 Yujun Tang
Yujun Tang Jiajia Liu
Jiajia Liu Dingyi Zhang
Dingyi Zhang Jinjun Ji
Jinjun Ji
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 July 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01708
This article is part of the Research Topic Coronavirus Disease (COVID-19): Pathophysiology, Epidemiology, Clinical Management and Public Health Response View all 400 articles
Severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2) is the pathogen that causes coronavirus disease 2019 (COVID-19). As of 25 May 2020, the outbreak of COVID-19 has caused 347,192 deaths around the world. The current evidence showed that severely ill patients tend to have a high concentration of pro-inflammatory cytokines, such as interleukin (IL)-6, compared to those who are moderately ill. The high level of cytokines also indicates a poor prognosis in COVID-19. Besides, excessive infiltration of pro-inflammatory cells, mainly involving macrophages and T-helper 17 cells, has been found in lung tissues of patients with COVID-19 by postmortem examination. Recently, increasing studies indicate that the “cytokine storm” may contribute to the mortality of COVID-19. Here, we summarize the clinical and pathologic features of the cytokine storm in COVID-19. Our review shows that SARS-Cov-2 selectively induces a high level of IL-6 and results in the exhaustion of lymphocytes. The current evidence indicates that tocilizumab, an IL-6 inhibitor, is relatively effective and safe. Besides, corticosteroids, programmed cell death protein (PD)-1/PD-L1 checkpoint inhibition, cytokine-adsorption devices, intravenous immunoglobulin, and antimalarial agents could be potentially useful and reliable approaches to counteract cytokine storm in COVID-19 patients.
In December 2019, an outbreak of a novel coronavirus-based disease was reported in Wuhan, China. On 11 February 2020, the World Health Organization (WHO) named this coronavirus “severe acute respiratory syndrome coronavirus 2” (SARS-CoV-2) and the disease that it caused “coronavirus disease 2019” (COVID-19). As of 25 May 2020, SARS-CoV-2 has affected over 212 countries, and about 5,529,195 cases have been confirmed around the world, of which 347,192 people have died.
The reason for these deaths is suspected to be the “cytokine storm” [also called “cytokine storm syndrome” (CSS)]. The International Classification of Diseases (ICD) does not include the cytokine storm or CSS. Cron and Behrens bring the current knowledge of CSS (1). They define that “cytokine storm” is an activation cascade of auto-amplifying cytokine production due to unregulated host immune response to different triggers. The triggers involved infections, malignancy, rheumatic disorders, etc. Another scholar described that cytokine storm is a systemic inflammatory response to infections and drugs and leads to excessive activation of immune cells and the generation of pro-inflammatory cytokines (2). A similar entity is termed “cytokine release syndrome” (CRS), which is not defined in the textbook of CSS (1). CRS is an acute systemic inflammatory syndrome characterized by multiple-organ dysfunction (MOD). It has been reported that chimeric antigen receptor (CAR)-T-cell therapy could help to distinguish CRS from a cytokine storm (2). Of note, the textbook described the criteria of CSS based on hemophagocytic lymphohistiocytosis (HLH) and secondary HLH (sHLH) associated with rheumatic disorders, such as macrophage activation syndrome (MAS) (1). Thus, it may be not applicable in COVID-19 because the COVID-19 is a contagious disease and relatively irrelevant to a genetic disorder. Up to date, there is still a lack of clinical and laboratory criteria to identify the cytokine storm. In this review, we referred COVID-19 associated cytokine storm as the patients who are severely ill along with a high concentration of pro-inflammatory cytokines.
For patients with COVID-19, the number of white blood cells, neutrophils, as well as levels of procalcitonin, C-reactive protein, and other inflammatory indices, are significantly higher in the intensive care unit (ICU) cases than in non-ICU cases (3, 4). Many studies showed that severely ill patients tended to have a higher concentration of pro-inflammatory cytokines, especially interleukin (IL) 6, than moderately ill patients in COVID-19 (5–9). The result of the bronchoalveolar lavage fluid (BALF) cells, which tested by transcriptome sequencing, reveals excessive chemokines releasing caused by SARS-CoV-2 infection, such as CXCL10 and CCL2 (10). The high level of cytokines also indicates a poor prognosis in COVID-19 (6, 11, 12). Furthermore, the pathology of postmortem examination of the lung, from who was died of COVID-19, demonstrated the existence of acute respiratory distress syndrome (ARDS) and T-cell overactivation (13). This phenomenon is due to an increase in the number of T-helper (Th) 17 cells and the high cytotoxicity of the CD8+ T cells (13). The innate and adaptive immune responses activated by SARS- CoV-2 infection lead to uncontrolled inflammatory responses and ultimately cause the cytokine storm (14). The cytokine storm can lead to apoptosis of epithelial cells and endothelial cells, and vascular leakage and, finally, result in ARDS, other severe syndromes, and even death (15).
To lower mortality due to cytokine storm, we summarized the clinical and pathology features of the coronavirus-related cytokine storm. We explored the efficacy and safety of potential treatments and their molecular mechanism. There is still lacking sufficient evidence supporting the regulation of cytokine expression may be beneficial to the mortality of COVID-19.
The early-stage clinical characteristics of MERS and SARS are influenza-like symptoms (16–18): pyrexia, sore throat, dry cough, myalgia, and dyspnea. Those symptoms are very similar to the characteristics of early COVID-19 and progress rapidly to pneumonia (3, 19, 20). It has been found that the regulation of several cytokines is disordered in the peripheral blood of SARS patients, as summarized by Chen and colleagues (21) and listed in Table 1. Table 1 shows an increase in levels of cytokines and chemokines and a decrease in levels of anti-inflammatory cytokines such as IL-10. Of note, the release of pro-inflammatory cytokines, especially interferon (IFN)-α and IFN-γ, is correlated with lethal SARS (22, 23). The cytokines with increased levels in fatal SARS are IL-6, IL-1β, IFN, and CXCL10. These cytokines are secreted mainly by dendritic cells (DCs) and macrophages, indicating that innate immunity plays a pivotal part in lethal SARS. CCR4+ CCR6+ Th17 cells have many chemokine receptors and may share the same mechanism and function in cell-cell interactions in SARS. Cytokines secreted by DCs and macrophages induce the infiltration and recruitment of pro-inflammatory Th17 cells. Analyses of lungs from SARS patients have revealed diffuse alveolar damage as a crucial feature. Histopathological studies have shown lung consolidation and edema with pleural effusions and focal hemorrhage, all of which resemble COVID-19 features (13, 24). Besides, the lungs of SARS patients are infiltrated extensively with neutrophils and macrophages, which are not observed in COVID-19. In peripheral blood, numbers of CD4+ and CD8+ T cells are reduced in cases of COVID-19 and SARS (13, 25) and are associated with death in the latter (25). Interestingly, unlike MERS and SARS, a high concentration of pro-inflammatory CC chemokine receptor (CCR)4+ CCR6+ Th17 cells are found in COVID-19 (13).
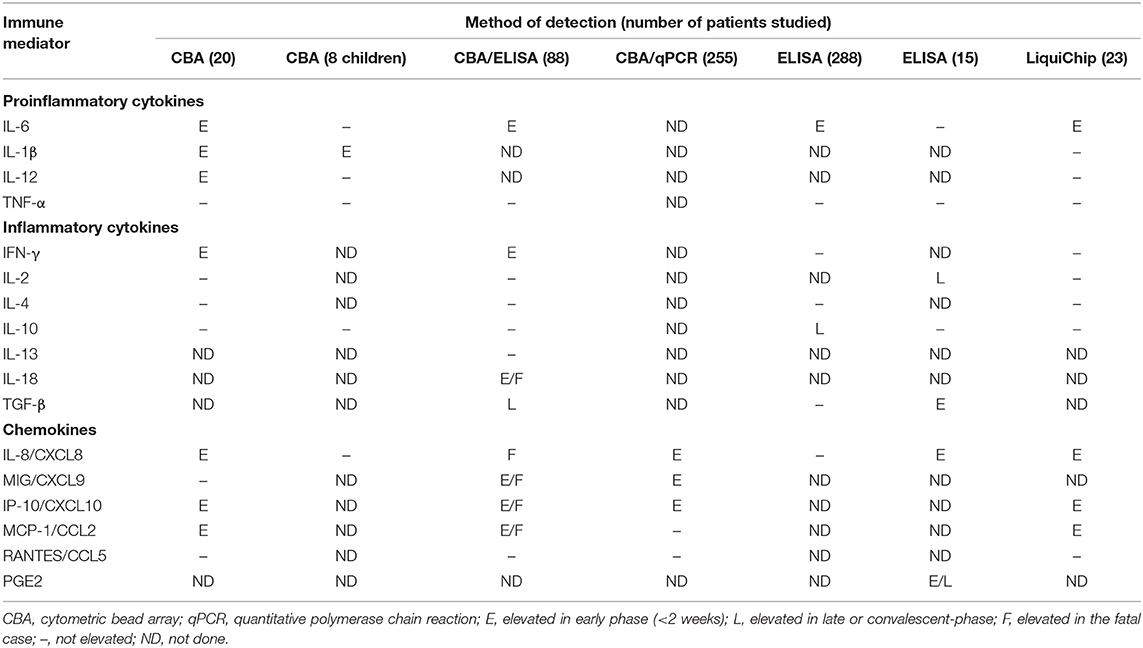
Table 1. Cytokine and chemokine responses detected in plasma or serum of SARS patients [adapted from Chen and Subbarao (21)].
The innate and adaptive immune system takes multiple measures to respond to virus infection. MERS-CoV infects human epithelial cells and leads to these cells inducing significant but delayed responses by IFN, pro-inflammatory cytokines (e.g., IL-1β, IL-6) and chemokines (e.g., IL-8) (26, 27). SARS-CoV infects airway epithelial cells and results in delayed release of chemokines such as CCL3, CCL5, CCL2, and CXCL10 (28). Besides, MERS-CoV infects hematopoietic cells such as monocytes, macrophages, and DCs, which is not seen in those cells upon SARS-CoV infection (29–32). MERS-CoV infects the cells mentioned above to induce delayed (but increased) levels of pro-inflammatory cytokines (e.g., IL-2) and chemokines (e.g., CCL2, CCL3) (27, 30). Although SARS-CoV is abortive in macrophages and DCs, the virus induces an increase in levels of pro-inflammatory cytokines and chemokines (31, 32). SARS-CoV and SARS-CoV-2 infect cells using the same receptor: angiotensin-converting enzyme-2 (33). Hence, it has been postulated that both viruses can affect the same spectrum of cells.
In the aspects of murine models of coronavirus, infection with SARS-CoV in BALB/c mice has been shown to induce an increase in the number of pathogenic inflammatory monocyte–macrophages (IMMs) (34). Through stimulation of IFN-α/β receptors, the accumulating IMMs produce monocyte chemokines (e.g., CCL2, CCL7, CCL12) and pro-inflammatory cytokines [e.g., tumor necrosis factor (TNF), IL-6, IL1-β], which results in further accumulation of pathogenic IMMs. Targeting of IFN signaling, IMMs, or pro-inflammatory cytokines could offer protection from lethal SARS-CoV infection. In this way, the chemokines (produced by activated monocytes and macrophages) lead to the recruitment of neutrophils, monocytes, and T cells into the lungs (28). After chemotaxis, activated effector T cells migrate to the lungs and destroy pneumocytes/permissive cells due to response to the virus infection (35). The damage caused by neutrophils, monocytes, and T cells results in lung-parenchyma changes, such as diffuse alveolar damage, which leads to ARDS (35).
In summary, the excessive cytokines and chemokines caused by lethal coronavirus infection involve mainly antigen-presenting cells (APCs) (such as macrophages) and T cells. However, cytokines secreted by immune cells are produced to eliminate viral infection, and deficiency of such cytokines may be harmful to the body. For example, virus titers are significantly higher in toll-like receptor (TLR)3−/−, TIR-domain-containing adapter-inducing interferon-β (TRIF)−/−, and IL-6−/− mice compared with their wild-type counterparts, and are associated with severe lung damage (36, 37).
In China, we classified the stage of COVID-19 according to the guidelines (38) issued by the National Health Commission of the People's Republic of China (NHC). According to the instructions, NHC defines severe illness of COVID-19 as one of the following conditions: respiratory rate ≥30 breaths/min in the resting state; Oxygen saturation ≤93%; arterial blood oxygen partial pressure (PaO2)/fraction of inspired oxygen concentration (FiO2) ≤300 mmHg. Critical illness as one of the following conditions: respiratory failure and requiring mechanical ventilation; shock; complication of other organ failures, and needs intensive care. The most common symptoms of COVID-19 were fever, cough, shortness of breath, fatigue, and myalgia (5, 7, 39, 40), and severe cases tend to be older with more basic diseases and suffer from dyspnea, more complications (5, 40). In COVID-19, 14% of patients progress to severe disease and 5% to critical illness (41). A prospective study reported that the computerized tomography (CT) of the lungs of COVID-19 (6). The lung lesions increase and the scope expands as the disease progresses, and ground-glass opacity coexisted with consolidation or striated shadow. Some severe patients showed diffuse lesions in both lungs.
Up to date, the inflammatory disorders (insufficient in chemokines) in COVID-19 have been reported in many clinical studies. The COVID-19 is inclined to cause a decrease of lymphocyte count and an increase of C reactive protein (CRP), especially in severely ill patients (5–7, 42–44). The major subsets of the T lymphocytes (T cell) (CD3+ CD4+ T cell and CD3+ CD8+ T cells) are reduced in the COVID-19 and are significantly lower in the severe cases (5, 12, 42, 43, 45, 46); however, controversial results are also reported in some studies (7, 40). The results of the other immune cells, the B cell and natural killer (NK) cell, have more inconsistency in recent researches. IL-6 was observed increased in all studies, and only one study show IL-10 was not elevated. About half of the studies we collected showed TNF-α was increased. Only Huang et al. (9) inspected the multiple types of chemokines and found that severe patients had higher levels of G-CSF, GM-CSF, IP-10, MCP-1, MIP-1a, MIP-1b, RANTES, and IL-8. The inflammatory disorders of COVID-19 were summarized in Table 2.
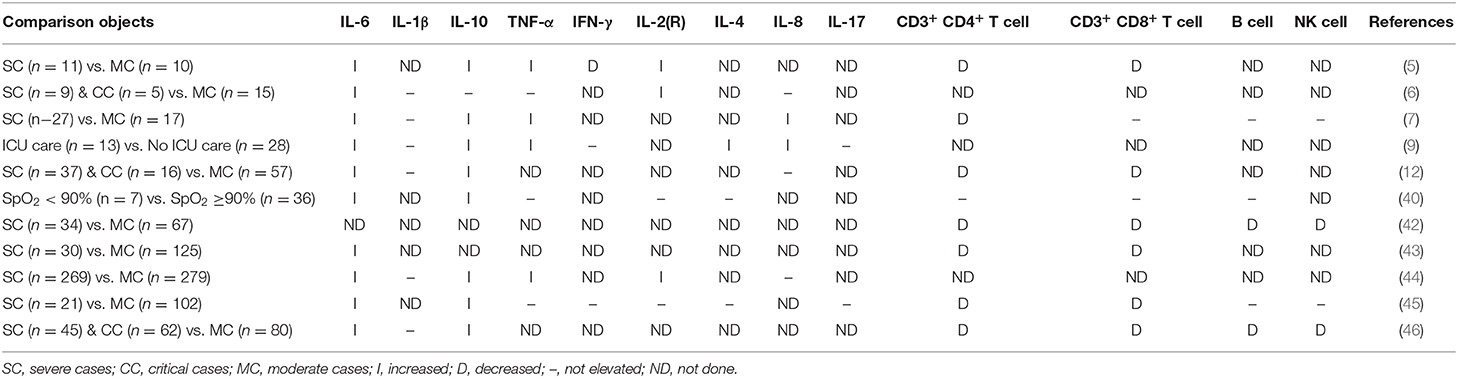
Table 2. Cytokine, chemokine, and leukomonocyte responses detected in COVID-19 patients.
The pathologic features of COVID-19 showed the lungs were infiltrated with excessive CCR6+ Th17 cells and high cytotoxicity of CD8+ T cells (13). But high cytotoxicity of CD8+ T cells does not mean they exert the normal function. The SARS-CoV-2 could lead to cytotoxic lymphocytes (mainly involving NK cells and CD8+ T cells) exhaustion, which is manifested as the upregulated exhaustion markers, such as NKG2. The exhaustion markers return to normal in patients who have recovered or are convalescent (47, 48). BALF cells were found extreme cytokine releases, such as CCL2, CXCL10, CCL3, and CCL4 (10). Furthermore, Xiong et al. (10) use the transcriptome dataset approach to discover that SARS-CoV-2 can activate apoptosis and P53 signaling pathway (one of the pathways responsible for the survival of the cell) in lymphocytes. These results could provide some reasons for the cause of patients' lymphopenia. Another team of Chen and his colleagues studied the mechanisms for lymphopenia (49). Their results demonstrate that SARS-CoV-2 infected the CD169+ macrophages in spleens and lymph nodes (LNs), and lead to lymphoid tissue damage, such as splenic nodule atrophy and lymph follicle depletion, etc. The CD169+ macrophages express high Fas and cause activation-induced cell death (AICD) through Fas/FasL interactions. Furthermore, SARS-CoV-2 selectively induced macrophages to produce IL-6, not TNF-α and IL-1β, to directly promotes lymphocyte necrosis. The analysis of peripheral blood mononuclear cells (PBMCs) revealed that non-structural protein (nsp) 9 and nsp10 of SARS-CoV-2 target NKRF (NF-κB repressor) to promote IL-6/IL-8 production (50). As a consequence, it recruits neutrophils and induces uncontrollable host inflammatory response.
Collectively, the clinical, immunological, and pathologic features of COVID-19 have something in common with SARS and MERS. For example, all the viruses can cause lymphopenia and influenza-like symptoms in the early stage. SARS and COVID-19 do not lead to the upgrade of TNF-α, but the increase of IL-6 and IL-10 is more prevalent in COVID-19. The IL-6 plays a crucial role in the pathologic of COVID-19, including the chemotaxis of neutrophils and lymphocyte necrosis. Importantly, COVID-19 is more able to cause cytotoxic lymphocytes exhaustion.
Tocilizumab (TCZ) is a recombinant humanized anti-human IL-6 receptor monoclonal antibody, preventing IL-6 binding to its receptor to exert the immunosuppression promoted by IL-6. Michot et al. (51) reported that 42-year-old male suffering from respiratory failure due to SARS-CoV-2 infection. After 4 days of TCZ treatment, the CRP decreased from 225 to 33 mg/L and ultimately clinically fully recovered. Similarly, some case reports showed TCZ is an efficacy and safety approach in COVID-19, even patients with other diseases combined, such as multiple myeloma, end-stage renal disease, and sickle cell disease (52–54). Recently, a retrospective study (55) found that TCZ decreased CRP in all patients (n = 15) rapidly, but three of them, who are critically ill, still dead. The dead patients show continuously rising of IL-6 even after the administration of TCZ and methylprednisolone, indicating that repeat doses of TCZ may be needed in COVID-19 patients who are critically ill. Another retrospective study (56) demonstrated that TCZ showed a quick control of severe COVID-19 manifestation, such as fever, respiratory function. All patients (n = 21, two were critically ill), have recovered and have been discharged from hospital, and no adverse event was reported during the treatment. A prospective open-label, multicenter single-arm study manifests the pilot results of the off-label application of TCZ in severe patients with COVID-19 (57). The study involved 63 patients with severe COVID-19, and TCZ succeeded in improving respiratory and laboratory parameters, such as Pa02, Fi02, consequently, increased the likelihood of survival (the death rate of the study is 11%). It is worth mentioning that a cautionary case report by Radbel et al. (58). Two patients were diagnosed with COVID-19 complicated by CRS and treated with TCZ. Unfortunately, both patients progressed to severe HLH, and one developed to viral myocarditis.
All the cytokines produced by immune cells are responsible for viral clearance. Suppression of cytokine release at an early stage of disease as treatment is controversial. Application of synthetic disease-modifying antirheumatic drugs (DMARDs) and biologic DMARDs to downregulate cytokine expression in RA increases the risk of infection (59, 60). The timing and the doses of the intervention still need to be inspected clearly. SARS-CoV-2 mainly causes a dramatic increase in IL-6 and does not remarkably promote other pro-inflammatory factors, such as IL-1β and IFN-γ. Although treating COVID-19 with TCZ is an off-label use, it may be relatively appropriate and safe in coping with COVID-19 associated cytokine storm basing on the current evidence. It still needs more large samples and high-quality studies to evaluate the exact efficacy and safety in COVID-19. The ongoing trials of potential treatments and other treatments focus on inflammatory disorders in COVID-19 are available in Supplementary Table 1.
Glucocorticoid therapy is used widely among critically ill patients with other coronavirus infections (e.g., SARS, MERS). Corticosteroids have been administered to ICU patients infected with SARS-CoV-2 (3, 4, 20). Glucocorticoids exhibit pharmacologic effects at any therapeutically relevant dose through classic genomic mechanisms. Some immunosuppressive effects are based on transactivation, and glucocorticoid induces gene transcription and protein synthesis of NF-κB inhibitors and lipocortin-1. Through inhibition of NF-κB signaling, glucocorticoids induce inhibition of synthesis of downstream proteins such as IL-1, IL-6, granulocyte-macrophage colony-stimulating factor, and inducible cyclooxygenase-2 (61, 62). Glucocorticoids reduce the proliferation, activation, differentiation, and survival of T cells and macrophages (63). Glucocorticoids proffer inhibitory actions on the transcription and action of various cytokines. The Th1 and macrophage-based pro-inflammatory cytokines IL-1β, IL-2, IL-6, TNF-α, and IL-17 are inhibited by glucocorticoids (63).
However, it is controversial whether corticosteroids are beneficial in the treatment of severe COVID-19 patients. A comment and a meta-analysis, which mainly bases on the evidence of SARS and MERS (64, 65), stated that corticosteroid would increase mortality and delayed clearance of viral in coronavirus infection diseases. Thus, the corticosteroids should not be administrated for the treatment of SARS-Cov-2 induced lung injury or shock. Newly published studies also indicate that the use of corticosteroids is not beneficial for COVID-19 patients (not severe cases), and high-dose corticosteroids are associated with mortality (44, 66, 67). Most COVID-19 patients discussed in these studies are not severe cases. Inspecting the studies included and analyzed by the meta-analysis, only one study (68) described the numbers of patients with corticosteroids and non-corticosteroids treatment in the severe group and non-severe group. The study demonstrated the benefit of corticosteroids use in severe SARS-Cov infection. Another comment (69), which was written by front-line physicians from China, showed corticosteroids might have some benefit for critically ill patients with COVID-19. Systematic corticosteroid therapy could promote oxygen saturation and PaO2/FiO2. However, corticosteroids might not improve mortality in critical COVID-19 patients.
Current evidence shows that SARS-Cov-2 induces an increase in a small range of cytokines. It might be overuse to administrate corticosteroids to counteract a wide range of cytokines. Furthermore, SARS-Cov-2 causes relatively serious lymphocytopenia and lymphocytes exhaustion. Glucocorticoid-mediated stimulation of the “hypothalamic-pituitary-adrenal axis” might also exacerbate lymphocytopenia (70). Thus, the use of corticosteroid is a double-edged sword in COVID-19. The dose, duration, and timing of corticosteroid therapy will be crucial if administrated to COVID-19 patients.
As stated above, lymphocytes exhaustion is one of the characteristics of COVID-19, and PD-1 checkpoint-inhibitor might some help in reversing the anergy of lymphocytes. Up to 4 May 2020, no study of PD-1 checkpoint-inhibitor has been reported in the Treatment of COVID-19. The pathway consisting of the receptor PD-1 and its ligands, PD-L1 and PD-L2, play crucial parts in the maintenance of peripheral tolerance. Treatments with antibodies targeting PD-1/PD-1 ligands have elicited an increased response in different cancer types and, in tandem with antibodies targeting cytotoxic-T-lymphocyte-associated antigen-4, have changed cancer therapy radically (71). Unfortunately, signaling regulated by the PD-1/PD-L pathway is also related to substantial inflammatory effects (e.g., sepsis), as this pathway plays a role in balancing protective immunity and immunopathology (72). Increased PD-L1 expression in monocytes is associated with mortality in patients with septic shock (73). A meta-analysis of checkpoint inhibitors showed that such therapy increased the chance of survival (74). Nivolumab (anti-PD-1) and BMS-936559 (anti-PD-L1) had completed phase-Ib randomized studies for severe sepsis. They revealed that giving a checkpoint inhibitor did not result in unexpected safety findings or indicate a cytokine storm (75, 76). Also, CD4+ and CD8+ T cells were hyperactivated, as revealed by the high proportions of human leukocyte antigen-DR isotype and CD38, in COVID-19; CD8+ T cells harbored high levels of cytotoxic granules in COVID-19 patients, in which the phenotype is similar to fatal H7N9 disease (13, 77). Those results suggest that lethal COVID, along with H7N9, may be related to defective activation and exhaustion of T cells, which also suggest that checkpoint-inhibitor administration may reverse this status.
Cytokine adsorption involves using a method, such as extracorporeal membrane oxygenation (ECMO), to filter harmful substances directly. An extracorporeal cytokine hemoadsorption device called Cytosorb® (Cytosorbents, Monmouth, NJ, USA) has been reported to capture and reduce inflammatory mediators. Bruenger and colleagues reported that the plasma level of IL-6 and procalcitonin decreased in one patient with severe ARDS after Treatment with ECMO using a hemoadsorption device (78). A 45-year-old patient with severe ARDS showed that venous arterial-ECMO combined with hemoadsorption therapy decreased plasma concentrations of IL-6 and IL-8. Moreover, hemodynamic stabilization, respiratory improvement, and a decline in capillary leakage can be achieved in combination therapy (79). Two trials employing hemoadsorption therapy for infection-related cytokine storm are ongoing (NCT04195126, NCT03685383).
A similar therapy involves dialysis. The mainly water-soluble mediators are removed from plasma, and the hemofilters can have additional adsorptive properties (80). Continuous venovenous hemofiltration and adsorption for severe septic shock are being tested in one clinical trial (NCT03974386).
Neutralizing excessive cytokines with hemoadsorption devices might be relatively effective. The disadvantage is like corticosteroids: a wide range of cytokines would be adsorbed. Thus, it would lead to the a lack of cytokines, which are at reasonable or even insufficient levels. We suggest treating the cytokine storm in COVID-19 should base on the laboratory results of cytokines and chemokines. Meanwhile, adjusting the parameters of the devices (e.g., treatment duration) for preventing overtreatment.
IVIG can elicit passive immunity, anti-inflammatory, and immunomodulatory effects that can improve treatment effects and increase survival in severe infection. An IgG molecule binds to a specific target antigen through the humoral and cellular arms of the immune system. For example, IgG molecule blocks the cell-cell interactions mediated by cell-surface receptors (such as CD95 and CD95 ligand), neutralize the autoantibodies by anti-idiotypic antibodies, expanse the regulatory T (Treg) cell populations via the blockade of immune complex binding to low-affinity Fcγ receptors (FcγRs), to exert the functions of immunomodulation (81). Ma and colleagues detailed a severe case of glandular fever treated with IVIG (82). Levels of Th1 cytokines (IFN-γ, IL-12, soluble tumor necrosis factor receptor 1 (sTNFR1), CXCL10, CXCL9, CCL3), and viral loads eventually recovered after the combination of prednisolone with IVIG. A multicenter, double-blind, randomized controlled trial for cases with severe influenza A (H1N1) infection demonstrated that IVIG reduced the serum concentration of cytokines, viral load, and reduced mortality (83). A meta-analysis of 17 studies (1,958 participants) found IgM-enriched polyclonal and standard Ig molecules decreased mortality in adults with severe sepsis or septic shock. However, a meta-analysis did not reveal a benefit in adult mortality with polyclonal IVIG using high-quality trials only (84).
Despite a lack of clinical evidence, the US gave emergency approval to HCQ, a member of antimalarial agents, in COVID-19 on 28 March (85). A meta-analysis included the studies up to 5 April 2020 (86) and showed that four clinical trials and three observational studies are eligible for the study. Unfortunately, the authors concluded that HCQ has no clinical effect on patients with COVID-19. However, a randomized clinical trial published on 24 April, which included the patients (n = 81) with critically ill COVID-19 (such as high respiratory rate, peripheral oxygen saturation lower than 90%, shock), indicated 15.0% patients (6 of 40) have died in the low-dosage group (i.e., 450 mg twice daily on day 1 and once daily for 4 days). The critically ill death rate is over 50%, as reported by WHO (87). Thus, low-dosage of HCQ could be beneficial for critically ill patients with COVID-19. The study also indicates high dosage HCQ might not be suitable for critically ill patients because of its potential safety hazards.
Traditional Chinese medicine (TCM) has an essential role in the latest SARS epidemic. Several studies (88–93) have shown that the add-on of TCM to Western medicine can shorten the duration of hospitalization, alleviate symptoms, reduce mortality (including for critically ill patients), and reduce the prevalence of adverse reactions in SARS. Compared with a control group (Western medicine only), a combination of TCM with Western medicine has shown advantages in terms of symptom alleviation and preventing COVID-19 (94–96). However, the quality of the studies must be improved. The administration of TCM in a standard manner worldwide is complicated because of the different decoctions used and the matching of herbs.
Artemisinin can be obtained from Artemisia annua, and one kind of antimalarial agents. Hou and colleagues showed that extracts from artemisinin-family drugs could regulate cells from the innate and adaptive immune system, and lead to anti-inflammatory and immunomodulatory actions (97). The scope of application for artemisinin-family medicines includes infectious disease and autoimmune diseases, and artemisinin-family shows a difference in immune regulation compared with hydroxychloroquine (98–100).
As stated above, ALI and AKI are crucial mortality factors in infectious diseases. Artesunate is a derivative of artemisinin and can lessen the pathologic changes and neutrophil infiltration in the lungs of ALI patients, and decrease sepsis-induced mortality (101). By inhibiting expression of NF-κB signaling and enhancing heme oxygense-1 expression, the artesunate can lower the concentrations of TNF-α and IL-6 in serum and bronchoalveolar lavage fluid. Huang and colleagues discovered that dihydroartemisinin could attenuate lipopolysaccharide (LPS)-induced ALI through suppressing NF-κB signaling in a nuclear factor erythroid 2-related factor 2 (Nrf2)-dependent fashion, thereby leading to a decrease in expression of the pro-inflammatory cytokines IL-1β, TNF-α, and IL-6 (102). Hu and colleagues explored a new and efficacious approach for ALI (103). “Artesunate liposomes” were prepared using film dispersion and then lyophilized to obtain liposomal artesunate dry powder inhalers (LADPIs). After treatment with LADPIs, a rapid reduction in accelerated inhalation, ALI syndromes, and levels of TNF-α and IL-6 has been observed in rats. Besides, kidney impairment in hospitalized COVID-19 patients is associated with a high risk of in-hospital death (104). Cheng et al. (105) observed that dihydroartemisinin lessened glomerular injury and relieving increases in the urine albumin: creatinine ratio and serum levels of creatinine.
Current evidence of pathologic changes of COVID-19 suggests the dysregulation of the cytokines involves mainly macrophages/monocytes. In a burn-based sepsis model BALB/c mice, concentrations of adhesion molecules and neutrophil infiltration in the lungs and heart, and mortality rate are significantly increased, but those phenotypes could be reversed by artemisinin (106). The authors discovered that artemisinin downregulates protein levels of NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) and caspase 1 in macrophages in burn-induced sepsis mice. Also, a reduction in levels of the pro-inflammatory cytokines IL-1β and IL-18 has been observed post-therapy. NLRP3 is a sensor component expressed mainly in macrophages and which undergoes transcription by NF-κB. NLRP3 is responsible for the maturation and secretion of IL-1β and IL-18 (107–109). NF-κB also increases the level of IL-10 in the macrophages infected by Plasmodium falciparum, and artemisinin could reduce IL-10 production in animal models (110), as well as in the clinic (111).
Two studies focused on the relationship among TLR, NF-κB, nucleotide-binding oligomerization domain-containing protein (NOD)2, and macrophages. TLR2 mainly locates outside the cell membrane of macrophages, DCs, and granulocytes, and recognizes bacteria (112). TLR2 induces NF-kB activation through recruitment of TIR Domain Containing Adaptor Protein (TIRAP) and myeloid differentiation primary response (MyD)88 in macrophages and DCs. In inflammatory monocytes, TLR2 is expressed within endosomes and induces the release of type-I IFNs via Interferon regulatory factor 3 (IRF3) and IRF7 in response to viruses (113). Artesunate increases survival of mice challenged with live Staphylococcus aureus/methicillin-resistant Staphylococcus aureus (MRSA) compared with antibiotics alone, and its protection may be associated with reductions in TNF-α levels. Artesunate reduces the expression of TLR2 mRNA and Nod2 mRNA that upregulated by S. aureus/MRSA and also inhibits the activation of NF-κB (114). Kuang and colleagues found that the artesunate attenuated the release of TNF-α and IL-6 from macrophages by inhibiting TLR4-mediated autophagic activation (115). TLR4 also locates in the endolysosomal compartment, can recognize Gram-negative bacteria and viruses (112), shares the same pathway as the activation of NF-κB, and induces the release of type-I IFNs via the TNF receptor-associated factor (TRAF3)- TANK Binding Kinase 1 (TBK1)-IRF3 axis (113). However, Kuang and co-workers discovered that artesunate attenuates the cytokine release by the TRAF6-beclin1- Class III phosphatidylinositol 3-kinase (PI3KC3) pathway. In a model of severe acute pancreatitis in rats, artesunate attenuates the release of IL-1β and IL-6 via the TLR4-NF-κB axis (116). In addition, dihydroartemisinin inhibited the activation of TLR4 and IRF3 in the spleen cells of systemic lupus erythematosus (SLE)-prone MRL/lpr mice, which lead to a decrease in levels of IFN-α and IFN-β (117).
The mitogen-activated protein kinase (MAPK) signaling pathway plays a vital part in the development, differentiation, proliferation, transformation, and apoptosis of cells (118). The extracellular signal-regulated kinase (ERK), JNK/Stress-activated protein kinases (SAPK), and p38 MAPK are the dominant members of the MAPK family. The cascades can be summarized as the ERK pathway (Raf-MEK-ERK), JNK pathway (TAK1-MKK-JNK), and p38 pathway (TAK1-MKK-p38). Pro-inflammatory cytokines such as IL-1 and TNF-α, IFNα, and IFNγ can induce activation of the p38 pathway, and p38 can regulate NF-κB-dependent transcription after its nuclear translocation. Meanwhile, NF-κB is a crucial transcriptor for IL-6, which could activate the IL-6-janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathways (119). Wang and colleagues (120) found that another artemisinin derivative, SM905, suppressed generation of nitric oxide, TNF-α, IL-1β, and IL-6 in LPS-induced macrophages. The underlying mechanism was that SM905 reduced activation of p38 and ERK, and JNK suppressed IκBα degradation. Furthermore, they observed that NF-κB was inhibited correspondingly in SM905-treated cells. In another LPS-induced macrophage model, artemisinin has a property of prohibiting STAT1 activation, and it leads to the reduction of NO (an inflammatory-cascade inducer) in macrophages (121). Except for STAT1, STAT3, and STAT5 in the splenocytes of SLE-prone MRL/lpr mice could be inhibited by SM934, an artemisinin derivative (122).
Artesunate therapy has been shown to improve the survival of mice infected with the herpes simplex virus. Artesunate can lower levels of IL-1β, IL-2, IL-6, IFN-γ, CCL2, CCL3, and CCL4 in these mice. These cytokines are produced primarily by APCs and Th1 cells. Previous studies have suggested that the artesunate can regulate Th cells in virus infections. Du and colleagues (123) demonstrated that the artesunate downregulated the Th1 response and reduced levels of IFN-γ, TNF-α, IL-12, IL-18, CCL2, CXCL9, and CXCL10 in an experimental model of cerebral malaria. RA is an autoimmune disease manifested by dysfunction of various immune cells (e.g., APCs, Th1, Th17), which leads to a high concentration of IL-1, IL-6, TNF-α, and chemokines in plasma and tissues (124). In the experimental models of RA, the proliferation of Th17 cells and the production of IL-17A and IL-6 are inhibited by SM905 therapy and, correspondingly, the expression of retinoic acid receptor-related orphan nuclear receptor gamma t (RORγt) (a specific transcription factor for Th17 cells) is also reduced (125). Fan et al. (126) demonstrated similar data and found that DC32 (an artemisinin derivative) can restore the Treg/Th17 balance and reduce transcription of CXCL12 and CX3CL1. Treg can be anti-inflammatory, secrete anti-inflammatory cytokines (e.g., IL-10), target Th17 cells and macrophages, as well as reduce the concentration of IL-1, IL-6, TNF-α, and IL-17 (127). The immunosuppressive mechanisms of artemisinin on T cells include inhibiting differentiation of Th17 cells by regulating the expression of RORγt and maybe also inhibition of activation of the ERK pathway (Ras-Raf1-ERK1/2) (128). In the model of RA-fibroblast-like synoviocytes (FLS), artesunate decreased the production of IL-6, IL-8, and IL-1β through preventing NF-κB translocation and IκBα degradation (129).
Artemisinin-family drugs have shown efficacy and safety in treating malaria. One study reported 32 patients with severe malaria caused by Plasmodium falciparum. Ten patients suffered renal failure, eight had cerebral malaria, and 14 had other causes of severe malaria. After artesunate treatment, concentrations of IL-6, and soluble IL-6 receptor in plasma were normalized within 24 h (130).
In recent years, artemisinin-family drugs have been shown to be beneficial against infection caused by the human cytomegalovirus, hepatitis-B virus, Ebola virus, and human immunodeficiency virus (131). Shapira and co-workers reported the first case of the Treatment of HCMV infection with artesunate (132). Germi and collaborators (133) reported that the artesunate led to an effective response in three cases with mild HCMV infection but was not efficacious in two patients with severe HCMV infection.
The elevations of IL-6 and IL-10 are highly consistent in COVID-19. IL-6 targets the IL-6 receptor, and the letter recruit JAK, which transit cascade signal to activate signal transducer and activator of transcription 3 (STAT3) (119). Some physicians suggest tofacitinib, a small molecule compound target JAK1 and JAK3, could be applied in the treatment of COVID-19, and tofacitinib success in treating a COVID-19 patient complicated with ulcerative colitis (134). IL-10, a cytokine with anti-inflammatory properties, could be secreted by virtually all immune cells, including macrophages, DCs, NK cells, T cells, and B cells (135). We might tend to regard the high levels of IL-10 as negative feedback of counteracting the increase of IL-6 because IL-10 can block the activity of NF-κB to downregulate the production of IL-6 (135). However, an abundance of IL-10 also inhibits the function and proliferation of immune cells (e.g., Th1, NK cells, and CD8 T cells), which delays the clearance of viruses (135). Therefore, a mass of IL-10 might be responsible for the normal levels (one study report low level) of IFN-γ (a cytokine for the clearance of viruses) and the exhaustion of lymphocytes. The IL-10 inhibitor in the treatment of COVID-19 also needs to be considered. Even the combination of IL-10 and IL-6 inhibitor could be designed in future prospective studies. When using any method to regulate the dysregulation of cytokines, we might better closely monitor the laboratory index for preventing over-treatment. For example, if we use TCZ to reduce the levels of IL-6, we could check IL-6 levels once every 2 days to keep it at a suitable concentration, which should be studied in the future. Also, the dose and duration would be illuminated.
The current evidence indicates that TCZ, an IL-6 inhibitor, is relatively effective and safe. Based on the therapeutic mechanisms, we classified the remaining therapies, corticosteroids, PD-1/PD-L1 checkpoint inhibition, cytokine-adsorption devices, intravenous immunoglobulin, and antimalarial agents, as “less potential treatments.” No literature of COVID-19 except for corticosteroids mentions the effectiveness and safety of the less potential treatments. The benefits, dose, duration, and timing of corticosteroids still in debate, and the other less potential treatments need clinical evidence to validate.
Although the experimental model of infectious disease (e.g., malaria and sepsis) and autoimmune disease (e.g., RA and SLE) indicates that artemisinin-family drugs could target the inflammatory networks to decrease the levels of cytokines (e.g., IL-6 and TNF-α) and chemokines (e.g., IL-8, CXCL10) (Figure 1). The effect and safety of antimalarial agents still need to be validated in the high-quality clinical studies and the SARS-Cov-2 infection disease model.
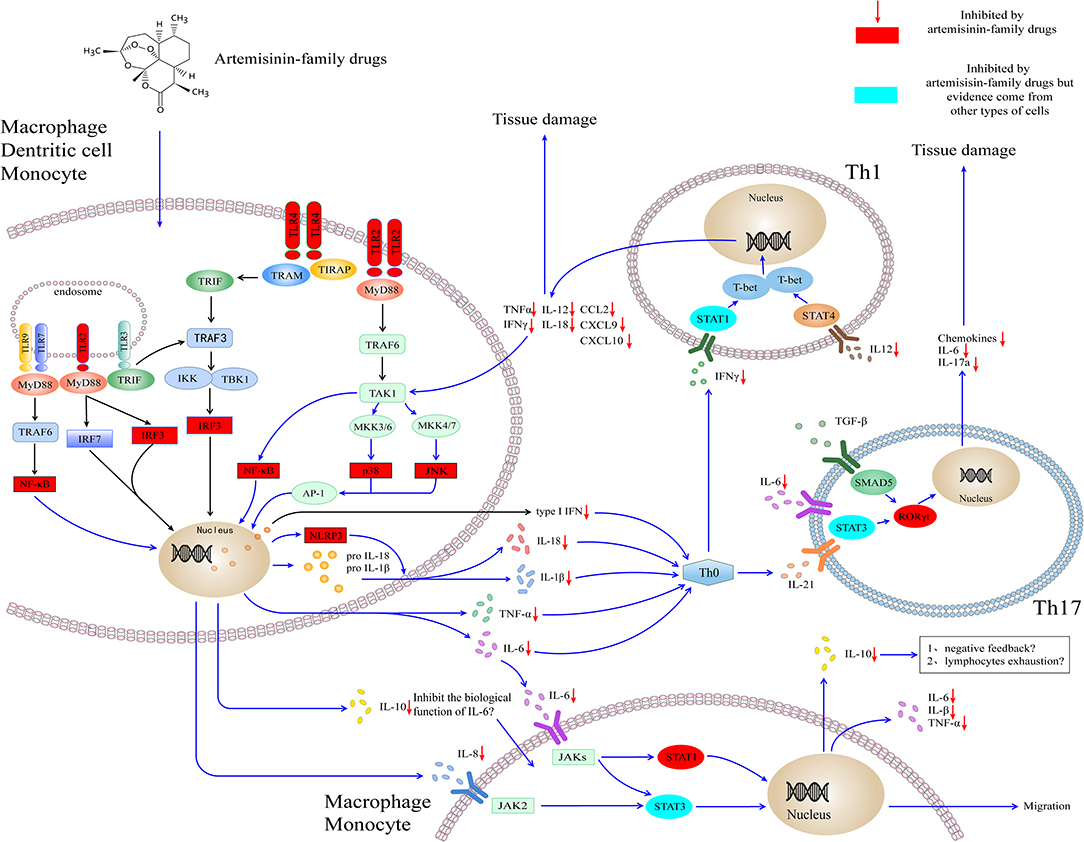
Figure 1. Artemisinin-family drugs for cytokine storm in COVID-19. The dysregulation of the cytokine storm involves mainly APCs. TLR2 and TLR4 locate mainly outside macrophages, DCs, and granulocytes. Also, they are expressed within endosomes, play a role in recognizing bacteria and viruses. Through MyD88-dependent or TRIF-dependent pathway, TLR2 and TLR4 transmit signals for the activation of IRF3 and NF-κB to induce the type I interferon and cytokines. Besides, TLR2 leads to the activation of AP-1, which is responsible for the transcription of inflammatory cytokines. The cytokines target at the naïve T helper cell, to result in the naïve T helper cell to differentiate to Th1 cell and Th17 cell, subsequently to secrete the inflammatory cytokines and chemokines. Moreover, the IL-6, IL-8, and IL-10 secreted by monocytes and macrophages could activate cytokines receptors (i.e., IL-6R, IL-8R), lead to the activation of JAK-STAT signaling pathways and cell migration. The artemisinin-family drugs target at a variety of molecules (red and blueness nodes) in the inflammatory networks, such as NF-κB, IRF3, ERK (not shown in the figure), and RORγt, which inhibit the differentiation of inflammatory cells and the production of cytokines and chemokines. IL-10 is an anti-inflammatory cytokine. It could be secreted by virtually all immune cells, including macrophages, DCs, NK cells, T cells, and B cells. At the moment, the high concentration of IL-10 in severely ill patients with COVID-19 is a mystery. On the one hand, it might play a role in antagonizing the biological function induced by IL-6. On the other hand, the high concentration of IL-10 might contribute to the lymphocytes exhaustion. AP-1, activating protein-1; CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand; IKK, IκB kinase; IFN, interferon; IRF3, interferon response factor 3; JAK, Janus kinase; JNK, Jun N-terminal kinase; MyD88, myeloid differentiation primary response protein 88; NF-κB, Nuclear factor κ B; NLPR3, NOD-, LRR- and pyrin domain-containing protein 3; MKK, Mitogen-activated protein kinase kinase; SMAD5, SMAD Family Member 5; RORγt, retinoic acid receptor-related orphan nuclear receptor gamma t; STAT, Signal transducer and activator of transcription; TAK1, TGFβ-activated kinase; T-bet, T-box transcription factor 21 (also known as TBX21); TLR, Toll-like receptor; TRAF, TNF receptor-associated factor; TRAM, TRIF-related adaptor molecule; TRIF, TIR domain–containing adaptor protein inducing interferon-β. IL, interleukin.
A precise definition of a cytokine storm is needed urgently. Mehta et al. (136) suggest that the criteria of sHLH could be applied. Moreover, the term needs to be placed in the ICD code. The ICD code would bring the standardization of disease names, the convenience of electronic medical records (EMR) management, and the efficiency in information sharing. For example, the characteristic of cytokine storm would be more accessible to be collected for a retrospective study.
YT: manuscript preparation and wrote the main part of the manuscript. JL: evidence collection, wrote the parts of the manuscript, and manuscript editing. DZ: helped to perform the analysis with constructive discussions. ZX: helped to revise the manuscript and gave many professional suggestions. JJ: ideas, formulation of overarching research goals, and aims. CW: critically reviewed the manuscript, project funding, and study initiation. All authors approved the final version of the manuscript.
This research was supported by Natural Science Foundation of Zhejiang Province Emergency Prevention and Treatment of COVID-19, under Grant No. LEZ20H190001.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Charlesworth Group (https://www.cwauthors.com.cn) for its linguistic assistance during the preparation of this manuscript. We thank Yujie Tang for the modification of the figures in the aesthetic view.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01708/full#supplementary-material
Supplementary Table 1. Clinical trial registration for inflammatory disorder with COVID-19.
1. Cron R, Behrens EM. Cytokine Storm Syndrome. 1 ed. Cham: Springer Nature Switzerland AG; Springer International Publishing (2019).
2. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
3. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
4. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
5. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. (2020) 130:2620–9. doi: 10.1101/2020.02.16.20023903
6. Chen L, Liu HG, Liu W, Liu J, Liu K, Shang J, et al. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. (2020) 43:203–8. doi: 10.3760/cma.j.issn.1001-0939.2020.0005
7. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. (2020). doi: 10.2139/ssrn.3541136. [Epub ahead of print].
8. Tan M, Liu Y, Zhou R, Deng X, Li F, Liang K, et al. Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. Immunology. (2020). doi: 10.1111/imm.13223. [Epub ahead of print].
9. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
10. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. (2020) 9:761–70. doi: 10.1080/22221751.2020.1747363
11. Zhong-yong C, Wei-bin Y, Qiang W, Guo-lin L. Clinical significance of serum hs-CRP, IL-6, and PCT in diagnosis and prognosis of patients with COVID-19. Drugs Clin. (2020) 35:417–20. doi: 10.7501/j.issn.1674-5515.2020.03.005
12. Guohua L, Ling L, Min H, Haibiao L, Peifeng K, Zishao Z, et al. Value of various inflammatory markers combined with lymphocyte subsets on clinical diagnosis of different clinical types of COVID-19. J Chong Med Univ. (2020). doi: 10.13406/j.cnki.cyxb.002465. [Epub ahead of print].
13. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
14. Cao X. COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol. (2020) 269–70. doi: 10.1038/s41577-020-0308-3
15. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
16. Arabi YM, Arifi AA, Balkhy HH, Najm H, Aldawood AS, Ghabashi A, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Internal Med. (2014) 160:389–97. doi: 10.7326/M13-2486
17. Al-Tawfiq JA, Hinedi K, Ghandour J, Khairalla H, Musleh S, Ujayli A, et al. Middle East respiratory syndrome coronavirus: a case-control study of hospitalized patients. Clin Infect Dis. (2014) 59:160–5. doi: 10.1093/cid/ciu226
18. Peiris JSM, Chu C-M, Cheng VC-C, Chan K, Hung I, Poon LL, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. (2003) 361:1767–72. doi: 10.1016/S0140-6736(03)13412-5
19. Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H, et al. First case of 2019 novel coronavirus in the United States. N Engl J Med. (2020) 382:929–36. doi: 10.1056/NEJMoa2001191
20. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
21. Chen J, Subbarao K. The immunobiology of SARS. Annu Rev Immunol. (2007) 25:443–72. doi: 10.1146/annurev.immunol.25.022106.141706
22. Cameron MJ, Ran L, Xu L, Danesh A, Bermejo-Martin JF, Cameron CM, et al. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol. (2007) 81:8692–706. doi: 10.1128/JVI.00527-07
23. Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, et al. An interferon-γ-related cytokine storm in SARS patients. J Med Virol. (2005) 75:185–94. doi: 10.1002/jmv.20255
24. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. (2005) 202:415–24. doi: 10.1084/jem.20050828
25. Li T, Qiu Z, Zhang L, Han Y, He W, Liu Z, et al. Significant changes of peripheral T lymphocyte subsets in patients with severe acute respiratory syndrome. J Infect Dis. (2004) 189:648–51. doi: 10.1086/381535
26. Menachery VD, Eisfeld AJ, Schäfer A, Josset L, Sims AC, Proll S, et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. MBio. (2014) 5:e01174–14. doi: 10.1128/mBio.01174-14
27. Lau SK, Lau CC, Chan K-H, Li CP, Chen H, Jin D-Y, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J General Virol. (2013) 94:2679–90. doi: 10.1099/vir.0.055533-0
28. Yen Y-T, Liao F, Hsiao C-H, Kao C-L, Chen Y-C, Wu-Hsieh BA. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J Virol. (2006) 80:2684–93. doi: 10.1128/JVI.80.6.2684-2693.2006
29. Tynell J, Westenius V, Rönkkö E, Munster VJ, Melén K, Österlund P, et al. Middle East respiratory syndrome coronavirus shows poor replication but significant induction of antiviral responses in human monocyte-derived macrophages and dendritic cells. J General Virol. (2016) 97(Pt 2):344. doi: 10.1099/jgv.0.000351
30. Zhou J, Chu H, Li C, Wong BH-Y, Cheng Z-S, Poon VK-M, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. (2014) 209:1331–42. doi: 10.1093/infdis/jit504
31. Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in sars-coronavirus–infected, monocyte-derived human dendritic cells. Blood. (2005) 106:2366–74. doi: 10.1182/blood-2004-10-4166
32. Cheung CY, Poon LL, Ng IH, Luk W, Sia S-F, Wu MH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. (2005) 79:7819–26. doi: 10.1128/JVI.79.12.7819-7826.2005
33. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
34. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
35. Tseng CT, Perrone LA, Zhu H, Makino S, Peters CJ. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol. (2005) 174:7977–85. doi: 10.4049/jimmunol.174.12.7977
36. Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, et al. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. (2012) 5:258–66. doi: 10.1038/mi.2012.2
37. Totura AL, Whitmore A, Agnihothram S, Schäfer A, Katze MG, Heise MT, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio. (2015) 6:e00638–15. doi: 10.1128/mBio.00638-15
38. National Health Commission of the People's Republic of China. Diagnosis and Treatment Protocol for COVID-19 Trial Version 7. (2020). Available online at: http://www.nhc.gov.cn/xcs/zhengcwj/202003/46c9294a7dfe4cef80dc7f5912eb1989.shtml
39. Ouyang Y, Yin J, Wang W, Shi H, Shi Y, Xu B, et al. Down-regulated gene expression spectrum and immune responses changed during the disease progression in COVID-19 patients. Clin Infect Dis. (2020). doi: 10.1093/cid/ciaa462. [Epub ahead of print].
40. Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin Infect Dis. (2020). doi: 10.1093/cid/ciaa272. [Epub ahead of print].
41. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the chinese center for disease control and prevention. JAMA. (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
42. Zilong L, Ruyuan H, Wenyang J, Tao F, Qing G. Clinical characteristics and immune function analysis of COVID-19. Med J Wuhan Univ. (2020) 41:529–32. doi: 10.14188/j.1671-8852.2020.0126
43. Jing X, Ming-feng H, Feng-de Z, Ting Z, Lei M. Clinical manifestations and sero-immunological characteristics of 155 patients with COVID-19. Chin J Nosocomiol. (2020) 30:2261–5. doi: 10.11816/cn.ni.2020-200577
44. Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. (2020) 146:110–8. doi: 10.1016/j.jaci.2020.04.006
45. Wan S, Yi Q, Fan S, Lv J, Zhang X, Guo L, et al. Relationships among lymphocyte subsets, cytokines, and the pulmonary inflammation index in coronavirus (COVID-19) infected patients. Br J Haematol. (2020) 189:428–37. doi: 10.1111/bjh.16659
46. Xu B, Fan C-y, Wang A-l, Zou Y-l, Yu Y-h, He C, et al. Suppressed T cell-mediated immunity in patients with COVID-19: a clinical retrospective study in Wuhan, China. J Infect. (2020) 81:e51–60. doi: 10.1016/j.jinf.2020.04.012
47. Chen X, Ling J, Mo P, Zhang Y, Jiang Q, Ma Z, et al. Restoration of leukomonocyte counts is associated with viral clearance in COVID-19 hospitalized patients. medRxiv [preprint]. (2020). doi: 10.1101/2020.03.03.20030437
48. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
49. Chen Y, Feng Z, Diao B, Wang R, Wang G, Wang C, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. medRxiv. (2020). doi: 10.1101/2020.03.27.20045427
50. Li J, Guo M, Tian X, Liu C, Wang X, Yang X, et al. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. bioRxiv. (2020). doi: 10.1101/2020.03.31.019216
51. Michot JM, Albiges L, Chaput N, Saada V, Pommeret F, Griscelli F, et al. Tocilizumab, an anti-IL6 receptor antibody, to treat Covid-19-related respiratory failure: a case report. Ann Oncol. (2020) 31:961–4. doi: 10.1016/j.annonc.2020.03.300
52. Zhang X, Song K, Tong F, Fei M, Guo H, Lu Z, et al. First case of COVID-19 in a patient with multiple myeloma successfully treated with tocilizumab. Blood Adv/. (2020) 4:1307–10. doi: 10.1182/bloodadvances.2020001907
53. Ferrey AJ, Choi G, Hanna RM, Chang Y, Tantisattamo E, Ivaturi K, et al. A case of novel coronavirus disease 19 in a chronic hemodialysis patient presenting with gastroenteritis and developing severe pulmonary disease. Am J Nephrol. (2020) 51:337–42. doi: 10.1159/000507417
54. Odievre MH, de Marcellus C, Ducou Le Pointe H, Allali S, Romain AS, Youn J, et al. Dramatic improvement after Tocilizumab of a severe COVID-19 in a child with sickle cell disease and acute chest syndrome. Am J Hematol. (2020). doi: 10.1002/ajh.25855. [Epub ahead of print].
55. Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J. Tocilizumab treatment in COVID-19: a single center experience. J Med Virol. (2020) 92:814–8. doi: 10.1002/jmv.25801
56. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci USA. (2020) 117:10970–5. doi: 10.1073/pnas.2005615117
57. Sciascia S, Apra F, Baffa A, Baldovino S, Boaro D, Boero R, et al. Pilot prospective open, single-arm multicentre study on off-label use of tocilizumab in severe patients with COVID-19. Clin Exp Rheumatol. (2020) 38:529–32.
58. Radbel J, Narayanan N, Bhatt PJ. Use of tocilizumab for COVID-19 infection-induced cytokine release syndrome: a cautionary case report. Chest. (2020) 158:e15–9. doi: 10.1016/j.chest.2020.04.024
59. Smolen JS, Landewe RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. (2020) 79:685–99. doi: 10.1136/annrheumdis-2019-216655
60. Sepriano A, Kerschbaumer A, Smolen JS, van der Heijde D, Dougados M, van Vollenhoven R, et al. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2019 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. (2020) 79:760–70. doi: 10.1136/annrheumdis-2019-216653
61. Almawi W, Melemedjian O. Negative regulation of nuclear factor-kappaB activation and function by glucocorticoids. J Mol Endocrinol. (2002) 28:69–78. doi: 10.1677/jme.0.0280069
62. Ristimäki A, Narko K, Hla T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J. (1996) 318:325–31. doi: 10.1042/bj3180325
63. Firestein GS, Budd R, Gabriel SE, McInnes IB, O'Dell JR. Kelley's Textbook of Rheumatology E-Book. Amsterdam: Elsevier Health Sciences (2012).
64. Yang Z, Liu J, Zhou Y, Zhao X, Zhao Q, Liu J. The effect of corticosteroid treatment on patients with coronavirus infection: a systematic review and meta-analysis. J Infect. (2020) 81:e13–20. doi: 10.1016/j.jinf.2020.03.062
65. Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. (2020) 395:473–5. doi: 10.1016/S0140-6736(20)30317-2
66. Liu K, Fang YY, Deng Y, Liu W, Wang MF, Ma JP, et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin Med J. (2020) 133:1025–31. doi: 10.1097/CM9.0000000000000744
67. Zha L, Li S, Pan L, Tefsen B, Li Y, French N, et al. Corticosteroid treatment of patients with coronavirus disease 2019 (COVID-19). Med J Austral. (2020) 212:416–20. doi: 10.5694/mja2.50577
68. Chen RC, Tang XP, Tan SY, Liang BL, Wan ZY, Fang JQ, et al. Treatment of severe acute respiratory syndrome with glucosteroids: the Guangzhou experience. Chest. (2006) 129:1441–52. doi: 10.1378/chest.129.6.1441
69. Zhou W, Liu Y, Tian D, Wang C, Wang S, Cheng J, et al. Potential benefits of precise corticosteroids therapy for severe 2019-nCoV pneumonia. Signal Transduct Target Ther. (2020) 5:18. doi: 10.1038/s41392-020-0127-9
70. Mahmud-Al-Rafat A, Majumder A, Taufiqur Rahman KM, Mahedi Hasan AM, Didarul Islam KM, Taylor-Robinson AW, et al. Decoding the enigma of antiviral crisis: does one target molecule regulate all? Cytokine. (2019) 115:13–23. doi: 10.1016/j.cyto.2018.12.008
71. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. (2016) 375:1767–78. doi: 10.1056/NEJMra1514296
72. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
73. Shao R, Fang Y, Yu H, Zhao L, Jiang Z, Li C-S. Monocyte programmed death ligand-1 expression after 3–4 days of sepsis is associated with risk stratification and mortality in septic patients: a prospective cohort study. Critical Care. (2016) 20:124. doi: 10.1186/s13054-016-1301-x
74. Busch LM, Sun J, Cui X, Eichacker PQ, Torabi-Parizi P. Checkpoint inhibitor therapy in preclinical sepsis models: a systematic review and meta-analysis. Intensive Care Med Exp. (2020) 8:7. doi: 10.1186/s40635-019-0290-x
75. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med. (2019) 45:1360–71. doi: 10.1007/s00134-019-05704-z
76. Hotchkiss RS, Colston E, Yende S, Angus DC, Moldawer LL, Crouser ED, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized, placebo-controlled, single ascending dose study of antiprogrammed cell death-ligand 1 antibody (BMS-936559). Crit Care Med. (2019) 47:632–42. doi: 10.1097/CCM.0000000000003685
77. Wang Z, Zhu L, Nguyen THO, Wan Y, Sant S, Quinones-Parra SM, et al. Clonally diverse CD38(+)HLA-DR(+)CD8(+) T cells persist during fatal H7N9 disease. Nat Commun. (2018) 9:824. doi: 10.1038/s41467-018-03243-7
78. Bruenger F, Kizner L, Weile J, Morshuis M, Gummert JF. First Successful Combination of ECMO With Cytokine Removal Therapy in Cardiogenic Septic Shock: A Case Report. SAGE Publications; Sage UK: London, England (2015).
79. Träger K, Schütz C, Fischer G, Schröder J, Skrabal C, Liebold A, et al. Cytokine reduction in the setting of an ARDS-associated inflammatory response with multiple organ failure. Case Rep Crit Care. (2016) 2016:9852073. doi: 10.1155/2016/9852073
80. Rimmelé T, Kellum JA. Clinical review: blood purification for sepsis. Crit Care. (2011) 15:205. doi: 10.1186/cc9411
81. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. (2013) 13:176–89. doi: 10.1038/nri3401
82. Ma C, Wong CK, Wong BC, Chan KC, Lun SW, Lee N, et al. Cytokine responses in a severe case of glandular fever treated successfully with foscarnet combined with prednisolone and intravenous immunoglobulin. J Med Virol. (2009) 81:99–105. doi: 10.1002/jmv.21383
83. Hung IFN, To KKW, Lee CK, Lee KL, Yan WW, Chan K, et al. Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A(H1N1) infection. Chest. (2013) 144:464–73. doi: 10.1378/chest.13-0571
84. Alejandria MM, Lansang MAD, Dans LF, Mantaring JB, 3rd. Intravenous immunoglobulin for treating sepsis, severe sepsis and septic shock. Cochrane Database Syst Rev. (2013) 2013:CD001090-CD. doi: 10.1002/14651858.CD001090.pub2
85. Lenzer J. Covid-19: US gives emergency approval to hydroxychloroquine despite lack of evidence. BMJ. (2020) 369:m1335. doi: 10.1136/bmj.m1335
86. Singh AK, Singh A, Singh R, Misra A. Hydroxychloroquine in patients with COVID-19: a systematic review and meta-analysis. Diabetes Metab Syndr. (2020) 14:589–96. doi: 10.1016/j.dsx.2020.05.017
87. World Health Organization. Coronavirus Disease 2019(COVID-19) Situation Report-41: Data as Reported by 10AM CET 01 March 2020. (2020). Available online at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200301-sitrep-41-covid-19.pdf?sfvrsn=6768306d_2
88. Hsu C-H, Hwang K-C, Chao C-L, Chang SGN, Ker C-C, Chien L-C, et al. The lesson of supplementary treatment with Chinese medicine on severe laboratory-confirmed SARS patients. Am J Chin Med. (2006) 34:927–35. doi: 10.1142/S0192415X06004405
89. Lau JTF, Leung PC, Wong ELY, Fong C, Cheng KF, Zhang SC, et al. The use of an herbal formula by hospital care workers during the severe acute respiratory syndrome epidemic in Hong Kong to prevent severe acute respiratory syndrome transmission, relieve influenza-related symptoms, and improve quality of life: a prospective cohort study. J Altern Complement Med. (2005) 11:49–55. doi: 10.1089/acm.2005.11.49
90. Lau TF, Leung PC, Wong ELY, Fong C, Cheng KF, Zhang SC, et al. Using herbal medicine as a means of prevention experience during the SARS crisis. Am J Chin Med. (2005) 33:345–56. doi: 10.1142/S0192415X05002965
91. Jinpan Z, Baojin H, Changhuai C, Guicheng X, Hao S, Yin W, et al. Clinical features of 42 patients with SARS and integrated Chinese and western medicine. Chin J Integr Tradition West Med Intensive Critical Care. (2003) 23:486–8.
92. Junhui P, Hui Y, Qinghe Y, Feng W, Zhinan Q, Shuqing Z, et al. Clinical study on 71 cases of SARS patients intervened with traditional chinese medicine. Chin J Integr Tradition West Med Intensive Crit Care. (2003) 10:204−7.
93. Xiaolin T, Aiguo L, Zhiyuan Z, Jun D, Xiaoguang C, Chuanjin H, et al. Clinical observation on 16 cases of infectious atypical pneumonia treated by traditional Chinese Medicine. J Tradition Chin Med. (2003) 44:506–7. doi: 10.13288/j.11-2166/r.2003.07.020
94. Tan W, Li S, Yiyang C, Yakun F, Wei Y, Xiaozheng D, et al. Clinical efficacy analysis of 50 cases of corona virus disease 2019 in traditional Chinese medicine. Jilin J Chin Med. (2020) 40:281–5. doi: 10.13463/j.cnki.jlzyy.2020.03.001
95. Kai-tao Y, Ming-yu L, Xin L, Ji-han H, Hong-bin C. Retrospective clinical analysis on treatment of novel coronavirus-infected pneumonia with traditional chinese medicine Lianhua Qingwen. Chin J Exp Tradition Med Formul. (2020) 26:8–12. doi: 10.13422/j.cnki.syfjx.20201099
96. Wen-guang X, Chang-qing A, Chan-juan Z, Ji-xian Z, Min H, Yu W, et al. Clinical study on 34 novel coronavirus pneumoniae treated with integrated traditional Chinese and Western Medicine. J Tradition Chin Med. (2020) 61:375–82. doi: 10.13288/j.11-2166/r.2020.05.002
97. Hou L, Huang H. Immune suppressive properties of artemisinin family drugs. Pharmacol Therap. (2016) 166:123–7. doi: 10.1016/j.pharmthera.2016.07.002
98. An J, Minie M, Sasaki T, Woodward JJ, Elkon KB. Antimalarial drugs as immune modulators: new mechanisms for old drugs. Annu Rev Med. (2017) 68:317–30. doi: 10.1146/annurev-med-043015-123453
99. Shi C, Li H, Yang Y, Hou L. Anti-inflammatory and immunoregulatory functions of artemisinin and its derivatives. Mediat Inflamm. (2015) 2015:435713. doi: 10.1155/2015/435713
100. Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Hydroxychloroquine: from malaria to autoimmunity. Clin Rev Allergy Immunol. (2012) 42:145–53. doi: 10.1007/s12016-010-8243-x
101. Cao T-h, Jin S-g, Fei D-s, Kang K, Jiang L, Lian Z-y, et al. Artesunate protects against sepsis-induced lung injury via heme oxygenase-1 modulation. Inflammation. (2016) 39:651–62. doi: 10.1007/s10753-015-0290-2
102. Huang XT, Liu W, Zhou Y, Hao CX, Zhou Y, Zhang CY, et al. Dihydroartemisinin attenuates lipopolysaccharideinduced acute lung injury in mice by suppressing NFkappaB signaling in an Nrf2dependent manner. Int J Mol Med. (2019) 44:2213–22. doi: 10.3892/ijmm.2019.4387
103. Hu Y, Li M, Zhang T, Jin Y. Preparation of liposomal artesunate dry powder inhalers and the effect on the acute lung injury of rats. Acta Pharm Sin. (2016) 51:1906–12. doi: 10.16438/j.0513-4870.2016-0848
104. Cheng Y, Luo R, Wang K, Zhang M, Wang Z, Dong L, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. (2020) 97:829–38. doi: 10.1016/j.kint.2020.03.005
105. Cheng Z, Qi R, Li L, Liu Q, Zhang W, Zhou X, et al. Dihydroartemisinin ameliorates sepsis-induced hyperpermeability of glomerular endothelium via up-regulation of occludin expression. Biomed Pharmacother. (2018) 99:313–8. doi: 10.1016/j.biopha.2018.01.078
106. Long H, Xu B, Luo Y, Luo K. Artemisinin protects mice against burn sepsis through inhibiting NLRP3 inflammasome activation. Am J Emerg Med. (2016) 34:772–7. doi: 10.1016/j.ajem.2015.12.075
107. Tao J-H, Zhang Y, Li X-P. P2X7R: a potential key regulator of acute gouty arthritis. Semin Arthritis Rheum. (2013) 43:376–80. doi: 10.1016/j.semarthrit.2013.04.007
108. Nakanishi H, Kawashima Y, Kurima K, Chae JJ, Ross AM, Pinto-Patarroyo G, et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci USA. (2017) 114:E7766–e75. doi: 10.1073/pnas.1702946114
109. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann NY Acad Sci. (2014) 1319:82–95. doi: 10.1111/nyas.12458
110. Bobade D, Khandare AV, Deval M, Shastry P, Deshpande P. Hemozoin-induced activation of human monocytes toward M2-like phenotype is partially reversed by antimalarial drugs-chloroquine and artemisinin. Microbiologyopen. (2019) 8:e00651. doi: 10.1002/mbo3.651
111. Idowu AO, Bhattacharyya S, Gradus S, Oyibo W, George Z, Black C, et al. Plasmodium falciparum treated with artemisinin-based combined therapy exhibits enhanced mutation, heightened cortisol and TNF-α Induction. Int J Med Sci. (2018) 15:1449–57. doi: 10.7150/ijms.27350
112. Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. (2013) 13:551–65. doi: 10.1038/nri3479
113. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
114. Li B, Li J, Pan X, Ding G, Cao H, Jiang W, et al. Artesunate protects sepsis model mice challenged with Staphylococcus aureus by decreasing TNF-α release via inhibition TLR2 and Nod2 mRNA expressions and transcription factor NF-κB activation. Int Immunopharmacol. (2010) 10:344–50. doi: 10.1016/j.intimp.2009.12.006
115. Kuang M, Cen Y, Qin R, Shang S, Zhai Z, Liu C, et al. Artesunate attenuates pro-inflammatory cytokine release from macrophages by inhibiting TLR4-mediated autophagic activation via the TRAF6-Beclin1-PI3KC3 pathway. Cell Physiol Biochem. (2018) 47:475–88. doi: 10.1159/000489982
116. Cen Y, Liu C, Li X, Yan Z, Kuang M, Su Y, et al. Artesunate ameliorates severe acute pancreatitis (SAP) in rats by inhibiting expression of pro-inflammatory cytokines and Toll-like receptor 4. Int Immunopharmacol. (2016) 38:252–60. doi: 10.1016/j.intimp.2016.06.007
117. Huang X, Xie Z, Liu F, Han C, Zhang D, Wang D, et al. Dihydroartemisinin inhibits activation of the Toll-like receptor 4 signaling pathway and production of type I interferon in spleen cells from lupus-prone MRL/lpr mice. Int Immunopharmacol. (2014) 22:266–72. doi: 10.1016/j.intimp.2014.07.001
118. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. (2002) 12:9–18. doi: 10.1038/sj.cr.7290105
119. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
120. Wang JX, Hou LF, Yang Y, Tang W, Li Y, Zuo JP. SM905, an artemisinin derivative, inhibited NO and pro-inflammatory cytokine production by suppressing MAPK and NF-kappaB pathways in RAW 264.7 macrophages. Acta Pharmacol Sin. (2009) 30:1428–35. doi: 10.1038/aps.2009.138
121. Park KH, Yoon YD, Han SB, Oh SJ, Yun J, Lee CW, et al. Artemisinin inhibits lipopolysaccharide-induced interferon-β production in RAW 264.7 cells: implications on signal transducer and activator of transcription-1 signaling and nitric oxide production. Int Immunopharmacol. (2012) 14:580–4. doi: 10.1016/j.intimp.2012.09.012
122. Hou LF, He SJ, Li X, Yang Y, He PL, Zhou Y, et al. Oral administration of artemisinin analog SM934 ameliorates lupus syndromes in MRL/lpr mice by inhibiting Th1 and Th17 cell responses. Arthritis Rheum. (2011) 63:2445–55. doi: 10.1002/art.30392
123. Du Y, Chen G, Zhang X, Yu C, Cao Y, Cui L. Artesunate and erythropoietin synergistically improve the outcome of experimental cerebral malaria. Int Immunopharmacol. (2017) 48:219–30. doi: 10.1016/j.intimp.2017.05.008
124. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers. (2018) 4:18001. doi: 10.1038/nrdp.2018.1
125. Wang JX, Tang W, Zhou R, Wan J, Shi LP, Zhang Y, et al. The new water-soluble artemisinin derivative SM905 ameliorates collagen-induced arthritis by suppression of inflammatory and Th17 responses. Br J Pharmacol. (2008) 153:1303–10. doi: 10.1038/bjp.2008.11
126. Fan M, Li Y, Yao C, Liu X, Liu X, Liu J. Dihydroartemisinin derivative DC32 attenuates collagen-induced arthritis in mice by restoring the Treg/Th17 balance and inhibiting synovitis through down-regulation of IL-6. Int Immunopharmacol. (2018) 65:233–43. doi: 10.1016/j.intimp.2018.10.015
127. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. (2011) 11:119–30. doi: 10.1038/nri2916
128. Wang JX, Tang W, Shi LP, Wan J, Zhou R, Ni J, et al. Investigation of the immunosuppressive activity of artemether on T-cell activation and proliferation. Br J Pharmacol. (2007) 150:652–61. doi: 10.1038/sj.bjp.0707137
129. Xu H, He Y, Yang X, Liang L, Zhan Z, Ye Y, et al. Anti-malarial agent artesunate inhibits TNF-alpha-induced production of proinflammatory cytokines via inhibition of NF-kappaB and PI3 kinase/Akt signal pathway in human rheumatoid arthritis fibroblast-like synoviocytes. Rheumatology. (2007) 46:920–6. doi: 10.1093/rheumatology/kem014
130. Wenisch C, Linnau KF, Looaresuwan S, Rumpold H. Plasma levels of the interleukin-6 cytokine family in persons with severe Plasmodium falciparum malaria. J Infect Dis. (1999) 179:747–50. doi: 10.1086/314630
131. D'Alessandro S, Scaccabarozzi D, Signorini L, Perego F, Ilboudo DP, Ferrante P, et al. The use of antimalarial drugs against viral infection. Microorganisms. (2020) 8:85. doi: 10.3390/microorganisms8010085
132. Shapira MY, Resnick IB, Chou S, Neumann AU, Lurain NS, Stamminger T, et al. Artesunate as a potent antiviral agent in a patient with late drug-resistant cytomegalovirus infection after hematopoietic stem cell transplantation. Clin Infect Dis. (2008) 46:1455–7. doi: 10.1086/587106
133. Germi R, Mariette C, Alain S, Lupo J, Thiebaut A, Brion JP, et al. Success and failure of artesunate treatment in five transplant recipients with disease caused by drug-resistant cytomegalovirus. Antiviral Res. (2014) 101:57–61. doi: 10.1016/j.antiviral.2013.10.014
134. Jacobs J, Clark-Snustad K, Lee S. Case report of a SARS-CoV-2 infection in a patient with ulcerative colitis on tofacitinib. Inflamm Bowel Dis. (2020) 26:e64. doi: 10.1093/ibd/izaa093
135. Rojas JM, Avia M, Martín V, Sevilla N. IL-10: a multifunctional cytokine in viral infections. J Immunol Res. (2017) 2017:6104054. doi: 10.1155/2017/6104054
Keywords: COVID-19, cytokine storm, treatment strategies, immunoregulation, tocilizumab, antimalarial agents
Citation: Tang Y, Liu J, Zhang D, Xu Z, Ji J and Wen C (2020) Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 11:1708. doi: 10.3389/fimmu.2020.01708
Received: 19 March 2020; Accepted: 26 June 2020;
Published: 10 July 2020.
Edited by:
Mario Clerici, University of Milan, ItalyReviewed by:
Vijayakumar Velu, Emory University, United StatesCopyright © 2020 Tang, Liu, Zhang, Xu, Ji and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinjun Ji, am9zaWFuQDEyNi5jb20=; Chengping Wen, d2VuZ2NwQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.