 Pusen Wang1†
Pusen Wang1† Hao Li
Hao Li Lin Zhong
Lin Zhong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 August 2020
Sec. Alloimmunity and Transplantation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01723
This article is part of the Research Topic New Insights on Liver Transplant and Tolerance View all 18 articles
Liver transplantation (LT) has become the best chance and a routine practice for patients with end-stage liver disease and small hepatocellular carcinoma. However, life-long immunosuppressive regimens could lead to many post-LT complications, including cancer recurrence, infections, dysmetabolic syndrome, and renal injury. Impeccable management of immunosuppressive regimens is indispensable to ensure the best long-term prognosis for LT recipients. This is challenging for these patients, who probably have a post-LT graft survival of more than 10 or even 20 years. Approximately 20% of patients after LT could develop spontaneous operational tolerance. They could maintain normal graft function and histology without any immunosuppressive regimens. Operational tolerance after transplantation has been an attractive and ultimate goal in transplant immunology. The liver, as an immunoregulatory organ, generates an immune hyporesponsive microenvironment under physiological conditions. In this regard, LT recipients may be ideal candidates for studies focusing on operative tolerance. Cell-based strategies are one of the most promising methods for immune tolerance induction, including chimerism induced by hematopoietic stem cells and adoptive transfer of regulatory T cells, regulatory dendritic cells, regulatory macrophages, regulatory B cells, and mesenchymal stromal cells. The safety and the efficacy of many cell products have been evaluated by prospective clinical trials. In this review, we will summarize the latest perspectives on the clinical application of cell-based strategies in LT and will address a number of concerns and future directions regarding these cell products.
Liver transplantation (LT) has become the best chance for patients with end-stage liver disease and small hepatocellular carcinoma with chronic liver disease since it was first performed in 1963 (1, 2). With the development of new immunosuppressive regimens and the improvement of surgical techniques, LT has become a routine practice and is increasingly conducted around the world. However, most recipients need open-ended and even lifelong immunosuppression to achieve ideal long-term outcomes. This open-ended immunosuppressive therapy can result in many post-LT complications, such as cancer recurrence, dysmetabolic syndrome, infections, and renal injury. Interestingly, some LT recipients who are taken off immunosuppression for different reasons accidently develop immune tolerance. Among highly selected LT recipients, some of them could discontinue all immunosuppression for more than 1 year while maintaining a stable allograft status, which is defined as “operational tolerance” (3, 4). For these reasons, tolerance of LT has been an attractive and ultimate goal in transplant immunology. Approximately 20% of recipients could become completely tolerant without any immunosuppressant drugs after LT (5–7), whereas such “operational tolerance” is reported only anecdotally in recipients of other organs (8).
The liver has been generally recognized as an immunoregulatory organ (9, 10). It consists in parenchymal and innate immune cells, including hepatocytes and cholangiocytes, liver sinusoidal endothelial cells, hepatic stellate cells, Kupffer cells, stromal cells, liver-derived dendritic cells, natural killer (NK) cells, natural killer T (NKT) cells, and so on (11). The complex interactions between these cells and immune cells contribute to the induction of immune tolerance in the liver (12). The liver receives blood from both the portal vein and the hepatic artery. In the portal vein, the liver confronts various antigens from digested food and the gut microbiome under physiological conditions (13). The immune reaction is tightly controlled and regulated, generating liver-protective immunity while these antigens pass through sinusoids. The responsible mechanisms are associated with various elements, including immature and non-professional antigen-presenting cells, exhausted lymphocytes, transforming growth factor β (TGFβ), and interleukin (IL)-10 in the cytokine milieu, and high proportions of regulatory cells (14). Compared to that in other solid organ transplantations, the incidence of chronic rejection in LT is lower (15). With this background, LT recipients may be ideal candidates for clinical trials studying operative tolerance. A reproducible strategy to induce stable transplant tolerance may achieve success first in LT.
Substantial progress has been made toward immune tolerance induction and in the study of relevant mechanisms in animal models. However, the translation of these strategies into clinical transplantation remains challenging. In addition to novel immunomodulatory drugs such as belatacept, the most promising strategy is cell-based therapy. A variety of cell products were tested to induce tolerance in preclinical experiments, but only a small part of them were evaluated in clinical studies, including induction of chimerism by hematopoietic stem cells and adoptive transfer of regulatory cells and mesenchymal stromal cells. There are many advantages of cell-based strategies, such as low toxicity and long-term efficacy. In addition, a cell-based strategy is expected to control many kinds of inflammatory cells and generate donor antigen-specific tolerance (16). In this review, we mainly focus on cell-based strategies of tolerance induction in LT by clarifying the translational potential of these strategies.
Since the first successful application of mixed chimerism in tolerance induction in human kidney transplantation in 2008 (17), mixed chimerism induced by donor hematopoietic stem cell (HSC) infusion remains one of the most effective treatments for tolerance induction. Chimerism can be defined as tissues from two genetically distinct organisms coexisting in one organism (18). Mixed chimerism, in which both donor and recipient HSCs coexist, leads to donor-specific transplantation tolerance and retains immunocompetence for primary immune responses (19–22). Additionally, mixed chimerism can be induced through a non-myeloablative conditioning protocol, which represents a lower risk and severity of graft-vs.-host disease (GVHD) than the fully allogeneic chimerism induced by myeloablative conditioning.
In LT, the application of hematopoietic chimerism to achieve graft tolerance has been studied by various groups. The St. Mary's Hospital group reported that full donor chimerism induced by HSC transplantation could maintain stable allograft tolerance without immunosuppressants. Two patients were immunosuppressant-free with normal liver function for 6 and 7 years (Table 1) (23). Donckier et al. reported two pilot studies in which donor stem cell infusion under non-myeloablative conditioning was used to induce tolerance in living donor liver transplantation (LDLT). Both patients, who were treated with pretransplant conditioning using cyclophosphamide and anti-thymocyte globulin (ATG), discontinued immunosuppressive therapies 90 and 28 days after transplantation without subsequent rejection episodes (Table 1) (24). In the other study, three prospectively enrolled patients were treated with post-transplant conditioning using high doses of ATG and donor CD34+ stem cell infusion (5.3–10 × 106 cells/kg) (Figure 1; Table 1). Two of the three recipients successfully discontinued immunosuppression early without subsequent graft deterioration. Of note is that both patients developed acute rejection during follow-up (25). These results are promising. However, among the four immunosuppression-free patients, Donckier et al. reported relatively short follow-ups from 270 days to 561 days. In another study conducted by the University of Miami group, unprocessed donor bone marrow cell infusion without conditioning therapy was investigated for tolerance induction (Figure 1; Table 1). A total of 104 patients of at least 3 years post-transplantation were enrolled, among which 45 patients received donor bone marrow cell infusions (5.94 ± 0.4 × 108 cells/kg) during the early post-operative period and 59 patients did not. Immunosuppressive therapies were tapered slowly over 3 years after their enrollment. Twenty patients, 10 from each group, were immunosuppression-free during follow-up without a significant difference (26). These data indicate that chimerism-based strategies can produce long-term tolerance, but conditioning therapy seems indispensable. In the application of mixed chimerism for tolerance induction in LT, any risk of acute or chronic GVHD should be avoided. Although many studies reported less frequent incidences of GVHD using non-myeloablative conditioning regimen (27), there is still a risk of GVHD with durable mixed chimerism (28, 29). Therefore, modifications of the conditioning protocol and maybe delayed HSC infusion might be important next steps for chimerism-induced tolerance in LT.
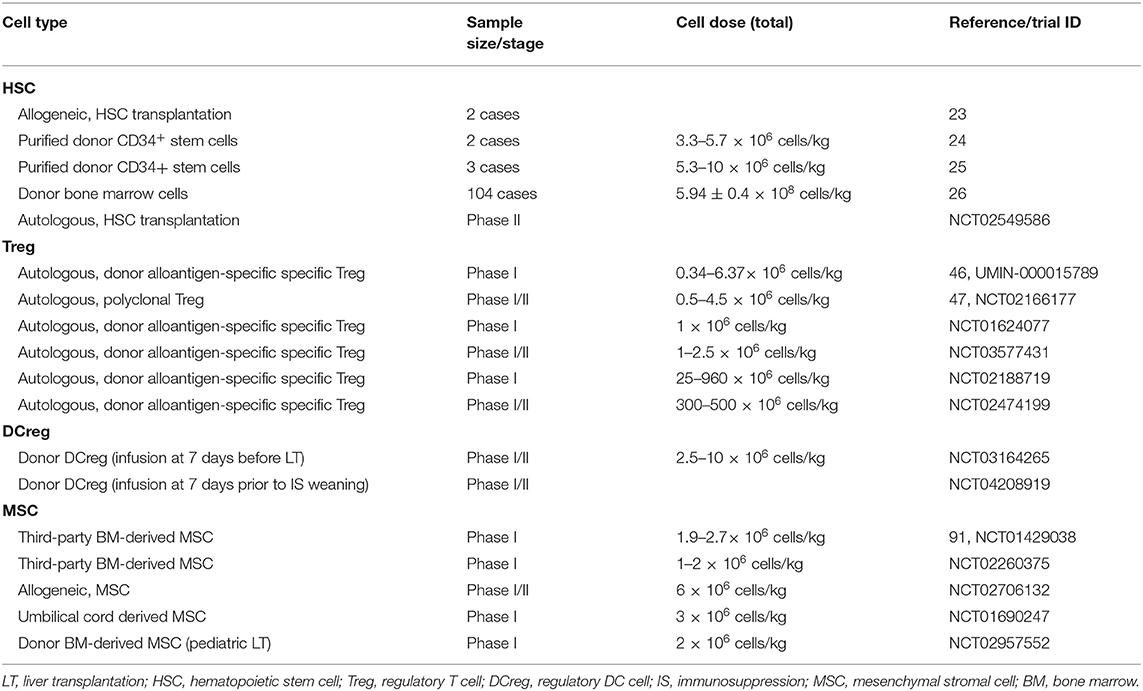
Table 1. Clinical studies/trials of cell-based strategies for tolerance induction in LT.
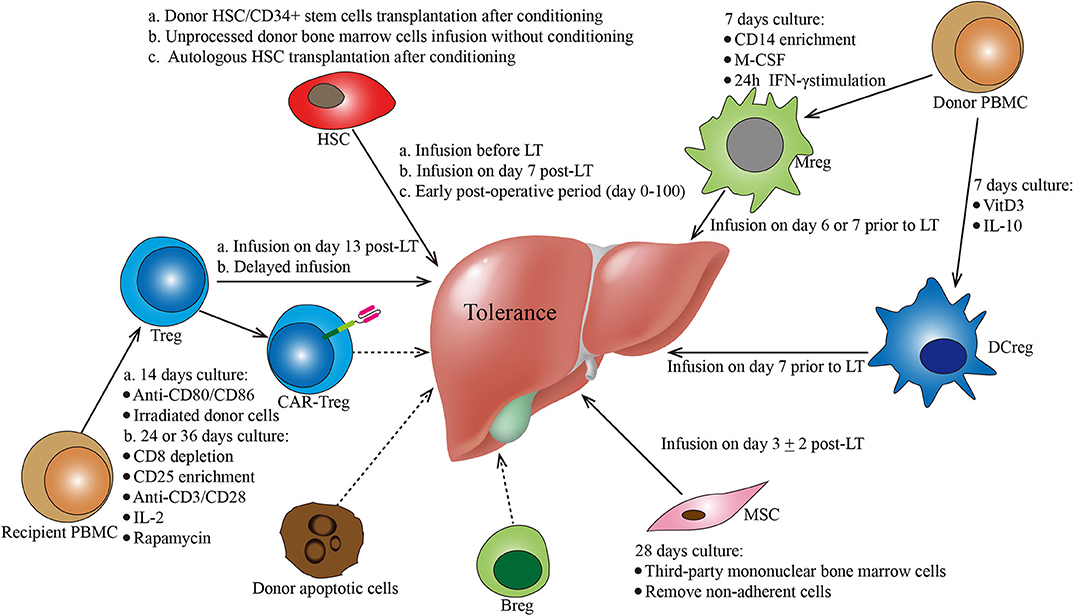
Figure 1. Protocols of various cell-based strategies for tolerance induction in liver transplantation. This figure summarizes the different cell-based therapies described in the manuscript. Cell types, culture conditions, dosages (i.e., cell number), and administration conditions are mentioned for each cell-based therapy. Breg, regulatory B cells; CAR, chimeric antigen receptor; DCreg, regulatory dendritic cells; HSC, hematopoietic stem cells; MSC, mesenchymal stroma cells; PBMC, peripheral blood mononuclear cells; Treg, regulatory CD4+ T cells.
In addition to donor HSC infusion, autologous hematopoietic stem cell transplantation (HSCT) has also been studied for tolerance induction. Autologous HSCT may reset the deregulated immune system into a tolerant status by regenerating new autotolerant T and B cells and increasing immune regulatory mechanisms (30–32). The therapeutic efficacy of autologous HSCT for various autoimmune diseases has been reported by clinical trials, such as trials in systemic and multiple sclerosis (33–35), Crohn's disease (36), and scleroderma (37). In LT, there is an ongoing clinical trial evaluating the ability of autologous HSCT to induce tolerance (NCT02549586) (Figure 1; Table 1). The researchers plan to recruit 10 liver transplant recipients. The purified HSCs will be infused following a chemotherapy- and ATG-based conditioning regimen. The immunosuppressive drugs will be withdrawn at 6 months post-HSCT.
Regulatory T cells (Tregs), commonly distinguished into natural Tregs (nTregs) and inducible Tregs (iTregs) (38), are a population of CD4+ cells that constitutively express the Forkhead box P3 (Foxp3) transcription factor. In 2001, human Tregs were initially identified as CD4+CD25+ T cells (39), which comprise 5–10% of total peripheral CD4+ T cells (40). Over the past two decades, Tregs have been studied and are known to be responsible for maintaining immune homeostasis and tolerance (41, 42). The adoptive transfer of Tregs has been successful and proven effective in murine (43, 44) and non-human primate transplantation models (45).
In the LT setting, tolerance induction by Treg infusion has been reported in two clinical trials to date (46, 47). Researchers from Hokkaido University reported a pilot study of tolerance induction with Treg-based cell therapy in living donor LT in 2016 (46). In this study, donor-antigen-specific iTregs were obtained ex vivo by coculturing recipient lymphocytes with irradiated donor cells and anti-CD80/CD86 mAbs for 2 weeks. At day 13 after LT, the expanded cells were administered to the recipients at a mean dose of 3.39 × 106/kg CD4+CD25+Foxp3+ cells (Figure 1; Table 1). This dose is much lower than the dose of nTregs for transplant cell therapy since donor-antigen-specific iTregs are considered to be more potent than nTregs (48, 49). The infusion caused no significant adverse events. After infusion, the immunosuppressive agent weaning program was initiated at 6 months post-LT and completely discontinued at 18 months. Among the 10 consecutively enrolled patients, seven completely stopped their immunosuppressive regimen for 16–33 months with normal graft function and histology. The other three recipients who had autoimmune liver diseases developed acute cellular rejection and resumed reduced doses of immunotherapy. This is the first study of successful operational tolerance induction using the adoptive transfer of Tregs in LT.
More recently, the King's College London group published the results of their phase I clinical trial, ThRIL, evaluating the safety and the efficacy profile of Treg therapy in LT recipients (47). In this trial, patients with an autoimmune disease were excluded. Tregs isolated from the recipients were expanded under polyclonal conditions ex vivo using anti-CD3/CD28 beads, IL-2, and rapamycin for 24 or 36 days. Three patients awaiting LT were enrolled and received an infusion of 1 × 106 Tregs/kg 83–110 days post-transplant, while the other six patients were recruited 6–12 months post-transplant and received an infusion of 4.5 × 106 Tregs/kg 112–151 days after enrollment (Figure 1; Table 1). Of note is that one of the patients who received an infusion of 4.5 × 106 Tregs developed a fever of >39°C associated with rigors, which was classified as a dose-limiting toxicity. In general, this autologous non-specific Treg transfer was considered to be safe and exerted potentially beneficial donor-specific immunosuppressive effects.
Treg-induced immune regulation is the best-studied and core mechanism of tolerance. Both preliminary clinical studies demonstrated the safety and the efficacy of Treg strategies in human LT, which has great potential for future clinical translation. Many other registered phase I/II clinical trials assessing the safety and the efficacy of Treg infusion are in progress (NCT01624077, NCT03577431, NCT02188719, and NCT02474199) (Table 1). However, multicenter studies with large sample sizes need to be conducted, and future studies should focus on the protocol of Treg infusion, such as cell dosage, timing/frequency of infusion, optimal immunosuppressive regimen, and its late complications. Additionally, some other approaches to generate antigen-specific Tregs can be promising in LT, such as chimeric antigen receptor (CAR) transduction (50). It was shown that the adoptive transfer of Tregs engineered with a CAR which targets HLA-A2 can suppress skin allograft rejection in humanized mouse models (51, 52). Therefore, these modified cells may have a great potential in LT.
Dendritic cells (DCs) were first identified by Steinman and Cohn in 1973 (53, 54) and have proved potent antigen-presenting cells linking the innate and the adaptive immune responses (55). Over the following years, based on their morphological features, ontogenies, locations, and functions, various DC subsets have been identified, including conventional DCs (cDCs), plasmacytoid DCs (pDCs), Langerhans cells (LCs), and inflammatory DCs (56, 57). It has been reported that DCs can be either immunogenic or tolerogenic in different states (58, 59). Of note is that the ablation of DCs could break the self-tolerance of CD4+ T cells and result in spontaneous fatal autoimmunity (60).
Tolerogenic DCs or regulatory DCs (DCregs) are characterized by a low expression of MHC gene products (MHC class I and II) and co-stimulatory molecules (CD80 and CD86) and a high expression of co-inhibitory ligands (PD-L1) and death-inducing ligands (FasL). In terms of functions, DCregs are resistant to maturation, able to produce anti-inflammatory cytokines (IL-10 and TGFβ), and impair T cell proliferation.
In LT, tolerance induction by DCregs is ongoing as phase I/II clinical trials conducted by the University of Pittsburgh group (61). Actually, the safety, the tolerability, and the feasibility of autologous DCreg infusion have been confirmed by various clinical studies in autoimmune disorders, such as rheumatoid arthritis (62, 63), type 1 diabetes (64), and Crohn's disease (65). In a preclinical non-human primate renal transplant model, it has been demonstrated that a single pretransplant DCreg infusion can prolong MHC-mismatched renal allograft survival (66–68). In an ongoing trial (NCT03164265), according to preclinical experience, DCregs are generated from monocytes of prospective living donors in the presence of VitD3 and IL-10. Then, the expanded cells are infused at a dose of 2.5–10 × 106/kg (54) into the respective recipients 7 days before transplantation (Figure 1; Table 1). Weaning and withdrawal of the immunosuppressive drugs begin at 12 and 18 months post-transplantation, respectively.
More recently, the University of Pittsburgh group registered another clinical trial evaluating the delayed infusion of DCreg for tolerance induction in LDLT (NCT04208919) (Table 1). In this clinical trial, recipients who are between 1 and 3 years after transplantation will be enrolled. Those enrolled patients will receive a single infusion of donor-derived DCreg. At 1 week after that, immunosuppression weaning will be initiated slowly. Since preclinical experience to date has proven that DCreg infusion is safe, the results of their clinical trials are awaited with great interest.
Macrophages are immune cells of hematopoietic origin that provide crucial innate immune defense and have tissue-specific functions in the regulation and the maintenance of organ homeostasis (69). Various macrophage subsets with distinct functions have been identified, including classically activated macrophages (M1 macrophages), alternatively activated macrophages (M2 macrophages), regulatory macrophages (Mregs), tumor-associated macrophages, and myeloid-derived suppressor cells (70–73).
Human Mregs reflect a unique state of macrophage differentiation, distinguished from macrophages in other activation states by their particular mode of derivation, robust phenotype, and potent T cell-suppressing function (74). According to the protocol from University Hospital Regensburg (74, 75), Mregs could be generated from CD14+ blood monocytes in the presence of M-CSF and a further 24-h stimulation with IFN-γ after 7 days of culture. In terms of cell surface phenotype, these cells are homogeneously CD14−/lowHLA-DR+CD80−/lowCD86+CD16−TLR2−CD163−/low (75). In this pilot study, these cells were administered 1 week prior to transplantation to two living-donor renal transplant recipients at doses of 7.1 × 106 and 8 × 106 cells/kg (75) (Figure 1). Both patients were weaned to low-dose tacrolimus monotherapy and maintained normal graft function. Of note is that the infused cells were 111In-labeled, which is different from that of other studies. The tracking results showed that the donor-derived Mregs remained viable for more than 30 days and migrated from the pulmonary vasculature via the blood to the liver, spleen, and bone marrow. The clinical trial of Mreg treatment for renal transplantation was registered as the One study, in which donor-derived Mregs (2.5–7.5 × 106 cells/kg) were infused 6–7 days before transplantation into recipients of a living donor renal transplant (NCT02085629). However, in LT, there is no registered clinical trial about Mregs to date.
Mesenchymal stromal cells (MSCs) are plastic-adherent non-hematopoietic multipotent cells that are able to differentiate into osteoblasts, adipocytes, and chondroblasts under standard in vitro differentiating conditions (76). In terms of cell surface phenotype as measured by flow cytometry, these cells express CD105, CD73, and CD90 and lack expression (<2% positive) of CD45, CD34, CD14, or CD11b, CD79a or CD19, and HLA class II (76, 77). Bone marrow was first and most commonly used to isolate MSCs for clinical applications since 2004 (78). Over the last few years, other sources of MSCs have been found and proposed for clinical use, including adipose tissue (79), dental tissues (80), placenta (81), umbilical cord tissue (82), and cord blood (83). MSC-induced immunosuppression targets both the innate (84) and the adaptive immune systems (85). A variety of studies documented the potent immunosuppressive capacity of MSCs in vitro and in vivo (86–88). The immunoregulatory effect of MSCs is dose dependent and seems independent of MHC (89). The effects of autologous MSC infusion have been evaluated in kidney transplantation in a randomized controlled trial with a large sample size (90). In this study, MSC infusion resulted in a lower incidence of acute rejection, a lower risk of opportunistic infection, and better renal function at 1 year.
In LT, MSC therapy has been evaluated in a phase I–II clinical study (91). In this study, MSCs were generated by isolating mononuclear BM cells with Ficoll and expanding them in a 4 week culture. Third-party unrelated MSCs were infused at a dose of 1.5–3 × 106/kg on post-transplantation day 3 ± 2 in 10 patients (Figure 1; Table 1). Compared with 10 control liver transplant recipients, patients who received MSC infusion did not have an impairment of organ functions or an increased rate of opportunistic infection or malignancies. Weaning and withdrawal of the immunosuppressive drugs were attempted from month 6 to 12 post-transplantation. Among nine MSC recipients, tacrolimus and mycophenolate mofetil withdrawal were successfully achieved in only one patient, while the other eight patients failed due to either graft rejection or a significant increase in transaminases.
The very fast tapering of immunosuppressive drugs within 3 months might explain the failure of tolerance induction using MSCs in this study (91, 92). MSC-induced immunoregulation might be disrupted by very fast drug discontinuation, which could promote effector T cell activation (92). In addition, lack of induction therapy, insufficient MSC dosage, timing of infusion, infusion routes, and different sources might also account for the failure. As far as we know, MSC infusion for tolerance induction in LT is being studied by various other registered clinical trials (NCT02260375, NCT02706132, NCT01690247, and NCT02957552) (Table 1), including that using donor-derived MSC and that using umbilical-cord-derived MSC. Indeed these variables should be studied in future studies to achieve better results.
Regulatory B cells (Bregs) are immunosuppressive cells that support immunological tolerance (93, 94). Bregs express immune-regulatory cytokines, including IL-10, TGF-β, and IL-35, through which Bregs can suppress the differentiation of various pro-inflammatory lymphocytes (93, 94).
The role of Bregs in transplant tolerance has been studied mostly in rodents using heart (95, 96) and islet (97, 98) graft models. Stable immune tolerance to solid organ and islet cell grafts can be induced using antibodies directed at CD45RB, and this tolerance is dependent on Bregs (95, 97, 98). Furthermore, graft rejection in B cell-deficient mice after islet transplantation could be reversed to tolerance by the adoptive transfer of B cells from tolerant mice (97). The mechanisms of Breg-induced tolerance remain unclear, and research is still active. Breg-dependent islet transplant tolerance also requires the functions of natural killer cells and Tregs (98). Although promising, many questions remain unsolved in Breg-based strategies, such as the method of ex vivo culture and the expansion and the ability of tolerance induction in large animals. The field of Breg-induced tolerance is very immature and has a long way to go before translation into clinical LT.
Apoptosis is a genetically programmed cell death mechanism occurring during the elimination of unwanted or dangerous cells. Apoptotic cells exhibit immunomodulatory properties through various mechanisms, including inhibiting pathogenic T or B cell responses and inducing pro-tolerogenic/regulatory cells (99). In addition to those specific cell populations, apoptotic cell-based therapy is another promising strategy for tolerance induction in transplantation. In many animal studies, apoptotic cells, mainly apoptotic splenocytes in transplantation setting, favor the engraftment of liver (100, 101), cardiac (102, 103), islet (104), and hematopoietic (105) allografts.
In LT, tolerance induction using apoptotic cells was studied in rats by two groups. Researchers from Zhejiang University reported that donor apoptotic cells can promote liver graft acceptance using a rat LT model (106). In this study, apoptotic splenic lymphocytes induced by ultraviolet-C (UVC) irradiation at a dosage of 5 × 107 cells/rat were infused intravenously at 7 days before LT. In terms of mechanism, they found increased peripheral blood Tregs in rats treated with UVC-irradiated lymphocytes. In another study performed by the Zhejiang University group, they showed that the combination of tolerogenic DCs and apoptotic lymphocytes alleviates rejection after LT (100). The other group, Zhejiang Cancer Hospital group, also reported that preinfusion of apoptotic lymphocytes can induce immune tolerance in a rat LT model (101). The apoptotic cells used in this study were obtained from donor peripheral blood. Lymphocytes irradiated by X-ray from an electron linear accelerator at an absorbed dose of 2.0 Gy were infused intravenously at a dosage of 5 × 107 cells/rat 7 days before operation. However, there are no clinical studies or studies using nonhuman primate transplantation models considering the immunoregulatory role of apoptotic cells in LT. This can be an important next step in the field.
There are many concerns for cell-based therapies. First, for allogenic/autologous HSC infusion, intense myeloablative or non-myeloablative conditioning therapy may not be tolerated by patients with end-stage liver disease. Of note is that, in the absence of conditioning therapy, donor HSC infusion may show no significant effect in LT (26). Various non-myeloablative conditioning regimens of reduced intensity are now being studied, revised, and improved to induce tolerance with less toxicity (27, 107–109). Second, protocols of these regulatory cell-based therapies are not clearly established. The optimal dose of regulatory cells could be highly important for those strategies using non-donor-specific therapies. Overdose may result in original disease recurrence, infection, and other immunosuppression-related complications. In addition, there is no current consensus regarding the method and the timing of administration. The potential influence of different protocols needs to be observed in a study with long follow-up. Third, the purity of in vitro-expanded cells represents another significant concern. After stimulation, culture, and expansion, cell products without classical confirmed markers cannot be purified. There are subpopulations in these cell products that have different functions, such as resting and activated Tregs, highly suppressive Tregs, and non-Treg FoxP3+ cells (110–113). Additionally, Tregs can acquire the expression of transcription factors associated with effector T cell programs (called Th-like Tregs), which is called Treg cell plasticity (114). These Th-like Tregs can be pro-inflammatory rather than suppressive, which play an opposite role in tolerance induction. Fourth, the stability of infused cells has not been determined. Except for one study (75), none of the studies thus far have used cell products that were radioactively labeled. Without genetic or radioactive labels, in vivo homing and the stability of infused cells are difficult to detect. Fifth, at least for now, another drawback of cell-based therapy is the cost. Basic researches, clinical trials, and products of these cell-based therapies are very costly. Last but not least, the selection of patients is one of the top issues. Allogenic/autologous HSC infusion may not be conducted in patients with end-stage organ disease considering the toxicity of recipient conditioning (115, 116). Regulatory cells may play a detrimental role in patients with hepatitis B infection (117, 118), hepatitis C infection (119, 120), or hepatocellular carcinoma (121, 122). Pilot studies or phase I/II clinical trials with small sample sizes are unable to uncover all potential risks. It is vital to conduct high-quality studies with large sample sizes to assess the safety and the efficacy of these cellular strategies in tolerance induction.
Undoubtedly, cell-based strategies have a great potential in tolerance induction in transplantation. In particular, LT provides a great opportunity to achieve this goal because of an immunoregulatory microenvironment in the liver. Compared to immunosuppressant drugs, cell products can generate donor-specific tolerance with low toxicity and long-term efficacy. Theoretically, they could maintain normal graft function without immunosuppression and keep the protective properties of the immune system intact. Various cell products using different infusion protocols are being evaluated in clinical studies (Figure 1). These preliminary clinical studies have demonstrated a promising breakthrough in tolerance induction using cell-based therapies, including HSCs, Tregs, Mregs, DCregs, and MSCs. Breg-induced tolerance has mostly been studied in animal models, and the field is still in its infancy. The clinical studies or trials exploring the safety and the efficacy of cell-based strategies for tolerance induction in LT are listed in Table 1. Many excellent translational results may show up in the next few years. However, various problems with these strategies remain challenging. These strategies are not equally effective in each patient, suggesting that diverse mechanisms of immune tolerance exist among different individuals. Identifying useful biomarkers of immune tolerance that could guide the gradual tapering of immunosuppression after cell-based therapies is necessary. In addition, the heterogeneity of immunological status and health state between patients makes tolerance induction difficult with a universal protocol. The combination of cell products and other therapies as well as individualized treatment might provide ideal results. However, more high-quality clinical studies focusing on these strategies need to be performed for practical translation from bench to bedside.
PW and ZJ wrote this manuscript and prepared the table and the figure. LZ and DX devised and supervised this project. CW, XL, and HL assisted in the literature search and manuscript editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell RW. Homotransplantation of the liver in humans. Surg Gynecol Obstet. (1963) 117:659–76.
2. Starzl TE. The long reach of liver transplantation. Nat Med. (2012) 18:1489–92. doi: 10.1038/nm.2927
3. Benitez C, Londono MC, Miquel R, Manzia TM, Abraldes JG, Lozano JJ, et al. Prospective multicenter clinical trial of immunosuppressive drug withdrawal in stable adult liver transplant recipients. Hepatology. (2013) 58:1824–35. doi: 10.1002/hep.26426
4. Feng S, Ekong UD, Lobritto SJ, Demetris AJ, Roberts JP, Rosenthal P, et al. Complete immunosuppression withdrawal and subsequent allograft function among pediatric recipients of parental living donor liver transplants. JAMA. (2012) 307:283–93. doi: 10.1001/jama.2011.2014
5. Adams DH, Sanchez-Fueyo A, Samuel D. From immunosuppression to tolerance. J Hepatol. (2015) 62(Suppl. 1):S170–85. doi: 10.1016/j.jhep.2015.02.042
6. Crispe IN, Giannandrea M, Klein I, John B, Sampson B, Wuensch S. Cellular and molecular mechanisms of liver tolerance. Immunol Rev. (2006) 213:101–18. doi: 10.1111/j.1600-065X.2006.00435.x
7. Feng S. Spontaneous and induced tolerance for liver transplant recipients. Curr Opin Organ Transplant. (2016) 21:53–8. doi: 10.1097/MOT.0000000000000268
8. Taner T. Liver transplantation: rejection and tolerance. Liver Transpl. (2017) 23:S85–8. doi: 10.1002/lt.24840
9. Demetris AJ, Bellamy CO, Gandhi CR, Prost S, Nakanuma Y, Stolz BD. Functional immune anatomy of the liver-as an allograft. Am J Transplant. (2016) 16:1653–80. doi: 10.1111/ajt.13749
10. Tiegs G, Lohse WA. Immune tolerance: what is unique about the liver. J Autoimmun. (2010) 34:1–6. doi: 10.1016/j.jaut.2009.08.008
11. Huang H, Lu Y, Zhou T, Gu G, Xia Q. Innate immune cells in immune tolerance after liver transplantation. Front Immunol. (2018) 9:2401. doi: 10.3389/fimmu.2018.02401
12. Zheng M, Tian Z. Liver-mediated adaptive immune tolerance. Front Immunol. (2019) 10:2525. doi: 10.3389/fimmu.2019.02525
13. Chaudhry S, Emond J, Griesemer A. Immune cell trafficking to the liver. Transplantation. (2019) 103:1323–37. doi: 10.1097/TP.0000000000002690
14. Feng S, Bucuvalas J. Tolerance after liver transplantation: where are we? Liver Transpl. (2017) 23:1601–14. doi: 10.1002/lt.24845
15. Wu SL, Pan EC. Tolerance and chimerism and allogeneic bone marrow/stem cell transplantation in liver transplantation. World J Gastroenterol. (2013) 19:5981–7. doi: 10.3748/wjg.v19.i36.5981
16. Ten Brinke A, Martinez-Llordella M, Cools N, Hilkens CMU, van Ham SM, Sawitzki B, et al. Ways forward for tolerance-inducing cellular therapies- an AFACTT perspective. Front Immunol. (2019) 10:181. doi: 10.3389/fimmu.2019.00181
17. Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med. (2008) 358:353–61. doi: 10.1056/NEJMoa071074
18. Wu S, Xu H, Ravindra K, Ildstad TS. Composite tissue allotransplantation: past, present and future-the history and expanding applications of CTA as a new frontier in transplantation. Transplant Proc. (2009) 41:463–5. doi: 10.1016/j.transproceed.2009.01.027
19. Ildstad ST, Sachs HD. Reconstitution with syngeneic plus allogeneic or xenogeneic bone marrow leads to specific acceptance of allografts or xenografts. Nature. (1984) 307:168–70. doi: 10.1038/307168a0
20. Ruedi E, Sykes M, Ildstad ST, Chester CH, Althage A, Hengartner H, et al. Antiviral T cell competence and restriction specificity of mixed allogeneic (P1 + P2—-P1) irradiation chimeras. Cell Immunol. (1989) 121:185–95. doi: 10.1016/0008-8749(89)90016-6
21. Mache CJ, Schwinger W, Spendel S, Zach O, Regauer S, Ring E. Skin transplantation to monitor clinical donor-related tolerance in mixed hematopoietic chimerism. Pediatr Transplant. (2006) 10:128–31. doi: 10.1111/j.1399-3046.2005.00412.x
22. Trivedi HL, Vanikar AV, Modi PR, Shah VR, Vakil JM, Trivedi VB, et al. Allogeneic hematopoietic stem-cell transplantation, mixed chimerism, and tolerance in living related donor renal allograft recipients. Transplant Proc. (2005) 37:737–42. doi: 10.1016/j.transproceed.2005.01.028
23. Kim SY, Kim DW, Choi JY, Kim DG, Min WS, Lee JW, et al. Full donor chimerism using stem-cell transplantation for tolerance induction in the human leukocyte antigen-matched liver transplant setting. Transplantation. (2009) 88:601–3. doi: 10.1097/TP.0b013e3181b164d5
24. Donckier V, Troisi R, Toungouz M, Colle I, Van Vlierberghe H, Jacquy C, et al. Donor stem cell infusion after non-myeloablative conditioning for tolerance induction to HLA mismatched adult living-donor liver graft. Transpl Immunol. (2004) 13:139–46. doi: 10.1016/j.trim.2004.05.004
25. Donckier V, Troisi R, Le Moine A, Toungouz M, Ricciardi S, Colle I, et al. Early immunosuppression withdrawal after living donor liver transplantation and donor stem cell infusion. Liver Transpl. (2006) 12:1523–8. doi: 10.1002/lt.20872
26. Tryphonopoulos P, Tzakis AG, Weppler D, Garcia-Morales R, Kato T, Madariaga JR, et al. The role of donor bone marrow infusions in withdrawal of immunosuppression in adult liver allotransplantation. Am J Transplant. (2005) 5:608–13. doi: 10.1111/j.1600-6143.2004.00743.x
27. Sasaki H, Oura T, Spitzer TR, Chen YB, Madsen JC, Allan J, et al. Preclinical and clinical studies for transplant tolerance via the mixed chimerism approach. Hum Immunol. (2018) 79:258–65. doi: 10.1016/j.humimm.2017.11.008
28. Peggs KS, Thomson K, Hart DP, Geary J, Morris EC, Yong K, et al. Dose-escalated donor lymphocyte infusions following reduced intensity transplantation: toxicity, chimerism, disease responses. Blood. (2004) 103:1548–56. doi: 10.1182/blood-2003-05-1513
29. Perruche S, Marandin A, Kleinclauss F, Angonin R, Fresnay S, Baron MH, et al. Association of mixed hematopoietic chimerism with elevated circulating autoantibodies and chronic graft-versus-host disease occurrence. Transplantation. (2006) 81:573–82. doi: 10.1097/01.tp.0000183878.53367.77
30. Swart JF, Delemarre EM, van Wijk F, Boelens JJ, Kuball J, van Laar JM, et al. Haematopoietic stem cell transplantation for autoimmune diseases. Nat Rev Rheumatol. (2017) 13:244–56. doi: 10.1038/nrrheum.2017.7
31. Muraro PA, Martin R, Mancardi GL, Nicholas R, Sormani MP, Saccardi R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat Rev Neurol. (2017) 13:391–405. doi: 10.1038/nrneurol.2017.81
32. Muraro PA, Robins H, Malhotra S, Howell M, Phippard D, Desmarais C, et al. T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest. (2014) 124:1168–72. doi: 10.1172/JCI71691
33. Burt RK, Oliveira MC, Shah SJ, Moraes DA, Simoes B, Gheorghiade M, et al. Cardiac involvement and treatment-related mortality after non-myeloablative haemopoietic stem-cell transplantation with unselected autologous peripheral blood for patients with systemic sclerosis: a retrospective analysis. Lancet. (2013) 381:1116–24. doi: 10.1016/S0140-6736(12)62114-X
34. Mancardi GL, Sormani MP, Gualandi F, Saiz A, Carreras E, Merelli E, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology. (2015) 84:981–8. doi: 10.1212/WNL.0000000000001329
35. Muraro PA, Pasquini M, Atkins HL, Bowen JD, Farge D, Fassas A, et al. Multiple sclerosis-autologous hematopoietic stem cell transplantation long-term outcomes study: long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. (2017) 74:459–69. doi: 10.1001/jamaneurol.2016.5867
36. Pineton de Chambrun G, Pariente B, Seksik P, Altwegg R, Vuitton L, Stefasnescu C, et al. Adalimumab for patients with Crohn's disease complicated by intra-abdominal abscess: a multicentre, prospective, observational cohort study. J Crohns Colitis. (2019) 13(Suppl. 1):S616. doi: 10.1093/ecco-jcc/jjz045
37. Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. (2018) 378:35–47. doi: 10.1056/NEJMoa1703327
38. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. (2012) 209:1713–22, S1–19. doi: 10.1084/jem.20120822
39. Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J Exp Med. (2001) 193:1303–10. doi: 10.1084/jem.193.11.1303
40. Wang YM, Ghali J, Zhang GY, Hu M, Wang Y, Sawyer A, et al. Development and function of Foxp3+ regulatory T cells. Nephrology. (2016) 21:81–5. doi: 10.1111/nep.12652
41. Sakaguchi S, Miyara M, Costantino CM, Hafler AD. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
42. Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. (2000) 101:455–8. doi: 10.1016/S0092-8674(00)80856-9
43. Chandrasekharan D, Issa F, Wood JK. Achieving operational tolerance in transplantation: how can lessons from the clinic inform research directions? Transpl Int. (2013) 26:576–89. doi: 10.1111/tri.12081
44. van der Net JB, Bushell A, Wood KJ, Harden NP. Regulatory T cells: first steps of clinical application in solid organ transplantation. Transpl Int. (2016) 29:3–11. doi: 10.1111/tri.12608
45. Ezzelarab MB, Zhang H, Guo H, Lu L, Zahorchak AF, Wiseman RW, et al. Regulatory T cell infusion can enhance memory T cell and alloantibody responses in lymphodepleted nonhuman primate heart allograft recipients. Am J Transplant. (2016) 16:1999–2015. doi: 10.1111/ajt.13685
46. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. (2016) 64:632–43. doi: 10.1002/hep.28459
47. Sanchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant. (2019) 20:1125–36. doi: 10.1111/ajt.15700
48. Tang Q, Bluestone AJ. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med. (2013) 3:a015552. doi: 10.1101/cshperspect.a015552
49. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. (2011) 3:83ra42. doi: 10.1126/scitranslmed.3002076
50. Raffin C, Vo LT, Bluestone AJ. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol. (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
51. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant. (2017) 17:917–30. doi: 10.1111/ajt.14175
52. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
53. Steinman RM, Cohn AZ. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. (1973) 137:1142–62. doi: 10.1084/jem.137.5.1142
54. Steinman RM, Cohn AZ. Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J Exp Med. (1974) 139:380–97. doi: 10.1084/jem.139.2.380
55. Mellman I. Dendritic cells: master regulators of the immune response. Cancer Immunol Res. (2013) 1:145–9. doi: 10.1158/2326-6066.CIR-13-0102
56. Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. (2007) 8:1217–26. doi: 10.1038/ni1522
57. Balan S, Saxena M, Bhardwaj N. Dendritic cell subsets and locations. Int Rev Cell Mol Biol. (2019) 348:1–68. doi: 10.1016/bs.ircmb.2019.07.004
58. Vander Lugt B, Riddell J, Khan AA, Hackney JA, Lesch J, DeVoss J, et al. Transcriptional determinants of tolerogenic and immunogenic states during dendritic cell maturation. J Cell Biol. (2017) 216:779–92. doi: 10.1083/jcb.201512012
59. Steinman RM, Hawiger D, Nussenzweig CM. Tolerogenic dendritic cells. Annu Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
60. Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med. (2009) 206:549–59. doi: 10.1084/jem.20082394
61. Thomson AW, Humar A, Lakkis FG, Metes MD. Regulatory dendritic cells for promotion of liver transplant operational tolerance: rationale for a clinical trial and accompanying mechanistic studies. Hum Immunol. (2018) 79:314–21. doi: 10.1016/j.humimm.2017.10.017
62. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med. (2015) 7:290ra87. doi: 10.1126/scitranslmed.aaa9301
63. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
64. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. (2011) 34:2026–32. doi: 10.2337/dc11-0472
65. Jauregui-Amezaga A, Cabezon R, Ramirez-Morros A, Espana C, Rimola J, Bru C, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory Crohn's disease: a phase I study. J Crohns Colitis. (2015) 9:1071–8. doi: 10.1093/ecco-jcc/jjv144
66. Ezzelarab MB, Zahorchak AF, Lu L, Morelli AE, Chalasani G, Demetris AJ, et al. Regulatory dendritic cell infusion prolongs kidney allograft survival in nonhuman primates. Am J Transplant. (2013) 13:1989–2005. doi: 10.1111/ajt.12310
67. Ezzelarab MB, Raich-Regue D, Lu L, Zahorchak AF, Perez-Gutierrez A, Humar A, et al. Renal allograft survival in nonhuman primates infused with donor antigen-pulsed autologous regulatory dendritic cells. Am J Transplant. (2017) 17:1476–89. doi: 10.1111/ajt.14182
68. Ezzelarab MB, Lu L, Guo H, Zahorchak AF, Shufesky WF, Cooper DK, et al. Eomesodermin(lo) CTLA4(hi) alloreactive CD8+ memory T cells are associated with prolonged renal transplant survival induced by regulatory dendritic cell infusion in CTLA4 immunoglobulin-treated nonhuman primates. Transplantation. (2016) 100:91–102. doi: 10.1097/TP.0000000000000871
69. Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. (2015) 15:731–44. doi: 10.1038/nri3920
70. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. (2008) 13:453–61. doi: 10.2741/2692
71. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
72. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
73. Murray PJ, Wynn AT. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
74. Hutchinson JA, Riquelme P, Geissler EK, Fandrich F. Human regulatory macrophages. Methods Mol Biol. (2011) 677:181–92. doi: 10.1007/978-1-60761-869-0_13
75. Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, et al. Cutting edge: immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. (2011) 187:2072–8. doi: 10.4049/jimmunol.1100762
76. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy. (2006) 8:315–7. doi: 10.1080/14653240600855905
77. Friedenstein AJ, Piatetzky S II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. (1966) 16:381–90.
78. Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. (2004) 363:1439–41. doi: 10.1016/S0140-6736(04)16104-7
79. Orbay H, Tobita M, Mizuno H. Mesenchymal stem cells isolated from adipose and other tissues: basic biological properties and clinical applications. Stem Cells Int. (2012) 2012:461718. doi: 10.1155/2012/461718
80. La Noce M, Paino F, Spina A, Naddeo P, Montella R, Desiderio V, et al. Dental pulp stem cells: state of the art and suggestions for a true translation of research into therapy. J Dent. (2014) 42:761–8. doi: 10.1016/j.jdent.2014.02.018
81. In 't Anker PS, Scherjon SA, Kleijburg-van der Keur C, de Groot-Swings GM, Claas FH, Fibbe WE, et al. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells. (2004) 22:1338–45. doi: 10.1634/stemcells.2004-0058
82. Sarugaser R, Lickorish D, Baksh D, Hosseini MM, Davies EJ. Human umbilical cord perivascular (HUCPV) cells: a source of mesenchymal progenitors. Stem Cells. (2005) 23:220–9. doi: 10.1634/stemcells.2004-0166
83. Flynn A, Barry F, O'Brien T. UC blood-derived mesenchymal stromal cells: an overview. Cytotherapy. (2007) 9:717–26. doi: 10.1080/14653240701584578
84. Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. (2012) 12:383–96. doi: 10.1038/nri3209
85. Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. (2005) 105:2214–9. doi: 10.1182/blood-2004-07-2921
86. Barry FP, Murphy JM, English K, Mahon PB. Immunogenicity of adult mesenchymal stem cells: lessons from the fetal allograft. Stem Cells Dev. (2005) 14:252–65. doi: 10.1089/scd.2005.14.252
87. Lazarus HM, Koc ON, Devine SM, Curtin P, Maziarz RT, Holland HK, et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cells and hematopoietic stem cells in hematologic malignancy patients. Biol Blood Marrow Transplant. (2005) 11:389–98. doi: 10.1016/j.bbmt.2005.02.001
88. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. (2002) 99:3838–43. doi: 10.1182/blood.V99.10.3838
89. Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringden O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol. (2003) 57:11–20. doi: 10.1046/j.1365-3083.2003.01176.x
90. Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA. (2012) 307:1169–77. doi: 10.1001/jama.2012.316
91. Detry O, Vandermeulen M, Delbouille MH, Somja J, Bletard N, Briquet A, et al. Infusion of mesenchymal stromal cells after deceased liver transplantation: a phase I-II, open-label, clinical study. J Hepatol. (2017) 67:47–55. doi: 10.1016/j.jhep.2017.03.001
92. Casiraghi F, Perico N, Remuzzi G. Mesenchymal stromal cells for tolerance induction in organ transplantation. Hum Immunol. (2018) 79:304–13. doi: 10.1016/j.humimm.2017.12.008
93. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
94. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
95. Deng S, Moore DJ, Huang X, Lian MM, Mohiuddin M, Velededeoglu E, et al. Cutting edge: transplant tolerance induced by anti-CD45RB requires B lymphocytes. J Immunol. (2007) 178:6028–32. doi: 10.4049/jimmunol.178.10.6028
96. Huang X, Moore DJ, Mohiuddin M, Lian MM, Kim JI, Sonawane S, et al. Inhibition of ICAM-1/LFA-1 interactions prevents B-cell-dependent anti-CD45RB-induced transplantation tolerance. Transplantation. (2008) 85:675–80. doi: 10.1097/TP.0b013e3181663422
97. Lee KM, Kim JI, Stott R, Soohoo J, O'Connor MR, Yeh H, et al. Anti-CD45RB/anti-TIM-1-induced tolerance requires regulatory B cells. Am J Transplant. (2012) 12:2072–8. doi: 10.1111/j.1600-6143.2012.04055.x
98. Schuetz C, Lee KM, Scott R, Kojima L, Washburn L, Liu L, et al. Regulatory B cell-dependent islet transplant tolerance is also natural killer cell dependent. Am J Transplant. (2017) 17:1656–62. doi: 10.1111/ajt.14265
99. Saas P, Daguindau E, Perruche S. Concise review: apoptotic cell-based therapies-rationale, preclinical results and future clinical developments. Stem Cells. (2016) 34:1464–73. doi: 10.1002/stem.2361
100. Peng Y, Ye Y, Jia J, He Y, Yang Z, Zhu X, et al. Galectin-1-induced tolerogenic dendritic cells combined with apoptotic lymphocytes prolong liver allograft survival. Int Immunopharmacol. (2018) 65:470–82. doi: 10.1016/j.intimp.2018.10.019
101. Feng JF, Chen F, Liu H, Liu J. Induction of immune tolerance by pre-infusion of apoptotic lymphocytes derived from peripheral blood of donor rats before liver transplantation. Minerva Chir. (2013) 68:183–9.
102. Sun E, Gao Y, Chen J, Roberts AI, Wang X, Chen Z, et al. Allograft tolerance induced by donor apoptotic lymphocytes requires phagocytosis in the recipient. Cell Death Differ. (2004) 11:1258–64. doi: 10.1038/sj.cdd.4401500
103. Wang Z, Larregina AT, Shufesky WJ, Perone MJ, Montecalvo A, Zahorchak AF, et al. Use of the inhibitory effect of apoptotic cells on dendritic cells for graft survival via T-cell deletion and regulatory T cells. Am J Transplant. (2006) 6:1297–311. doi: 10.1111/j.1600-6143.2006.01308.x
104. Mougel F, Bonnefoy F, Kury-Paulin S, Borot S, Perruche S, Kantelip B, et al. Intravenous infusion of donor apoptotic leukocytes before transplantation delays allogeneic islet graft rejection through regulatory T cells. Diabetes Metab. (2012) 38:531–7. doi: 10.1016/j.diabet.2012.08.008
105. Bittencourt MC, Perruche S, Contassot E, Fresnay S, Baron MH, Angonin R, et al. Intravenous injection of apoptotic leukocytes enhances bone marrow engraftment across major histocompatibility barriers. Blood. (2001) 98:224–30. doi: 10.1182/blood.V98.1.224
106. Cheng J, Zhou L, Qin YS, Wang Y, Xie HY, Feng XW, et al. Donor apoptotic lymphocyte transfusion-induced liver allograft tolerance by up-regulation of CD4+CD25+ regulatory T cells in peripheral blood. Transplant Proc. (2009) 41:3893–7. doi: 10.1016/j.transproceed.2009.05.033
107. Kawai T, Poncelet A, Sachs DH, Mauiyyedi S, Boskovic S, Wee SL, et al. Long-term outcome and alloantibody production in a non-myeloablative regimen for induction of renal allograft tolerance. Transplantation. (1999) 68:1767–75. doi: 10.1097/00007890-199912150-00022
108. Leventhal JR, Mathew JM, Salomon DR, Kurian SM, Friedewald JJ, Gallon L, et al. Nonchimeric HLA-identical renal transplant tolerance: regulatory immunophenotypic/genomic biomarkers. Am J Transplant. (2016) 16:221–34. doi: 10.1111/ajt.13416
109. Scandling JD, Busque S, Shizuru JA, Lowsky R, Hoppe R, Dejbakhsh-Jones S, et al. Chimerism, graft survival, and withdrawal of immunosuppressive drugs in HLA matched and mismatched patients after living donor kidney and hematopoietic cell transplantation. Am J Transplant. (2015) 15:695–704. doi: 10.1111/ajt.13091
110. Miyara M, Chader D, Sage E, Sugiyama D, Nishikawa H, Bouvry D, et al. Sialyl lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci USA. (2015) 112:7225–30. doi: 10.1073/pnas.1508224112
111. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
112. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
113. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/JCI81187
114. Shi H, Chi H. Metabolic control of treg cell stability, plasticity, tissue-specific heterogeneity. Front Immunol. (2019) 10:2716. doi: 10.3389/fimmu.2019.02716
115. Fehr T, Sykes M. Clinical experience with mixed chimerism to induce transplantation tolerance. Transpl Int. (2008) 21:1118–35. doi: 10.1111/j.1432-2277.2008.00783.x
116. Mazariegos GV. Immunosuppression withdrawal after liver transplantation: what are the next steps? Transplantation. (2011) 91:697–9. doi: 10.1097/TP.0b013e31820c85a3
117. Liu C, Xu L, Xia C, Long Y, Liu C, Lu S, et al. Increased proportion of functional subpopulations in circulating regulatory T cells in patients with chronic hepatitis B. Hepatol Res. (2019) 50:439–52. doi: 10.1111/hepr.13472
118. Pal S, Nandi M, Dey D, Chakraborty BC, Shil A, Ghosh S, et al. Myeloid-derived suppressor cells induce regulatory T cells in chronically HBV infected patients with high levels of hepatitis B surface antigen and persist after antiviral therapy. Aliment Pharmacol Ther. (2019) 49:1346–59. doi: 10.1111/apt.15226
119. Carpentier A, Conti F, Stenard F, Aoudjehane L, Miroux C, Podevin P, et al. Increased expression of regulatory Tr1 cells in recurrent hepatitis C after liver transplantation. Am J Transplant. (2009) 9:2102–12. doi: 10.1111/j.1600-6143.2009.02743.x
120. Utsumi M, Takaki A, Umeda Y, Koike K, Napier SC, Watanabe N, et al. Frequency of regulatory T-cell and hepatitis C viral antigen-specific immune response in recurrent hepatitis C after liver transplantation. Transpl Immunol. (2014) 31:33–41. doi: 10.1016/j.trim.2014.05.006
121. Lan YT, Fan XP, Fan YC, Zhao J, Wang K. Change in the Treg/Th17 cell imbalance in hepatocellular carcinoma patients and its clinical value. Medicine (Baltimore). (2017) 96:e7704. doi: 10.1097/MD.0000000000007704
Keywords: immune tolerance, liver transplantation, hematopoietic stem cells, regulatory cells, mesenchymal stromal cells
Citation: Wang P, Jiang Z, Wang C, Liu X, Li H, Xu D and Zhong L (2020) Immune Tolerance Induction Using Cell-Based Strategies in Liver Transplantation: Clinical Perspectives. Front. Immunol. 11:1723. doi: 10.3389/fimmu.2020.01723
Received: 13 February 2020; Accepted: 29 June 2020;
Published: 18 August 2020.
Edited by:
Ye Htun Oo, University of Birmingham, United KingdomReviewed by:
Nicole Andrea Mifsud, Monash University, AustraliaCopyright © 2020 Wang, Jiang, Wang, Liu, Li, Xu and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dingyin Xu, eHVkeTIwMDRAc2luYS5jb20=; Lin Zhong, emhvbmdsXzMyODRAaG90bWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.