 Vitaliy Davidov
Vitaliy Davidov Garrett Jensen
Garrett Jensen Sunny Mai2
Sunny Mai2 Shu-Hsia Chen
Shu-Hsia Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 September 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01842
This article is part of the Research Topic Roles of Tumor-Recruited Myeloid Cells in Immune Evasion in Cancer View all 13 articles
Tumor-mediated regulation of the host immune system involves an intricate signaling network that results in the tumor's inherent survival benefit. Myeloid cells are central in orchestrating the mechanisms by which tumors escape immune detection and continue their proliferative programming. Myeloid cell activation has historically been classified using a dichotomous system of classical (M1-like) and alternative (M2-like) states, defining general pro- and anti-inflammatory functions, respectively. Explosions in bioinformatics analyses have rapidly expanded the definitions of myeloid cell pro- and anti-inflammatory states with different combinations of tissue- and disease-specific phenotypic and functional markers. These new definitions have allowed researchers to target specific subsets of disease-propagating myeloid cells in order to modify or arrest the natural progression of the associated disease, especially in the context of tumor-immune interactions. Here, we discuss the myeloid cell contribution to solid tumor initiation and maintenance, and strategies to reprogram their phenotypic and functional fate, thereby disabling the network that benefits tumor survival.
In recent decades the traditional view of tumor development and metastasis has evolved to include new and emerging cell types, extrinsic to the tumor itself. Over time it has become apparent that tumors are composed of many cell types from different origins, all with varying functions. By defining the tumor as a distinct organ, cell populations can be broadly separated into two categories: parenchymal tumor cells and stromal tumor-associated cells. Tumor-associated cells can originate either from the tissue in which the malignancy arises, or they can migrate from the periphery and infiltrate the tumor after it forms. The tumor itself and the tumor-associated cells together comprise what is termed the tumor microenvironment (1). When the tumor microenvironment being discussed relates to the influx and function of the immune system, it is termed the tumor immune microenvironment (TIME) (2). Therapies targeting different components of the tumor microenvironment, such as neovascularization, cellular proliferation, growth factors, extracellular matrix proteins, and more, have all been utilized to regulate tumor growth, each with various levels of success (3). More recently, targeting the immune component of the malignancy, deemed immunotherapy, has shown great promise and curative potential in several tumors (4).
Fundamentally, the goal of immunotherapy is to modulate the mechanisms that tumors use to suppress the immune response. The ability of a tumor to evade immune mediated killing is one of the hallmarks of cancer development, highlighting the importance of the immune response in preventing cancer formation (1). Classically, the immune system is divided into two branches: adaptive and innate. The innate division determines how to respond to danger by sensing the environment with an array of pattern recognition receptors and cytokine receptors that allow them to sense tissue damage, pathogens, and inflammation. The defining feature of the adaptive branch is its ability to respond in an antigen specific manner and memory responses (5). The importance of the innate immune system in regulating malignancies has come into sharper focus with the discovery of immunomodulatory myeloid cells residing within and around tumors. These myeloid cells are known to play a central role in suppressing adaptive immunity and are comprised of diverse clusters that fulfill various roles in promoting the viability of the developing malignancy. Two central groups of suppressive myeloid cells are the tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) (6). Initially called natural suppressor cells, these cells were shown to inhibit cytotoxic lymphocyte activity and support tumor growth (7). A body of work has shown that tumor development frequently causes defects in the differentiation and activity of myeloid cells, ultimately leading to a functional state that favors the tumor progression. Given the massive heterogeneity of infiltrating leukocytes found in tumors, and the striking difference in the TIME seen between different tumor types, there is a need to better understand the mechanisms contributing to this overall immune suppressive environment at the single cell and high-dimensional level. Advances in single-cell RNA sequencing (scRNAseq) and mass cytometry have enabled these types of studies and comparisons and are giving rise to new generations of data that may provide greater understanding of the mechanisms leading to immune suppression, TAM and MDSC polarization, and immune evasion.
Studies testing the potential of modulating the TIME via altering cellular recruitment, differentiation, proliferation, and survival are currently underway. These are reviewed elsewhere (8–11). Here, we discuss tumor associated suppressive myeloid cells, analyze recent findings obtained through high resolution dissection of their phenotypes, and highlight potential reprogramming strategies to orient cells toward anti-tumor functionality.
In the 1960's, it was first observed that tumor bearing mice developed a leukemoid reaction with expanded myeloid cell populations in both the circulation and in the tumor. This correlated with enhanced tumor growth and these cells were subsequently shown to suppress cytotoxic T cell activity (7). Over time additional research has demonstrated that these myeloid cells exist as two separate populations: TAMs and MDSCs (12). Studies seeking to understand the factors that led to the differentiation of these populations demonstrated that tumor-associated macrophages develop from both tissue resident and circulating monocyte populations (13). New myeloid cells recruited from the bone marrow exhibit different programming from embryonically derived tissue resident macrophages (TRMs) (14), and commonly represent the definition of “tumor-associated macrophage” populations (15, 16), albeit not without debate, depending on tumor model (17–19).
Correctly identifying MDSCs in vivo remains challenging despite decades of intense study. MDSCs are commonly identified in tumor bearing mice by the Gr-1 surface marker, and recently, CD84 has arrived into the spotlight as another potential marker in murine models. There is potential for application of CD84 to differentiate MDSCs from conventional myeloid cells in human studies, but this has yet to be validated (20). Despite shortcomings in MDSC phenotypic definitions, several surface markers are employed in the literature with varying degrees of success and have been discussed elsewhere (12, 21). Thus, the gold standard and only reliable method to correctly identify MDSCs is to evaluate their ability to suppress CD3-mediated T cell activation and function in vitro (22–24).
MDSC recruitment and maintenance within the tumor tissue is thought to be more complex than that for TAMs, in part because of the hypothesized signaling required to maintain MDSCs in an immature state. This is thought to be accomplished by a combination of multiple growth factors and polyunsaturated fatty acids (25). Supplementary inflammatory signals generated by the tumor traps these immature cells in a pathogenic suppressive state (25, 26). A combination of TLR4/IFNγ/GM-CSF signaling and activation of intracellular STAT3 is needed to control the development and function of MDSCs (27–30). MDSCs are typically replenished by bone marrow precursors and the spleen functions as their reservoir (31), but it is unclear as to how extramedullary hematopoiesis contributes to their replenishment.
A growing body of evidence supports that MDSCs retain some ability to polarize to a cell displaying more characteristics of typical monocytes (32, 33). Genetic and pharmacologic methods can promote maturation or polarization in MDSCs, with multiple groups reporting that M-MDSCs can be functionally characterized into not only suppressive states, but also into reactive states (32, 34, 35). Transcriptional programs initiated by c-EBPβ, STAT3, PU.1, IRF8, and RORC1, among others, regulate the suppressive activities of MDSCs (36). Blocking these programs to force MDSCs into an activating, rather than suppressive role, is a potential therapeutic strategy, with several mechanisms to do so (37). MDSCs represent just one of the suppressive populations in the TIME; quantifying the phenotypes and functional states of the environment at the single cell level will offer more clues for therapeutic applications.
Tumor-associated macrophages comprise the macrophage populations located in and around a solid tumor (38). Originating from both tissue resident macrophages and circulating monocytes, TAMs are also known to perform a prominent role in modulating immune responses to tumors (39). TAMs can arise from peripheral monocytes in response to a combination of CCL2 and CSF1 produced by the tumor (27–30, 40, 41). Once monocytes reach the tumor site, they follow a maturation course that leads to their TAM finale (42, 43), under the influence of tumor factors, local cytokine milieu, and integrin signaling (44). Other than replenishment of TAM populations, the role of undifferentiated monocytes within the TIME has not been clearly defined at the single-cell level. Additional important signaling pathways resulting in macrophage recruitment and subsequent TAM differentiation include VEGF, IL-4, CCL2, CCL18, and CCL9 (45). TAMs further mobilize additional TAMs to the tumor niche by signaling to the bone marrow via CCL8 (14) to replenish and maintain their populations, although an undefined mechanism for the transition of TRMs to TAMs has been observed (15). Through a combination of TLR and cytokine signaling, infiltrating MDSCs can also differentiate into TAMs and function as a source of TAM replenishment (46–49).
TAMs are identified and distinguished from MDSCs by the presence of characteristic surface markers that are shared with mature macrophages (22). Frequently described as M2-like macrophages, TAMs have distinct phenotypic and transcriptional characteristics that can be used to distinguish them from conventional M2-macrophages. Additionally, TAMs demonstrate marked immunosuppressive functionality not seen in the M2 macrophage population (50).
TRMs have an interesting role in tumorigenesis. Because they develop with the tissue, they are present long before any noticeable malignancy, but are thought to contribute to the early stages of tumor development (2). The contribution of various myeloid cell ontologies to tumor development and immunosuppression is highly debated (51), although myeloid cells recruited from the periphery seemingly have a more important role in propagating the growth and invasiveness of malignancies (17, 52). However, this might be a tumor-specific phenomenon, as evidence from breast cancer patients and murine models shows proliferating resident macrophages in the tumor contributing to the bulk of the myeloid compartment (19). Interestingly, there is some evidence that both populations may also play distinct roles in supporting tumor growth, and their origins bias their transcriptional networks (53). Therefore, it is possible that the developing tumor modulates both the tissue resident and infiltrating myeloid cell populations concurrently.
The mechanisms of immunosuppression employed by TAMs and MDSCs are targeted toward inhibiting the activity of the adaptive immune system, namely T-cells, and NK cells. Suppressive myeloid cells do so by either direct cell-cell interaction with target cells, or through secreted factors. The mechanisms to suppress anti-tumor immune responses in vivo and have been extensively reviewed elsewhere (45, 50, 54–56). Briefly, they utilize four distinct functions to suppress T-cell mediated immunity: (1) signaling via the stereotypical inhibitory receptors PD-1 and CTLA-4 mediate leukocyte apoptosis and anergy (57–61); (2) depriving the local environment of nutrients necessary for T-cell activation and function (62–67); (3) generation of nitric oxygen and reactive nitrogen species, by iNOS expression, that induce T-cell exhaustion (12, 23, 62, 68, 69); (4) production of reactive oxygen species (12, 70). These mechanisms ultimately lead to a decrease in the effect and numbers of anti-tumor T-cells while enhancing the populations of tumor supporting regulatory T-cells (23, 24, 71, 72).
Stereotypically, STAT and PPAR signaling pathways are independently responsible for programming that drives suppressive functionality of myeloid cells (73, 74), but there are studies that describe their joint interaction in programming as well (75). STAT3 signaling in myeloid cells can be initiated by tumor derived factors, including IL-10 and lactate. Activation of STAT3 typically results in activation of SOCS to block intracellular inflammation cascades and initiate an “M2-like” state, complete with functional and phenotypical markers, such as ARG1 and CD206. More importantly, STAT3 activation also results in the production of factors that benefit tumor viability and invasiveness, such as VEGF, matrix metalloproteases, and IDO (76–78). With respect to MDSCs, STAT3 has been identified as a crucial factor for both their development and function. STAT3 is capable of modulating gene expression of anti-apoptotic proteins Bcl-xL, c-Myc, Cyclin D1, and others to promote cell survival. STAT3 also engages programs that prevent monocytic lineages from terminal differentiation to maintain an immature phenotype, a hallmark of M-MDSCs (27). Supporting the central role STAT3 plays in MDSC function, inhibition or deletion of STAT3 abrogates the function and development of MDSCs in vivo (79, 80).
STAT6 signaling also promotes a suppressive program in myeloid cells. IL-4 and IL-13 induce a cascade of phosphorylation events that eventually lead to phosphorylation and homodimerization of STAT6, translocation to the nucleus, and binding to the promoters for various “M2-like” genes, such as ARG1 and CD206. As is the case for STAT3, STAT6 can also bind to IFNγ-induced activation sites and repress the transcription of associated genes (81). One of the transcriptional targets of STAT6 is PPARγ, which augments the effect of the suppressive programming set in place by STAT6 (75, 82). Moreover, PPARγ also increases oxidative pathways that result in increased ROS production (83), among other suppressive pathways (84).
GCN2, an intracellular nutrient sensor, also regulates macrophage function and promotes the pro-tumorigenic phenotype of both TAMs and MDSCs by enhancing translation of the CREB-2/ATF4 transcriptional factor responsible for promoting their differentiation (64). Fundamentally the changes induced by these altered differentiation pathways results in a pro-tumorigenic response rather than mediating tumor elimination.
In addition to their role in aiding tumor immune evasion, TAMs and MDSCs also help orchestrate tumor progression. MDSCs remodel the extracellular matrix and promote blood flow to increase nutrient delivery via the production of various metalloproteases, cathepsins, and pro-angiogenic factors (24, 69). M-CSF can promote recruitment of peripheral myeloid cells to the tumor site and differentiate them into directors of angiogenesis (85). This distinct proangiogenic TAM subset, identified by surface TIE2 expression, secretes classic proangiogenic factors, such as VEGF proteins and SEMA4D (86, 87). These factors simultaneously retain anti-inflammatory functionality via autocrine and paracrine signaling through TIE2 (88). The combination of neovascularization and immune suppression can promote early dissemination of malignant cells (89), potentially through the breakdown of cadherin junctions between vascular endothelial cells (90). In some cases, the mobilization of TIE2+ macrophages is initialized as a response to chemotherapy, highlighting the complex systemic reaction to therapy.
Myeloid cell support of tumoral fitness isn't limited to the primary site of malignancy, as subsets of patrolling monocytes have been found to increase angiogenesis to distal metastatic sites (19). MDSCs can serve a similar role and “fertilize the soil” in pre-metastatic sites for malignant cells to settle. Through undefined mechanisms, MDSCs can be recruited to a premetastatic niche before TAMs and establish a nutrient-rich, vascularized, and immunosuppressive environment for tumors to seed (91, 92). Along the same lines, a subset of CCR2+ myeloid cells has also been associated with primary tumor recurrence (19), or re-fertilizing the soil for any remaining local or circulating tumor cells to grow.
Identification of cells implicated in facilitating cancer growth is imperative for several reasons. Despite established knowledge that TAM/MDSC infiltration is associated with worse prognosis (93), it is clear that not all myeloid cells in the tumor microenvironment directly benefit the growing malignancy. Finding a defined population specifically associated with tumor aggressiveness or invasiveness can serve as a prognostic marker. Furthermore, chemotherapy is not a “silver-bullet” to diminish or deplete malignant cells. It results in changes to the local and distant environment that are not easy to predict without studying the effects in vivo or ex vivo (40). Beneficial off-target effects are possible, such as concurrently depleting myeloid cells from the tumor microenvironment (94). Some therapies, however, can exacerbate the suppressive actions of TAMs, MDSCs, and other local myeloid cells, reducing their in vivo efficacy (95–97). It is also unclear as to which myeloid cell subsets are most affected by the therapy. Defining the myeloid cell subsets that are resistant, or even retaliatory, to a particular therapy is crucial for response prediction. Lastly, defining the myeloid suppressive phenotype that is most associated with malignancy and most associated with therapy resistance brings therapeutic efforts one step closer to targeting a specific cell cluster that contributes to several requirements of the hallmarks of cancer (1, 98, 99).
Historically, identification of stromal contribution was achieved with immunohistochemistry and staining for a limited set of markers on serial sections. This practice, however, can be quite wasteful of precious biological specimens and data due to the limited number of concurrent stains that can be performed. As the definitions of all of the players in the tumor microenvironment are expanding exponentially, an expanded panel of markers must be employed to adequately study the TIME. Tissue analysis at single-cell resolution is allowing for discoveries of distinct myeloid cell phenotypes and connecting their gene and protein expression patterns to immunosuppressive and tumor-promoting mechanisms (98, 100). The myeloid compartment has vast heterogeneity in itself, even within monocyte/macrophage subsets (43). Commonly identified subsets are TAMs, monocytes, TRMs, and MDSCs. TAMs and MDSCs are the most interesting populations, as they seem to have the highest correlation to tumor progression and are typically present in the greatest quantities, compared to other immune cells (69). Within these populations are even more complex subsets. Technologies such as scRNAseq (101) and mass cytometry (102, 103) have created new definitions for these populations that highlight heterogeneity previously unappreciated by conventional flow cytometry, allowing for discoveries of rare cell populations. These technologies have also effectively outdated the standard classification scheme of M1- vs. M2-like phenotypes for macrophages. Standard M1/M2-like phenotypic markers should not be applied with absolute exclusivity, as many of the stereotypic genes that represent classical or alternative activation states can be co-expressed and even correlated with each other (43). Therefore, it is crucial to perform deeper statistical analyses to identify these smaller subsets that are more closely associated with the initiation, progression, and maintenance of the malignant niche, in addition to patient outcomes.
In defining the PD-1/PD-L1 (104) interaction and CTLA-4/CD80/86 (105), the search for novel immune checkpoints broadened into identifying new mechanisms that keep the adaptive immune cell out of the tumor environment and immunologically ignorant (2). More recently, myeloid cells in and around the tumor microenvironment have been recognized, as their utility for prognostication becomes more delineated. Generally speaking, TAMs, and MDSCs perform the same task of nurturing tumor growth among all cancers (106). The subset of culprit cells and the mechanisms by which they cloak or support the cancer can range. The surface markers of TAMs and MDSCs are not easily defined. Some markers of alternative activation are shared among TAMs and MDSCs, such as CD163+, CD68+ (40), or CD206+ (107), the same cells can also express markers of classical activation, such as CD169 and CD163 (107). Additionally, TAMs and MDSCs of different malignancies have different phenotypes, indicating differences in mechanisms of suppression, albeit with minimal conservation. Below, we highlight breast, lung, and central nervous system malignancies to address the myeloid cell heterogeneity, as these are the tumor models that have sufficient studies defining single-cell immune populations. For quick reference, immunosuppressive mechanisms discussed throughout the text are summarized in Figure 1 and Table 1. We have also summarized outstanding myeloid cell populations discussed in the text in Table 2.
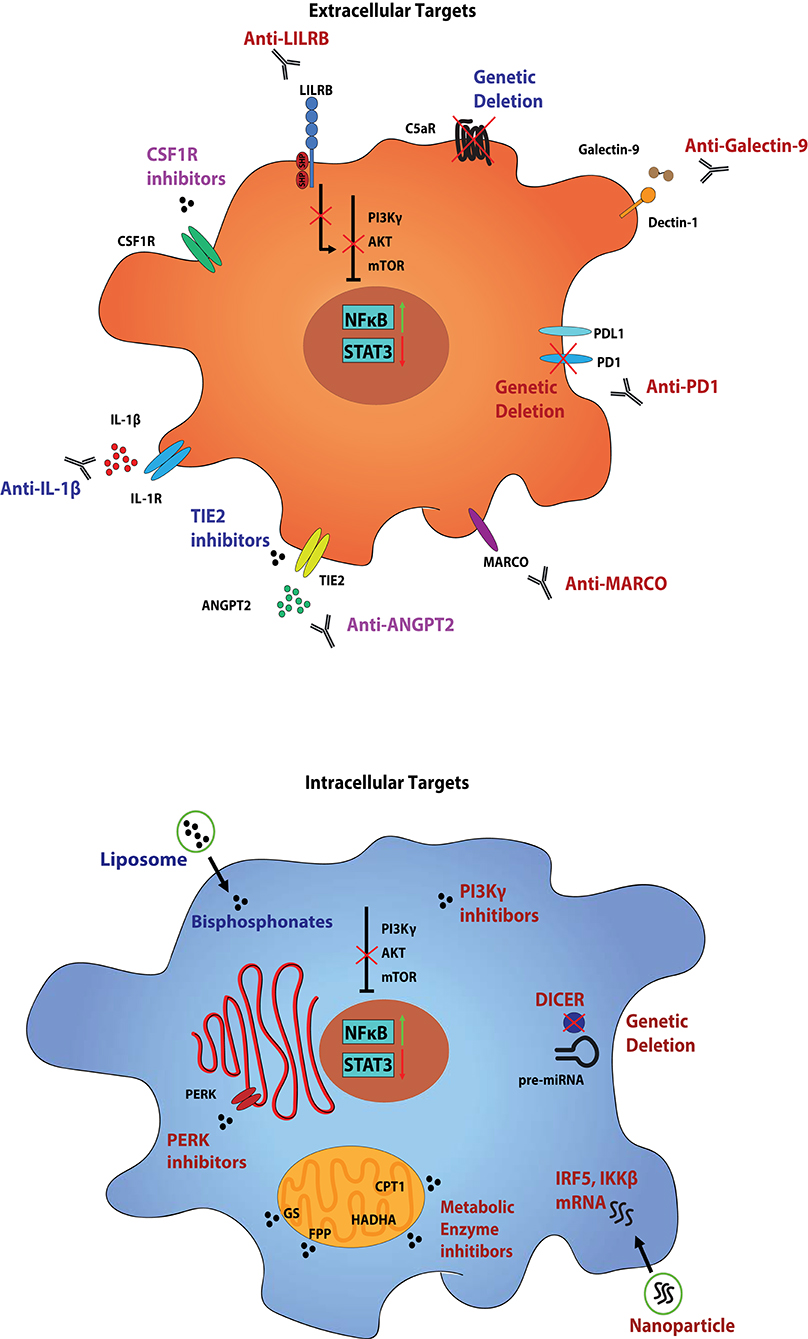
Figure 1. Myeloid cell populations within the TIME and strategies to target them. Tumor-infiltrating myeloid cells, such as MDSCs and TAMs, perform a variety of functions in order to keep the TIME a hospitable niche for tumor growth and progression. Mechanisms include direct secretion of growth factors to support the viability of the tumor, in addition to maintaining an immunosuppressive environment to prevent recognition by cytotoxic immune cells. These functional states are controlled by the growing tumor itself through various combinations of ligand-receptor interactions, and can be propagated by the tumor-associated myeloid cells. Several markers, both surface and intracellular, can be used to not only identify the individual populations of tumor-associated myeloid cells, but also as therapeutic targets. Therapies aimed at these targets generally serve to either deplete the individual clusters of cells from the TIME, or to reprogram them from pro- to anti-tumor states. Presented are conserved targets on MDSCs and TAMs across tumor types, although they exist in different combinations amongst various tumor-associated clusters. The simplified cell diagram on the top presents various surface targets to reprogram (red), deplete (blue), or a combination of both (purple), tumor-associated myeloid populations, and the simplified diagram on the bottom presents intracellular targets. While only a single cell diagram is portrayed, these strategies represent individualized therapies in targeting specific tumor-associated myeloid cell populations. While some receptors may overlap between populations, we hypothesize that a multifactorial approach is imperative to abolish myeloid cell support of tumor growth.
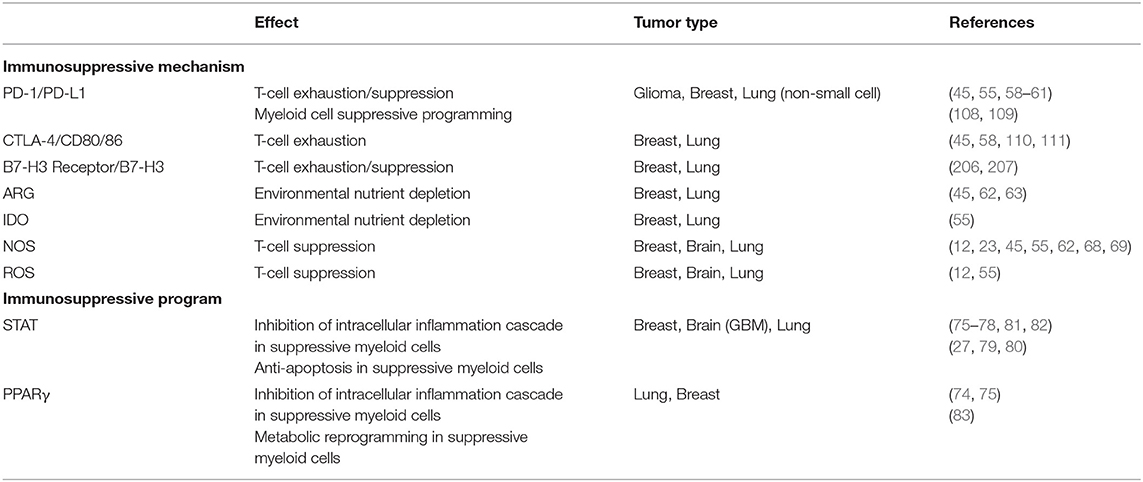
Table 1. Immunosuppressive mechanisms employed by MDSCs and TAMs, as well as stereotypic programming that regulate the mechanisms.
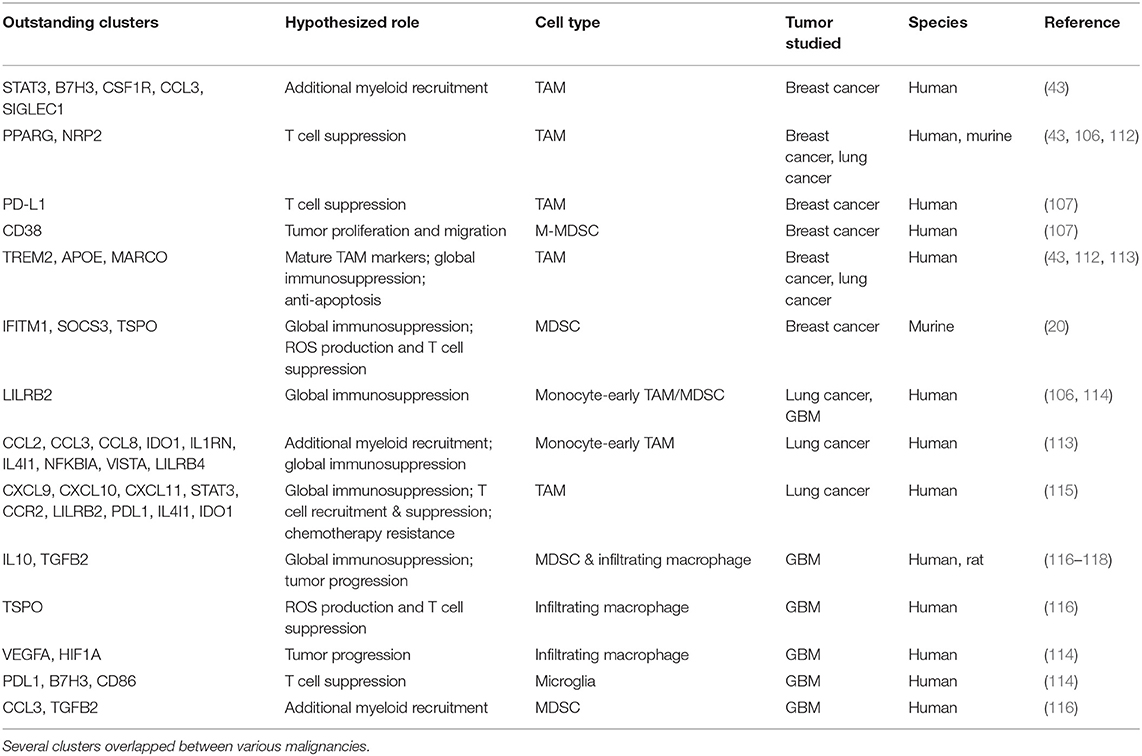
Table 2. Specific clusters of myeloid cells highlighted in the text are summarized here.
Without stratifying by breast cancer subtypes or stages, the myeloid landscape presented by different studies shows similarities. Notably, individual TAMs co-express both M1-like and M2-like associated genes along the same positive correlation trajectory (43, 107). Azizi et al. (43) identified TAM populations from human samples that expressed both classical and alternative activation markers, such as CCL3 and MARCO, respectively, in addition to enrichment of signaling networks that are associated with each of the activation states. Highlighting a potential role for further recruitment of additional TAMs to the malignant site, one TAM cluster in the study by Azizi et al. (43) had distinctly enhanced expression of STAT3, B7H3, CSF1R, and CCL3. This same cluster also had upregulated SIGLEC1, which can serve as an independent predictor of poor prognosis [(14, 43), Supp.]. A separate TAM cluster in the same study was enriched in PPARG and NRP2, indicating distinct functional properties as a potential suppressor of T-cell activity through NRP2 (43, 119). Azizi et al. (43) further validated the individuality of the clusters and rejected the null hypothesis of unimodality across components that explain their variation.
Using scRNAseq information, Wagner et al. (107) detailed TAMs and MDSCs present in human breast cancer. A unique population of PD-L1+ TAMs and a population of MDSCs with high expression of CD38 is also identifiable among breast cancer samples (107). Notably, CD38 has been found to aid the proliferation and migration of tumor cells and is also independently associated with the establishment of an immunosuppressive environment, even when expressed on M-MDSCs isolated from peripheral blood (120–122). As a note of caution, studying peripheral blood immune cells as biomarkers for diseases comes with its own challenges, as PBMC phenotypes don't necessarily agree with tumor-infiltrated immune cells (106). The complexity and heterogeneity of intra-tumoral myeloid cell populations is not well-represented by peripheral myeloid cells, possibly due to the effect of local tumor-associated signaling, therefore care must be taken when associating peripheral cells to the local disease. However, locally expressed CD38 can bypass disinhibition from PD-1/PD-L1 targeted therapy (123).
The TAM population in breast cancer studies seems to be the most mature cell population, defined by a signature defined by several factors, such as TREM2, APOE, and MARCO (43). All can be used as phenotypic markers of mature myeloid populations, such as macrophages, but TREM2 can serve as a functional marker of an anti-apoptotic state (124). Similar populations of TAMs are described in other cancers later (14, 112). Several studies showed the presence of undifferentiated monocyte populations within the breast TIME. Azizi et al. (43) described several monocyte populations with no enrichment of immune gene sets in addition to several other populations that are on track to dendritic cell differentiation. Likewise, Wagner et al. (107) described a border of monocytes to wall off the TAMs within the tumor core.
In murine models of breast cancer by Alshetaiwi et al. (20) MDSCs can be distinguished with scRNAseq. However, their identification presents a sizable challenge, as they do generally do not form distinct clusters by standard informatics analyses. With deeper analysis, they are distinguishable from other myeloid cell populations by their own transcriptional signature (20). Most notable in their transcriptional signature is the dramatic upregulation of IFITM1 and SOCS3, marking their suppressive programming, in addition to TSPO (translocator protein) when compared to other myeloid cell clusters, highlighting their functional role in the TIME. TSPO is a mitochondrial membrane protein that, when activated, results in a respiratory burst and generates reactive oxygen species from myeloid cells, subsequently causing inhibition of T-cell activity (125). Unfortunately, no studies to date have evaluated the phenotypes of individual MDSC clusters to differentiate their functional roles in the TIME, although it is hypothesized that distinct clusters do exist (126).
Taken together, phenotypically distinct populations of TAMs/MDSCs have different functional responsibilities within the TIME in breast cancer. Notably, the majority of these suppressive cells are more mature TAMs, rather than MDSCs. Yet to be determined is the ontogeny of TAMs, i.e., whether they are the product of MDSC maturation or monocyte differentiation.
Normal lung tissue is rich in immune cells responsible for eliminating foreign bodies and infections, therefore it is important to segregate TAM/MDSC populations from the normal lung myeloid populations for correct analysis. In adenocarcinoma, TAMs may have expression networks that make them more readily identifiable from normal myeloid cells, but deeper analyses like scRNAseq is required in order to differentiate their signatures and identify distinct populations (112, 113). TAMs in a later stage of macrophage differentiation within lung adenocarcinoma are distinguishable from resident myeloid cells via concurrent expression of TREM2, MARCO, and APOE, as mentioned earlier. As in other tumors, the TAMs from early lung adenocarcinoma express M1- and M2-like markers, including HLA-DR and CD163, respectively. Importantly, subsets of TAM populations in non-squamous cell lung cancer (NSCLC) show an enrichment of PPARG expression that can initiate anti-inflammatory transcriptional networks that propagate immune ignorance (127, 128), differentiating them from both normal lung macrophages and peripheral myeloid cells (106, 112, 115, 129). Zilionis et al. (106) also describe a population of tumor-infiltrating monocytes that express anti-inflammatory-like markers, such as LILRB2, a potent activator of the STAT6 signaling network. As this population has comparably low CD14 expression, we speculate that this population of monocytes could represent newly-trafficked cells [(106), Supp.] that display immunosuppressive functionality early in the TAM differentiation process. This supports the notion that tumoral recruitment of suppressive cells happens early and at a systemic level. Our group has shown that the murine homolog to LILRB2, PIRB, can regulate the entire network of suppressive functionality of myeloid cells, making the expression of LILRB2 an interesting therapeutic target (130). Additionally, we have shown that targeted therapy against LILRB2 on tumor-infiltrating myeloid cells can reverse their suppressive fate initiated by the malignancy and diminish lung cancer tumor burden in murine models (131).
Tumor associated myeloid cells in lung cancer have the ability to further recruit new myeloid cells, as seen in other cancer types. Lambrechts et al. (113) describe the heterogeneity of immune cells within NSCLC, and describe a particular myeloid cell compartment that is enriched in several genes that recruit more immune cells to its location, such as CCL2, CCL3, and CCL8, in addition to IDO1, IL1RN (132). The same cluster exhibits high expression of IL4I1, NFKBIA, VISTA, and LILRB4. Like LILRB2, LILRB4-mediated ITIM signaling has a strong effect on the anti-inflammatory phenotype of myeloid cells (133), and we hypothesize that LILRB4 could act as a central regulator of the immunosuppressive cascade network in this myeloid cluster, as Deng et al. (134) showed a significant decrease of NFKBIA (IκB) at the protein level, following genetic ablation of LILRB4 in myeloid cells (135). The additional correlation to VISTA within the same cluster is of particular importance, as VISTA is proving to be an attractive target to prevent inhibition of T-cell cytotoxicity (136). In concert, this network would presumably directly program newly recruited myeloid cells to a suppressive state and add to the immunosuppressive border surrounding the growing malignancy.
Clusters of suppressive myeloid cells can incorporate other cell types to augment their effect. A population of macrophages has been shown to induce T-regulatory cells to further fortify the immune barrier to cancer recognition (115). This macrophage cluster expresses markers of T-cell recruitment, such as CXCL9, CXCL10, and CXCL11, but the cluster is also enriched for anti-inflammatory-like genes, such as STAT3, CCR2, and LILRB2 [(115), Supp.]. Most importantly, the same cluster is extraordinarily enriched for PDL1, IL4I1, and IDO1—genes heavily implicated in suppression of cytotoxic T-cell activity and induction of T-regulatory cell programming (137–142). According to Maynard et al. (115), this cell population is expanded in patients that show progression of malignancy on therapy, highlighting a crucial mechanism for therapy failure that corroborates previous work (143). This demonstrates another role of myeloid cells in tumoral viability—creating a hospitable environment for recurrence. While the entire population of myeloid cells is frequently targeted for cancer therapeutics (144), it's clear that more efficient strategies are needed. From the study by Lambrechts et al. [(113), Supp.], there does not appear to be any one particular myeloid cluster that has outstanding expression of PD-L1, PD-1, or B7-H3, underscoring the relevance of the other strategies employed by TAMs to keep the adaptive immunity at bay.
In summary, the lung cancer studies show off the power of deep analysis of the tumor microenvironment. Even in the case of the TAM compartment, which is frequently depicted as a single cell type, there is substantial heterogeneity in cell types that seemingly assume different roles to protect and contribute to the tumor growth. This also underlines a key aspect of immunotherapy targeted against the tumor microenvironment: it is unlikely a single therapeutic would have the capability to transform or reprogram all involved cells—in this case, TAMs/MDSCs. While targets such as PD-1/PD-L1 or CTLA4/CD80 are important, these mechanisms address just one mechanism of TAM-mediated suppression, and a downstream effector, which could explain the limited clinical benefit.
Central nervous system (CNS) malignancies account for a small percentage of all diagnosed cancers (145), but they are frequently associated with abysmal prognoses. The resident immune system of the CNS, namely the microglia, are established contributors to CNS malignancies (146), but there are several other phagocytic myeloid cell populations in the CNS that are also, if not more so, implicated in a poor prognosis of the most aggressive form of CNS malignancy, glioblastoma multiforme (GBM). Perivascular, meningeal, and choroid plexus macrophages of the CNS have generally been overlooked as contributors to GBM (147, 148), but the involvement of bone marrow-derived myeloid cells has recently been established, and even positively correlated, to poor outcomes in GBM models (116, 149). As seen in the previous cancer studies, GBM TAMs co-express M1- and M2-associated markers, again making simple surface phenotyping of cells rather difficult, and creating the need for mechanism and pathway analysis (116). Invading peripheral myeloid cells show a greater suppressive potential than do microglia, marked by increased expression of IL10 and TGFB2—potent inducers of T-regulatory cells (12, 43)—compared to the resident immune cells (116, 117). Likewise, peripheral myeloid cells were also enriched in genes involved in the citric acid cycle and TSPO compared to the resident microglia, resembling TAMs that we speculate to directly inhibit T-cell functionality mentioned previously in the Breast Cancer section [(116), Supp.].
Unfortunately, current scRNA-seq studies of the TIME in GBM use consensus clustering only to distinguish the roles of microglia and peripheral macrophages. This method limits the resolution and only allows for the evaluation of two myeloid cell clusters. Despite this, Muller et al. (116) describe myeloid cell heterogeneity that is the result of their spatial relationship with the malignancy, suggesting that suppressive myeloid cells perform different roles according to their physical location. Likewise, Darmanis et al. (114) show that macrophages make up the majority of myeloid cells within the tumor core and microglia make up the myeloid population of the surrounding stroma. The macrophages in the core seemingly contribute more to the overall viability of the tumor via their expression of VEGFA and HIF1A, while the juxtatumoral microglia serve as the main masqueraders of the malignancy with increased expression of PDL1, B7H3, CD80, and CD86 (114). The myeloid cells within the tumor core are also the main source of LILRB2 expression, offering a selective target for reprogramming a significant cell population for maintaining tumoral viability. Also interesting is that the majority of LILRB2-expressing myeloid cells do not co-express MARCO, a pattern recognition receptor enriched on TAMs (150); we speculate that these cells could be MDSCs (114). Most GBM-associated myeloid cell populations are involved in recruiting additional immunosuppressive myeloid cells, marked by exorbitant expression of CCL3 and TGFB2 in numerous GBM specimens (116). Combined expression of CCL3 and TGFB2 in a variety of bulk tumor samples from tissues of different origin is strongly associated with the local presence of MDSCs, despite the difficulty in their identification (151). More importantly, high expression of the combination is associated with a worse overall median survival in high grade glioma, referenced in multiple data repositories (152).
While there are limited studies that recognize the presence of MDSCs, and specifically analyze heterogeneity of MDSCs, in models of CNS malignancy, it is imperative that we discuss them in this context. MDSCs have been detected in the tumor microenvironment and play a significant role in tumor progression (153). They do not exist in healthy CNS tissue outside of the context of malignancy (149, 153). Alban et al. (118) use MDSC infiltration in GBM as prognostic markers and indicate a hazard ratio of 4.7 (1.69–13.4) when comparing overall survival of patients with high MDSC GBM infiltration to low infiltration. Under the assumption that all M-MDSC populations that infiltrate GBMs are programmed into the same functional state, their role is to secrete IL-10 and TGF-β, just like their macrophage counterparts. The presence of these cytokines is correlated to overall stage of the malignancy [(118, 149), Supp., (154)], indicating that there is most likely a dose effect as a greater amount of MDSCs in the local environment is correlated to staging as well.
In addition to the local involvement of suppressive myeloid cells, the peripheral differential cell count offers insight to prognosis of GBM patients (118, 149). MDSCs in the periphery are heavily implicated in higher grade, more aggressive CNS malignancies. Peripheral MDSCs have a strong positive correlation with worse prognoses in GBM patients, and the converse is true as well. Alban et al. (118) showed that, after surgical resection of GBMs, patients with increasing fractions of MDSC populations had inferior survival time, compared to those of decreasing MDSC fractions. A cohort of newly diagnosed patients in the study received standard-of-care adjuvant therapy (155), but the expansion of M-MDSCs were variable, indicating a potential difference in activation of myeloid cells following chemo- or radiotherapy (156, 157). Additionally, a number of studies show increased peripheral MDSC counts in subsets of patients who received dexamethasone perioperatively, indicating a potential confounder, or contributor, in correlating overall survival with MDSC levels (118, 148, 149).
GBM is well-known to be an extraordinarily heterogeneous malignancy, making it very difficult to target with “off-the-shelf” therapy. However, it is striking to see that even across the heterogeneity of malignancies from different patients, the myeloid cell clustering, and signaling networks seem to remain conserved (114). Manipulating the programming of both bone marrow-derived myeloid cells and resident microglia is important in regulating the entire network of immune suppression and pro-tumor functionality. While microglia appear to be attractive targets for the popular therapies targeting PD-L1 or B7 family of proteins, involvement of the peripheral immune system within the tumor microenvironment is more closely associated to prognoses and should also be considered for immunomodulation. Whether the infiltrating TAMs, the malignancy itself, or a combination of both is causing the suppressive programing of the microglia remains to be determined. Table 1 details the pathways and receptors that mediate immunosuppression along with the specific effect and tumors impacted by the signaling pathway.
Currently, there are two main strategies for manipulating tumor associated myeloid cells: depletion and reprogramming. Depletion involves broad, systemic targeting of myeloid cells, although newer, more specific approaches are aimed at depleting only the myeloid cells that are specifically involved with the malignancy (40). The therapeutic strategies are summarized in Table 3, along with recent clinical trial information.
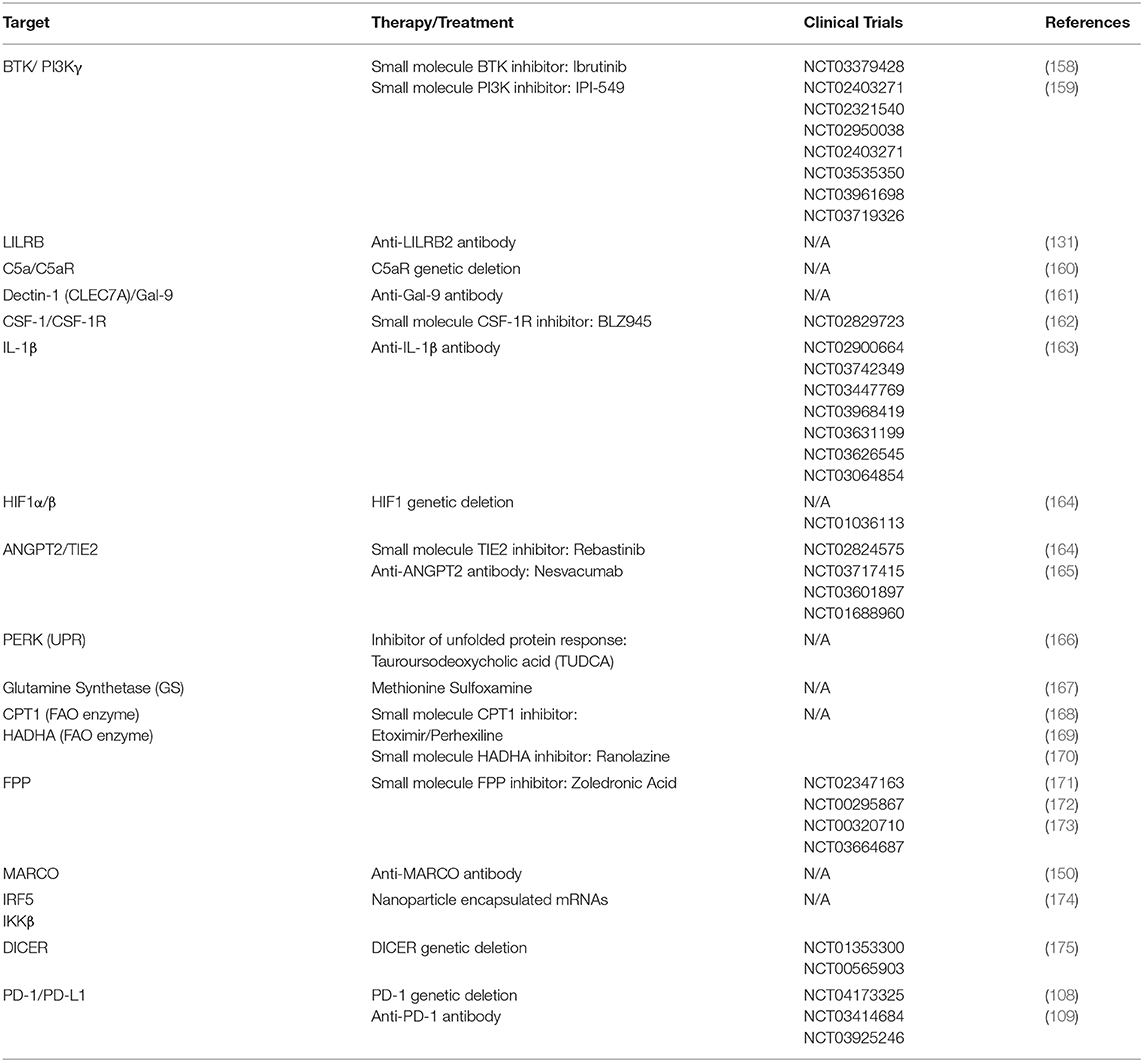
Table 3. Therapies used to reprogram tumor associated macrophages and MDSCs.
Strategies to deplete myeloid cells from the TME include mechanisms to prevent myeloid cell trafficking to the malignancy or initiate apoptosis. Tumoral recruitment and expansion of bone marrow-derived myeloid cells occurs through a CCR2-CCL2–dependent signal and, along with increasing serum levels of CCL2, is independently associated with worse prognosis. Disruption of CCR2 signaling prevents the recruitment and development of suppressive myeloid cells, while suppressing tumor metastasis and prolongs survival across several cancer models (16, 176, 177). Importantly, disrupting CCR2 signaling also reduces TAM/MDSC recruitment to premetastatic niches (16).
Antagonizing the CSF1–CSF1R axis is an interesting approach as it disrupts several mechanisms for therapeutic effect. Blocking the axis can disrupt localization of suppressive TAMs to the site of malignancy (178) as well as reprogram TAMs for anti-tumor activity (162), in addition to preventing the conversion of M-MDSCs to TAMs (12). JNJ-28312141, a CSF1R inhibitor, depleted F4/80+ TAMs in a subcutaneous H460 human lung tumor xenograft model and increased plasma CSF1, a potential biomarker in CSF1R inhibition (179). Biologics have also been studied in this regard—RG7155, a monoclonal CSF1R antibody, greatly reduced F4/80+ TAMs in animal models of colon cancer. RG7155 showed promise in human applications as well, as it induced apoptosis of CSF1R+CD163+ macrophages in patients with diffuse type giant cell tumor tissue (Dt-GCT) (178). However, as CSFR1 blockage with pexidartinib has proven to be ineffective in patients, targeting the CSF1–CSF1R signaling axis might have limited applications (180). Combination therapy of CSF1R blockade with immune checkpoint blockade is currently ongoing in a solid malignancy clinical trial (Trial # NCT02713529).
Targeting CD38 is proving to be a good strategy for antibody-mediated depletion in some cancer models. CD38+ MDSC populations are expanded in cancer patients and can even serve as an escape mechanism after PD-1/PD-L1 therapy. Daratumumab, a CD38 antagonist antibody, can deplete immunosuppressive myeloid cells from circulation, as well as serve as an independent therapy for CD38+ myelomas. CD38 antibody therapy initiates apoptosis via antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Other suppressive cell types, such as T-regs, are also sensitive to anti-CD38 treatment (122, 123, 181).
Liposomal delivery of dichloromethylene biphosphonates is another effective method to deplete tumor associated myeloid cells, as it deposits its payload directly into the intracellular space. Liposomes are enclosed multifunctional structures that consist of one or more phospholipid bilayers surrounding a hydrophilic core. This organization allows for hydrophobic therapies to associate with the lipid bilayer, and hydrophilic therapies, including genetic material such as RNA, DNA or siRNA, to be carried in the core. Clodronate and other bisphosphonates are a class of drugs typically used for the treatment of osteolytic bone disease and osteoporosis by inhibiting bone resorption, as they specifically target the phagocytic cells involved (182). By encapsulating clodronate in liposomes, clodronate can be delivered to the tumor site where it is phagocytosed by macrophages, ultimately initiating apoptosis. However, these effects have only been shown in vitro and animal models (183–185).
An interesting, albeit controversial, aspect of MDSCs in the TIME is the effect of chemotherapies on MDSC quantities and suppressive programming. 5-fluorouracil (5-FU) and gemcitabine were able to induce apoptosis and deplete MDSCs in both spleens and tumors in 4T1 murine breast cancer model. Moreover, both 5-FU and gemcitabine can activate caspase-1 and induce IL-1β production via the NLRP3 inflammasome pathway (186, 187). Evidence points to conflicting effects of IL-1β with respect to the TIME. While some studies show a beneficial effect of increased IL-1β in the TIME (64), others show that blockade of IL-1β signaling can prevent immunosuppressive cell recruitment (163). Other secondary effects of chemotherapy on the immune system are discussed in depth elsewhere (40, 54).
The tumor microenvironment can polarize TAMs to an immunosuppressive M2-like functional state, leading to enhanced tumor growth, progression, and metastasis. Besides depleting TAMs and MDSCs, myeloid cells can be reprogrammed toward a pro-inflammatory state by direct intervention via small molecules and antibodies targeting key receptors. Two reprogramming strategies can be used—blocking a receptor that normally transduces an inhibitory intracellular signal, or using an exogenous ligand to activate a receptor that stimulates pro-inflammatory intracellular cascades (188). Despite its success in diminishing tumor burden, pro-inflammatory agonist therapy is frequently associated with systemic toxicity (189, 190), therefore, we will discuss the former strategy.
In some cases where disruption of the CSF1-CSF1R signaling axis is unsuccessful in depleting TAMs, antagonism of CSF1R signaling can reprogram TAMs away from an M2-like state. Using glioma xenograft models, Pyonteck et al. (162) describe how CSF1R antagonism did not decrease TAM numbers nor did it alter their CSF1R expression pattern. However, inhibition of AKT phosphorylation and M2-related gene expression, such as ARG1 and CD206, indicated that CSF1R antagonism initiated a functional shift to a pro-inflammatory state to block glioma progression (162).
In a murine pancreatic ductal adenocarcinoma (PDAC) model, crosstalk between B-cells and FcRγ+ TAMs resulted in an M2-like phenotype through Bruton's tyrosine kinase (BTK) activation in a PI3Kγ-dependent manner. Using the BTK inhibitor ibrutinib, PI3Kγ inhibition in PDAC tumor-bearing mice reprogrammed TAMs toward an M1-like state and increased CD8+ T-cell cytotoxicity to slow PDAC tumor growth (158). PI3K is a critical switch to promote suppressive activity in macrophages, as PI3K signaling via AKT and mTOR inhibits NFκB to promote M2-like functionality in TAMs. Conversely, inhibiting PI3K prevents C/EBPβ activation and disinhibits NFκB to induce a pro-inflammatory phenotype. Combining PI3K blockade with anti-PD-1 therapy can promote tumoral T-cell infiltration to slow tumor growth and enhance survival in tumor-bearing mice (159).
Signaling pathways that activate NFκB to initiate pro-inflammatory functionality represent valuable therapeutic strategies. Our group found that PIRB/LILRB signaling pathways can function as crucial regulators of NFκB activity. Ablation of PIRB in MDSCs forced a transition to an M1-like phenotype, resulting in decreased suppressive function, T-reg activation, tumor growth, and metastasis (130). PIRB−/− monocytes expressed stereotypic markers of inflammatory functionality, such as increased iNOS, TNFα, with decreased IL-10 and ARG1 when compared to WT monocytes. PIRB−/− MDSCs also demonstrated increased ERK, MAPK, and NFκB activation upon LPS stimulation, and enhanced IFNγ-related inflammatory responses. LILRB2—the human ortholog to murine PIRB—blockade via monoclonal antibodies favored the activation of NF-κB and STAT1 and the inhibition of STAT6 activation by IL-4. In vitro, we observed decreased levels of CD14, CD163, CD16, and DC-SIGN in A549-derived macrophages cultured in the presence of αLILRB2 antagonist antibodies. Humanized MISTRG (M-CSFhi, IL-3/GM-CSFhi, and TPOhi) mice treated with αLILRB2 antibodies to reprogram human macrophages to a M1-like classically activated phenotype. Our group has also generated BAC-transgenic mice expressing LILRB2 for various studies. Recently we showed that αLILRB2 antibody therapy had a synergistic effect when combined with αPD-1 therapy to diminish tumor burden in a lung cancer model with BAC-transgenic LILRB2 mice, while simultaneously suppressing MDSC and T-reg infiltration into the tumor site (131).
High Dectin-1 and the novel Dectin-1 agonist Galectin-9 expression were found in the TME of PDAC bearing mice. Dectin-1 is a c-type Lectin expressed mainly on macrophages and other myeloid-monocytic lineage cells. It is postulated that Dectin-1 ligation in TAMs leads to immunosuppression, thereby promoting PDAC growth. Dectin-1 does not have direct pro-tumorigenic effect on transformed PDAC but its deletion in tumor infiltrating macrophages induced immunogenic reprogramming. Similar to the outcome of Dectin-1 deletion, Galectin 9 neutralization enhanced intra-tumoral T-cell activation in PDAC (161).
MARCO (macrophage receptor with collagenous structure) is a scavenger receptor found on M2 immunosuppressive TAMs. We discussed the presence of this receptor in TAMs across multiple tumor types. Conditioned medium from cultured B16 melanoma cells and IL-10 stimulated culture resulted MARCO expression on M0 bone marrow derived macrophages (BMDM). Treatment with anti-MARCO antibodies decreased tumor sizes, increased M1-like, and decreased M2-like TAM populations in the TIME in 4T1 breast cancer and B16 melanoma mouse models. The TIME displayed decreased immature macrophages, increased CD4/T-reg cell ratio, and an upregulation of M1-like genes such as TNF, IL-1β, NOS2, and a downregulation of IL-10 suggesting polarization of TAMs to a more inflammatory phenotype (150).
Last but not least, the PD-1/PD-L1 axis is one the best studied and most clinically successful checkpoint inhibitors. In cancer, the PD-1/PD-L1 axis is best known for T cell regulation. Previously, macrophages were known to express PD-1 during pathogenic infections (191, 192). Since then, it was discovered that TAMs can also express high levels of PD-1, with increasing levels over time in murine models and higher expression in increasing human cancer disease stage. PD-1/PD-L1 blockade in vivo increased PD-1+ macrophage phagocytosis activity and reduced tumor growth in murine colon carcinoma models (108). A more recent study showed that PD-1 ablation or blockade with monoclonal antibodies prevented the accumulation of granulocyte/macrophage progenitors under cancer driven emergency myelopoiesis. Interestingly, PD-1 deficient myeloid progenitors also had increased cholesterol synthesis which is required for the differentiation of inflammatory macrophages. Additionally, PD-1 ablation on myeloid cells decreased tumor growth more effectively than T-cell specific PD-1 ablation in a murine fibrosarcoma and melanoma models (109). Cumulatively, PD-1/PD-L1 blockade or ablation on myeloid cells promotes phagocytosis in macrophages, reprogramming of myeloid progenitors and even furthers myeloid differentiation via metabolic pathways. None of the aforementioned single-cell studies show exceptional levels of PD-1 on myeloid cells, but that does not exclude it from being a potential target for diminishing immunosuppressive phenotypes of myeloid cells.
C5a is a protein fragment released from cleavage of complement C5 that may be involved with PMN-MDSC recruitment. In one study, C5a was found to enhance tumor growth and inhibit CD8 T-cell mediated cytotoxicity by recruiting PMN-MDSC (CD11b+Gr1+) to the tumor microenvironment. C5a also enhanced PMN-MDSC's suppressive capacity by increasing the production of reactive oxygen (ROS) and nitrogen species (RNS) which inhibits CD8+ T cell response (193). Ablation of C5aR reduced the ratio of PMN-MDSC to M-MDSC in tumor bearing mice compared to wild type mice. C5aR blockade is a potential strategy to modulate the tumor microenvironment by preventing the recruitment of immunosuppressive PMN-MDSC (160).
IL-1β, a proinflammatory cytokine, is a potential target for macrophage reprogramming because it impacts CSF1/CSF1R signaling. In early tumor progression models using 4T1 cells in Balb/c mice, IL-1β acts as a master cytokine, exhibiting both pro- and anti-tumoral functionality (163). IL-1β recruited CCR2+ inflammatory monocytes to the tumor site through the induction of CCL2 but also promoted the differentiation of these monocytes into immunosuppressive macrophages by inducing CSF1. IL-1β deficient mice displayed significant reduction in inflammatory monocytes recruitment and macrophage differentiation. Combination therapy of αIL-1β and αPD-1 completely abrogated breast tumor progression (163).
Besides cell surface receptors, cytokines and chemokines, the oxygen level in the tumor also affects the microenvironment. Tissue hypoxia develops as tumor cells proliferate until oxygen demand overwhelms the supply. To restore oxygen to the microenvironment, malignant cells initiate a hypoxic response to drive a more aggressive phenotype, promoting angiogenesis, cell proliferation, self-renewal, and other pro-tumoral programs. Two master regulators of hypoxia in cells are HIF1α and HIF2α. TAMs within this hypoxic environment are more strongly associated with M2-like functionality (194), and HIF2α ablation in TAMs resulted in a more favorable outcome in models of hepatocellular carcinoma and colitis associated colon carcinoma (164).
Just as oxygen levels affect the TIME, tumor vascularization also plays a role. The angiopoietin (ANGPT2)/TIE2 kinase signaling axis is essential to angiogenesis. TIE2 can be found on a subset of pro-angiogenic macrophages (TIE2+ macrophages) and promote tumor angiogenesis and tumor metastasis. Rebastinib, a TIE2 kinase inhibitor, suppressed the infiltration of TIE2+ macrophages to the tumor site in the PyMT mouse model of breast cancer (195). Another study showed that vascular endothelial production of ANGPT2 recruited TIE2+ macrophages to the tumor and the inhibition of ANGPT2 binding suppressed TAM recruitment (165).
Recent studies have attempted to explain why tumor-associated immunosuppressive myeloid cells cannot simply be binned in an M1/M2-like dichotomy, and Mohamed et al. (166) describe ER stress as a potential mechanism. Undefined tumoral signaling causes an upregulation in the unfolded protein response of MDSCs, leading to an activation of the intermediate media PERK and NRF2 drive the immunoregulatory phenotype. PERK ablation led to a reprogramming of MDSC functionality, specifically, to initiate a type I interferon anti-tumor response. More importantly, similar anti-tumor effects can be achieved with the exogenous administration of PERK inhibitors (166).
Recent studies have shown that immunometabolism plays a very important role in the regulation of macrophage function in the tumor microenvironment. The metabolic profile of TAMs determines their status as pro- or anti-tumoral effector cells. M1-like macrophage metabolism is generally characterized with increased glycolysis, fatty acid synthesis, and a truncated TCA cycle whereas M2-like macrophage metabolism is skewed toward fatty acid oxidation (FAO) and the TCA cycle (196–200). For example, tumor-derived lactate induces an M2-like state in macrophages, measured by the induction of the M2-related genes VEGF, RELMA, MGL1, and MGL2. Lactate can also promote the expression of ARG1 and stabilize HIF1α–key functional elements of a suppressive macrophage (201). Preventing the metabolic profile initiated by lactate using a small molecule inhibitor may reduce the presence of immunosuppressive myeloid cells (77). Similarly, methionine sulfoxamine, a potent inhibitor of glutamine synthetase, skewed M2-polarized macrophages toward an M1-like state characterized by reduced intracellular glutamine and increased succinate to promote glycolysis (167).
However, promoting glycolysis in macrophages of the TIME is a risky endeavor as cancer cells also preferentially use glycolysis as an energy source, according to the Warburg effect. Therefore, targeting a metabolic pathway that inhibits tumor progression while simultaneously promoting an anti-tumor immune response would be an attractive strategy. FAO is one potential pathway, as it is the defining metabolic program of M2-like macrophages. FAO inhibition can impair the proliferation of leukemia cells (168) and reduced cellular ATP and viability in glioma (169). In multiple tumor models, tumor infiltrating MDSC were found to have increased fatty acid uptake and activated FAO (170). Etomoxir, a pharmacologic inhibitor of FAO, decreased the overall metabolic activity of MDSCs, their ability to prevent T-cell proliferation, and production of critical cytokines that maintain the induction and differentiation of MDSCs. Tumor-bearing mice treated with etomoxir and a related inhibitor, ranolazine, showed delayed tumor growth attributable to increased T-cell mediated cytotoxicity (170).
As previously mentioned, liposomal delivery of bisphosphonates can be used to deplete macrophages via apoptosis. Zoledronic acid (ZA) is a bisphosphonate containing a double nitrogen group. It inhibits the active site of the enzyme farnesyl pyrophosphate synthase in the mevalonate pathway, which is critical for isoprenoid and cholesterol synthesis (171). ZA also has a direct proapoptotic effect on tumor cells and reduces their metastatic potential (172). TAMs were significantly reduced in a TUBO cell murine mammary tumor model. Peritoneal macrophages and TAMs in ZA-treated mice displayed enhanced M1-like markers, shown by nuclear translocation of NFκB, NOS expression, and NO production (173).
Gene therapy is a unique strategy to polarize TAMs. Zhang et al. (174) describe using in vitro-transcribed mRNA encoding IRF5 and its activating kinase IKKβ encapsulated in nanoparticles to reprogram TAMs in models of ovarian cancer, melanoma, and GBM. The nanoparticles were engineered with D-mannose on the surface to efficiently and specifically target the mannose receptor CD206+ on TAMs. Upon mRNA uptake, TAMs adopted a tumor-clearing, pro-inflammatory profile (174).
Similarly, endogenous RNA processing mechanisms can be exploited to reprogram TAMs. MicroRNAs (miRNA) are a class of small non-coding RNAs that negatively regulate RNA transcription and transcript levels through a sequence dependent mechanism. Normally, DICER, an RNAse-III enzyme, processes hairpin-shaped precursor miRNAs into mature miRNAs (202). Baer et al. (175) describe conditional deletion of DICER in TAMs to prevent maturation of miRNAs that otherwise inhibit M1-like functionality, rewiring the cells toward a pro-inflammatory state characterized by the activation of IFNγ and STAT1 signaling. Moreover, DICER-deficient TAMs promoted the recruitment of cytotoxic T cells that completely eradicated tumors in mouse models when combined with PD-1 checkpoint blockade (175). A summary of these methods, specific targets, and ongoing clinical trials to target them is provided in Table 3.
Emerging techniques such as scRNAseq and mass cytometry have allowed for enhanced analyses of previously uncharacterized cell subsets in the tumor immune microenvironment, offering new avenues for discovering potential novel therapeutic targets and pathways that support tumor progression. Although these have not translated into the clinic yet, there is optimism that greater understanding of the tumor immune microenvironment and associated immunomodulatory mechanisms will allow for targeted therapeutic strategies to improve patient survival. While the tumor-associated myeloid cell population collectively functions to support the growing malignancy, subsets of the population are driven by assorted environmental cues that induce different functional programs. Different subsets of tumor-associated myeloid cells can directly contribute to the viability of the tumor, prohibit recognition of the tumor by the adaptive immune system, and drive chemotherapy or immunotherapy resistance. The questions left to be answered are: what combinations of signals cause the heterogeneity within the microenvironment and do they originate from the parenchyma, stroma, or both? What effect does chemotherapy or immunomodulation have on the various populations? Is there a specific population that is correlated with local or distal recurrence? For any of these cases, is one subset enough to drive any of these phenomena, or is the collection of these subsets necessary? Is there a combination of therapies that would be most effective in eradicating these detrimental subsets?
Defining previously uncharacterized subsets of immune cells by single-cell analyses is crucial to the understanding of tumor biology, but in situ cell relationships also require attention. Loss of tissue architecture is a major limitation to suspension-cell-based assays, such as scRNAseq and suspension mass cytometry, thereby discounting important spatial information that comes from delineating cell-cell interactions. Several of the studies referenced above underscored heterogeneity of myeloid cell phenotypes based on their physical orientation to the tumor—within the tumor or surrounding the periphery of the tumor (18, 107, 116, 203). The location in which cells are found also dictates their functional role in the development of the malignancy, as juxtatumoral immune cells most likely serve as a suppressive barrier to cloak the malignancy, while intratumoral immune cells directly contribute to the viability of the growing tumor (114). Techniques that incorporate spatial information also offer the ability to determine direct cell-cell interaction using Cell Neighborhood Analysis (204) and predict the roles of immune cells (205). These functional states can serve as additional prognostication metrics, as several studies to date have already defined the presence of bulk TAMs and MDSCs in tumor parenchyma vs. stroma in terms of patient outcomes (19, 40, 100, 206, 207). Further work in associating the added dimension of space to the tumor immune microenvironment is required to fully understand the complex interplay between myeloid cells and malignancies.
VD, GJ, SM, S-HC, and P-YP contributed to the intellectual content of the manuscript and contributed to the drafting of the manuscript. All authors read and approved of the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Texas A&M Open Access to Knowledge Fund (OAKFund), by the University Libraries (to VD), by NIH R01 CA127483, R01 CA204191, and R01 CA208703 (to S-HC), and by NIH R01 CA188610 (to P-YP). S-HC is the Emily Hermann Chair in Immunology Research and this work was supported in part by that endowment. This work was also supported in part by grants from the National Cancer Institute, Methodist Innovative Cancer Research fund from the Cancer Center of Excellence, and the Houston Methodist Research Institute to S-HC and P-YP.
TIME, tumor-immune microenvironment; TAM, tumor-associated macrophage; MDSC, myeloid-derived suppressor cell; M-MDSC, monocytic myeloid-derived suppressor cell; scRNAseq, single-cell RNA sequencing; TRM, tissue resident macrophages; TLR, toll-like receptor; IFN, interferon; GM-CSF, granulocyte-macrophage colony-stimulating factor; STAT, signal transducer and activator of transcription; C-EBP, CCAAT/enhancer-binding protein; IRF, interferon regulatory factor; ROR, retinoic acid-related orphan receptor; CCL, C-C motif chemokine ligand; M-CSF/CSF1, colony stimulating factor 1; VEGF, vascular endothelial growth factor; IL, interleukin; PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; CTLA, cytotoxic lymphocyte antigen; iNOS, inducible nitric oxide synthase; PPAR, peroxisome proliferator activated receptor; SOCS, suppressor of cytokine signaling; ARG1, arginase 1; IDO, indoleamine-pyrrole 2,3-dioxygenase; Bcl-xL, B-cell Lymphoma-extra large; ROS, reactive oxygen species; GCN2, general control non-derepressible 2; CREB, CAMP response element-binding protein; ATF, activating transcription factor; TGF, transforming growth factor; SPARC, secreted protein acidic and rich in cysteine, osteonectin; CCR, C-C motif chemokine receptor; MARCO, macrophage receptor with collagenous structure; NRP2, neuropilin 2; APOE, apolipoprotein E; IFITM1, interferon-induced transmembrane protein; TSPO, translocator protein; NSCLC, non-small-cell lung cancer; LILRB, leukocyte immunoglobulin like receptor B; PIRB, paired immunoglobulin-like receptor B; IL1RN, interleukin 1 receptor antagonist; NFKBIA, nuclear factor κ B inhibitor α, I κ B α; VISTA, V-domain Ig suppressor of T cell activation; CNS, central nervous system; GBM, glioblastoma multiforme; HIF, hypoxia-induced factor; PDAC, pancreatic ductal adenocarcinoma; TME, tumor microenvironment; PMN-MDSC, polymorphonuclear myeloid-derived suppressor cell; α-, anti-.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
3. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. (2019) 20:840. doi: 10.3390/ijms20040840
4. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. (2018) 175:313–26. doi: 10.1016/j.cell.2018.09.035
5. Murphy K, Weaver C. Janeway's Immunobiology. 9th ed. New York, NY: Garland Science/Taylor & Francis Group, LLC (2016). p. 904. doi: 10.1201/9781315533247
6. Sica A, Porta C, Morlacchi S, Banfi S, Strauss L, Rimoldi M, et al. Origin and functions of tumor-associated myeloid cells (TAMCs). Cancer Microenviron. (2012) 5:133–49. doi: 10.1007/s12307-011-0091-6
7. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
8. Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, et al. Tuning the tumor myeloid microenvironment to fight cancer. Front Immunol. (2019) 10:1611. doi: 10.3389/fimmu.2019.01611
9. Kowal J, Kornete M, Joyce JA. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy. (2019) 11:677–89. doi: 10.2217/imt-2018-0156
10. Salmaninejad A, Valilou SF, Soltani A, Ahmadi S, Abarghan YJ, Rosengren RJ, et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol (Dordr). (2019) 42:591–608. doi: 10.1007/s13402-019-00453-z
11. Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol. (2018) 9:2629. doi: 10.3389/fimmu.2018.02629
12. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. (2015) 125:3365–76. doi: 10.1172/JCI80006
13. Laviron M, Boissonnas A. Ontogeny of tumor-associated macrophages. Front Immunol. (2019) 10:1799. doi: 10.3389/fimmu.2019.01799
14. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. (2019) 35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009
15. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
16. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
17. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. (2015) 17:170–82. doi: 10.1038/ncb3090
18. Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. (2018) 215:2536–53. doi: 10.1084/jem.20180534
19. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van Overmeire E, Van Ginderachter JA. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim Biophys Acta. (2016) 1865:23–34. doi: 10.1016/j.bbcan.2015.06.009
20. Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol. (2020) 5:eaay6017. doi: 10.1126/sciimmunol.aay6017
21. Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. (2012) 61:1155–67. doi: 10.1007/s00262-012-1294-5
22. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
23. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother. (2019) 68:687–97. doi: 10.1007/s00262-019-02302-2
24. Ochando JC, Chen SH. Myeloid-derived suppressor cells in transplantation and cancer. Immunol Res. (2012) 54:275–85. doi: 10.1007/s12026-012-8335-1
25. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
26. Ribechini E, Hutchinson JA, Hergovits S, Heuer M, Lucas J, Schleicher U, et al. Novel GM-CSF signals via IFN-gammaR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv. (2017) 1:947–60. doi: 10.1182/bloodadvances.2017006858
27. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. (2011) 32:19–25. doi: 10.1016/j.it.2010.10.002
28. Conti I, Rollins BJ. CCL2 (monocyte chemoattractant protein-1) and cancer. Semin Cancer Biol. (2004) 14:149–54. doi: 10.1016/j.semcancer.2003.10.009
29. Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. (2017) 23:137–48. doi: 10.1158/1078-0432.CCR-16-0870
30. Mondini M, Loyher PL, Hamon P, Gerbé de Thoré M, Laviron M, Berthelot K, et al. CCR2-dependent recruitment of tregs and monocytes following radiotherapy is associated with TNFα-mediated resistance. Cancer Immunol Res. (2019) 7:376–87. doi: 10.1158/2326-6066.CIR-18-0633
31. Jordan KR, Kapoor P, Spongberg E, Tobin RP, Gao D, Borges VF, et al. Immunosuppressive myeloid-derived suppressor cells are increased in splenocytes from cancer patients. Cancer Immunol Immunother. (2017) 66:503–13. doi: 10.1007/s00262-016-1953-z
32. Yang W-C, Ma G, Chen SH, Pan PY. Polarization and reprogramming of myeloid-derived suppressor cells. J Mol Cell Biol. (2013) 5:207–9. doi: 10.1093/jmcb/mjt009
33. Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res. (2015) 21:4073–85. doi: 10.1158/1078-0432.CCR-14-2742
34. Zhou J, Donatelli SS, Gilvary DL, Tejera MM, Eksioglu EA, Chen X, et al. Therapeutic targeting of myeloid-derived suppressor cells involves a novel mechanism mediated by clusterin. Sci Rep. (2016) 6:29521. doi: 10.1038/srep29521
35. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. (2008) 83:1136–44. doi: 10.1189/jlb.0907611
36. Sica A, Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J Autoimmun. (2017) 85:117–25. doi: 10.1016/j.jaut.2017.07.010
37. Jayaraman P, Parikh F, Newton JM, Hanoteau A, Rivas C, Krupar R, et al. TGF-β1 programmed myeloid-derived suppressor cells (MDSC) acquire immune-stimulating and tumor killing activity capable of rejecting established tumors in combination with radiotherapy. Oncoimmunology. (2018) 7:e1490853. doi: 10.1080/2162402X.2018.1490853
38. Franklin RA, Li MO. Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer. (2016) 2:20–34. doi: 10.1016/j.trecan.2015.11.004
39. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci USA. (2012) 109:2491–6. doi: 10.1073/pnas.1113744109
40. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. (2016) 16:447–62. doi: 10.1038/nrc.2016.54
41. Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, et al. A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Rep. (2018) 23:1239–48. doi: 10.1016/j.celrep.2018.04.007
42. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell. (2018) 175:1014–30.e19. doi: 10.1016/j.cell.2018.09.030
43. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. (2018) 174:1293–308.e36. doi: 10.1016/j.cell.2018.05.060
44. Song Q, Hawkins GA, Wudel L, Chou PC, Forbes E, Pullikuth AK, et al. Dissecting intratumoral myeloid cell plasticity by single cell RNA-seq. Cancer Med. (2019) 8:3072–85. doi: 10.1002/cam4.2113
45. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
46. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
47. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
48. Trellakis S, Bruderek K, Hütte J, Elian M, Hoffmann TK, Lang S, et al. Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer. Innate Immun. (2013) 19:328–36. doi: 10.1177/1753425912463618
49. Mishalian I, Bayuh R, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol Immunother. (2013) 62:1745–56. doi: 10.1007/s00262-013-1476-9
50. Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. (2017) 9:289–302. doi: 10.2217/imt-2016-0135
51. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–38.e6. doi: 10.1016/j.immuni.2017.07.014
52. Parney IF, Waldron JS, Parsa AT. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J Neurosurg. (2009) 110:572–82. doi: 10.3171/2008.7.JNS08475
53. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
54. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
55. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
56. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
57. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
58. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
59. Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer. (2019) 7:305. doi: 10.1186/s40425-019-0770-2
60. Tang F, Zheng P. Tumor cells versus host immune cells: whose PD-L1 contributes to PD-1/PD-L1 blockade mediated cancer immunotherapy? Cell Biosci. (2018) 8:34. doi: 10.1186/s13578-018-0232-4
61. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. (2016) 5:e1247135. doi: 10.1080/2162402X.2016.1247135
62. Raber P, Ochoa AC, Rodriguez PC. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest. (2012) 41:614–34. doi: 10.3109/08820139.2012.680634
63. Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. (2016) 165:153–64. doi: 10.1016/j.cell.2016.02.035
64. Halaby MJ, Hezaveh K, Lamorte S, Ciudad MT, Kloetgen A, MacLeod BL, et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci Immunol. (2019) 4:eaax8189. doi: 10.1126/sciimmunol.aax8189
65. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
66. Van de Velde LA, Guo XJ, Barbaric L, Smith AM, Oguin TH III, Thomas PG, et al. Stress kinase GCN2 controls the proliferative fitness and trafficking of cytotoxic T cells independent of environmental amino acid sensing. Cell Rep. (2016) 17:2247–58. doi: 10.1016/j.celrep.2016.10.079
67. Hu H, Tian M, Ding C, Yu S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. (2018) 9:3083. doi: 10.3389/fimmu.2018.03083
68. Xiong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, et al. Anti-PD-L1 treatment results in functional remodeling of the macrophage compartment. Cancer Res. (2019) 79:1493–506. doi: 10.1158/0008-5472.CAN-18-3208
69. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
70. Yarosz EL, Chang CH. The role of reactive oxygen species in regulating T cell-mediated immunity and disease. Immune Netw. (2018) 18:e14. doi: 10.4110/in.2018.18.e14
71. Bruger AM, Dorhoi A, Esendagli G, Barczyk-Kahlert K, van der Bruggen P, Lipoldova M, et al. How to measure the immunosuppressive activity of MDSC: assays, problems and potential solutions. Cancer Immunol Immunother. (2019) 68:631–44. doi: 10.1007/s00262-018-2170-8
72. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
73. Yu T, Gan S, Zhu Q, Dai D, Li N, Wang H, et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat Commun. (2019) 10:4353. doi: 10.1038/s41467-019-12384-2
74. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. (1998) 391:79–82. doi: 10.1038/34178
75. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. (2010) 33:699–712. doi: 10.1016/j.immuni.2010.11.009
76. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
77. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. (2018) 17:428–38. doi: 10.1080/15384101.2018.1444305
78. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
79. Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+ HLA-DR−/low cells in melanoma patients are Stat3hi and Overexpress CD80, CD83, and DC-Sign. Cancer Res. (2010) 70:4335–45. doi: 10.1158/0008-5472.CAN-09-3767
80. Rosborough BR, Mathews LR, Matta BM, Liu Q, Raïch-Regué D, Thomson AW, et al. Cutting edge: Flt3 ligand mediates STAT3-independent expansion but STAT3-dependent activation of myeloid-derived suppressor cells. J Immunol. (2014) 192:3470–3. doi: 10.4049/jimmunol.1300058
81. Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. (2013) 33:1135–44. doi: 10.1161/ATVBAHA.113.301453
82. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
83. Srivastava N, Kollipara RK, Singh DK, Sudderth J, Hu Z, Nguyen H, et al. Inhibition of cancer cell proliferation by PPARgamma is mediated by a metabolic switch that increases reactive oxygen species levels. Cell Metab. (2014) 20:650–61. doi: 10.1016/j.cmet.2014.08.003
84. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
85. Eubank TD, Galloway M, Montague CM, Waldman WJ, Marsh CB. M-CSF induces vascular endothelial growth factor production and angiogenic activity from human monocytes. J Immunol. (2003) 171:2637–43. doi: 10.4049/jimmunol.171.5.2637
86. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. (2017) 17:457–74. doi: 10.1038/nrc.2017.51
87. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. (2017) 8:15081. doi: 10.1038/ncomms15081
88. Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer. (2010) 10:575–85. doi: 10.1038/nrc2894
89. Linde N, Casanova-Acebes M, Sosa MS, Mortha A, Rahman A, Farias E, et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun. (2018) 9:21. doi: 10.1038/s41467-017-02481-5
90. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. (2015) 5:932–43. doi: 10.1158/2159-8290.CD-15-0012
91. Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. (2010) 70:6139–49. doi: 10.1158/0008-5472.CAN-10-0706
92. Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. (2019) 10:172. doi: 10.3389/fimmu.2019.00172
93. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
94. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. (2013) 23:249–62. doi: 10.1016/j.ccr.2013.01.008
95. Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med. (2013) 210:2873–86. doi: 10.1084/jem.20130875
96. Takeuchi S, Baghdadi M, Tsuchikawa T, Wada H, Nakamura T, Abe H, et al. Chemotherapy-derived inflammatory responses accelerate the formation of immunosuppressive myeloid cells in the tissue microenvironment of human pancreatic cancer. Cancer Res. (2015) 75:2629–40. doi: 10.1158/0008-5472.CAN-14-2921
97. Wu C, Tan X, Hu X, Zhou M, Yan J, Ding C. Tumor microenvironment following gemcitabine treatment favors differentiation of immunosuppressive Ly6C(high) myeloid cells. J Immunol. (2020) 204:212–23. doi: 10.4049/jimmunol.1900930
98. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. (2006) 313:1960–4. doi: 10.1126/science.1129139
99. Wang Q, Guldner IH, Golomb SM, Sun L, Harris JA, Lu X, et al. Single-cell profiling guided combinatorial immunotherapy for fast-evolving CDK4/6 inhibitor-resistant HER2-positive breast cancer. Nat Commun. (2019) 10:3817. doi: 10.1038/s41467-019-11729-1
100. Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, et al. The single-cell pathology landscape of breast cancer. Nature. (2020) 578:615–20. doi: 10.1038/s41586-019-1876-x
101. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. (2018) 50:96. doi: 10.1038/s12276-018-0071-8
102. Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. (2014) 11:417–22. doi: 10.1038/nmeth.2869
103. Ornatsky O, Bandura D, Baranov V, Nitz M, Winnik MA, Tanner S. Highly multiparametric analysis by mass cytometry. J Immunol Methods. (2010) 361:1–20. doi: 10.1016/j.jim.2010.07.002
104. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
105. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. (2001) 19:565–94. doi: 10.1146/annurev.immunol.19.1.565
106. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. (2019) 50:1317–34.e10. doi: 10.1016/j.immuni.2019.03.009
107. Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. (2019) 177:1330–45.e18. doi: 10.1016/j.cell.2019.03.005
108. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
109. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol. (2020) 5:eaay1863. doi: 10.1126/sciimmunol.aay1863
110. Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. (2016) 22:3425–31. doi: 10.1158/1078-0432.CCR-15-2428.
111. Chen C, Shen Y, Qu QX, Chen XQ, Zhang XG, Huang JA. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp Cell Res. (2013) 319:96–102. doi: 10.1016/j.yexcr.2012.09.006.
112. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. (2017) 169:750–65.e17. doi: 10.1016/j.cell.2017.04.014
113. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
114. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. (2017) 21:1399–410. doi: 10.1016/j.celrep.2017.10.030
115. Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr L, et al. Heterogeneity and targeted therapy-induced adaptations in lung cancer revealed by longitudinal single-cell RNA sequencing. bioRxiv. (2019) doi: 10.1101/2019.12.08.868828
116. Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. (2017) 18:234. doi: 10.1186/s13059-017-1362-4
117. Gieryng A, Pszczolkowska D, Bocian K, Dabrowski M, Rajan WD, Kloss M, et al. Immune microenvironment of experimental rat C6 gliomas resembles human glioblastomas. Sci Rep. (2017) 7:17556. doi: 10.1038/s41598-017-17752-w
118. Alban TJ, Alvarado AG, Sorensen MD, Bayik D, Volovetz J, Serbinowski E, et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight. (2018) 3:e122264. doi: 10.1172/jci.insight.122264
119. Roy S, Bag AK, Dutta S, Polavaram NS, Islam R, Schellenburg S, et al. Macrophage-derived neuropilin-2 exhibits novel tumor-promoting functions. Cancer Res. (2018) 78:5600–17. doi: 10.1158/0008-5472.CAN-18-0562
120. Deaglio S, Vaisitti T, Aydin S, Bergui L, D'Arena G, Bonello L, et al. CD38 and ZAP-70 are functionally linked and mark CLL cells with high migratory potential. Blood. (2007) 110:4012–21. doi: 10.1182/blood-2007-06-094029
121. Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An immune atlas of clear cell renal cell carcinoma. Cell. (2017) 169:736–49.e18. doi: 10.1016/j.cell.2017.04.016
122. Karakasheva TA, Dominguez GA, Hashimoto A, Lin EW, Chiu C, Sasser K, et al. CD38+ M-MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI Insight. (2018) 3:e97022. doi: 10.1172/jci.insight.97022
123. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. (2018) 8:1156–75. doi: 10.1158/2159-8290.CD-17-1033
124. Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC, et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med. (2015) 212:681–97. doi: 10.1084/jem.20141732
125. Tanimoto Y, Onishi Y, Sato Y, Kizaki H. Benzodiazepine receptor agonists modulate thymocyte apoptosis through reduction of the mitochondrial transmembrane potential. Jpn J Pharmacol. (1999) 79:177–83. doi: 10.1254/jjp.79.177
126. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. (2014) 134:2853–64. doi: 10.1002/ijc.28622
127. Heming M, Gran S, Jauch SL, Fischer-Riepe L, Russo A, Klotz L, et al. Peroxisome Proliferator-activated receptor-gamma modulates the response of macrophages to lipopolysaccharide and glucocorticoids. Front Immunol. (2018) 9:893. doi: 10.3389/fimmu.2018.00893
128. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. (2007) 6:137–43. doi: 10.1016/j.cmet.2007.06.010
129. Mould KJ, Jackson ND, Henson PM, Seibold M, Janssen WJ. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight. (2019) 4:e126556. doi: 10.1172/jci.insight.126556
130. Ma G, Pan PY, Eisenstein S, Divino CM, Lowell CA, Takai T, et al. Paired immunoglobin-like receptor-B regulates the suppressive function and fate of myeloid-derived suppressor cells. Immunity. (2011) 34:385–95. doi: 10.1016/j.immuni.2011.02.004
131. Chen HM, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest. (2018) 128:5647–62. doi: 10.1172/JCI97570
132. Suarez-Lopez L, Sriram G, Kong YW, Morandell S, Merrick KA, Hernandez Y, et al. MK2 contributes to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis. Proc Natl Acad Sci USA. (2018) 115:E4236–44. doi: 10.1073/pnas.1722020115
133. Brown DP, Jones DC, Anderson KJ, Lapaque N, Buerki RA, Trowsdale J, et al. The inhibitory receptor LILRB4 (ILT3) modulates antigen presenting cell phenotype and, along with LILRB2 (ILT4), is upregulated in response to salmonella infection. BMC Immunol. (2009) 10:56. doi: 10.1186/1471-2172-10-56
134. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. (2018) 562:605–9. doi: 10.1038/s41586-018-0615-z
135. Gui X, Deng M, Song H, Chen Y, Xie J, Li Z, et al. Disrupting LILRB4/APOE interaction by an efficacious humanized antibody reverses T-cell suppression and blocks AML development. Cancer Immunol Res. (2019) 7:1244–57. doi: 10.1158/2326-6066.CIR-19-0036
136. Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci USA. (2019) 116:1692–7. doi: 10.1073/pnas.1811067116
137. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. (2009) 183:2475–83. doi: 10.4049/jimmunol.0900986
138. Molinier-Frenkel V, Mestivier D, Castellano F. Alterations of the immunosuppressive IL4I1 enzyme activity induced by naturally occurring SNP/mutations. Genes Immun. (2016) 17:148–52. doi: 10.1038/gene.2015.55
139. Cousin C, Aubatin A, Le Gouvello S, Apetoh L, Castellano F, Molinier-Frenkel V. The immunosuppressive enzyme IL4I1 promotes FoxP3+ regulatory T lymphocyte differentiation. Eur J Immunol. (2015) 45:1772–82. doi: 10.1002/eji.201445000
140. Yue Y, Huang W, Liang J, Guo J, Ji J, Yao Y, et al. IL4I1 is a novel regulator of m2 macrophage polarization that can inhibit T cell activation via L-tryptophan and arginine depletion and IL-10 production. PLoS ONE. (2015) 10:e0142979. doi: 10.1371/journal.pone.0142979
141. Santarlasci V, Maggi L, Mazzoni A, Capone M, Querci V, Rossi MC, et al. IL-4-induced gene 1 maintains high Tob1 expression that contributes to TCR unresponsiveness in human T helper 17 cells. Eur J Immunol. (2014) 44:654–61. doi: 10.1002/eji.201344047
142. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a gemini of immune checkpoints. Cell Mol Immunol. (2018) 15:447–57. doi: 10.1038/cmi.2017.143
143. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
144. Ding AS, Routkevitch D, Jackson C, Lim M. Targeting myeloid cells in combination treatments for glioma and other tumors. Front Immunol. (2019) 10:1715. doi: 10.3389/fimmu.2019.01715
145. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
146. Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci USA. (2019) 116:997–1006. doi: 10.1073/pnas.1721434116
147. Koizumi T, Kerkhofs D, Mizuno T, Steinbusch HWM, Foulquier S. Vessel-associated immune cells in cerebrovascular diseases: from perivascular macrophages to vessel-associated microglia. Front Neurosci. (2019) 13:1291. doi: 10.3389/fnins.2019.01291
148. Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. ELife. (2020) 9:e52176. doi: 10.7554/eLife.52176
149. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. (2016) 1:e85841. doi: 10.1172/jci.insight.85841
150. Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Ostling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
151. Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. (2017) 8:14979. doi: 10.1038/ncomms14979
152. Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer. (2014) 14:970. doi: 10.1186/1471-2407-14-970
153. Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol. (2013) 13:206–18. doi: 10.1038/nri3391
154. Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol. (2018) 9:697. doi: 10.3389/fimmu.2018.00697
155. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
156. Gholamin S, Youssef OA, Rafat M, Esparza R, Kahn S, Shahin M, et al. Irradiation or temozolomide chemotherapy enhances anti-CD47 treatment of glioblastoma. Innate Immun. (2020) 26:130–7. doi: 10.1177/1753425919876690
157. Daniel P, Sabri S, Chaddad A, Meehan B, Jean-Claude B, Rak J, et al. Temozolomide induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol. (2019) 9:41. doi: 10.3389/fonc.2019.00041
158. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
159. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
160. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–35. doi: 10.1038/ni.1655
161. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. (2017) 23:556–67. doi: 10.1038/nm.4314
162. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
163. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci USA. (2019) 116:1361–9. doi: 10.1073/pnas.1812266115
164. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. (2010) 120:2699–714. doi: 10.1172/JCI39506
165. Wu X, Giobbie-Hurder A, Liao X, Connelly C, Connolly EM, Li J, et al. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol Res. (2017) 5:17–28. doi: 10.1158/2326-6066.CIR-16-0206
166. Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity. (2020) 52:668–82.e7. doi: 10.1016/j.immuni.2020.03.004
167. Palmieri EM, Menga A, Martin-Perez R, Quinto A, Riera-Domingo C, De Tullio G, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. (2017) 20:1654–66. doi: 10.1016/j.celrep.2017.07.054
168. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. (2010) 120:142–56. doi: 10.1172/JCI38942
169. Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta. (2011) 1807:726–34. doi: 10.1016/j.bbabio.2010.10.022
170. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
171. Roelofs AJ, Thompson K, Gordon S, Rogers MJ. Molecular mechanisms of action of bisphosphonates: current status. Clin Cancer Res. (2006) 12(20 Pt 2):6222s−30s. doi: 10.1158/1078-0432.CCR-06-0843
172. Stresing V, Daubine F, Benzaid I, Monkkonen H, Clezardin P. Bisphosphonates in cancer therapy. Cancer Lett. (2007) 257:16–35. doi: 10.1016/j.canlet.2007.07.007
173. Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med. (2010) 14:2803–15. doi: 10.1111/j.1582-4934.2009.00926.x
174. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun. (2019) 10:3974. doi: 10.1038/s41467-019-11911-5
175. Baer C, Squadrito ML, Laoui D, Thompson D, Hansen SK, Kiialainen A, et al. Suppression of microRNA activity amplifies IFN-gamma-induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol. (2016) 18:790–802. doi: 10.1038/ncb3371
176. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
177. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. (2018) 17:887–904. doi: 10.1038/nrd.2018.169
178. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
179. Manthey CL, Johnson DL, Illig CR, Tuman RW, Zhou Z, Baker JF, et al. JNJ-28312141, a novel orally active colony-stimulating factor-1 receptor/FMS-related receptor tyrosine kinase-3 receptor tyrosine kinase inhibitor with potential utility in solid tumors, bone metastases, and acute myeloid leukemia. Mol Cancer Ther. (2009) 8:3151–61. doi: 10.1158/1535-7163.MCT-09-0255
180. Bercovici N, Guerin MV, Trautmann A, Donnadieu E. The remarkable plasticity of macrophages: a chance to fight cancer. Front Immunol. (2019) 10:1563. doi: 10.3389/fimmu.2019.01563
181. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. (2016) 128:384–94. doi: 10.1182/blood-2015-12-687749
182. Ghinoi V, Brandi ML. Clodronate: mechanisms of action on bone remodelling and clinical use in osteometabolic disorders. Expert Opin Pharmacother. (2002) 3:1643–56. doi: 10.1517/14656566.3.11.1643
183. van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods. (1996) 193:93–9. doi: 10.1016/0022-1759(96)00056-7
184. Rooijen NV, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. (1994) 174:83–93. doi: 10.1016/0022-1759(94)90012-4
185. Sousa S, Auriola S, Monkkonen J, Maatta J. Liposome encapsulated zoledronate favours M1-like behaviour in murine macrophages cultured with soluble factors from breast cancer cells. BMC Cancer. (2015) 15:4. doi: 10.1186/s12885-015-1005-7
186. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
187. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. (2013) 19:57–64. doi: 10.1038/nm.2999
188. van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother. (2017) 66:1079–87. doi: 10.1007/s00262-017-2023-x
189. Feng Y, Mu R, Wang Z, Xing P, Zhang J, Dong L, et al. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat Commun. (2019) 10:2272. doi: 10.1038/s41467-019-10354-2
190. Mullins SR, Vasilakos JP, Deschler K, Grigsby I, Gillis P, John J, et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J Immunother Cancer. (2019) 7:244. doi: 10.1186/s40425-019-0724-8
191. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci USA. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
192. Shen L, Gao Y, Liu Y, Zhang B, Liu Q, Wu J, et al. PD-1/PD-L pathway inhibits M.tb-specific CD4+ T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci Rep. (2016) 6:38362. doi: 10.1038/srep38362
193. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. (2004) 172:989–99. doi: 10.4049/jimmunol.172.2.989
194. Murdoch C, Lewis CE. Macrophage migration and gene expression in response to tumor hypoxia. Int J Cancer. (2005) 117:701–8. doi: 10.1002/ijc.21422
195. Harney AS, Karagiannis GS, Pignatelli J, Smith BD, Kadioglu E, Wise SC, et al. The selective Tie2 inhibitor rebastinib blocks recruitment and function of Tie2(Hi) macrophages in breast cancer and pancreatic neuroendocrine tumors. Mol Cancer Ther. (2017) 16:2486–501. doi: 10.1158/1535-7163.MCT-17-0241
196. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289. doi: 10.3389/fimmu.2017.00289
197. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
198. Ho PC, Liu PS. Metabolic communication in tumors: a new layer of immunoregulation for immune evasion. J Immunother Cancer. (2016) 4:4. doi: 10.1186/s40425-016-0109-1
199. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
200. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
201. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
202. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014) 15:509–24. doi: 10.1038/nrm3838
203. Tirosh I, Izar B, Prakadan SM, Wadsworth MH II, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. (2016) 352:189–96. doi: 10.1126/science.aad0501
204. Schapiro D, Jackson HW, Raghuraman S, Fischer JR, Zanotelli VRT, Schulz D, et al. histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat Methods. (2017) 14:873–6. doi: 10.1038/nmeth.4391
205. Schulz D, Zanotelli VRT, Fischer JR, Schapiro D, Engler S, Lun XK, et al. Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst. (2018) 6:25–36.e5. doi: 10.1016/j.cels.2017.12.001
206. Rohan TE, Xue X, Lin HM, D'Alfonso TM, Ginter PS, Oktay MH, et al. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J Natl Cancer Inst. (2014) 106:dju136. doi: 10.1093/jnci/dju136
207. Najjar YG, Rayman P, Jia X, Pavicic PG Jr, Rini BI, Tannenbaum C, et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1beta, IL8, CXCL5, and Mip-1alpha. Clin Cancer Res. (2017) 23:2346–55. doi: 10.1158/1078-0432.CCR-15-1823
Keywords: MDSC, TAM, TIME, sc-RNAseq, reprogram
Citation: Davidov V, Jensen G, Mai S, Chen S-H and Pan P-Y (2020) Analyzing One Cell at a TIME: Analysis of Myeloid Cell Contributions in the Tumor Immune Microenvironment. Front. Immunol. 11:1842. doi: 10.3389/fimmu.2020.01842
Received: 15 May 2020; Accepted: 09 July 2020;
Published: 02 September 2020.
Edited by:
Sergei Kusmartsev, University of Florida, United StatesReviewed by:
Paul R. Dominguez-Gutierrez, University of Florida, United StatesCopyright © 2020 Davidov, Jensen, Mai, Chen and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-Hsia Chen, c2NoZW4zQGhvdXN0b25tZXRob2Rpc3Qub3Jn; Ping-Ying Pan, cHBhbkBob3VzdG9ubWV0aG9kaXN0Lm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.