 Mie K. Jakobsen
Mie K. Jakobsen Morten F. Gjerstorff
Morten F. Gjerstorff
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Immunol. , 02 September 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01568
The discovery that cancer cells are recognized and targeted by the immune system has recently increased interest in cancer immunotherapies. Currently, the most widely used immunotherapy is immune checkpoint blockade using monoclonal antibodies to block intrinsic downregulators of immunity, which are often over-expressed in cancer. Although beneficial to some patients, the majority do not respond to this treatment (1), highlighting the need for new, novel, strategies. One new approach is T-cell therapy with genetically engineered T cells to generate an effective anti-tumor immune response through ex vivo manipulation of the T-cell tumor-specificity. This can be accomplished by gene transfer of T-cell receptors (TCRs) or chimeric antigen receptors (CARs) into autologous T cells before reinfusing them into the patient. CAR T-cell therapy has shown excellent results in the treatment of some B-cell malignancies, but the lack of suitable antigens presents a challenge to transferring this therapy to other malignancies, including solid cancers (2). Here, we review data on cancer/testis (CT) antigens as targets for CAR T-cell therapy and present a strategy to upregulate CT antigen expression on tumor cells via epigenetic treatment to sensitize cancer cells to CAR T-cell therapy (Figure 1).
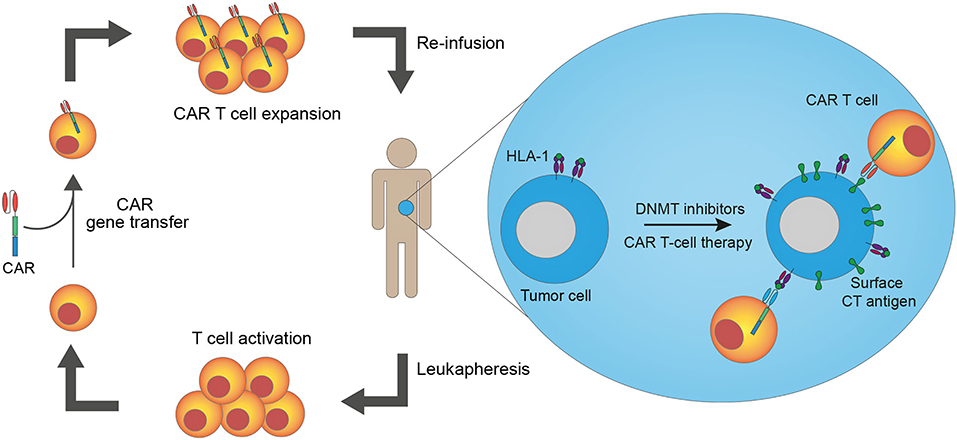
Figure 1. Epigenetic pre-treatment sensitizes cancer cells to cancer/testis antigen-directed CAR T-cell therapy. T cells from a patient are harvested through leukapheresis. The T cells are activated and genetically modified to express chimeric antigen receptors (CARs) ex vivo before they are expanded and reinfused into the patient. Pre-treatment of patients with epigenetic drugs, such as DNA methyltransferase (DNMT) inhibitors, increases cancer/testis (CT) antigen and HLA-I expression on cancer cells, leading to enhanced recognition of cancer cells by CAR T cells. Both CAR T cells made from conventional antibodies and TCR mimic antibodies (recognizing antigen peptides presented in complex with HLA-I) are available strategies.
CAR T cells are T cells genetically engineered to express artificial receptors, called CARs, on their cell surface, facilitating enhanced recognition of specific tumor antigens and thereby killing of cancer cells (3). CARs are comprised of an extracellular antigen-recognition domain, often derived from a monoclonal antibody, and an intracellular signal-transduction domain resembling that on TCRs, containing CD3ζ and up to two costimulatory domains, such as CD28 or 4-1BB. The extracellular domain provides specificity to the CAR and directs the T cell toward cancer cells through recognition of antigens on the cell surface (4). TCRs recognize epitopes presented by HLA-I molecules on the surface of tumor cells. Since antigen processing and presentation are complex, the identification of epitopes for TCRs is laborious (5). CARs, unlike normal TCRs, recognize antigens independent of HLA-antigen processing and presentation and thereby circumvent the challenges with TCR epitope identification. Also, CARs can recognize tumor cells with downregulated HLA expression or decreased protosomal processing, which are mechanisms that contribute to antigen escape by TCR-mediated immunity (6). Correspondingly, suitable targets for CARs need to be located on the cell surface for the receptor to recognize them, which makes the potential target pool smaller compared to potential targets for TCRs (2, 7). Furthermore, the HLA-unrestricted recognition of antigens by CARs makes available CAR constructs useful for patients with all HLA subtypes and facilitates an off-the-shelf strategy (8). The safety profiles of TCR and CAR T-cell therapy are similar, but there may be important differences. On-target/off-tumor toxicity is a risk of both TCR and CAR T-cell therapy and occurs when the targeted antigen is also present on healthy tissue, but CAR or affinity-maturated TCR constructs may require lower levels of antigen expression in target cells. Similarly, the risk for off-target/off-tumor toxicities, where T cells with engineered receptors cross-react with other antigens, is enhanced with CARs or affinity-maturated TCRs or may result from the combination of native and engineered TCR chains (9, 10).
The major breakthrough for CAR T-cell therapy came with the CD19-specific CAR targeting the cardinal B-cell antigen, CD19. This therapy has shown excellent results in treating multiple B-cell malignancies, e.g., Maude et al. reported 90% complete remission in patients with relapsed or refractory ALL for up to 2 years after treatment with autologous CD19 CAR T-cell therapy (11–14). Despite the great success of the CD19 CAR, relapses due to antigen loss still occur, and new targets are needed to treat these patients (15, 16). New targets are also needed for hematological cancers that do not express CD19. The success of CAR T cells in treating hematological cancers has led to this therapy receiving a large amount of attention as a treatment for solid cancers, but so far, the clinical efficacy is limited (17–19). Where target identification and antigen loss are the major obstacles in CAR T-cell therapy of hematological cancers, multiple factors contribute to the low clinical efficacy in the treatment of solid cancers. CD19 is a linage B-cell antigen also expressed on normal B cells in the early stages of B-cell development. Targeting CD19, therefore, leads to B-cell aplasia, which is clinically manageable. Targeting linage antigens on solid tumors is not an option due to severe toxicities (2, 20). The ideal target for CAR T-cell therapy should be ubiquitously expressed on all tumor cells and be completely absent from healthy tissue to avoid complications such as on-target/off-tumor toxicities, as described above. However, these antigens have proven difficult to find. Often, a heterogeneous expression pattern within the tumor tissue and/or expression in healthy tissue limits the clinical potential of the target (21). Tumor-specific antigens include neoantigens, viral antigens, and CT antigens. CT antigens have remained unrecognized as targets for CAR T-cell therapy so far, but due to the restricted expression pattern toward tumor tissue and especially the possibility of upregulating their expression by epigenetic drugs, these antigens may represent promising candidates for CAR T-cell therapy.
In solid cancers, the CAR T cells must overcome the tumor microenvironment (TME) in order to reach the tumor cells. Initially, there is the physical barrier of the stroma that may prevent T cell entry into the tumor. Next, T cells that have successfully entered the tumor may be functionally repressed by immunosuppressive factors (like PD-L1 and CTLA-4), inhibitory cytokines (e.g., TGFβ, IL-4, and IL-10) and inhibitory cells (e.g., regulatory T cells and tumor-associated macrophages) (22).
CT antigens are a unique set of antigens expressed in germ cells of the testis and various malignancies of different histological origin but not in healthy somatic tissues (23). Their cancer-restricted expression pattern, along with their immunogenic properties, make CT antigens ideal targets for immunotherapy (24, 25). Methylation of promoter-regions controlling CT antigen genes is a well-known silencer of gene expression in healthy somatic tissue (26). Malignant transformation is often associated with global DNA hypomethylation, which leads to the induction of CT antigen gene expression in some tumors. However, CT antigens show a very heterogeneous expression pattern within tumors (27), most likely reflecting epigenetic variation and plasticity among tumor cells. This may give rise to immune-escape variants in the form of tumor cells not expressing the antigen, creating an obstacle when targeting CT antigens with immunotherapy. Results from us and others show that treatment with epigenetic drugs, such as DNA methyltransferase inhibitors (DNMTis), specifically upregulates CT antigen expression within tumors, thereby inducing a more ubiquitous expression pattern of the antigens (28, 29). Therefore, epigenetic treatment can be used to sensitize cancer cells for immunotherapy, such as CAR T-cell therapy, and lead to increased elimination of cancer cells (30–34). For instance, in an immunocompetent murine breast cancer model, epigenetic priming of tumors combined with adoptive transfer was demonstrated to control metastatic spread (35).
CT antigen expression is known to be mainly intracellular, which excludes many of the antigens as CAR targets, but CT antigens with a surface localization have been identified (36–38). A recent study scrutinized 3,700 different genes predicted to encode proteins located on the surface of human cells and found 22 genes with restricted expression in testis (36), many of which were upregulated in multiple hematological and solid malignancies. These genes represent highly promising targets for CAR T-cell therapy, and further investigations should be performed to elucidate the inducibility of the antigens with DNMTis, identify surface epitopes, and explore options for CAR-targeting. Other, more well-described, CT antigens have been tested as potential CAR targets. MAGE-A1 was investigated as a possible target in the treatment of lung adenocarcinoma (LUAD) after immunostaining revealed a surface epitope of the antigen (37). A MAGE-A1-specific CAR showed cytotoxic activity both in vitro and in vivo, where it was able to infiltrate MAGE-A1-positive tumors and specifically target and inhibit LUAD xenograft growth in nude mice. Further studies are ongoing to evaluate the potential of MAGE-A1-specific CAR T cells in the treatment of LUAD. Because MAGE-A1 is expressed in multiple other cancers and can be upregulated by treatment with epigenetic modulators, MAGE-A1-specific CARs could present an attractive option for the treatment of these diseases (31, 39–41). PRAME is another well-described CT antigen that has been tested as a target in several immunotherapeutic strategies. Because PRAME was previously recognized as an intracellular protein, and therefore non-targetable by traditional antibodies or CARs, a TCR mimic antibody with the same specificity as a TCR was developed (42). This molecule specifically recognized a PRAME peptide presented in complex with HLA-A2 and provided proof-of-concept that such antibodies can recognize and generate an immune response against intracellular antigens that are otherwise only targetable with engineered TCRs. TCR mimic antibodies can be engineered into alternative formats, such as CARs or bispecific T-cell engagers (BiTEs), which may mediate effective T-cell responses against tumor cells in an HLA-restricted manner. Such strategies were pursued for the NY-ESO-1 CT antigen, demonstrating that HLA-A2/NY-ESO-1 peptide-specific CARs could mediate tumor recognition, which opens up an exciting potential for broadening the repertoire of CAR T-cell targets (43, 44). Recently, a computational transmembrane analysis predicted an extracellular region of the PRAME protein that could be specifically targeted by a conventional PRAME-specific antibody on multiple solid and hematological cancer cell lines in vitro and in vivo, thereby presenting new opportunities for additional CAR strategies targeting this protein (38). These results, and the fact that PRAME is overexpressed in many malignancies, indicate that PRAME is a promising target for CAR T-cell therapy (45, 46). Other CT antigens with a proposed surface localization include CT83, SP17, SLCO6A1, and PLAC1 (47–50). SP17 is overexpressed in multiple cancer types (48, 51–56), and expression is upregulated by DNMTis (57). SP17 is highly immunogenic (58), but SP17 expression in human ciliated cells of various normal tissues brings into question its suitability as an immunotherapeutic target (51, 59, 60). Nonetheless, SP17 vaccination of humans has been shown to be well-tolerated, with no side-effects regarding expression in normal cells (61). Further investigations must be performed to elucidate the potential of targeting SP17 by immunotherapy. CT83 is also expressed in multiple cancers, such as breast, gastric, and lung cancers, and the expression can be upregulated by DNMTis (62–67). CT83 as a target for TCR-based therapies has shown promising results (64, 68), but the potential of the antigen as a target for CAR T-cell therapy remains unexplored. Similarly, CAR T-cell therapy or alternative antibody therapy has not been pursued for SLO6A1 and PLAC1 despite interesting potential.
CARs with alternative antigen-binding domains are also being investigated to overcome the challenges with target identification. Although not a CT antigen in a strict sense, the IL13-type receptor IL13RA2 is mainly expressed in testis among healthy tissues (69). This receptor recognizes IL13 with higher affinity than the ubiquitously expressed IL13RA1 (69, 70). IL13RA2 is often overexpressed in glioblastoma multiforme (GBM), and expression is correlated with poor patient outcome. New treatment strategies for GBM are much needed, and therefore IL13RA2 is being investigated as a new therapeutic target (71). CARs with antigen-binding domains composed of IL13 mutants, with increased affinity for IL13RA2 and lowered affinity for IL13RA1, have been developed. Preclinical studies investigating these CARs (69, 70, 72), show anti-tumor efficacy and low on-target/off-tumor toxicity after intracranial delivery, due to the abscence of IL13RA2 expression in normal brain tissue. Clinical studies (NCT00730613, NCT01082926, NCT02208362) confirm these promising results and only show manageable side-effects. For example, complete remission was observed in a single patient with recurrent GBM for 7.5 months after several rounds of intracranial delivery of a second-generation IL13RA2 CAR (73). IL13RA2 is also expressed by different immune cells, and IL13RA2 expression in these cells is correlated with immune inhibition. Eradication of cells expressing IL13RA2 by CAR T cells can therefore also increase anti-tumor immunity (69).
To date, the most common immunotherapy is immune checkpoint blockade, which unleashes the activity of T cells by blocking the immune checkpoint molecules PD-1 and CTLA-4 (74). The clinical response to immune checkpoint blockade is generally most significant in patients with tumors that carry a high mutational burden, such as melanoma and non-small-cell lung cancer, but even in these cancer types, the response varies among patients (75). CAR T-cell therapy is an attractive option for non-responders to immune checkpoint blockade and for patients with less immunogenic tumors, such as breast, pancreatic, and some hematological cancers such as AML and ALL (2, 76). However, common obstacles to CAR T-cell therapy must be overcome to ensure an effective clinical response, including the identification of appropriate CAR targets. CT antigens show a restricted expression pattern toward testis and tumor cells, and combining CT antigen-specific CAR T cells with epigenetic therapy, such as DNMTis, can diminish the heterogeneous expression of these antigens within tumors. One might speculate that epigenetic modulators also induce CT antigen expression in healthy tissue to cause serious side effects, but the induction by DNMTis seems to be tumor-specific (29). This may be due to differences in chromatin organization and epigenetic control of gene expression, leaving cancer cells more susceptible to epigenetic enhancement of CT antigen expression than normal cells, but the subject needs further clarification. The safety of combining epigenetic enhancement of antigen presentation with adoptive transfer was further validated in a murine model, where no adverse effects were reported (35).
Even after upregulation of antigen expression by epigenetic modulators, antigen-escape variants, in the form of antigen-negative cells, may be present in tumors. Also, antigen escape can occur as a consequence of the highly selective pressure from mono-specific CAR T cells. Thus, targeting multiple antigens simultaneously may be required for complete responses. Different strategies to achieve this are now being investigated, e.g., pooled uni-specific CAR T cells, bi-specific CAR T cells, and tandem CAR T cells, and are showing promising results in decreasing antigen escape by tumor cells and increasing anti-tumor efficacy (77–80); for example, CAR T-cell therapy using a tandem CAR redirected against both IL13RA2 and HER2 was able to mitigate antigen escape in a murine glioblastoma model compared to uni-specific CARs (81).
Another obstacle to CAR T-cell therapy in regard to solid tumors is the observed low T-cell trafficking to tumor tissue and the hostile TME surrounding the tumor cells. It is now clear that, apart from upregulating CT antigens, DNMTis upregulate a series of immune pathways that augment tumor recognition and elimination by T cells, such as interferon signaling pathways, cytokine and chemokine signaling, inflammation, and genes in the antigen presentation and processing machinery (82–84). DNMTis cause hypomethylation of repeat elements of DNA, leading to increased activation of these regions and increased expression of endogenous retroviral dsRNA in the cytosol. The increased amount of dsRNA in the cytosol triggers a dsRNA sensing pathway as a viral defense mechanism, causing increased release of proinflammatory cytokines and interferons. These molecules act on cells in the nearby environments, leading to inhibition of cellular proliferation and release of chemokines, such as CXCL9/10, that attract cytotoxic T cells to the TME (85, 86). The molecules also have an effect on immune cells in the TME, initiating an innate immune response and increased anti-tumor immunity. Thus, DNMTis may change the hostile TME toward a more T-cell supportive state, which can augment the effect of immunotherapy when used in combination. A side-effect of DNMTis, observed in tumors, is increased expression of PD-L1 and CTLA-4 due to decreased methylation of adjacent promotor regions (87, 88). This increased expression provides the rationale for triple combination therapy of CT antigen-specific CAR T cells, epigenetic drugs, and immune checkpoint blockade.
In conclusion, epigenetic treatment can augment the clinical efficacy of CT antigen-specific CAR T-cell therapy by increasing surface CT antigen expression and diminishing the inhibitory state of the TME, and the therapeutic benefits of combining the two should be pursued through preclinical and clinical testing.
MG and MJ contributed equally to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Velux Foundation, the Danish Cancer Society (R146-A9213-16-S2), the Academy of Geriatric Cancer Research (AgeCare), the Novo Nordisk Foundation (NNF18OC0052303), the Danish Research Council for Independent Research (6108-00372A), the A.P Møller Foundation, Fabrikant Einar Willumsens Foundation, the Danish Cancer Research Foundation, and Læge Sofus Carl Emil Friis og Hustru Olga Doris Friis Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank M. K. Occhipinti for editorial assistance.
1. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. (2018) 50:165. doi: 10.1038/s12276-018-0191-1
2. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. (2017) 545:423–31. doi: 10.1038/nature22395
3. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
4. Hughes-Parry HE, Cross RS, Jenkins MR. The evolving protein engineering in the design of chimeric antigen receptor T cells. Int J Mol Sci. (2019) 21:204. doi: 10.3390/ijms21010204
5. Sharma G, Rive CM, Holt RA. Rapid selection and identification of functional CD8(+) T cell epitopes from large peptide-coding libraries. Nat Commun. (2019) 10:4553. doi: 10.1038/s41467-019-12444-7
6. Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med. (2018) 10:eaar3342. doi: 10.1126/scitranslmed.aar3342
7. Zhao L, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front Immunol. (2019) 10:2250. doi: 10.3389/fimmu.2019.02250
8. Filley AC, Henriquez M, Dey M. CART immunotherapy: development, success, and translation to malignant gliomas and other solid tumors. Front Oncol. (2018) 8:453. doi: 10.3389/fonc.2018.00453
9. Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. (2013) 39:49–60. doi: 10.1016/j.immuni.2013.07.002
10. Yang JC. Toxicities associated with adoptive T-Cell transfer for cancer. Cancer J. (2015) 21:506–9. doi: 10.1097/PPO.0000000000000157
11. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
12. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
13. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-Cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
14. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
15. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 Immunotherapy. Cancer Discov. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
16. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. (2018) 24:1504–6. doi: 10.1038/s41591-018-0146-z
17. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183
18. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
19. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor Receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-Positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
20. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2019) 17:147–67. doi: 10.1038/s41571-019-0297-y
21. Abbott RC, Cross RS, Jenkins MR. Finding the keys to the CAR: identifying novel target antigens for T cell redirection immunotherapies. Int J Mol Sci. (2020) 21:515. doi: 10.3390/ijms21020515
22. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics. (2016) 3:16006. doi: 10.1038/mto.2016.6
23. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. (2005) 5:615–25. doi: 10.1038/nrc1669
24. Fratta E, Coral S, Covre A, Parisi G, Colizzi F, Danielli R, et al. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol. (2011) 5:164–82. doi: 10.1016/j.molonc.2011.02.001
25. Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. (2015) 6:15772–87. doi: 10.18632/oncotarget.4694
26. De Smet C, Lurquin C, Lethe B, Martelange V, Boon T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol Cell Biol. (1999) 19:7327–35. doi: 10.1128/MCB.19.11.7327
27. Sigalotti L, Fratta E, Coral S, Tanzarella S, Danielli R, Colizzi F, et al. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylation-regulated and functionally reverted by 5-aza-2'-deoxycytidine. Cancer Res. (2004) 64:9167–71. doi: 10.1158/0008-5472.CAN-04-1442
28. Gjerstorff M, Burns JS, Nielsen O, Kassem M, Ditzel H. Epigenetic modulation of cancer-germline antigen gene expression in tumorigenic human mesenchymal stem cells: implications for cancer therapy. Am J Pathol. (2009) 175:314–23. doi: 10.2353/ajpath.2009.080893
29. Coral S, Covre A, Nicolay HJ, Parisi G, Rizzo A, Colizzi F, et al. Epigenetic remodelling of gene expression profiles of neoplastic and normal tissues: immunotherapeutic implications. Br J Cancer. (2012) 107:1116–24. doi: 10.1038/bjc.2012.361
30. Li B, Zhu X, Sun L, Yuan L, Zhang J, Li H, et al. Induction of a specific CD8+ T-cell response to cancer/testis antigens by demethylating pre-treatment against osteosarcoma. Oncotarget. (2014) 5:10791–802. doi: 10.18632/oncotarget.2505
31. Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. (2010) 116:1908–18. doi: 10.1182/blood-2009-11-249474
32. Gang AO, Frosig TM, Brimnes MK, Lyngaa R, Treppendahl MB, Gronbaek K, et al. 5-Azacytidine treatment sensitizes tumor cells to T-cell mediated cytotoxicity and modulates NK cells in patients with myeloid malignancies. Blood Cancer J. (2014) 4:e197. doi: 10.1038/bcj.2014.14
33. Toor AA, Payne KK, Chung HM, Sabo RT, Hazlett AF, Kmieciak M, et al. Epigenetic induction of adaptive immune response in multiple myeloma: sequential azacitidine and lenalidomide generate cancer testis antigen-specific cellular immunity. Br J Haematol. (2012) 158:700–11. doi: 10.1111/j.1365-2141.2012.09225.x
34. Kunert A, van Brakel M, van Steenbergen-Langeveld S, da Silva M, Coulie PG, Lamers C, et al. MAGE-C2-Specific TCRs combined with epigenetic drug-enhanced antigenicity yield robust and tumor-selective T cell responses. J Immunol. (2016) 197:2541–52. doi: 10.4049/jimmunol.1502024
35. Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, et al. De novo induction of a cancer/testis antigen by 5-aza-2'-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. (2006) 66:1105–13. doi: 10.1158/0008-5472.CAN-05-3020
36. da Cunha JP, Galante PA, de Souza JE, de Souza RF, Carvalho PM, Ohara DT, et al. Bioinformatics construction of the human cell surfaceome. Proc Natl Acad Sci USA. (2009) 106:16752–7. doi: 10.1073/pnas.0907939106
37. Mao Y, Fan W, Hu H, Zhang L, Michel J, Wu Y, et al. MAGE-A1 in lung adenocarcinoma as a promising target of chimeric antigen receptor T cells. J Hematol Oncol. (2019) 12:106. doi: 10.1186/s13045-019-0793-7
38. Pankov D, Sjostrom L, Kalidindi T, Lee SG, Sjostrom K, Gardner R, et al. In vivo immuno-targeting of an extracellular epitope of membrane bound preferentially expressed antigen in melanoma (PRAME). Oncotarget. (2017) 8:65917–31. doi: 10.18632/oncotarget.19579
39. Noh ST, Lee HS, Lim SJ, Kim SW, Chang HK, Oh J, et al. MAGE-A1-6 expression in patients with head and neck squamous cell carcinoma: impact on clinical patterns and oncologic outcomes. Int J Clin Oncol. (2016) 21:875–82. doi: 10.1007/s10147-016-0989-6
40. Srdelic S, Kuzmic-Prusac I, Spagnoli GC, Juretic A, Capkun V. MAGE-A4 and MAGE-A1 Immunohistochemical Expression in High-grade Endometrial Cancer. Int J Gynecol Pathol. (2019) 38:59–65. doi: 10.1097/PGP.0000000000000470
41. Wang D, Wang J, Ding N, Li Y, Yang Y, Fang X, et al. MAGE-A1 promotes melanoma proliferation and migration through C-JUN activation. Biochem Biophys Res Commun. (2016) 473:959–65. doi: 10.1016/j.bbrc.2016.03.161
42. Chang AY, Dao T, Gejman RS, Jarvis CA, Scott A, Dubrovsky L, et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J Clin Invest. (2017) 127:2705–18. doi: 10.1172/JCI92335
43. Maus MV, Plotkin J, Jakka G, Stewart-Jones G, Riviere I, Merghoub T, et al. An MHC-restricted antibody-based chimeric antigen receptor requires TCR-like affinity to maintain antigen specificity. Mol Ther Oncolytics. (2016) 3:1–9. doi: 10.1038/mto.2016.23
44. Maruta M, Ochi T, Tanimoto K, Asai H, Saitou T, Fujiwara H, et al. Direct comparison of target-reactivity and cross-reactivity induced by CAR- and BiTE-redirected T cells for the development of antibody-based T-cell therapy. Sci Rep. (2019) 9:13293. doi: 10.1038/s41598-019-49834-2
45. Ding K, Wang XM, Fu R, Ruan EB, Liu H, Shao ZH. PRAME gene expression in acute leukemia and its clinical significance. Cancer Biol Med. (2012) 9:73–6. doi: 10.3969/j.issn.2095-3941.2012.01.013
46. Oberthuer A, Hero B, Spitz R, Berthold F, Fischer M. The tumor-associated antigen PRAME is universally expressed in high-stage neuroblastoma and associated with poor outcome. Clin Cancer Res. (2004) 10:4307–13. doi: 10.1158/1078-0432.CCR-03-0813
47. Ye Z, Liang Y, Ma Y, Lin B, Cao L, Wang B, et al. Targeted photodynamic therapy of cancer using a novel gallium (III) tris (ethoxycarbonyl) corrole conjugated-mAb directed against cancer/testis antigens 83. Cancer Med. (2018) 7:3057–65. doi: 10.1002/cam4.1601
48. Lim SH, Wang Z, Chiriva-Internati M, Xue Y. Sperm protein 17 is a novel cancer-testis antigen in multiple myeloma. Blood. (2001) 97:1508–10. doi: 10.1182/blood.V97.5.1508
49. Lee SY, Williamson B, Caballero OL, Chen YT, Scanlan MJ, Ritter G, et al. Identification of the gonad-specific anion transporter SLCO6A1 as a cancer/testis (CT) antigen expressed in human lung cancer. Cancer Immun. (2004) 4:13.
50. Silva WA Jr, Gnjatic S, Ritter E, Chua R, Cohen T, et al. PLAC1, a trophoblast-specific cell surface protein, is expressed in a range of human tumors and elicits spontaneous antibody responses. Cancer Immun. (2007) 7:18.
51. Straughn JM Jr, Shaw DR, Guerrero A, Bhoola SM, Racelis A, et al. Expression of sperm protein 17 (Sp17) in ovarian cancer. Int J Cancer. (2004) 108:805–11. doi: 10.1002/ijc.11617
52. Li FQ, Liu Q, Han YL, Wu B, Yin HL. Sperm protein 17 is highly expressed in endometrial and cervical cancers. BMC Cancer. (2010) 10:429. doi: 10.1186/1471-2407-10-429
53. Grizzi F, Gaetani P, Franceschini B, Di Ieva A, Colombo P, Ceva-Grimaldi G, et al. Sperm protein 17 is expressed in human nervous system tumours. BMC Cancer. (2006) 6:23. doi: 10.1186/1471-2407-6-23
54. Gupta G, Sharma R, Chattopadhyay TK, Gupta SD, Ralhan R. Clinical significance of sperm protein 17 expression and immunogenicity in esophageal cancer. Int J Cancer. (2007) 120:1739–47. doi: 10.1002/ijc.22463
55. Mirandola L, Pedretti E, Figueroa JA, Chiaramonte R, Colombo M, Chapman C, et al. Cancer testis antigen Sperm Protein 17 as a new target for triple negative breast cancer immunotherapy. Oncotarget. (2017) 8:74378–90. doi: 10.18632/oncotarget.20102
56. Gjerstorff MF, Pohl M, Olsen KE, Ditzel HJ. Analysis of GAGE, NY-ESO-1 and SP17 cancer/testis antigen expression in early stage non-small cell lung carcinoma. BMC Cancer. (2013) 13:466. doi: 10.1186/1471-2407-13-466
57. Zhang Z, He Q, Tao Y, Guo J, Xu F, Wu LY, et al. Decitabine treatment sensitizes tumor cells to T-cell-mediated cytotoxicity in patients with myelodysplastic syndromes. Am J Transl Res. (2017) 9:454–65.
58. Lea IA, Adoyo P, O'Rand MG. Autoimmunogenicity of the human sperm protein Sp17 in vasectomized men and identification of linear B cell epitopes. Fertil Steril. (1997) 67:355–61. doi: 10.1016/S0015-0282(97)81923-1
59. Gjerstorff MF, Ditzel HJ. Limited SP17 expression within tumors diminishes its therapeutic potential. Tissue Antigens. (2012) 80:523–7. doi: 10.1111/tan.12015
60. Grizzi F, Chiriva-Internati M, Franceschini B, Bumm K, Colombo P, Ciccarelli M, et al. Sperm protein 17 is expressed in human somatic ciliated epithelia. J Histochem Cytochem. (2004) 52:549–54. doi: 10.1177/002215540405200414
61. Dadabayev AR, Wang Z, Zhang Y, Zhang J, Robinson WR, Lim SH. Cancer immunotherapy targeting Sp17: when should the laboratory findings be translated to the clinics? Am J Hematol. (2005) 80:6–11. doi: 10.1002/ajh.20415
62. Kondo Y, Fukuyama T, Yamamura R, Futawatari N, Ichiki Y, Tanaka Y, et al. Detection of KK-LC-1 protein, a cancer/testis antigen, in patients with breast cancer. Anticancer Res. (2018) 38:5923–8. doi: 10.21873/anticanres.12937
63. Cohen AS, Khalil FK, Welsh EA, Schabath MB, Enkemann SA, Davis A, et al. Cell-surface marker discovery for lung cancer. Oncotarget. (2017) 8:113373–402. doi: 10.18632/oncotarget.23009
64. Paret C, Simon P, Vormbrock K, Bender C, Kolsch A, Breitkreuz A, et al. CXorf61 is a target for T cell based immunotherapy of triple-negative breast cancer. Oncotarget. (2015) 6:25356–67. doi: 10.18632/oncotarget.4516
65. Shida A, Futawatari N, Fukuyama T, Ichiki Y, Takahashi Y, Nishi Y, et al. Frequent high expression of kita-kyushu lung cancer Antigen-1 (KK-LC-1) in gastric cancer. Anticancer Res. (2015) 35:3575–9.
66. Fukuyama T, Hanagiri T, Takenoyama M, Ichiki Y, Mizukami M, So T, et al. Identification of a new cancer/germline gene, KK-LC-1, encoding an antigen recognized by autologous CTL induced on human lung adenocarcinoma. Cancer Res. (2006) 66:4922–8. doi: 10.1158/0008-5472.CAN-05-3840
67. Chen Z, Zuo X, Pu L, Zhang Y, Han G, Zhang L, et al. Hypomethylation-mediated activation of cancer/testis antigen KK-LC-1 facilitates hepatocellular carcinoma progression through activating the Notch1/Hes1 signalling. Cell Prolif. (2019) 52:e12581. doi: 10.1111/cpr.12581
68. Marcinkowski B, Stevanovic S, Helman SR, Norberg SM, Serna C, Jin B, et al. Cancer targeting by TCR gene-engineered T cells directed against Kita-Kyushu Lung Cancer Antigen-1. J Immunother Cancer. (2019) 7:229. doi: 10.1186/s40425-019-0678-x
69. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. (2014) 16:1304–12. doi: 10.1093/neuonc/nou045
70. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
71. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. (2004) 64:9160–6. doi: 10.1158/0008-5472.CAN-04-0454
72. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Ralpha2-Targeted Chimeric antigen receptor t cells for improved anti-tumor efficacy against glioblastoma. Mol Ther. (2018) 26:31–44. doi: 10.1016/j.ymthe.2017.10.002
73. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-Cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
74. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
75. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
76. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
77. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. (2013) 21:2087–101. doi: 10.1038/mt.2013.185
78. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. (2019) 568:112–6. doi: 10.1038/s41586-019-1054-1
79. Ormhoj M, Scarfo I, Cabral ML, Bailey SR, Lorrey SJ, Bouffard AA, et al. Chimeric Antigen Receptor T Cells Targeting CD79b Show Efficacy in Lymphoma with or without Cotargeting CD19. Clin Cancer Res. (2019) 25:7046–57. doi: 10.1158/1078-0432.CCR-19-1337
80. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. (2019) 129:3464. doi: 10.1172/JCI131246
81. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. (2016) 126:3036–52. doi: 10.1172/JCI83416
82. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. (2014) 5:587–98. doi: 10.18632/oncotarget.1782
83. Siebenkas C, Chiappinelli KB, Guzzetta AA, Sharma A, Jeschke J, Vatapalli R, et al. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE. (2017) 12:e0179501. doi: 10.1371/journal.pone.0179501
84. Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun. (2018) 9:248. doi: 10.1038/s41467-017-02630-w
85. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. (2017) 169:361. doi: 10.1016/j.cell.2017.03.036
86. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. (2015) 162:961–73. doi: 10.1016/j.cell.2015.07.056
87. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. (2014) 28:1280–8. doi: 10.1038/leu.2013.355
Keywords: CAR T cell, cancer/testis antigen, DNA methyltransferase inhibitor, cancer immunotherapy, T cell engineering
Citation: Jakobsen MK and Gjerstorff MF (2020) CAR T-Cell Cancer Therapy Targeting Surface Cancer/Testis Antigens. Front. Immunol. 11:1568. doi: 10.3389/fimmu.2020.01568
Received: 20 February 2020; Accepted: 15 June 2020;
Published: 02 September 2020.
Edited by:
Yoshihiko Hirohashi, Sapporo Medical University, JapanReviewed by:
Zong Sheng Guo, School of Medicine, University of Pittsburgh, United StatesCopyright © 2020 Jakobsen and Gjerstorff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Morten F. Gjerstorff, bWdqZXJzdG9yZmZAaGVhbHRoLnNkdS5kaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.