
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 August 2020
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01595
This article is part of the Research Topic Autoimmunity and Chronic Inflammation in Early Life View all 13 articles
 Xiaoxiao Sun1,2†
Xiaoxiao Sun1,2† Haipeng Pang1,2†Jiaqi Li1,2Shuoming Luo1,2
Haipeng Pang1,2†Jiaqi Li1,2Shuoming Luo1,2 Gan Huang1,2Xia Li1,2
Gan Huang1,2Xia Li1,2 Zhiguo Xie1,2*
Zhiguo Xie1,2* Zhiguang Zhou1,2*
Zhiguang Zhou1,2*The NLRP3 (nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3) inflammasome is a protein complex expressed in cells. It detects danger signals and induces the production of active caspase-1 and the maturation and release of IL (interleukin)-33, IL-18, IL-1β and other cytokines. T1DM (type 1 diabetes mellitus) is defined as a chronic autoimmune disorder characterized by the autoreactive T cell-mediated elimination of insulin-positive pancreatic beta-cells. Although the exact underlying mechanisms are obscure, researchers have proposed that both environmental and genetic factors are involved in the pathogenesis of T1DM. Furthermore, immune responses, including innate and adaptive immunity, play an important role in this process. Recently, the NLRP3 inflammasome, a critical component of innate immunity, was reported to be associated with T1DM. Here, we review the assembly and function of the NLRP3 inflammasome. In addition, the activation and regulatory mechanisms that enhance or attenuate NLRP3 inflammasome activation are discussed. Finally, we focus on the relationship between the NLRP3 inflammasome and T1DM, as well as its potential value for clinical use.
The inflammatory response is a common mechanism of many diseases. However, the clinical manifestations caused by the combination of a certain microenvironment and a variety of stimuli from common or specific pathways are different. Currently, many chronic diseases, particularly diabetes, are serious threats to human health. Many researchers have recognized that inflammatory immune factors induce many chronic diseases. Innate immune cells induce a series of inflammatory responses by detecting various PAMPs (pathogen-associated molecular patterns) or DAMPs (damage-associated molecular patterns) through innate sensors (1). With a relative molecular mass of approximately 700,000 Da, the NLRP3 inflammasome is a polyprotein complex that plays a critical role in the course of inflammatory responses (2). The NLRP3 inflammasome is comprised of NLRP3, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and procaspase-1 (3, 4). It is the most well-studied inflammasome and functions as a site for the activation of caspase-1 (3, 5). Based on emerging evidence, activated caspase-1 causes the maturation of IL-1 (6, 7). Because the NLRP3 inflammasome may trigger the release of IL-1β after stimulation with various danger signals, it represents a potentially effective target to regulate the onset and development of various autoimmune diseases, such as T1DM.
T1DM is defined as an organ-specific autoimmune disorder characterized by the autoreactive T cell-mediated elimination of insulin-producing pancreatic beta-cells (8). Although the exact underlying mechanisms are still unknown, a combination of environmental and genetic elements are involved in the pathophysiological process of T1DM (9–11). Both innate immunity and adaptive immunity are involved in the progression of T1DM (12–14). Innate immunity, which serves as the first line of defense against an exogenous attack by bacteria, viruses, and fungi, is a relatively conserved immune response compared with adaptive immunity (15, 16). Previous studies have confirmed that the innate immune system exerts its effect via highly conserved PRRs (pattern-recognition receptors) to initiate innate inflammatory responses to both exogenous and endogenous trigger factors and further activate adaptive immunity (16–18). Upon the recognition of DAMPs and PAMPs, which are associated with cellular stress and microbial pathogens, PRRs promote the secretion of proinflammatory cytokines by inducing either non-transcriptional or transcriptional innate immune responses (19, 20). NLRP3 is a PRR, and the NLRP3 inflammasome is a component of the innate immune system that plays a key role in the inflammatory response. In this review, we discuss the components and functions of the NLRP3 inflammasome and the activation mechanisms and regulatory mechanisms that potentiate or limit NLRP3 inflammasome activation. In addition, we describe the function of the NLRP3 inflammasome in T1DM to provide a potential treatment target for the prevention and improvement of this disorder.
The NLRP3 inflammasome is a protein complex that includes procaspase-1, ASC and NLRP3 (21). NLRP3 is a member of the NLR (Nod like-receptor) protein family, which is widely expressed in macrophages, monocytes, and dendritic cells and has the function of recognizing pathogens. NLRP3 has a characteristic NLR protein family LRR (leucine-rich repeat) domain at the C-terminus (22). The middle region of NLRP3 is called the NBD (nucleotide-binding domain), also known as NOD or NACHT. The NBD belongs to the NTPase superfamily and hydrolyzes ATP into GTP. The N-terminus contains a PYD (pyrin domain), which is also called the CARD (caspase recruitment domain) or BIR (baculovirus IAP repeat) domain; this domain participates in multiple inflammatory responses by binding molecules with the same domain. For example, ASC is bound via the PYD-PYD interaction. ASC is the adapter protein of the NLRP3 inflammasome. The N-terminus of ASC contains a PYD domain that is the same as the PYD domain in NLRP3, whereas the C-terminus contains a CARD recruitment domain that is the same as the CARD domain in procaspase-1. Therefore, ACS functions as a dual adapter protein molecule that binds to both NLRP3 and procaspase-1 through PYD-PYD and CARD-CARD domain interactions. Caspase-1, also called IL-1β-converting enzyme, is an effector protein of the inflammasome that cleaves inactive proinflammatory cytokines, including pro-IL-1β and pro-IL-18, to produce activated IL-1β and IL-18 (Figure 1) (23, 24).
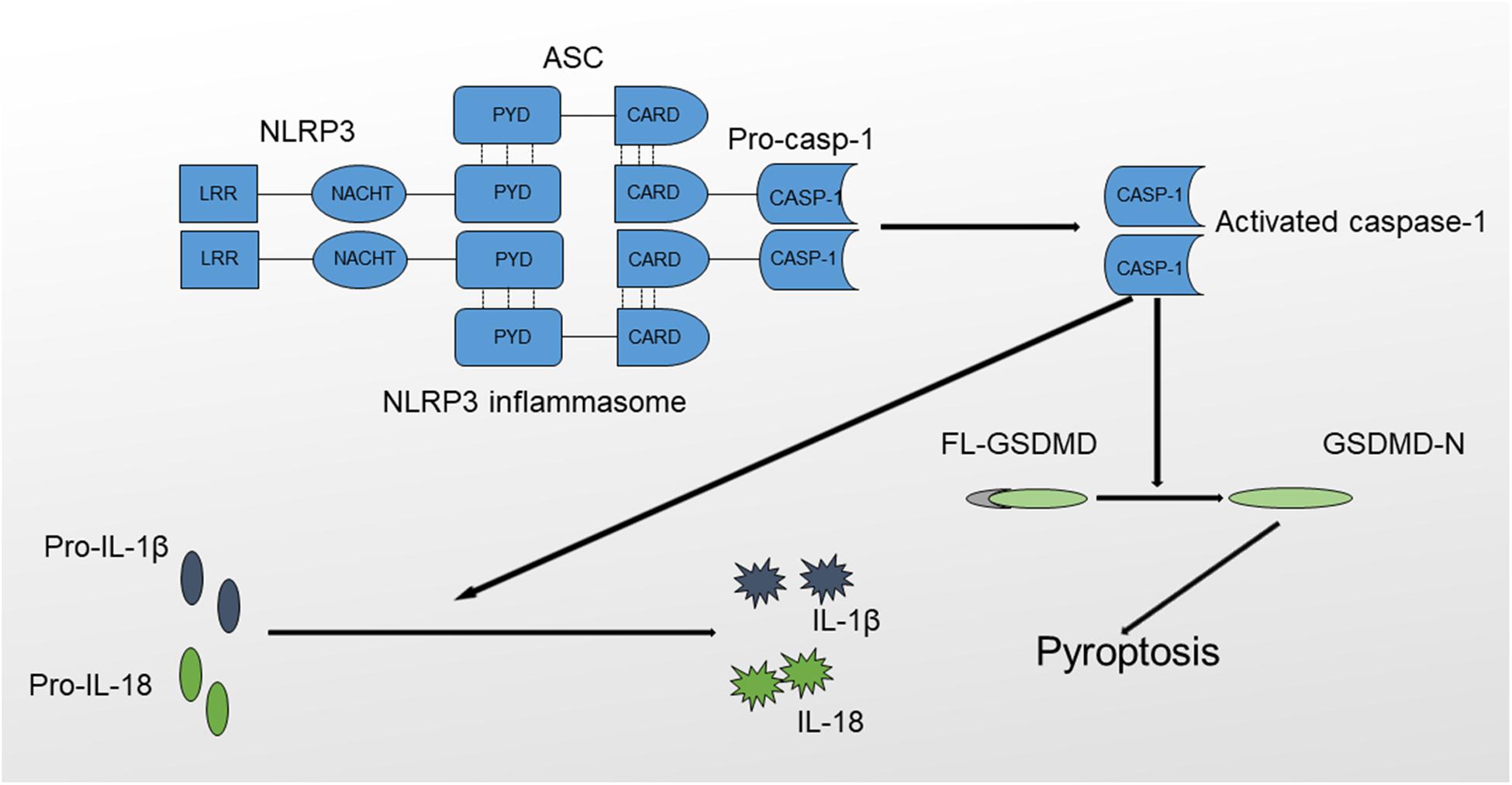
Figure 1. Structure and function of the NLRP3 inflammasome. The NLRP3 inflammasome comprises NLRP3, ASC and procaspase-1. The formation of the NLRP3 inflammasome results in the activation of caspase-1 through the self-cleavage of procaspase-1. Activated caspase-1 causes the maturation of IL-1β and IL-18 and triggers inflammatory cell death mediated by GSDMD, also termed pyroptosis.
NLRP3 activation results in the oligomerization and recruitment of ASC and procaspase-1, which increase the autocleavage and maturation of procaspase-1. Active caspase-1 cleaves pro-IL-1β to produce mature IL-1β, which, when released, recruits other inflammatory cells and exerts direct cytotoxic effects. In addition, the NLRP3 inflammasome mediates a special type of programmed cell death named pyroptosis, which is inherently inflammatory and is triggered by pathological stimuli through the activation of caspase-1 (25, 26). The process of pyroptosis is mediated by Gasdermin (GSDMD), consisting of an amino-terminal cell death region, a carboxy-terminal autoinhibitory region, and a central linker domain (27). GSDMD is activated by caspase-1 through the removal of its carboxyl inhibitory terminus, and activated GSDMD induces cell death characterized by plasma membrane rupture, DNA cleavage and cell lysis by binding to the inner leaflet of the cell membrane, oligomerizing and forming a pore containing 16 symmetrical promoters (28). Based on the results of in vitro studies, activated GSDMD possesses a bactericidal property, but the exact mechanisms remain obscure (29). In addition, GSDMD-dependent pyroptosis promotes IL-1β and IL-18 release via a non-conventional pathway (30, 31). In conclusion, caspase-1 activation will result in the production of activated proinflammatory cytokines and lead to rapid cell death (32, 33).
NLRP3 is activated by a number of pathogens, as well as many PAMPs and DAMPs, which are structurally diverse, and environmental irritants. NLRP3 oligomerizes via homotypic interactions between NACHT domains to form a high-molecular-weight complex that triggers procaspase-1 activation when it is stimulated (22). The pathogenic agents that activate the NLRP3 inflammasome include (1) the fungi Saccharomyces cerevisiae and Candida albicans that function via the Syk signaling pathway (34); (2) a pore-forming toxin-producing bacteria (35); and (3) viruses, including the influenza virus, adenovirus, and the Sendai virus (36, 37).
The NLRP3 inflammasome is activated by a wide range of stimuli. For example, the NLRP3 inflammasome detects signals produced by metabolism, such as increased extracellular glucose levels, which is an essential manifestation of diabetes (38). However, given their structural and chemical dissimilarity, NLRP3 is not likely to be activated through a direct interaction with its stimuli (39). Researchers have speculated that different agonists will lead to a common cellular event that ultimately activates the NLRP3 inflammasome.
When NLRP3 is not activated, the LRR domain interacts with HSP90 (heat-shock protein 90) and the ubiquitin ligase-associated protein SGT1HSP90, which are likely to maintain NLRP3 in an inactive but signaling-competent state (40). Two types of signals are needed to activate the NLRP3 inflammasome (41). First, a ligand binds to TLR4 on the membrane to provide the first signal that induces the expression of NLRP3, IL-1β, and IL-18 by triggering the NF-kB signaling pathway (42). In addition, TLR4 may provide the first signal via an unknown mechanism through the proteins myD88 and IRAK1. A low level of TLR4 stimulation is sufficient for the ATP activation pathway, and this pathway does not require the synthesis of new proteins (43–45). Because the expression of endogenous NLRP3 in immune cells is not sufficient to activate the NLRP3 inflammasome, the activation of NF-kB is necessary for the sufficient production of NLPR3. The second signal is the appearance of the activator of the NLRP3 inflammasome. The NLRP3 inflammasome begins to assemble when it is stimulated (46).
The mechanisms underlying the activation of the NLRP3 inflammasome are still not completely understood and may be associated with ROS (reactive oxygen species) production, lysosomal damage, P2X7R (purinergic ligand-gated ion channel 7 receptor) activation, and K+ efflux. To date, three models explaining the activation of the NLRP3 inflammasome have been acknowledged by most researchers.
The first model concerns the efflux of K+, which is the most common mechanism of NLRP3 inflammasome activation. A decreasing cytosolic level of K+ induced by NLRP3 stimuli, ATP, or nigericin mediates IL-1β activation and release in mouse macrophages and human monocytes (47, 48). Moreover, the efflux of K+ alone results in the activation of NLRP3, and a high extracellular K+ concentration inhibits NLRP3 activation (49). Therefore, the intracellular hypokalemia that induces mitochondrial damage and the subsequent release of ROS and mtDNA (mitochondrial DNA) is sufficient to activate the NLRP3 inflammasome (49, 50). In addition, K+ efflux is necessary to activate NLRP3 in caspase-11-mediated non-canonical inflammasome signaling (6, 51).
Three explanations for K+ efflux have been proposed. First, bacterial toxins destroy the integrity of the cell membrane, and thus K+ flows out along the ion concentration gradient (52). Second, the combination of extracellular ATP and the pyrogenic P2X7 ATP-gated ion channel (53) triggers K+ efflux. Another type of channel, pannexin-1, also participates in the activation of NLRP3 via other ATP-dependent pathways (54–56). Third, microbial molecules are delivered to the cytosol in a pannexin 1-independent manner (54). The generation of pores disrupts the intracellular K+ concentration gradient and transports bacterial molecules to the cytosol, which may help clarify how bacteria that do not exist in the cytosol activate cytosolic sensors.
Although K+ efflux has been considered the most common mechanism to activate the NLRP3 inflammasome, recent reports have indicated that some small molecules, including CL097 and GB111-NH2, activate NLRP3 independently of K+ efflux (57). Moreover, an NLRP3 mutant leads to inflammasome activation induced by lipopolysaccharides in the absence of K+ efflux in mouse macrophages (58). In conclusion, K+ efflux is sufficient, but not unique, in activating this inflammasome. Further investigations are needed to elucidate the underlying mechanisms by which NLRP3 senses alterations in the intracellular K+ concentration.
The second model concerns lysosomal damage. Particulate matter, such as MSU, and adjuvants including alum (59, 60) activate the NLRP3 inflammasome in macrophages. The phagocytosis of specific particulate structures and crystalline structures results in lysosomal membrane disintegration and damage and the cytosolic release of lysosomal contents, which are sensed by the NLRP3 inflammasome to some extent.
Lysosomal disruption triggered by Leu-Leu-OMe activates the NLRP3 inflammasome (61). However, the exact mechanisms by which lysosomal damage contributes to the activation of NLRP3 remain obscure. Currently, two factors, lysosomal acidification and cathepsins, have been identified to be associated with the activation mechanisms. An H+ ATPase inhibitor blocks NLRP3 inflammasome activation induced by particulate matter in macrophages (61). Additionally, both in vitro and in vivo experiments suggest that inhibitors of lysosomal acidification suppress IL-1β production (62). In fact, the acidic conditions tend to cause Na+ release and increase cellular osmolarity and subsequent water influx, resulting in intracellular hypokalemia (62). Moreover, lysosomal rupture leads to enzyme release and the activation of the NLRP3 inflammasome. These proteases suppress the activation of negative regulators and increase the activation of NLRP3 through proteolytic reactions, which lead to inflammasome assembly (63). Lysosomal protease CTSB (cathepsin B) plays an important role in the model. CTSB inhibitors attenuate NLRP3 activation in macrophages treated with particulate matter (6). Furthermore, lysosomal CTSB release is required for IL-1β secretion, indicating the participation of CTSH in NLRP3 activation (64). Therefore, the cytosolic release of lysosomal contents is another mechanism of NLRP3 inflammasome activation. However, CTSB-deficient mouse macrophages show normal caspase-1 activation and IL-1β maturation induced by particulate NLRP3 agonists, suggesting that some undefined mechanisms exist (65). Further research is required to solve the existing conflicts and clarify the actual role of lysosomal damage in NLRP3 inflammasome activation.
The third model concerns the generation of ROS. In this model, all agonists of NLRP3 induce ROS production, and this collective pathway involves the NLRP3 inflammasome (66–68). All NLRP3 agonists that have been confirmed, including particulate activators and ATP, induce ROS production, and chemical scavengers that block ROS generation inhibit inflammasome activation (34, 66–69). Consistent with the role of ROS production, the activation of caspase-1 by asbestos is suppressed in NAC (N-acetyl cysteine)-treated cells, in which NAC inhibits ROS generation (67). The source of ROS is currently unknown, but NADPH oxidases may be associated with their production, as in vitro studies indicate that inhibition of the common p22 subunit, which plays a critical role in ROS formation, suppresses inflammasome activation (67). However, the genetic or pharmacological blockade of NADPH oxidase does not affect NLRP3 activation in both mouse and human cells. The tissue-specific role of ROS may explain the differences in the activation of NLRP3 inflammasome. NOX2 (NADPH oxidase 2) knockout mice were recently shown to display decreased production, assembly and activation of the NLRP3 inflammasome in the injured cerebral cortex, but not in the umbilical vein endothelium.
The mechanisms underlying ROS-dependent NLRP3 inflammasome activation remain to be revealed in more detail. Recently, a ROS-sensitive NLRP3 ligand, TXNIP/VDUP1 (thioredoxin-interacting protein), was shown to be involved in NLRP3 activation (38, 70). When cellular phagocytosis is dysfunctional, activators such as uric acid crystals increase ROS production and simultaneously trigger the dissociation of TXNIP from TRX (thioredoxin). TXNIP has been identified as a common binding partner of TRX (71). TXNIP decreases the reductase activity of TRX by directly interacting with the redox-active part of TRX. A yeast two-hybrid screen using the LRRs of NLRP3 as bait revealed that TXNIP is also a potential binding partner of NLRP3 (72, 73). Overexpressed TXNIP and endogenous TXNIP interact with the LRR region of NLRP3, and the nucleotide-binding NACHT domain of NLRP3 also interacts with TXNIP. NLRP3 detects the presence of ROS, the production of which in cells is directly or indirectly induced by activators of the NLRP3 inflammasome. The complex formed by TXNIP and TRX senses increasing amounts of ROS and causes the dissociation of the complex. Subsequently, the interaction of TXNIP and NLRP3 activates NLRP3, recruits ASC and procaspase-1, and leads to the assembly of the active NLRP3 inflammasome. Intriguingly, accumulating evidence indicates that TXNIP is associated with glucose metabolism and diabetes (74). In pancreatic beta-cells, the expression of TXNIP is downregulated by insulin and is consistently increased in patients diagnosed with T2DM (type 2 diabetes mellitus) (74). Additionally, mutations in TXNIP are associated with reduced plasma glucose levels and hypertriglyceridemia (75). Published data that have been confirmed suggest that the expression of TXNIP is substantially upregulated by exposure to high glucose concentrations in pancreatic islet cells (76, 77). Although the ROS model is supported by many studies, many questions still remain and need to be resolved. For example, researchers have not clarified whether the mechanism by which superoxide directly inhibits caspase-1 activity by regulating redox-sensitive cysteines (78) provides dose- or temporal-dependent negative feedback to limit the function of caspase-1 triggered by a ROS-dependent NLRP3 pathway.
In recent years, mitochondria have been shown to play an essential role in the activation of the NLRP3 inflammasome (79, 80). Mitochondria are an ideal platform to assemble the NLRP3 inflammasome. On the other hand, NLRP3 is directly affected by molecules from mitochondria, such as mitochondrial ROS (mtROS), mtDNA, and cardiolipin.
NLRP3 promotes the secretion of IL-33, IL-18, and IL-1β, which are very important molecules that control pathological infections. However, the excessive production of cytokines exerts a deleterious effect on the body. For instance, the excessive activation of proinflammatory cytokines, including TNF-α, IL-1β, and IFNs, is associated with autoimmune diseases, such as T1DM. Therefore, the activation of the NLRP3 inflammasome must be strictly regulated to maintain the balance of the internal environment and homeostasis. Four negative regulatory mechanisms of the NLRP3 inflammasome have been identified.
The first mechanism is associated with negative regulatory molecules. A group of small proteins that consist of either a PYD or a CARD domain have emerged as key regulators of the inflammasome. As two types of endogenous dominant-negative proteins, both COPs (CARD-only proteins) and POPs (PYD-only proteins) decrease the activity of the NLRP3 inflammasome in response to tissue injury and pathogen infection (81).
POPs, such as POP1 and POP2, which display 64 and 37% homology with the PYD subunit of ASC, respectively, prevent ASC recruitment to NLRP3 by interacting with ASC in a PYD-dependent manner and replacing other proteins that interact with ASC (82). In vitro overexpression models confirm that POP1 and POP2 bind to ASC and block the interaction between NLRP3 and ASC (83). Moreover, in vivo studies using transgenic mice expressing POP2 have revealed decreased inflammatory cytokine levels in response to LPS, and the animals tend to resist bacterial infections compared with wild-type mice (84). To date, five proteins belonging to the COP family have been identified, including Iceberg, Nod2-S, caspase-12s, COP1/pseudo-ICE and INCA (27). COPs, which are extremely similar to the CARD subunit of procaspase-1, function as decoy proteins by isolating caspase-1 through CARD domain interactions and preventing its binding to activating adaptors (83). Because the expression of Iceberg is increased in the inflammatory environment, this protein appears to function as a negative feedback regulator that inhibits systemic inflammation. Notably, our understanding of the regulatory effects of POPs and COPs on NLRP3 activation is limited because these molecules are not expressed in mice. However, the development of transgenic mice provides a great opportunity to further analyze these proteins.
The second mechanism involves certain cells and cytokines. Various immunocytes and proinflammatory cytokines participate in the downregulation of inflammasome activation. For example, human-derived activated memory T cells negatively regulate the P2X7R signaling pathway, leading to the inhibition of the NLRP3 inflammasome (85). C5aR2 (C5a receptor 2), which is expressed on the T cell surface, inhibit NLRP3 inflammasome assembly by inversely modulating C5 activation and stimulating C5aR1 (C5a receptor 1) (85, 86). IL-1β signaling promotes the recruitment of neutrophils; in turn, increased neutrophil apoptosis results in the resolution of the inflammatory response (87). The type I interferon signaling pathway represses the activity of the NLRP3 inflammasome and inhibits the maturation of IL-1β through the STAT1 transcription factor (88).
The third mechanism is associated with autophagy. Autophagy, also referred to as macroautophagy, is a conserved process involving the transport of the cytoplasmic content to lysosomes via autophagosomes, and the substrates degraded by this process mainly include long-lived proteins, intracellular pathogens and organelles. Autophagy is involved in the pathogenesis of many inflammatory disorders and modulates many aspects of the immune response, including inflammation (89, 90).
Autophagy inhibits the formation of the NLRP3 inflammasome by degrading impaired mitochondria, decreasing mtROS production and disaggregating the ASC complex (91). In vitro experiments using macrophages from mouse models indicate that the depletion of beclin 1 and LC3B, proteins that are associated with autophagy, increases the activation of caspase-1 and release of IL-18 and IL-1β by impairing mitochondrial homeostasis (50, 92, 93). Additionally, autophagy dysfunction, whether it is caused by autophagy deficiency or mitochondrial inhibitors, increases the activation of the NLRP3 inflammasome via the production of mtROS (92). Consistent with the result obtained from cell-based experiments, in vivo studies suggest that LC3B-dificient mice produce caspase-1-depedent cytokines at higher levels and exhibit a greater susceptibility to LPS (92). Therefore, autophagy is closely associated with the well-being of mitochondria, and autophagy and the mitochondria modulate the NLRP3-dependent inflammatory response together.
Finally, the other mechanisms, including the production of NO (nitric oxide) and CO (carbon monoxide), negatively regulate the NLRP3 inflammasome. NO regulates multiple physiological responses and defends against pathogens. Endogenous NO downregulates NLRP3 inflammasome activation and stabilizes mitochondria. According to the results of in vitro experiments, NO blocks NLRP3-mediated caspase-1 and IL-1β activation in mice and human myeloid cells (94). Additionally, decreased production of NO caused by iNOS (inducible NO synthase) elimination or pharmacological blockade leads to increased cytokine production induced by the activated NLRP3 inflammasome and the accumulation of dysfunctional mitochondria in vivo (94). CO is toxic and damages the respiratory system. However, endogenous CO possesses anti-inflammatory properties. CO inhibits the production of IL-1β induced by inflammasomes and suppresses the activation of the NLRP3 inflammasome in bone marrow-derived macrophages; furthermore, CO inhibits mtROS generation, preserves the integrity of mitochondrial membrane, and prevents mtDNA translocation into cytosol (95, 96). Therefore, we conclude that NO and CO negatively modulate the activation of the NLRP3 inflammasome mainly by stabilizing mitochondria.
The NLRP3 inflammasome has consistently been shown to participate in the pathogenesis of many autoimmune disorders, including MS (multiple sclerosis), EAE (experimental autoimmune encephalomyelitis), IBD (inflammatory bowel disease) and T1DM (97–100). T1DM is a metabolic disease characterized by an absolute deficiency in insulin and subsequent hyperglycemia resulting from an autoimmune assault. Autoreactive T cells infiltrate pancreatic islets and induce insulitis, causing beta-cell death (101). In addition to adaptive immunity, innate immunity plays an important role in the pathogenesis of T1DM. Based on accumulating evidence, among all components of the innate immune system, the NLRP3 inflammasome and its downstream cytokines, particularly IL-1β, are involved in the development of T1DM (Figure 2) (102, 103).
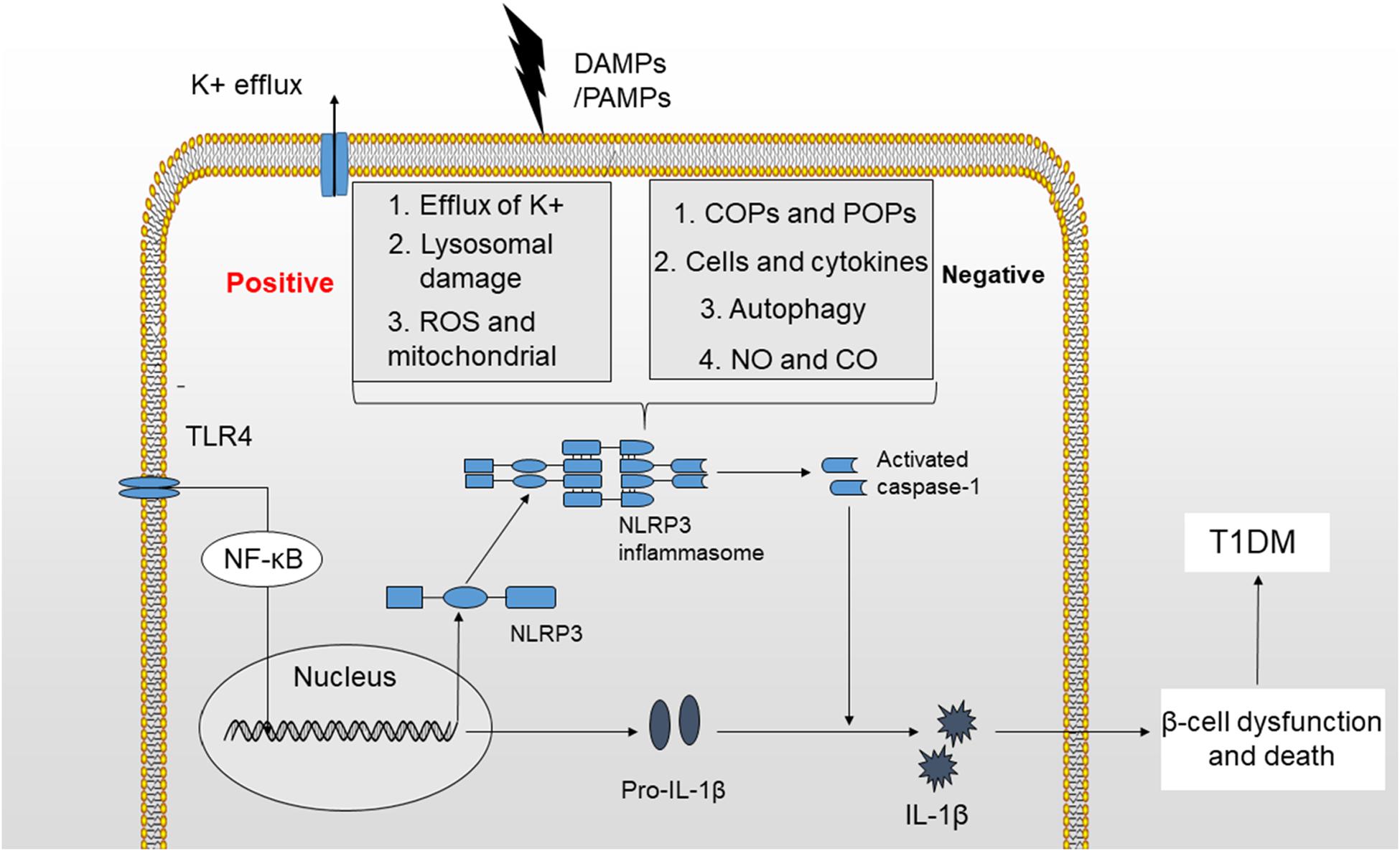
Figure 2. Mechanisms of NLRP3 inflammasome activation. The activation of the NLRP3 inflammasome requires two signals. (1) TLR4 stimulation increases the production of NLRP3 and pro-IL-1β by activating the NF-κB signaling pathway. (2) K+ efflux, cathepsin release by ruptured lysosomes and ROS generation provide a second signal that may activate the NLRP3 inflammasome and produce activated caspase-1, leading to the maturation of IL-1β. The activation of NLRP3 inflammasome is potentially negatively regulated by small molecules, such as COPs and POPs, cells and cytokines, autophagy, NO, and CO. Finally, IL-1β induces beta-cell dysfunction and death, which will ultimately lead to the development of T1DM.
IL-1β induces the migration of proinflammatory cells into pancreatic islets, mediates cytokine-induced beta-cell apoptosis, exerts direct cytotoxic effects on beta-cells, and may function as inflammatory signal in the early stage of T1DM (102, 104, 105). IL-1β levels are increased in both patients with a new T1DM diagnosis and patients with chronic T1DM compared with healthy controls, and after treatment, IL-1β levels are decreased in patients who have been newly diagnosed with T1DM (106–108). Furthermore, the levels of IL-1 receptor antagonist (IL-1RA), which inhibits the interaction between IL-1β and its receptor and blocks downstream signaling, are decreased in islets from non-diabetic donors exposed to sera from patients with T1DM, and decreased expression of IL-1RA not only results in insulin-producing beta-cell dysfunction and death but also IL-1β production, thus further affecting beta-cells (109). Additionally, NOD mice pretreated with IL-1RA exhibit reduced chemical-induced hyperglycemia, but not islet inflammation (110). Based on these findings, some new treatment strategies aiming to suppress IL-1β activity, such as synthetic IL-1RA and IL-1β traps, have been developed to reverse or ameliorate autoimmune diseases. Indeed, patients with T1DM have decreased requirements for insulin and similar HbA1c (hemoglobin A1c) levels after IL-1RA treatment (102, 111). However, some discrepancies remain to be solved. Animal experiments with NOD caspase-1(–/–) mice revealed reduced IL-1 levels, but an unchanged incidence of diabetes and sensitivity to streptozotocin compared with wild type NOD mouse models (112). At a minimum, caspase-1-mediated IL-1β production is not indispensable for diabetes development in NOD mice. Another in vivo experiment also indicates that CD4 + T cell-induced beta-cell death and diabetes is independent of IL-1 and IL-18 in NOD mice (113). Moreover, the larger, multicenter preclinical studies did not observe synergistic effects of IL-1 blockade and anti-CD3 therapy on new-onset autoimmune diabetes in NOD mouse models (114). In conclusion, the role of IL-1β in T1DM pathogenesis is not completely understood and further research is required before its clinical application.
Because IL-1β exerts a systemic effect on immunological intolerance and plays a potential role in T1DM development, its upstream activator, the NLRP3 inflammasome, is a functionally plausible complex contributing to the pathogenesis of T1DM. that the expression of the NLRP3 inflammasome in human pancreatic islets is regulated by LPS (115). Interestingly, recent genetic association studies have indicated a potential association between polymorphisms in inflammasome genes and an increased risk of developing T1DM. As shown in our previous study, SNPs (single-nucleotide polymorphisms) in the NLRP1 gene are associated with T1DM, as well as the age of onset in Chinese Han patients with T1DM (100). More importantly, two SNPs within NLRP3 were found to be associated with an increased risk of T1DM and celiac disease in a separate study. An association between T1DM and a risk SNP (rs10754558) within NLRP3 in the northeastern Brazilian population was identified in a human study (116). However, the contribution of this mutation to the genetic predisposition should be further confirmed in other populations and its resulting function requires further study. In addition, the diabetogenic role of NLRP3 has been observed in animal experiments. NLRP3 was recently shown to play an important role in the immune development of T1DM in NOD mice (103). NLRP3 deficiency affects the activation and maturation of T cells, and more importantly, the elimination of NLRP3 alters the migration of T cells to pancreatic islets, which is a critical pathogenic process that causes beta-cell damage. Furthermore, NLRP3 knockout downregulates the expression of the chemokines CCL5 (C-C motif ligand 5) and CXCL10 (C-X-C motif ligand 10) in pancreatic islet cells in an IRF-1-dependent manner, suggesting that it regulates chemotaxis (103). Moreover, mtDNA-mediated NLRP3 activation induces caspase-1-dependent IL-1β production and contributes to STZ (streptozotocin)-induced T1DM in a murine model, directly indicating a diabetogenic effect of NLRP3-caspase-1-IL-1β signaling (117). Recently, mtDNA was also shown to be involved in the vascular dysfunction in individuals with T1DM, highlighting the association of the NLRP3 inflammasome with diabetic complications (118). Other studies have confirmed that glibenclamide and metformin, both of which are typical treatments for diabetes, have the potential to inhibit the activation of the NLRP3 inflammasome, indirectly indicating that the NLRP3 inflammasome is associated with T1DM. In summary, all of the evidence mentioned above indicates a close relationship between T1DM and the NLRP3 inflammasome.
However, some questions regarding the NLRP3 inflammasome remain to be explored and resolved. The inflammasome exerts a protective effect on maintaining immune homeostasis (119–121). More importantly, the expression of the NLRP3 inflammasome is downregulated in patients with SLE compared with healthy controls and is negatively correlated with disease activity, indicating a protective effect of the inflammasome on SLE (122). Consistent with these findings, another study examining T1DM also indicated that downregulated NLRP3 inflammasome signaling participates in the early stage of autoimmune diabetes (123). Further studies are required to investigate whether the downregulation of the NLRP3 inflammasome is an outcome or cause of the progression of autoimmune disorders. Moreover, some inflammasome-independent pathways activate IL-1β and are potentially involved in development of T1DM (124, 125), indicating that the onset of T1DM is caused by complex networks rather than a single pathway. In summary, a better understanding of the NLRP3 inflammasome is still needed to completely ascertain its effect on the pathogenesis of T1DM and develop new treatment strategies.
Researchers have improved their knowledge of the NLRP3 inflammasome. We have identified many different activators and a close relationship with inflammatory diseases, including T1DM. However, the regulatory mechanisms and their function in the development of the disease must be further clarified. The discovery of the NLRP3 inflammasome has provided a new opportunity to explore the pathogenesis of inflammation-related diseases. In-depth research into the NLRP3 inflammasome, which is a regulator of IL-18 and IL-1β, will provide a new strategy for the treatment and prevention of inflammatory diseases.
Currently, the pathogenesis of T1DM is not completely understood. However, both environmental and genetic factors are involved in the onset and development of T1DM. The overactivation of the immune system, including innate immune responses, resulting from predisposing genetic mutations is the main cause of the loss and dysfunction of beta-cells. The NLRP3 inflammasome is much more likely to predispose an individual to the onset of T1DM. However, the mechanisms of NLRP3 inflammasome activation and regulation remain obscure, and the exact role of this inflammasome in the pathogenesis of T1DM requires further investigation. We propose that immunotherapy targeting the NLRP3 inflammasome is a promising approach to fight T1DM.
XS performed the literature search and wrote the first draft of the manuscript. HP generated the figures and revised the text. JL, SL, GH, and XL critically revised the text and provided substantial scientific contributions. ZZ and ZX proposed the project and revised the manuscript. All authors approved the final version of the manuscript.
This review was supported by grants from the National Natural Science Foundation of China (81873634 and 81400783), the National Key R&D Program of China (2016YFC1305000, 2016YFC1305001, and 2018YFC1315603), the Science and Technology Major Project of Hunan Province (2017SK1020), and Hunan Province Natural Science Foundation of China (2018JJ2573 and 2020JJ2053).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Daskalaki, MG, Tsatsanis, C, Kampranis, SC. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J Cell Physiol. (2018). 233:6495–507. doi: 10.1002/jcp.26497
2. Zhang, C, Boini, KM, Xia, M, Abais, JM, Li, X, Liu, Q, et al. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension. (2012) 60:154–62. doi: 10.1161/HYPERTENSIONAHA.111.189688
3. Song, N, Li, T. Regulation of NLRP3 inflammasome by phosphorylation. Front Immunol. (2018) 9:2305. doi: 10.3389/fimmu.2018.02305
4. Moossavi, M, Parsamanesh, N, Bahrami, A, Atkin, SL, Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol Cancer. (2018) 17:158. doi: 10.1186/s12943-018-0900-3
5. Stutz, A, Golenbock, DT, Latz, E. Inflammasomes: too big to miss. J Clin Invest. (2009) 119:3502–11. doi: 10.1172/JCI40599
6. Kelley, N, Jeltema, D, Duan, Y, He, Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
7. Yu, JW, Lee, MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res. (2016) 39:1503–18. doi: 10.1007/s12272-016-0827-4
8. DiMeglio, LA, Evans-Molina, C, Oram, RA. Type 1 diabetes. Lancet. (2018) 391:2449–62. doi: 10.1016/S0140-6736(18)31320-5
9. Esposito, S, Toni, G, Tascini, G, Santi, E, Berioli, MG, Principi, N. Environmental factors associated with type 1 diabetes. Front Endocrinol. (2019) 10:592. doi: 10.3389/fendo.2019.00592
10. Wang, Z, Xie, Z, Lu, Q, Chang, C, Zhou, Z. Beyond genetics: what causes type 1 diabetes. Clin Rev Allergy Immunol. (2017) 52:273–86. doi: 10.1007/s12016-016-8592-1
11. Xia, Y, Xie, Z, Huang, G, Zhou, Z. Incidence and trend of type 1 diabetes and the underlying environmental determinants. Diabetes Metab Res Rev. (2019) 35:e3075. doi: 10.1002/dmrr.3075
12. Gao, S, Wolanyk, N, Chen, Y, Jia, S, Hessner, MJ, Wang, X. Investigation of coordination and order in transcription regulation of innate and adaptive immunity genes in type 1 diabetes. BMC Med Genomics. (2017) 10:7. doi: 10.1186/s12920-017-0243-8
13. Cabrera, SM, Henschel, AM, Hessner, MJ. Innate inflammation in type 1 diabetes. Transl Res. (2016) 167:214–27. doi: 10.1016/j.trsl.2015.04.011
14. Huang, J, Xiao, Y, Xu, A, Zhou, Z. Neutrophils in type 1 diabetes. J Diabetes Investig. (2016) 7:652–63. doi: 10.1111/jdi.12469
15. Medzhitov, R, Janeway, C Jr. Innate immunity. N Engl J Med. (2000) 343:338–44. doi: 10.1056/NEJM200008033430506
16. Kubelkova, K, Macela, A. Innate immune recognition: an issue more complex than expected. Front Cell Infect Microbiol. (2019) 9:241. doi: 10.3389/fcimb.2019.00241
17. Chu, H, Mazmanian, SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. (2013) 14:668–75. doi: 10.1038/ni.2635
18. Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature. (2007) 449:819–26. doi: 10.1038/nature06246
19. Brubaker, SW, Bonham, KS, Zanoni, I, Kagan, JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
20. Patin, EC, Thompson, A, Orr, SJ. Pattern recognition receptors in fungal immunity. Semin Cell Dev Biol. (2019) 89:24–33. doi: 10.1016/j.semcdb.2018.03.003
21. Hamilton, C, Anand, PK. Right place, right time: localisation and assembly of the NLRP3 inflammasome. F1000Res. (2019) 8:F1000 Faculty Rev-676. doi: 10.12688/f1000research.18557.1
22. Schroder, K, Tschopp, J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
23. Eisenbarth, SC, Colegio, OR, O’Connor, W, Sutterwala, FS, Flavell, RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. (2008) 453:1122–6. doi: 10.1038/nature06939
24. Qiu, Z, Lei, S, Zhao, B, Wu, Y, Su, W, Liu, M, et al. NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev. (2017) 2017:9743280. doi: 10.1155/2017/9743280
25. Wang, S, Yuan, YH, Chen, NH, Wang, HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int Immunopharmacol. (2019) 67:458–64. doi: 10.1016/j.intimp.2018.12.019
26. Yu, ZW, Zhang, J, Li, X, Wang, Y, Fu, YH, Gao, XY. A new research hot spot: the role of NLRP3 inflammasome activation, a key step in pyroptosis, in diabetes and diabetic complications. Life Sci. (2020) 240:117138. doi: 10.1016/j.lfs.2019.117138
27. Swanson, KV, Deng, M, Ting, JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
28. Shi, J, Zhao, Y, Wang, K, Shi, X, Wang, Y, Huang, H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
29. Liu, X, Zhang, Z, Ruan, J, Pan, Y, Magupalli, VG, Wu, H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
30. Evavold, CL, Ruan, J, Tan, Y, Xia, S, Wu, H, Kagan, JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. (2018) 48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013
31. Monteleone, M, Stanley, AC, Chen, KW, Brown, DL, Bezbradica, JS, von Pein, JB, et al. Interleukin-1beta maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. (2018) 24:1425–33. doi: 10.1016/j.celrep.2018.07.027
32. Brennan, MA, Cookson, BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. (2000) 38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x
33. Fink, SL, Cookson, BT. Pillars article: caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006. 8: 1812-1825. J Immunol. (2019) 202:1913–26. doi: 10.1111/j.1462-5822.2006.00751.x
34. Gross, O, Poeck, H, Bscheider, M, Dostert, C, Hannesschlager, N, Endres, S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. (2009) 459:433–6. doi: 10.1038/nature07965
35. Mariathasan, S, Weiss, DS, Newton, K, McBride, J, O’Rourke, K, Roose-Girma, M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. doi: 10.1038/nature04515
36. Kanneganti, TD, Body-Malapel, M, Amer, A, Park, JH, Whitfield, J, Franchi, L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. (2006) 281:36560–8. doi: 10.1074/jbc.M607594200
37. Muruve, DA, Petrilli, V, Zaiss, AK, White, LR, Clark, SA, Ross, PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. (2008) 452:103–7. doi: 10.1038/nature06664
38. Zhou, R, Tardivel, A, Thorens, B, Choi, I, Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. (2010) 11:136–40. doi: 10.1038/ni.1831
39. Lamkanfi, M, Kanneganti, TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol. (2010) 42:792–5. doi: 10.1016/j.biocel.2010.01.008
40. Mayor, A, Martinon, F, De Smedt, T, Petrilli, V, Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol. (2007) 8:497–503. doi: 10.1038/ni1459
41. Ding, S, Xu, S, Ma, Y, Liu, G, Jang, H, Fang, J. Modulatory mechanisms of the nlrp3 inflammasomes in diabetes. Biomolecules. (2019) 9:850. doi: 10.3390/biom9120850
42. Kesavardhana, S, Kanneganti, TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol. (2017) 29:201–10. doi: 10.1093/intimm/dxx018
43. Fernandes-Alnemri, T, Kang, S, Anderson, C, Sagara, J, Fitzgerald, KA, Alnemri, ES. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol. (2013) 191:3995–9. doi: 10.4049/jimmunol.1301681
44. Juliana, C, Fernandes-Alnemri, T, Kang, S, Farias, A, Qin, F, Alnemri, ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. (2012) 287:36617–22. doi: 10.1074/jbc.M112.407130
45. Schroder, K, Sagulenko, V, Zamoshnikova, A, Richards, AA, Cridland, JA, Irvine, KM, et al. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology. (2012) 217:1325–9. doi: 10.1016/j.imbio.2012.07.020
46. Jo, EK, Kim, JK, Shin, DM, Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. (2016) 13:148–59. doi: 10.1038/cmi.2015.95
47. Perregaux, D, Gabel, CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. (1994) 269:15195–203.
48. Walev, I, Reske, K, Palmer, M, Valeva, A, Bhakdi, S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. (1995) 14:1607–14. doi: 10.1002/j.1460-2075.1995.tb07149.x
49. Munoz-Planillo, R, Kuffa, P, Martinez-Colon, G, Smith, BL, Rajendiran, TM, Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
50. Zhou, R, Yazdi, AS, Menu, P, Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
51. Ruhl, S, Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. (2015) 45:2927–36. doi: 10.1002/eji.201545772
52. Sutterwala, FS, Ogura, Y, Zamboni, DS, Roy, CR, Flavell, RA. NALP3: a key player in caspase-1 activation. J Endotoxin Res. (2006) 12:251–6. doi: 10.1179/096805106X118771
53. Kahlenberg, JM, Dubyak, GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. (2004) 286:C1100–8. doi: 10.1152/ajpcell.00494.2003
54. Kanneganti, TD, Lamkanfi, M, Kim, YG, Chen, G, Park, JH, Franchi, L, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. (2007) 26:433–43. doi: 10.1016/j.immuni.2007.03.008
55. Pelegrin, P, Barroso-Gutierrez, C, Surprenant, A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol. (2008) 180:7147–57. doi: 10.4049/jimmunol.180.11.7147
56. Pelegrin, P, Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. (2006) 25:5071–82. doi: 10.1038/sj.emboj.7601378
57. Gross, CJ, Mishra, R, Schneider, KS, Medard, G, Wettmarshausen, J, Dittlein, DC, et al. K(+) efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. (2016) 45:761–73. doi: 10.1016/j.immuni.2016.08.010
58. Meng, G, Zhang, F, Fuss, I, Kitani, A, Strober, W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. (2009) 30:860–74. doi: 10.1016/j.immuni.2009.04.012
59. Sharp, FA, Ruane, D, Claass, B, Creagh, E, Harris, J, Malyala, P, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci USA. (2009) 106:870–5. doi: 10.1073/pnas.0804897106
60. Franchi, L, Nunez, G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol. (2008) 38:2085–9. doi: 10.1002/eji.200838549
61. Hornung, V, Bauernfeind, F, Halle, A, Samstad, EO, Kono, H, Rock, KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. (2008) 9:847–56. doi: 10.1038/ni.1631
62. Schorn, C, Frey, B, Lauber, K, Janko, C, Strysio, M, Keppeler, H, et al. Sodium overload and water influx activate the NALP3 inflammasome. J Biol Chem. (2011) 286:35–41. doi: 10.1074/jbc.M110.139048
63. Bruchard, M, Mignot, G, Derangere, V, Chalmin, F, Chevriaux, A, Vegran, F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. (2013) 19:57–64. doi: 10.1038/nm.2999
64. Weber, K, Schilling, JD. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem. (2014) 289:9158–71. doi: 10.1074/jbc.M113.531202
65. Dostert, C, Guarda, G, Romero, JF, Menu, P, Gross, O, Tardivel, A, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One. (2009) 4:e6510. doi: 10.1371/journal.pone.0006510
66. Cassel, SL, Eisenbarth, SC, Iyer, SS, Sadler, JJ, Colegio, OR, Tephly, LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci USA. (2008) 105:9035–40. doi: 10.1073/pnas.0803933105
67. Dostert, C, Petrilli, V, Van Bruggen, R, Steele, C, Mossman, BT, Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. (2008) 320:674–7. doi: 10.1126/science.1156995
68. Cruz, CM, Rinna, A, Forman, HJ, Ventura, AL, Persechini, PM, Ojcius, DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. (2007) 282:2871–9. doi: 10.1074/jbc.M608083200
69. Petrilli, V, Papin, S, Dostert, C, Mayor, A, Martinon, F, Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. (2007) 14:1583–9. doi: 10.1038/sj.cdd.4402195
70. Martinon, F. Signaling by ROS drives inflammasome activation. Eur J Immunol. (2010) 40:616–9. doi: 10.1002/eji.200940168
71. Nishiyama, A, Matsui, M, Iwata, S, Hirota, K, Masutani, H, Nakamura, H, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. (1999) 274:21645–50. doi: 10.1074/jbc.274.31.21645
72. Kim, SY, Suh, HW, Chung, JW, Yoon, SR, Choi, I. Diverse functions of VDUP1 in cell proliferation, differentiation, and diseases. Cell Mol Immunol. (2007) 4:345–51.
73. Kaimul, AM, Nakamura, H, Masutani, H, Yodoi, J. Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radic Biol Med. (2007) 43:861–8. doi: 10.1016/j.freeradbiomed.2007.05.032
74. Chen, J, Saxena, G, Mungrue, IN, Lusis, AJ, Shalev, A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes. (2008) 57:938–44. doi: 10.2337/db07-0715
75. van Greevenbroek, MM, Vermeulen, VM, Feskens, EJ, Evelo, CT, Kruijshoop, M, Hoebee, B, et al. Genetic variation in thioredoxin interacting protein (TXNIP) is associated with hypertriglyceridaemia and blood pressure in diabetes mellitus. Diabet Med. (2007) 24:498–504. doi: 10.1111/j.1464-5491.2007.02109.x
76. Minn, AH, Hafele, C, Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology. (2005) 146:2397–405. doi: 10.1210/en.2004-1378
77. Turturro, F, Friday, E, Welbourne, T. Hyperglycemia regulates thioredoxin-ROS activity through induction of thioredoxin-interacting protein (TXNIP) in metastatic breast cancer-derived cells MDA-MB-231. BMC Cancer. (2007) 7:96. doi: 10.1186/1471-2407-7-96
78. Meissner, F, Molawi, K, Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol. (2008) 9:866–72. doi: 10.1038/ni.1633
79. Liu, Q, Zhang, D, Hu, D, Zhou, X, Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. (2018) 103:115–24. doi: 10.1016/j.molimm.2018.09.010
80. Piantadosi, CA. Mitochondrial DNA, oxidants, and innate immunity. Free Radic Biol Med. (2020) 152:455–61. doi: 10.1016/j.freeradbiomed.2020.01.013
81. Stehlik, C, Dorfleutner, A. COPs and POPs: modulators of inflammasome activity. J Immunol. (2007) 179:7993–8. doi: 10.4049/jimmunol.179.12.7993
82. Stehlik, C, Krajewska, M, Welsh, K, Krajewski, S, Godzik, A, Reed, JC. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochem J. (2003) 373(Pt 1):101–13. doi: 10.1042/bj20030304
83. Indramohan, M, Stehlik, C, Dorfleutner, A. COPs and POPs patrol inflammasome activation. J Mol Biol. (2018) 430:153–73. doi: 10.1016/j.jmb.2017.10.004
84. Periasamy, S, Porter, KA, Atianand, MK, T Le, H, Earley, S, Duffy, EB, et al. Pyrin-only protein 2 limits inflammation but improves protection against bacteria. Nat Commun. (2017) 8:15564. doi: 10.1038/ncomms15564
85. Beynon, V, Quintana, FJ, Weiner, HL. Activated human CD4+CD45RO+ memory T-cells indirectly inhibit NLRP3 inflammasome activation through downregulation of P2X7R signalling. PLoS One. (2012) 7:e39576. doi: 10.1371/journal.pone.0039576
86. Arbore, G, West, EE, Spolski, R, Robertson, AAB, Klos, A, Rheinheimer, C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science. (2016) 352:aad1210. doi: 10.1126/science.aad1210
87. Mitroulis, I, Kambas, K, Ritis, K. Neutrophils, IL-1beta, and gout: is there a link? Semin Immunopathol. (2013) 35:501–12. doi: 10.1007/s00281-013-0361-0
88. Guarda, G, Braun, M, Staehli, F, Tardivel, A, Mattmann, C, Forster, I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. (2011) 34:213–23. doi: 10.1016/j.immuni.2011.02.006
89. Saitoh, T, Fujita, N, Jang, MH, Uematsu, S, Yang, BG, Satoh, T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. (2008) 456:264–8. doi: 10.1038/nature07383
90. Matsuzawa-Ishimoto, Y, Hwang, S, Cadwell, K. Autophagy and inflammation. Annu Rev Immunol. (2018) 36:73–101. doi: 10.1146/annurev-immunol-042617-053253
91. Sharma, D, Kanneganti, TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. (2016) 213:617–29. doi: 10.1083/jcb.201602089
92. Nakahira, K, Haspel, JA, Rathinam, VA, Lee, SJ, Dolinay, T, Lam, HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
93. Shimada, K, Crother, TR, Karlin, J, Dagvadorj, J, Chiba, N, Chen, S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
94. Mao, K, Chen, S, Chen, M, Ma, Y, Wang, Y, Huang, B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. (2013) 23:201–12. doi: 10.1038/cr.2013.6
95. Jung, SS, Moon, JS, Xu, JF, Ifedigbo, E, Ryter, SW, Choi, AM, et al. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L1058–67. doi: 10.1152/ajplung.00400.2014
96. Wegiel, B, Larsen, R, Gallo, D, Chin, BY, Harris, C, Mannam, P, et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest. (2014) 124:4926–40. doi: 10.1172/JCI72853
97. Barclay, W, Shinohara, ML. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. (2017) 27:213–9. doi: 10.1111/bpa.12477
98. Tourkochristou, E, Aggeletopoulou, I, Konstantakis, C, Triantos, C. Role of NLRP3 inflammasome in inflammatory bowel diseases. World J Gastroenterol. (2019) 25:4796–804. doi: 10.3748/wjg.v25.i33.4796
99. Soares, JL, Oliveira, EM, Pontillo, A. Variants in NLRP3 and NLRC4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult Scler Relat Disord. (2019) 29:26–34. doi: 10.1016/j.msard.2019.01.023
100. Sun, X, Xia, Y, Liu, Y, Wang, Y, Luo, S, Lin, J, et al. Polymorphisms in NLRP1 gene are associated with Type 1 diabetes. J Diabetes Res. (2019) 2019:7405120. doi: 10.1155/2019/7405120
101. Zheng, Y, Wang, Z, Zhou, Z. miRNAs: novel regulators of autoimmunity-mediated pancreatic beta-cell destruction in type 1 diabetes. Cell Mol Immunol. (2017) 14:488–96. doi: 10.1038/cmi.2017.7
102. Grishman, EK, White, PC, Savani, RC. Toll-like receptors, the NLRP3 inflammasome, and interleukin-1beta in the development and progression of type 1 diabetes. Pediatr Res. (2012) 71:626–32. doi: 10.1038/pr.2012.24
103. Hu, C, Ding, H, Li, Y, Pearson, JA, Zhang, X, Flavell, RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci USA. (2015) 112:11318–23. doi: 10.1073/pnas.1513509112
104. Vives-Pi, M, Rodriguez-Fernandez, S, Pujol-Autonell, I. How apoptotic beta-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis. (2015) 20:263–72. doi: 10.1007/s10495-015-1090-8
105. Pang, H, Luo, S, Huang, G, Xia, Y, Xie, Z, Zhou, Z. Advances in knowledge of candidate genes acting at the beta-cell level in the pathogenesis of T1DM. Front Endocrinol. (2020) 11:119. doi: 10.3389/fendo.2020.00119
106. Bradshaw, EM, Raddassi, K, Elyaman, W, Orban, T, Gottlieb, PA, Kent, SC, et al. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol. (2009) 183:4432–9. doi: 10.4049/jimmunol.0900576
107. Dogan, Y, Akarsu, S, Ustundag, B, Yilmaz, E, Gurgoze, MK. Serum IL-1beta, IL-2, and IL-6 in insulin-dependent diabetic children. Mediators Inflamm. (2006) 2006:59206. doi: 10.1155/MI/2006/59206
108. Kaizer, EC, Glaser, CL, Chaussabel, D, Banchereau, J, Pascual, V, White, PC. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab. (2007) 92:3705–11. doi: 10.1210/jc.2007-0979
109. Maedler, K, Sergeev, P, Ehses, JA, Mathe, Z, Bosco, D, Berney, T, et al. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci USA. (2004) 101:8138–43. doi: 10.1073/pnas.0305683101
110. Schwarznau, A, Hanson, MS, Sperger, JM, Schram, BR, Danobeitia, JS, Greenwood, KK, et al. IL-1beta receptor blockade protects islets against pro-inflammatory cytokine induced necrosis and apoptosis. J Cell Physiol. (2009) 220:341–7. doi: 10.1002/jcp.21770
111. Sumpter, KM, Adhikari, S, Grishman, EK, White, PC. Preliminary studies related to anti-interleukin-1beta therapy in children with newly diagnosed type 1 diabetes. Pediatr Diabetes. (2011) 12:656–67. doi: 10.1111/j.1399-5448.2011.00761.x
112. Schott, WH, Haskell, BD, Tse, HM, Milton, MJ, Piganelli, JD, Choisy-Rossi, CM, et al. Caspase-1 is not required for type 1 diabetes in the NOD mouse. Diabetes. (2004) 53:99–104. doi: 10.2337/diabetes.53.1.99
113. Wen, L, Green, EA, Stratmann, T, Panosa, A, Gomis, R, Eynon, EE, et al. In vivo diabetogenic action of CD4+ T lymphocytes requires Fas expression and is independent of IL-1 and IL-18. Eur J Immunol. (2011) 41:1344–51. doi: 10.1002/eji.201041216
114. Gill, RG, Pagni, PP, Kupfer, T, Wasserfall, CH, Deng, S, Posgai, A, et al. A preclinical consortium approach for assessing the efficacy of combined anti-CD3 plus IL-1 blockade in reversing new-onset autoimmune diabetes in NOD mice. Diabetes. (2016) 65:1310–6. doi: 10.2337/db15-0492
115. Lebreton, F, Berishvili, E, Parnaud, G, Rouget, C, Bosco, D, Berney, T, et al. NLRP3 inflammasome is expressed and regulated in human islets. Cell Death Dis. (2018) 9:726. doi: 10.1038/s41419-018-0764-x
116. Pontillo, A, Brandao, L, Guimaraes, R, Segat, L, Araujo, J, Crovella, S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity. (2010) 43:583–9. doi: 10.3109/08916930903540432
117. Carlos, D, Costa, FR, Pereira, CA, Rocha, FA, Yaochite, JN, Oliveira, GG, et al. Mitochondrial DNA activates the NLRP3 inflammasome and predisposes to type 1 diabetes in murine model. Front Immunol. (2017) 8:164. doi: 10.3389/fimmu.2017.00164
118. Pereira, CA, Carlos, D, Ferreira, NS, Silva, JF, Zanotto, CZ, Zamboni, DS, et al. Mitochondrial DNA promotes NLRP3 inflammasome activation and contributes to endothelial dysfunction and inflammation in type 1 diabetes. Front Physiol. (2019) 10:1557. doi: 10.3389/fphys.2019.01557
119. Zaki, MH, Lamkanfi, M, Kanneganti, TD. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. (2011) 32:171–9. doi: 10.1016/j.it.2011.02.002
120. Zaki, MH, Boyd, KL, Vogel, P, Kastan, MB, Lamkanfi, M, Kanneganti, TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. (2010) 32:379–91. doi: 10.1016/j.immuni.2010.03.003
121. Hirota, SA, Ng, J, Lueng, A, Khajah, M, Parhar, K, Li, Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. (2011) 17:1359–72. doi: 10.1002/ibd.21478
122. Yang, Q, Yu, C, Yang, Z, Wei, Q, Mu, K, Zhang, Y, et al. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J Rheumatol. (2014) 41:444–52. doi: 10.3899/jrheum.130310
123. Liu, H, Xu, R, Kong, Q, Liu, J, Yu, Z, Zhao, C. Downregulated NLRP3 and NLRP1 inflammasomes signaling pathways in the development and progression of type 1 diabetes mellitus. Biomed Pharmacother. (2017) 94:619–26. doi: 10.1016/j.biopha.2017.07.102
124. Netea, MG, Simon, A, van de Veerdonk, F, Kullberg, BJ, Van der Meer, JW, Joosten, LA. IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog. (2010) 6:e1000661. doi: 10.1371/journal.ppat.1000661
Keywords: T1DM, NLRP3 inflammasome, IL-1β, innate immunity, autoimmune disorder
Citation: Sun X, Pang H, Li J, Luo S, Huang G, Li X, Xie Z and Zhou Z (2020) The NLRP3 Inflammasome and Its Role in T1DM. Front. Immunol. 11:1595. doi: 10.3389/fimmu.2020.01595
Received: 15 March 2020; Accepted: 16 June 2020;
Published: 27 August 2020.
Edited by:
Michael Zemlin, Saarland University Hospital, GermanyReviewed by:
Helen Thomas, The University of Melbourne, AustraliaCopyright © 2020 Sun, Pang, Li, Luo, Huang, Li, Xie and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Xie, eGllemhpZ3VvQGNzdS5lZHUuY24=; Zhiguang Zhou, emhvdXpoaWd1YW5nQGNzdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.