 Jeffrey O. Zhou
Jeffrey O. Zhou Hussain A. Zaidi
Hussain A. Zaidi Therese Ton2
Therese Ton2 Daniela Fera
Daniela Fera
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 17 July 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01529
Understanding affinity maturation of antibodies that can target many variants of HIV-1 is important for vaccine development. While the antigen-binding site of antibodies is known to mutate throughout the co-evolution of antibodies and viruses in infected individuals, the roles of the mutations in the antibody framework region are not well understood. Throughout affinity maturation, the CH103 broadly neutralizing antibody lineage, from an individual designated CH505, altered the orientation of one of its antibody variable domains. The change in orientation was a response to insertions in the variable loop 5 (V5) of the HIV envelope. In this study, we generated CH103 lineage antibody variants in which residues in the variable domain interface were mutated, and measured the binding to both autologous and heterologous HIV-1 envelopes. Our data show that very few mutations in an early intermediate antibody of the lineage can improve binding toward both autologous and heterologous HIV-1 envelopes. We also crystallized an antibody mutant to show that framework mutations alone can result in a shift in relative orientations of the variable domains. Taken together, our results demonstrate the functional importance of residues located outside the antigen-binding site in affinity maturation.
A well-recognized aspect of antibody affinity maturation is the somatic hypermutation (SHM) of the antigen binding regions, i.e., the complementarity determining regions, or CDRs (1). This is even observed in antibodies that target rapidly evolving pathogens, such as HIV-1 and influenza. Antibodies target the viral spike, the envelope (Env) or hemagglutinin (HA), in the case of HIV-1 and flu, respectively (2, 3). In certain cases, antibodies that can neutralize a broad spectrum (>50%) of viral variants can be achieved and these antibodies typically target certain conserved regions of the viral spike. In the case of HIV-1, such broadly neutralizing antibodies (bnAbs) arise in ~10–20% of chronically infected patients after about 5 years of infection. The long time frame is partly due to the fact that the virus rapidly develops escape mutations to avoid antibody recognition (4, 5). This results in bnAbs with high mutation frequencies.
Several bnAb lineages have been identified that target HIV-1 Env or flu HA and they each use different strategies during the affinity maturation process to alter their CDRs to bind the viral spike. In the CAP256-VRC26 bnAb lineage, a disulfide bond was introduced within the heavy chain CDR loop 3 (HCDR3) that rigidified and properly oriented it for binding HIV-1 Env, thereby leading to breadth (6, 7). Rigidification of the HCDR3 was also important in the development of breadth of the CH65–CH67 lineage against flu HA (8). In other cases, structural changes of the paratope were not observed. Instead, there were mutations within the CDRs that introduced residues or rotamers of residues necessary for interacting with Env. This was observed in the DH270 and CH235 antibody lineages where improbable mutations were necessary in interacting with key components of Env and/or Env glycans (9–12). Deletions and/or insertions in the CDRs could also alter the angle of approach of antibodies with Env, allowing them to accommodate glycans on Env, as observed in the PGT121 bnAb lineage (13–16).
While CDRs are responsible for the majority of the direct contacts made with an antigen, SHM does also occur in antibodies outside of their CDRs, in the intervening framework regions (FWRs). While the FWRs help stabilize the antigen-binding site and define the conformations of the CDR loops, the roles of mutations in the FWR are not well-understood. Reports have shown that FWR mutations can be responsible for the thermal destabilization of HIV-1 bnAbs, but not weakly neutralizing HIV-1 antibodies (17, 18). FWR mutations have also been shown to increase the dynamics of antibodies, leading to neutralization breadth (17, 19, 20).
The CH103 bnAb lineage, derived from patient CH505, produced several bnAbs including CH103, which target the CD4 receptor binding site (CD4bs) of the HIV-1 Env (21). Crystal structures of antibody fragments from this lineage showed that the orientation of the heavy chain variable domain (VH) changed relative to the light chain variable domain (VL) in the transition from intermediate antibody I3.2 to I2 during affinity maturation, potentially through FWR mutations (22). While the CD4bs is relatively conserved, rapid mutations in the nearby Env variable loop 5 (V5) can lead to resistance against CD4bs antibodies (23). In the CH505 patient, Env insertion mutations in V5 occurred throughout virus evolution, which reduced the potency of CH103 lineage bnAb precursors through steric interference (22). In response to these insertions, over the course of affinity maturation the antibody VL domain shifted away from VH and the Env V5 loop to accommodate the V5 loop insertions. Different VL orientations relative to VH were observed in the structures of intermediate antibody I3.2 and of the chimeric antibody, I3.1. These antibodies both contain the VHDJH of I3, but this is paired with either the VLJL of the unmutated common ancestor (UCA) in I3.2 or with the VLJL of the more mature intermediate antibody I2 in I3.1. Thus, this revealed that the shift in VL was mainly attributed by the identity of the antibody's light chain.
To determine if FWR mutations, specifically ones in the antibody light chain at the VH-VL interface, were responsible for the shift observed in the CH103 lineage and breadth development, we performed binding measurements to show that these mutations improved affinity toward an autologous (strain specific) HIV-1 Env and heterologous (different strain) HIV-1 Env with longer V5 loops. We also determined the crystal structure of a mutant, which shows that two residues in the FWR at the VH-VL interface could lead to a large part of the observed shift seen in the VLJL of the UCA and I2 between intermediate antibodies I3.2 and I2. Thermal melts showed that stability was not altered. Our analysis suggests that few mutations can have a large impact on antigen binding, and that antigen distal FWR mutations occurring during antibody maturation can lead to the structural properties necessary to improve affinity toward an antigen without altering stability.
During infection of the CH505 donor, insertion mutations were introduced in the V5 loop of the CH505 HIV-1 envelopes as they evolved (21). While the UCA and early intermediate antibodies had a structure whose VL domain, specifically the antibody DE loop residues 65L-67L, would clash with V5 loops that were longer than that of the CH505 transmitted founder (T/F) Env, the relative shift in VL with respect to VH appeared to accommodate these V5 loop insertions as well as the longer V5 loops found in heterologous HIV-1 Envs (Figure 1A). As a result, autologous CH505 Envs with longer V5 loops displayed differential binding to different members of the CH103 antibody lineage (i.e., they bound better to later members of the lineage than earlier members), whereas the CH505 T/F Env bound with similar affinity to all CH103 lineage members (Table 1) (22).
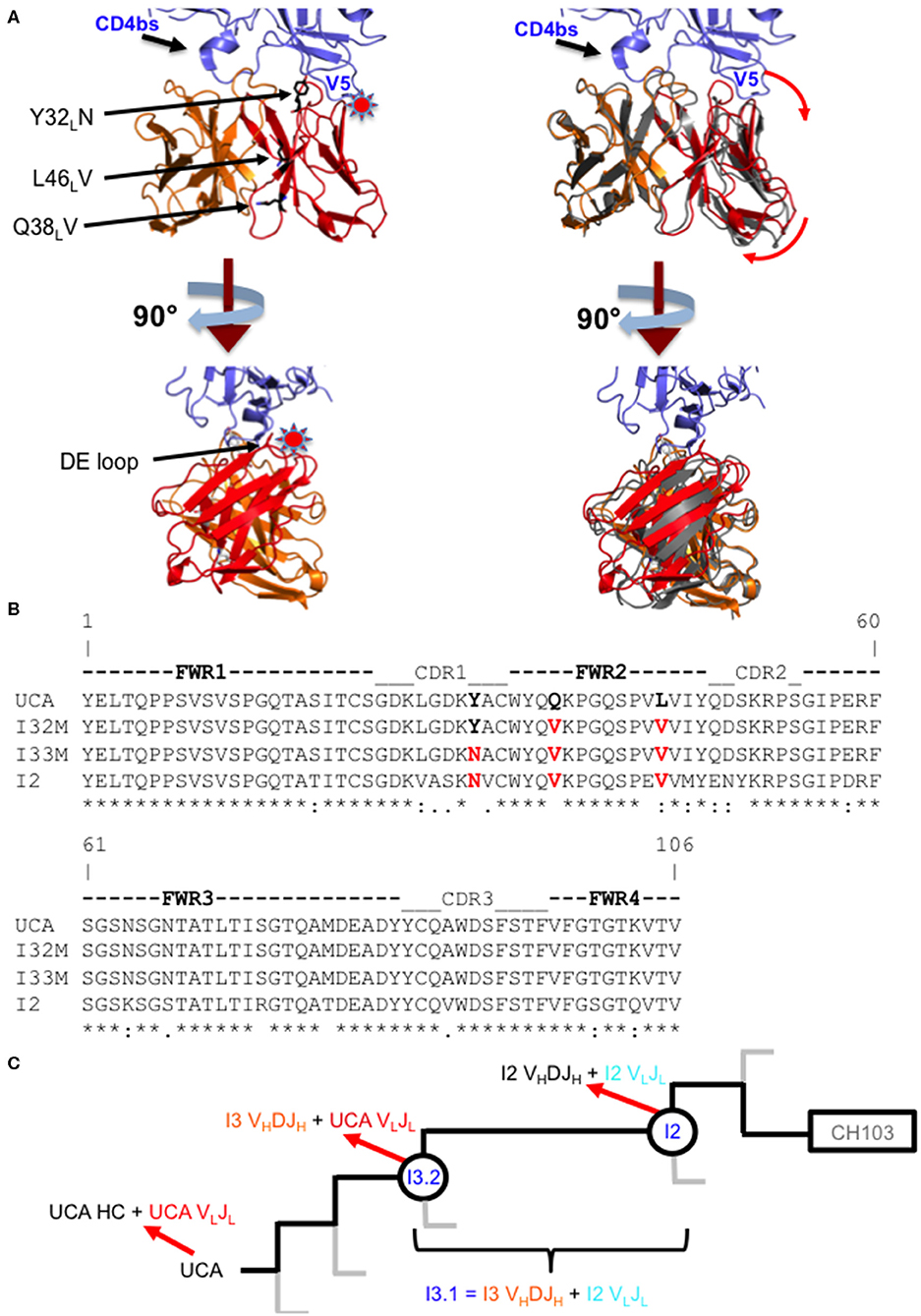
Figure 1. Comparison of wild-type and mutant Fabs. (A) Superposition of the I3.2 Fab with the CH103/gp120 outer domain complex. Two orthogonal views of the variable region of the I3.2 Fab (heavy chain VH in orange, light chain VL in red) superposed onto the variable region of the CH103 Fab (gray) in complex with the gp120 outer domain (blue) from ZM176.66 [Protein Data Bank (PDB) ID code 4JAN]. The V5 and CD4 binding site loops of gp120 are marked. A star indicates location of steric clash. Interface mutations introduced in this study are shown as black sticks. Image created in PyMOL. (B) Sequence alignment of the variable light chains of I3 wild-type and mutant Fabs. Mutations analyzed in this study are highlighted in red. (*) fully conserved residue, (:) strong conservation of properties, (.) weak conservation of properties, a blank space indicates little to no conservation of properties. (C) CH103 bnAb clonal lineage tree. The part of the CH103 lineage under study is shown. Gray lines represent other branches in the lineage. The UCA is shown on the left, the intermediates of interest are circled, and the CH103 bnAb has a rectangle around it. Heavy and light chain compositions are indicated with red arrows. The chimeric antibody is also defined based on its heavy and light chain compositions.
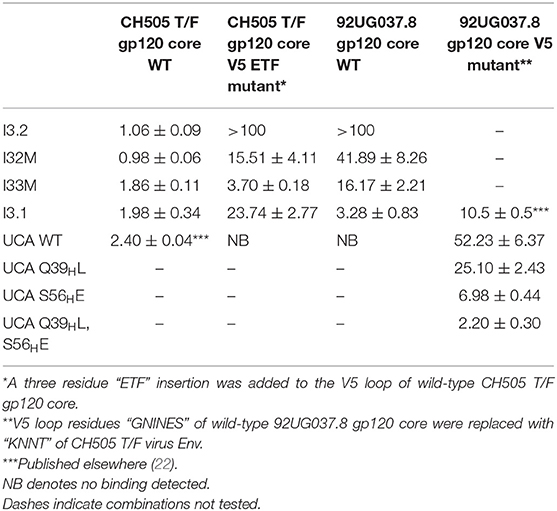
Table 1. Dissociation constants (KD, in μM) between CH103 lineage Fabs and gp120 core variants.
To assess the role of residues at the VH-VL interface on binding Env, we generated an Fab mutant, “I32M”, which consists of two interface mutations in the light chain of I3.2 (i.e., the UCA VLJL). The light chain mutations we introduced, Q38LV and L46LV, were based on residues found in the mature members of the lineage (Figures 1B,C). To test binding to HIV-1 Env, we performed biolayer interferometry (BLI) with gp120 core, the monomeric form of the HIV-1 Env ectodomain. Because we wanted to identify differential binding capabilities, we tested binding to an autologous CH505 gp120 T/F core with a three-residue “ETF” V5 loop insertion, which occurred during the natural evolution of the virus in the CH505 patient. I32M bound significantly better than I3.2 to this CH505 gp120 core and with a similar KD as the chimeric antibody I3.1, which was previously shown to have a VL shifted relative to VH (22) (Figure 2A, Table 1), demonstrating the importance to binding of the two mutations at the VH-VL interface, Q38LV and L46LV. The CH505 T/F wild-type gp120 core did not bind differentially to CH103 lineage members, as expected, nor to our mutant, I32M, which had similar KD (Table 1). These results suggest that the mutations we introduced did not disrupt binding to Env and that they improve binding to autologous Envs with significantly longer V5 loops.
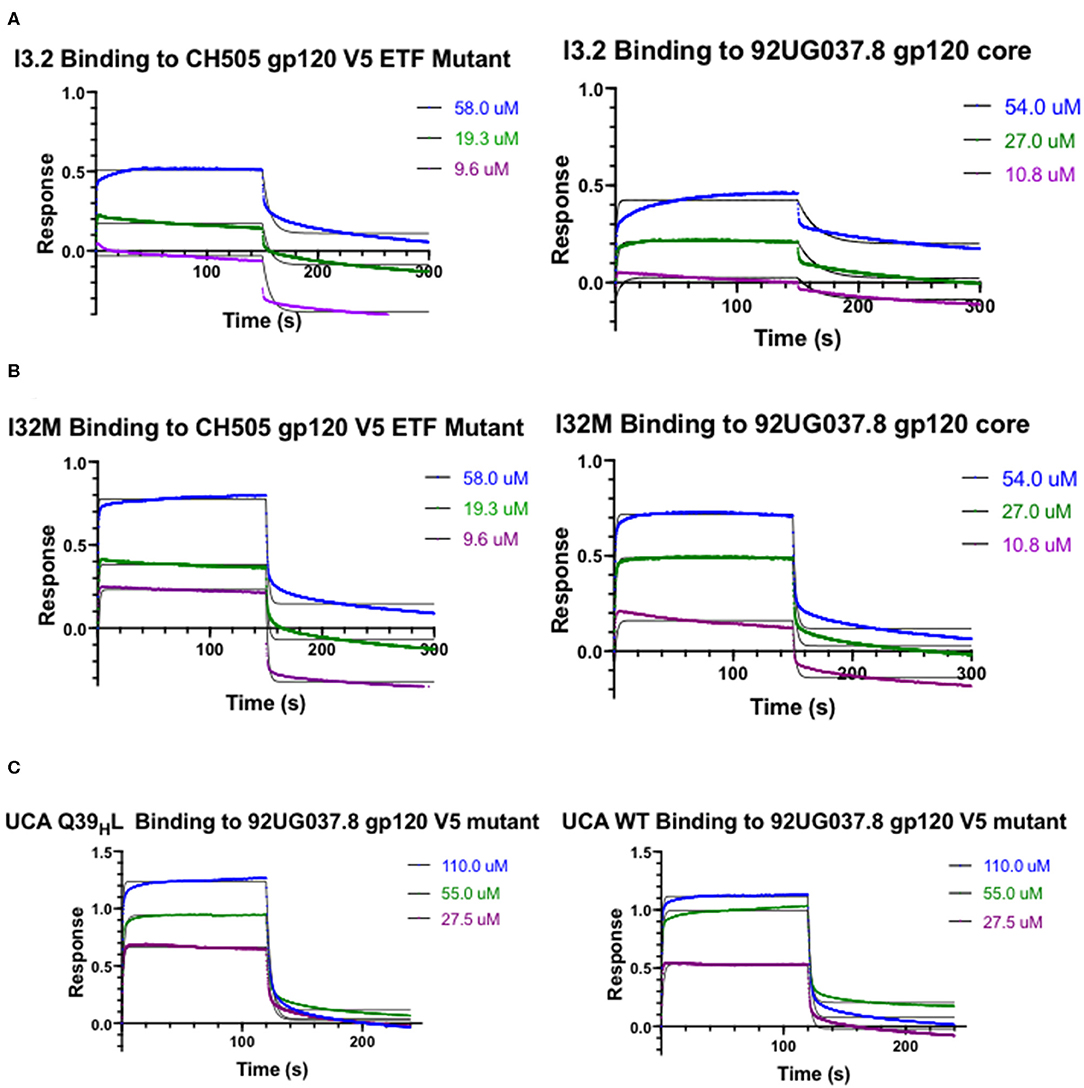
Figure 2. Kinetic Studies by biolayer interferometry. Curves of I3.2 and I32M binding to (A) CH505 gp120 core with an “ETF” insertion in the V5 loop and (B) 92UG037.8 gp120 core. (C) Curves of the UCA and the UCA Q39HL mutant binding to 92UG037.8 gp120 core with a V5 mutation. The V5 loop of CH505 T/F Env was grafted in the place of the V5 loop of 92UG037.8 Env. In each case, the Fab was immobilized onto an anti-human Fab-CH1 biosensor, and gp120 constructs were introduced at three or more different concentrations, ranging from low micromolar to high micromolar, depending on the mutant tested.
A hallmark of breadth is the ability of antibodies to bind and neutralize heterologous virus strains. To determine if our I32M mutant could also display improved binding toward heterologous Envs, we tested binding to HIV-1 strain 92UG037.8, whose sequence in the V5 loop differs and is two amino acids longer than that of the CH505 T/F Env. Analysis by BLI shows that I32M binds to this Env with a significantly smaller KD than its wild-type counterpart I3.2, but not as well as the chimeric antibody I3.1 (Figure 2B, Table 1). These results suggest that two mutations alone in the VH-VL interface improve binding to both autologous and heterologous Envs with longer V5 loops, and thereby contribute to breadth against different HIV-1 strains, however, additional light chain mutations may further improve binding to heterologous Envs.
The UCA is the ancestor of an antibody lineage that is produced against the autologous virus that causes infection. In the UCA of the CH103 lineage, Q39H in the heavy chain forms a reciprocal side-chain hydrogen bond with the light chain Q38L at the VH-VL interface (22). Moreover, the I3.2 early intermediate antibody has 18 mutations, only in its heavy chain, when compared to the UCA (Supplementary Figure 1). To determine whether mutations at the VH-VL interface can improve binding to the progenitor antibody, we introduced the Q39HL mutation in its heavy chain and tested binding by BLI. Since the UCA of the CH103 lineage binds with a similar affinity to the CH505 T/F gp120 core as other members of the lineage and it cannot bind to CH505 or heterologous gp120 cores with longer V5 loops (Table 1), we tested a 92UG037.8 gp120 core whose V5 loop was replaced with a shorter one from the CH505 T/F Env (22). While binding is fairly weak, comparison to the binding of the wild-type UCA shows that the introduction of this single mutation can improve binding by a factor of two (Figure 2C, Table 1). To ensure that the rapid association and dissociation rates observed were not due to bulk shifts at such high gp120 core concentrations, we also tested binding to the VRC01 bnAb, which recognizes the CD4bs but with a different approach angle (24, 25). This bnAb displayed slower on and off rates, distinguishing its binding kinetics from those of CH103 lineage members and their corresponding mutants (Supplementary Table 1, Supplementary Figure 2). This result suggests that the disruption of the VH-VL interface Q39H-Q38L reciprocal side-chain hydrogen bond is important in affinity maturation and can even improve binding of the earliest antibody lineage member.
Because binding to the UCA Q39HL mutant was still relatively weak (KD = 25.10 μM), we wanted to see if any mutations could further significantly improve binding to the heterologous Env. The published CH103 bnAb-gp120 structure shows that E56H of the CH103 bnAb heavy chain HCDR2 loop engages in hydrogen bonding contacts with the backbone of the conserved gp120 CD4bs (21). Thus, we introduced the S56HE mutation into the UCA Q39HL mutant and tested binding to the 92UG037.8 gp120 core V5 mutant. The UCA Q39HL, S56HE double mutant could bind to the 92UG037.8 gp120 core V5 mutant with low micromolar affinity (KD = 2.20 μM) (Table 1). This data suggests that while the loss of that VH-VL interface hydrogen bond is important even for the UCA in gaining breadth against different viruses, the contribution from CDRs is still important.
Because Q38L and L46L of the light chain are involved in significant interactions at the VH-VL interface and mutating these residues improves binding to Envs, we wanted to determine what structural changes these mutations cause. To aid in crystallization, we added an additional mutation, Y32LN, in the light chain of I32M to create “I33M.” Y32LN occurs during affinity maturation of the CH103 lineage and N32L first appears in the VLJL of intermediate antibody I2 (Figures 1B,C). In later members of the lineage, N32L of the light chain CDR loop 1 (LCDR1) was observed to hydrogen bond with the LCDR2 and HCDR3 loops, stabilizing the conformation of the latter and so we hypothesized the same would happen in I33M.
We determined the crystal structure of I33M to a resolution of 2.36 Å (Table 2) and compared it to those of the wild-type intermediate I3.2, the CH103 bnAb, and chimeric I3.1 Fabs (Figure 3, Supplementary Figure 3). The VH of I33M was superposed on that of the wild-type Fabs, since the VH contacts the CD4bs on Env (Figure 1), which is invariant throughout the evolution of the virus and thus expected to bind antibodies in the same way throughout antibody affinity maturation. Superimposed VH domains of I3.2, I33M, and I3.1 demonstrate that I33M has a VL orientation intermediate between that of I3.2 and I3.1 (Figures 3A–C), though it is much more similar to that of I3.1 and to mature members of the antibody lineage (Figure 3B, Supplementary Figure 3A). Additionally, the DE loop of the antibody, from which residues 65L-67L clash with the Env V5 loop in the case of I3.2 and the UCA of the lineage, is shifted for I33M in a way that would eliminate the clash, although it is not shifted to the same extent as I3.1 and the CH103 bnAb (Figure 3D, Supplementary Figure 3B).
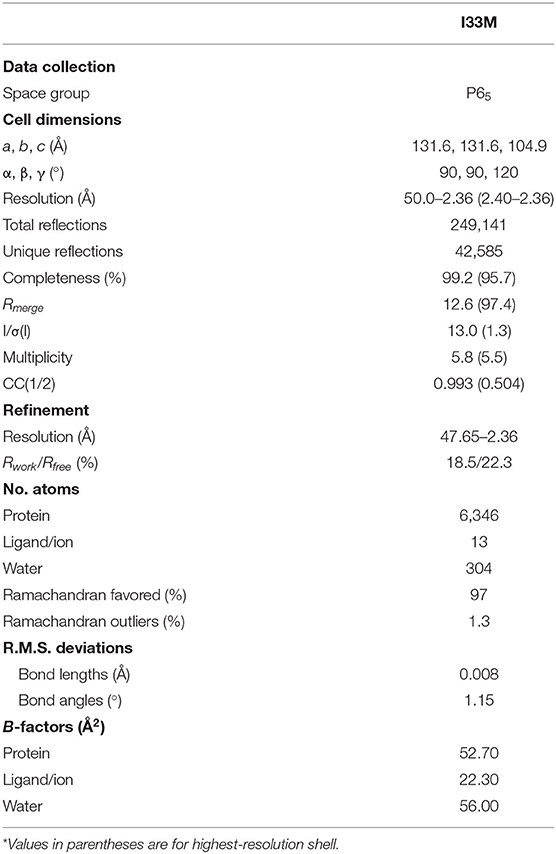
Table 2. Data collection and refinement statistics.
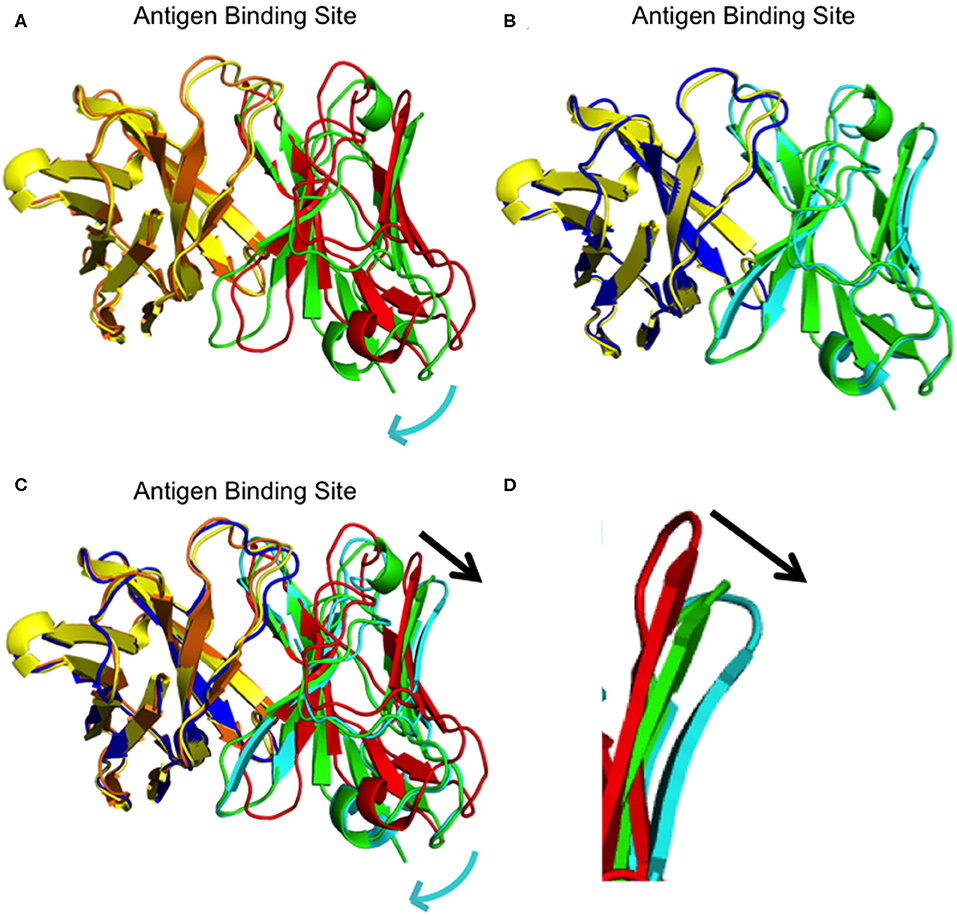
Figure 3. Superpositions of I3.2, I33M, and chimeric I3.1. (A) Overall superposition of mutant Fab I33M onto I3.2. The domains are colored: I33M VH (yellow) and VL (green), and I3.2 VH (orange) and VL (red). (B) Overall superposition of mutant Fab I33M onto I3.1. The domains are colored: I33M VH (yellow) and VL (green), and I3.1 VH (blue) and VL (cyan). (C) Overall superposition of mutant Fab I33M onto I3.2 and I3.1. Domains are colored as in panels (A,B). The constant domains are not shown for clarity. Relative shifts are indicated with a cyan arrow. The black arrow shows the shift in the antibody DE loop. (D) Zoomed in view of the DE loops of I3.2 (red), I33M (green), and I3.1 (cyan).
As expected, the Q38LV mutation in I33M disrupted the reciprocal side-chain hydrogen bond interaction with Q39H, resulting in more compact hydrophobic packing seen in the mature CH103 lineage Fabs (Figure 4, Supplementary Figure 3). Moreover, in the case of the UCA and I3.2, L46L is involved in hydrophobic packing by bearing on the F100DH ring, which is a bridge between P96H and Y49L at the domain interface. Replacing leucine with the shorter valine at this position in I33M causes F100DH (Y100DH in the case of the CH103 bnAb) to swing closer to Y49L to maintain hydrophobic interactions, while P96H appears unaffected. This is similar to what was observed in I3.1 and the CH103 bnAb (Figure 4, Supplementary Figure 3C) (22). Thus, Q38LV and L46LV contribute to VH-VL packing.
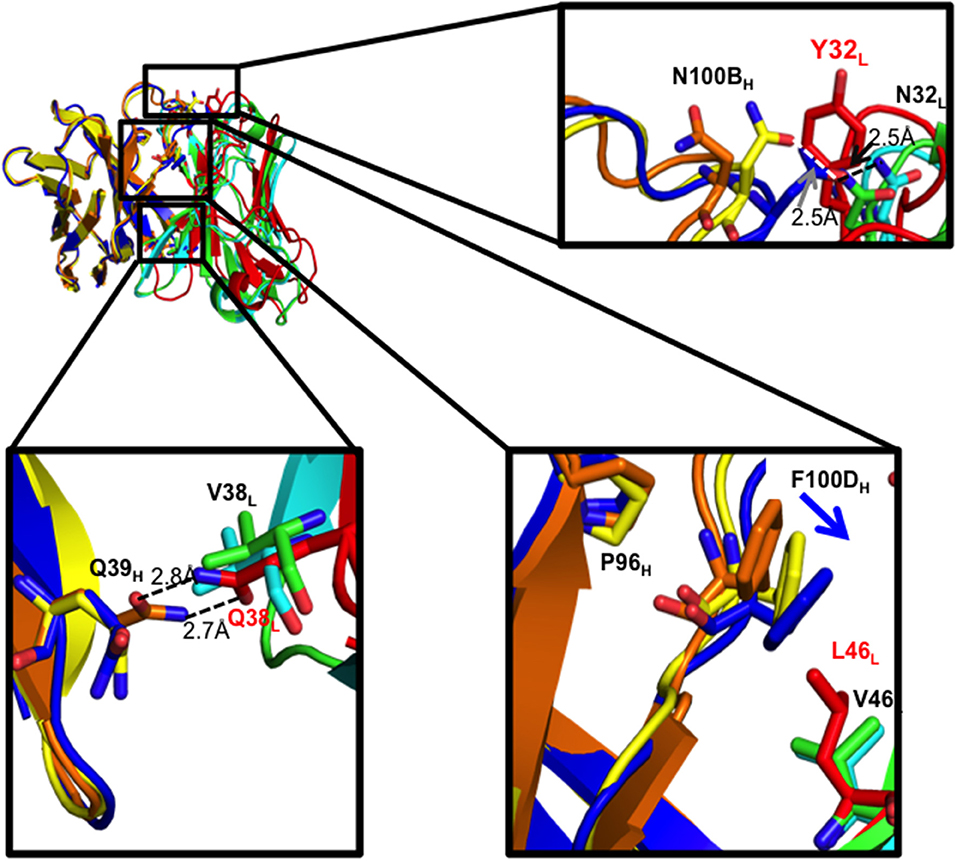
Figure 4. VH-VL interface residues and their effects. Zoomed in views of residues that were mutated are shown as sticks for the three Fabs. Domains for the different Fabs are colored as follows: I33M VH (yellow) and VL (green), I3.2 VH (orange) and VL (red), and I3.1 VH (blue) and VL (cyan). Hydrogen bond distances are given.
We further analyzed the structures to see if N32L made similar contacts in the different antibodies despite the different extents of orientation shifts of their VL domains relative to VH. As with the chimeric I3.1 and mature CH103 bnAb, N32L in I33M could form the same hydrogen bond with the HCDR3 loop, specifically with N100BH, which is conserved in all members of the lineage (Figure 4, Supplementary Figures 1, 3C). Of note, the distances between the backbones of light chain residue 32 (Y32L in the case of the early CH103-lineage members UCA and I3.2 and N32L for later members and I33M) and heavy chain residue 100B are very similar for the three Fabs: 6.4 Å for I33M, 6.2 Å for I3.1, and 6.4 Å for I3.2, suggesting that the Y32LN mutation does not alter the VH-VL domain interface spacing and that the Q38LV and L46LV mutations are the sole determinants of the observed shift.
To determine what effect Y32LN has on binding Envs, we performed BLI with I33M and found that it bound with a stronger affinity to the CH505 gp120 T/F core ETF mutant and 92UG037.8 wild-type gp120 core than I32M but with the same affinity to CH505 gp120 T/F wild-type gp120 core (Table 1). This suggests that the hydrogen bond that N32L makes with the HCDR3 loop, which binds to the CD4bs, contributes to Env binding.
To determine what, if any, effects the VH-VL interface mutations and structural changes have on thermostability, we measured the melting temperature (Tm) of the mutants we generated using circular dichroism thermal melts (Figure 5). Tm values for our mutants are similar and fall within the wide reported range of Fab melting temperatures (65–90°C) (26, 27). Comparison to the melting temperature of I3.2 shows that the introduction of mutations in the light chain at the VH-VL interface, or Y32LN in the CDR, has no significant effect on thermostability. We also determined the Tm for the CH103 bnAb. CH103 had a similar melting temperature as I3.2, consistent with the reported value of 65.5°C and Tm trend determined by differential scanning calorimetry by others (18).
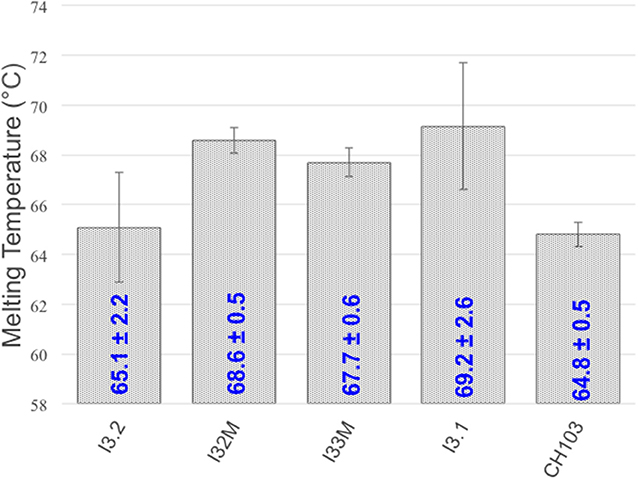
Figure 5. Melting temperatures of Fabs. Tm's of various Fabs and mutants are shown. Standard deviations are from three or more replicates.
HIV-1 bnAbs that evolve naturally in infected individuals have high levels of mutations in the CDRs and FWR. While many mutations in the CDR can impact binding to antigen, the roles of the FWR mutations are usually less clear. It has been shown that FWR mutations can be important for the development of breadth (18, 28). In this study, we identified two FWR residues at the VH-VL interface, Q38LV and L46LV, that were important for improving binding to HIV-1 Envs with longer V5 loops. These two mutations alone were sufficient for the significant shift of the orientation of the VL domain with respect to VH over the course of affinity maturation of the CH103 bnAb lineage. Moreover, these mutations did not alter the stability of the antibodies consistent with reports elsewhere (18).
Q38LV, distal to the Env binding site, eliminates hydrogen bonding with Q39H. Moreover, the Q39HL heavy chain mutation first appeared in I2 of the lineage and so I3.2 has a Q39H (Supplementary Figure 1). Our structure of I33M shows that a relative shift in orientation of the domains can occur in the absence of the Q39HL mutation, since this mutant only consisted of light chain mutations in I3.2 (Figure 4). Moreover, mutation of just one of these residues alone, which disrupts the variable interface hydrogen bond, is sufficient for improving binding to a heterologous HIV-1 Env (Figure 2C, Table 1), an important feature in the development of breadth. L46LV, also within the VH-VL interface, alters hydrophobic packing. Thus, the mutations Q38LV and L46LV in the antibody light chain brought the relative orientation of the VH and VL domains of the antibodies closer to that of the mature members of the CH103 lineage to accommodate Envs with larger V5 loops. Of note, the I32M double mutant bound to the CH505 gp120 core “ETF” V5 loop mutant with a similar dissociation constant (Table 1) as a later intermediate, I2 (22) and the chimeric I3.1. This suggests that all the mutations accrued in late CH103 lineage members may not be necessary for antibody affinity maturation, at least against autologous Envs. These results further demonstrate that it is important to consider more than just the CDRs for vaccine design.
Our work did also identify a few critical CDR mutations that occurred throughout affinity maturation that greatly improved antibody binding to Env. A significant improvement in KD was observed when we introduced the S56HE mutation into the wild-type UCA (~7-fold) or the UCA Q39HL mutant (~3-fold; Table 1). Another example of a single CDR mutation enhancing affinity was demonstrated by our I33M mutant, which had the Y32LN mutation in the LCDR1. The I33M mutant bound to the 92UG037.8 gp120 core with a ~3-fold lower KD than that of I32M (Table 1). A likely consequence of the Y32LN mutation in the LCDR1 of I33M and other mature CH103-lineage members is the stabilization of the configuration of the HCDR3 loop, important for contacting the conserved CD4bs of the CH505 HIV-1 Env. The HCDR3 loop was disordered in the UCA, but not in later members of the CH103 lineage (22). The hydrogen bonding we observed in I33M between N32L and N100BH (Figure 4) is reminiscent of what is observed in I2 and the bnAbs from this lineage. This illustrates that very few mutations were necessary for the correct configuration of the HCDR3 loop. While FWR mutations have been shown to increase antibody flexibility in some cases (17, 28), results from our crystal structure are consistent with more standard trends observed, i.e., that affinity maturation results in a more rigid antibody structure, specifically in the HCDR3 loop (8, 29–33).
In the CH103 lineage, a significant increase in both heterologous binding and heterologous neutralization was observed during the transition from I3.2 to I2, without much change thereafter (21). Additionally, a shift in the orientation of the VL domain relative to that of the VH was also observed between these two antibodies, and the CH103 bnAb had a similar VL orientation as I2 and the chimeric I3.1 with respect to VH (22). Furthermore, it was shown through structures and binding data, that CH103-lineage Fabs whose VL domain had shifted relative to the VH domain had higher binding affinity toward HIV Envs with longer V5 loops than antibodies from the lineage in which this shift had not yet occurred. Because many other HIV Envs tend to have V5 loops that are longer than that of the CH505 T/F, this suggests there may be a correlation between a shift in VL and heterologous neutralization breadth in the case of the CH103 lineage. Since our I3.2 mutant also had a shift in VL, we expect that I32M and I33M have broader neutralization capabilities than the wild-type I3.2, though this has yet to be confirmed experimentally.
A challenge in eliciting antibodies with great breadth via HIV-1 vaccination is the fact that bnAbs take years to develop, requiring high mutation frequencies. Thus, a deep understanding of antibody affinity maturation, paired with virus evolution, is needed to determine key steps in these processes and to identify which stages to target by vaccination. The relative shift in the VH and VL domains was demonstrated to be important and here we show that very few mutations in the antibody variable domain interface are necessary for this structural change. Thus, exposure to an antigen that would lead to such a shift may lead to a more rapid antibody response than one in which many mutations in the antigen combining site are necessary. Our results also have implications for the design of therapeutic antibodies. Alterations at the VH-VL interface can alter the antigen-combining site in a favorable way to lead to more potent and broad antibodies.
Complex evolutionary trajectories are followed in different HIV-1 bnAb lineages, therefore, structural analyses at multiple time points during the maturation pathway could reveal new phenomena. Since the shift in orientation of VL relative to VH that we observed in our mutant is similar to but not exactly the same as that observed in the mature CH103 lineage members, there may be additional mutations that would result in a greater shift and should be explored. By comparing available structures of the wild-type, chimeric, and mutant antibodies (22), we find that there are no other mutations in the antibody light chain at the VH-VL interface that change identity, rotamers, or amino acid binding partners throughout affinity maturation of this particular lineage. Thus, it is possible that there are mutations in other regions of the FWR or in the CDRs that affect the shift in VL relative to VH. Additionally, a high-resolution structure of the CH103 UCA in complex with an Env has not yet been determined. Such a structure is necessary to better understand antibody-virus co-evolution in this lineage as well as properties of HIV-1 Env that triggered the lineage. While the CH103 UCA can only bind the CH505 T/F Env, it binds poorly to an Env stabilized in a closed conformation (data not shown). Our work shows that introducing the S56HE mutation in the UCA HCDR2 loop alone significantly improves its binding to a heterologous Env (Table 1). This provides a means for determining an atomic resolution structure of the UCA with Env to provide greater insights for HIV-1 vaccine development.
Site-directed mutagenesis was performed using manufacturer's protocols (Stratagene) to introduce mutations into the UCA heavy and light chains. Fabs were expressed in HEK293F suspension adapted cells via transient transfection using linear polyethylenimine (PEI) following the manufacturer's suggested protocol. For 200 mL of suspension cell culture, 100 μg of total plasmid was used. After 5 days of expression, the supernatant was clarified by centrifugation. Each His-tagged CH103-lineage Fab was loaded onto Ni-NTA superflow resin (Qiagen) pre-equilibrated with Buffer A (10 mM Tris, pH 7.5, 200 mM NaCl), washed with Buffer A then Buffer A + 10 mM imidazole, and finally, eluted with Buffer A + 350mM imidazole. Each Fab was then purified by gel filtration chromatography using a SuperdexTM 200 Increase 10/300 GL column (GE Healthcare) in buffer of 10 mM Tris, pH 7.5, 100 mM NaCl. Fractions of interest were further concentrated.
Plasmids containing the CMVR VRC01 Ig heavy and light chains were obtained from the NIH Aids Reagents Program. The protein was expressed in 293F cells as described for Fabs above. The clarified supernatant was diluted twofold using 1× PBS buffer and purified using protein A agarose resin (Pierce), according to manufacturer's protocols. The eluent was concentrated and further purified by gel filtration chromatography in buffer of 10 mM Tris, pH 7.5, 100 mM NaCl using a SuperdexTM 200 Increase 10/300 GL column (GE Healthcare). The VRC01 Fab was obtained by digesting the Ig using papain (Pierce) and by running the digested products through protein A resin (Pierce) according to manufacturer's protocol. The Fab was then purified by gel filtration chromatography again as described above.
The codon-optimized synthetic constructs of the CH505 T/F HIV-1 subtype C gp120 containing amino acid (a.a.) 41–492 (HXB2 numbering) ΔV123 (core) was described previously (22). Briefly, the same pVRC-8400 expression vector as described for Fabs was used and the construct contained an N-terminal 6 × -histidine tag. The expression construct also contained a leader sequence encoding the tissue plasminogen activator signal sequence. The IRES-puro vector containing 92UG037.8 HIV-1 subtype A gp120 a.a. residues 1–492 (HXB2 numbering) ΔV123 (core) with a 6 × -histidine tag inserted between residues 40 and 41 and V5 mutations was also described previously (22). Recombinant Env glycoproteins were expressed in HEK 293T cells and purified as described for His-tagged Fabs.
Kinetic measurements of Fab binding to HIV-1 Env gp120 cores were carried out using the fortéBIO BLItz instrument using the same buffer all of our proteins were purified in (10 mM Tris, pH 7.5, 100mM NaCl) to minimize bulk shift effects. Fabs at concentration 0.2 mg/mL were immobilized on Dip and ReadTM Anti-Human Fab-CH1 second Generation Biosensors until saturation was reached (3 min). Env constructs were tested at concentrations ranging from 9.6 to 110 μM, depending on the particular construct used and its solubility, as well as based on its binding affinity to each Fab tested. The CH505 T/F gp120 wild-type core was tested at concentrations of 6.8, 22.0, and 66.0 μM, the CH505 T/F gp120 core ETF mutant was tested at concentrations 9.6, 19.3, and 58.0 μM, the 92UG037.8 gp120 core was tested at concentrations 10.8, 27, and 54.0 μM, and the 92UG037.8 gp120 core V5 mutant was tested at concentrations 27.5, 55.0, and 110.0 μM. Association and dissociation were each monitored for 2–2.5 min. Step corrections, at the start of association and at the start of dissociation, were performed manually using Microsoft Excel. Analyses were then performed using a global fit of at least three measurements using nonlinear regression curve fitting using the Graphpad Prism software, version 8.
CD spectra were recorded over the range of 260–210 nm to confirm the presence of a β-sheet signal at 216 nm (34) using an AVIV Model 435 Circular Dichroism Spectrometer. Fabs were loaded into 700 μL quartz crystal cuvettes at a concentration of 0.02 mg/mL in PBS buffer. Thermal melts were done from 20 to 90°C. CD signals at 216 nm were monitored every 0.5°C as the temperature of the samples increased at a ramp rate of 0.5°C/min. The samples were allowed to equilibrate for 5 s at each temperature before CD signals were recorded at an averaging time of 15 s. Data was analyzed with OriginLab using signal smoothing and assuming a two-state model with constant ΔH (35). In order to obtain the best fits, the approximated starting and ending baselines (which were assumed to be linear), melting temperature, and enthalpy of unfolding were adjusted. Curves were fit to a chi-squared tolerance value of 1 × 10−9.
I33M was concentrated to 11.24 mg/mL prior to crystallization. I33M crystallized in 96-well-tray format, set up with a TTPLabTech Mosquito® HTS in 1.5 M AmSO4, 100 mM Hepes, pH 7.0, 5% PEG 400 and was optimized in 24-well-format in which the pH of the solution was adjusted to 7.23.
X-ray diffraction data was collected using beamlines at the Advanced Photon Source at Argonne National Laboratory. I33M data were collected at beam line 24-ID-E using a 0.98 Å wavelength. Data set from an individual crystal was processed with HKL2000 (36). Molecular replacement calculations for the Fab were carried out with PHASER (37) using published I3.2 [Protein Data Bank (PDB) ID 4QHL] as the search model. The Fab was broken up into the variable and constant domains in order to perform two separate searches. I33M contained two molecules in the asymmetric subunit.
Refinement was carried out with PHENIX (38), and all model modifications were carried out with Coot (39) and PyMOL. During refinement, maps were generated from combinations of positional, group B-factor, and TLS (translation/libration/screw) refinement algorithms. All refinements used secondary structure and NCS restraints. Structure validations were performed periodically during refinement using the MolProbity server (40). The final refinement statistics are summarized in Table 2.
The structures analyzed in this study were superposed by least squares fitting in Coot.
The datasets generated for this study can be found in the Protein Data Bank accession code 6VPY.
JZ and DF coordinated and designed the study, analyzed and evaluated the data, and wrote and edited the manuscript and figures. JZ, HZ, TT, and DF performed experiments: TT, JZ, and DF expressed and purified proteins. TT and HZ performed BLI experiments. JZ collected CD spectra and performed thermal melts. JZ and DF crystallized proteins and determined structures. All authors contributed to the article and approved the submitted version.
JZ was supported by the Meyer Davidson '57 Summer Research Fellowship Grant. DF acknowledges support from the Mathilde Krim Fellowship in Basic Biomedical Research (109502-61-RKVA) from amfAR and Swarthmore College Startup and Faculty Research Funds. This work is based upon research conducted at NE-CAT beamlines, which are funded by the NIGMS from the NIH (P30 GM124165). The Eiger 16M detector on the 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank staff at Northeastern Collaborative Access Team (NE-CAT) X-Ray beam lines 24 ID-C/E (Advanced Photon Source). We thank Liliya Yatsunyk and Deondre Jordan for advice on the CD experiments. We thank the laboratory of Stephen Harrison for providing plasmids.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01529/full#supplementary-material
1. Mishra AK, Mariuzza RA. Insights into the structural basis of antibody affinity maturation from next-generation sequencing. Front Immunol. (2018) 9:117. doi: 10.3389/fimmu.2018.00117
2. Binley JM, Lybarger EA, Crooks ET, Seaman MS, Gray E, Davis KL, et al. Profiling the specificity of neutralizing antibodies in a large panel of plasmas from patients chronically infected with human immunodeficiency virus type 1 subtypes B and C. J Virol. (2008) 82:11651–68. doi: 10.1128/JVI.01762-08
3. Wu NC, Wilson IA. Structural insights into the design of novel anti-influenza therapies. Nat Struct Mol Biol. (2018) 25:115–21. doi: 10.1038/s41594-018-0025-9
4. Richman DD, Wrin T, Little SJ, Petropoulos CJ. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci U S A. (2003) 100:4144–9. doi: 10.1073/pnas.0630530100
5. Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, et al. Antibody neutralization and escape by HIV-1. Nature. (2003) 422:307–12. doi: 10.1038/nature01470
6. Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature. (2014) 509:55–62. doi: 10.1038/nature13036
7. Davenport TM, Gorman J, Joyce MG, Zhou T, Soto C, Guttman M, et al. Somatic hypermutation-induced changes in the structure and dynamics of HIV-1 broadly neutralizing antibodies. Structure. (2016) 24:1346–57. doi: 10.1016/j.str.2016.06.012
8. Schmidt AG, Xu H, Khan AR, O'Donnell T, Khurana S, King LR, et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci U S A. (2013) 110:264–9. doi: 10.1073/pnas.1218256109
9. Bonsignori M, Kreider EF, Fera D, Meyerhoff RR, Bradley T, Wiehe K, et al. Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci Transl Med. (2017) 9:eaai7514. doi: 10.1126/scitranslmed.aai7514
10. Fera D, Lee MS, Wiehe K, Meyerhoff RR, Piai A, Bonsignori M, et al. HIV envelope V3 region mimic embodies key features of a broadly neutralizing antibody lineage epitope. Nat Commun. (2018) 9:1111. doi: 10.1038/s41467-018-03565-6
11. Saunders KO, Wiehe K, Tian M, Acharya P, Bradley T, Alam SM, et al. Targeted selection of HIV-specific antibody mutations by engineering B cell maturation. Science. (2019) 366:eaay7199. doi: 10.1126/science.aay7199
12. Wiehe K, Bradley T, Meyerhoff RR, Hart C, Williams WB, Easterhoff D, et al. Functional relevance of improbable antibody mutations for HIV broadly neutralizing antibody development. Cell Host Microbe. (2018) 23:759–65.e6. doi: 10.1016/j.chom.2018.04.018
13. Sok D, Laserson U, Laserson J, Liu Y, Vigneault F, Julien JP, et al. The effects of somatic hypermutation on neutralization and binding in the PGT121 family of broadly neutralizing HIV antibodies. PLoS Pathog. (2013) 9:e1003754. doi: 10.1371/journal.ppat.1003754
14. Jardine JG, Sok D, Julien JP, Briney B, Sarkar A, Liang CH, et al. Minimally mutated HIV-1 broadly neutralizing antibodies to guide reductionist vaccine design. PLoS Pathog. (2016) 12:e1005815. doi: 10.1371/journal.ppat.1005815
15. Garces F, Lee JH, de Val N, de la Pena AT, Kong L, Puchades C, et al. Affinity maturation of a potent family of HIV antibodies is primarily focused on accommodating or avoiding glycans. Immunity. (2015) 43:1053–63. doi: 10.1016/j.immuni.2015.11.007
16. Garces F, Sok D, Kong L, McBride R, Kim HJ, Saye-Francisco KF, et al. Structural evolution of glycan recognition by a family of potent HIV antibodies. Cell. (2014) 159:69–79. doi: 10.1016/j.cell.2014.09.009
17. Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. (2013) 153:126–38. doi: 10.1016/j.cell.2013.03.018
18. Henderson R, Watts BE, Ergin HN, Anasti K, Parks R, Xia S-M, et al. Selection of immunoglobulin elbow region mutations impacts interdomain conformational flexibility in HIV-1 broadly neutralizing antibodies. Nat Commun. (2019) 10:654. doi: 10.1038/s41467-019-08415-7
19. Koenig P, Lee CV, Walters BT, Janakiraman V, Stinson J, Patapoff TW, et al. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc Natl Acad Sci U S A. (2017) 114:E486–E95. doi: 10.1073/pnas.1613231114
20. Ovchinnikov V, Louveau JE, Barton JP, Karplus M, Chakraborty AK. Role of framework mutations and antibody flexibility in the evolution of broadly neutralizing antibodies. Elife. (2018) 7:e33038. doi: 10.7554/eLife.33038
21. Liao HX, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. (2013) 496:469–76. doi: 10.1038/nature12053
22. Fera D, Schmidt AG, Haynes BF, Gao F, Liao HX, Kepler TB, et al. Affinity maturation in an HIV broadly neutralizing B-cell lineage through reorientation of variable domains. Proc Natl Acad Sci U S A. (2014) 111:10275–80. doi: 10.1073/pnas.1409954111
23. Pietzsch J, Scheid JF, Mouquet H, Klein F, Seaman MS, Jankovic M, et al. Human anti-HIV-neutralizing antibodies frequently target a conserved epitope essential for viral fitness. J Exp Med. (2010) 207:1995–2002. doi: 10.1084/jem.20101176
24. Li Y, O'Dell S, Walker LM, Wu X, Guenaga J, Feng Y, et al. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J Virol. (2011) 85:8954–67. doi: 10.1128/JVI.00754-11
25. Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. (2010) 329:811–7. doi: 10.1126/science.1192819
26. McConnell AD, Zhang X, Macomber JL, Chau B, Sheffer JC, Rahmanian S, et al. A general approach to antibody thermostabilization. MAbs. (2014) 6:1274–82. doi: 10.4161/mabs.29680
27. Wozniak-Knopp G, Stadlmayr G, Perthold JW, Stadlbauer K, Gotsmy M, Becker S, et al. An antibody with Fab-constant domains exchanged for a pair of CH3 domains. PLoS One. (2018) 13:e0195442. doi: 10.1371/journal.pone.0195442
28. Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. (2011) 333:1633–7. doi: 10.1126/science.1207227
29. Eisen HN, Chakraborty AK. Evolving concepts of specificity in immune reactions. Proc Natl Acad Sci U S A. (2010) 107:22373–80. doi: 10.1073/pnas.1012051108
30. Foote J, Milstein C. Conformational isomerism and the diversity of antibodies. Proc Natl Acad Sci U S A. (1994) 91:10370–4. doi: 10.1073/pnas.91.22.10370
31. Worn A, Pluckthun A. Mutual stabilization of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry. (1998) 37:13120–7. doi: 10.1021/bi980712q
32. Julian MC, Li L, Garde S, Wilen R, Tessier PM. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci Rep. (2017) 7:45259. doi: 10.1038/srep45259
33. Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. (1997) 276:1665–9. doi: 10.1126/science.276.5319.1665
34. Greenfield NJ, Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc (2006) 1:2876-90. doi: 10.1038/nprot.2006.202
35. Ramsay GD, Eftnik MR. Analysis of multidimensional spectroscopic data to monitor unfolding of proteins. Methods Enzymol. (1994) 240:615–45. doi: 10.1016/S0076-6879(94)40066-0
36. Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. (1997) 276:307–26. doi: 10.1016/S0076-6879(97)76066-X
37. McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Cryst. (2007) 63:32–41. doi: 10.1107/S0907444906045975
38. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. (2010) 66:213–221. doi: 10.1107/S0907444909052925
39. Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. (2004) 60:2126–32. doi: 10.1107/S0907444904019158
Keywords: human immunodeficiency virus (HIV), antibody, evolution, crystal structure, somatic hypermutation, framework
Citation: Zhou JO, Zaidi HA, Ton T and Fera D (2020) The Effects of Framework Mutations at the Variable Domain Interface on Antibody Affinity Maturation in an HIV-1 Broadly Neutralizing Antibody Lineage. Front. Immunol. 11:1529. doi: 10.3389/fimmu.2020.01529
Received: 11 February 2020; Accepted: 10 June 2020;
Published: 17 July 2020.
Edited by:
Zhiwei Wu, Nanjing Medical University, ChinaReviewed by:
Davide Angeletti, University of Gothenburg, SwedenCopyright © 2020 Zhou, Zaidi, Ton and Fera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Fera, ZGZlcmExQHN3YXJ0aG1vcmUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.