and mitochondrial transfer between iPSC-MSCs and airway cells. Therefore, transfer of mitochondria from IPSC-MSC cells to replace damaged mitochondria by oxidative stress may present a novel approach to treating conditions such as COPD.
As an important component of air pollution, ozone has been closely related to the development of COPD. The link comes from two sides: on the one hand, chronic exposure to ozone in murine model of lung inflammation and emphysema and on the other, long-term exposure to ambient air pollutant such as ozone, has been associated with increases in emphysema evaluated by computed tomographic imaging with chronic airflow obstruction. The mechanisms of ozone-induced lung and airway changes are the release of inflammatory factors such as IL-1α, IL-6, IL-8, CXCL-12, CCL2, ICAM-1, KEAP-1, and MIF, the activation of intracellular pathways such as the MAPK pathway, TLR, cell death pathways, NLRP3 inflammasome, and NF-κB, the induction of oxidative stress through a decrease in the antioxidative response and an increase in the production of ROS, with a detrimental effect on mitochondrial function such as increased mitochondrial ROS, decreased ATP content and abnormal ETC complex (Figure 1). Other mechanisms include the disruption of airway epithelial barrier, the development of AHR and emphysema and the state of CS insensitivity. Inhibitors of mitochondrial ROS, NLRP3 inflammasome, DNA sensor, cell death pathways and IL-1, including hydrogen sulfide, apocynin, mitochondrial-targeted antioxidants such as MitoQ and MitoTEMPO, mucolytic/antioxidant agents such as NAC, VX765, and iPS-MSCs mitochondrial transfer represent novel therapeutic options for treating oxidative stress-induced COPD.
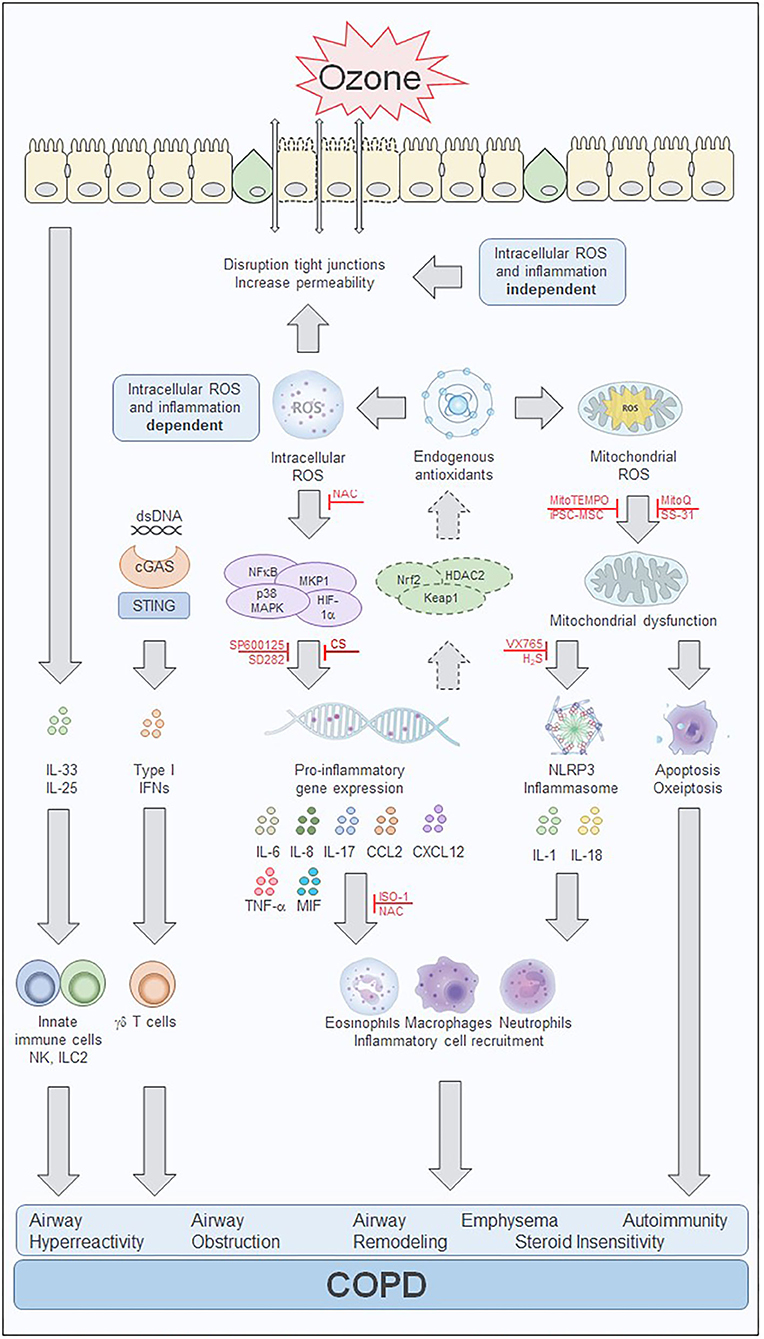
Figure 1. Ozone-mediated effects on intracellular pathways involved in cell injury and inflammation. Overview of the effects of ozone in in vitro and in vivo models. The biphasic response to ozone starts with an immediate intracellular reactive oxygen species (ROS) and inflammation independent phase that is induced by extracellular oxidative stress. ROS induces membrane damage with changes in cell membrane integrity, disruption of tight junctions, epithelial cell stress, and death. Inflammatory mediators including IL-25 and IL-33 are released and attract innate immune cells such as natural killer (NK) cells and innate lymphoid type 2 (ILC2) cells. Cell death and release of dsDNA can induce an interferon (IFN) inflammation response via the cGAS/STING pathway. Processes activated during the intracellular ROS and inflammation-dependent phase of the effects of ozone include transcription factor-mediated inflammatory response and the activation of the antioxidant defense mechanism. Several inhibitors have been shown to prevent or treat the pro-inflammatory gene expression and subsequently inhibit the inflammatory response, including the JNK inhibitor, SP600125, and the p38 MAPK inhibitor, SD282. In addition, activation of the endogenous antioxidant defense system involving HADC2, Keap1, and Nrf2 may be sufficient to counteract the oxidant stress during acute ozone exposure but may be overwhelmed during chronic ozone exposure. Treatment with the antioxidant N-acetylcysteine (NAC) also reduces the inflammatory response, by scavenging of intracellular ROS with subsequent reduction of cytokine and chemokine production. Mitochondrial oxidative stress and mitochondrial dysfunction contribute to apoptotic processes and the activation of the NLRP3 inflammasome further enhancing the inflammatory response. Several treatment strategies targeting the mitochondria have been able to reduce or prevent the mitochondrial oxidative-induced dysfunction. These include several mitochondrial-targeted antioxidants such as MitoQ, MitoTEMPO, and SS-31. In addition, stem cell therapy with induced pluripotent stem cell-derived mesenchymal stem cells (iPSC-MSCs) prevented ozone-induced mitochondrial dysfunction and inflammation which may result from direct interaction and mitochondrial transfer between the iPSC-MSCs and airway cells. Treatment with the caspase-1 inhibitor VX765 and hydrogen sulfide (H2S) prevent the activation of the inflammasome and reduces inflammation and mitochondrial oxidative stress. The MIF inhibitor ISO-1 blocks the ozone exposure induced inflammation and airway hyperreactivity and might have an impact on the corticosteroid insensitivity present in chronic ozone exposed lungs. Corticosteroids reduce inflammation induced by acute ozone exposures but fail to affect these processes in the steroid insensitive chronic ozone exposed lung. The ozone exposure driven intracellular processes contribute to the inflammatory cytokine and chemokine production, immune cell recruitment, and eventually the development of airway hyperreactivity, airway obstruction, airway remodeling, emphysema, autoimmunity, and steroid insensitivity which are hallmarks of COPD. cGAS, cyclic GMP-AMP synthase; COPD, chronic obstructive pulmonary disease; dsDNA, double stranded DNA; H2S, hydrogen sulfide; HDAC2, histone deacetylase 2; HIF-1α, hypoxia inducible factor subunit 1α; IFN, interferon; IL, interleukin; ILC2, innate lymphoid cells type 2; iPSC-MSC, induced pluripotent stem cell-derived mesenchymal stem cell; ISO-1, (S,R)3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester; Keap1, Kelch-like ECH associated protein 1; MAPK, mitogen activated kinase; MKP-1, mitogen-activated protein kinase phosphatase-1; MIF, macrophage migration inhibitory factor; MKP-1, mitogen-activated protein kinase phosphatase-1; NAC, N-acetylcysteine; NFκB, nuclear factor κB; NK, natural killer cell; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; Nrf2, nuclear factor (erythroid-derived 2)-like 2; ROS, reactive oxygen species; SD282, p38 MAPK inhibitor; SP600125, c-jun NH2 terminal kinase (JNK) inhibitor; SS-31, d-Arg-2', 6'-dimethyltyrosine-Lys-Phe-NH2 mitochondrial antioxidant; STING, stimulator of interferon genes; TNF-α, tumor necrosis factor -α; VX765, Belnacasan, caspase 1 inhibitor.
CW, FL, DT, and KC wrote the review. KC and BR inspired the work and corrected the manuscript. All authors have read the contribution.
This work was supported by Center National de la Recherche Scientifique (CNRS), European Regional Development Fund (FEDER) (N 2016-00110366 and EX005756), Respir Ozone (N 2014-00091905), The Wellcome Trust (KC), the Key International (Regional) Cooperative Project of the National Natural Science Foundation of China (Grant No. 51420105010), the project of the National Natural Science Foundation of China (Grant No. 81870031), and the STAR Project from Shanghai Jiao Tong University (YG2019ZDA24). LF was supported by the National Nature Science Foundation of China (81870031).
DT was employed by the company ArtImmune SAS.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
16HBE, human bronchial epithelial cell line; AHR, airways hyperreactivity; AIFM1, mitochondrial apoptosis inducing factor 1; AP-1, activator protein 1; ASMC, airway smooth muscle cell; ATP, adenosine triphosphate; BAL, bronchoalveolar lavage; cGAS, cyclic GMP-AMP synthase; CBS, cystathionine-beta-synthetase; CCL2, C-C motif chemokine ligand 2; CGL, cystathionine-gamma-lyase; CLDN, claudin; COPD, chronic obstructive pulmonary disease; CS, corticosteroid; CSM, cigarette smoke medium; CXCL1, chemokine (C-X-C) ligand 1; ΔΨm, mitochondrial membrane potential; DRP1, dynamin-related protein 1; ERK, extracellular signal-regulated kinase; ETC, electron transport chain; GR, glucocorticoid receptor; H2O2, hydrogen peroxide; H2S, hydrogen sulfide; HDAC2, histone deacetylase 2; HIF-1α, hypoxia inducible factor subunit 1α; ICAM1, intracellular adhesion molecule 1; IFNγ, interferon gamma; IL, interleukin; ILC2, innate lymphoid cells type 2; iNOS, inducible nitric oxide synthase; iPSC-MSC, induced pluripotent stem cell-derived mesenchymal stem cell; ISO-1, (S,R)3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester; JNK, c-jun NH2 terminal kinase; KEAP1, Kelch-like ECH associated protein 1; MAPK, mitogen activated kinase; MFF, mitochondrial fission factor; MFN2, mitofusin 2; MIF, macrophage migration inhibitory factor; MIP-2, macrophage inflammatory protein 2; MKP-1, mitogen-activated protein kinase phosphatase-1; MMP-12, matrix metalloproteinase 12; MPST, 3-mercaptypyruvate sulfurtransferase; MyD88, myeloid differentiation marker 88; NAC, N-acetylcysteine; NADPH, nicotinamide adenine dinucleotide phosphate; NaHS, sodium hydrosulfide; NET, neutrophil extracellular trap; NFκB, nuclear factor κB; NK, natural killer cell; NLRP3, NACHT, LRR and PYD domains-containing protein 3; Nrf2, nuclear factor (erythroid-derived 2)-like 2; OVA, ovalbumin; PAF, platelet-activating factor; PGAM5, phosphoglycerate mutase family, member 5; PGE2, prostaglandin E2; ppm, parts per million; ROS, reactive oxygen species; SS-31, d-Arg-2′,6′-dimethyltyrosine-Lys-Phe-NH2 mitochondrial antioxidant; STING, stimulator of interferon genes; TIRAP, Toll-Interleukin 1 receptor (TIR) domain containing adaptor protein; TLR, toll-like receptor; TNF-α, tumor necrosis factor-α; TRPC6, canonical transient receptor potential channel 6; TSLP, thymic stromal lymphopoietin; VEGF, vascular endothelial growth factor.
1. Bell ML, McDermott A, Zeger SL, Samet JM, Dominici F. Ozone and short-term mortality in 95 US urban communities, 1987-2000. JAMA. (2004) 292:2372–8. doi: 10.1001/jama.292.19.2372
2. Vicedo-Cabrera AM, Sera F, Liu C, Armstrong B, Milojevic A, Guo Y, et al. Short term association between ozone and mortality: global two stage time series study in 406 locations in 20 countries. BMJ. (2020) 368:m108. doi: 10.1136/bmj.m108
3. Gao H, Wang K, Au WW, Zhao W, Xia ZL. A systematic review and meta-analysis of short-term ambient ozone exposure and COPD hospitalizations. Int J Environ Res Public Health. (2020) 17:2130. doi: 10.3390/ijerph17062130
4. Halonen JI, Lanki T, Yli-Tuomi T, Kulmala M, Tiittanen P, Pekkanen J. Urban air pollution, and asthma and COPD hospital emergency room visits. Thorax. (2008) 63:635–41. doi: 10.1136/thx.2007.091371
5. Paulin LM, Gassett AJ, Alexis NE, Kirwa K, Kanner RE, Peters S, et al. Association of long-term ambient ozone exposure with respiratory morbidity in smokers. JAMA Intern Med. (2020) 180:106–15. doi: 10.1001/jamainternmed.2019.5498
6. Wang M, Aaron CP, Madrigano J, Hoffman EA, Angelini E, Yang J, et al. Association between long-term exposure to ambient air pollution and change in quantitatively assessed emphysema and lung function. JAMA. (2019) 322:546–56. doi: 10.1001/jama.2019.10255
7. Bromberg PA. Mechanisms of the acute effects of inhaled ozone in humans. Biochim Biophys Acta. (2016) 1860:2771–81. doi: 10.1016/j.bbagen.2016.07.015
8. Triantaphyllopoulos K, Hussain F, Pinart M, Zhang M, Li F, Adcock I, et al. A model of chronic inflammation and pulmonary emphysema after multiple ozone exposures in mice. Am J Physiol Lung Cell Mol Physiol. (2011) 300:L691–700. doi: 10.1152/ajplung.00252.2010
9. Agusti A, Hogg JC, Singh D, Agusti A, Anzueto A, Barnes PJ, et al. Update on the pathogenesis of chronic obstructive pulmonary disease global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. N Engl J Med. (2019) 381:1248–56. doi: 10.1056/NEJMra1900475
10. Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. (2008) 31:1334–56. doi: 10.1183/09031936.00018908
11. Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet. (2009) 374:733–43. doi: 10.1016/S0140-6736(09)61303-9
12. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
13. Koo HK, Vasilescu DM, Booth S, Hsieh A, Katsamenis OL, Fishbane N, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study chronic cough as a novel phenotype of chronic obstructive pulmonary disease. Lancet Respir Med. (2018) 6:591–602. doi: 10.1016/S2213-2600(18)30196-6
14. McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. (2011) 365:1567–75. doi: 10.1056/NEJMoa1106955
15. Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. (2000) 343:269–80. doi: 10.1056/NEJM200007273430407
16. Scichilone N, Battaglia S, La Sala A, Bellia V. Clinical implications of airway hyperresponsiveness in COPD. Int J Chron Obstruct Pulmon Dis. (2006) 1:49–60. doi: 10.2147/copd.2006.1.1.49
17. Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol. (2003) 29:88–97. doi: 10.1165/rcmb.2002-0228OC
18. Tan WSD, Shen HM, Wong WSF. Dysregulated autophagy in COPD: a pathogenic process to be deciphered. Pharmacol Res. (2019) 144:1–7. doi: 10.1016/j.phrs.2019.04.005
19. Caramori G, Ruggeri P, Di Stefano A, Mumby S, Girbino G, Adcock IM, et al. Autoimmunity and COPD: clinical implications. Chest. (2018) 153:1424–31. doi: 10.1016/j.chest.2017.10.033
20. Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, et al. Inflammaging and oxidative stress in human diseases: from molecular mechanisms to novel treatments. Int J Mol Sci. (2019) 20:4472. doi: 10.3390/ijms20184472
21. Chung KF, Marwick JA. Molecular mechanisms of oxidative stress in airways and lungs with reference to asthma and chronic obstructive pulmonary disease. Ann N Y Acad Sci. (2010) 1203:85–91. doi: 10.1111/j.1749-6632.2010.05600.x
22. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 184:796–802. doi: 10.1164/rccm.201010-1605OC
23. Zhou JS, Li ZY, Xu XC, Zhao Y, Wang Y Chen HP, et al. Cigarette smoke-initiated autoimmunity facilitates sensitisation to elastin-induced COPD-like pathologies in mice. Eur Respir J. (2020). doi: 10.1183/13993003.00404-2020. [Epub ahead of print].
24. Byrne R, Todd I, Tighe PJ, Fairclough LC, Caramori G, Ruggeri P, et al. Autoantibodies in chronic obstructive pulmonary disease: a systematic review autoimmunity and COPD: clinical implications. Immunol Lett. (2019) 214:8–15. doi: 10.1016/j.imlet.2019.08.007
25. Wen L, Krauss-Etschmann S, Petersen F, Yu X. Autoantibodies in chronic obstructive pulmonary disease. Front Immunol. (2018) 9:66. doi: 10.3389/fimmu.2018.00066
26. Mudway IS, Kelly FJ. Ozone and the lung: a sensitive issue. Mol Aspects Med. (2000) 21:1–48. doi: 10.1016/S0098-2997(00)00003-0
27. Pryor WA. Mechanisms of radical formation from reactions of ozone with target molecules in the lung. Free Radic Biol Med. (1994) 17:451–65. doi: 10.1016/0891-5849(94)90172-4
28. Becker S, Madden MC, Newman SL, Devlin RB, Koren HS, Devlin RB, et al. Modulation of human alveolar macrophage properties by ozone exposure in vitro. Toxicol Appl Pharmacol. (1991) 110:403–15. doi: 10.1016/0041-008X(91)90042-D
29. Devlin RB, McKinnon KP, Noah T, Becker S, Koren HS. Ozone-induced release of cytokines and fibronectin by alveolar macrophages and airway epithelial cells. Am J Physiol. (1994) 266:L612–9. doi: 10.1152/ajplung.1994.266.6.L612
30. Devlin RB, Folinsbee LJ, Biscardi F, Hatch G, Becker S, Maden MC, et al. Inflammation and cell damage induced by repeated exposure of humans to ozone. Inhal Toxicol. (1997) 9:211–2325. doi: 10.1080/089583797198222
31. Mumby S, Chung KF, Adcock IM. Transcriptional effects of ozone and impact on airway inflammation. Front Immunol. (2019) 10:1610. doi: 10.3389/fimmu.2019.01610
32. Tovar A, Smith GJ, Thomas JM, Crouse WL, Harkema JR, Kelada SNP. Transcriptional profiling of the murine airway response to acute ozone exposure. Toxicol Sci. (2020) 173:114–30. doi: 10.1093/toxsci/kfz219
33. Manzer R, Wang J, Nishina K, McConville G, Mason RJ. Alveolar epithelial cells secrete chemokines in response to IL-1beta and lipopolysaccharide but not to ozone. Am J Respir Cell Mol Biol. (2006) 34:158–66. doi: 10.1165/rcmb.2005-0205OC
34. Manzer R, Dinarello CA, McConville G, Mason RJ. Ozone exposure of macrophages induces an alveolar epithelial chemokine response through IL-1alpha. Am J Respir Cell Mol Biol. (2008) 38:318–23. doi: 10.1165/rcmb.2007-0250OC
35. Chen QZ, Zhou YB, Zhou LF, Fu ZD, Wu YS, Chen Y, et al. TRPC6 modulates adhesion of neutrophils to airway epithelial cells via NF-kappaB activation and ICAM-1 expression with ozone exposure. Exp Cell Res. (2019) 377:56–66. doi: 10.1016/j.yexcr.2019.02.013
36. Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol. (2014) 229:434–42. doi: 10.1002/jcp.24461
37. Gibon J, Tu P, Bohic S, Richaud P, Arnaud J, Zhu M, et al. The over-expression of TRPC6 channels in HEK-293 cells favours the intracellular accumulation of zinc. Biochim Biophys Acta. (2011) 1808:2807–18. doi: 10.1016/j.bbamem.2011.08.013
38. Ding Y, Winters A, Ding M, Graham S, Akopova I, Muallem S, et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J Biol Chem. (2011) 286:31799–809. doi: 10.1074/jbc.M111.248344
39. Finney-Hayward TK, Popa MO, Bahra P, Li S, Poll CT, Gosling M, et al. Expression of transient receptor potential C6 channels in human lung macrophages. Am J Respir Cell Mol Biol. (2010) 43:296–304. doi: 10.1165/rcmb.2008-0373OC
40. Chen Q, Zhou Y, Zhou L, Fu Z, Yang C, Zhao L, et al. TRPC6-dependent Ca(2+) signaling mediates airway inflammation in response to oxidative stress via ERK pathway. Cell Death Dis. (2020) 11:170. doi: 10.1038/s41419-020-2678-7
41. Ge MQ, Kokalari B, Flayer CH, Killingbeck SS, Redai IG, MacFarlane AW, et al. Cutting edge: role of NK cells and surfactant protein D in dendritic cell lymph node homing: effects of ozone exposure. J Immunol. (2016) 196:553–7. doi: 10.4049/jimmunol.1403042
42. Harkema JR, Wagner JG. Innate lymphoid cell-dependent airway epithelial and inflammatory responses to inhaled ozone: a new paradigm in pathogenesis. Toxicol Pathol. (2019) 47:993–1003. doi: 10.1177/0192623319873872
43. Ong CB, Kumagai K, Brooks PT, Brandenberger C, Lewandowski RP, Jackson-Humbles DN, et al. Ozone-induced type 2 immunity in nasal airways. Development and lymphoid cell dependence in mice. Am J Respir Cell Mol Biol. (2016) 54:331–40. doi: 10.1165/rcmb.2015-0165OC
44. Kumagai K, Lewandowski RP, Jackson-Humbles DN, Buglak N, Li N, White K, et al. Innate lymphoid cells mediate pulmonary eosinophilic inflammation, airway mucous cell metaplasia, and type 2 immunity in mice exposed to ozone. Toxicol Pathol. (2017) 45:692–704. doi: 10.1177/0192623317728135
45. Williams AS, Nath P, Leung SY, Khorasani N, McKenzie AN, Adcock IM, et al. Modulation of ozone-induced airway hyperresponsiveness and inflammation by interleukin-13. Eur Respir J. (2008) 32:571–8. doi: 10.1183/09031936.00121607
46. Liang L, Li F, Bao A, Zhang M, Chung KF, Zhou X. Activation of p38 mitogen-activated protein kinase in ovalbumin and ozone-induced mouse model of asthma. Respirology. (2013) 18 (Suppl. 3):20–9. doi: 10.1111/resp.12189
47. Wiegman CH, Li F, Clarke CJ, Jazrawi E, Kirkham P, Barnes PJ, et al. A comprehensive analysis of oxidative stress in the ozone-induced lung inflammation mouse model. Clin Sci. (2014) 126:425–40. doi: 10.1042/CS20130039
48. Li Z, Potts-Kant EN, Garantziotis S, Foster WM, Hollingsworth JW. Hyaluronan signaling during ozone-induced lung injury requires TLR4, MyD88, and TIRAP. PLoS ONE. (2011) 6:e27137. doi: 10.1371/journal.pone.0027137
49. Williams AS, Leung SY, Nath P, Khorasani NM, Bhavsar P, Issa R, et al. Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J Appl Physiol. (2007) 103:1189–95. doi: 10.1152/japplphysiol.00172.2007
50. Bauer AK, Rondini EA, Hummel KA, Degraff LM, Walker C, Jedlicka AE, et al. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: a role for heat-shock protein 70. Environ Health Perspect. (2011) 119:1091–7. doi: 10.1289/ehp.1003326
51. Michaudel C, Couturier-Maillard A, Chenuet P, Maillet I, Mura C, Couillin I, et al. Inflammasome, IL-1 and inflammation in ozone-induced lung injury. Am J Clin Exp Immunol. (2016) 5:33–40.
52. Li F, Zhang M, Hussain F, Triantaphyllopoulos K, Clark AR, Bhavsar PK, et al. Inhibition of p38 MAPK-dependent bronchial contraction after ozone by corticosteroids. Eur Respir J. (2011) 37:933–42. doi: 10.1183/09031936.00021110
53. Xu M, Wang L, Wang M, Wang H, Zhang H, Chen Y, et al. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radic Res. 53:780–90. doi: 10.1080/10715762.2019.1630735
54. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. (2018) 281:8–27. doi: 10.1111/imr.12621
55. Michaudel C, Maillet I, Fauconnier L, Quesniaux V, Chung KF, Wiegman C, et al. Interleukin-1alpha mediates ozone-induced myeloid differentiation factor-88-dependent epithelial tissue injury and inflammation. Front Immunol. (2018) 9:916. doi: 10.3389/fimmu.2018.00916
56. Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. (2009) 179:903–13. doi: 10.1164/rccm.200808-1274OC
57. Franklin BS, Mangan MS, Latz E. Crystal formation in inflammation. Annu Rev Immunol. (2016) 34:173–202. doi: 10.1146/annurev-immunol-041015-055539
58. Michaudel C, Mackowiak C, Maillet I, Fauconnier L, Akdis CA, Sokolowska M, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J Allergy Clin Immunol. (2018) 142:942–58. doi: 10.1016/j.jaci.2017.11.044
59. Kim BG, Lee PH, Lee SH, Park CS, Jang AS. Impact of ozone on claudins and tight junctions in the lungs. Environ Toxicol. (2018) 33:798–806. doi: 10.1002/tox.22566
60. Kosmider B, Loader JE, Murphy RC, Mason RJ. Apoptosis induced by ozone and oxysterols in human alveolar epithelial cells. Free Radic Biol Med. (2010) 48:1513–24. doi: 10.1016/j.freeradbiomed.2010.02.032
61. Pulfer MK, Taube C, Gelfand E, Murphy RC. Ozone exposure in vivo and formation of biologically active oxysterols in the lung. J Pharmacol Exp Ther. (2005) 312:256–64. doi: 10.1124/jpet.104.073437
62. McKinnon KP, Madden MC, Noah TL, Devlin RB. In vitro ozone exposure increases release of arachidonic acid products from a human bronchial epithelial cell line. Toxicol Appl Pharmacol. (1993) 118:215–23. doi: 10.1006/taap.1993.1027
63. Wright DT, Adler KB, Akley NJ, Dailey LA, Friedman M. Ozone stimulates release of platelet activating factor and activates phospholipases in guinea pig tracheal epithelial cells in primary culture. Toxicol Appl Pharmacol. (1994) 127:27–36. doi: 10.1006/taap.1994.1135
64. Madden MCF, Dailey LA, Samet JM. Inhibition of arachidonic acid esterification in human airway epithelial cells exposed to ozone in vitro. Inhal Toxicol. (1998) 10:795–811. doi: 10.1080/089583798197466
65. Sokolowska M, Quesniaux VFJ, Akdis CA, Chung KF, Ryffel B, Togbe D. Acute respiratory barrier disruption by ozone exposure in mice. Front Immunol. (2019) 10:2169. doi: 10.3389/fimmu.2019.02169
66. Michaudel C, Fauconnier L, Jule Y, Ryffel B. Functional and morphological differences of the lung upon acute and chronic ozone exposure in mice. Sci Rep. (2018) 8:10611. doi: 10.1038/s41598-018-28261-9
67. Pinart M, Zhang M, Li F, Hussain F, Zhu J, Wiegman C, et al. IL-17A modulates oxidant stress-induced airway hyperresponsiveness but not emphysema. PLoS ONE. (2013) 8:e58452. doi: 10.1371/journal.pone.0058452
68. Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell. (2018) 44:542–53. doi: 10.1016/j.devcel.2018.01.019
69. Benmerzoug S, Ryffel B, Togbe D, Quesniaux VFJ. Self-DNA sensing in lung inflammatory diseases. Trends Immunol. (2019) 40:719–34. doi: 10.1016/j.it.2019.06.001
70. Benmerzoug S, Rose S, Bounab B, Gosset D, Duneau L, Chenuet P, et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat Commun. (2018) 9:5226. doi: 10.1038/s41467-018-07425-1
71. Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. (2018) 19:130–40. doi: 10.1038/s41590-017-0013-y
72. Scaturro P, Pichlmair A. Oxeiptosis-a cell death pathway to mitigate damage caused by radicals. Cell Death Differ. (2018) 25:1191–3. doi: 10.1038/s41418-018-0134-3
73. Scaturro P, Pichlmair A. Oxeiptosis: a discreet way to respond to radicals. Curr Opin Immunol. (2019) 56:37–43. doi: 10.1016/j.coi.2018.10.006
74. Paludan SR, Reinert LS, Hornung V. DNA-stimulated cell death: implications for host defence, inflammatory diseases and cancer. Nat Rev Immunol. (2019) 19:141–53. doi: 10.1038/s41577-018-0117-0
75. Bialas AJ, Sitarek P, Milkowska-Dymanowska J, Piotrowski WJ, Gorski P. The role of mitochondria and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary disease. Oxid Med Cell Longev. (2016) 2016:7808576. doi: 10.1155/2016/7808576
76. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest. (2017) 152:618–26. doi: 10.1016/j.chest.2017.03.020
77. Mustafa MG, Cross CE. Effects of short-term ozone exposure on lung mitochondrial oxidative and energy metabolism. Arch Biochem Biophys. (1974) 162:585–94. doi: 10.1016/0003-9861(74)90219-7
78. Zychlinski L, Raska-Emery P, Balis JU, Montgomery MR. Age-related difference in bioenergetics of lung and heart mitochondrial from rats exposed to ozone. J Biochem Toxicol. (1989) 4:251–4. doi: 10.1002/jbt.2570040408
79. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2015) 136:769–80. doi: 10.1016/j.jaci.2015.01.046
80. Servais S, Boussouar A, Molnar A, Douki T, Pequignot JM, Favier R. Age-related sensitivity to lung oxidative stress during ozone exposure. Free Radic Res. (2005) 39:305–16. doi: 10.1080/10715760400011098
81. Mustafa MG, DeLucia AJ, York GK, Arth C, Cross CE. Ozone interaction with rodent lung. II effects on oxygen consumption of mitochondria. J Lab Clin Med. (1973) 82:357–65.
82. Li F, Xu M, Wang M, Wang L, Wang H, Zhang H, et al. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir Res. (2018) 19:230. doi: 10.1186/s12931-018-0931-8
83. Li X, Michaeloudes C, Zhang Y, Wiegman CH, Adcock IM, Lian Q, et al. Mesenchymal stem cells alleviate oxidative stress-induced mitochondrial dysfunction in the airways. J Allergy Clin Immunol. (2018) 141:1634–45.e1635. doi: 10.1016/j.jaci.2017.08.017
84. Bao W, Zhang Y, Zhang M, Bao A, Fei X, Zhang X, et al. Effects of ozone repeated short exposures on the airway/lung inflammation, airway hyperresponsiveness and mucus production in a mouse model of ovalbumin-induced asthma. Biomed Pharmacother. (2018) 101:293–303. doi: 10.1016/j.biopha.2018.02.079
85. Kierstein S, Krytska K, Sharma S, Amrani Y, Salmon M, Panettieri RA Jr, et al. Ozone inhalation induces exacerbation of eosinophilic airway inflammation and hyperresponsiveness in allergen-sensitized mice. Allergy. (2008) 63:438–46. doi: 10.1111/j.1398-9995.2007.01587.x
86. Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. (2008) 205:385–93. doi: 10.1084/jem.20071507
87. Lee LY, Kwong K, Lin YS, Gu Q. Hypersensitivity of bronchopulmonary C-fibers induced by airway mucosal inflammation: cellular mechanisms. Pulm Pharmacol Ther. (2002) 15:199–204. doi: 10.1006/pupt.2002.0338
88. Bhalla DK. Ozone-induced lung inflammation and mucosal barrier disruption: toxicology, mechanisms, and implications. J Toxicol Environ Health B Crit Rev. (1999) 2:31–86. doi: 10.1080/109374099281232
89. Li F, Wiegman C, Seiffert JM, Zhu J, Clarke C, Chang Y, et al. Effects of N-acetylcysteine in ozone-induced chronic obstructive pulmonary disease model. PLoS ONE. (2013) 8:e80782. doi: 10.1371/journal.pone.0080782
90. Mathews JA, Kasahara DI, Ribeiro L, Wurmbrand AP, Ninin FM, Shore SA. γδ T cells are required for M2 macrophage polarization and resolution of ozone-induced pulmonary inflammation in mice. PLoS ONE. (2015) 10:e0131236. doi: 10.1371/journal.pone.0131236
91. Salmon M, Koto H, Lynch OT, Haddad EB, Lamb NJ, Quinlan GJ, et al. Proliferation of airway epithelium after ozone exposure: effect of apocynin and dexamethasone. Am J Respir Crit Care Med. (1998) 157:970–7. doi: 10.1164/ajrccm.157.3.9704067
92. Zhang P, Li F, Wiegman CH, Zhang M, Hong Y, Gong J, et al. Inhibitory effect of hydrogen sulfide on ozone-induced airway inflammation, oxidative stress, and bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. (2015) 52:129–37. doi: 10.1165/rcmb.2013-0415OC
93. Li F, Zhang P, Zhang M, Liang L, Sun X, Li M, et al. Hydrogen sulfide prevents and partially reverses ozone-induced features of lung inflammation and emphysema in mice. Am J Respir Cell Mol Biol. (2016) 55:72–81. doi: 10.1165/rcmb.2015-0014OC
94. Russell KE, Chung KF, Clarke CJ, Durham AL, Mallia P, Footitt J, et al. The MIF antagonist ISO-1 attenuates corticosteroid-insensitive inflammation and airways hyperresponsiveness in an ozone-induced model of COPD. PLoS ONE. (2016) 11:e0146102. doi: 10.1371/journal.pone.0146102
95. Alexis NE, Lay JC, Haczku A, Gong H, Linn W, Hazucha MJ, et al. Fluticasone propionate protects against ozone-induced airway inflammation and modified immune cell activation markers in healthy volunteers. Environ Health Perspect. (2008) 116:799–805. doi: 10.1289/ehp.10981
96. Chung KF, Adcock IM. Induction of nuclear factor-kappa B by exposure to ozone and inhibition by glucocorticoids. Meth Enzymol. (2000) 319:551–62. doi: 10.1016/S0076-6879(00)19052-4
97. Haddad EB, Salmon M, Koto H, Barnes PJ, Adcock I, Chung KF. Ozone induction of cytokine-induced neutrophil chemoattractant (CINC) and nuclear factor-kappa b in rat lung: inhibition by corticosteroids. FEBS Lett. (1996) 379:265–8. doi: 10.1016/0014-5793(95)01524-8
98. Haddad EB, Liu SF, Salmon M, Robichaud A, Barnes PJ, Chung KF. Expression of inducible nitric oxide synthase mRNA in brown norway rats exposed to ozone: effect of dexamethasone. Eur J Pharmacol. (1995) 293:287–90. doi: 10.1016/0926-6917(95)00032-1
99. Haddad EB, Salmon M, Sun J, Liu S, Das A, Adcock I, et al. Dexamethasone inhibits ozone-induced gene expression of macrophage inflammatory protein-2 in rat lung. FEBS Lett. (1995) 363:285–8. doi: 10.1016/0014-5793(95)00333-5
100. Pinart M, Hussain F, Shirali S, Li F, Zhu J, Clark AR, et al. Role of mitogen-activated protein kinase phosphatase-1 in corticosteroid insensitivity of chronic oxidant lung injury. Eur J Pharmacol. (2014) 744:108–14. doi: 10.1016/j.ejphar.2014.10.003
101. Adcock IM, Marwick J, Casolari P, Contoli M, Chung KF, Kirkham P, et al. Mechanisms of corticosteroid resistance in severe asthma and chronic obstructive pulmonary disease (COPD). Curr Pharm Des. (2010) 16:3554–73. doi: 10.2174/138161210793797889
102. Chung KF, Marwick JA, Chung KF, Adcock IM. Molecular mechanisms of oxidative stress in airways and lungs with reference to asthma and chronic obstructive pulmonary disease multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Ann N Y Acad Sci. (2010) 1203:85–91.
103. Chung KF. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest. (2011) 139:1470–9. doi: 10.1378/chest.10-1914
104. Perridon BW, Leuvenink HG, Hillebrands JL, van Goor H, Bos EM. The role of hydrogen sulfide in aging and age-related pathologies. Aging. (2016) 8:2264–89. doi: 10.18632/aging.101026
105. Xu D, Jin H, Wen J, Chen J, Chen D, Cai N, et al. Hydrogen sulfide protects against endoplasmic reticulum stress and mitochondrial injury in nucleus pulposus cells and ameliorates intervertebral disc degeneration. Pharmacol Res. (2017) 117:357–69. doi: 10.1016/j.phrs.2017.01.005
106. Zheng J, Zhao T, Yuan Y, Hu N, Tang X. Hydrogen sulfide (H2S) attenuates uranium-induced acute nephrotoxicity through oxidative stress and inflammatory response via Nrf2-NF-κB pathways. Chem Biol Interact. (2015) 242:353–62. doi: 10.1016/j.cbi.2015.10.021
107. Chung KF. Hydrogen sulfide as a potential biomarker of asthma. Expert Rev Respir Med. (2014) 8:5–13. doi: 10.1586/17476348.2014.856267
108. Perry MM, Tildy B, Papi A, Casolari P, Caramori G, Rempel KL, et al. The anti-proliferative and anti-inflammatory response of COPD airway smooth muscle cells to hydrogen sulfide. Respir Res. (2018) 19:85. doi: 10.1186/s12931-018-0788-x
109. Sun Y, Wang K, Li MX, He W, Chang JR, Liao CC, et al. Metabolic changes of H2S in smokers and patients of COPD which might involve in inflammation, oxidative stress and steroid sensitivity. Sci Rep. (2015) 5:14971. doi: 10.1038/srep14971
110. Lin F, Liao C, Sun Y, Zhang J, Lu W, Bai Y, et al. Hydrogen sulfide inhibits cigarette smoke-induced endoplasmic reticulum stress and apoptosis in bronchial epithelial cells. Front Pharmacol. (2017) 8:675. doi: 10.3389/fphar.2017.00675
111. Guan R, Cai Z, Wang J, Ding M, Li Z, Xu J, et al. Hydrogen sulfide attenuates mitochondrial dysfunction-induced cellular senescence and apoptosis in alveolar epithelial cells by upregulating sirtuin 1. Aging. (2019) 11:11844–64. doi: 10.18632/aging.102454
112. Rogliani P, Matera MG, Page C, Puxeddu E, Cazzola M, Calzetta L. Efficacy and safety profile of mucolytic/antioxidant agents in chronic obstructive pulmonary disease: a comparative analysis across erdosteine, carbocysteine, and N-acetylcysteine. Respir Res. (2019) 20:104. doi: 10.1186/s12931-019-1078-y
113. Williams AS, Issa R, Leung SY, Nath P, Ferguson GD, Bennett BL, et al. Attenuation of ozone-induced airway inflammation and hyper-responsiveness by c-Jun NH2 terminal kinase inhibitor SP600125. JPharmacol ExpTher. (2007) 322:351–9. doi: 10.1124/jpet.107.121624
114. Williams AS, Issa R, Durham A, Leung SY, Kapoun A, Medicherla S, et al. Role of p38 mitogen-activated protein kinase in ozone-induced airway hyperresponsiveness and inflammation. EurJPharmacol. (2008) 600:117–22. doi: 10.1016/j.ejphar.2008.09.031
Keywords: ozone, oxidative stress, inflammation, empyema, model, COPD
Citation: Wiegman CH, Li F, Ryffel B, Togbe D and Chung KF (2020) Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front. Immunol. 11:1957. doi: 10.3389/fimmu.2020.01957
Received: 30 March 2020; Accepted: 20 July 2020;
Published: 02 September 2020.
Edited by:
Takayuki Yoshimoto, Tokyo Medical University, JapanReviewed by:
Min-Jong Kang, Yale University, United StatesCopyright © 2020 Wiegman, Li, Ryffel, Togbe and Chung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Coen H. Wiegman, Yy53aWVnbWFuQGltcGVyaWFsLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.