 Shuzhong Zhang1
Shuzhong Zhang1 Chandra L. Shrestha1Benjamin L. Wisniewski1,2Hanh Pham2Xucheng Hou3Wenqing Li3
Chandra L. Shrestha1Benjamin L. Wisniewski1,2Hanh Pham2Xucheng Hou3Wenqing Li3 Yizhou Dong3
Yizhou Dong3 Benjamin T. Kopp1,2*
Benjamin T. Kopp1,2*- 1Center for Microbial Pathogenesis, The Abigail Wexner Research Institute at Nationwide Children's Hospital, Columbus, OH, United States
- 2Division of Pulmonary Medicine, Nationwide Children's Hospital, Columbus, OH, United States
- 3Pharmaceutics and Pharmacology, The Ohio State University, Columbus, OH, United States
Macrophage dysfunction is fundamentally related to altered immunity in cystic fibrosis (CF). How genetic deficits in the cystic fibrosis transmembrane conductance regulator (CFTR) lead to these defects remains unknown. Rapid advances in genomic editing such as the clustered regularly interspaced short palindromic repeats associated protein 9 (CRISPR/Cas9) system provide new tools for scientific study. We aimed to create a stable CFTR knockout (KO) in human macrophages in order to study how CFTR regulates macrophage function. Peripheral blood monocytes were isolated from non-CF healthy volunteers and differentiated into monocyte-derived macrophages (MDMs). MDMs were transfected with a CRISPR Cas9 CFTR KO plasmid. CFTR KO efficiency was verified and macrophage halide efflux, phagocytosis, oxidative burst, apoptosis, and cytokine functional assays were performed. CFTR KO in human MDMs was efficient and stable after puromycin selection. CFTR KO was confirmed by CFTR mRNA and protein expression. CFTR function was abolished in CFTR KO MDMs. CFTR KO recapitulated known defects in human CF MDM (CFTR class I/II variants) dysfunction including (1) increased apoptosis, (2) decreased phagocytosis, (3) reduced oxidative burst, and (4) increased bacterial load. Activation of the oxidative burst via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase assembly was diminished in CFTR KO MDMs (decreased phosphorylated p47phox). Cytokine production was unchanged or decreased in response to infection in CFTR KO MDMs. In conclusion, we developed a primary human macrophage CFTR KO system. CFTR KO mimics most pathology observed in macrophages obtained from persons with CF, which suggests that many aspects of CF macrophage dysfunction are CFTR-dependent and not just reflective of the CF inflammatory milieu.
Introduction
Cystic fibrosis (CF) is a multi-system disorder characterized by chronic polymicrobial pulmonary infections that lead to progressive morbidity (1). CF pathophysiology results from variants in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which lead to absent or dysfunctional CFTR protein expression. CFTR is present in epithelial-lined organs throughout the body, as well as in immune cells including macrophages (2, 3). While the persistence of pathogens in CF is multi-factorial, recent evidence suggest that alterations in the innate immune system are critically related to both acute and chronic infections in CF. (2, 4–17). However, the overall relationship between CFTR deficits and immune function remains poorly defined.
Macrophages have an important role in CF immune dysfunction due to multiple defects including reduced phagocytosis (2, 18–20) and a defective oxidative burst (4) among others (21, 22). How genetic alterations in CFTR contribute to these defects remains unknown. Recent advances in genomic editing such as the clustered regularly interspaced short palindromic repeats associated protein 9 (CRISPR/Cas9) system have provided unique tools for scientific study. However, there are limited strategies to allow reliable and efficient gene editing in human macrophages due to their terminally differentiated state. Although new viral vectors, polymers, or lipid chemical complexes can improve genetic editing approaches, many can induce untoward immune responses (23, 24). Thus, we aimed to create a stable CFTR knockout (KO) in human macrophages to study how CFTR regulates macrophage function. We hypothesized that macrophage effector functions are directly dependent on CFTR and therefore, a CFTR KO would reflect the functional macrophage deficits observed in primary human CF macrophages with CFTR (class I/II) variants that result in little or no CFTR protein expression.
Materials and Methods
Human Subjects
Thirty-two healthy non-CF, non-smoking, adult controls were recruited with approval by the Institutional Review Board of Nationwide Children's Hospital (IRB16-01020). All participants provided written informed consent. Male (40%) and females (60%) were recruited, with an average age of 36 years (range 22–66 years). Research procedures were done according to local and international guidelines and regulations for human research. Subject information was stored in a REDCap database.
Macrophage Isolation
Blood samples in Ethylenediaminetetraacetic acid (EDTA) tubes (Becton Dickinson 367861) were obtained from non-CF healthy controls per prior methods (2). In brief, monocytes were isolated from whole blood using Lymphocyte Separation Medium (Corning, 25-072-CV). Isolated monocytes were cultured in Roswell Park Memorial Institute (RPMI) media (Gibco, 22400-089) plus 20% human AB serum (Corning, 35-060-Cl) and differentiated for 5 days at 37°C into MDMs as previously described (6, 25). MDM purity and integrity was confirmed by microscopy and flow cytometry. MDMs were plated in a monolayer culture with fresh RPMI, rested for 4 days, and then infected with a bacterial multiplicity of infection (MOI) ranging from 2 to 50.
Bacterial Strains and Colony Forming Unit Assay
Macrophage infections occurred with multi-drug resistant CF clinical isolates. The Pseudomonas aeruginosa isolate was obtained from a CF patient's sputum. The Burkholderia cenocepacia k56-2 strain is an isolate of the epidemic clinical strain from the Edinburgh-Toronto (ET12) lineage (26). Bacteria were grown in Luria-Bertani (LB) media for 24 h prior to use. Colony forming unit (CFU) analysis was performed as previously described (8). Quantification of bacteria was performed by plating serial dilutions on LB agar plates.
CRISPR-Cas9 CFTR Knockout
We optimized our prior brief description of CRISPR-Cas9 CFTR KO (2). Monocytes were differentiated for 5 days into MDMs and then 10 million MDMs were suspended in 0.85 ml of antibiotic-free RPMI medium with 10% AB serum and kept on ice for use. A mixture of N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP, Roche 11202375001) liposomal transfection reagent and CFTR double nickase plasmid (NIC, Santa Cruz sc-400653) was then prepared according to the manufacturer's instructions. Briefly, a plasmid solution (A) was prepared by diluting 2.5 μg of plasmid to a final volume of 50 μl in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffered saline pH 7.4 (HBS, Bioworld 40820012) in a sterile polystyrene tube. A transfection solution (B) was prepared by mixing 50 μl DOTAP with HBS buffer to a final volume of 100 μl. Solution A was added dropwise directly to solution B and mixed gently by pipetting and incubated for 15 min at room temperature. The DOTAP/plasmid mixture was added directly to the MDMs and gently mixed by repeated pipetting. The MDM/plasmid suspension was transferred to a sterile standard vial (Savilex 200-015-12) and incubated for 14 h at 37°C. MDMs were then plated with new media and incubated for another 20 h. Finally, the supernatant was aspirated and replaced with fresh media containing puromycin at 2 μg/ml for additional 24 h before harvesting transfected MDMs for protein or RNA extraction. RNA was extracted from MDMs using the Total RNA Purification Kit (Norgen Biotek, 17200). Semi-quantitative reverse transcription polymerase chain reaction (qRT-PCR) was then performed by synthesizing complementary DNA (cDNA) by reverse transcriptase (Santa Cruz, sc43500) and then using nested PCR via two pairs of CFTR primer (CFTR (h)-PR, Santa Cruz, sc35054-PR) for determining CFTR transcription level. 18S rRNA (Forward: 5′-GGTGAAATTCTTGGACCGGC-3′; Reverse: 5′-GACTTTGGTTTCCCGGAAGC-3′) was used as an internal control.
Immunoblotting
MDM supernatants were removed post treatment and the cells were washed twice with phosphate buffered saline (PBS, Corning 21030-CV). MDMs were lysed in lysis buffer (10 mM HEPES, 5 mM MgCl2, 1 mM EGTA, 140 mM KCL,1% NP-40) with a protease inhibitor (Roche Applied Science, 10-519-978-001). Then 30 μg of protein was denatured in Laemmli sample buffer for 10 min at 95°C, and then separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Membranes were immunoblotted for β-actin (Cell Signaling, 8H10D10), anti-CFTR (Abcam CF3 2784), anti-CFTR (596) obtained from the CFTR Antibody Distribution Program https://www.cff.org/Research/Researcher-Resources/Tools-and-Resources/CFTR-Antibodies-Distribution-Program/, phospho-p47phox (donated by Jamel El-Benna), and total p47phox (Life Technologies, A16636). Protein bands were detected with HRP-conjugated secondary antibodies and visualized using enhanced chemiluminescence reagents (Life Sciences, RPN2106). Membrane and cytosolic fractionations were prepared via manufacturer kit instructions (Thermo Fisher, 78840). Bands were quantified by densitometric scanning of the films and analyzed using ImageJ.
CFTR-Mediated Halide Efflux
Non-CF and CFTR KO MDMs were plated in a 96-well plate at 1 × 106/100 μl/well and rested for 48–72 h prior to use. Media was then removed and cells were washed with efflux solution (mM):135 NaNO3, 1 CaSO4, 1 MgSO4, 2.4 K2HPO4, 0.6 KH2PO4, 10 HEPES, and 10 Glucose. A fluorescent indicator of intracellular Cl− (N-Ethoxycarbonylmethyl-6-Methoxyquinolinium Bromide [MQAE], Thermofisher E3101) was loaded with a hypotonic buffer (0.56 ml 36 mM MQAE stock with 1 ml of filtered deionised water and 0.44 ml of efflux solution) for 5 min at 37°C in the dark. MDMs were then washed with efflux solution to remove extraneous MQAE and incubated in 100 μl/well of warmed halide efflux solution for 5 min at 37°C. The maximum fluorescence was then recorded on a plate reader. The halide efflux solution was removed and intracellular MQAE was quenched with warmed NaI (Sigma, 409286) buffer (135 mM NaI in efflux buffer, 100 μl/well) at 15 min at 37°C. NaI buffer was removed and halide efflux solution added back for 5 min and basal fluorescence measured. Cells were then incubated with forskolin (20 μM, Sigma F6886) and 3-isobutyl-1-methylxanthine (IBMX, 100 μM, Sigma 15869) or CFTR inhibitor 172 (CFTRinh172, 10 μM, Simga C2992) at 37°C and fluorescence measured every 5 min for 30 min. The minimum fluorescence was obtained by exposing cells to the quenching buffer (150 mM KSCN, 5 μM valinomycin in NaI buffer) for 30 min at 37°C. CFTR-dependent chloride efflux was calculated as maximum fluorescence after forskolin stimulation.
Apoptosis Assay
Apoptosis was performed per prior methods (2). Briefly, MDMs were plated at a density of 1 × 106/ml in 12 well plates. Apoptosis was measured by flow cytometry and fluorescence-activated cell sorting (FACS) analysis using APC Annexin V (Biolegend, 640920) and propidium iodide (BioLegend 421301). MDMs were detached by Accutase solution, collected, washed, re-suspended in 100 μl of Annexin V Binding Buffer (Biolegend 422201), and then stained with 5 μl of Annexin V and 10 μl propidium iodide for 15 min at room temperature in the dark. The percentage of viable and apoptotic cells was calculate using flow cytometry (BD LSR II Flow Cytometer).
Phagocytosis Assay
Phagocytosis was performed per prior methods (2). Briefly, SpheroTM Ultra Rainbow Fluorescent beads (bead diameter 3–3.4 μM, Spherotech Inc.) were opsonized with human serum at 37°C for 45 min and layered onto MDMs in a ratio of 50:1 (beads to cells) for 18 h, washed twice with PBS, re-suspended in Hank's balanced salt solution and then imaged with an inverted fluorescence microscope or detached by Accutase (Sigma 16964) for FACS analysis. For the bacterial phagocytosis assay, RFP-expressing B. cenocepacia were fixed using 4% paraformaldehyde for 30 min at room temperature. They were then washed five times with PBS and re-suspended in PBS containing 10% AB serum for 60 min at 37°C. Serum-opsonized B. cenocepacia were incubated with MDMs at 50:1 (bacteria to cells) for 40 min at 37°C and then washed with PBS. Last, MDMs were detached and the percent uptake of RFP-expressing B. cenocepacia was detected by FACS analysis.
Reactive Oxygen Species (ROS) Assay
The oxidative burst was measured by a 2′,7′-dichlorofluorescein (DCF) assay (Life Technologies, D399) using relative fluorescent units (RFUs). MDMs were adhered to 96 well plates at 4 × 106 cells/well in duplicate for 2 h, and repleted in Dulbecco's PBS with 10 mM HEPES and 1 mg/ml human serum albumin plus 0.1% glucose (DPBS-HHG). After a 30-min incubation at 37°C, 10% DCF was added to the wells for 30 min at 37°C. A phorbol 12-myristate 13-acetate (PMA) st imulus was added to MDMs and fluorescence measured at a 485 nm excitation wavelength and a 515 nm emission wavelength every 2 min for 2 h. In preliminary experiments, PMA was used at varying concentrations, with 200 μM determined to be optimal for DCF experiments to activate protein-kinase C (PKC)-mediated NADPH oxidase ROS production.
Cytokine Production
Cytokines were measured per prior methods (2). MDM supernatants were collected at baseline or during infection with B. cenocepacia for 18 h, harvested, and stored at −20°C until assayed. Cytokine levels were measured using a cytometric Bead Array (CBA) Human Inflammatory cytokine kit (BD Biosciences, 551811) according to the manufacturer's instructions.
Macrophage Polarization
MDMs were cultured in 12-well plates and exposed to an M1 polarization stimulus using E. coli lipopolysaccharide (LPS) (Sigma, 20 ng/ml) plus recombinant human interferon (rh-IFN)-γ (Fisher, 20 ng/ml) and into M2a using rhIL-4 (20 ng/ml) for 48 h per prior methods (2, 27). Resident cells without any stimulation were used for baseline comparison. Aliquots incubated in human Fc-γ Block (Biolegend, 422301) for 30 min on ice and then stained using phycoerythrin (PE) anti-human CD80 (Biolegend, 305208, Clone 2D10) and phycoerythrin cyanine tandem conjugate (PE/Cy7) anti-human CD11b (Biolegend, 101216, Clone M1/70) or allophycocyanine (APC) anti-human CD163 (Biolegend, 326510, Clone RM3/1) and allophycocyanin cyanine 7 tandem conjugate (APC/Cy7) anti-human CD206 (Biolegend, 321120, Clone 15-2). Next, FITC anti-human CD68 (Biolegend, 333806, Clone Y1/82A) intracellular staining was performed post-fixation and permeabilization according to the manufacture's recommendations. Stained cells were gated via viable CD11b+subsets and identified by CD68CD80 (M1) and CD163CD206 (M2) markers using FlowJo (Tree Star).
Statistical Analysis
Statistical analyses were calculated with GraphPad Prism software (version 8.2). Two sample unpaired t-tests were used for independent sample comparisons. One-way ANOVA was used for denistometry and cytokine analysis with post-hoc Tukey tests. Statistical significance was defined as a p < 0.05. Each individual served as their own internal age- and gender-matched control during experiments when possible (KO vs. no treatment). However, due to cell recovery differences, this was not possible for all replicates and therefore paired t-tests were not performed.
Results
CFTR KO Model
CFTR KO was optimized in non-CF MDMs as described in the methods. The methods section details the timing of transfection, length of incubation, puromycin selection of transfected cells, and use of DOTAP as a transfection media. We found that KO was more effective in differentiated macrophages compared to monocytes during preliminary experiments (data not shown). CFTR KO was first determined visually through identification of flow cytometry sorted GFP+ MDMs prior to puromycin selection. Mean initial transfection efficiency was 30% (Figure 1A). After puromycin selection, CFTR KO efficiency was visually determined at >85% (Figure 1B). To confirm CFTR KO, we measured CFTR expression via qRT-PCR. CFTR expression was significantly reduced in CFTR KO MDMs compared to control MDMs as shown in Figure 2A and summed quantification in Figure 2B. CFTR protein expression was also significantly reduced via western blot (representative blot in Figure 2C and summed densitometry in Figure 2D). All western blots can be found in (Supplementary Figure 1), which also includes an example comparison of different CFTR antibodies. We further confirmed the specificity of CFTR KO by measuring CFTR function via CFTR-dependent chloride efflux. CFTR function was abolished in CFTR KO MDMs compared to untreated vector control non-CF MDMs (Figure 2E). Combined, these results supported robust CFTR KO via our new model as measured through multiple techniques.
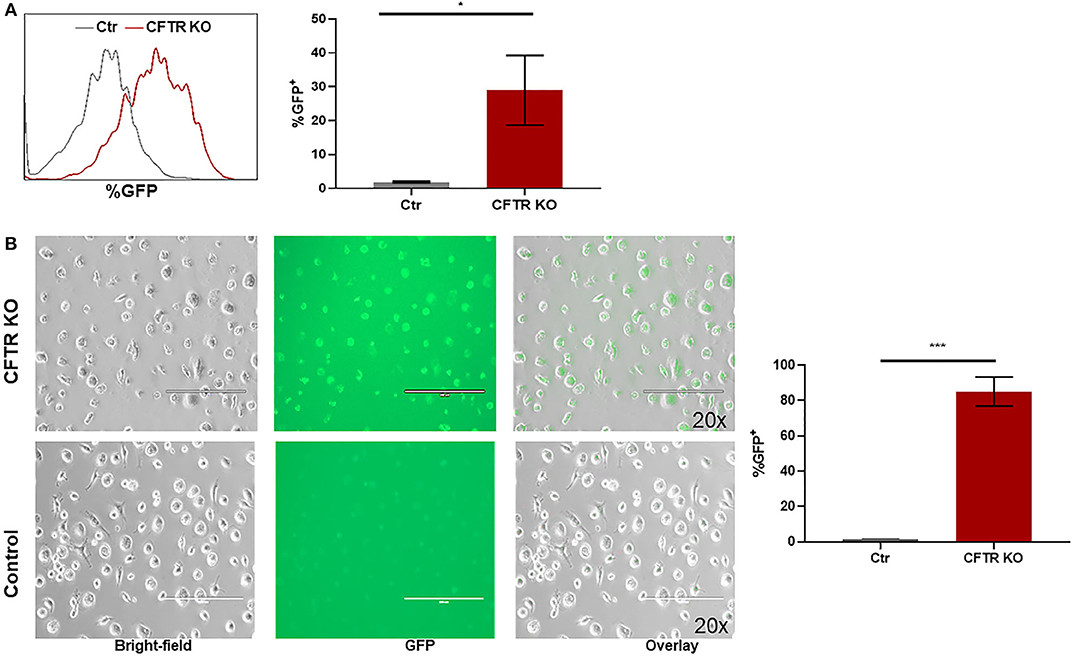
Figure 1. Setup of CFTR KO model. (A) Representative flow cytometry histogram and corresponding summary of GFP detection of CFTR KO MDMs prior to puromycin selection. *p < 0.05 via unpaired t-test, n = 3, mean ± SD. (B) Representative GFP fluorescence images of hMDMs transfected by CFTR Crispr Double Nickase plasmid in DOTAP with corresponding summary of GFP detection, n = 3. (Top) 48 h selection by puromycin. (Bottom) hMDMs in DOTAP only as control. 200 μm scale bar. Bright-field, GFP+, and overlay images shown in left to right order. ***p < 0.0001.
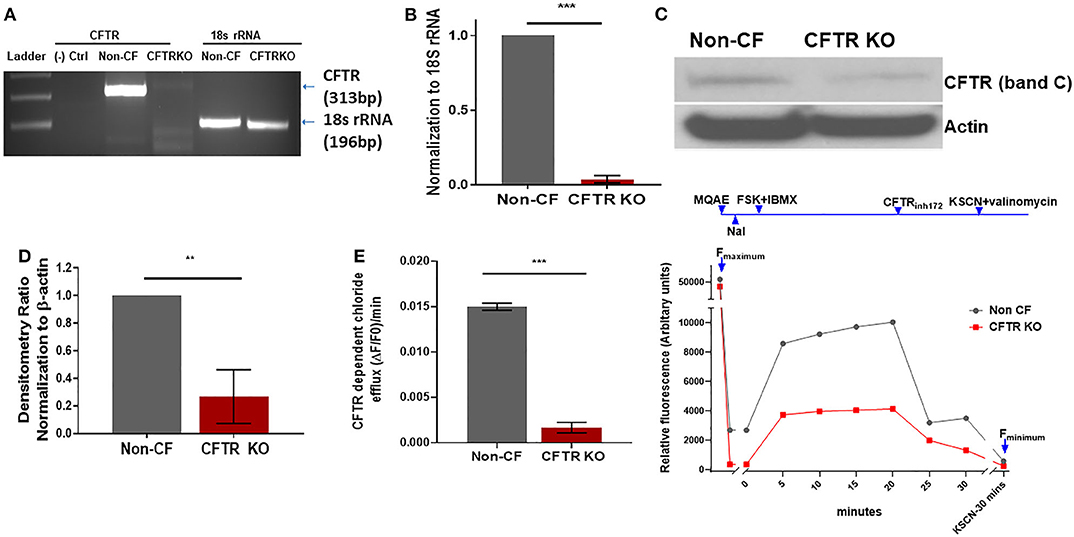
Figure 2. CFTR changes in CFTR KO model. (A) Representative semi-quantitative RT-PCR for CFTR and (B) corresponding quantitative summary of CFTR RT-PCR in CFTR KO and non-CF MDMs, n = 3, mean ± SD. (C) Representative CFTR western blot (Abcam CF3 2784) and (D) summed densitometry of CFTR KO western blots in CFTR KO and non-CF MDMs, n = 3, mean ± SD. (E) MQAE halide efflux assay of CFTR dependent chloride efflux as measure of CFTR function in non-CF and CFTR KO MDMs, n = 6. Maximal fluorescence after forskolin stimulation shown. Representative time course also shown with annotations for forskolin plus IBMX stimulation (FSK + IBMX), CFTR inhibition (CFTRinh172), and quenching with KSCN plus valinomycin. **p < 0.01, ***p < 0.001, via unpaired t-test, mean ± SD.
CFTR KO Alters Macrophage Stability and ROS Production
Next, we sought to determine whether previously observed deficits in human CF macrophages (2, 4) were recapitulated by CFTR KO. We measured baseline apoptosis of unstimulated non-CF and CFTR KO MDMs in culture via annexin V staining. CFTR KO MDMs had 3–4 times higher apoptosis compared to non-CF MDMs (Figure 3A), which is similar to our previous data in primary cells (2). We then measured ROS production in response to PMA stimulus. CFTR KO MDMs demonstrated a significant reduction in max endpoint ROS production in response to PMA (Figure 3B), also similar to our findings in primary CF MDMs (4). ROS production was also higher in non-CF MDMs at initial recording with continued separation over time as shown in the representative time course (Figure 3B). As a result of the deficit in ROS production, we measured expression of p47phox, a critical component necessary for NADPH oxidase activation and subsequent ROS generation. Total p47phox expression was high, and phosphorylated p47phox was low in response to PMA in CFTR KO MDMs, in contrast to non-CF MDMs (Figure 3C). We then measured cytosolic and membrane-bound fractions of phosphorylated p47phox as recruitment of phosphorylated p47phox to the plasma membrane is necessary for functional NADPH oxidase assembly. CFTR KO MDMs showed decreased phosphorylated p47phox in the membrane-bound fraction (Figures 3C–E), again similar to prior findings in human CF MDMs (4). Together, these results suggest that loss of CFTR results in reduced macrophage stability and altered assembly of the NADPH oxidase complex, with resulting decreased ROS production.
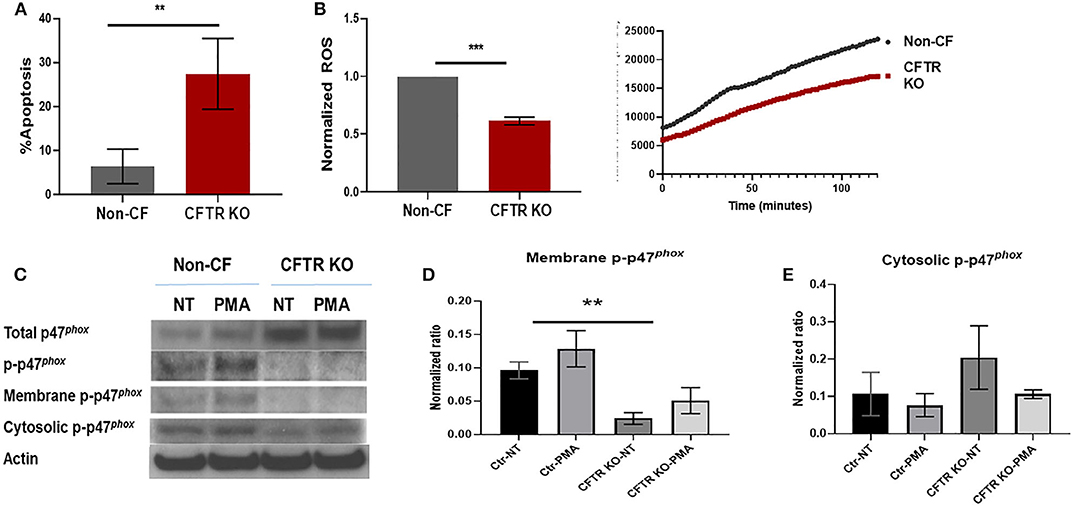
Figure 3. CFTR KO alters macrophage stability and ROS production. (A) Summed apoptosis as measured by flow cytometric detection of Annexin V in CFTR KO and non-CF MDMs, n = 4, mean ± SD. (B) Summed end-point analysis of maximum 2 h ROS production in response to PMA. ROS shown as ratio of max ROS normalized to non-CF. Representative time course of ROS production also shown. n = 3, mean ± SD. (C) Western blots of total, phosphorylated, and membrane and cytosolic fractions of phosphorylated p47phox in non-CF and CFTR KO MDM lysates in response to no treatment (NT) or PMA exposure. Actin shown as loading control. Summed densitometry of western blots for (D) membrane, n = 3 and (E) cytosolic fractions of phosphorylated p47phox, n = 4, in non-CF and CFTR KO MDMs stimulated with PMA, mean ± SD. **p < 0.01, ***p < 0.001, via unpaired t-test for (A,B) and one-way ANOVA for (D,E).
CFTR KO Reduces Bacterial Phagocytosis and Killing
Deficits in bacterial phagocytosis by CF monocytes and macrophages have been reported by numerous groups (2, 18–20, 28). However, a recent study demonstrated no difference between CF and non-CF monocyte phagocytosis of opsonized Escherichia coli (29), although this study was done in whole blood. To clarify these discrepancies, we measured phagocytosis in non-CF and CFTR KO MDMs with opsonized beads. CFTR KO was associated with a significant decrease in phagocytosis of beads compared to non-CF MDMs (Figures 4A–C). We also measured phagocytosis of RFP-labeled B. cenocepacia. CFTR KO was associated with an approximately 40% reduction in bacterial phagocytosis compared to non-CF (Figures 4D,E). Both bead and bacterial results were comparable to our prior data in primary human CF MDMs (2). To determine if these deficits in phagocytosis were associated with decreased killing of bacteria in the CFTR KO model, we measured killing of clinical isolates of three important CF pathogens, Burkholderia cenocepacia, and Pseudomonas aeruginosa. CFTR KO was associated with significantly higher bacterial burden of both B. cenocepacia and P. aeruginosa (Figure 4F, log scale) compared to non-CF MDMs. Together, these results demonstrate that CFTR is necessary for macrophage phagocytosis and subsequent killing of bacteria.
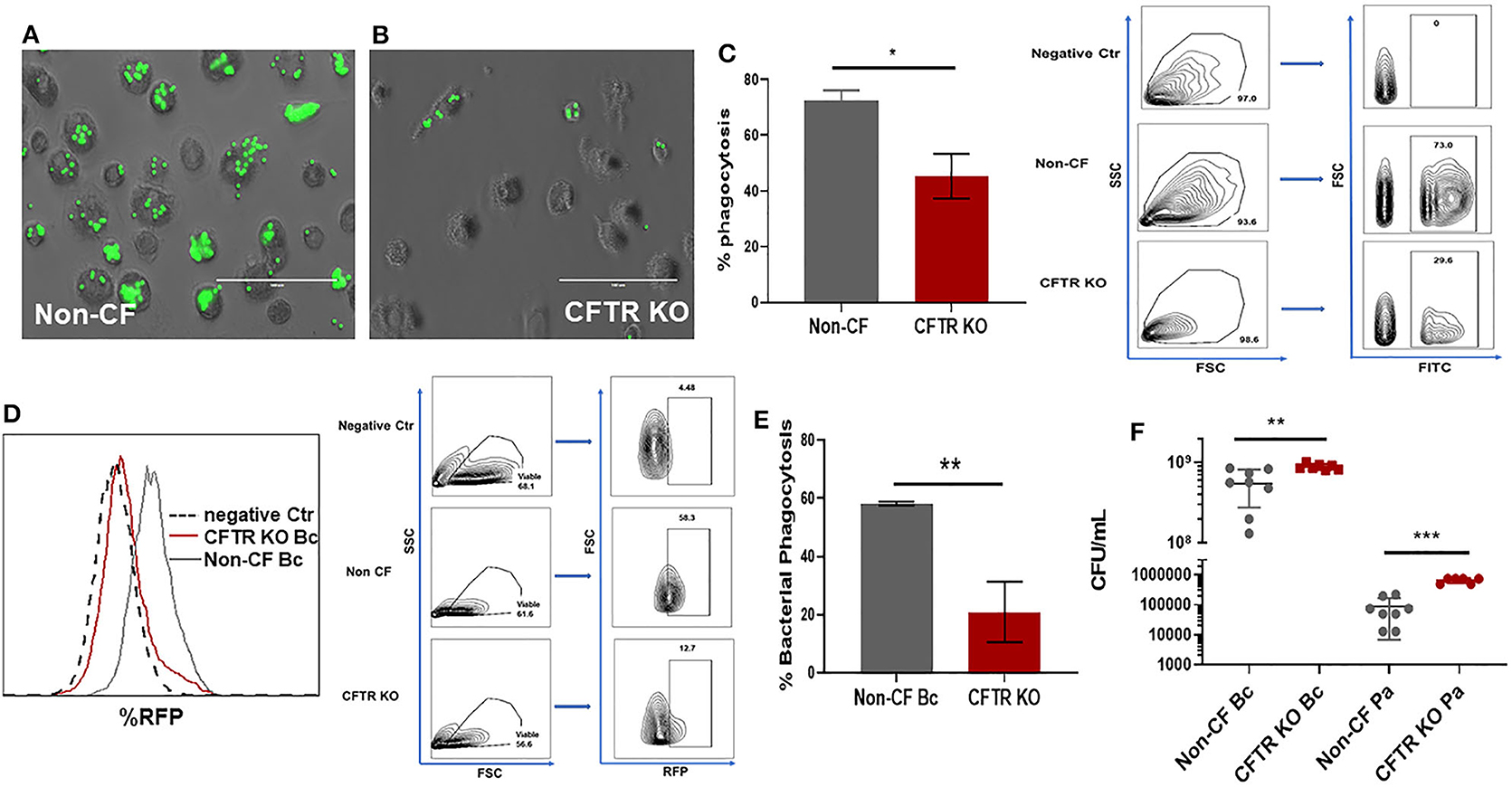
Figure 4. CFTR KO reduces bacterial phagocytosis and killing. Phagocytosis of FITC-labeled beads for (A) non-CF and (B) CFTR KO MDMs, 100 μm scale bar. Non-phagocytosed beads are washed away before imaging. (C) Summed % phagocytosis and representative FACS gating strategy for (A,B), n = 3 donors, mean ± SD. Gating on FSC, SSC, and FITC. Phagocytosis shown as percent of cells with internalized beads. (D) Representative flow cytometry histogram and FACS gating strategy for bacterial phagocytosis assay for non-CF and CFTR KO MDMs. Histogram with overlay difference in uptake of RFP-labeled B. cenocepacia (Bc). The dashed black line (negative Ctr) were non-CF MDMs without infection. FACS gating on FSC, SSC, and RFP. (E) Summed %phagocytosis of RFP-expressing B. cenocepacia, n = 3/group, MOI 50, mean ± SD. (F) CFU assay in non-CF and CFTR KO MDMs infected with B. cenocepacia (Bc) or P. aeruginosa (Pa), log scale, n = 6–8 donors, mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, via unpaired t-test.
CFTR KO Is Not Associated With Increased Cytokine Production
CF is characterized by heightened inflammation, and in particular, increased macrophage cytokine production during infection or other stimuli (2, 6, 22, 30–34). To determine if CFTR KO alters macrophage cytokine production, we measured production of 5 cytokines (IL-8, IL-1β, IL-6, IL-10, and TNF-α) at baseline and in response to infection with B. cenocepacia. At baseline there were no differences in cytokine production between non-CF MDMs in media alone (DOTAP), when transfected with an empty vector (Ctr plasmid), or during CFTR KO (Crispr) (Figure 5A). There were significant reductions in IL-8, IL-6, and TNF-α production during infection in CFTR KO MDMs when compared to media alone or empty vector transfection (Figure 5A). There was also no significant difference in IL-1β or IL-10 production between CFTR KO MDMs compared to media or empty vector, although all CFTR KO MDMs had less IL-1β production compared to empty vector alone. These results suggest that CFTR KO alone does not recapitulate the increased cytokine production observed in human CF MDMs.
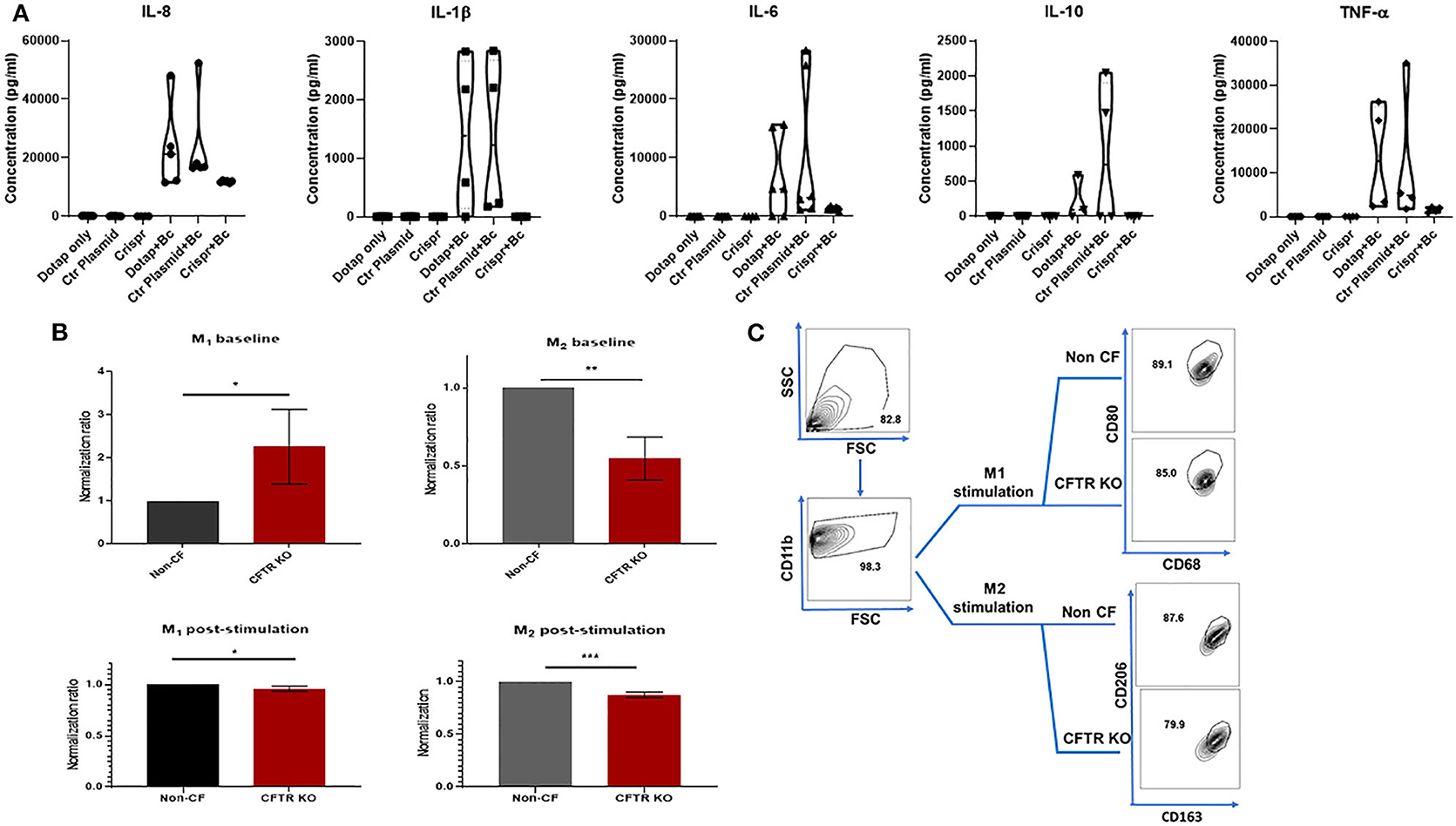
Figure 5. CFTR KO is associated with decreased cytokine and polarization responses. (A) Violin plots for IL-8, IL-1β, IL-6, IL-10, and TNF-α cytokine production. Cytokines were measured in macrophage supernatants at baseline for three conditions (DOTAP medium alone, transfection with an empty vector [Ctr plasmid], or CFTR KO [Crispr]). Cytokines were also measured during infection with B. cenocepacia (Bc) for the same three conditions as occurred at baseline. Significance determined via one-way ANOVA for baseline or infected conditions. n = 4–7 per group. (B) M1 (%CD68+ CD11b+ CD80+) and M2 (%CD11b+ CD163+ CD206+) polarization of non-CF and CFTR KO MDMs without a polarizing stimulus (top, baseline), and after 48 h exposure to a polarizing stimulus (bottom, post-stimulation). Data were normalized to non-CF MDMs. n = 4, mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, via unpaired t-test. (C) FACS gating strategy for 5B post-stimulation studies. M1 & M2 MDM subsets were sequentially gated on FSC/SSC, CD11b, and CD68+CD80+ (M1) or CD163+CD206+(M2). Gating for M1 and M2 baseline from 5B was the same except for lack of M1/M2 stimulus.
CFTR KO Has a Minimal Effect on Macrophage Polarization
Macrophages can exist in different polarized states such as M1 (inflammatory) or M2 (anti-inflammatory/reparative) subsets. Previously reported alterations in CF macrophage polarization phenotypes differ depending on individual factors such as age or medication use, experimental conditions, and use of monocytes or differentiated macrophages (2, 20, 35–37). Because of these prior discrepancies and the unexpected cytokine findings from Figure 5A, we measured changes in macrophage polarization in response to CFTR KO. In unstimulated states, CFTR KO was associated with increased M1 MDMs and decreased M2 MDMs compared to non-CF MDMs (Figures 5B,C). However, in response to M1 or M2a polarizing stimuli, there were small but significantly decreased proportions of both M1 and M2a MDMs after CFTR KO compared to non-CF MDMs (Figures 5B,C). These findings correlate with the decreased cytokine responses from CFTR KO MDMs in Figure 5A.
Discussion
CFTR dysfunction leads to the production of dehydrated mucus, impaired immunity, and subsequent chronic infections that cause progressive lung damage. We now recognize the role of underlying deficits in macrophage and overall innate immune responses that allow bacteria to establish in the CF lung (5, 12, 15, 32, 38–43). It remains unclear whether deficits in CF immune function are a result of intrinsic defects in CFTR (2, 32, 44), a result of the inflammatory CF lung milieu [reviewed in (21, 22)], or more likely a combination of both factors. Through generation of a CFTR KO model in human macrophages we demonstrated several aspects of macrophage dysfunction that are dependent on the presence of CFTR. Conversely, other aberrations observed in primary macrophages, such as increased cytokine production, may be a result of local environmental influences or epigenetic changes over time (22). These findings have implications for future approaches to improve immune responses to infections in CF.
In this study we found that CFTR KO resulted in an increased propensity for macrophages to undergo apoptosis. This finding is consistent with our previous study of CF macrophage (CFTR class I/II/III variants) responses to CFTR modulators, where primary human CF MDMs had increased apoptotic rates that could be reduced with ivacaftor treatment (2). However, primary CF sputum macrophages demonstrate low rates of apoptosis, despite a high likelihood of being macrophages recruited from the peripheral blood (45). These differences in apoptosis may reflect macrophage adaptations to a more stressful lung environment, or may simply be indicative of relative differences in apoptosis between macrophages and other immune cell populations (e.g., neutrophils) when studied simultaneously. Although our findings are consistent with increased apoptosis in alveolar macrophages that have decreased CFTR after treatment with CFTR specific small interfering RNA (CFTR-siRNA) (46), we believe that adaptation to the CF lung likely influences CF macrophage responses. Future studies need to examine if therapies that decrease CF macrophage apoptosis allow for improved functional capabilities, whether increased CF macrophage apoptosis contributes to the accumulation of inflammatory cellular debris in the lung, and how CF macrophage apoptosis influences macrophage extracellular trap formation that may also be involved in pathogen clearance or sterile inflammation.
While increased apoptosis could have a variety of impacts upon CF macrophage responses, we also found that CFTR KO decreased two important aspects of macrophage host defenses, phagocytosis and ROS production. These decreased defense mechanisms likely create a two-pronged insult that contributes to the increased bacterial loads observed with CFTR KO. CF macrophages are first unable to effectively phagocytose bacteria, and if successful at phagocytosis are subsequently unable to mount effective intracellular killing responses. CF macrophages have been previously shown to have several deficits related to phagocytosis, including deregulated transient receptor potential cation channel subfamily V member 2 (TRPV2)-mediated calcium influx, (19) reduced Ezrin-mediated toll-like receptor-4 (TLR4) signaling (47), and reduced complement-mediated phagocytosis (28). Additionally, ROS responses are altered in both human CF macrophages (4) and CF neutrophils (17). We are currently investigating the mechanism whereby CFTR dysfunction results in defective NADPH oxidase assembly (as shown by decreased phosphorylation of cytosolic components), and whether this deficit is dependent on phagocytosis-mediated signaling.
In contrast to most findings in this study, changes in cytokine production with CFTR KO did not reflect alterations previously observed in human CF macrophages (2, 6, 21, 22). Surprisingly, CFTR KO was associated with decreased production of several inflammatory cytokines in response to infection. Decreased cytokine production was in contrast to a prior study of siRNA-silencing of CFTR in alveolar macrophages (46), although the authors only examined basal IL-8 production and not responses to infection. Our findings may be related to the small but significant decrease in the ability of CFTR KO macrophages to respond to a pro-inflammatory M1 stimulus, despite a higher proportion of M1 macrophages after CFTR KO in the absence of infection. The observed deficits in CFTR KO macrophage responses to M1 or M2 polarization stimuli are similar to what we observed in primary CF macrophages (2), as well as decreased CF macrophage M2 polarization responses shown by other groups (20). Other studies have shown that medications such as azithromycin can alter CF macrophage polarization (36), but our studies were performed in the absence of any in vitro medication exposures. It is also possible that other laboratory conditions influence the polarization status such as the type of media, serum, or polarizing stimuli (48). However, because we did not have an increase in M2 polarized macrophages after CFTR KO, we do not think our conditions would be related to decreased inflammatory cytokine production in CFTR KO only. We suggest that all the findings combined support the assertion that CF macrophages are not inherently hyper-inflammatory, but are metabolically re-programmed to a hyper-inflammatory state in CF as originally postulated by Wilson and Fudenberg (49) and supported by recent studies (31). The concept of induced innate immune memory (50) also needs further investigation in CF and could contribute to hyper-inflammatory macrophage signaling. However, early and sustained use of new CFTR modulators may help reverse potential alterations in macrophage inflammatory responses that adversely impact CF outcomes. Prior studies of first-generation CFTR modulators have not shown sustained reductions in sputum inflammatory markers (51, 52). New investigations into early immune responses in CF are the focus of several ongoing studies around the globe.
This study was limited to the use of MDMs and did not include alveolar, sputum, or tissue-resident macrophages, which may respond differently to CFTR KO based on distinct transcriptional profiles in CF (53). Alveolar macrophages have become more difficult to obtain since the recent release of elexacaftor/tezacaftor/ivacaftor, which has decreased rates of expectorated sputum and clinical indications for flexible bronchoscopies. MDMs are recruited to sites throughout the body (including the lungs) and remain an easily accessible source of CF macrophages, lending utility to the current model for future studies in the era of highly effective CFTR modulator therapy. The CFTR KO model could also be used to test new therapeutic strategies in patients with class I CFTR variants, as this model closely resembles a clinical state where no CFTR protein is expressed. However, it will remain important for future studies to compare the model to primary patient macrophages of different CFTR variants as well as use CFTR channel inhibition with chemical inhibitors to look at the role of ion signaling. It is also possible that the cytokine differences observed in our KO model compared to primary human CF macrophages are an inherent limitation of the KO model. Although our control samples with transfection media alone or empty vehicle did not demonstrate altered cytokine responses, these studies can be further validated in future studies looking at long-term primary macrophage responses to highly effective CFTR modulators.
In summary, we developed a CRISPR-Cas9 CFTR KO model to study the impact of CFTR on macrophage function. CFTR KO recapitulated alterations in primary CF macrophage functions including apoptosis, phagocytosis, ROS production, and bacterial killing. CFTR KO was not associated with increased inflammatory cytokine production. This study suggests that many aspects of CF macrophage dysfunction are CFTR-dependent, which is important as we develop new approaches to regulate immune responses to infection in CF.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Nationwide Children's Hospital IRB (IRB16-01020). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
SZ performed scientific experiments, contributed to study design and analysis, and edited the manuscript. CS assisted with scientific experiments and edited the manuscript. BW assisted with experiments and edited the manuscript. HP collected patient samples and edited the manuscript. XH, WL, and YD assisted in implementation and optimization of the CRISPR-Cas9 KO model and edited the manuscript. BK designed the experiments, recruited participants, analyzed data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by CF Foundation grant KOPP16I0 (BK) and NIH 1K08AI108792 (BK). Macrophages for this work were supplied by the Cure CF Columbus Immune Core (C3IC) at Nationwide Children's Hospital. C3IC was supported by the Division of Pediatric Pulmonary Medicine and the C3 Translational Core. C3IC Grant support provided by The Ohio State University Center for Clinical and Translational Science (National Center for Advancing Translational Sciences, Grant UL1TR002733) and by the Cystic Fibrosis Foundation [Research Development Program, Grant MCCOY19RO– Core C (BK)].
Conflict of Interest
BK has previously served on the US CF Advisory Board for Vertex Pharmaceuticals to provide advice on clinical trial development.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the participants and their families for their contributions. Thank you to Dr. Jamel El-Benna for the phospho-p47phox antibody.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01871/full#supplementary-material
Abbreviations
APC, allophycocyanine; APC/Cy7, allophycocyanin cyanine 7 tandem conjugate; CFTRinh172, CFTR inhibitor 172; CRISPR-Cas9, clustered regularly interspaced short palindromic repeats associated protein 9; cDNA, complementary DNA; CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; CFU, Colony forming unit; DCF, 2′,7′-dichlorofluorescein; EDTA, Ethylenediaminetetraacetic acid; ET12, Edinburgh-Toronto epidemic lineage strain; FACS, fluorescence-activated cell sorting; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; HBS, HEPES buffered saline; IBMX, 3-isobutyl-1-methylxanthine; KO, knockout; LPS, lipopolysaccharide; LB, Luria-Bertani; MDMs, monocyte-derived macrophages; MOI, multiplicity of infection; DOTAP, N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate; MQAE, N-Ethoxycarbonylmethyl-6-Methoxyquinolinium Bromide; NADPH, nicotinamide adenine dinucleotide phosphate; PE, phycoerythrin conjugate; PECy7, phycoerythrin cyanine 7 tandem conjugate; PKC, protein-kinase C; PBS, phosphate buffered saline; PMA, phorbol 12-myristate 13-acetate; ROS, reactive oxygen species; RFUs, relative fluorescent units; rhIFN-γ, recombinant human interferon gamma; RPMI, Roswell Park Memorial Institute; qRT-PCR, semi-quantitative reverse transcription polymerase chain reaction; siRNA, small-interfering RNA; TLR4, toll-like receptor-4; TRPV2, transient receptor potential cation channel subfamily V member 2.
References
2. Zhang S, Shrestha CL, Kopp BT. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci Rep. (2018) 8:17066. doi: 10.1038/s41598-018-35151-7
3. Sorio C, Buffelli M, Angiari C, Ettorre M, Johansson J, Vezzalini M, et al. Defective CFTR expression and function are detectable in blood monocytes: development of a new blood test for cystic fibrosis. PLoS ONE. (2011) 6:e22212. doi: 10.1371/journal.pone.0022212
4. Assani K, Shrestha CL, Robledo-Avila F, Rajaram MV, Partida-Sanchez S, Schlesinger LS, et al. Human cystic fibrosis macrophages have defective calcium-dependent protein kinase c activation of the NADPH oxidase, an effect augmented by burkholderia cenocepacia. J Immunol. (2017) 198:1985–94. doi: 10.4049/jimmunol.1502609
5. Assani K, Tazi MF, Amer AO, Kopp BT. IFN-gamma stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS ONE. (2014) 9:e96681. doi: 10.1371/journal.pone.0096681
6. Kopp BT, Abdulrahman BA, Khweek AA, Kumar SB, Akhter A, Montione R, et al. Exaggerated inflammatory responses mediated by Burkholderia cenocepacia in human macrophages derived from Cystic fibrosis patients. Biochem Biophys Res Commun. (2012) 424:221–7. doi: 10.1016/j.bbrc.2012.06.066
7. Kotrange S, Kopp B, Akhter A, Abdelaziz D, Abu Khweek A, Caution K, et al. Burkholderia cenocepacia O polysaccharide chain contributes to caspase-1-dependent IL-1beta production in macrophages. J Leukoc Biol. (2011) 89:481–8. doi: 10.1189/jlb.0910513
8. Shrestha CL, Assani KD, Rinehardt H, Albastroiu F, Zhang S, Shell R, et al. Cysteamine-mediated clearance of antibiotic-resistant pathogens in human cystic fibrosis macrophages. PLoS ONE. (2017) 12:e0186169. doi: 10.1371/journal.pone.0186169
9. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. (2011) 7:1359–70. doi: 10.4161/auto.7.11.17660
10. Tazi MF, Dakhlallah DA, Caution K, Gerber MM, Chang SW, Khalil H, et al. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy. (2016) 12:2026–37. doi: 10.1080/15548627.2016.1217370
11. Forrest OA, Ingersoll SA, Preininger MK, Laval J, Limoli DH, Brown MR, et al. Frontline science: pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J Leukoc Biol. (2018) 104:665–75. doi: 10.1002/JLB.5HI1117-454RR
12. Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am J Respir Cell Mol Biol. (2009) 40:295–304. doi: 10.1165/rcmb.2008-0170OC
13. Riquelme SA, Hopkins BD, Wolfe AL, DiMango E, Kitur K, Parsons R, et al. Cystic fibrosis transmembrane conductance regulator attaches tumor suppressor PTEN to the membrane and promotes anti pseudomonas aeruginosa immunity. Immunity. (2017) 47:1169–81.e1167. doi: 10.1016/j.immuni.2017.11.010
14. Shah VS, Meyerholz DK, Tang XX, Reznikov L, Abou Alaiwa M, Ernst SE, et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science. (2016) 351:503–7. doi: 10.1126/science.aad5589
15. Sorio C, Montresor A, Bolomini-Vittori M, Caldrer S, Rossi B, Dusi S, et al. Mutations of cystic fibrosis transmembrane conductance regulator gene cause a monocyte-selective adhesion deficiency. Am J Respir Crit Care Med. (2016) 193:1123–33. doi: 10.1164/rccm.201510-1922OC
16. Bartlett JA, Ramachandran S, Wohlford-Lenane CL, Barker CK, Pezzulo AA, Zabner J, et al. Newborn cystic fibrosis pigs have a blunted early response to an inflammatory stimulus. Am J Respir Crit Care Med. (2016) 194:845–54. doi: 10.1164/rccm.201510-2112OC
17. Robledo-Avila FH, Ruiz-Rosado JD, Brockman KL, Kopp BT, Amer AO, McCoy K, et al. Dysregulated calcium homeostasis in cystic fibrosis neutrophils leads to deficient antimicrobial responses. J Immunol. (2018) 201:2016–27. doi: 10.4049/jimmunol.1800076
18. Barnaby R, Koeppen K, Nymon A, Hampton TH, Berwin B, Ashare A, et al. Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L432–8. doi: 10.1152/ajplung.00461.2017
19. Leveque M, Penna A, Le Trionnaire S, Belleguic C, Desrues B, Brinchault G, et al. Phagocytosis depends on TRPV2-mediated calcium influx and requires TRPV2 in lipids rafts: alteration in macrophages from patients with cystic fibrosis. Sci Rep. (2018) 8:4310. doi: 10.1038/s41598-018-22558-5
20. Tarique AA, Sly PD, Holt PG, Bosco A, Ware RS, Logan J, et al. CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J Cyst Fibros. (2017) 16:475–82. doi: 10.1016/j.jcf.2017.03.011
21. Bruscia EM, Bonfield TL. Innate and adaptive immunity in cystic fibrosis. Clin Chest Med. (2016) 37:17–29. doi: 10.1016/j.ccm.2015.11.010
22. Bruscia EM, Bonfield TL. Cystic fibrosis lung immunity: the role of the macrophage. J Innate Immun. (2016) 6:550–63. doi: 10.1159/000446825
23. Goncalves GAR, Paiva RMA. Gene therapy: advances, challenges and perspectives. Einstein(São Paulo). (2017) 15:369–75. doi: 10.1590/s1679-45082017rb4024
24. Ramamoorth M, Narvekar A. Non viral vectors in gene therapy- an overview. J Clin Diagn Res. (2015) 9:GE01–6. doi: 10.7860/JCDR/2015/10443.5394
25. Schlesinger LS. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol. (1993) 150:2920–30.
26. Mahenthiralingam E, Coenye T, Chung JW, Speert DP, Govan JR, Taylor P, et al. Diagnostically and experimentally useful panel of strains from the Burkholderia cepacia complex. J Clin Microbiol. (2000) 38:910–3. doi: 10.1128/JCM.38.2.910-913.2000
27. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio. (2013) 4:e00264–13. doi: 10.1128/mBio.00264-13
28. van de Weert-van Leeuwen PB, van Meegen MA, Speirs JJ, Pals DJ, Rooijakkers SH, van der Ent CK, et al. Optimal complement-mediated phagocytosis of Pseudomonas aeruginosa by monocytes is cystic fibrosis transmembrane conductance regulator-dependent. Am J Respir Cell Mol Biol. (2013) 49:463–70. doi: 10.1165/rcmb.2012-0502OC
29. Leuer L, Krill A, Wilkens H, Wagenpfeil G, Bischoff M, Meier C, et al. The phagocytosis of blood leukocytes from cystic fibrosis patients is not impaired in general. Lung. (2019) 198:235–9. doi: 10.1007/s00408-019-00290-9
30. Luly FR, Leveque M, Licursi V, Cimino G, Martin-Chouly C, Theret N, et al. MiR-146a is over-expressed and controls IL-6 production in cystic fibrosis macrophages. Sci Rep. (2019) 9:16259. doi: 10.1038/s41598-019-52770-w
31. Lara-Reyna S, Scambler T, Holbrook J, Wong C, Jarosz-Griffiths HH, Martinon F, et al. Metabolic reprograming of cystic fibrosis macrophages via the IRE1α arm of the unfolded protein response results in exacerbated inflammation. Front Immunol. (2019) 10:1789. doi: 10.3389/fimmu.2019.01789
32. Paemka L, McCullagh BN, Abou Alaiwa MH, Stoltz DA, Dong Q, Randak CO, et al. Monocyte derived macrophages from CF pigs exhibit increased inflammatory responses at birth. J Cyst Fibros. (2017) 16:471–4. doi: 10.1016/j.jcf.2017.03.007
33. Leveque M, Le Trionnaire S, Del Porto P, Martin-Chouly C. The impact of impaired macrophage functions in cystic fibrosis disease progression. J Cyst Fibros. (2017) 16:443–53. doi: 10.1016/j.jcf.2016.10.011
34. Lubamba BA, Jones LC, O'Neal WK, Boucher RC, Ribeiro CM. X-box-binding protein 1 and innate immune responses of human cystic fibrosis alveolar macrophages. Am J Respir Crit Care Med. (2015) 192:1449–61. doi: 10.1164/rccm.201504-0657OC
35. Tarique AA, Sly PD, Cardenas DG, Luo L, Stow JL, Bell SC, et al. Differential expression of genes and receptors in monocytes from patients with cystic fibrosis. J Cyst Fibros. (2019) 18:342–8. doi: 10.1016/j.jcf.2018.07.012
36. Cory TJ, Birket SE, Murphy BS, Hayes D Jr, Anstead MI, Kanga JF, et al. Impact of azithromycin treatment on macrophage gene expression in subjects with cystic fibrosis. J Cyst Fibros. (2014) 13:164–71. doi: 10.1016/j.jcf.2013.08.007
37. Murphy BS, Bush HM, Sundareshan V, Davis C, Hagadone J, Cory TJ, et al. Characterization of macrophage activation states in patients with cystic fibrosis. J Cyst Fibros. (2010) 9:314–22. doi: 10.1016/j.jcf.2010.04.006
38. Yonker LM, Cigana C, Hurley BP, Bragonzi A. Host-pathogen interplay in the respiratory environment of cystic fibrosis. J Cyst Fibros. (2015) 14:431–9. doi: 10.1016/j.jcf.2015.02.008
39. Peters-Hall JR, Brown KJ, Pillai DK, Tomney A, Garvin LM, Wu X, et al. Quantitative proteomics reveals an altered cystic fibrosis in vitro bronchial epithelial secretome. Am J Respir Cell Mol Biol. (2015) 53:22–32. doi: 10.1165/rcmb.2014-0256RC
40. Tang AC, Turvey SE, Alves MP, Regamey N, Tummler B, Hartl D. Current concepts: host-pathogen interactions in cystic fibrosis airways disease. Eur Respir Rev. (2014) 23:320–32. doi: 10.1183/09059180.00006113
41. Rieber N, Hector A, Carevic M, Hartl D. Current concepts of immune dysregulation in cystic fibrosis. Int J Biochem Cell Biol. (2014) 52:108–12. doi: 10.1016/j.biocel.2014.01.017
42. Keiser NW, Birket SE, Evans IA, Tyler SR, Crooke AK, Sun X, et al. Defective innate immunity and hyper-inflammation in newborn CFTR-knockout ferret lungs. Am J Respir Cell Mol Biol. (2015) 52:683–94. doi: 10.1165/rcmb.2014-0250OC
43. Zhang PX, Cheng J, Zou S, D'Souza AD, Koff JL, Lu J, et al. Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat Commun. (2015) 6:6221. doi: 10.1038/ncomms7221
44. Ng HP, Jennings S, Wellems D, Sun F, Xu J, Nauseef WM, et al. Myeloid CFTR loss-of-function causes persistent neutrophilic inflammation in cystic fibrosis. J Leukoc Biol. (2020). doi: 10.1002/JLB.3A0520-193RR. [Epub ahead of print].
45. Hisert KB, Liles WC, Manicone AM. A flow cytometric method for isolating cystic fibrosis airway macrophages from expectorated sputum. Am J Respir Cell Mol Biol. (2019) 61:42–50. doi: 10.1165/rcmb.2018-0236MA
46. Xu Y, Krause A, Hamai H, Harvey BG, Worgall TS, Worgall S. Proinflammatory phenotype and increased caveolin-1 in alveolar macrophages with silenced CFTR mRNA. PLoS ONE. (2010) 5:e11004. doi: 10.1371/journal.pone.0011004
47. Di Pietro C, Zhang PX, O'Rourke TK, Murray TS, Wang L, Britto CJ, et al. Ezrin links CFTR to TLR4 signaling to orchestrate anti-bacterial immune response in macrophages. Sci Rep. (2017) 7:10882. doi: 10.1038/s41598-017-11012-7
48. Paoletti A, Rohmer J, Ly B, Pascaud J, Riviere E, Seror R, et al. Monocyte/macrophage abnormalities specific to rheumatoid arthritis are linked to miR-155 and are differentially modulated by different TNF inhibitors. J Immunol. (2019) 203:1766–75. doi: 10.4049/jimmunol.1900386
49. Wilson GB, Fudenberg HH. Does a primary host defense abnormality involving monocytes-macrophages underlie the pathogenesis of lung disease in cystic fibrosis? Med Hypotheses. (1982) 8:527–42. doi: 10.1016/0306-9877(82)90014-7
50. Dominguez-Andres J, Joosten LA, Netea MG. Induction of innate immune memory: the role of cellular metabolism. Curr Opin Immunol. (2019) 56:10–6. doi: 10.1016/j.coi.2018.09.001
51. Harris JK, Wagner BD, Zemanick ET, Robertson CE, Stevens MJ, Heltshe SL, et al. Changes in airway microbiome and inflammation with ivacaftor treatment in patients with cystic fibrosis and the G551D mutation. Ann Am Thorac Soc. (2020) 17:212–20. doi: 10.1513/AnnalsATS.201907-493OC
52. Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. (2014) 190:175–84. doi: 10.1164/rccm.201404-0703OC
Keywords: cystic fibrosis, macrophage, bacteria, CRISPR, CFTR
Citation: Zhang S, Shrestha CL, Wisniewski BL, Pham H, Hou X, Li W, Dong Y and Kopp BT (2020) Consequences of CRISPR-Cas9-Mediated CFTR Knockout in Human Macrophages. Front. Immunol. 11:1871. doi: 10.3389/fimmu.2020.01871
Received: 01 June 2020; Accepted: 13 July 2020;
Published: 18 August 2020.
Edited by:
Alexandre Corthay, Oslo University Hospital, NorwayReviewed by:
Luigia Grazia Fresu, University of Eastern Piedmont, ItalyBettina C. Schock, Queen's University Belfast, United Kingdom
Susan E. Birket, University of Alabama at Birmingham, United States
Copyright © 2020 Zhang, Shrestha, Wisniewski, Pham, Hou, Li, Dong and Kopp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin T. Kopp, YmVuamFtaW4ua29wcEBuYXRpb253aWRlY2hpbGRyZW5zLm9yZw==