 Francesco Manfredi1,2
Francesco Manfredi1,2 Beatrice Claudia Cianciotti2,3
Beatrice Claudia Cianciotti2,3 Alessia Potenza2,4Elena Tassi2Maddalena Noviello2Andrea Biondi5Fabio Ciceri1,2Chiara Bonini1,2*†Eliana Ruggiero2*†
Alessia Potenza2,4Elena Tassi2Maddalena Noviello2Andrea Biondi5Fabio Ciceri1,2Chiara Bonini1,2*†Eliana Ruggiero2*†- 1Vita-Salute San Raffaele University, Milan, Italy
- 2Experimental Hematology Unit, Division of Immunology, Transplantation and Infectious Diseases, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 3Fondazione Centro San Raffaele, Milan, Italy
- 4School of Medicine and Surgery, University of Milano – Bicocca, Milan, Italy
- 5Clinica Pediatrica Università degli Studi di Milano Bicocca, Fondazione MBBM, Monza, Italy
Adoptive T cell therapy (ACT) is a rapidly evolving therapeutic approach designed to harness T cell specificity and function to fight diseases. Based on the evidence that T lymphocytes can mediate a potent anti-tumor response, initially ACT solely relied on the isolation, in vitro expansion, and infusion of tumor-infiltrating or circulating tumor-specific T cells. Although effective in a subset of cases, in the first ACT clinical trials several patients experienced disease progression, in some cases after temporary disease control. This evidence prompted researchers to improve ACT products by taking advantage of the continuously evolving gene engineering field and by improving manufacturing protocols, to enable the generation of effective and long-term persisting tumor-specific T cell products. Despite recent advances, several challenges, including prioritization of antigen targets, identification, and optimization of tumor-specific T cell receptors, in the development of tools enabling T cells to counteract the immunosuppressive tumor microenvironment, still need to be faced. This review aims at summarizing the major achievements, hurdles and possible solutions designed to improve the ACT efficacy and safety profile in the context of liquid and solid tumors.
Introduction
Adoptive T cell therapy for cancer (ACT) is a branch of cancer immunotherapy that relies on the ability to redirect T cell specificity to selectively target tumor antigens. ACT stemmed from two remarkable clinical observations: (i) The magnitude of T cells infiltrating tumor masses often correlates with response to treatment (1) and (ii) Allogeneic donor T cells infused in the context of hematopoietic stem cell transplantation promote clinical response in hematological malignancies (2). Initially, ACT solely relied on tumor-specific T cells isolated from the tumor masses and expanded in vitro (3). This approach was limited to resectable tumors from which enough T cells could be harvested and expanded. The development of gene engineering technologies dramatically changed the landscape of the ACT field, rapidly making this treatment accessible to an unprecedented number of patients and tumor types. By inserting an exogeneous T cell receptor (TCR) into cells, T cells specificity could be precisely redirected toward selected tumor antigens (Figure 1). This new opportunity shifted the research focus and raised some novel questions: the main issue was no more how to harvest a sufficient number of tumor-specific T cells from each single patient, but how to isolate and harness high-avidity tumor-specific TCRs, and how to proficiently generate and expand the most fit engineered T cells. The flexibility of the genetic modification tools offered the chance to insert and/or remove different genes in T cells and to permanently express, in the therapeutic products, entirely synthetic molecules. A striking deliverable produced by these efforts is represented by T cells expressing Chimeric Antigen Receptors (CARs), that generated astonishing clinical results against blood malignancies (4–9).
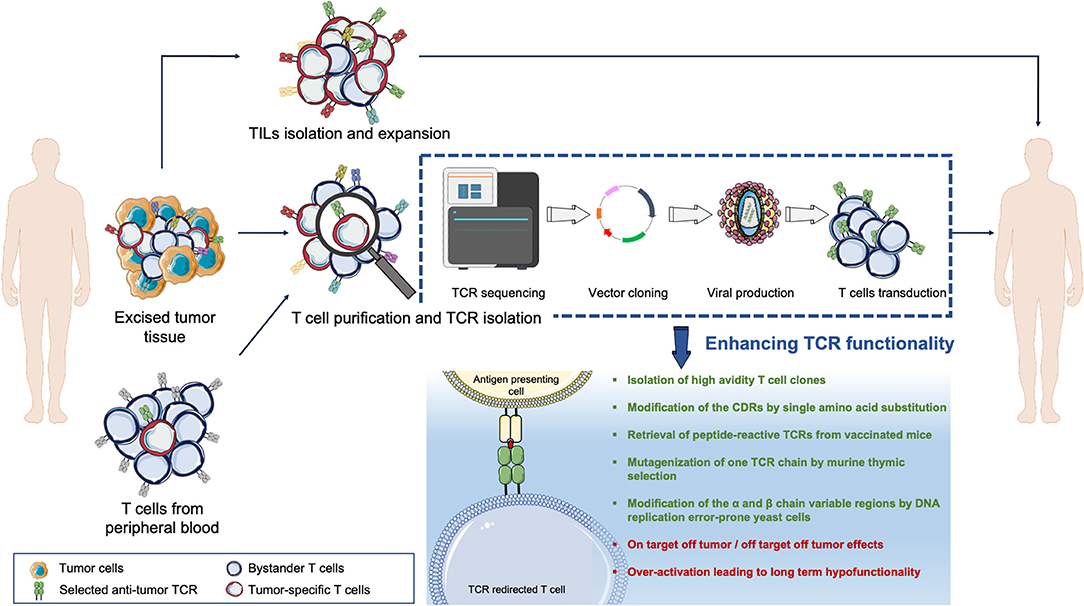
Figure 1. Overview of the TCR adoptive T cell therapy. Tumor-reactive lymphocytes can be isolated from either the tumor mass (tumor-infiltrating lymphocytes, TILs) or from the T cell pool circulating in patients' peripheral blood. T cells can be expanded in vitro and then re-infused back into the patient such as in TILs therapy. Else, the tumor-reactive T cell Receptor (TCR) genes can be isolated, sequenced, and transferred into acceptor T cells via vectors to redirect T cell specificities against tumor epitopes.
The outcomes of the first ACT clinical trials contributed to further elucidate the complex interplay between the immunosuppressive tumor microenvironment and the cellular players of immunity. Results suggested that modulation of ex vivo T cell expansion protocols and additional engineering of the T cell genome could be used to tweak T cell qualities, improving persistence and functionality of the therapeutic products. Several strategies were proposed to improve engineered T cell persistence, homing ability to the tumor site, capacity to recognize and eliminate tumor cells, and represent today's intense research lines. The possibility to modulate TCR affinity and T cell costimulatory and inhibitory signal pathways opens up novel therapeutic scenarios. The following review has the scope to summarize the cornerstones and the most relevant hurdles and efforts currently pursued to improve ACT.
From Allogeneic Stem Cell Transplantation to Adoptive T Cell Therapy
Allogeneic Hematopoietic Stem cell transplantation (Allo-HSCT) is a therapeutic modality relying on the infusion of hematopoietic stem and progenitor cells, harvested from a healthy donor, to a patient previously conditioned with high-doses chemo-radiotherapy. Although initially developed to regenerate the bone marrow of patients with genetic diseases or with hematological malignancies requiring strong myeloablative chemotherapy (10, 11), Allo-HSCT proved able to control malignant cells largely through an immunological mechanism, as delineated by two major observations. Firstly, T-lymphocyte depleted grafts had a decreased efficacy in eradicating malignant diseases, suggesting that the donor to host immune response, and in particular the activity of allogeneic T cells, had per se an effect in abating the risk of relapse after transplant (2, 12, 13). Secondly, the infusion of circulating mature lymphocytes harvested from the donor (donor lymphocyte infusion, DLI) (14) correlated with the anti-leukemic effect (graft vs. leukemia, GvL) in a dose-dependent fashion (15). The efficacy of Allo-HSCT and DLI in restoring a state of disease remission represents one of the first compelling evidences of the potential of adoptive T cell therapy. Unfortunately, the benefits of allogeneic transplant and DLI against cancer are counterbalanced by toxicities, mainly due to the presence of a heterogeneous TCR repertoire with unknown specificities in the infused T cell population. Indeed, it's been calculated that ~10% of the T cell repertoire circulating in healthy donors is alloreactive (16). The most common manifestation of such toxicities is graft vs. host disease (GvHD), an immune reaction against the host's healthy tissues, occurring with varying degrees of severity but potentially fatal. The efforts to reduce toxicity while preserving the efficacy of DLI, and to export this therapeutic opportunity beyond the HSCT context, were the driving forces in promoting innovative ACT approaches.
The first ACT strategies tested with autologous T lymphocytes were based on the isolation of T cells infiltrating primary lesions resected from patients with melanoma (tumor-infiltrating lymphocytes, TILs), followed by their in vitro expansion with high-doses of interleukin-2 (IL-2) (17). The infusion of these cellular products, composed of an oligoclonal T cell repertoire incorporating CD4+ and CD8+ T cells, mediated potent anti-tumor responses with no toxicities in cell types other than melanocytes (3, 18, 19). The Objective Response Rate (ORR) observed was 41% across various clinical trials for patients with metastatic melanoma (20). Based on these encouraging results, the approach was widened and offered to patients affected by other solid tumors with variable outcomes, promising in some settings [e.g., sarcoma (21), cervical and ovarian cancer (22, 23)] but rather modest in others [e.g., renal (24), metastatic renal (25) and, colorectal (26, 27) cancer, Table 1]. The inconsistent efficacy of TILs may be linked to various causes: (i) the technical difficulties in isolating T cells from immune-cold tumors (44); (ii) the poor reactivity of the screened T cells, especially in tumors characterized by a low mutational burden (45, 46), and (iii) the overall low frequency of tumor-specific T cells infiltrating cancer lesions when compared to bystander T cells (47, 48). The high success rate of TILs therapy in melanoma can be in fact explained by the melanoma cells high tumor mutational burden, resulting in a heightened immunogenicity and a consequent enrichment of tumor-specific T lymphocytes (49).
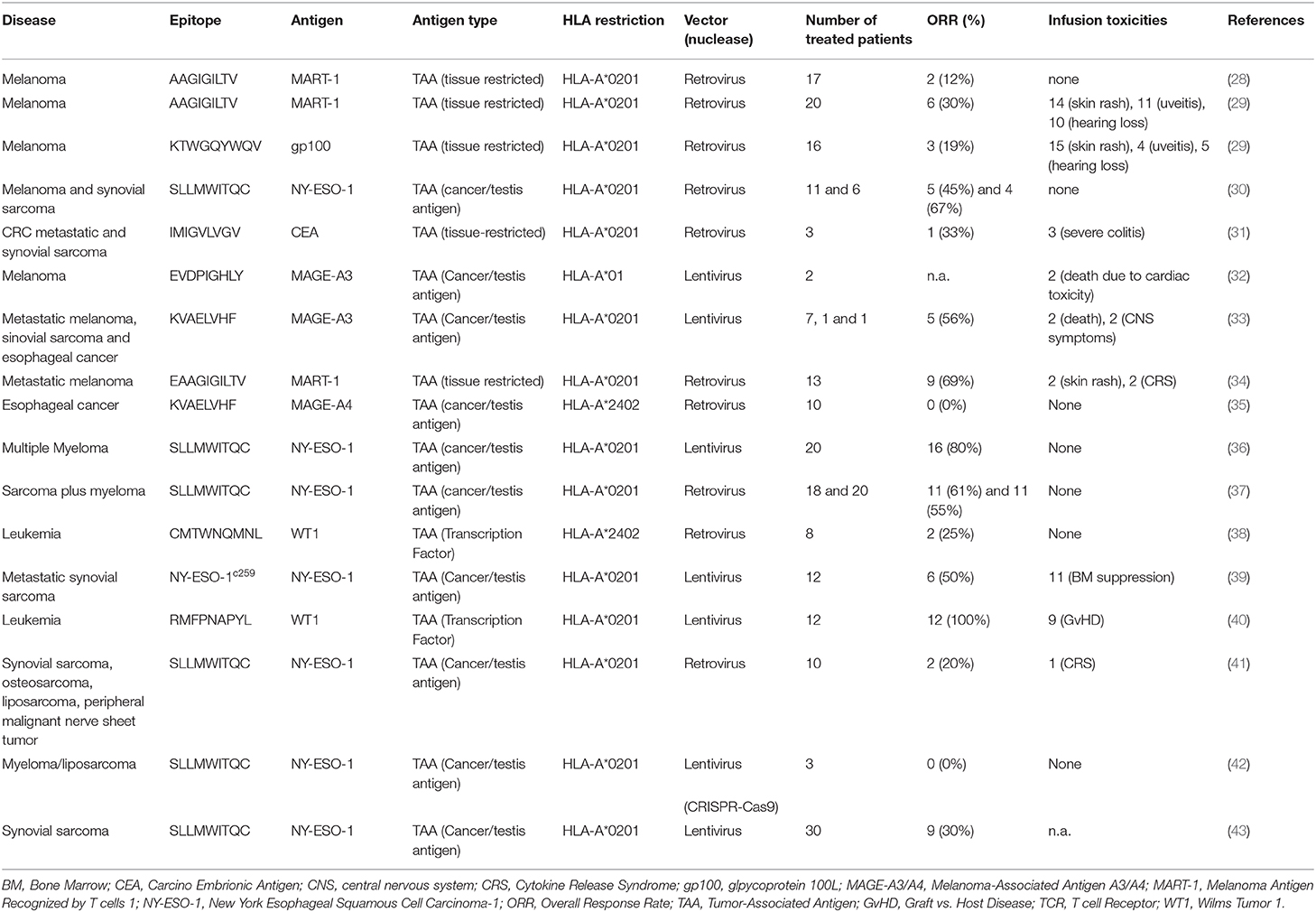
Table 1. Overview of TCR-engineered T cell-based clinical trials.
To expand the beneficial effect of TILs while overcoming the hurdles intrinsically associated with this therapy, the use of circulating T cells, harvested from patients and stimulated in vitro with immunogenic cancer epitopes, was proposed. This approach promotes the selective expansion of the tumor-specific T cell fraction, in numbers sufficient to enable their re-infusion to patients, resulting in clinical benefits (50, 51). Nonetheless, the use of TILs and circulating T cells lead to the generation of a T cell population for which the affinity and functionality of the TCR could not be predicted a priori and whose ability to effectively induce clinical responses was tightly linked to the expansion potential of harvested cells.
Gene editing and gene transfer technologies greatly boosted the ACT field, allowing modification of the T cell genome and redirection of T lymphocytes specificities by inserting highly functional, tumor-specific TCRs (52) into patients' T cells, that could be subsequently expanded in vitro. In the 90s the discovery that the Fab region of an antibody could be efficiently fused to the CD3 zeta chain and to other costimulatory intracellular domains to create Chimeric Antigen Receptors (CARs), further revolutionized the T cell-based immunotherapy field. CARs are able to activate T cells upon binding to a surface receptor expressed by the target tumor cell (53) and their use proved instrumental in widening the therapeutic window of blood tumor treatment in otherwise poor survivors (4–9), thus confirming TCR-engineered T cells as a new therapeutic.
Making Tumor Specific T Cells: From TCR Gene Transfer to TCR Gene Editing
The ability of T cells to respond to a wide spectrum of foreign antigens relies on the high variety of TCRs, heterodimeric glycoproteins composed of one α and one β chain associated to the CD3 complex (54), able to specifically interact with antigenic peptides bound to human leukocyte antigen (HLA) restriction elements (55). A series of genetic rearrangements in the α and β chain genes occur slightly differently in every single cell, thus creating a heterogeneous TCR repertoire that can recognize a vast epitope array. Hence, to fully characterize the T cell specificity it is necessary to determine the rearranged α and β chain sequences.
In the 80s, the progression of genomics allowed the isolation of TCR genes (56, 57) and the study in detail of their sequences. The advent of next generation sequencing technologies rendered feasible a comprehensive identification of tumor specific TCR sequences that are today used to genetically engineer T lymphocytes in adoptive T cell therapy studies.
Gene Transfer at the Service of ACT
Most TCR-based gene therapy approaches rely on the ex-vivo transduction of T cells with viral vectors. The first vectors used in gene therapy were adenoviruses (58), vectors endowed with high cargo capacity (up to 30 kb) but unable to foster transgene integration in the host genome. This feature reduced the adenoviruses utility for ACT: since T cells robustly proliferate upon antigen encounters, integration of the transferred TCR genes in their genome is critical to the preservation of transgenic specificity in T cell progeny. Furthermore, the immunogenicity of adenoviral proteins, highlighted by the high incidence of adenovirus-specific neutralizing antibodies in humans, potentially leads to viral inactivation (59) or to life-threatening inflammatory responses (60), thus limiting their exploitation. Retroviral vectors (RV), instead, have been broadly used because of their wide cell tropism (61, 62), good integration capacity, and for the high and stable gene expression they convey. Cell division is required for RV transduction, but this limitation does not impact their use since T cells are highly proliferating in vitro. RV have been widely used to deliver a variety of molecules, including suicide genes (63–65), TCRs (28, 66), and CARs (53) in T lymphocytes. Lentiviral vectors (LV) gained interest more recently, particularly for their efficiency profile and their capacity for transducing dividing as well as non-dividing cells, a feature particularly relevant for the genetic manipulation of stem cells (67). The safety profile of RV and LV is guaranteed by a vector design ensuring replication incompetence (68, 69) and has been proven in human trials (70–72). Adeno-Associate Viruses (AAV) (73) have been widely used in cancer gene therapy and proven to be well-tolerated and safe. Still, the need to synthesize the complementarity strand to promote transgene integration represents a limitation. To circumvent the process, both strands can be packaged as a single molecule to pair and form a dsDNA as a self-complementary AAV vector (scAAV). While this technological advancement allowed AAV to be independent from host cell complementary strand synthesis (74), it almost halved the vector packaging capacity. Nonetheless, scAAV outperformed conventional AAVs in terms of efficacy in preclinical models (75, 76).
Integrating viral vectors insert the genetic cassette semi-randomly into the host genome, thus potentially leading to unwanted insertions in exons, that leads to disruption of the gene hit, or in enhancer regions, potentially altering gene regulation. Theoretically, viral integrations in oncogene regulatory elements or in tumor suppressor genes may contribute to oncogenic transformation (77), a rare event that, most importantly, has never been reported in engineered T cells. Nevertheless, several strategies have been implemented to increase the safety profile of integrating vectors. These include the elimination of viral genes responsible for virulence (78), splitting packaging genes into different plasmids (79), and the introduction of inactivation switches in the vectors constructs (80). In addition, chromatin insulator elements can be added to the flanking regions of the insertion cassette, acting as physical barriers to hamper the interactions between viral enhancers and other regulatory elements (81). To increase the safety profile of viral-mediated gene delivery, it is nowadays possible to instruct viral vectors to integrate into specific “safe harbors,” genomic regions distant from transcribed genes, enhancers, regulatory RNA, or microRNA regions to minimize the risk of perturbing gene expressions (82, 83). Identified safe harbors are either housekeeping genes, e.g., AAVS1 (84, 85) and ROSA26 (86), or specific loci not affecting gene expression and identified by mapping the viral vectors integration sites (87).
In addition to viral vectors, a variety of non-viral gene transfer methods have been explored to transfer transgenes into T cells (Figure 2). Transposons are mobile elements composed of a transposase gene flanked by inverted terminal repeats (ITRs) (88). For the purpose of gene therapy, two plasmids are transfected together, one encoding for the transposase and the other one containing the expression cassette flanked by ITRs; upon entry, the transposase integrates the gene of interest in the genome. The so-called “Sleeping Beauty” transposon system gained the widest application, being able to transfer up to 6 kilobases into mammalian cells (89). This system may be considered as efficient as viral gene transfer, at least in vitro, if the transposon/transposase ratio is tightly controlled (90) to avoid the formation of functionally inactive transposase oligomers (91). In general, transient expression of the transposase is usually preferred, and ensured by mRNA electroporation (92). Transposons have an acceptable production cost and a low immunogenicity potential. Nonetheless, gene transfer efficiency varies according to the target cell and is sensitive to the size of the expression cassette. In the ACT field, transposons have been efficiently utilized to express functional CARs (9, 93–95) and TCRs (96–98) but their exploitation in clinical practice is still limited.
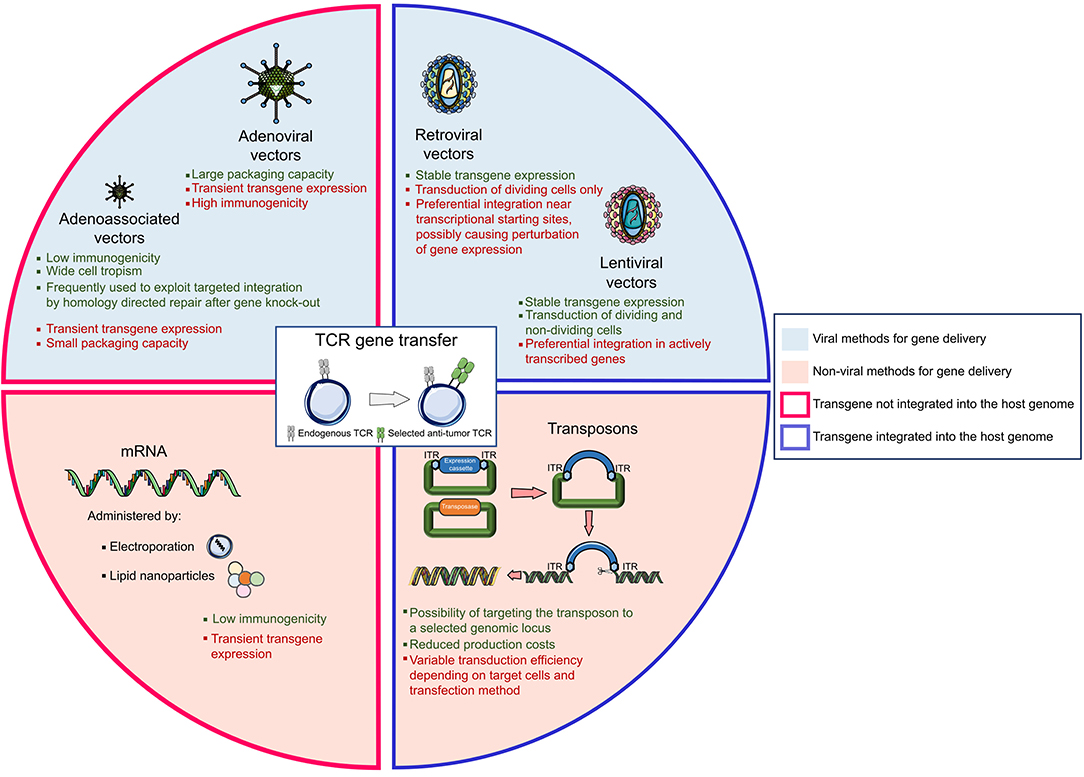
Figure 2. The landscape of gene delivery methods. The genetic transfer of an exogeneous T cell receptor (TCR) into a donor T cell can be obtained with different vectors, the most widely used being viral vectors, mRNA, and transposons systems. Strengths and weaknesses are listed for each technology.
Messenger RNA-based gene transfer, usually achieved by mRNA electroporation or by enclosing the mRNA into lipid nanoparticles (99), is devoid of insertional mutagenesis risk. However, mRNA can convey only transient transgene expression and in vitro transcribed mRNAs could trigger cellular inflammatory reactions, whose incidence can be mitigated by introducing base modifications in the synthetic RNA (100). In ACT studies, CAR-T cells have been generated using mRNA gene transfer; still, multiple administrations of the engineered T cells were necessary to mediate tumor regression. Of interest, in a phase I clinical trial, CAR-T cells targeting mesothelin generated upon mRNA electroporation and administered to patients with advanced cancers proved safe and mediated anti-tumor activity despite transient persistence (101).
TCR Gene Transfer
In 1986, a murine exogenous α and β TCR gene pair was successfully transferred into another cytotoxic T cell, endowing the recipient cell with a new TCR specificity (52). The efficacy of the TCR gene transfer was tested in immortalized T cells, where the cDNA of a MART-1-specific TCR isolated from melanoma TILs was stably expressed (102) and, shortly after, on primary human T cells, granting recipient T cells cytolytic activities specifically toward their target epitope. The encouraging safety and efficacy pre-clinical results observed by targeting MART-1 (103, 104), the murine MDM2 oncoprotein (105) and the EBV-associated LMP protein (106), prompted the approval of TCR gene transfer in human clinical trials. In the context of metastatic melanoma, TCR-transferred T cells successfully induced tumor regression in two out of 15 patients and persisted in vivo for at least 2 months after infusion (28). The safety profile of TCR-transferred T cells specific for MART-1 or gp100 was similar to that of TILs, with on-target off-tumor toxicities toward skin and eye melanocytes (107). Thanks to these seminal results, the TCR transfer clinical application was widened to other relevant targets, such as the New York esophageal squamous cell carcinoma (NY-ESO)-1, expressed in Melanoma and Synovial Sarcoma (30, 43) and the carcinoembryonic antigen (CEA), expressed in colorectal cancer (31).
The broader use of TCR transfer, however, underlined some of the limitations of this new technology. Firstly, endogenous and exogenous TCRs competed for assembly with the CD3 subunits (108), thus resulting in suboptimal surface expression of the transferred receptor. Secondly, α and β chains from exogenous and endogenous TCRs could mis-pair, further diluting the expression of the correctly paired tumor-specific receptor and introducing new specificities, potentially leading to unwanted toxic reactivities (109). TCR mispairing has been described in vitro by using human cells (109) and was associated to immune-mediated toxicities in murine models (110). So far, no events potentially associated to TCR mispairings have been reported in clinical trials. Nonetheless, to address this safety issue and to increase the expression level of the exogenous TCR, several strategies have been proposed: (i) the replacement of the human TCR constant region, essential for pairing, with a murine-derived sequence (111), (ii) the introduction of cysteine residues to stabilize proper pairing of the TCR chains via disulphide bonds (112, 113), (iii) the generation of a human TCR incorporating the CD3ζ chain (114), (iv) the swapping of TCR constant domains between the α and β chains (115), and (v) the incorporation in the vector cassette of small interfering RNA sequences able to reduce the expression of the endogenous TCR genes (116). To overcome the limitations of TCR gene transfer, nascent genome editing technology has been exploited to develop the TCR gene editing approach (117).
Genome Editing in the Service of ACT
The use of artificially modified nucleases enables the disruption of the genes encoding α and β chains of the endogenous TCR, thus completely and permanently avoiding the risk of TCR mispairing and the mutual dilution effect resulting from the expression of four TCR chains in a single cell. Artificial nucleases bind DNA in selected genomic regions, in which they mediate a DNA double-stranded break (DSB), either repaired by the high-fidelity homologous direct repair (HDR) system or by the mutagenic non-homologous end joining repair machinery (NHEJ). HDR uses a DNA template, usually the sister allele, to correct the break and restore gene function, while NHEJ introduces or erases a variable number of nucleotides upon repair, with the chance of creating premature stop codons and frameshift mutations. Both repair mechanisms can be exploited for gene therapy purposes, with different aims: HDR is suitable for gene correction when an exogenous donor DNA template is delivered with the nuclease (118), while NHEJ is preferred if a gene has to be disrupted (119).
The zinc fingers nucleases (ZFNs), among the first efficient gene editing tools developed, are large multimeric molecules, each monomer targeting a 3-4 DNA base pair sequence, linked to the FokI endonuclease (120). While the multimers confer ZFNs specificity, that can be increased even by elongating the length of the multimers, the endonuclease mediates DNA cleavage. This gene editing tool supported the first genome editing clinical applications (121) and the first TCR gene editing approach. In fact, ZFN-mediated disruption of the endogenous TCR has been combined with LV gene transfer to efficiently generate WT1-specific TCR-edited T cells that outperformed TCR gene transferred T cells in safety, specificity, and efficacy in vitro and in vivo (117). Despite these encouraging results, the first protocol reported required 40 days of manufacturing to be completed and multiple manipulation steps. To improve feasibility, a single-editing strategy, based on the sole disruption of the TCR α chain gene was proposed resulting in optimal expression of a NY-ESO-1-specific TCR and efficient tumor rejection in animal models, in the absence of adverse events (122).
An alternative to the ZFNs system is represented by transcription activator-like effector nucleases (TALENs), small (33–35 amino acids) transcription factors fused with an endonuclease domain (123). TALENs specificity is modified by mutating the two hyper-variable residues that bind the DNA helix. The nucleotide sequence recognized by TALENs is fairly short, increasing the likelihood of off-target binding sites throughout the genome and potentially leading to unwanted DNA breaks. To overcome this limitation, the DNA binding regions can be elongated by multiplying the hyper-variable residues, hence increasing TALENs specificity at the expense of a more complicated protein design. In the ACT context, TALENs have been proficiently used to disrupt endogenous TCR genes in preclinical models and in clinical trials (124, 125).
Meganucleases represent alternative genome editing tools originating from naturally occurring endonucleases that directly bind DNA (126). Meganucleases present some advantages, such as the generation of a 3′overhang at the cleavage site that favors HDR when compared with 5′overhang, and their overall small size, suitable for several delivery methods (127). Still, the difficulty in separating the endonuclease cleavage domains from the DNA binding site limits the number of DNA sequences that can be targeted. To circumvent this obstacle, chimeric proteins have been generated by fusing meganucleases with ZFNs and/or TALENs DNA binding domains, at the expense of increased manufacturing complexity (128, 129).
The introduction of the CRISPR/Cas9 nucleases, bacterial proteins adapted to excise phage DNA fragments (130), completely revolutionized the genome editing field. While ZFNs and TALENs recognize the target DNA sequence via protein-DNA interaction, the CRISPR/Cas9 system relies on a short RNA sequence (single guide RNA, sgRNA). The RNA interacts with the Cas9, conferring the binding specificity and guiding the nuclease activity (131). The CRISPR/Cas9 platform is highly efficient and versatile, since the specific DNA binding is entirely mediated by the sgRNA, short enough to be easily synthetized in vitro but long enough to ensure high specificity. Compared to the previously developed nucleases, the CRISPR/Cas9 system provides three major advantages: (i) rapid and relatively inexpensive manufacturing, (ii) the possibility of multiplex genome engineering obtained by simultaneously targeting several genes, and (iii) compliance with several delivery systems adapted to different cell types (132).
Multiplex genome engineering is a remarkable feature of CRISPR/Cas9, not easily achieved with other nucleases. The possibility of disrupting genes in a single step streamlined different editing procedures (133) and had a direct impact on ACT manufacturing processes, where the synchronous disruption of the α and β TCR chains (42, 134) can sensibly decrease the in vitro manipulation time. In addition, a template strand can be delivered together with the CRISPR/Cas9 system, allowing the integration of the genetic material exactly at the cleavage site (135).
Different tools can be employed to deliver CRISPR/Cas9 complexes into cells. Plasmid delivery has been used (136), but with suboptimal efficiency and with an increased risk of plasmid integration in the host genome. Furthermore, the expression of the Cas9 protein is retained for a fairly long amount of time, increasing the likelihood of adverse immune responses or off-target gene editing. An alternative approach depends on delivering the sgRNA together with the in vitro transcribed Cas9 mRNA (137), ensuring transient Cas9 expression but posing the risk of decreased cleavage efficiency. Lastly, the native Cas9 protein can be pre-assembled in vitro with sgRNA in a ribonucleoprotein complex and then electroporated into the target cells (138). This transfer method overcomes the need for transcription/translation and the risk of intracellular degradation of the free sgRNA (139), thus improving safety and reducing off-target mutagenesis risks.
As for ZFNs (140–142) and TALENs (143, 144), a side effect of the CRISPR/Cas9 system is the risk of editing off-target genes. The nuclease activity can potentially cause DNA strand breaks in other genomic regions, knocking-down unwanted genes or promoting genome translocations (145, 146). Off-target editing can also affect the RNA transcriptome, with toxic consequences for the cell (147). Three methods have been employed to minimize off-targets while increasing on-target activity when using CRISPR/Cas9: (i) the use of modified sgRNAs with higher specificity for the target site, (ii) the titration of the sgRNA and Cas9 ratio (148), and (iii) the introduction of a single point mutation in the Cas9 (149). Furthermore, additional enzymes have been incorporated in the system to mediate base editing without affecting the transcriptome (150).
Several techniques have recently been optimized to map off-target cleavage sites. Mutation detection assays using T7 endonuclease followed by deep sequencing of the resulting amplicons have been initially used, but their sensitivity is limited, especially when dealing with large deletions (151). The tendency of integrase-defective lentiviral vectors to incorporate into DSBs can be exploited to barcode regions of Cas9 activity (152). BLESS [direct in situ Breaks Labeling, Enrichment on Streptavidin and next-generation Sequencing (153)] can map double-strand breaks by using biotinylated linkers that are incorporated at the DSB site; the biotinylated DNA regions are then purified and the captured DNA fragments sequenced. One of the most sensitive off-target detection assay is GUIDE-seq (genome-wide, unbiased identification of DSBs enabled by sequencing), where short phosphorylated double-stranded oligo-deoxynucleotides are incorporated into DSBs to detect Cas9 cleavage sites (154). More rarely, nucleases can cause chromosomal translocation that can be detected using high-throughput, genome-wide translocation sequencing (HTGTS) methods (155), chromatin immunoprecipitation sequencing (ChIP-seq) (156), and digenome-seq or the recently proposed CIRCLE-Seq (157).
All described genome editing technologies have been employed for the modification of either hematopoietic stem cells or T cells. As summarized in Figure 3, genome editing has been exploited with several purposes in the ACT field: to completely redirect T cell specificity (42, 117, 124, 158–160), to avoid the risk of GvHD or fratricide effects mediated by CAR-T cells (161–165), to make adoptively transferred T cells resistant to the immunosuppressive environment (42, 166–173) or to lymphodepleting drugs (174, 175). The high efficiency of gene editing and the overall flexibility of the CRISPR/Cas9 system makes it the most relevant tool to precisely and rapidly edit high numbers of T cells to be used in ACT. Nonetheless, issues remain to be addressed regarding the manufacturing, the delivery, and the broad accessibility of genome editing products to patients (176).
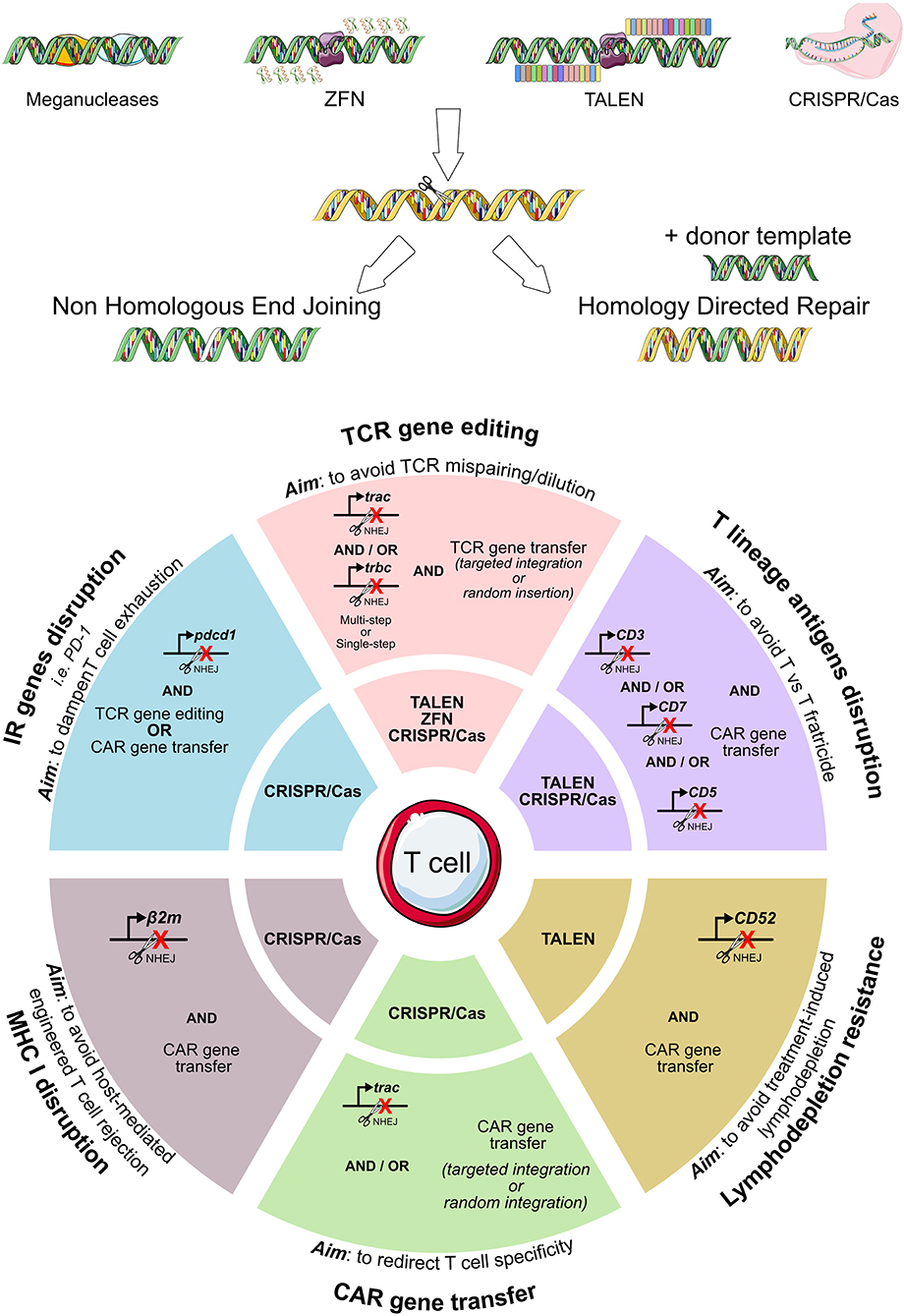
Figure 3. Genome editing exploitation for adoptive T cell therapy. To eliminate the expression of T cell genes, meganucleases, transcription activator-like effector nucleases (TALENs), and zinc-finger nucleases (ZFNs), the CRISPR/Cas9 system can be employed. A summary of the genes edited in the context of adoptive T cell therapy (TCR- or CAR-T cell immunotherapies) is reported, together with the specific nuclease system used. CAR, chimeric antigen receptor; TCR, T cell receptor.
Antigen Identification and TCR Gene Hunting
Different Classes of Target Antigens
One of the major questions in today's adoptive T cell gene therapy is the choice of the target antigen. Theoretically, the ideal candidate should be (i) expressed on tumor cells and not on healthy tissues (to avoid toxicities), (ii) expressed on cancer stem cells (to promote tumor eradication), (iii) associated with the oncogenic process (to reduce the risks of tumor immune evasion), (iv) able to elicit an immune response, and (v) efficiently processed and presented in the context of a common HLA allele (177). Unlike CAR-T cells, TCR-redirected T cells can target antigens independently of their intracellular localization, as soon as they are processed and presented by HLA molecules. For the majority of cancer types, the ideal antigen is yet to be identified, and the search is proving more difficult than expected, particularly for those tumors with undefined clonal evolution and not fully understood in terms of molecular pathogenesis.
The two major classes of antigens in the context of ACT are tumor-associated antigens (TAAs) and neoantigens. TAAs are epitopes originated from endogenous wild-type proteins whose expression is increased in tumors and limited in magnitude or in spatial expression in healthy tissues; neoantigens are instead epitopes derived from somatic DNA alterations.
Different TAAs have been investigated for their potential therapeutic relevance (178): cancer/testis antigens such as melanoma-associated antigen (MAGE)-A3 (179, 180), MAGE-A4 (35, 181), and New York esophageal squamous cell carcinoma (NY-ESO)-1 (182); oncogenes/oncosuppressors such as WT1 (50) and p53 (183); tissue-restricted/differentiation antigens such as MART-1 (28, 34), gp100 (29), or CEA (31). The toxicity and safety profile of T cell therapies targeting TAAs seem to be heterogeneous, depending on the chosen epitope. Several TAA-specific TCRs showed important side effects: cardiovascular and neurological toxicities [with MAGE-3 specific T cells (32, 33)], undesired recognition of melanocytes (with MART-1 specific T cells) (28), and severe transient colitis (with CEA specific T cells) (31). TCR targeting NY-ESO-1, instead, conveyed no toxicities but limited clinical response in a small cohort of patients affected by melanoma and synovial sarcoma (30) and of sarcomas/nerve sheet tumors (41). Its application appeared promising in various pre-clinical tumor models, both in terms of efficacy and safety [bladder (184), ovarian (185), esophageal (186) and prostate cancers (187), multiple myeloma (188), medulloblastoma (189), non-small cell lung carcinoma (190) mesenchymal tumors (191), and breast cancer (192)] but clinical studies are needed to validate these results. The expected good toxicity profile may reside in the limited NY-ESO-1 expression in healthy tissues, essentially restricted to the gonads, an immune-privileged site.
Focusing on neoantigens appears to be another efficient choice for cancer immunotherapy (193). Neoantigens represent the main target of autologous T cell responses in patients treated with TILs (194). Since neoantigens peptides are not presented to thymocytes, T cells specific for those epitopes are not deleted by central tolerance mechanisms, thus the chance of retrieving high affinity TCRs is enhanced. However, the clinical exploitation of neoantigens in TCR-mediated ACT is hindered by their intrinsic qualities: (i) neoantigen-forming mutations tend to differ among patients, making difficult the development of a widely applicable immunotherapeutic product, and (ii) neoantigen expression might be heterogeneous across the tumor tissue. This is particularly true for passenger mutations, random alterations caused by genome instability and not homogeneously spread in the tumor mass (45). In addition, the mutational rate is highly variable in different tumors. In fact, the likelihood of identifying neoantigens from tumors with a low mutational load is poor and their relevance as therapeutic targets is limited (195). Despite these limitations, initial reports highlighting the occurrence of immunogenic neoantigens widely shared in tumor cells and among patients are emerging (196). Additionally, thanks to their broad expression in cancer cells and to their involvement in oncogenesis, founder mutations may be considered promising ACT candidate targets (197). However, epitopes arising from founder mutations may be poorly immunogenic or differ among patients, thus reducing their appeal.
Overall, the interest in neoantigens has increased in recent years, and more than 100 clinical trials exploiting these candidates are currently in progress. However, the ongoing trials largely rely on in vivo peptide vaccination rather than on engineered T cell infusions, further underlining the difficulties in validating a proper target for adoptive T cell therapy (198).
A third possible choice of targets is represented by Minor Histocompatibility antigens (MiHA), peptides derived by polymorphic intracellular proteins, potentially overexpressed by tumor cells. The most known examples are HA-1 and HB-1 (199–201), expressed selectively by hematopoietic cells and by different liquid cancers, thus constituting highly relevant targets (202, 203). Since MiHA derive from coding regions of polymorphic genomic sites, coupling HA-1-directed T cell therapy with Allo-HSCT from a recipient not harboring the same single nucleotide polymorphism (SNP) could provide a valuable therapeutic strategy, able to spare the donor hematopoietic stem cell population while eradicating cancer. So far, HA-1-specific engineered T cells have been tested in vitro and in preclinical models, showing an optimal efficacy and safety profile (204). However, MiHA targeting requires specific combination of SNPs and HLA allele between the donor and recipients, which reduces the broad applicability of this approach.
A fourth source of antigens are proteins encoded by oncogenic viruses. Being involved in tumorigenesis, viral epitopes are shared uniformly by the tumor mass and, due to their nature of foreign molecules, they're potentially highly immunogenic. Hereby, the isolation of high-avidity viral-specific T cells is easier than with other antigen classes, making these targets particularly appealing for ACT. Unfortunately, few tumors have a clear viral pathogenesis, thus this antigen source is currently mostly limited to some HPV and EBV-associated malignancies (205, 206).
The Challenges in Selecting Tumor Antigens and Tumor-Specific TCRs
Apart from choosing the antigen class of interest, tumor screening for immunologically relevant epitopes is a particularly laborious process. The most used and standardized strategy relies on paired whole-exome sequencing or, alternatively, the comparative RNA sequencing analysis of tumor and of healthy tissues to determine differently expressed genes. Once the most promising hits are identified, in silico assessment of HLA presentation and binding (207) is required. In this regard, prediction algorithms are still suboptimal (208), not always accurate and they very often lead to false positive results (209). Thus, extensive validation of the results is always required. The analysis of the tumor ligandome can now be considered a good alternative approach. This technology is based on mass spectrometry typing of all the peptides eluted from the HLAs of a specific tumor type or tumor cell line (210). Here too, the amino-acidic sequences retrieved need to be validated for their relevance in the tumor setting. Despite the great potential of this methodology, some limitations still remain, namely the high number of tumor cells needed, an endpoint is not always attainable when using primary tumor samples, and the low number of epitopes retrieved upon in silico and in vitro screening (211, 212).
Finding the perfect immunogenic epitope is only half of the issue, since the typing of epitope reactive TCRs may prove challenging as well. To isolate a tumor-reactive T cell, a proper source must be selected, the clone of interest must ideally harbor a high avidity TCR and reach a significant level of frequency and purity. These prerequisites are fundamental for the successful retrieval of functional TCR αβ nucleotide sequences.
T cell isolation is particularly challenging especially for poorly immunogenic tumors and for those in which the T cell infiltrate is scant. Recent works showed that, in different cancers, tumor-reactive TILs represent only a minimal fraction of the total T cell subpopulation infiltrating the tumor (47) and that reinvigoration of tumor immunity is associated with recruitment of new T cell clones (213). These observation lead to the hypothesis that selecting T cell clones on the basis of their abundance in the tumor may be misleading. In addition, the poor efficiency of the T cell ex vivo expansion procedures may be taken in consideration as a limiting factor in the retrieval of tumor-specific T cells, especially when studying lymphocytes originated from tumors characterized by a microenvironment known to blunt T cell proliferation (214).
When tumor specific T cells are retrieved, the greatest challenge of the TCR sequencing step is the correct pairing of the cancer-specific α and β TCR chains for each T cell clone. The initial approach for the identification of the TCR repertoire was based on single cell cloning coupled with Sanger sequencing (215, 216), which makes it difficult to estimate the overall repertoire diversity. The field greatly benefited from the introduction of high-throughput sequencing technologies which enabled researchers to profile the diversity of millions of TCR molecules in the analyzed samples. With this approach a complete overview of the TCR sequences constituting the repertoire of the sample is obtained (217–219). However, to successfully characterize and select the TCR αβ pair of interest among the numerous sequences retrieved, the specimen needs to be enriched in anti-tumor specificities in order to obtain an oligoclonal population. To this aim sequencing can be preceded by enriching steps, such as co-culturing TILs with tumor cells harvested ex vivo or performing serial stimulations with professional antigen-presenting cells pulsed with the peptide (or peptide library) of interest. In the latter case, an entire protein sequence can be screened by epitope scanning (220).
An interesting approach that avoids the ex vivo enrichment step is PAIRseq: the sample is split into parallel PCR runs, each one tagged with a specific barcode, and then the results are deconvoluted to identify proper TCR pairs (221). An evolution of this laborious setting is perhaps single cell RNA sequencing, where the use of a cell-specific oligo-DNA barcode allows researchers to retrieve TCRs at single cell resolution (222). The typical output of these systems is generally hundreds or thousands of TCR pairs, questioning whether or not faster sequencing translates to a more laborious validation phase. Alternatively, recent advances in the proteogenomic field may speed up the selection of tumor-specific TCRs. In fact, it is now possible to combine single-cell resolution TCR sequencing with barcoded multimers loaded with a specific HLA molecule and with a selected tumor epitope (222). This approach is extremely helpful in characterizing, in a single step, both the tumor specific TCR sequence and its epitope specificity. This new technology may greatly speed up the isolation of TCRs directly from human samples, avoiding any enriching steps and jumping directly to the functional validation of newly retrieved TCR sequences in vitro.
Another point of discussion is the choice of the ideal specimen to be used for the isolation of tumor-reactive T cell clones. Hunting for tumor-specific T cell receptors directly from the tumor site in patients has historically been the most straightforward choice (17) since anti-tumor reactivities can be intuitively more abundant in the tumor mass. However, the development of a tumor implies an escape from immune surveillance, suggesting either that the infiltrating T cells present at the tumor site were not efficient enough to eradicate the disease, thus questioning their use, or that these highly specific cells were blunted in their activity by the tumor microenvironment (223). In the latter case, the anti-tumor efficacy of the tumor-specific T lymphocytes assessed by in vitro functional assays upon isolation may not be informative, but the TCR is worth isolating and employing in TCR transfer approaches. Recent reports (224, 225) also demonstrated that the exhaustion signature could be exploited, defining a T cell subset enriched with neoantigen-specific T cells.
Since tumor-reactive T cells also circulate throughout the body, patients' peripheral blood and lymph nodes from tumor patients are suitable sites to harvest these cells, and often the only available sites for tumors that cannot be excised. Tumor-specific T cells have been found in tumor-draining lymph nodes, and efficiently used for ACT (226). Melanoma-specific T cells have been enriched and expanded ex vivo starting from peripheral blood (227). However, at least in some tumor types, the low frequency of circulating tumor-reactive T cell clones (228) might impair their retrieval.
Trading Toxicity With Efficacy
In the process of hunting for new TCR specificities, the aim is to define the TCR sequences most efficiently mediating tumor lytic functions. These highly promising TCRs can be collected and used for off-the-shelf immunotherapeutic approaches readily accessible for each candidate patient. The leading TCRs are the ones with the highest binding affinity and avidity (229) toward the HLA-peptide complex, the fastest association rate and the slowest dissociation speed (230, 231). According to this concept, isolated TCRs were screened for strong and fast killing efficacy and the most suitable ones were further developed. In addition, TCRs were modified in their complementary-determining regions (CDRs) in vitro to artificially increase their affinity for the target, thus overcoming the barrier of thymic selection, that deletes thymocytes harboring autoreactive high avidity TCRs. Several approaches have been recently exploited with the ultimate aim of generating high affinity TCRs: (i) mutations in complementarity determining regions by sequential single amino acid substitutions (232–234), (ii) vaccination of mice and consequent retrieval of peptide-reactive TCRs (235), (iii) murine thymic selection to mutagenize one of the TCR chains (236), (iv) transfer of the entire human TCR αβ gene loci into mice to educate T cells against human self-antigens (237, 238), and (v) DNA replication error-prone yeast cells to modify the α and β chain variable regions (239). These techniques have been particularly useful when dealing with TAAs, since highly avid T cell clones recognizing these antigens in a specific and efficient manner have been difficult to isolate.
The advantages of TCR affinity enhancement have been demonstrated in vitro (240–242) and in clinical trials (30, 36, 37, 39). The opposite side of the coin, though, is the risk of enabling engineered T cells to respond to tissues displaying low antigen expression, fostering on- or off-target off-tumor toxicities (243). A clinical trial with an artificially enhanced TCR directed against MAGE-A3 proved highly efficient in eradicating tumor cells but was also endowed with a remarkable off-target off-tumor cardiac toxicity (32, 244), leading to the suspension of the trial. On the same line, an artificially-enhanced TCR directed against CEA was associated with the occurrence of on-target off-tumor reactions and strong systemic inflammation, underlining the limitation of procedures aimed at enhancing TCR avidity when targeting TAAs (31). As a matter of fact, it proved very challenging to predict any possible cross-reactivity of engineered T cells against human tissues. The most commonly used techniques, in vitro testing and epitope alanine scanning, have been further refined in recent years (241, 244) and extended to scan all the possible amino acid substitutions in the target epitope (245). Nevertheless, concerns about affinity enhancing techniques still persist. In this context it might prove safe to introduce a kill switch in engineered T cells, enabling their ablation if necessary (63, 246). Otherwise, the conditioning regimen prior to T cell infusion can be modulated, reducing therapy-induced tissue damage and antigen spreading, two phenomena potentially fostering off-target reactions.
In addition to the adverse events observed in clinical trials, the modification of TCR affinity may also convey excessive activation signals to T cells, that could lead to hypo-functionality and/or premature T cell death. An extensive and continuous activation is indeed detrimental for T cell function (247–249). Reports in the context of TCR engineering are still scant (40) but new insights from the field of CAR-T cell therapy have highlighted this issue (250). Since TCRs is even more sensitive to antigen density variations than CARs (251), it's reasonable to suppose that this mechanism could be relevant in the context of TCR engineering. The picture might even be more complex with TCRs, since costimulatory signals are more tightly involved in the immunological synapse (252) than in CAR-T cells, a feature that provides more flexibility but that requires greater attention to signal tuning.
Persistence of Adoptively Transferred T Cells and Clinical Responses
Nowadays, it's still unclear which variables impact long-term persistence in adoptively transferred T cells the most. This scientific question is particularly relevant because T cell persistence is a fundamental requisite for durable immunosurveillance. Whether immunosurveillance is required for the maintenance of clinical remission is still a matter of debate. However, reports indicate that the sustained and prolonged in vivo expansion of engineered T cells correlates with relapse-free survival and tumor control (39, 40). Several measures have been implemented to foster ACT persistence, including the use of preconditioning regimens and the choice of manufacturing protocols able to enrich memory cells. The use of a lymphodepleting conditioning regimen prior to ACT inhibits host immune cells, including regulatory T cells (Tregs), and favors the accumulation of homeostatic cytokines (253), critical in sustaining engineered T cell engraftment and expansion (254, 255).
The administration of low-doses IL-2 can also sustain adoptively transferred T cell proliferation in vivo (17, 19) and has been included in several ACT protocols. The use of IL-2, however, conveys the risk of toxic reactions related to the activation of bystander host cells. Furthermore, prolonged administration of this cytokine was shown to preferentially expand Tregs (256).
The cellular composition, in terms of subsets and differentiation of the therapeutic product, has a direct impact on efficacy. Long telomeres (257), CD27, and CD28 co-expression (258) on TILs were associated to clinical responses in initial ACT trials. With engineered T cell products, the co-infusion of CD4 and CD8 cells fosters T cell persistence (8). In some ACT applications, manufacturing includes a selection step to enrich the product in CD8 central memory (TCM) lymphocytes (259). The polyfunctionality of adoptively transferred engineered T cells correlated with clinical responses in several clinical trials, targeting NY-ESO-1 (36), MART-1 (29), and WT1 (38). The relevance of the intrinsic qualities of the infused T cells was further highlighted by the observation that even low numbers (105) of highly fit engineered T cells were sufficient to mediate anti-tumor responses (260, 261). Based on these observations and with the final aim of improving the fitness of the infused T cell products, different T cell expansion protocols have been developed and compared. T cell activation with phytohaemagglutinin (PHA) was shown to promote T cell expansion, but also T cell terminal differentiation (262), whereas stimulation with an anti-CD3 monoclonal antibody coupled with high doses of IL-2 reduced the TCR repertoire diversity and enhanced apoptosis (263). The combination of TCR triggering with co-stimulation, obtained thanks to the use of anti-CD3 and anti-CD28 antibodies, followed by the culture of the T cells in the presence of high-doses of IL-2, improved the fitness of the cellular products (264).
Despite showing potent tumor killing abilities in vitro, effector T cells were paradoxically less effective than early differentiated T cells when transferred in tumor-bearing mice (265). These results can be explained by the progressive model of mature T lymphocyte differentiation (266). Upon antigen encounter naïve T cells differentiate into stem cells memory T cells (TSCM), TCM and subsequently in effector memory and terminally differentiated cells, progressively losing proliferating and persistence ability. Recent studies demonstrated that TSCM, originating directly from naïve T cells (267, 268), are endowed with stem cell-like properties and with the ability to persist for decades in vivo (269, 270). The persistence capacity of TSCM was confirmed in patients treated with genetically engineered T cells, in the context of both malignant (271) and non-malignant (272) diseases. In the attempt to preserve this early-differentiated T cell subset, the in vitro protocol used for TILs expansion was shortened (273) and the anti-CD3/anti-CD28 antibodies were conjugated to cell-size beads (274) or nanomatrixes (275). The cytokine cocktail used to sustain T cell expansion in vitro also plays a major role in determining the fitness of cellular products. The introduction of Interleukin-21 in the culture medium promotes a TCM phenotype (276) while Interleukin-7 and Interleukin-15 supplementation, in the absence of IL-2, expands the TSCM pool (277, 278).
As already mentioned, the intrinsic T cell fitness has an impact on the persistence and thus on the efficacy of the T cells used in ACT. In CAR-T cell therapy trials, CAR-T cells isolated from poor responders expressed genes associated with effector memory differentiation and apoptosis, a glycolytic metabolism, and hypo-functionality. Conversely, efficient anti-tumor activity was associated with an early-memory differentiation signature, expression of CD27, and absence of the exhaustion marker PD-1 (259, 279, 280). Similar observations were reported in TCR-based studies. The transfer of a WT1-specific TCR into Epstein-Barr virus-specific donor CD8 T cells has been exploited to generate functional, memory-like cellular products (281). Using this manufacturing procedure, high levels of engraftment and long-term persistence were observed in humans (40). Furthermore, in an ACT trial with TCR engineered T cells, the extent of cytokine release was associated with anti-tumor activity (107).
Once adoptively transferred, T cells interact with the host immune system. Competition of infused and unmodified T cells for proliferative signal accessibility may decrease cell survival. Furthermore, engineered T cells may be recognized and rejected by the host immune system, thus abrogating ACT efficacy. In the autologous setting, rejection could be due to an immune response against the transgene products, as observed in preclinical models (282). An immune response against the murine-derived CD19 CAR was described before and after cell therapy. In the JULIET study (283), the majority of treated patients showed detectable levels of pre-existing anti-murine CD19-specific antibodies, that further increased upon CAR-T cells infusion. Nonetheless, the kinetics of engraftment was unmodified, and rejection barely detected, questioning the relevance of these markers in predicting CAR-T cells persistence. T cell mediated immune responses against Herpes Simplex Virus-derived Thymidine Kinase (TK) epitopes were described in patients treated with TK-DLI, often leading to the elimination of genetically engineered T cells (284). The immunogenicity of TK could be overcome in the HSCT context by infusing transduced T cells during the immunosuppressive phase that follows transplantation (285). For ACT applications that do not involve HSCT, the minimization of transgene immunogenicity remains a desirable and relevant goal.
Overcoming Barriers to T Cell Homing at Tumor Site
The efficacy of ACT is strictly dependent on the ability of the infused product to infiltrate neoplastic lesions. This is particularly difficult in solid tumors, often characterized by a dense stromal architecture, an abnormal vessel structure, and by alteration of chemo-attractants that impinge T cell homing (286–288). In recent years, different strategies have been developed to counteract these factors and hence, increase the ability of T cells to migrate inside neoplastic lesions, where they can properly exert their anti-tumor activity.
Interfering With Cancer Metabolism and Chemokines to Increase ACT Infiltration
The connection between metabolism and oncogenesis is well-documented. Metabolic reprogramming, a hallmark of cancer, does not only impact on cancer cell survival and proliferation, but also on the immunological microenvironment. The presence of reactive nitrogen species (RNS), produced by several human tumors, can induce nitration of different proteins present in the tumor microenvironment (TME) with consequences on T cell functions (289). As an example, the nitration of the CCL2 chemokine decreases its binding affinity for CCR2, thus reducing T cell recruitment. In mouse models, preconditioning of the tumor microenvironment with small molecules blocking RNS production increased the CCL2-mediated recruitment of adoptively transferred tumor-specific CD8 T cells (290), making it an interesting target for further therapeutic development.
Tumors can alter the fucosylation of T cell surface glycoproteins (291), again impinging T cell homing at tumor sites. The ex vivo glycoprotein fucosylation increases in vivo migration and cytotoxic abilities of tumor-specific T cells in leukemia, breast cancer, and melanoma models (292).
The CXCL12/CXCR4 is an additional relevant axis activated by neoplastic cells and cancer-associated fibroblasts in several human tumors. CXCR4 expression correlates with desmoplasia, metastases formation, and immunosuppression (293–298). In murine models of leukemia, melanoma and ovarian cancer, CXCR4 inhibition, obtained with blocking antibodies or with the CXCR4 antagonist AMD3100, increased the effector to Tregs ratio at the tumor site and reduced tumor growth (298–301). Stemming from these observations, several clinical trials are now exploring the efficacy of CXCR4 blockade in solid tumors (302).
Exploiting Cancer Vasculature to Foster ACT Infiltration
Tumor neo-angiogenesis involves the formation of a disorganized network of irregular and leaky vessels, inefficient in delivering oxygen, drugs, and immune cells to the neoplastic microenvironment. This process is largely orchestrated by the vascular-endothelial growth factor (VEGF) and results in tumor growth promotion and altered inflammation (303). VEGF inhibitors are currently used in the treatment of several cancers (304) owing, in particular, to their ability to increase T cell tumor homing (305–309). By promoting vessel maturation, VEGF inhibitors positively impact on immunotherapy and ACT (310). Vascular-targeting peptides represent additional effective tools for the precise delivery of small molecules capable of inducing tumor vessels normalization and increased T cells infiltration. In mouse models, tumor-necrosis factor-targeted (TFN) delivery to the tumor vasculature by linking the TNF protein with the CNGRCG angiogenic vessel-homing peptide (NGR-TNF fusion protein) enhanced the local production of immunomodulatory cytokines, favoring the extravasation of immune cells and improving ACT therapeutic activity (311). The fusion of the TNF superfamily member LIGHT with a vascular targeting peptide (LIGHT-VTP) (312, 313) and the specific delivery of IFN-γ and TNF-α by the homing peptide TCP-1 (314) enhanced endothelial permeability and T cell infiltration in mouse models. The cyclic peptide iRGD (315) and the targeting of the vascular integrity regulator VE-cadherin by CD5-2, a specific inhibitor, facilitated T cell homing to the TME in tumor-bearing mice treated with ACT (316).
Although only some of these therapies have been tested in association with ACT, their effect in increasing T cell migration into tumors strongly suggests that strategies aimed at normalizing the neoplastic vasculature could potentially increase ACT efficacy.
Enforcing Chemokine Receptor Expression in T Cells
The interaction of specific chemokines and cytokines with their receptors is a key determinant of immune cell migration, and a mismatch between the chemokines secreted by neoplastic or stromal cells and the receptors expressed by T lymphocytes strongly limits T cell homing in tumors (317–320).
In recent years, attempts have been made to correct this mismatch by engineering T cells with receptors for chemokines or cytokines abundant in the TME, showing preliminary encouraging results. In TRAMP mice with metastatic prostate adenocarcinoma expressing high levels of CCL2, the expression of CCR2 in SV40 Tag-specific CD8 lymphocytes increased T cell homing to the tumor (321). In xenograft models, T cell transduction with a RV encoding CX3CR1 enhanced migration toward human cell lines expressing Fractalkine, the CX3CL1 ligand, and inhibited tumor growth (322). In a lymphoma murine model, adoptively transferred CD8 T cells, overexpressing CXCR4, were preferentially recruited by CXCL12 expressing cells in the bone marrow, to promote tumor control (323). Anchoring IL-4 receptors to the membrane of adoptively transferred T cells increased in vivo tumor homing, cytokine secretion, and the killing of melanoma (324). Lentiviral T cell transduction to express CXCR2 enhanced in vivo homing toward human melanoma in xenograft models, by exploiting the high IL-8/CXCL8 secretion levels (325).
Classical chemo-radiotherapy is also able to modulate T cell recruitment at the tumor site, suggesting that its use with ACT can have additive effects. In murine models, chemo-radiotherapy has been shown to increase the local release of CCL5, CXCL9, and CXCL11 in the TME, improving ACT-induced tumor growth control (326). Treatment with doxorubicin in mouse models bearing either murine or human melanoma induced CXCL9 and CXCL10 expression by neoplastic cells and increased the infiltration of adoptively transferred T lymphocytes. The effect was further enhanced if ACT followed a combined treatment with doxorubicin and IL-2 (327). Unfortunately, the involved chemokine receptor pattern was not deeply investigated, leaving undemonstrated a causal relation between the increased chemokine secretion by tumor cells and the increased T cell homing.
Overall, these studies prompt further investigation and possible exploitation of chemokine receptors in the context of ACT. Interestingly, a large analysis of 142 patients enrolled in ACT trials revealed the association of genetically determined alterations in chemokine receptors expression with response to therapy (328), underlining the knowledge gap on the impact of chemokines in ACT.
Generating Genetically Engineered T Resident-Memory Cells
Among TILs, T resident-memory (TRM) cells are permanent tissue-resident T cells able to mount an immune reaction; they've been proposed to be key determinants in the magnitude of anti-tumor immune responses, and their presence in different human cancers correlates with survival (329–336). It is tempting to assume that the induction and/or manipulation of this T cell subset might improve anti-tumor immunity and disease control. Indeed, in murine models, the administration of anti-cancer vaccines through routes that enhance the induction of TRM is associated to tumor growth inhibition and, importantly, provides protection also at distant sites (330, 337–339). In a melanoma mouse model, ACT with T cells lacking RUNX3, a transcription factor essential for TRM development, strongly reduced TILs accumulation and treatment efficacy (340). These results suggest that the association of ACT with strategies directed at promoting tissue residency of the infused cells might be beneficial to improve tumor control.
Surviving the Tumor Microenvironment (TME)
As already demonstrated in several disease contexts, a long-lasting protective memory is a critical requirement for a successful ACT. Despite improvement in manufacturing protocols and fitness of the final cellular products, adoptively transferred T cells have to face a harsh environment while interacting with cancer cells (Figure 4). Different immunosuppressive mechanisms act at the tumor site, with the potential to reduce or even dampen the curative action of ACT. If a T cell encounters the cognate antigen in an immune active environment with plenty of additional co-stimuli, the resultant immune reaction is most likely to be efficient in clearing antigen-bearing cells. However, at the tumor site a variety of signals make T cells chronically exposed to antigenic stimulation in the absence of appropriate co-stimuli. Different TME resident cells and tumor cells are responsible for this, either by directly interacting with T cells or by releasing soluble factors. As a consequence, T cell depletion, anergy, exhaustion, and accumulation of Tregs (341–345) promote tumor immune escape (214).
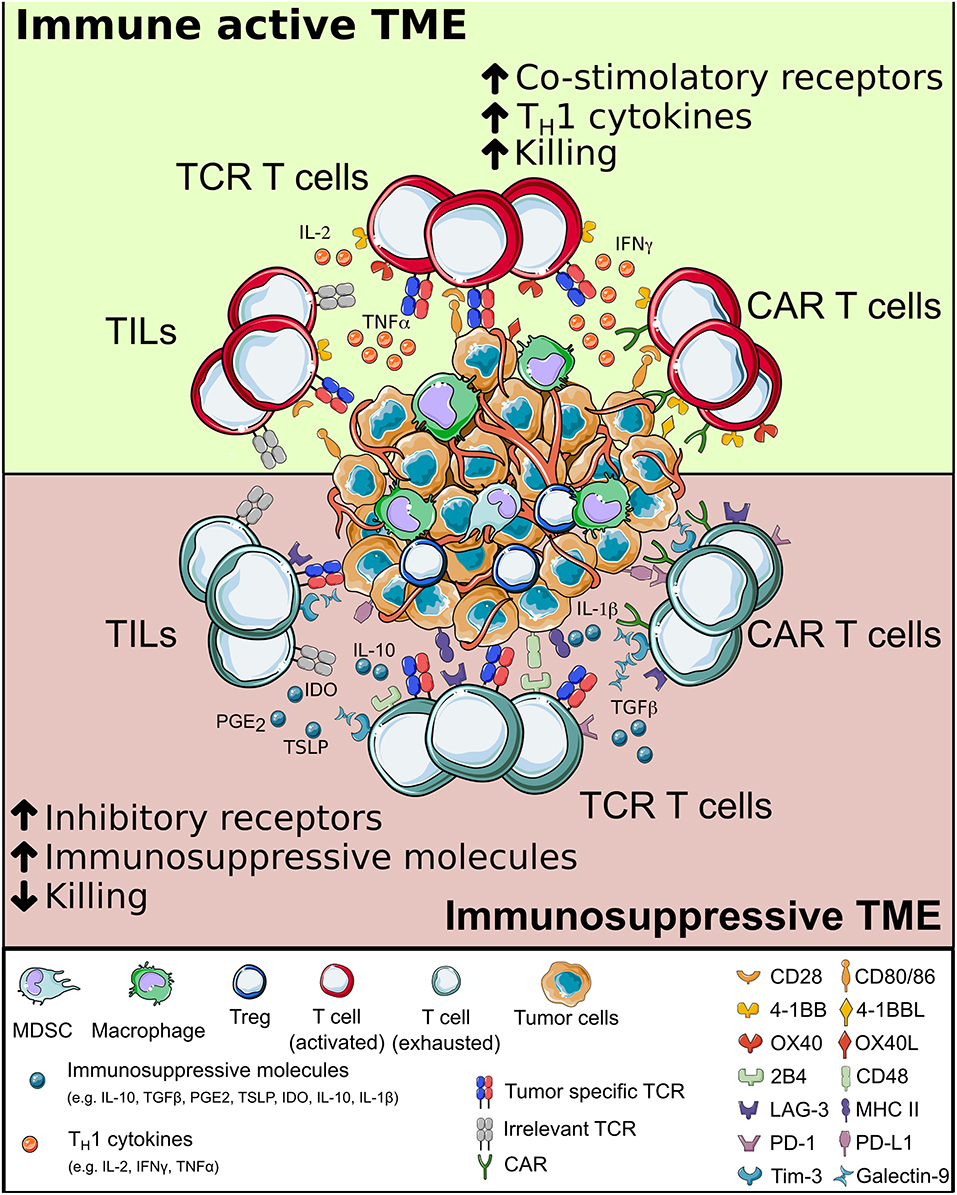
Figure 4. The interplay between T cells and tumor microenvironment. When adoptively transferred T cells (either tumor-infiltrating lymphocytes, Chimeric Antigen Receptor or T cell Receptor-redirected T cells) infiltrate the tumor, they interact with a complex environment, in which a combination of intracellular signals compete. When inflammatory signals dominate, T cells can perform effector functions and potentially eradicate cancer cells; else, they may become exhausted, have limited survival and fail in killing tumor cells. CAR, chimeric antigen receptor; TCR, T cell receptor.
Checkpoint Blockade and ACT
Tumor-specific T cells infused to patients and chronically exposed to tumor antigens often enter a dysfunctional state defined as T cell exhaustion that hampers the anti-tumor response and fosters tumor escape (346).
T cell exhaustion is a process of progressive and hierarchical loss of effector functions (e.g., chemokines production and cytolytic activities), resistance to activation through TCR engagement, metabolic deregulation, and failure to acquire an antigen-independent memory state (347). The sustained co-expression of multiple inhibitory receptors (IRs) such as PD-1, CTLA-4, LAG-3, Tim-3, 2B4, CD39, CD160, BTLA, and TIGIT was identified as a hallmark of exhausted T cells (TEX) (249) in solid and hematological tumors (e.g., melanoma, leukemia, breast, prostate, ovarian, renal, lung, and hepatocellular carcinoma (348–358). Interestingly, the pattern of inhibitory receptors expressed by TEX significantly varies among different tumor types. This indicates that exhaustion mechanisms are differentially shaped by various tumor microenvironments (248) and suggests that ACT approaches need to be tailored according to the specific features of each tumor.
Blocking the interactions between the IRs expressed by tumor-reactive T cells and their cognate ligands leads to the reversal of T cell exhaustion (359, 360). Monoclonal antibodies impeding the PD-1/PD-L1 binding (361) and affecting the CTLA-4 axis (362, 363) are able to restore tumor T cell recognition and tumor regression in a relevant subset of terminally ill patients. Due to their efficacy, the use of monoclonal antibodies in association with ACT could greatly benefit T cell resistance toward the tumor microenvironment. Although still exploratory, this combinatory approach showed encouraging preclinical results in the context of CAR-T cells (364).
The use of an immune checkpoint blockade is associated with significant toxicity, indicating that the indiscriminate blockage of inhibitory receptors on the entire T cell repertoire may be deleterious. Indeed, immune-related adverse events (irAEs) occurred in up to 80% of treated patients, and were life threatening in a significant fraction of cases (365). irAEs mainly occurred because potential autoreactive T cells were unleashed and Tregs functionality was dampened (366). In order to maintain the benefit of inhibitory receptor blockade while reducing toxicities, it may be beneficial to counteract inhibitory axes selectively on tumor-specific T cells. This is currently one of the focuses in the T cell-based immunotherapy field. The increased anti-tumor activity of CAR- and TCR-engineered T cells in which an inhibitory receptor gene has been disrupted was shown in different preclinical models (161, 166, 171, 367). Recently, the results of the first-in-human phase I trial with PD-1 disrupted TCR-edited autologous T cells in patients with refractory tumors have been published, highlighting the feasibility and safety of multiplex gene-editing in tumor-specific T cells (42).
T cell exhaustion can also be exploited for tumor-reactive T cell isolation purposes. In fact, in the context of melanoma, it was shown that PD-1 expressing TILs are enriched in melanoma-specific T cells. These cells, despite being exhausted, could be isolated and their function restored (224). In addition, circulating PD-1 positive T cells showed to be enriched in neoantigen-specificities (368) when compared to the PD-1 negative counterparts.
Counteracting Immune Suppressive Cells Accumulating in TME
Besides T cell exhaustion, the presence of immunosuppressive cell subpopulations or soluble cytokines may also dampen anti-tumor T cells responses. In tumors, monocytes have been described to preferentially polarize into M2-tumor associated macrophages (M2-TAM) (369) or Tie-2-expressing monocytes (TEM) (370). M2-TAM and TEM sustain tumor survival and blunt immune reactions. Myeloid-derived suppressor cells (371) and different components of the stroma have been implicated as well in tumor progression, through different mechanisms: (i) the expression of inhibitory receptor ligands (372), (ii) the production of metabolites or soluble factors [e.g., indoleamine 2,3-dioxygenase (IDO) (373), Interleukin-1β and thymic-stromal lymphopoietin (TSLP) (374, 375), and prostaglandin E2 (376)], or (iii) alteration of pH and oxygen levels (377–380). Among the soluble cytokines, the role of TGF-β gained particular attention. TGF-β is released by neoplastic cells of different origins (381) and its secretion is linked to common cancer genetic mutations (382). At the tumor site, TGF-β acts as a local immunosuppressor, thus reducing the effect of immunotherapy on cancer cell growth. In ACT models, the infusion of CD8 T cells genetically manipulated to resist TGF-β outperformed TGF-β sensitive cells in mediating tumor control (383–387). Interestingly, in a recent clinical study 4 out of 8 patients with Hodgkin lymphoma treated with tumor-specific T cells engineered to express a dominant negative form of TGF-β receptor type II (388) experienced an objective clinical response (389).
To help engineered T cells in counteracting the immunosuppressive microenvironment, different strategies are currently under scrutiny. CAR-T cells have been genetically modified to secrete Interleukin-12 or Interleukin-18 (TRUCK cells) upon CAR engagement (390). A deeper understanding of the expression profile associated to functional CAR-T cells has been now translated in new manipulation processes, involving the overexpression of transcription factors, such as c-Jun (391), or the deletion of inhibitory molecules such as REGNASE-1 (392). These newly proposed strategies could lead to the generation of T cell products endowed with early differentiated phenotypes and enhanced anti-tumor functionality.
Final Remarks
Adoptive T cell therapy represents a unique and innovative therapeutic pillar for cancer treatment. T cells couple the ability to circulate and home at different sites, to sense and respond to the surrounding environment and to persist long-term, thus providing immunosurveillance against residual malignant cells. Each of these characteristics, intrinsic to T cell biology, is however challenged by several immune escape mechanisms active in cancer patients.
Table 1 summarizes the efficacy and toxicity profiles of TCR-redirected T cells reported in clinical trials. Results demonstrate the feasibility of the approach, indicating its therapeutic potential, but also underline the challenges that TCR-based ACT needs to face. Firstly, a suboptimal efficacy of TCR-based studies has currently been observed in patients and no clinical results have been published yet with engineered T cells targeting neoantigens or MiHA. Both evidences underline the difficulty in isolating high affinity tumor-specific TCRs that might be exploited for treating a large number of patients. Secondly, extensive in vitro and in vivo assays are necessary to lower the incidence of adverse events and increase the safety profile of the infused T cell products.
An interesting point of discussion is whether CAR-T cell therapy should be preferred to TCR-based T cell therapy or vice versa. The answer likely lies in the middle and might envisage the alternated or even the combined use of TCR- and CAR-T cells in different clinical settings, according to the antigenic profile of each tumor type. Combinations could exploit the strength of both strategies. Despite CAR-T cells can count on the extensive level of knowledge acquired on cancer cell phenotypes, testable targets remain few. It's possible that this effect underlies the intrinsic limitation of CAR-T cells, able to target surface proteins but at present unable to recognize all the intracellular mutated or overexpressed proteins in cancer cells. An engineered TCR, instead, has the ability to virtually recognize every tumor antigen, independently of intracellular localization, including mutated molecules, intracytoplasmic proteins, and transcription factors. Hence, finding suitable targets for TCR-engineered T cells is theoretically easier. However, the HLA-restriction of TCR-based immunotherapies needs to be taken into consideration.
In terms of efficacy, CAR-T cells showed optimal results in the context of relapsed/refractory ALL. TCR therapy seems promising in liquid tumors, but both CAR and TCR-engineered T cell therapies showed less than satisfactory results in solid tumors other than melanoma when compared to TILs therapy.
In terms of signaling, TCRs are sensitive to much smaller epitope densities than CARs (393) and T cell activation is finely tuned by the affinity and the avidity for the ligand itself. These differences may prove important when dealing with low antigen density and also in promoting immunological memory while avoiding T cell exhaustion. Even if the precise role of the different signals conveyed in the immunological synapse are still not completely understood, TCR signaling might be more rewarding in the ability to balance T cell activation, possibly solving the limitation in T cell persistence and functionality reported in ACT with CARs.
The possibility of genetically engineering T cells by redirecting their specificity toward cancer by employing CARs or TCRs has already produced relevant clinical results in patients affected by selected tumor types. By combining T cell therapy with alternative therapeutic approaches (i.e., checkpoint blockades) and by implementing multiple genetic manipulation (i.e., by genome editing) in T cells, the efficacy of cancer immunotherapy is further increasing, and we might envisage its successful extension to a larger range of tumor types. As the field progresses, several challenges, including manufacturing complexity, regulatory issues, and sustainability will need to be faced, with the ultimate aim of offering this new therapeutic tool to all patients who could benefit.
Author Contributions
AP, BC, ET, ER, FM, and MN did the primary research and wrote the manuscript. AP, BC, and FM edited the figures. FM and ER oversaw the preparation of the manuscript. FC, AB, CB, and ER edited the final draft. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CB received funding from AIRC (Ig 18458) and AIRC 5 per Mille, Rif. 22737, Italian Ministry of Research and University (PRIN 2017WC8499), Italian Ministry of Health (Research project on CAR-T cells for hematological malignancies and solid tumors). This work was partially supported by Ministero della Salute (Ricerca Finalizzata _Grant No. GR-2016-02364847) to ER, by an AIRC Fellowship to BC and by RCR-2019-23669115 grant to AB. Figures were created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License.
References
1. Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell–inflamed versus Non-T cell–inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. (2018) 6:990–1000. doi: 10.1158/2326-6066.CIR-18-0277
2. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. (1990) 75:555–62. doi: 10.1182/blood.V75.3.555.bloodjournal753555
3. Topalian SL, Solomon D, Avis FP, Chang AE, Freerksen DL, Linehan WM, et al. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: a pilot study. J Clin Oncol. (1988) 6:839–53. doi: 10.1200/JCO.1988.6.5.839
4. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
5. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
6. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
7. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
8. Turtle CJ, Hanafi L, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
9. Biondi A, Magnani CF, Tettamanti S, Gaipa G, Biagi E. Redirecting T cells with Chimeric Antigen Receptor (CAR) for the treatment of childhood acute lymphoblastic leukemia. J Autoimmun. (2017) 85:141–52. doi: 10.1016/j.jaut.2017.08.003
10. Thomas ED, Lochte HL, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. (1957) 257:491–6. doi: 10.1056/NEJM195709122571102
11. Thomas E, Buckner C, Banaji M, Clift R, Fefer A, Flournoy N, et al. One hundred patients with acute leukemia treated by chemotherapy, total body irradiation, and allogeneic marrow transplantation. Blood. (1977) 49:511–33. doi: 10.1182/blood.V49.4.511.bloodjournal494511
12. Marmont AM, Horowitz MM, Gale RP, Sobocinski K, Ash RC, Van Bekkum DW, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood. (1991) 78:2120–30. doi: 10.1182/blood.V78.8.2120.bloodjournal7882120
13. Apperley JF, Mauro FR, Goldman JM, Gregory W, Arthur CK, Hows J, et al. Bone marrow transplantation for chronic myeloid leukaemia in first chronic phase: Importance of a graft-versus-leukaemia effect. Br J Haematol. (1988) 69:239–45. doi: 10.1111/j.1365-2141.1988.tb07628.x
14. Kolb HJ, Mittermüller J, Clemm C, Holler E, Ledderose G, Brehm G, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. (1990) 76:2462–5.
15. Simula MP, Marktel S, Fozza C, Kaeda J, Szydlo RM, Nadal E, et al. Response to donor lymphocyte infusions for chronic myeloid leukemia is dose-dependent: the importance of escalating the cell dose to maximize therapeutic efficacy. Leukemia. (2007) 21:943–8. doi: 10.1038/sj.leu.2404641
16. Lechler RI, Lombardi G, Richard Batchelor J, Reinsmoen N, Bach FH. The molecular basis of alloreactivity. Immunol Today. (1990) 11:83–8. doi: 10.1016/0167-5699(90)90033-6
17. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. (1986) 233:1318–21. doi: 10.1126/science.3489291
18. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
19. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. (2008) 26:5233–39. doi: 10.1200/JCO.2008.16.5449
20. Dafni U, Michielin O, Lluesma SM, Tsourti Z, Polydoropoulou V, Karlis D, et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Ann Oncol. (2019) 30:1902–13. doi: 10.1093/annonc/mdz398
21. Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother. (2008) 31:742–51. doi: 10.1097/CJI.0b013e31818403d5
22. Hilders CGJM, Ras L, van Eendenburg JDH, Nooyen Y, Fleuren GJ. Isolation and characterization of tumor-infiltrating lymphocytes from cervical carcinoma. Int J Cancer. (1994) 57:805–13. doi: 10.1002/ijc.2910570608
23. Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. (2015) 33:1543–50. doi: 10.1200/JCO.2014.58.9093
24. Linna T, Moody D, Feeney L, Okarma T, Tso C, Belldegrun A. Tumor microenvironment and immune effector cells: isolation, large scale propagation and characterization of CD8+ tumor infiltrating lymphocytes from renal cell carcinomas. Immunol Ser. (1994) 61:175–8. doi: 10.1201/9781003067245-19
25. Figlin RA, Thompson JA, Bukowski RM, Vogelzang NJ, Novick AC, Lange P, et al. Multicenter, randomized, phase III trial of CD8 + tumor-infiltrating lymphocytes in combination with recombinant interleukin-2 in metastatic renal cell carcinoma. J Clin Oncol. (1999) 17:2521. doi: 10.1200/JCO.1999.17.8.2521
26. Gardini A, Ercolani G, Riccobon A, Ravaioli M, Ridolfi L, Flamini E, et al. Adjuvant, adoptive immunotherapy with tumor infiltrating lymphocytes plus interleukin-2 after radical hepatic resection for colorectal liver metastases: 5-year analysis. J Surg Oncol. (2004) 87:46–52. doi: 10.1002/jso.20066
27. Turcotte S, Gros A, Hogan K, Tran E, Hinrichs CS, Wunderlich JR, et al. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol. (2013) 191:2217–25. doi: 10.4049/jimmunol.1300538
28. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
29. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
30. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
31. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
32. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. (2013) 122:863–72. doi: 10.1182/blood-2013-03-490565
33. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following Anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
34. Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. (2014) 20:2457–65. doi: 10.1158/1078-0432.CCR-13-3017
35. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res. (2015) 21:2268–77. doi: 10.1158/1078-0432.CCR-14-1559
36. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. (2015) 21:914–21. doi: 10.1038/nm.3910
37. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
38. Tawara I, Kageyama S, Miyahara Y, Fujiwara H, Nishida T, Akatsuka Y, et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood. (2017) 130:1985–94. doi: 10.1182/blood-2017-06-791202
39. D'angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1c259T cells in synovial sarcoma. Cancer Discov. (2018) 8:944–57. doi: 10.1158/2159-8290.CD-17-1417
40. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
41. Nowicki TS, Berent-Maoz B, Cheung-Lau G, Huang RR, Wang X, Tsoi J, et al. A pilot trial of the combination of transgenic NY-ESO-1–reactive adoptive cellular therapy with dendritic cell vaccination with or without ipilimumab. Clin Cancer Res. (2019) 25:2096–108. doi: 10.1158/1078-0432.CCR-18-3496
42. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) 7365:eaba7365. doi: 10.1126/science.aba7365
43. Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J Immunother Cancer. (2019) 7:1–14. doi: 10.1186/s40425-019-0762-2
44. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. doi: 10.1126/science.aaa8172
45. Lawrence MS, Stojanov P, Polak P, Kryukov G V, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. (2013) 499:214–8. doi: 10.1038/nature12213
46. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
47. Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. (2019) 25:89–94. doi: 10.1038/s41591-018-0266-5
48. Hammerl D, Massink MPG, Smid M, van Deurzen CHM, Meijers-Heijboer HEJ, Waisfisz Q, et al. Clonality, antigen recognition, and suppression of CD8+ T cells differentially affect prognosis of breast cancer subtypes. Clin Cancer Res. (2020) 26:505–17. doi: 10.1158/1078-0432.CCR-19-0285
49. Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun. (2017) 8:1738. doi: 10.1038/s41467-017-01460-0
50. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. (2013) 5:174ra27. doi: 10.1126/scitranslmed.3004916
51. Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. (2008) 358:2698–703. doi: 10.1056/NEJMoa0800251
52. DembiC Z, Haas W, Weiss S, McCubrey J, Kiefer H, von Boehmer H, et al. Transfer of specificity by murine α and β T-cell receptor genes. Nature. (1986) 320:232–8. doi: 10.1038/320232a0
53. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
54. Flier JS, Underhill LH, Acuto O, Reinherz EL. The human T-cell receptor. N Engl J Med. (1985) 312:1100–11. doi: 10.1056/NEJM198504253121706
55. Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class i and MHC class II antigen presentation. Nat Rev Immunol. (2011) 11:823–36. doi: 10.1038/nri3084
56. Yanagi Y, Yoshikai Y, Leggett K, Clark SP, Aleksander I, Mak TW. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature. (1984) 308:145–9. doi: 10.1038/308145a0
57. Hedrick SM, Nielsen EA, Kavaler J, Cohen DI, Davis MM. Sequence relationships between putative T-cell receptor polypeptides and immunoglobulins. Nature. (1984) 308:153–8. doi: 10.1038/308153a0
58. Douglas JT. Adenoviral vectors for gene therapy. Mol Biotechnol. (2007) 36:71–80. doi: 10.1007/s12033-007-0021-5
59. Hendrickx R, Stichling N, Koelen J, Kuryk L, Lipiec A, Greber UF. Innate immunity to adenovirus. Hum Gene Ther. (2014) 25:265–84. doi: 10.1089/hum.2014.001
60. Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. (2003) 80:148–58. doi: 10.1016/j.ymgme.2003.08.016
61. Ferrari G, Rossini S, Giavazzi R, Maggioni D, Nobili N, Soldati M, et al. An in vivo model of somatic cell gene therapy for human severe combined immunodeficiency. Science. (1991) 251:1363–6. doi: 10.1126/science.1848369
62. Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay J-P, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. (2002) 346:1185–93. doi: 10.1056/NEJMoa012616
63. Bonini C, Ferrari G, Verzelletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. (1997) 276:1719–24. doi: 10.1126/science.276.5319.1719
64. Ciceri F, Bonini C, Stanghellini MTL, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. (2009) 10:489–500. doi: 10.1016/S1470-2045(09)70074-9
65. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
66. Dossett ML, Teague RM, Schmitt TM, Tan X, Cooper LJN, Pinzon C, et al. Adoptive immunotherapy of disseminated leukemia with TCR-transduced, CD8+ T cells expressing a known endogenous TCR. Mol Ther. (2009) 17:742–9. doi: 10.1038/mt.2008.300
67. Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. (1996) 272:263–7. doi: 10.1126/science.272.5259.263
68. Yu SF, von Ruden T, Kantoff PW, Garber C, Seiberg M, Rüther U, et al. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc Natl Acad Sci USA. (1986) 83:3194–8. doi: 10.1073/pnas.83.10.3194
69. Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. (1998) 72:8150–7. doi: 10.1128/JVI.72.10.8150-8157.1998
70. Thornhill SI, Schambach A, Howe SJ, Ulaganathan M, Grassman E, Williams D, et al. Self-inactivating gammaretroviral vectors for gene therapy of x-linked severe combined immunodeficiency. Mol Ther. (2008) 16:590–8. doi: 10.1038/sj.mt.6300393
71. Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. (2013) 341:1233158. doi: 10.1126/science.1233158
72. Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-aldrich syndrome. Science. (2013) 341:1233151. doi: 10.1126/science.1233151
73. Kotin RM, Siniscalcot M, Samulskit RJ, Zhu X, Huntert L, Laughlin CA, et al. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci USA. (1990) 87:2211–5. doi: 10.1073/pnas.87.6.2211
74. McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. (2001) 8:1248–54. doi: 10.1038/sj.gt.3301514
75. Koeberl DD, Pinto C, Sun B, Li S, Kozink DM, Benjamin DK, et al. AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. Mol Ther. (2008) 16:665–72. doi: 10.1038/mt.2008.15
76. Wu JQ, Zhao WH, Li Y, Zhu B, Yin KS. Adeno-associated virus mediated gene transfer into lung cancer cells promoting CD40 ligand-based immunotherapy. Virology. (2007) 368:309–16. doi: 10.1016/j.virol.2007.07.006
77. Baum C, Modlich U, Göhring G, Schlegelberger B. Concise review: Managing genotoxicity in the therapeutic modification of stem cells. Stem Cells. (2011) 29:1479–84. doi: 10.1002/stem.716
78. Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. (1997) 15:871–5. doi: 10.1038/nbt0997-871
79. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A Third-generation lentivirus vector with a conditional packaging system. J Virol. (1998) 72:8463–71. doi: 10.1128/JVI.72.11.8463-8471.1998
80. Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. (1998) 72:9873–80. doi: 10.1128/JVI.72.12.9873-9880.1998
81. Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. (2006) 7:703–13. doi: 10.1038/nrg1925
82. Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. (2012) 12:51–8. doi: 10.1038/nrc3179
83. Papapetrou EP, Schambach A. Gene insertion into genomic safe harbors for human gene therapy. Mol Ther. (2016) 24:678–84. doi: 10.1038/mt.2016.38
84. Kotin RM, Linden RM, Berns KI. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. (1992) 11:5071–8. doi: 10.1002/j.1460-2075.1992.tb05614.x
85. Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. (2014) 510:235–40. doi: 10.1038/nature13420
86. Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA βgeo 26 gene trap strain leads to widespread expression of β-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci USA. (1997) 94:3789–94. doi: 10.1073/pnas.94.8.3789
87. Mitchell RS, Beitzel BF, Schroder ARW, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. (2004) 2:e234. doi: 10.1371/journal.pbio.0020234
88. Hua-Van A, Le Rouzic A, Maisonhaute C, Capy P. Abundance, distribution and dynamics of retrotransposable elements and transposons: similarities and differences. Cytogenet Genome Res. (2005) 110:426–40. doi: 10.1159/000084975
89. Ivics ZN, Hackett PB, Plasterk RH, Izsvá Z. Molecular reconstruction of sleeping beauty, a Tc1-like transposon from fish, and its transposition in human cells its original location and promotes its reintegration else- where in the genome (Plasterk, 1996). Autonomous Members of a transposon. Cell. (1997) 91:501–10. doi: 10.1016/S0092-8674(00)80436-5
90. Izsvák Z, Ivics Z. Sleeping Beauty transposition: biology and applications for molecular therapy. Mol Ther. (2004) 9:147–56. doi: 10.1016/j.ymthe.2003.11.009
91. Grabundzija I, Irgang M, Mátés L, Belay E, Matrai J, Gogol-Döring A, et al. Comparative analysis of transposable element vector systems in human cells. Mol Ther. (2010) 18:1200–9. doi: 10.1038/mt.2010.47
92. Wilber A, Frandsen JL, Geurts JL, Largaespada DA, Hackett PB, McIvor RS. RNA as a source of transposase for sleeping beauty-mediated gene insertion and expression in somatic cells and tissues. Mol Ther. (2006) 13:625–30. doi: 10.1016/j.ymthe.2005.10.014
93. Huang X, Guo H, Kang J, Choi S, Zhou TC, Tammana S, et al. Sleeping beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther. (2008) 16:580–9. doi: 10.1038/sj.mt.6300404
94. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase i trials using sleeping beauty to generate CD19-specific CAR-T cells. J Clin Invest. (2016) 126:3363–76. doi: 10.1172/JCI86721
95. Ivics Z. Potent CAR-T cells engineered with Sleeping Beauty transposon vectors display a central memory phenotype. Gene Ther. (2020). doi: 10.1038/s41434-020-0138-8
96. Peng PD, Cohen CJ, Yang S, Hsu C, Jones S, Zhao Y, et al. Efficient nonviral Sleeping Beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther. (2009) 16:1042–9. doi: 10.1038/gt.2009.54
97. Field AC, Vink C, Gabriel R, Al-Subki R, Schmidt M, Goulden N, et al. Comparison of lentiviral and sleeping beauty mediated αβ T cell receptor gene transfer. PLoS One. (2013) 8:e68201. doi: 10.1371/journal.pone.0068201
98. Deniger DC, Pasetto A, Tran E, Parkhurst MR, Cohen CJ, Robbins PF, et al. Stable, nonviral expression of mutated tumor neoantigen-specific T-cell receptors using the sleeping beauty transposon/transposase system. Mol Ther. (2016) 24:1078–89. doi: 10.1038/mt.2016.51
99. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. (2019) 27:710–28. doi: 10.1016/j.ymthe.2019.02.012
100. Youn H, Chung J-K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin Biol Ther. (2015) 15:1337–48. doi: 10.1517/14712598.2015.1057563
101. Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. (2010) 70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880
102. Cole DJ, Weil DP, Shilyansky J, Custer M, Kawakami Y, Rosenberg SA, et al. Characterization of the functional specificity of a cloned T-cell receptor heterodimer recognizing the MART-1 melanoma antigen. Cancer Res. (1995) 55:748–52.
103. Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. (1999) 163:507–13.
104. Hughes MS, Yu YYL, Dudley ME, Zheng Z, Robbins PF, Li Y, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. (2005) 16:457–72. doi: 10.1089/hum.2005.16.457
105. Stanislawski T, Voss RH, Lotz C, Sadovnikova E, Willemsen RA, Kuball J, et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol. (2001) 2:962–70. doi: 10.1038/ni1001-962
106. Orentas RJ, Roskopf SJ, Nolan GP, Nishimura MI. Retroviral transduction of a T cell receptor specific for an Epstein-Barr virus-encoded peptide. Clin Immunol. (2001) 98:220–8. doi: 10.1006/clim.2000.4977
107. Ahmadzadeh M, Johnson L A, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen – specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
108. Heemskerk MHM, Hagedoorn RS, Van Der Hoorn MAWG, Van Der Veken LT, Hoogeboom M, Kester MGD, et al. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood. (2007) 109:235–43. doi: 10.1182/blood-2006-03-013318
109. Van Loenen MM, De Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci USA. (2010) 107:10972–7. doi: 10.1073/pnas.1005802107
110. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, De Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. (2010) 16:565–70. doi: 10.1038/nm.2128
111. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. (2006) 66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450
112. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. (2007) 109:2331–8. doi: 10.1182/blood-2006-05-023069
113. Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. (2007) 67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986
114. Sebestyén Z, Schooten E, Sals T, Zaldivar I, San José E, Alarcón B, et al. Human TCR that incorporate CD3ζ induce highly preferred pairing between TCRα and β chains following gene transfer. J Immunol. (2008) 180:7736–46. doi: 10.4049/jimmunol.180.11.7736
115. Bethune MT, Gee MH, Bunse M, Lee MS, Gschweng EH, Pagadala MS, et al. Domain-swapped t cell receptors improve the safety of TCR gene therapy. Elife. (2016) 5:1–24. doi: 10.7554/eLife.19095
116. Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. (2009) 69:9003–11. doi: 10.1158/0008-5472.CAN-09-1450
117. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. (2012) 18:807–15. doi: 10.1038/nm.2700
118. Song KY, Schwartz F, Maeda N, Smithies O, Kucherlapati R. Accurate modification of a chromosomal plasmid by homologous recombination in human cells. Proc Natl Acad Sci USA. (1987) 84:6820–4. doi: 10.1073/pnas.84.19.6820
119. Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. (2012) 47:497–510. doi: 10.1016/j.molcel.2012.07.029
120. Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA. (1996) 93:1156–60. doi: 10.1073/pnas.93.3.1156
121. Carroll D. Genome engineering with zinc-finger nucleases. Genetics. (2011) 188:773–82. doi: 10.1534/genetics.111.131433
122. Mastaglio S, Genovese P, Magnani Z, Ruggiero E, Landoni E, Camisa B, et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood. (2017) 130:606–18. doi: 10.1182/blood-2016-08-732636
123. Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. (2011) 29:143–50. doi: 10.1038/nbt.1755
124. Berdien B, Mock U, Atanackovic D, Fehse B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. (2014) 21:539–48. doi: 10.1038/gt.2014.26
125. Rasaiyaah J, Georgiadis C, Preece R, Mock U, Qasim W. TCRαβ/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight. (2018) 3:1–13. doi: 10.1172/jci.insight.99442
126. Rosen LE, Morrison HA, Masri S, Brown MJ, Springstubb B, Sussman D, et al. Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucleic Acids Res. (2006) 34:4791–4800. doi: 10.1093/nar/gkl645
127. Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther. (2011) 11:11–27. doi: 10.2174/156652311794520111
128. Kleinstiver BP, Wolfs JM, Kolaczyk T, Roberts AK, Hu SX, Edgell DR. Monomeric site-specific nucleases for genome editing. Proc Natl Acad Sci USA. (2012) 109:8061–6. doi: 10.1073/pnas.1117984109
129. Boissel S, Jarjour J, Astrakhan A, Adey A, Gouble A, Duchateau P, et al. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. (2013) 42:2591–601. doi: 10.1093/nar/gkt1224
130. Ishino Y, Shinagawa H, Makino K, Amemura M, Nakatura A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isoenzyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. (1987) 169:5429–33. doi: 10.1128/JB.169.12.5429-5433.1987
131. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. (2013) 339:823–6. doi: 10.1126/science.1232033
132. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. (2018) 9:1911. doi: 10.1038/s41467-018-04252-2
133. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. (2013) 154:1370–9. doi: 10.1016/j.cell.2013.08.022
134. Stadtmauer EA, Faitg TH, Lowther DE, Badros AZ, Chagin K, Dengel K, et al. Long-term safety and activity of NY-ESO-1 SPEAR T cells after autologous stem cell transplant for myeloma. Blood Adv. (2019) 3:2022–34. doi: 10.1182/bloodadvances.2019000194
135. Bak RO, Porteus MH. CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Rep. (2017) 20:750–6. doi: 10.1016/j.celrep.2017.06.064
136. Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. (2015) 520:186–91. doi: 10.1038/nature14299
137. Hu X, Chang N, Wang X, Zhou F, Zhou X, Zhu X, et al. Heritable gene-targeting with gRNA/Cas9 in rats. Cell Res. (2013) 23:1322–5. doi: 10.1038/cr.2013.141
138. Kim S, Kim D, Cho SW, Kim J-S, Kim J-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. (2014) 24:1012–9. doi: 10.1101/gr.171322.113
139. Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. (2015) 33:985–9. doi: 10.1038/nbt.3290
140. Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. (2003) 300:763. doi: 10.1126/science.1078395
141. Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, Joung JK, et al. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther. (2008) 16:352–8. doi: 10.1038/sj.mt.6300357
142. Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. (2011) 29:816–23. doi: 10.1038/nbt.1948
143. Mussolino C, Morbitzer R, Lütge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. (2011) 39:9283–93. doi: 10.1093/nar/gkr597
144. Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, Joung JK, et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods. (2014) 11:429–35. doi: 10.1038/nmeth.2845
145. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. (2013) 31:822–6. doi: 10.1038/nbt.2623
146. Jacinto F V, Link W, Ferreira BI. CRISPR/Cas9-mediated genome editing: from basic research to translational medicine. J Cell Mol Med. (2020) 24:1–13. doi: 10.1111/jcmm.14916
147. Grünewald J, Zhou R, Garcia SP, Iyer S, Lareau CA, Aryee MJ, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. (2019) 569:433–7. doi: 10.1038/s41586-019-1161-z
148. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. (2018) 36:765–71. doi: 10.1038/nbt.4192
149. Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med. (2018) 24:1216–24. doi: 10.1038/s41591-018-0137-0
150. Grünewald J, Zhou R, Iyer S, Lareau CA, Garcia SP, Aryee MJ, et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat Biotechnol. (2019) 37:1041–8. doi: 10.1038/s41587-019-0236-6
151. Sentmanat MF, Peters ST, Florian CP, Connelly JP, Pruett-Miller SM. A survey of validation strategies for CRISPR-Cas9 editing. Sci Rep. (2018) 8:1–8. doi: 10.1038/s41598-018-19441-8
152. Wang X, Wang Y, Wu X, Wang J, Wang Y, Qiu Z, et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol. (2015) 33:175–9. doi: 10.1038/nbt.3127
153. Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods. (2013) 10:361–5. doi: 10.1038/nmeth.2408
154. Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. (2014) 32:569–76. doi: 10.1038/nbt.2908
155. Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. (2015) 33:179–88. doi: 10.1038/nbt.3101
156. O'Geen H, Henry IM, Bhakta MS, Meckler JF, Segal DJ. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. (2015) 43:3389–404. doi: 10.1093/nar/gkv137
157. Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. (2017) 14:607–14. doi: 10.1038/nmeth.4278
158. Osborn MJ, Webber BR, Knipping F, Lonetree C, Tennis N, DeFeo AP, et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol Ther. (2016) 24:570–81. doi: 10.1038/mt.2015.197
159. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR-T cells. Mol Ther. (2017) 25:949–61. doi: 10.1016/j.ymthe.2017.02.005
160. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
161. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR-T cells resistant to PD1 inhibition. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
162. Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. (2018) 32:1970–83. doi: 10.1038/s41375-018-0065-5
163. Raikar SS, Fleischer LC, Moot R, Fedanov A, Paik NY, Knight KA, et al. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology. (2018) 7:e1407898. doi: 10.1080/2162402X.2017.1407898
164. Georgiadis C, Rasaiyaah J, Gkazi SA, Preece R, Etuk A, Christi A, et al. Universal' fratricide-resistant CAR-T Cells against T cell leukemia generated by coupled & uncoupled deamination mediated base editing. Blood. (2019) 134:3219. doi: 10.1182/blood-2019-130057
165. Gehrke JM, Edwards A, Murray RC, Shaw A, Poh Y-C, Smith S, et al. Highly efficient multiplexed base editing with minimized off-targets for the development of universal CAR-T cells to treat pediatric T-ALL. Blood. (2019) 134:5127. doi: 10.1182/blood-2019-130562
166. Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. (2016) 6:1–14. doi: 10.1038/srep20070
167. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR-T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
168. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:737. doi: 10.1038/s41598-017-00462-8
169. Zhao Z, Shi L, Zhang W, Han J, Zhang S, Fu Z, et al. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget. (2018) 9:5208–15. doi: 10.18632/oncotarget.23730
170. Zhang C, Peng Y, Hublitz P, Zhang H, Dong T. Genetic abrogation of immune checkpoints in antigen-specific cytotoxic T-lymphocyte as a potential alternative to blockade immunotherapy. Sci Rep. (2018) 8:5549. doi: 10.1038/s41598-018-23803-7
171. Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, et al. Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front Pharmacol. (2018) 9:1118. doi: 10.3389/fphar.2018.01118
172. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR-T cell effector functions. Cancer Immunol Immunother. (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
173. Zhang W, Shi L, Zhao Z, Du P, Ye X, Li D, et al. Disruption of CTLA-4 expression on peripheral blood CD8 + T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother Pharmacol. (2019) 83:911–20. doi: 10.1007/s00280-019-03800-x
174. Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies. Cancer Res. (2015) 75:3853–64. doi: 10.1158/0008-5472.CAN-14-3321
175. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR-T cells. Sci Transl Med. (2017) 9:1–10. doi: 10.1126/scitranslmed.aaj2013
176. Porteus MH. A new class of medicines through DNA editing. N Engl J Med. (2019) 380:947–59. doi: 10.1056/NEJMra1800729
177. Van der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, et al. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev. (2002) 188:51–64. doi: 10.1034/j.1600-065X.2002.18806.x
178. Liu CC, Yang H, Zhang R, Zhao JJ, Hao DJ. Tumour-associated antigens and their anti-cancer applications. Eur J Cancer Care. (2017) 26:12446. doi: 10.1111/ecc.12446
179. van der Bruggen P, Bastin J, Gajewski T, Coulie PG, Boël P, De Smet C, et al. A peptide encoded by human gene MAGE-3 and presented by HLA-A2 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE-3. Eur J Immunol. (1994) 24:3038–43. doi: 10.1002/eji.1830241218
180. Yao X, Lu YC, Parker LL, Li YF, El-Gamil M, Black MA, et al. Isolation and characterization of an HLA-DPB1*04:01-restricted MAGE-A3 T-cell receptor for cancer immunotherapy. J Immunother. (2016) 39:191–201. doi: 10.1097/CJI.0000000000000123
181. Zhang Y, Stroobant V, Russo V, Boon T, Van der Bruggen P. A MAGE-A4 peptide presented by HLA-B37 is recognized on human tumors by cytolytic T lymphocytes. Tissue Antigens. (2002) 60:365–71. doi: 10.1034/j.1399-0039.2002.600503.x
182. Valmori D, Dutoit V, Liénard D, Rimoldi D, Pittet MJ, Champagne P, et al. Naturally occurring human lymphocyte antigen-A2 restricted CD8+ T-cell response to the cancer testis antigen NY-ESO-1 in melanoma patients. Cancer Res. (2000) 60:4499–506.
183. Barfoed AM, Petersen TR, Kirkin AF, Thor Straten P, Claesson MH, Zeuthen J. Cytotoxic T-lymphocyte clones, established by stimulation with the HLA- A2 binding p5365-73 wild type peptide loaded on dendritic cells in vitro, specifically recognize and lyse HLA-A2 tumour cells overexpressing the p53 protein. Scand J Immunol. (2000) 51:128–33. doi: 10.1046/j.1365-3083.2000.00668.x
184. Sharma P, Gnjatic S, Jungbluth AA, Williamson B, Herr H, Stockert E, et al. Frequency of NY-ESO-1 and LAGE-1 expression in bladder cancer and evidence of a new NY-ESO-1 T-cell epitope in a patient with bladder cancer. Cancer Immun Arch. (2003) 3:19.
185. Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. (2003) 63:6076–83.
186. Akcakanat A, Kanda T, Koyama Y, Watanabe M, Kimura E, Yoshida Y, et al. NY-ESO-1 expression and its serum immunoreactivity in esophageal cancer. Cancer Chemother Pharmacol. (2004) 54:95–100. doi: 10.1007/s00280-004-0768-3
187. Fosså A, Berner A, Fosså SD, Hernes E, Gaudernack G, Smeland EB. NY-ESO-I protein expression and humoral immune responses in prostate cancer. Prostate. (2004) 59:440–7. doi: 10.1002/pros.20025
188. Van Rhee F, Szmania SM, Zhan F, Gupta SK, Pomtree M, Lin P, et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood. (2005) 105:3939–44. doi: 10.1182/blood-2004-09-3707
189. Oba-Shinjo SM, Caballero OL, Jungbluth AA, Rosemberg S, Old LJ, Simpson AJG, et al. Cancer-testis (CT) antigen expression in medulloblastoma. Cancer Immun. (2008) 8:1–7.
190. Kim SH, Lee S, Lee CH, Lee MK, Kim YD, Shin DH, et al. Expression of cancer-testis antigens MAGE-A3/6 and NY-ESO-1 in non-small-cell lung carcinomas and their relationship with immune cell infiltration. Lung. (2009) 187:401–11. doi: 10.1007/s00408-009-9181-3
191. Lai JP, Rosenberg AZ, Miettinen MM, Lee CCR. NY-ESO-1 expression in sarcomas a diagnostic marker and immunotherapy target. Oncoimmunology. (2012) 1:1409–10. doi: 10.4161/onci.21059
192. Ademuyiwa FO, Bshara W, Attwood K, Morrison C, Edge SB, Ambrosone CB, et al. NY-ESO-1 cancer testis antigen demonstrates high immunogenicity in triple negative breast cancer. PLoS ONE. (2012) 7:e38783. doi: 10.1371/annotation/5cdf6105-2a52-497a-86b3-db8f4a4e439c
193. Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. (2013) 19:747–52. doi: 10.1038/nm.3161
194. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. (2015) 125:3981–91. doi: 10.1172/JCI82416
195. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
196. Perumal D, Imai N, Lagana A, Finnigan J, Melnekoff D, Leshchenko VV, et al. Mutation-derived Neoantigen-specific T-cell responses in multiple myeloma. Clin Cancer Res. (2020) 26:450–64. doi: 10.1158/1078-0432.CCR-19-2309
197. Kessler JH, Bres-Vloemans SA, van Veelen PA, de Ru A, Huijbers IJG, Camps M, et al. BCR-ABL fusion regions as a source of multiple leukemia-specific CD8+ T-cell epitopes. Leukemia. (2006) 20:1738–50. doi: 10.1038/sj.leu.2404354
198. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217–21. doi: 10.1038/nature22991
199. de Bueger M, Bakker A, Van Rood JJ, Van der Woude F, Goulmy E. Tissue distribution of human minor histocompatibility antigens. Ubiquitous versus restricted tissue distribution indicates heterogeneity among human cytotoxic T lymphocyte-defined non-MHC antigens. J Immunol. (1992) 149:1788–94.
200. Goulmy E. Human minor histocompatibility antigens. Curr Opin Immunol. (1996) 8:75–81. doi: 10.1016/S0952-7915(96)80108-7
201. Den Haan JMM, Meadows LM, Wang W, Pool J, Blokland E, Bishop TL, et al. The minor histocompatibility antigen HA-1: a diallelic gene with a single amino acid polymorphism. Science. (1998) 279:1054–57. doi: 10.1126/science.279.5353.1054
202. Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. (2011) 89:396–407. doi: 10.1038/icb.2010.124
203. Meij P, Jedema I, van der Hoorn MAWG, Bongaerts R, Cox L, Wafelman AR, et al. Generation and administration of HA-1-specific T-cell lines for the treatment of patients with relapsed leukemia after allogeneic stem cell transplantation: a pilot study. Haematologica. (2012) 97:1205–8. doi: 10.3324/haematol.2011.053371
204. Dossa RG, Cunningham T, Sommermeyer D, Medina-Rodriguez I, Biernacki MA, Foster K, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood. (2018) 131:108–20. doi: 10.1182/blood-2017-07-791608
205. Roskrow MA, Suzuki N, Gan YJ, Sixbey JW, Ng CYC, Kimbrough S, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of patients with EBV-positive relapsed Hodgkin's disease. Blood. (1998) 91:2925–34. doi: 10.1182/blood.V91.8.2925.2925_2925_2934
206. Draper LM, Kwong MLM, Gros A, Stevanović S, Tran E, Kerkar S, et al. Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin Cancer Res. (2015) 21:4431–9. doi: 10.1158/1078-0432.CCR-14-3341
207. Rammensee HG, Bachmann J, Emmerich NPN, Bachor OA, Stevanović S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics. (1999) 50:213–9. doi: 10.1007/s002510050595
208. Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE. (2007) 2:1–13. doi: 10.4016/4651.01
209. Kalaora S, Barnea E, Merhavi-Shoham E, Qutob N, Teer JK, Shimony N, et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget. (2016) 7:5110–7. doi: 10.18632/oncotarget.6960
210. Singh-Jasuja H, Emmerich NPN, Rammensee HG. The Tübingen approach: Identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol Immunother. (2004) 53:187–95. doi: 10.1007/s00262-003-0480-x
211. Haen SP, Rammensee HG. The repertoire of human tumor-associated epitopes - identification and selection of antigens and their application in clinical trials. Curr Opin Immunol. (2013) 25:277–83. doi: 10.1016/j.coi.2013.03.007
212. Bilich T, Nelde A, Bichmann L, Roerden M, Salih HR, Kowalewski DJ, et al. The HLA ligandome landscape of chronic myeloid leukemia delineates novel T-cell epitopes for immunotherapy. Blood. (2019) 133:550–65. doi: 10.1182/blood-2018-07-866830
213. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. (2019) 25:1251–9. doi: 10.1038/s41591-019-0522-3
214. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
215. Sant'Angelo DB, Lucas B, Waterbury PG, Cohen B, Brabb T, Goverman J, et al. A molecular map of T cell development. Immunity. (1998) 9:179–86. doi: 10.1016/S1074-7613(00)80600-7
216. Correia-Neves M, Waltzinger C, Mathis D, Benoist C. The shaping of the T cell repertoire. Immunity. (2001) 14:21–32. doi: 10.1016/S1074-7613(01)00086-3
217. Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor β-chain diversity in αβ T cells. Blood. (2009) 114:4099–107. doi: 10.1182/blood-2009-04-217604
218. Linnemann C, Heemskerk B, Kvistborg P, Kluin RJC, Bolotin DA, Chen X, et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med. (2013) 19:1534–41. doi: 10.1038/nm.3359
219. Ruggiero E, Nicolay JP, Fronza R, Arens A, Paruzynski A, Nowrouzi A, et al. High-resolution analysis of the human T-cell receptor repertoire. Nat Commun. (2015) 6:8081. doi: 10.1038/ncomms9081
220. Rodda SJ. Peptide libraries for T cell epitope screening and characterization. J Immunol Methods. (2002) 267:71–7. doi: 10.1016/S0022-1759(02)00141-2
221. Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, et al. High-throughput pairing of T cell receptor α and β sequences. Sci Transl Med. (2015) 7:aac5624. doi: 10.1126/scitranslmed.aac5624
222. Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol. (2016) 34:1037–45. doi: 10.1038/nbt.3662
223. Molldrem JJ, Lee PP, Kant S, Wieder E, Jiang W, Lu S, et al. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J Clin Invest. (2003) 111:639–47. doi: 10.1172/JCI200316398
224. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific infiltrating human tumors. J Clin Invest. (2014) 124:2246–59. doi: 10.1172/JCI73639
225. Gros A, Tran E, Parkhurst MR, Ilyas S, Pasetto A, Groh EM, et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J Clin Invest. (2019) 129:4992–5004. doi: 10.1172/JCI127967
226. Yoshizawa H, Chang AE, Shu S. Specific adoptive immunotherapy mediated by tumor-draining lymph node cells sequentially activated with anti-CD3 and IL-2. J Immunol. (1991) 147:729–37.
227. Stevens EJ, Jacknin L, Robbins PF, Kawakami Y, el Gamil M, Rosenberg SA, et al. Generation of tumor-specific CTLs from melanoma patients by using peripheral blood stimulated with allogeneic melanoma tumor cell lines. Fine specificity and MART-1 melanoma antigen recognition. J Immunol. (1995) 154:762–71.
228. Strønen E, Toebes M, Kelderman S, Buuren Van MM, Yang W, Rooij Van N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. (2016) 2288:1–11. doi: 10.1126/science.aaf2288
229. Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. (1996) 273:104–6. doi: 10.1126/science.273.5271.104
230. Hombrink P, Raz Y, Kester MGD, De Boer R, Weißbrich B, Von dem Borne PA, et al. Mixed functional characteristics correlating with TCR-ligand koff-rate of MHC-tetramer reactive T cells within the naive T-cell repertoire. Eur J Immunol. (2013) 43:3038–50. doi: 10.1002/eji.201343397
231. Nauerth M, Weissbrich B, Busch DH. The clinical potential for koff-rate measurement in adoptive immunotherapy. Expert Rev Clin Immunol. (2013) 9:1151–3. doi: 10.1586/1744666X.2013.855609
232. Manning TC, Schlueter CJ, Brodnicki TC, Parke EA, Speir JA, Garcia KC, et al. Alanine scanning mutagenesis of an αβ T cell receptor: mapping the energy of antigen recognition. Immunity. (1998) 8:413–25. doi: 10.1016/S1074-7613(00)80547-6
233. Manning TC, Parke EA, Teyton L, Kranz DM. Effects of complementarity determining region mutations on the affinity of an α/β t cell receptor: measuring the energy associated with CD4/CD8 repertoire skewing. J Exp Med. (1999) 189:461–70. doi: 10.1084/jem.189.3.461
234. Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. (2008) 180:6116–31. doi: 10.4049/jimmunol.180.9.6116
235. Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, et al. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res. (2009) 15:169–80. doi: 10.1158/1078-0432.CCR-08-1638
236. Schmitt TM, Aggen DH, Ishida-Tsubota K, Ochsenreither S, Kranz DM, Greenberg PD. Generation of higher affinity T cell receptors by antigen-driven differentiation of progenitor t cells in vitro. Nat Biotechnol. (2017) 35:1188–95. doi: 10.1038/nbt.4004
237. Li LP, Lampert JC, Chen X, Leitao C, Popović J, Müller W, et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med. (2010) 16:1029–34. doi: 10.1038/nm.2197
238. Obenaus M, Leitão C, Leisegang M, Chen X, Gavvovidis I, Van Der Bruggen P, et al. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat Biotechnol. (2015) 33:402–7. doi: 10.1038/nbt.3147
239. Kieke MC, Sundberg E, Shusta EV, Mariuzza RA, Wittrup KD, Kranz DM. High affinity T cell receptors from yeast display libraries block T cell activation by superantigens. J Mol Biol. (2001) 307:1305–15. doi: 10.1006/jmbi.2001.4560
240. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. (2005) 23:349–54. doi: 10.1038/nbt1070
241. Sanderson JP, Crowley DJ, Wiedermann GE, Quinn LL, Crossland KL, Tunbridge HM, et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology. (2020) 9:1682381. doi: 10.1080/2162402X.2019.1682381
242. Docta RY, Ferronha T, Sanderson JP, Weissensteiner T, Pope GR, Bennett AD, et al. Tuning T-cell receptor affinity to optimize clinical risk-benefit when targeting alpha-fetoprotein–positive liver cancer. Hepatology. (2019) 69:2061–75. doi: 10.1002/hep.30477
243. Zhong S, Malecek K, Johnson LA, Yu Z, De Miera EVS, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci USA. (2013) 110:6973–8. doi: 10.1073/pnas.1221609110
244. Cameron BJ, Gerry AB, Dukes J, Harper J V, Kannan V, Bianchi FC, et al. Identification of a titin-derived HLA-A1 – presented peptide as a cross-reactive target for engineered MAGE A3 – directed T cells. Sci Transl Med. (2013) 5:197ra103. doi: 10.1126/scitranslmed.3006034
245. Karapetyan AR, Chaipan C, Winkelbach K, Wimberger S, Jeong JS, Joshi B, et al. TCR fingerprinting and off-target peptide identification. Front Immunol. (2019) 10:2501. doi: 10.3389/fimmu.2019.02501
246. Thomis DC, Marktel S, Bonini C, Traversari C, Gilman M, Bordignon C, et al. A Fas-based suicide switch in human T cells for the treatment of graft-versus-host disease. Blood. (2001) 97:1249–57. doi: 10.1182/blood.V97.5.1249.h8001249_1249_1257
247. Zajac A J, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. (1998) 188:2205–13. doi: 10.1084/jem.188.12.2205
248. Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
249. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
250. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR-T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
251. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol. (2018) 200:1088–100. doi: 10.4049/jimmunol.1700236
252. Stone JD, Harris DT, Soto CM, Chervin AS, Aggen DH, Roy EJ, et al. A novel T cell receptor single-chain signaling complex mediates antigen-specific T cell activity and tumor control. Cancer Immunol Immunother. (2014) 63:1163–76. doi: 10.1007/s00262-014-1586-z
253. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. (2005) 26:111–7. doi: 10.1016/j.it.2004.12.003
254. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol. (2016) 34:2389–97. doi: 10.1200/JCO.2016.66.7220
255. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. (2002) 298:850–4. doi: 10.1126/science.1076514
256. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
257. Zhou J, Dudley ME, Rosenberg SA, Robbins PF. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J Immunother. (2005) 28:53–62. doi: 10.1097/00002371-200501000-00007
258. Powell DJ Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27. Blood. (2005) 105:241–50. doi: 10.1182/blood-2004-06-2482
259. Sheih A, Voillet V, Hanafi LA, DeBerg HA, Yajima M, Hawkins R, et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat Commun. (2020) 11:1–13. doi: 10.1038/s41467-019-13880-1
260. Schumacher TNM. T-cell-receptor gene therapy. Nat Rev Immunol. (2002) 2:512–9. doi: 10.1038/nri841
261. Kessels HWHG, Wolkers MC, van den Boom MD, van den Valk MA, Schumacher TNM. Immunotherapy through TCR gene transfer. Nat Immunol. (2001) 2:957–61. doi: 10.1038/ni1001-957
262. Duarte RF, Chen FE, Lowdell MW, Potter MN, Lamana ML, Prentice HG, et al. Functional impairment of human T-lymphocytes following PHA-induced expansion and retroviral transduction: Implications for gene therapy. Gene Ther. (2002) 9:1359–68. doi: 10.1038/sj.gt.3301807
263. Ferrand C, Robinet E, Contassot E, Certoux JM, Lim A, Hervé P, et al. Retrovirus-mediated gene transfer in primary T lymphocytes: influence of the transduction/selection process and of ex vivo expansion on the T cell receptor β chain hypervariable region repertoire. Hum Gene Ther. (2000) 11:1151–64. doi: 10.1089/10430340050015202
264. Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. (1990) 128:189–201. doi: 10.1016/0022-1759(90)90210-M
265. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. (2005) 115:1616–26. doi: 10.1172/JCI24480
266. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia a. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. (1999) 401:708–12. doi: 10.1038/44385
267. Cieri N, Oliveira G, Greco R, Forcato M, Taccioli C, Cianciotti B, et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood. (2015) 125:2865–74. doi: 10.1182/blood-2014-11-608539
268. Roberto A, Castagna L, Zanon V, Bramanti S, Crocchiolo R, McLaren JE, et al. Role of naïve-derived T memory stem cells in T cell reconstitution following allogeneic transplantation. Blood. (2015) 125:2855–65. doi: 10.1182/blood-2014-11-608406
269. Fearon DT, Manders P, Wagner SD. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science. (2001) 293:248–50. doi: 10.1126/science.1062589
270. Gattinoni L, Lugli E. A human memory T-cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
271. Oliveira G, Ruggiero E, Stanghellini MTL, Cieri N, DAgostino M, Fronza R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med. (2015) 7:317ra198. doi: 10.1126/scitranslmed.aac8265
272. Biasco L, Pellin D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L, et al. In vivo Tracking of human hematopoiesis reveals patterns of clonal dynamics during early and steady-state reconstitution phases. Cell Stem Cell. (2015) 19:107–19. doi: 10.1016/j.stem.2016.04.016
273. Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8+ enriched “Young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. (2010) 16:6122–31. doi: 10.1158/1078-0432.CCR-10-1297
274. Bondanza A, Valtolina V, Magnani Z, Ponzoni M, Fleischhauer K, Bonyhadi M, et al. Suicide gene therapy of graft-versus-host disease induced by central memory human T lymphocytes. Blood. (2006) 107:1828–36. doi: 10.1182/blood-2005-09-3716
275. Arcangeli S, Falcone L, Camisa B, De Girardi F, Biondi M, Giglio F, et al. Next-generation manufacturing protocols enriching TSCM CAR-T cells can overcome disease-specific T cell defects in cancer patients. Front Immunol. (2020) 11:1217. doi: 10.3389/fimmu.2020.01217
276. Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. (2008) 111:5326–33. doi: 10.1182/blood-2007-09-113050
277. Kaneko S, Mastaglio S, Bondanza A, Ponzoni M, Sanvito F, Aldrighetti L, et al. IL-7 and IL-15 allow the generation of suicide gene modified alloreactive self-renewing central memory human T lymphocytes. Blood. (2009) 113:1006–15. doi: 10.1182/blood-2008-05-156059
278. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. (2013) 121:573–84. doi: 10.1182/blood-2012-05-431718
279. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
280. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. (2018) 558:307–12. doi: 10.1038/s41586-018-0178-z
281. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. (2008) 118:294–305. doi: 10.1172/JCI32103
282. van der Leun AM, Thommen DS, Schumacher TN. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. (2020) 20:218–32. doi: 10.1038/s41568-019-0235-4
283. Awasthi R, Pacaud L, Waldron E, Tam CS, Jäger U, Borchmann P, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. (2020) 4:560–72. doi: 10.1182/bloodadvances.2019000525
284. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. (2006) 107:2294–302. doi: 10.1182/blood-2005-08-3503
285. Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F, et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. (2007) 109:4708–15. doi: 10.1182/blood-2006-04-015230
286. Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. (2014) 4:522–6. doi: 10.1158/2159-8290.CD-13-0985
287. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. (2015) 15:669–82. doi: 10.1038/nri3902
288. Newick K, O'Brien S, Moon E, Albelda SM. CAR-T cell therapy for solid tumors. Annu Rev Med. (2017) 68:139–52. doi: 10.1146/annurev-med-062315-120245
289. De Sanctis F, Sandri S, Ferrarini G, Pagliarello I, Sartoris S, Ugel S, et al. The emerging immunological role of post-translational modifications by reactive nitrogen species in cancer microenvironment. Front Immunol. (2014) 5:69. doi: 10.3389/fimmu.2014.00069
290. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
291. Miyoshi E, Moriwaki K, Nakagawa T. Biological function of fucosylation in cancer biology. J Biochem. (2008) 143:725–9. doi: 10.1093/jb/mvn011
292. Alatrash G, Qiao N, Zhang M, Zope M, Perakis AA, Sukhumalchandra P, Garber HR, Kerros C, St John LS, et al. Fucosylation enhances the efficacy of adoptively transferred antigen-specific cytotoxic T lymphocytes. Clin Cancer Res. (2019) 25:2610–20. doi: 10.1158/1078-0432.CCR-18-1527
293. Maréchal R, Demetter P, Nagy N, Berton A, Decaestecker C, Polus M, et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br J Cancer. (2009) 100:1444–51. doi: 10.1038/sj.bjc.6605020
294. Iwasa S, Yanagawa T, Fan J, Katoh R. Expression of CXCR4 and its ligand SDF-1 in intestinal-type gastric cancer is associated with lymph node and liver metastasis. Anticancer Res. (2009) 29:4751–8.
295. Matsusue R, Kubo H, Hisamori S, Okoshi K, Takagi H, Hida K, et al. Hepatic stellate cells promote liver metastasis of colon cancer cells by the action of SDF-1/CXCR4 axis. Ann Surg Oncol. (2009) 16:2645–53. doi: 10.1245/s10434-009-0599-x
296. Izumi D, Ishimoto T, Miyake K, Sugihara H, Eto K, Sawayama H, et al. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin β1 clustering and invasiveness in gastric cancer. Int J Cancer. (2016) 138:1207–19. doi: 10.1002/ijc.29864
297. Guo ZJ, Yang L, Qian F, Wang YX, Yu X, Ji CD, et al. Transcription factor RUNX2 up-regulates chemokine receptor CXCR4 to promote invasive and metastatic potentials of human gastric cancer. Oncotarget. (2016) 7:20999–1012. doi: 10.18632/oncotarget.8236
298. Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci USA. (2019) 116:4558–66. doi: 10.1073/pnas.1815515116
299. Mitsui H, Shibata K, Suzuki S, Umezu T, Mizuno M, Kajiyama H, et al. Functional interaction between peritoneal mesothelial cells and stem cells of ovarian yolk sac tumor (SC-OYST) in peritoneal dissemination. Gynecol Oncol. (2012) 124:303–10. doi: 10.1016/j.ygyno.2011.10.006
300. Jin C-H, Li Y, Xia J, Li Y, Chen M, Hu Z, et al. CXCR4 blockade improves leukemia eradication by allogeneic lymphocyte infusion. Am J Hematol. (2018) 93:786–93. doi: 10.1002/ajh.25099
301. Saxena R, Wang Y, Mier JW. CXCR4 inhibition modulates the tumor microenvironment and retards the growth of B16-OVA melanoma and Renca tumors. Melanoma Res. (2020) 30:14–25. doi: 10.1097/CMR.0000000000000639
302. Daniel SK, Seo YD, Pillarisetty VG. The CXCL12-CXCR4/CXCR7 axis as a mechanism of immune resistance in gastrointestinal malignancies. Semin Cancer Biol. (2020). 65:176–188. doi: 10.1016/j.semcancer.2019.12.007
303. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. (2018) 9:978. doi: 10.3389/fimmu.2018.00978
304. De Aguiar RB, De Moraes JZ. Exploring the immunological mechanisms underlying the anti-vascular endothelial growth factor activity in tumors. Front Immunol. (2019) 10:1023. doi: 10.3389/fimmu.2019.01023
305. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. (2010) 70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153
306. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA. (2012) 109:17561–6. doi: 10.1073/pnas.1215397109
307. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J. (2015) 29:227–38. doi: 10.1096/fj.14-250985
308. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. (2016) 7:1–8. doi: 10.1038/ncomms12624
309. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 with Ramucirumab strongly impacts effector/ activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J Immunother Cancer. (2018) 6:1–14. doi: 10.1186/s40425-018-0403-1
310. Peske JD, Thompson ED, Gemta L, Baylis RA, Fu YX, Engelhard VH. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun. (2015) 6:7114. doi: 10.1038/ncomms8114
311. Calcinotto A, Grioni M, Jachetti E, Curnis F, Mondino A, Parmiani G, et al. Targeting TNF-α to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol. (2012) 188:2687–94. doi: 10.4049/jimmunol.1101877
312. Johansson-Percival A, He B, Li ZJ, Kjellén A, Russell K, Li J, et al. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol. (2017) 18:1207–17. doi: 10.1038/ni.3836
313. He B, Johansson-Percival A, Backhouse J, Li J, Lee GYF, Hamzah J, et al. Remodeling of metastatic vasculature reduces lung colonization and sensitizes overt metastases to immunotherapy. Cell Rep. (2020) 30:714–24.e5. doi: 10.1016/j.celrep.2019.12.013
314. Shen J, Li ZJ, Li LF, Lu L, Xiao ZG, Wu WKK, et al. Vascular-targeted TNFα and IFNγ inhibits orthotopic colorectal tumor growth. J Transl Med. (2016) 14:1–13. doi: 10.1186/s12967-016-0944-3
315. Ding N, Zou Z, Sha H, Su S, Qian H, Meng F, et al. iRGD synergizes with PD-1 knockout immunotherapy by enhancing lymphocyte infiltration in gastric cancer. Nat Commun. (2019) 10:1336. doi: 10.1038/s41467-019-09296-6
316. Zhao Y, Ting KK, Li J, Cogger VC, Chen J, Johansson-Percival A, et al. Targeting vascular endothelial-cadherin in tumor-associated blood vessels promotes T-cell–mediated immunotherapy. Cancer Res. (2017) 77:4434–47. doi: 10.1158/0008-5472.CAN-16-3129
317. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8 + T-CeII recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
318. De Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, et al. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res. (2011) 71:6391–9. doi: 10.1158/0008-5472.CAN-11-0952
319. Zsiros E, Duttagupta P, Dangaj D, Li H, Frank R, Garrabrant T, et al. The ovarian cancer chemokine landscape is conducive to homing of vaccine-primed and CD3/CD28-Costimulated T cells prepared for adoptive therapy. Clin Cancer Res. (2015) 21:2840–50. doi: 10.1158/1078-0432.CCR-14-2777
320. Sackstein R, Schatton T, Barthel SR. T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab Investig. (2017) 97:669–97. doi: 10.1038/labinvest.2017.25
321. Garetto S, Sardi C, Martini E, Roselli G, Morone D, Angioni R, et al. Tailored chemokine receptor modification improves homing of adoptive therapy T cells in a spontaneous tumor model. Oncotarget. (2016) 7:43010–26. doi: 10.18632/oncotarget.9280
322. Siddiqui I, Erreni M, Van Brakel M, Debets R, Allavena P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: Importance of the chemokine gradient. J Immunother Cancer. (2016) 4:1–12. doi: 10.1186/s40425-016-0125-1
323. Khan AB, Carpenter B, Santos E, Sousa P, Pospori C, Khorshed R, Griffin J, et al. Redirection to the bone marrow improves T cell persistence and antitumor functions. J Clin Invest. (2018) 128:2010–24. doi: 10.1172/JCI97454
324. Gunassekaran GR, Hong CM, Vadevoo SMP, Chi L, Guruprasath P, Ahn BC, et al. Non-genetic engineering of cytotoxic T cells to target IL-4 receptor enhances tumor homing and therapeutic efficacy against melanoma. Biomaterials. (2018) 159:161–73. doi: 10.1016/j.biomaterials.2018.01.013
325. Idorn M, Skadborg SK, Kellermann L, Halldórsdóttir HR, Holmen Olofsson G, Met Ö, et al. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology. (2018) 7:e1450715. doi: 10.1080/2162402X.2018.1450715
326. Lai JZ, Zhu YY, Ruan M, Chen L, Zhang QY. Local irradiation sensitized tumors to adoptive T cell therapy via enhancing the cross-priming, homing, and cytotoxicity of antigen-specific CD8 T cells. Front Immunol. (2019) 10:2857. doi: 10.3389/fimmu.2019.02857
327. Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G, et al. T-cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors. Clin Cancer Res. (2018) 24:2920–34. doi: 10.1158/1078-0432.CCR-17-1365
328. Bedognetti D, Spivey TL, Zhao Y, Uccellini L, Tomei S, Dudley ME, et al. CXCR3/CCR5 pathways in metastatic melanoma patients treated with adoptive therapy and interleukin-2. Br J Cancer. (2013) 109:2412–23. doi: 10.1038/bjc.2013.557
329. Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. (2017) 18:940–50. doi: 10.1038/ni.3775
330. Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun. (2017) 8:15221. doi: 10.1038/ncomms15221
331. Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. (2018) 24:986–93. doi: 10.1038/s41591-018-0078-7
332. Dumauthioz N, Labiano S, Romero P. Tumor resident memory T cells: New players in immune surveillance and therapy. Front Immunol. (2018) 9:2076. doi: 10.3389/fimmu.2018.02076
333. Molodtsov A, Turk MJ. Tissue resident CD8 memory T cell responses in cancer and autoimmunity. Front Immunol. (2018) 9:2810. doi: 10.3389/fimmu.2018.02810
334. Amsen D, Van Gisbergen KPJM, Hombrink P, Van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. (2018) 19:538–46. doi: 10.1038/s41590-018-0114-2
335. Egelston CA, Avalos C, Tu TY, Rosario A, Wang R, Solomon S, et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight. (2019) 4:1–15. doi: 10.1172/jci.insight.130000
336. Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, et al. Tissue-resident memory CD8 + T cells promote melanoma-immune equilibrium in skin. Nature. (2019) 565:366–71. doi: 10.1038/s41586-018-0812-9
337. Murray T, Marraco SAF, Baumgaertner P, Bordry N, Cagnon L, Donda A, et al. Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol. (2016) 7:573. doi: 10.3389/fimmu.2016.00573
338. Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martýnez-Cano S, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat Commun. (2017) 8:1–11. doi: 10.1038/ncomms16073
339. Gálvez-Cancino F, López E, Menares E, Díaz X, Flores C, Cáceres P, et al. Vaccination-induced skin-resident memory CD8+ T cells mediate strong protection against cutaneous melanoma. Oncoimmunology. (2018) 7:e1442163. doi: 10.1080/2162402X.2018.1442163
340. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature. (2017) 552:253–7. doi: 10.1038/nature24993
341. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. (2003) 9:606–12.
342. Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer. (2003) 98:1089–99. doi: 10.1002/cncr.11618
343. Ormandy L, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. (2005) 65:2457–64. doi: 10.1158/0008-5472.CAN-04-3232
344. Schaefer C, Kim GG, Albers A, Hoermann K, Myers EN, Whiteside TL. Characteristics of CD4+CD25+ regulatory T cells in the peripheral circulation of patients with head and neck cancer. Br J Cancer. (2005) 92:913–20. doi: 10.1038/sj.bjc.6602407
345. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. (2006) 12:5423–34. doi: 10.1158/1078-0432.CCR-06-0369
346. Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. (2018) 33:547–62. doi: 10.1016/j.ccell.2018.03.012
348. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci USA. (2010) 107:7875–80. doi: 10.1073/pnas.1003345107
349. Zhang Y, Huang S, Gong D, Qin Y, Shen Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell Mol Immunol. (2010) 7:389–95. doi: 10.1038/cmi.2010.28
350. Nunes C, Wong R, Mason M, Fegan C, Man S, Pepper C. Expansion of a CD8 +PD-1 + replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin Cancer Res. (2012) 18:678–87. doi: 10.1158/1078-0432.CCR-11-2630
351. Muenst S, Schaerli AR, Gao F, Däster S, Trella E, Droeser RA, et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat. (2014) 146:15–24. doi: 10.1007/s10549-014-2988-5
352. Gatalica Z, Snyder C, Maney T, Ghazalpour A, Holterman DA, Xiao N, et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. (2014) 23:2965–70. doi: 10.1158/1055-9965.EPI-14-0654
353. Zheng Z, Bu Z, Liu X, Zhang L, Li Z, Wu A, et al. Level of circulating PD-L1 expression in patients with advanced gastric cancer and its clinical implications. Chinese J Cancer Res. (2014) 26:104–11. doi: 10.3978/j.issn.1000-9604.2014.02.08
354. Severson JJ, Serracino HS, Mateescu V, Raeburn CD, McIntyre RC, Sams SB, et al. PD-1+Tim-3+ CD8+ T lymphocytes display varied degrees of functional exhaustion in patients with regionally metastatic differentiated thyroid cancer. Cancer Immunol Res. (2015) 3:620–30. doi: 10.1158/2326-6066.CIR-14-0201
355. Chauvin J, Korman AJ, Zarour HM, Chauvin J, Pagliano O, Fourcade J, et al. TIGIT and PD-1 impair tumor antigen – specific CD8 + T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. doi: 10.1172/JCI80445
356. Tassi E, Grazia G, Vegetti C, Bersani I, Bertolini G, Molla A, et al. Early effector T lymphocytes coexpress multiple inhibitory receptors in primary non-small cell lung cancer. Cancer Res. (2017) 77:851–61. doi: 10.1158/0008-5472.CAN-16-1387
357. Fucikova J, Rakova J, Hensler M, Kasikova L, Belicova L, Hladikova K, et al. TIM-3 dictates functional orientation of the immune infiltrate in ovarian cancer. Clin Cancer Res. (2019) 25:4820–31. doi: 10.1158/1078-0432.CCR-18-4175
358. Noviello M, Manfredi F, Ruggiero E, Perini T, Oliveira G, Cortesi F, et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat Commun. (2019) 10:1065. doi: 10.1038/s41467-019-08871-1
359. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
360. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
361. Hodi FS. Improved survival with Ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
362. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. (2003) 100:8372–7. doi: 10.1073/pnas.1533209100
363. Sanderson K, Scotland R, Lee P, Liu D, Groshen S, Snively J, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. (2005) 23:741–50. doi: 10.1200/JCO.2005.01.128
364. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
365. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. (2016) 54:139–48. doi: 10.1016/j.ejca.2015.11.016
366. June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles' heel of cancer immunotherapy? Nat Med. (2017) 23:540–7. doi: 10.1038/nm.4321
367. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. (2017) 11:554–62. doi: 10.1007/s11684-017-0543-6
368. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. (2016) 22:433–8. doi: 10.1038/nm.4051
369. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. (2005) 23:344–6. doi: 10.1016/j.immuni.2005.10.001
370. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. (2013) 23:277–86. doi: 10.1016/j.ccr.2013.02.013
371. de Haas N, de Koning C, Spilgies L, de Vries IJM, Hato SV. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology. (2016) 5:1–11. doi: 10.1080/2162402X.2016.1196312
372. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:1–9. doi: 10.1038/cddis.2015.162
373. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
374. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. (2011) 208:469–78. doi: 10.1084/jem.20101876
375. Pedroza-Gonzalez A, Xu K, Wu TC, Aspord C, Tindle S, Marches F, et al. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J Exp Med. (2011) 208:479–90. doi: 10.1084/jem.20102131
376. Zelenay S, Van Der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. (2015) 162:1257–70. doi: 10.1016/j.cell.2015.08.015
377. Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
378. Speiser DE, Ho P, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. (2016) 16:599–611. doi: 10.1038/nri.2016.80
379. Zarour HM. Reversing T-cell dysfunction and exhaustion in cancer. Clin Cancer Res. (2016) 22:1856–64. doi: 10.1158/1078-0432.CCR-15-1849
380. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
381. Bierie B, Moses HL. Tumour microenvironment - TGF?: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. (2006) 6:506–20. doi: 10.1038/nrc1926
382. Zdanov S, Mandapathil M, Eid RA, Adamson-Fadeyi S, Wilson W, Qian J, et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol Res. (2016) 4:354–65. doi: 10.1158/2326-6066.CIR-15-0241
383. Zhang Q, Yang X, Pins M, Javonovic B, Kuzel T, Kim SJ, et al. Adoptive transfer of tumor-reactive transforming growth factor-β-insensitive CD8+ T cells: eradication of autologous mouse prostate cancer. Cancer Res. (2005) 65:1761–9. doi: 10.1158/0008-5472.CAN-04-3169
384. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor Activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-b receptor. J Immunother. (2008) 31:500–5. doi: 10.1097/CJI.0b013e318177092b
385. Wang L, Wen W, Yuan J, Helfand B, Li Y, Shi C, et al. Immunotherapy for human renal cell carcinoma by adoptive transfer of autologous transforming growth factor β-insensitive CD8+ T cells. Clin Cancer Res. (2010) 16:164–73. doi: 10.1158/1078-0432.CCR-09-1758
386. Lin R, Chen L, Chen G, Hu C, Jiang S, Sevilla J, et al. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. J Clin Invest. (2014) 124:5352–67. doi: 10.1172/JCI76561
387. Principe DR, DeCant B, Mascariñas E, Wayne EA, Diaz AM, Akagi N, et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. (2016) 76:2525–39. doi: 10.1158/0008-5472.CAN-15-1293
388. Bollard CM, Rössig C, Julia Calonge M, Helen Huls M, Wagner HJ, Massague J, et al. Adapting a transforming growth factor β-related tumor protection strategy to enhance antitumor immunity. Blood. (2002) 99:3179–87. doi: 10.1182/blood.V99.9.3179
389. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-specific t-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol. (2018) 36:1128–39. doi: 10.1200/JCO.2017.74.3179
390. Chmielewski M, Abken H. TRUCKs: The fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
391. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR-T cells induces exhaustion resistance. Nature. (2019) 576:293–300. doi: 10.1038/s41586-019-1805-z
392. Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. (2019) 576:471–6. doi: 10.1038/s41586-019-1821-z
Keywords: TCR - T cell receptor, genetic engineering, cancer immunotherapy, adoptive T cell immunotherapy, cancer immunoediting
Citation: Manfredi F, Cianciotti BC, Potenza A, Tassi E, Noviello M, Biondi A, Ciceri F, Bonini C and Ruggiero E (2020) TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 11:1689. doi: 10.3389/fimmu.2020.01689
Received: 08 April 2020; Accepted: 24 June 2020;
Published: 03 September 2020.
Edited by:
Cristina Maccalli, Sidra Medicine, QatarReviewed by:
Bart Vandekerckhove, Ghent University, BelgiumSiok Tey, The University of Queensland, Australia
Copyright © 2020 Manfredi, Cianciotti, Potenza, Tassi, Noviello, Biondi, Ciceri, Bonini and Ruggiero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Bonini, bonini.chiara@hsr.it; Eliana Ruggiero, ruggiero.eliana@hsr.it
†These authors have contributed equally to this work