 Chi Li
Chi Li Howard Donninger
Howard Donninger John Eaton
John Eaton Kavitha Yaddanapudi
Kavitha Yaddanapudi- 1Experimental Therapeutics Group, James Graham Brown Cancer Center, University of Louisville, Louisville, KY, United States
- 2Department of Medicine, University of Louisville, Louisville, KY, United States
- 3Immuno-Oncology Group, James Graham Brown Cancer Center, University of Louisville, Louisville, KY, United States
- 4Division of Immunotherapy, Department of Surgery, University of Louisville, Louisville, KY, United States
- 5Department of Microbiology and Immunology, University of Louisville, Louisville, KY, United States
Extracellular vesicles (EVs) are a heterogenous group of membrane-surrounded structures. Besides serving as a harbor for the unwanted material exocytosed by cells, EVs play a critical role in conveying intact protein, genetic, and lipid contents that are important for intercellular communication. EVs, broadly comprised of microvesicles and exosomes, are released to the extracellular environment from nearly all cells either via shedding from the plasma membrane or by originating from the endosomal system. Exosomes are 40–150 nm, endosome-derived small EVs (sEVs) that are released by cells into the extracellular environment. This review focuses on the biological properties of immune cell-derived sEVs, including composition and cellular targeting and mechanisms by which these immune cell-derived sEVs influence tumor immunity either by suppressing or promoting tumor growth, are discussed. The final section of this review discusses how the biological properties of immune cell-derived sEVs can be manipulated to improve their immunogenicity.
Introduction
In the first marathon, run solo by Philippides, both the messenger and the message were targeted (although sadly the messenger died having delivered the message). Such precision targeting would be a desirable feature for successful cancer immunotherapy and here we argue that small vesicles spontaneously released by cells might be just the ticket.
Extracellular vesicles (EVs) are cell-derived nanovesicles that exhibit immunomodulatory properties and show promise as potential therapeutic agents (1, 2). In vitro and in vivo studies suggest that cell to cell communication is facilitated via an acellular EV-mediated process, leading to intercellular transfer of molecules (3). Importantly, EVs can transfer proteins, mRNA, and microRNA, thus, facilitating the genetic exchange between cells (4). Despite significant strides made in delineating biogenesis (5) and protein/lipid composition (6), the in vivo biological relevance of EVs in cancer-bearing hosts remains largely unclear. Early pre-clinical studies provide evidence that EVs can operate as therapeutic agents. EVs derived from antigen presenting cells (APCs) that are loaded with either peptide or whole protein antigens are reported to induce anti-tumor immunity in animal models but show only modest improvements in cancer patients (2, 7–9). These observations support the proposal that nano-sized EVs can be used as carriers to deliver soluble antigens in tumor models (10). The currently expanding knowledge about the biological effects of EVs provides clues about the pros and cons of using EVs in cancer therapy. The initial part of this review focuses on the nomenclature and biogenesis of EVs. The initial part of this review describes the composition and mechanisms by which immune cell-derived EVs interact with and influence host cells. The final part of this review describes how the biological properties of these immune cell-derived EVs can be engineered to amplify their immunogenicity as novel anti-cancer immunotherapeutic agents.
Nomenclature of Extracellular Vesicles (EVs)
EVs is an umbrella term that encompasses different types of vesicles including microparticles and exosomes released from eukaryotic cells. Accumulating evidence suggests that cells release EVs of different sizes and subcellular origin. The heterogeneity of EVs and the existence of non-vesicular extracellular nanoparticles creates confusion with respect to nomenclature. This also increases the complexity of defining the composition and functional properties of these very diverse secreted components. Until recently, parameters such as size, presence of unique proteins, subcellular origin, and isolation techniques that have been used to characterize the different vesicles have led to confusion rather than clarity in the field. One such example is the finding that EVs originating from late endosomes (exosomes) and vesicles originating from the plasma membrane (ectosomes/microparticles) (11, 12) share common molecular signatures and markers [e.g., TSG101and Alix (1, 13)]. In 2018, the International Society on Extracellular Vesicles (ISEV) endorsed EV as the generic term to be used for particles of any cellular origin that lack a nucleus and are delimited by a lipid bilayer (14). Additionally, the ISEV documented the “Minimal Information for Studies of Extracellular Vesicles (MISEV) guidelines” (15); additional findings have led to more recent updates to these guidelines (14). To counter the existing contradictions in the field of EVs, these guidelines recommend critical experimentation and reporting requirements pertaining to EV isolation, composition, characterization, and functional studies. One such class of characterization parameters include: (1) Size of EVs—small EVs (100–200 nm), large EVs (200–1,000 nm); (2) Sedimentation or density of EVs—low, middle, or high; (3) Marker expression—e.g., CD63, CD81, or Annexin A1-expressing EVs; (4) Types of cells—e.g., EVs-derived from heat-stressed cells, immune cells, apoptotic cells or hypoxic tumor cells; and (5) Biogenesis—e.g., plasma membrane or endosome.
Exosomes are 40–150 nm, endosome-derived small EVs that are released by cells into the extracellular environment. This process involves the fusion of endosomes with the plasma membrane (1). In contrast to exosomes (small EVs), microvesicles are large EVs (lEVs) and are generated via a process of shedding from the plasma membrane (16, 17).
Biogenesis of Exosomes
Exosomes are small EVs (sEVs). sEVs are formed intracellularly by inward budding of the endosomal membrane resulting in sequestration of RNA, DNA, proteins, and lipids into intraluminal vesicles (ILVs) within the lumen of multivesicular bodies (MVBs) (17). Fusion of MVBs with the plasma membrane leads to release of ILVs which are then termed sEVs; this budding event during sEV formation occurs in a reverse membrane orientation (17). Little is known about the molecules and the cytosolic machinery involved in the modulation of the sEV secretion.
The release of sEVs into the extracellular milieu involves fusion of the MVB with the plasma membrane. Several proteins loaded into sEVs originate from the MVB membrane. Some of these proteins include the major histocompatibility complex (MHC) and costimulatory molecules that ultimately participate in sEV-mediated regulation of immune responses. The cargoes of sEVs originate from the golgi apparatus or from the plasma membrane and are sorted into MVBs before being released as ILVs. This implies that cargoes that recycle back to the plasma membrane are typically not enriched in vesicles, however, a disruption to this recycling process can occur as seen in the case of transferrin receptor in reticulocytes (18). Also, the transport of cargo from golgi to endosomes implies that any disruption to the endosomal recycling or conditions that allow “retrograde transport” from endosomes to golgi will adversely affect the sorting of cargoes into sEVs (17).
Several sorting machineries participate in successful generation of sEVs (17). In an initial step for sEV generation, membrane-associated proteins and lipids are clustered in distinct microdomains of the limiting membrane of the MVB. The process of sorting proteins into MVBs is facilitated by a set of proteins called “endosomal sorting complex required for transport (ESCRT).” The process of binding of the cargo to the endosomes is initiated by ESCRT-0. Tamai et al. demonstrated convincingly that hepatocyte growth factor-regulated tyrosine kinase substrate (HRS; an ESCRT-0 protein) is required for sEV generation and release (19). Following this initiation by ESCRT-0, ESCRTs-I, -II, and -III complexes act in tandem to help the cargo accumulate on the endosomal membrane (19). VPS4, an AAA-type ATPase, disrupts the ESCRT complexes and the membrane with its cargo then gets embedded into the endosome to produce an MVB (19). MVBs utilize the intercellular membrane traffic system that aids in the release of ILVs as sEVs by direct fusion with the plasma membrane (5). Members of the Rab GTPase family (Rab5, Rab11, Rab27a, and Rab27b) (20, 21) have been shown to be involved in the secretion of sEVs (22, 23). Recent research suggests that the different cellular pathways involved in sEV biogenesis depend on the properties of the producing cell. For example, sEV biogenesis in T cells occurs at the plasma membrane and exploits a unique molecular machinery that is typically associated with endosomal biogenesis of ILVs (13, 20). Additionally, sEV generation and cargo composition can be altered by interfering with the components of the ESCRT pathway. One such approach is to target ESCRT accessory protein ALG-2 interacting protein X (ALIX) (21) to disrupt the release of ILVs. This is based on the fact that ALIX protein is involved in bridging protein cargoes with other subunits of the ESCRT protein sorting machinery (21). Such methods can be utilized to engineer cargoes to be secreted as sEVs.
sEVs can also be generated and secreted in an ESCRT-independent manner (24). Recent studies have revealed that ILVs loaded with CD63 can be formed even when the functional components of the four ESCRT complexes are depleted (24). One such ESCRT-independent mechanism for generation of sEVs is dependent on ceramide (23). Ceramide, which is generated by a type II sphingomyelinase, induces the formation of membrane microdomains. Ceramide has been shown to be converted to sphingosine 1-phosphate, which then activates Gi-protein-coupled sphingosine 1-phosphate receptor, a key player that sorts cargoes into exosomal ILVs (25). One other ESCRT-independent mechanism involves proteins of the tetraspanin family (CD81, CD82, CD9, and CD63) (5, 26, 27). These proteins route various sEV cargoes by forming clusters with each other as well as with other proteins (both transmembrane and cytosolic). They also facilitate the formation of microdomains that eventually bud into cargo carrying sEVs (5, 26, 27). For example, cone-shaped clusters of tetraspanin CD81 induce inward budding of microdomains enriched with CD81 protein (28). Based on these studies, it is apparent that the transmembrane cargoes can be sorted into the sEVs via both ESCRT-dependent and -independent mechanisms. For example, sorting of MHC Class II molecules into immune cell-derived sEVs relies on both ESCRT-dependent and ESCRT-independent mechanisms. Several different cytosolic proteins become part of the cargo protein repertoire. Chaperones such as heat shock proteins and heat shock cognate proteins (HSP70 and HSC70) play an important role in co-sorting these cytosolic proteins into ILVs in cells, including immune cells (29, 30).
Additional details and other characteristics of sEVs (exosomes) have been reviewed elsewhere (16, 17, 31, 32). The following sections are focused on composition and functional properties of sEVs derived from various immune cell types that exhibit either immune stimulatory or immune suppressive functionality in cancer.
Composition of Immune Cell-derived sEVs
Immune cell-derived sEVs contain ubiquitous and cell type-specific proteins that are involved in both immune stimulatory and suppressive functions (3, 33). Various cargoes including lipids and nucleic acids are selectively incorporated into sEVs (17). Moreover, several proteins present in the sEVs are conserved in the different parent cell types (3). A recent study by Jeppesen et al. shows that sEVs are devoid of any cytosolic glycolytic enzymes, cytoskeletal proteins, or double-stranded DNA (34). This study also identifies Annexin A1 as a specific marker of microvesicles (34).
With regards to composition and function, sEVs from antigen-presenting cells (APCs), which include Dendritic cells (DCs) and B cells, have been well-characterized. These APC-derived sEVs participate in antigen presentation via MHC Class I and MHC Class II molecules that are loaded with antigenic peptides. Antigens involved in this process are typically tubulin, actin, signal transduction protein kinases, metabolic enzymes, and heat shock proteins (3, 35, 36). Immune cell-derived sEVs are enriched in tetraspanins, a family of ubiquitous proteins, which include CD9, CD63, CD81, and CD82. These tetraspanins on sEVs have been shown to interact with other proteins such as MHC molecules and integrins expressed on the target cells, consequently leading to organization of membrane subdomains (37, 38).
Besides proteins, sEVs released from immune cells also exhibit unique lipid composition profiles. Sphingomyelin, phosphatidylcholine, and phosphatidylethanolamine are some of the lipids that are present in significant quantities in these sEVs (6, 39, 40). Additionally, sphingomyelin, cholesterol, and other lipids present in immune cell-derived sEVs are critical for the maintenance of rafts (41). sEVs mediate exchange of lipids between different cells. In contrast to lipoproteins that contain a phospholipid monolayer, sEVs have a bilayer membrane surrounding cytosolic material and therefore transport lipids using a distinct mechanism. These sEVs display increased “trans-bilayer flip-flop movements of phospholipids.” These flip-flop movements facilitate the fusion of sEVs to target cell membrane (6). Immune cell-derived sEVs also harbor a unique set of enzymes that participate in lipid metabolism (phospholipase A2, C, and D) (39, 42). Intriguingly, immune cell-derived sEVs possess a phospholipid composition that is different from that found in the parent cells. DC-derived sEVs (dexosomes) have higher sphingomyelin and lower phosphatidylcholine levels when compared to that observed in the parent cells. Phospholipase D2 is highly expressed in sEVs and this enrichment allows these vesicles to interact with target cells. This process is dependent on the fusogenic properties of its signaling messenger—phosphatidic acid (PA) (43). PA can trigger membrane fusion in the presence of calcium (44) that subsequently facilitates interdigitation of lipid molecules between different membranes (41).
Recent technological advances such as next generation sequencing have paved the way for identification of the nucleic acid cargo (DNA and RNA) harbored in sEVs (4, 45). Immune cell-derived sEVs have been shown to harbor miRNAs that influence both innate (monocytes) and adaptive immunity (T and B cells) in cancer. sEVs also harbor long non-coding (lnc) RNAs that are then transferred into target cells (46). Several different lncRNAs (MALAT-1, linc-POU3F3, ZFAS1, and GAS5) have been identified (47–49). sEVs harboring GAS5 are also known to regulate innate and adaptive immune responses (50–52). A better understanding of the molecular events within the parent cell that are involved in the sorting and processing of miRNA and lncRNA into sEVs is key toward developing novel biomarkers and sEV-based therapeutics.
Additional details of the cargo that participate in the immune stimulatory or suppressive functions of the immune cell-derived sEVs are discussed below.
Isolation of sEVs and Their Interaction With Target Cells
Differential ultracentrifugation is a commonly employed procedure used to isolate sEVs released into the extracellular milieu. One limitation of this widely used procedure is that it does not allow discriminating between sEVs and other vesicle-like structures. This limitation can be overcome by using sucrose gradient floatation (53). Since sEVs float on sucrose gradients, sEVs can be separated from other contaminants. However, repeated ultracentrifugation can induce irreversible damage to the vesicles and reduce yields (54). An alternative to ultracentrifugation is concentration of sEV-containing solution using ultrafiltration devices or size-exclusion chromatography (53). Particle yield and purity depend on the type of isolation method used and the source of the sEV-containing sample (e.g., sEV isolation from cell-culture supernatant vs. sEVs from human plasma). Robust and high-throughput methods that maximize isolation of homogeneous sEVs are critical to establish biological applications involving sEVs. In a recent study, Robert Coffey's group has employed a two-step method to isolate pure sEVs: step (1) high-resolution iodixanol density gradient fractionation to separate sEVs from other contaminating vesicles; followed by step (2) direct immunoaffinity capture (DIC) with capture beads targeting sEV tetraspanins (34). This two-step method allows for the separation of a low-density fraction (cup-shaped vesicles with morphology and size consistent with sEVs) and a high-density fraction (pool of non-vesicular components) (34).
sEVs in the extracellular milieu can transfer their contents to a target cell and influence its function and phenotype. This transfer process involves a sequence of events that include docking of sEVs at the plasma membrane, surface receptor activation/signaling by the target cell, and endocytosis of the vesicle or its fusion with target cells. Details of the molecular events that are part of this delivery process, including uptake and intercellular trafficking, are yet to be unraveled. The events are likely to rely on multiple variables such as the origin of sEVs as well as identity of recipient cells.
It is very likely that the recognition between sEVs and the target cell involve proteins present at the cell surface of sEVs (55, 56). Immune cell-derived sEVs have been shown to communicate with target cells via expression of a series of cell-specific transmembrane proteins (e.g., DC-specific α and β chains of integrins, B cell-specific ICAM-1 and CD54 proteins), cells surface peptidases [e.g., aminopeptidase N (CD13) expressed on mast cells], and the integrin-interacting protein lactadherin (3, 35, 57, 58). Tetraspanins expressed on sEVs have also been reported to interact with integrins and facilitate sEV uptake by target cells (59–62). CD44 is a cell surface glycoprotein expressed on immune cells and has been reported to be involved in docking and uptake of sEVs (63). Cargoes delivered by sEVs can elicit signaling events in the target cells. These events have been well-documented for sEVs derived from B cells and dexosomes which when internalized are able to present antigens to T cells and activate antigen-specific T cell-mediated immune responses (8, 64). Cargoes released from sEVs into target immune cells (including APCs) are processed in the endocytic compartment in a manner similar to that used for processing other internalized antigens. Thus, cargoes delivered by sEVs can directly influence immune modulatory functions of the target cells (55) (discussed further in the sections below).
Immunostimulatory Effects of Immune Cell-derived sEVs in Cancer
Exosomal sEVs derived from DCs, B cells, T cells, NK cells, and mastocytes have been widely characterized and are known to exhibit diverse functional properties (Figure 1). In the case of sEVs derived from APCs (B cells and DCs), immune stimulation requires interaction of antigen-loaded MHC-I and MHC-II molecules expressed on sEVs with TCRs expressed on T cells. Alternatively, T cell stimulation may be via an indirect route that mainly occurs through a process of internalization of antigen-loaded exosomes by APCs that then present the processed antigen as MHC-restricted peptides to T cells. APC-derived sEVs carry pro-inflammatory cytokines and chemokines that can influence T cell migration and expansion. Apart from antigen presentation, sEVs isolated from immune cells such as NK cells express cytolytic proteins and granules that can perform effector functions and induce cytotoxic cell lysis and elimination of tumors and, in some cases, activated immune cells. Likewise, CD8+ CTL-derived sEVs can produce cytotoxic cytokines and can target tumor cells for cytotoxic elimination. Mastocyte-derived sEVs are known to carry effector cytokines IL-2 and IFN-γ, induce B cell and T cell proliferation, and induce DC maturity (65). Cumulative research over the last decade suggest that sEV-mediated immune stimulation is dependent on functional interactions between several different immune cell populations. For example, induction of CD8+ T cell-mediated anti-tumor immunity by B cell-derived sEVs is reliant on cross-talk with the T helper cell arm (CD4+ T cell) and innate cell arm (NK cells) of the immune system (66). Along similar lines, sEVs from DCs loaded with proteins (but not peptides) are efficient activators of the immune responses and this immune activation is dependent on help from CD4+ T cells and B cells (67). In this section, the immune stimulatory properties (composition of cargo, target cell recognition and cargo release) of sEVs derived from immune cell types (mentioned above) are discussed in detail (sections Dendritic Cell-Derived sEVs, B Cell-Derived sEVs, Natural Killer (NK) Cell-Derived sEVs, T Cell-Derived sEVs, and Mast Cell-Derived sEVs).
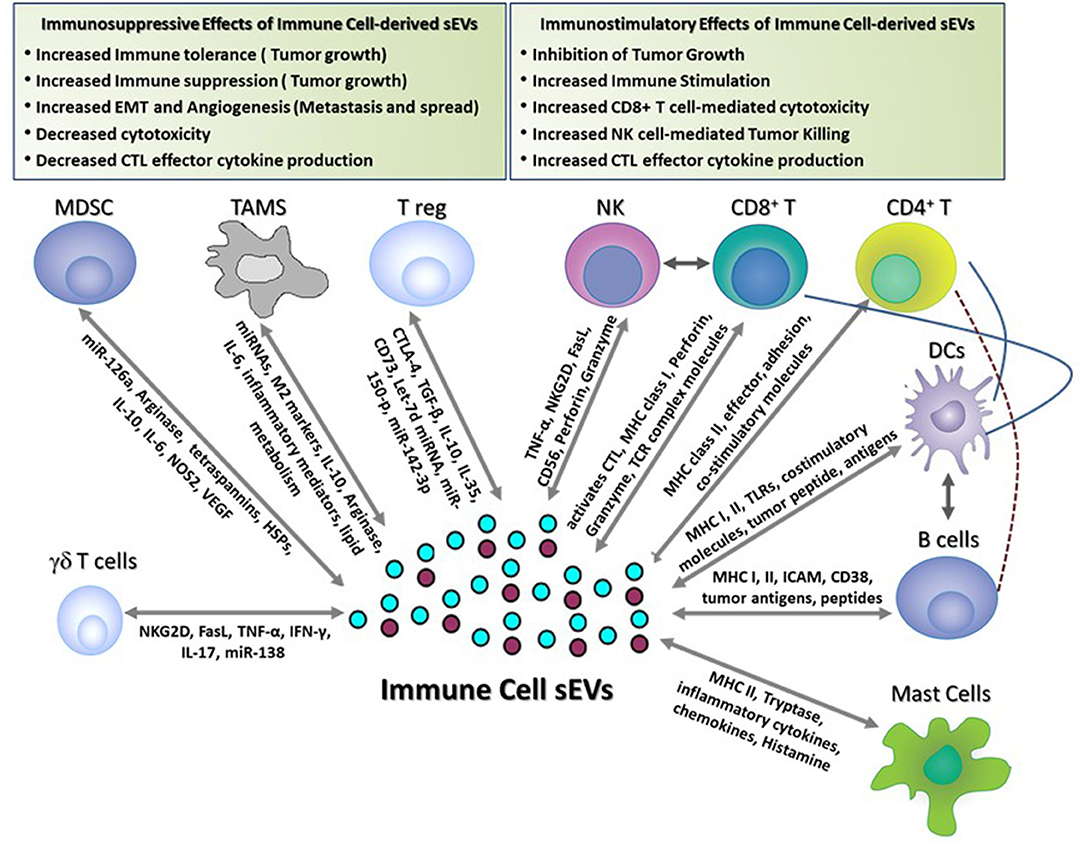
Figure 1. Immune-stimulatory and suppressive effects of immune cell-derived sEVs in cancer. This figure shows the different functions of immune cell-derived sEVs that have been reported to play a role in modulating immune responses in tumor-bearing hosts. These sEVs activate anti-tumor immune responses and inhibit tumor growth by (1) inducing Dendritic cell- and B cell-mediated antigen presentation, (2) promoting CD4+ T cell helper responses, (3) activating CD8+ T cell- and NK cell-mediated effector responses and cytotoxicity, and (4) inducing mast cell-mediated secretion of immune regulatory cytokines and chemokines. Conversely, immune cell-derived sEVs suppress the immune responses and promote tumor growth, invasion, and metastasis by (1) suppressing T cell-mediated anti-tumor effector responses and cytotoxicity, (2) promoting angiogenesis and EMT, (3) inducing FasL-mediated T cell apoptosis, and (4) producing immune suppressive soluble mediators and cytokines.
Dendritic Cell-Derived sEVs
Dexosomes (sEVs released from Dendritic cells) have received attention as anti-cancer immunotherapeutic agents since they harbor functional immune activating cargo (68, 69). Dendritic cells (DCs) are attracted to the site of dying tumor cells and play a key role in driving tumor antigen-specific T cell-mediated anti-tumor responses. The stability, ease of storage/shipping and more efficient uptake into target cells (when compared to that of soluble molecules) make dexosome-based vaccines a viable therapeutic strategy against cancer (7, 8, 10, 70). Immunostimulatory effects of dexosomes have been tested in pre-clinical mouse and ex vivo human models of cancer as well as in clinical trials. Dexosomes loaded with α-fetoprotein [AFP; (71)] are effective against hepatocellular carcinoma in mice. Immunization with AFP-containing sEVs results in a Th1-mediated anti-tumor CD8+ T cell infiltration into the tumors that is accompanied with a significant reduction in intra-tumoral T regulatory cells (71). In another example, mice immunized with sEVs from DCs loaded with cervical cancer-associated antigenic peptide (HPV early antigen 7 peptide) were protected against cervical cancer (72). Murine dexosomes have been shown to be able to stimulate melanoma antigen-specific CD4+ and CD8+ T cells in vitro and generate anti-cancer immunity in vivo (8, 73–75). In a HLA-A2 transgenic mouse model, sEVs pulsed with tumor peptides efficiently prime MART1-specific CTLs (76). Murine dexosomes indirectly loaded with either tumor cell lysate or total tumor RNA have been tested against aggressive tumors that respond poorly to therapies involving single tumor antigens. DC-derived sEVs loaded with tumor lysate enriched with molecular chaperone family of proteins such as calreticulin and heat shock protein 70 and 90 are reported to present a superior source of tumor antigens and immune responses in vivo than that offered with sEVs loaded with lysates obtained from freezing/thawing of tumor cells (77). DC-derived sEVs that have been indirectly loaded with a combination of common antigen ovalbumin and immunogenic invariant NK ligand α-galactosylceramide (αGC) have been shown to impart immunostimulatory effects in vivo in mouse models of cancer (78).
The maturation status of the DCs as well as the nature of the maturation stimuli have been shown to dictate the immunostimulatory efficacy of the DC-derived sEVs (79). DC maturation is best achieved upon treatment with TLR3, TLR4, or TLR9 ligands [poly(I:C), LPS, and CpG-B oligonucleotide; (79)]. EVs derived from DCs exposed to hyperthermia and stress (80, 81) contain increased amounts of heat shock proteins and chemokines (CCL2, CCL5, and CCL20) and as such, promote infiltration of tumor-attacking T cells and DCs into the tumor microenvironment of mice treated with these EVs (82). In addition to inducing CD4+ T and CD8+ T cell as well as NK cell-mediated responses, DC-derived antigen-loaded sEVs can induce B cell activation and IgG secretion which play an important helper role in activation of adaptive anti-tumor T cell responses (67, 75, 83, 84). Human DC-derived sEVs have been reported to be involved in activation and proliferation of NK cells and this activation process involves proinflammatory cytokines family of ligands including TNF and IL-15Rα (85, 86).
Although, in pre-clinical mouse and ex vivo human models of cancer, dexosome-based therapy is efficacious in inducing sufficient anti-tumor immunity, it has had no demonstrable success in clinical trials of cancer. In clinical trials with melanoma and lung cancer patients using sEVs from autologous monocyte derived-DC cultures, only modest anti-tumor responses were observed (7, 9, 87). This failure to obtain robust anti-tumor immune responses has been attributed to diminished NK cell function (9, 87). One limitation of this immunotherapeutic strategy could be that the dexosomes lack co-stimulatory signals required to mediate activation of CD8-mediated T effector cells that are equipped to recognize and destroy tumor cells. Also, the antigenic drift and a highly immune suppressive tumor microenvironment present in the non-T cell inflamed tumors could be important contributing factors for the therapeutic failure.
B Cell-Derived sEVs
B cells are a central component of the humoral arm of the immune system and are involved in presentation of antigens and production of immunoglobulins and pro-inflammatory soluble mediators. Receptors expressed on the B cells capture extracellular antigens and toxins which are then translocated and processed via the endosomal pathway for antigen presentation or harbored into vesicles and released into the extracellular milieu (88–90). sEVs isolated from both murine and human B cells contain MHC class II molecules and human leukocyte antigens (HLA) along with co-stimulatory and adhesion molecules that are part of the antigen presentation machinery of B cells; high levels of expression of these molecules present as cargo can induce antigen-specific immunostimulatory responses against cancer (37, 64, 91). sEVs derived from B cells that are loaded with MHC class II molecules have been shown to stimulate CD4+ T cells in vitro (64). B cell-derived sEVs also harbor functional integrins that then facilitate high-affinity interactions with other cells, including cytokine-activated fibroblasts (92).
B cell-derived sEVs, akin to other immune cell-derived sEVs, interact with other immune cells including DCs, macrophages, T cells, and influence their functionality; these associations occur in different immune and non-immune compartments that include blood, spleen, lymph nodes, and tumors (93). In this context, B cell-derived sEVs are known to migrate across tissue barriers by forging interactions with extracellular matrix proteins (ECM) for delivering the cargo to recipient effector cells which involves specific receptor-ligand interactions (92). B cell-derived sEVs have been shown to harbor functional integrins and ICAM-1 that facilitate high-affinity interactions with ECM which paves way for delivery of cargo to T cells. This cargo transfer is facilitated via the interaction of ICAM-1 with leukocyte function-associated antigen (LFA)-1 molecules expressed on T cells. In some cases, B cell-derived sEVs are known to deliver cargo to pro-inflammatory cytokine-activated fibroblasts (92).
Despite the high levels of expression of MHC class I molecules, antigen-loaded B cell-derived sEV-mediated CD8+ T cell immunogenicity relies on its complex interactions with other immune cells including host APCs (DCs/macrophages), CD4+ T cells and NK cells. Data from in vivo mouse studies show that the antigen presentation to CD8+ T cells requires MHC I on host APCs and is independent of sEV MHC I (94, 95), reiterating the importance of host immune cell interactions in B cell sEV-mediated CTL induction, a key step to enhance anti-tumor immunity. Likewise, another mechanism of induction of T cell immunity can be via DC-mediated cross-presentation of B cell-derived sEV antigens to T cells in vivo. Saunderson et al. showed that Langerin+CD8α+ DCs residing in the spleen take part in the cross-presentation of B cell-derived sEV antigens to CD8+ cytotoxic T cells following immunization into mice (66).
On their surface, B cell-derived sEVs carry CD38, an active glycoprotein enzyme, that associates with signaling complexes HSC-70, Lyn, and CD81; this is akin to the associations of CD38 with signaling complexes in the membrane rafts during TCR engagement. CD38 ligation-induced signaling cascade can potentially act as intercellular messengers of T cell activation (96). Papp et al. have reported that B cells covalently fix the complement C3 fragments on their cell membrane and these fragments are then released onto the B cell-derived sEVs. Antigen-loaded B cells release C3-carrying sEVs which then interact with G protein coupled receptors on T cells. This interaction lowers the antigenic stimulation threshold required for inducing T cell activation (97). In the tumor microenvironment as well as in draining lymph nodes, pathological activation of the complement pathway and interplay between immune cells and tumor cells can lead to release of sEVs bound with C3 fragment, ensuing immune modulation. In contrast to the primary B cell-derived sEVs, several B cell-derived lymphoblastoid cell-lines have been shown to release sEVs that harbor FasL molecules. These FasL-bound sEVs are capable of inducing apoptosis in CD4+ T cells and dampening the host immune responses (98).
Natural Killer (NK) Cell-Derived sEVs
NK cells are large lymphocytes that can efficiently kill oncogenic transformed tumor cells, cells that are infected with certain viruses, and cells that are devoid of MHC class I antigen expression. This innate killing activity is not governed by any antigen specific recognition, prior activation or immunization. NK cells also take part in the immune regulation of the adaptive arm of the immune system via secretion of proinflammatory cytokines and chemokines (99, 100). Seminal research has identified several different immune check points that participate in anti-tumor effector functions of NK cells (101, 102). These findings have led to the emergence of NK cells as important targets for cancer immunotherapy. NK cell-based therapeutic efficacy can be enhanced significantly by increasing its ability to home in into the tumors. The nano-size, diffusion and retention capabilities of NK cell-derived sEVs, and the leaky vasculature of the solid tumors (103, 104), provide an environment where tumor-killing effector molecules can be delivered directly to the tumor site, and thus, overcome the homing deficiency of whole NK cell-based therapies. sEVs released from human NK cells have been shown to contain prototype NK cells markers (CD56, CD16). These sEVs can induce target cell death in the following ways: interactions of death receptor ligand FasL (on sEVs) with its receptor Fas on target cells; and releasing lytic granules perforin upon fusion with the target cells. In one of the first studies, Lugini et al. showed that sEVs purified from resting human NK cells exhibit cytotoxic activity against hematologic cell lines but do not kill solid tumor cells (105). However, sEVs isolated from activated human NK cells contain higher levels of cytotoxic effector proteins granzyme A/B and perforin, thereby mediating significant ex vivo cytotoxic activity against a variety of solid tumor lines including neuroblastoma (106).
In an in vivo study with a mouse model of melanoma, intra-tumoral injection of NK cell-derived sEVs caused inhibition of tumor growth (107). These sEVs isolated from NK-92 cells express cytolytic proteins FasL, TNF-α, and granules perforin and are able to induce cytolytic activity against melanoma cells (107). In a subsequent study, NK cell-derived sEVs were tested against glioblastoma xenograft tumors in mice. In this model, NK cell-derived sEVs administered via intravenous route are able to cross the blood-brain barrier, accumulate specifically at the tumor site, persist for several days, and suppress the growth of neuroblastoma (108). NK-cell derived sEVs also participate in maintaining immune homeostasis by acting against over-reactive immune cell expansion. In fact, circulating NK cell-derived sEVs can be isolated from healthy donor plasma. Such sEVs contain classical NK markers CD56, perforin, and activating receptor NKG2D but lack FasL expression suggesting that circulating sEVs from resting NK cells can participate in immune regulation in a paracrine fashion (109). Apart from plasma membrane fusion and receptor-ligand interactions, NK cell-derived sEVs can induce target cell cytotoxicity in an activated caspase pathway-mediated or granulysin-mediated mechanisms (110).
One question that remains to be answered is: how do NK cell-derived sEVs recognize and show specificity toward tumor cells or activated immune cells? NK cells rely on a “self-missing” mechanism to recognize and kill target cells; however, it is not clear if such a phenomenon applies to NK cell-derived sEVs. One suggested mechanism is that once the sEVs reach the tumors, the acidic microenvironment promotes the fusion of sEVs with the tumor cells or with the stomal immune cells which then results in uptake and activation of sEV-mediated biological functions.
T Cell-Derived sEVs
Activated CD8+ T cells are indispensable for tumor cell destruction and elimination. T cells perform these activities by direct interaction with target cells presenting various tumor antigens along with MHC class I molecules (111). In addition to tumor cell killing, activated CD8+ T cells are known to execute tumor cell elimination using several different indirect methods, one of which is via sEVs. CD8+ T cell-derived sEVs express receptors that directly recognize target cell antigens presented in the context of MHC class I at the immunological synapse, contain cytotoxic effector cytokines (e.g., IFN-γ, TNF-α), and proteins (e.g., perforin, Granzyme B) that induce target cell lysis. sEVs carrying these molecules cumulatively participate in linking the cytotoxic T cells with the target cells (tumors cells in case of cancer) and mediate CTL-mediated destruction of the target cells (72). TCR stimulation (not mitogenic stimulation) induces release of sEVs from both in vitro cultured T cell clones and T cell blasts circulating in the peripheral blood of healthy volunteers (112). The cargo in these endocytic sEVs are composed of TCR complex molecules (CD3ε, ζ chain, and TCR β), CD63, CD81, adhesion molecules (CD2 and LFA-1), Src family of tyrosine kinases, c-Cbl, and chemokine receptors (112). Apart from these proteins, T cell-derived sEVs harbor heat shock proteins, enolase, integrins, and proteins of antigen processing machinery (MHC class I and β2-microglobulin).
In an in vitro study, the authors show that sEVs from CD3+ T cells activated with TCR stimulation in the presence of IL-2 stimulate autologous proliferation and cytokine and chemokine production in resting T cells (113). Seo et al. recently reported that sEVs from activated T cells, when injected into the tumor microenvironment in mice, are engulfed by mesenchymal stromal cells. This activity leads to depletion of the stromal cells that include PDGFRα+ CD140a+ mesenchymal stem cells and α-SMA+ cancer-associated fibroblasts that ultimately results in reduction of tumor growth, invasion, and metastasis (114). CD63-expressing sEVs from T cells are reported to polarize toward the immunological synapse formed during cognate APC-T cell interactions. This interaction facilitates the unidirectional transfer of miRNA from T cell-EVs to APCs such as B cells. Such miRNAs (e.g., miR-335) play a functional role in regulating gene expression and activation in B cells (115). The sumoylated heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) binds specifically to miRNAs and is reported to control the loading of miRNAs into vesicles (116). sEVs released from activated T cells are known to carry cargo that interact with endothelial cells and play an active role in promoting angiogenesis. sEVs from activated T cells carrying miR-142-3p upon interaction with endothelial cells induce endothelial permeability. CD47-expressing T cell-derived sEVs are reported to induce VEGF-dependent angiogenic activity by promoting endothelial cell expansion and tube formation (117).
Much like CD8+ T cells, CD4+ T helper cells activated by TCR stimulation in the presence of co-stimulatory signals release plasma membrane sEVs into immune synapses and exhibit immune regulatory properties (112, 118). van der Vlist et al. used a flow cytometry-based method to analyze sEVs. They find that a heterogenous pool of sEVs (based on density fractionation) are released from activated CD4+ T cells. This heterogeneity relies on the strengths of TCR activation and costimulatory signals (119). In fact, resting CD4+ T cells preferentially uptake sEVs released from circulating IL-2 activated CD4+ T cells. This process can induce proliferation and expansion in these resting cells. Such an autologous cellular tropism of circulating CD4+ T cell-derived sEVs can have important implications in disease conditions such as cancer and HIV (120). Along similar lines, mitogen-stimulated CD4+ T cells take up OVA-MHC Class I+ DC-derived sEVs and express acquired molecules MHC class I and OVA. These modified T cells can perform the role of APCs and stimulate antigen-specific CD4+ T-dependent CD8+ T cell proliferation, expansion and anti-tumor effector responses in vivo (73, 121). In a reciprocal interaction, unilateral transfer of mitochondrial DNA from T cell-derived sEVs to DCs primes activation of the cGAS/STING/DNA-sensing immune signaling pathway and expression of IRF3-responsive interferon genes that protects the DCs from infections and other insults (122).
A recent report shows that sEVs derived from follicular T helper cells (fTh) participate in modulating B cell function and differentiation (123). Given that fTh play a vital role in the differentiation of B cells into plasma cells, fTh-derived vesicles can perform similar functions in influencing B cell differentiation and affinity maturation in cancer.
Mast Cell-Derived sEVs
Mast cells are innate immune cells located in the mucous membrane and connective tissue. These are the major effectors involved in allergy responses. Activated mucosal and connective tissue mast cells play an important role in expulsion of parasites by releasing biologically active granules (histamine), proinflammatory lipid mediators, and Th2-type cytokines (IL-4, IL-10) (124). Mast cells express Toll like receptors (TLR) and display immune regulatory activity by producing cytokines and chemokines (IL-5, IL-6, IL-10, IL-13, and TNF-α) that influence differentiation and biological functions of adaptive immune cells (DCs, T, and B cells) (125–127). Important stimuli for activation of mast cells are evoked by synergistic allergen-mediated engagement of FcϵRI (receptors for the Ig Fc portion) and TLR signaling. This leads to activation of transcription factors that are involved in secretion of soluble mediators, cytokines, and chemokines (128). During inflammation and other pathological disease conditions, mast cells accumulate at various primary and secondary immune organ interfaces (skin, lung, gut, tonsils, and lymph nodes) and orchestrate immune responses (129, 130). It appears that mast cells induce both pro- and anti-tumorigenic responses and these functions are dictated by type and stage of cancer. Mast cells accumulate in the blood as well as in the tumor microenvironment and support tumor progression in certain cancer types. In other settings, they regulate DC and T cell functions and contribute to anti-tumor immune responses (131).
Mast cell-derived sEVs are reported to carry 200–400 different proteins, many of which are yet to be identified. Few identified proteins include Plasminogen Activator Inhibitor Type I, proinflammatory cytokines, secretory granules, α and γ subunits of FcεRI, mast cell-related G protein-coupled receptor family member X2 (MRGX2), tryptase, and MHC class II molecules which are found exclusively on the sEV membrane (132, 133). Initial research suggests that sEVs in mast cells are contained within either endosomal (type I) or secretory lysosomal compartments (type II). sEVs from type I compartment colocalize with mannose-6-phosphate receptors and lamp I/II. sEVs from type II colocalize with serotonin. sEVs within these two compartments carry different protein/mRNA cargo and thus, exhibit differential immune regulation properties (65). Later studies confirmed that, activated mast cells release sEVs through two secretory routes: constitutive secretion [e.g., mast cells pre-treated with IL-4; (65)] and exocytosis [mast cells activated via FcεRI crosslinking; (134, 135)]. Carroll-Portillo et al. (133), using immunoprecipitation and electron microscopy studies, showed that mast cell-derived sEVs carry intact FcεRI. Furthermore, “right-side out” orientation of these vesicles exposes the FcεRI–IgE complexes on their cell surface. This facilitates continued recycling of cross-linked antigens and amplification of immune response. In this context, uptake of IgE and antigen loaded mast cell-derived sEVs by APCs (B cells and DCs) can result in efficient presentation of antigenic peptides to T cells. A novel functional role of mast cell-derived sEVs is their ability to package mRNA and microRNA into their lumen and transfer these shuttle RNA (esRNA) to recipient cells that engulf these vesicles (136, 137). This can then result in initiation of protein translation in recipient cells, which in essence facilitates the intercellular communication between mast cells and other immune or tumor cells (136). Mast cell-derived sEVs were reported to carry endocytosed exogenous antigens that associate with chaperone proteins (heat shock proteins 60 and 70) and elicit adjuvant-like immunity. Also, sEVs from mast cells are capable of inducing in vivo IgG1- and IgG2a-mediated antibody responses. Upon exposure to mast cell-derived sEVs (but not B cell- or macrophage-derived sEVs), immature DCs upregulate maturation markers (MHC class II, CD40, CD80, CD86) and produce IL-12. These activated DCs can cross-present antigens from mast cell-derived sEVs to T cells suggesting that mast cells sEVs can induce functional activation of DCs (65). Given the plasticity of DCs, it appears that the interactions between mast cell-derived sEVs and DCs are likely to vary depending on the tissue type, e.g., skin vs. tumor microenvironment.
In addition to immune-stimulatory functionality, mast cell-derived sEVs can also promote angiogenesis and immune tolerance. Mast cell activation can lead to release of sEVs loaded with tryptase, which can induce the proliferation and migration of endothelial cells and promote angiogenesis (138). Also, sEVs released from mast cells that are pre-exposed to oxidative stress carry mRNA that are capable of inducing oxidative stress tolerance in target cells; this functional property differs from that present in sEVs obtained from non-stressed parental cells (136). In a recent study, Liang et al. used bioinformatic tools to compare and contrast the gene expression profile of mast cells and mast cell-derived sEVs. This analyses reveal that several genes are differentially expressed in mast cell-derived sEVs (139).
Immunosuppressive Effects of Immune Cell-derived sEVs in Cancer
The tumor microenvironment is comprised not only of malignant cells, but also non-malignant cells such as immune cells, fibroblasts, and vascular and lymphatic cells. The dynamic interplay between the tumor cells and these stromal cells plays a central role in establishing an immunosuppressive microenvironment (140). While many of the immunosuppressive immune cells recruited to the tumor microenvironment mediate their suppressive function through secretion of soluble mediators, there is now emerging evidence that immune cell-derived sEVs also contribute to their immunosuppressive activity (Figure 1). Virtually every immune cell releases sEVs with cargo that represents the molecular expression of the parental cell. Secretion of immunosuppressive sEVs greatly amplifies a given cell's influence in the TME by allowing it to impact a number of target cells. Identification of the specific immunosuppressive cargo and mechanisms mediated by immune cell-derived sEVs can lead to new therapeutic targets and strategies which may improve the efficacy of anti-cancer therapy.
sEVs-Derived From MDSCs
Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells that suppress T cells and NK cells activity and dampen the tumor-killing capabilities of these cells (141). MDSCs accumulate in late-stage cancer patients and contribute significantly to immunotherapeutic resistance in cancer (141). MDSCs also contribute to immune suppression in other disease settings such as chronic infections (142). Recently, one of the areas of research in the immuno-oncology field has been focused on pharmacological inhibition of MDSC activity and combining these drugs with other known immunotherapeutic agents to improve therapeutic responses in cancer patients. A recent report suggested that common exosomal proteins (e.g., annexins, tetraspanins, cytoskeletal proteins, heat shock proteins) as well as several unique proteins are present in both MDSCs and MDSC-derived sEVs (143, 144). These sEV-derived immune suppressive proteins participate in immune regulation in cancer-bearing hosts (143, 144). MDSC-derived sEVs are also implicated in therapeutic resistance in cancer-bearing hosts (145–147). Chemotherapy induces MDSC numbers in tumor-bearing hosts that results in attenuation of the anti-cancer efficacy of the chemotherapy (148–150). Notably, in tumor-bearing mice treated with doxorubicin, MDSC-derived sEVs directly accelerate the proliferation and metastasis of tumor cells, and this is mediated by miR-126a (151). Furthermore, sEVs from MDSCs mediate the induction of angiogenesis, enhance Th2 cell responses, and inhibit T cell proliferation, thereby promoting an immunosuppressive microenvironment (151). Additional research with these MDSC-derived sEVs will shed light on their interactions with other tumor-fighting immune cells/sEVs and as such, their influence on anti-cancer immunotherapeutic response. It will also be interesting to explore the possibility of harnessing the immune suppressive properties of MDSC-derived sEVs as a therapeutic option in autoimmune diseases to mitigate the uncontrolled immune responses.
sEVs Derived From Tumor-Associated Macrophages (TAMs)
In the tumor microenvironment TAMS are the most represented population of immune cells (152). TAMS promote angiogenesis, invasion and metastasis, and modulate immunosuppression (153–156). There is emerging evidence that TAMS are able to produce sEVs (157) which impact both malignant and non-malignant cells in the TME. TAMS have essentially two opposing phenotypes; M1 which is anti-tumorigenic and M2, which is pro-tumorigenic. It appears that the immunosuppressive activity of sEVs secreted from TAMs is predominantly from M2 polarized macrophages. sEVs derived from M2 TAMS promote tumor cell migration and invasion (158, 159). TAM-derived sEVs have been reported to mediate the interaction of TAMS with T cells in the TME through the transfer of specific miRNAs, which induces a Treg/Th17 imbalance and facilitates tumor progression and metastasis in ovarian cancer (160). Furthermore, TAM-derived sEVs confer therapeutic resistance to ovarian cancer cells, again via the transfer of miRNAs (161). In contrast to these findings, in preclinical models of colon and breast cancer, TAM-sEVs have been shown to have a molecular profile more indicative of an M1 polarization signature, and their cargo is enriched in markers of inflammation and lipid metabolism (162). These sEVs promote T cell proliferation and activation, and thus may have the potential to stimulate anti-tumor immunity (162). The ability of TAM-derived sEVs to potentiate the immunosuppressive function of TAMS or promote anti-tumor immunity may thus be context- and tumor type-dependent, and will require additional studies to clarify. Nevertheless, targeting TAM-derived sEVs, or the specific miRNAs that are critical for the immunosuppressive activities of TAMs, may be a potential therapeutic approach to facilitate current therapeutic strategies.
sEVs Derived From γδ T Cells
γδ T cells are a distinct subgroup of T cells containing T cell receptors γ and δ (163, 164). These cells represent only a minor lymphocyte population making up ~0.5–16% of total CD3+ T cells in the peripheral blood, but they predominate in the skin and intestine (165). Due to their unique biology and well-established role in cancer immunosurveillance, γδ T cells are gaining considerable attention. γδ T cells have a somewhat dichotomous function in that they can be both anti-tumorigenic and pro-tumorigenic.
Several features of γδ T cells makes them potential suitable candidates for anti-tumor immunotherapy: (i) their ability to recognize tumor antigens independently of MHC restriction and co-stimulation (166); (ii) production of effector cytokines (TNF-α and IFN-γ) and conferring cytotoxicity against tumor cells both directly and indirectly by stimulating macrophages and DCs (167–169); and (iii) activated γδ T cells acquire the phenotype of antigen presenting cells (APCs) and induce CD4+ and CD8+ T cell proliferation and cytotoxicity (170). The pro-tumorigenic properties of γδ T cells are largely driven by IL-17A, whose expression in γδ T cells is increased in preclinical models for several cancers (171–176). IL-17A from γδ T cells binds to IL-17 receptors and promotes cancer progression via several downstream effects on malignant cells as well as other immune cells (177). γδ T cells stimulate endothelial cells to promote angiogenesis (171), promote the recruitment of pro-angiogenic macrophages to tumors (175), and are one of the main chemoattractants for the recruitment of MDSCs (178). These pro-tumorigenic properties of γδ T cells translate into the clinic since IL-17-producing γδ T cells are associated with poor survival in several cancers (179–181).
Currently γδ T cell-derived sEVs are understudied. Since sEVs derived from other immune cells exhibit the characteristics and carry the cargo of their parental cells (109, 182, 183), it is reasonable to assume that sEVs derived from γδ T cells will inherit the characteristics and functions of the parental cells. Indeed, typical sEVs can be derived from expanded γδ T cells ex vivo and these sEVs express several of the cytotoxic markers (including NKG2D, FasL, TNF-α, and IFN-γ) of the parental cells and are able to inhibit tumor growth (184). Interestingly, overexpression of miR-138, a miRNA with tumor suppressor and immunoenhancing properties in γδ T cells resulted in a concomitant increase of miR-138 in sEVs derived from these γδ T cells. These miR-138 rich sEVs effectively stimulate anti-tumor immunity and exhibit a more potent cytotoxic effect on tumor cells (184). Since γδ T cell-derived sEVs inherit the cytotoxicity capacity of the parental cells, this raises the possibility that these sEVs can serve as an effective drug delivery system.
The pro-tumorigenic properties of γδ T cells are largely IL-17 driven. IL-17-producing γδ T cells are rarely found in healthy individuals, however these cells accumulate in inflammatory disease, such as cancer (179, 180, 185, 186). This may, in part, explain the lack of studies on the role of γδ T cell-derived sEVs on tumor growth and immunosuppression. To the best of our knowledge, there have been no reports on the immunomodulatory effects of IL-17-expressing sEVs from γδ T cells in cancer. Of note though, IL-17-containing sEVs were found at much higher levels in patients with moderate to severe psoriasis compared to those with mild psoriasis (187), suggesting that IL-17-containing sEVs may correlate with disease progression. Unfortunately, the cell type of origin for these IL-17-containing sEVs is unknown. The exact role of IL-17-expressing γδ T cell-derived sEVs in cancer has yet to be determined, but one can predict that IL-17 containing sEVs from γδ T cells would have a similar effect on the tumor microenvironment as the parental cells. Further investigation is required to determine the exact impact of these IL-17-expressing sEVs on the tumor microenvironment and immune modulation.
sEVs Derived From T Regulatory Cells (Tregs)
Tregs are an integral component of the adaptive immune system that contribute to maintaining immune tolerance and preventing autoimmune disease (188, 189). Tregs are a highly immunosuppressive subset of T cells that are characterized by the expression of the transcription factor FOXP3 (190–192). Tregs are recruited to the TME where their interaction with other immune cells creates an immunosuppressive environment (193), and are recognized as a major cause of reduced efficacy or failure of cancer immunotherapy (194). Tregs exert their immunosuppressive activity through several mechanisms: (1) Tregs consume excess amounts of IL-2, thereby limiting the availability of this cytokine to effector T cells (195); (2) Tregs suppress APC function through constitutive expression of CTLA4, thereby inhibiting the activation of effector T cells (196, 197); (3) Tregs express immunosuppressive cytokines such as TGF-β, IL-10, and IL-35 (198–201); (4) Tregs are instrumental in the conversion of ATP to the immunomodulatory metabolite adenosine which prevents T cell activation (202); and (5) they secrete granzyme and perforin to destroy effector cells (203).
Tregs also secrete sEVs which may arguably be critical for the immunosuppressive activity of the parental cells. Treg-derived sEVs inhibit CD8+ CTL responses and anti-tumor activity by suppressing T cell proliferation, modification of APCs and through CD73-mediated production of adenosine (204–206). Of particular importance in Treg-derived sEV immunomodulation is the transfer of miRNA from Tregs to target cells, which appears to be central for Treg function. sEV mediated transfer of Let-7d miRNA from Tregs to Th1 cells suppresses Th1 cell proliferation and IFN-γ production (207). Similarly, sEV-mediated transfer of miR-150-p and miR-142-3p from Tregs to DCs induced a more tolerogenic phenotype in the DCs characterized by an altered cytokine profile (208).
The immunosuppressive effects of Treg-derived sEVs in cancer are clearly detrimental, however, in other settings these effects may prove useful. For example, in the case of organ transplantation, Tregs can be engineered to express specific proteins and miRNAs, which accumulate in sEVs from these modified Tregs. These modified sEVs inhibit T cell alloreactivity and induce immune tolerance in transplantation and may be a useful therapeutic option to manipulate the immune system in patients undergoing organ transplantation (209).
The study of Treg-derived sEVs is relatively new. There is no doubt, however, that Treg-derived sEVs contribute significantly to Treg cell function and therefore identifying and targeting the immunosuppressive cargo of Treg-derived sEVs may serve as a potential therapeutic option for cancer. Conversely, since the cargo of sEVs can be altered by genetically modifying the parental cells, “designer” Treg-derived sEVs have the potential to act as therapeutic agents in patients with autoimmune disorders or who are receiving transplants.
Bioengineering of Immune Cell-derived sEVs
Immune cell-derived sEVs can either activate or suppress the immune responses and these contrasting outcomes are dictated by (1) composition of the cargo, (2) activation and maturation status of the immune cells, (3) identity of target cells, and (4) disease setting and host microenvironment. T cell- and activated NK cell-derived sEVs are reported to express death ligands FasL, Apo2L, and TRAIL. These death ligands, expressed in a membrane-bound form, can interact with death receptors and induce apoptosis or immune tolerance in target cells (e.g., autologous T cells and DCs) (210). Also, sEVs released from immature and mature DCs impart opposing effects on the antigen-specific immune responses. When compared with sEVs from mature DCs, sEVs from immature DCs express sub-optimal levels of MHC class II and co-stimulator molecules (CD80, CD40, CD86, and ICAM I) and thus, are unable to drive immunostimulatory responses, instead dampen in vivo immunity and promote immune tolerance (69, 211–213).
In the past decade, several research groups have focused on the study of modulation of the immunogenicity of sEVs. The cargo expressed by immune cell-derived sEVs can be manipulated to overcome the immune suppressive properties of sEVs. As mentioned earlier, sEVs from T cells activated with IL-2 or IL-12 can directly stimulate proliferation of bystander resting T cells; this activation does not require the presence of antigens or APCs (112, 113). The effects of modified dexosomal vaccine formulations including adjuvants that induce DC maturation and activation have been reported. Modified poly(I:C)- and OVA antigen-expressing dexosomes stimulate OVA-specific anti-tumor T effector responses and reduce tumor growth (79). DC-derived sEVs have also been modified to harbor the iNKT ligand α-galactosylceramide; this modification enables the dexosomes to activate the invariant natural killer T cells (iNKT) that results in stronger immune effector responses in vivo (78). Dexosomes from α-fetoprotein (AFP)-expressing DCs have been shown to exhibit efficient anti-tumor activity in transplantable, orthotopic and carcinogen-induced hepatocellular carcinoma (HCC) mouse models (71).
The tumor-destroying capabilities of NK cell-derived sEVs can be utilized to treat “cold tumors” that are non-T cell inflamed and contain immune suppressive cells. Blood-brain crossing capabilities of NK cell-derived sEVs can be exploited to treat brain tumors. As with the endogenous NK cells, it is possible that tumors can develop various inhibitory mechanisms to shield themselves from sEV-mediated killing. However, genetic engineering tools (e.g., blocking inhibitory receptor expression) and ex vivo NK cell activation can be used to reshape the cargo of the NK cell-derived sEVs and influence their immune stimulatory activity.
Several different approaches to load exogenous proteins into sEVs have been explored. Transfection of cells with exogenous transmembrane proteins such as G protein-coupled receptors (CCR7, CCR2, and CXCR4) can lead to secretion of sEVs expressing these exogenous proteins (214, 215). Such modified sEVs can be used as a valuable tool in drug development studies. B cell-derived sEVs that exogenously express C3 complement proteins can interact with G protein coupled receptors on T cells and induce immune activation (97).
A second approach to modify the protein content in sEVs involves the use of a chimeric protein consisting of the sEV surface expressed LAMP-2b protein fused to a RGV peptide obtained from a rabies virus glycoprotein. DCs are then transfected with the chimeric protein, and the sEVs obtained from these cells are incorporated with siRNA targeting mutant Huntington mRNA (216). These modified sEVs are able to cross the blood-brain barrier and are capable of actively blocking the expression of the Huntington protein in neurons in vivo in mice (216). However, LAMP-2b fusion proteins need to be further modified to protect against lysosomal degradation (217). One other method of loading proteins into sEVs involves the use of the C1C2 (lipid-binding) domains of the human lactadherin protein to generate protein chimeras that bind to phosphatidylserine in the sEV membranes in a non-covalent manner (218, 219). Such engineered sEVs are biologically and functionally active.
Recent studies have explored the use of artificial EVs that are pre-loaded with immunostimulatory cargo. These artificial EVs overcome at least two major short comings of primary immune cell-derived EVs: (1) Primary immune cells are difficult to isolate to 100% purity and cannot be homogenously activated (the isolated EVs are often contaminated with tolerogenic/immunosuppressive EVs); and (2) Production and expansion of large quantities of primary autologous clinical grade human cells is expensive and labor intensive. Therefore, artificial EVs, which are basically liposomes coated with immunostimulatory molecules/ligands, can be a practical and cost-effective substitute for primary immune cell-derived EVs (220).
Discussion
sEVs can be manipulated to promote immunogenicity against cancer, and such bioengineered sEVs can function as efficient cancer vaccines. Immunostimulatory sEVs derived from immune cells, particularly from DCs, hold promise as therapeutic agents against cancer, as they initiate effective anti-tumor immunity when compared to that of other cell-free therapeutic strategies. Advantages of sEVs include high bio-availability, bio-stability, and lower costs. However, significant questions pertaining to sEVs' potential use as an immune-prophylactic agent or therapeutic for cancer treatment remain unanswered: (1) What is the precise mechanism of action? (2) Which specific cancers are sensitive to this therapeutic strategy? (3) Which membrane vesicle characteristics define immunostimulatory or immunosuppressive properties of sEVs? (4) Can sEV-mediated immunostimulatory effects be reproduced with different batches of sEV preparations? and (5) Can the stimulatory potential of immune cell-derived sEVs counter the immunosuppressive milieu created by factors/molecules secreted by tumor cells? Finding answers to these questions will be key for the success of sEV-based anti-cancer therapies. Currently it is not known exactly what fraction of the total circulating sEVs are immune cell-derived, which would indicate the relative importance of these immune cell-derived sEVs to disease pathology. However, immune cell-derived sEVs play a critical role in tumor progression either negatively or positively. Determining the exact fraction of immune cell-derived sEVs in the total population of circulating EVs will require extensive and accurate characterization of the total circulating sEV population.
Thus, while sEVs have been shown to activate anti-tumor immune responses in pre-clinical studies, clinical data in cancer patients have yielded only modest benefits. Recent advances in deciphering the molecular composition and functional characteristics of immune cell-derived sEVs can lead to next-generation agents with potential for use in anti-cancer immunotherapy. sEVs derived from specific cell sources, such as the MDSCs, Tregs and TAMS may contain additional molecules which can be harnessed as targets for inducing greater levels of host immune responses against cancers.
Author Contributions
KY, CL, and HD wrote the paper. JE edited the manuscript and contributed to the scientific discussion in this manuscript. All authors approved the manuscript for publication.
Funding
This study was supported by NIH Grant R21CA198249 (KY), Free to Breathe Grant (KY), NIH Grant R01CA175003 (CL), and JE was supported by the Commonwealth of Kentucky Research Challenge Trust Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
2. Gehrmann U, Naslund TI, Hiltbrunner S, Larssen P, Gabrielsson S. Harnessing the exosome-induced immune response for cancer immunotherapy. Semin Cancer Biol. (2014) 28:58–67. doi: 10.1016/j.semcancer.2014.05.003
3. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. (2002) 2:569–79. doi: 10.1038/nri855
4. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
5. Buschow SI, Nolte-'t Hoen EN, van Niel G, Pols MS, ten Broeke T, Lauwen M, et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic. (2009) 10:1528–42. doi: 10.1111/j.1600-0854.2009.00963.x
6. Laulagnier K, Motta C, Hamdi S, Roy S, Fauvelle F, Pageaux JF, et al. Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem J. (2004) 380:161–71. doi: 10.1042/bj20031594
7. Viaud S, Thery C, Ploix S, Tursz T, Lapierre V, Lantz O, et al. Dendritic cell-derived exosomes for cancer immunotherapy: what's next? Cancer Res. (2010) 70:1281–5. doi: 10.1158/0008-5472.CAN-09-3276
8. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. (1998) 4:594–600. doi: 10.1038/nm0598-594
9. Escudier B, Dorval T, Chaput N, Andre F, Caby MP, Novault S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. (2005) 3:10. doi: 10.1186/1479-5876-3-10
10. Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A, et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. (2008) 68:1228–35. doi: 10.1158/0008-5472.CAN-07-3163
11. Stein JM, Luzio JP. Ectocytosis caused by sublytic autologous complement attack on human neutrophils. The sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem J. (1991) 274(Pt 2):381–6. doi: 10.1042/bj2740381
12. Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. (2015) 25:364–72. doi: 10.1016/j.tcb.2015.01.004
13. Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. Exosomes and HIV gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol. (2006) 172:923–35. doi: 10.1083/jcb.200508014
14. Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
15. Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles. (2014) 3:26913. doi: 10.3402/jev.v3.26913
16. Mathieu M, Martin-Jaular L, Lavieu G, Thery C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. (2019) 21:9–17. doi: 10.1038/s41556-018-0250-9
17. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
18. Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. (1983) 97:329–39. doi: 10.1083/jcb.97.2.329
19. Tamai K, Tanaka N, Nakano T, Kakazu E, Kondo Y, Inoue J, et al. Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT-0 protein. Biochem Biophys Res Commun. (2010) 399:384–90. doi: 10.1016/j.bbrc.2010.07.083
20. Gerber PP, Cabrini M, Jancic C, Paoletti L, Banchio C, von Bilderling C, et al. Rab27a controls HIV-1 assembly by regulating plasma membrane levels of phosphatidylinositol 4,5-bisphosphate. J Cell Biol. (2015) 209:435–52. doi: 10.1083/jcb.201409082
21. Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. (2012) 14:677–85. doi: 10.1038/ncb2502
22. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. (2010) 12:19–30. doi: 10.1038/ncb2000
23. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. (2008) 319:1244–7. doi: 10.1126/science.1153124
24. Stuffers S, Brech A, Stenmark H. ESCRT proteins in physiology and disease. Exp Cell Res. (2009) 315:1619–26. doi: 10.1016/j.yexcr.2008.10.013
25. Kajimoto T, Okada T, Miya S, Zhang L, Nakamura S. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat Commun. (2013) 4:2712. doi: 10.1038/ncomms3712
26. van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, et al. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell. (2011) 21:708–21. doi: 10.1016/j.devcel.2011.08.019
27. Charrin S, Jouannet S, Boucheix C, Rubinstein E. Tetraspanins at a glance. J Cell Sci. (2014) 127:3641–8. doi: 10.1242/jcs.154906
28. Zimmerman B, Kelly B, McMillan BJ, Seegar TCM, Dror RO, Kruse AC, et al. Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell. (2016) 167:1041–51.e11. doi: 10.1016/j.cell.2016.09.056
29. Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J, et al. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. (2001) 166:7309–18. doi: 10.4049/jimmunol.166.12.7309
30. Geminard C, De Gassart A, Blanc L, Vidal M. Degradation of AP2 during reticulocyte maturation enhances binding of hsc70 and Alix to a common site on TFR for sorting into exosomes. Traffic. (2004) 5:181–93. doi: 10.1111/j.1600-0854.2004.0167.x
31. Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. (2017) 27:172–88. doi: 10.1016/j.tcb.2016.11.003
32. Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci. (2017) 42:42–56. doi: 10.1016/j.tibs.2016.08.016
33. Stoorvogel W, Kleijmeer MJ, Geuze HJ, Raposo G. The biogenesis and functions of exosomes. Traffic. (2002) 3:321–30. doi: 10.1034/j.1600-0854.2002.30502.x
34. Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, et al. Reassessment of exosome composition. Cell. (2019) 177:428–45.e18. doi: 10.1016/j.cell.2019.02.029
35. Chaput N, Taieb J, Andre F, Zitvogel L. The potential of exosomes in immunotherapy. Expert Opin Biol Ther. (2005) 5:737–47. doi: 10.1517/14712598.5.6.737
36. Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. (1999) 147:599–610. doi: 10.1083/jcb.147.3.599
37. Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. (1998) 273:20121–7. doi: 10.1074/jbc.273.32.20121
38. Perez-Hernandez D, Gutierrez-Vazquez C, Jorge I, Lopez-Martin S, Ursa A, Sanchez-Madrid F, et al. The intracellular interactome of tetraspanin-enriched microdomains reveals their function as sorting machineries toward exosomes. J Biol Chem. (2013) 288:11649–61. doi: 10.1074/jbc.M112.445304
39. Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M, et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J Lipid Res. (2010) 51:2105–20. doi: 10.1194/jlr.M003657
40. Llorente A, Skotland T, Sylvanne T, Kauhanen D, Rog T, Orlowski A, et al. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim Biophys Acta. (2013) 1831:1302–9. doi: 10.1016/j.bbalip.2013.04.011
41. Record M, Carayon K, Poirot M, Silvente-Poirot S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim Biophys Acta. (2014) 1841:108–20. doi: 10.1016/j.bbalip.2013.10.004
42. Esser J, Gehrmann U, D'Alexandri FL, Hidalgo-Estevez AM, Wheelock CE, Scheynius A, et al. Exosomes from human macrophages and dendritic cells contain enzymes for leukotriene biosynthesis and promote granulocyte migration. J Allergy Clin Immunol. (2010) 126:1032–40, 1040.e1–4. doi: 10.1016/j.jaci.2010.06.039
43. Laulagnier K, Grand D, Dujardin A, Hamdi S, Vincent-Schneider H, Lankar D, et al. PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. FEBS Lett. (2004) 572:11–4. doi: 10.1016/j.febslet.2004.06.082
44. Blackwood RA, Smolen JE, Transue A, Hessler RJ, Harsh DM, Brower RC, et al. Phospholipase D activity facilitates Ca2+-induced aggregation and fusion of complex liposomes. Am J Physiol. (1997) 272:C1279–85. doi: 10.1152/ajpcell.1997.272.4.C1279
45. Pinto R, De Summa S, Petriella D, Tudoran O, Danza K, Tommasi S. The value of new high-throughput technologies for diagnosis and prognosis in solid tumors. Cancer Biomark. (2014) 14:103–17. doi: 10.3233/CBM-130328
46. Silva A, Bullock M, Calin G. The clinical relevance of long non-coding RNAs in cancer. Cancers. (2015) 7:2169–82. doi: 10.3390/cancers7040884
47. Lang HL, Hu GW, Chen Y, Liu Y, Tu W, Lu YM, et al. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur Rev Med Pharmacol Sci. (2017) 21:959–72. doi: 10.3892/or.2017.5742
48. Pan L, Liang W, Fu M, Huang ZH, Li X, Zhang W, et al. Exosomes-mediated transfer of long noncoding RNA ZFAS1 promotes gastric cancer progression. J Cancer Res Clin Oncol. (2017) 143:991–1004. doi: 10.1007/s00432-017-2361-2
49. Zhang R, Xia Y, Wang Z, Zheng J, Chen Y, Li X, et al. Serum long non coding RNA MALAT-1 protected by exosomes is up-regulated and promotes cell proliferation and migration in non-small cell lung cancer. Biochem Biophys Res Commun. (2017) 490:406–14. doi: 10.1016/j.bbrc.2017.06.055
50. Carpenter S. Determining the function of long noncoding RNA in innate immunity. Methods Mol Biol. (2016) 1390:183–95. doi: 10.1007/978-1-4939-3335-8_12
51. Carpenter S. Long noncoding RNA: novel links between gene expression and innate immunity. Virus Res. (2016) 212:137–45. doi: 10.1016/j.virusres.2015.08.019
52. Satpathy AT, Chang HY. Long noncoding RNA in hematopoiesis and immunity. Immunity. (2015) 42:792–804. doi: 10.1016/j.immuni.2015.05.004
53. Lobb RJ, Becker M, Wen SW, Wong CS, Wiegmans AP, Leimgruber A, et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles. (2015) 4:27031. doi: 10.3402/jev.v4.27031
54. Lamparski HG, Metha-Damani A, Yao JY, Patel S, Hsu DH, Ruegg C, et al. Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods. (2002) 270:211–26. doi: 10.1016/S0022-1759(02)00330-7
55. Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. (2004) 104:3257–66. doi: 10.1182/blood-2004-03-0824
56. Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. (2009) 9:581–93. doi: 10.1038/nri2567
57. Palmisano G, Jensen SS, Le Bihan MC, Laine J, McGuire JN, Pociot F, et al. Characterization of membrane-shed microvesicles from cytokine-stimulated beta-cells using proteomics strategies. Mol Cell Proteomics. (2012) 11:230–43. doi: 10.1074/mcp.M111.012732
58. Kim HS, Choi DY, Yun SJ, Choi SM, Kang JW, Jung JW, et al. Proteomic analysis of microvesicles derived from human mesenchymal stem cells. J Proteome Res. (2012) 11:839–49. doi: 10.1021/pr200682z
59. Nazarenko I, Rana S, Baumann A, McAlear J, Hellwig A, Trendelenburg M, et al. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. (2010) 70:1668–78. doi: 10.1158/0008-5472.CAN-09-2470
60. Zech D, Rana S, Buchler MW, Zoller M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun Signal. (2012) 10:37. doi: 10.1186/1478-811X-10-37
61. Rana S, Claas C, Kretz CC, Nazarenko I, Zoeller M. Activation-induced internalization differs for the tetraspanins CD9 and Tspan8: Impact on tumor cell motility. Int J Biochem Cell Biol. (2011) 43:106–19. doi: 10.1016/j.biocel.2010.10.002
62. Rana S, Yue S, Stadel D, Zoller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. (2012) 44:1574–84. doi: 10.1016/j.biocel.2012.06.018
63. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. (2015) 523:177–82. doi: 10.1038/nature14581
64. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. (1996) 183:1161–72. doi: 10.1084/jem.183.3.1161
65. Skokos D, Botros HG, Demeure C, Morin J, Peronet R, Birkenmeier G, et al. Mast cell-derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo. J Immunol. (2003) 170:3037–45. doi: 10.4049/jimmunol.170.6.3037
66. Saunderson SC, McLellan AD. Role of lymphocyte subsets in the immune response to primary B cell-derived exosomes. J Immunol. (2017) 199:2225–35. doi: 10.4049/jimmunol.1601537
67. Naslund TI, Gehrmann U, Qazi KR, Karlsson MC, Gabrielsson S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol. (2013) 190:2712–9. doi: 10.4049/jimmunol.1203082
68. Andre F, Chaput N, Schartz NE, Flament C, Aubert N, Bernard J, et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol. (2004) 172:2126–36. doi: 10.4049/jimmunol.172.4.2126
69. Segura E, Amigorena S, Thery C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cells Mol Dis. (2005) 35:89–93. doi: 10.1016/j.bcmd.2005.05.003
70. Pitt JM, Andre F, Amigorena S, Soria JC, Eggermont A, Kroemer G, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest. (2016) 126:1224–32. doi: 10.1172/JCI81137
71. Lu Z, Zuo B, Jing R, Gao X, Rao Q, Liu Z, et al. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol. (2017) 67:739–48. doi: 10.1016/j.jhep.2017.05.019
72. Chen S, Lv M, Fang S, Ye W, Gao Y, Xu Y. Poly(I:C) enhanced anti-cervical cancer immunities induced by dendritic cells-derived exosomes. Int J Biol Macromol. (2018) 113:1182–7. doi: 10.1016/j.ijbiomac.2018.02.034
73. Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol. (2002) 3:1156–62. doi: 10.1038/ni854
74. Segura E, Nicco C, Lombard B, Veron P, Raposo G, Batteux F, et al. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood. (2005) 106:216–23. doi: 10.1182/blood-2005-01-0220
75. Naslund TI, Gehrmann U, Gabrielsson S. Cancer immunotherapy with exosomes requires B-cell activation. Oncoimmunology. (2013) 2:e24533. doi: 10.4161/onci.24533
76. Chaput N, Schartz NE, Andre F, Taieb J, Novault S, Bonnaventure P, et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. (2004) 172:2137–46. doi: 10.4049/jimmunol.172.4.2137
77. Bu N, Wu H, Zhang G, Zhan S, Zhang R, Sun H, et al. Exosomes from dendritic cells loaded with chaperone-rich cell lysates elicit a potent T cell immune response against intracranial glioma in mice. J Mol Neurosci. (2015) 56:631–43. doi: 10.1007/s12031-015-0506-9
78. Gehrmann U, Hiltbrunner S, Georgoudaki AM, Karlsson MC, Naslund TI, Gabrielsson S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with alpha-galactosylceramide on exosomes. Cancer Res. (2013) 73:3865–76. doi: 10.1158/0008-5472.CAN-12-3918
79. Damo M, Wilson DS, Simeoni E, Hubbell JA. TLR-3 stimulation improves anti-tumor immunity elicited by dendritic cell exosome-based vaccines in a murine model of melanoma. Sci Rep. (2015) 5:17622. doi: 10.1038/srep17622
80. Chen TS, Arslan F, Yin Y, Tan SS, Lai RC, Choo AB, et al. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J Transl Med. (2011) 9:47. doi: 10.1186/1479-5876-9-47
81. Zhong H, Yang Y, Ma S, Xiu F, Cai Z, Zhao H, et al. Induction of a tumour-specific CTL response by exosomes isolated from heat-treated malignant ascites of gastric cancer patients. Int J Hyperthermia. (2011) 27:604–11. doi: 10.3109/02656736.2011.564598
82. Tao SC, Guo SC, Zhang CQ. Modularized extracellular vesicles: the dawn of prospective personalized and precision medicine. Adv Sci. (2018) 5:1700449. doi: 10.1002/advs.201700449
83. Qazi KR, Gehrmann U, Domange Jordo E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood. (2009) 113:2673–83. doi: 10.1182/blood-2008-04-153536
84. Colino J, Snapper CM. Exosomes from bone marrow dendritic cells pulsed with diphtheria toxoid preferentially induce type 1 antigen-specific IgG responses in naive recipients in the absence of free antigen. J Immunol. (2006) 177:3757–62. doi: 10.4049/jimmunol.177.6.3757
85. Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS ONE. (2009) 4:e4942. doi: 10.1371/journal.pone.0004942
86. Munich S, Sobo-Vujanovic A, Buchser WJ, Beer-Stolz D, Vujanovic NL. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology. (2012) 1:1074–83. doi: 10.4161/onci.20897
87. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. (2005) 3:9. doi: 10.1186/1479-5876-3-9
88. van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. (2012) 64:676–705. doi: 10.1124/pr.112.005983
89. McLellan AD. Exosome release by primary B cells. Crit Rev Immunol. (2009) 29:203–17. doi: 10.1615/CritRevImmunol.v29.i3.20
90. Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J. (2007) 26:4263–72. doi: 10.1038/sj.emboj.7601842
91. Arita S, Baba E, Shibata Y, Niiro H, Shimoda S, Isobe T, et al. B cell activation regulates exosomal HLA production. Eur J Immunol. (2008) 38:1423–34. doi: 10.1002/eji.200737694
92. Clayton A, Turkes A, Dewitt S, Steadman R, Mason MD, Hallett MB. Adhesion and signaling by B cell-derived exosomes: the role of integrins. FASEB J. (2004) 18:977–9. doi: 10.1096/fj.03-1094fje
93. Hood JL. The association of exosomes with lymph nodes. Semin Cell Dev Biol. (2017) 67:29–38. doi: 10.1016/j.semcdb.2016.12.002
94. Saunderson SC, Dunn AC, Crocker PR, McLellan AD. CD169 mediates the capture of exosomes in spleen and lymph node. Blood. (2014) 123:208–16. doi: 10.1182/blood-2013-03-489732
95. Saunderson SC, Schuberth PC, Dunn AC, Miller L, Hock BD, MacKay PA, et al. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J Immunol. (2008) 180:8146–52. doi: 10.4049/jimmunol.180.12.8146
96. Zumaquero E, Munoz P, Cobo M, Lucena G, Pavon EJ, Martin A, et al. Exosomes from human lymphoblastoid B cells express enzymatically active CD38 that is associated with signaling complexes containing CD81, Hsc-70 and Lyn. Exp Cell Res. (2010) 316:2692–706. doi: 10.1016/j.yexcr.2010.05.032
97. Papp K, Vegh P, Prechl J, Kerekes K, Kovacs J, Csikos G, et al. B lymphocytes and macrophages release cell membrane deposited C3-fragments on exosomes with T cell response-enhancing capacity. Mol Immunol. (2008) 45:2343–51. doi: 10.1016/j.molimm.2007.11.021
98. Klinker MW, Lizzio V, Reed TJ, Fox DA, Lundy SK. Human B cell-derived lymphoblastoid cell lines constitutively produce fas ligand and secrete MHCII+FasL+ killer exosomes. Front Immunol. (2014) 5:144. doi: 10.3389/fimmu.2014.00144
99. Shimasaki N, Campana D. Engineering of natural killer cells for clinical application. Methods Mol Biol. (2020) 2097:91–105. doi: 10.1007/978-1-0716-0203-4_6
100. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
101. Woan KV, Miller JS. Harnessing natural killer cell antitumor immunity: from the bench to bedside. Cancer Immunol Res. (2019) 7:1742–7. doi: 10.1158/2326-6066.CIR-19-0404
102. Wu J, Lanier LL. Natural killer cells and cancer. Adv Cancer Res. (2003) 90:127–56. doi: 10.1016/S0065-230X(03)90004-2
103. Gyorgy B, Hung ME, Breakefield XO, Leonard JN. Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu Rev Pharmacol Toxicol. (2015) 55:439–64. doi: 10.1146/annurev-pharmtox-010814-124630
104. Armstrong JP, Holme MN, Stevens MM. Re-engineering extracellular vesicles as smart nanoscale therapeutics. ACS Nano. (2017) 11:69–83. doi: 10.1021/acsnano.6b07607
105. Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. (2012) 189:2833–42. doi: 10.4049/jimmunol.1101988
106. Jong AY, Wu CH, Li J, Sun J, Fabbri M, Wayne AS, et al. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J Extracell Vesicles. (2017) 6:1294368. doi: 10.1080/20013078.2017.1294368
107. Zhu L, Kalimuthu S, Gangadaran P, Oh JM, Lee HW, Baek SH, et al. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics. (2017) 7:2732–45. doi: 10.7150/thno.18752
108. Zhu L, Oh JM, Gangadaran P, Kalimuthu S, Baek SH, Jeong SY, et al. Targeting and therapy of glioblastoma in a mouse model using exosomes derived from natural killer cells. Front Immunol. (2018) 9:824. doi: 10.3389/fimmu.2018.00824
109. Fais S. NK cell-released exosomes: natural nanobullets against tumors. Oncoimmunology. (2013) 2:e22337. doi: 10.4161/onci.22337
110. Wen C, Seeger RC, Fabbri M, Wang L, Wayne AS, Jong AY. Biological roles and potential applications of immune cell-derived extracellular vesicles. J Extracell Vesicles. (2017) 6:1400370. doi: 10.1080/20013078.2017.1400370
111. Hinrichs CS, Gattinoni L, Restifo NP. Programming CD8+ T cells for effective immunotherapy. Curr Opin Immunol. (2006) 18:363–70. doi: 10.1016/j.coi.2006.03.009
112. Blanchard N, Lankar D, Faure F, Regnault A, Dumont C, Raposo G, et al. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J Immunol. (2002) 168:3235–41. doi: 10.4049/jimmunol.168.7.3235
113. Wahlgren J, Karlson Tde L, Glader P, Telemo E, Valadi H. Activated human T cells secrete exosomes that participate in IL-2 mediated immune response signaling. PLoS ONE. (2012) 7:e49723. doi: 10.1371/journal.pone.0049723
114. Seo N, Shirakura Y, Tahara Y, Momose F, Harada N, Ikeda H, et al. Activated CD8+ T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat Commun. (2018) 9:435. doi: 10.1038/s41467-018-02865-1
115. Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. (2011) 2:282. doi: 10.1038/ncomms1285
116. Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. (2013) 4:2980. doi: 10.1038/ncomms3980
117. Kaur S, Singh SP, Elkahloun AG, Wu W, Abu-Asab MS, Roberts DD. CD47-dependent immunomodulatory and angiogenic activities of extracellular vesicles produced by T cells. Matrix Biol. (2014) 37:49–59. doi: 10.1016/j.matbio.2014.05.007
118. Choudhuri K, Llodra J, Roth EW, Tsai J, Gordo S, Wucherpfennig KW, et al. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature. (2014) 507:118–23. doi: 10.1038/nature12951
119. van der Vlist EJ, Arkesteijn GJ, van de Lest CH, Stoorvogel W, Nolte-'t Hoen EN, Wauben MH. CD4+ T cell activation promotes the differential release of distinct populations of nanosized vesicles. J Extracell Vesicles. (2012). 1. doi: 10.3402/jev.v1i0.18364. [Epub ahead of print].
120. Hong X, Schouest B, Xu H. Effects of exosome on the activation of CD4+ T cells in rhesus macaques: a potential application for HIV latency reactivation. Sci Rep. (2017) 7:15611. doi: 10.1038/s41598-017-15961-x
121. Hao S, Yuan J, Xiang J. Nonspecific CD4+ T cells with uptake of antigen-specific dendritic cell-released exosomes stimulate antigen-specific CD8+ CTL responses and long-term T cell memory. J Leukoc Biol. (2007) 82:829–38. doi: 10.1189/jlb.0407249
122. Torralba D, Baixauli F, Villarroya-Beltri C, Fernandez-Delgado I, Latorre-Pellicer A, Acin-Perez R, et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat Commun. (2018) 9:2658. doi: 10.1038/s41467-018-05077-9
123. Yang J, Bi L, He X, Wang Z, Qian Y, Xiao L, et al. Follicular helper T cell derived exosomes promote B cell proliferation and differentiation in antibody-mediated rejection after renal transplantation. Biomed Res Int. (2019) 2019:6387924. doi: 10.1155/2019/6387924
124. Metz M, Maurer M. Mast cells–key effector cells in immune responses. Trends Immunol. (2007) 28:234–41. doi: 10.1016/j.it.2007.03.003
125. Supajatura V, Ushio H, Nakao A, Akira S, Okumura K, Ra C, et al. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest. (2002) 109:1351–9. doi: 10.1172/JCI0214704
126. Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. (2005) 23:749–86. doi: 10.1146/annurev.immunol.21.120601.141025
127. Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. (2005) 6:135–42. doi: 10.1038/ni1158
128. Qiao H, Andrade MV, Lisboa FA, Morgan K, Beaven MA. FcepsilonR1 and toll-like receptors mediate synergistic signals to markedly augment production of inflammatory cytokines in murine mast cells. Blood. (2006) 107:610–8. doi: 10.1182/blood-2005-06-2271
129. Kashiwakura J, Yokoi H, Saito H, Okayama Y. T cell proliferation by direct cross-talk between OX40 ligand on human mast cells and OX40 on human T cells: comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J Immunol. (2004) 173:5247–57. doi: 10.4049/jimmunol.173.8.5247
130. Bachelet I, Levi-Schaffer F. Mast cells as effector cells: a co-stimulating question. Trends Immunol. (2007) 28:360–5. doi: 10.1016/j.it.2007.06.007
131. Derakhshani A, Vahidian F, Alihasanzadeh M, Mokhtarzadeh A, Lotfi Nezhad P, Baradaran B. Mast cells: a double-edged sword in cancer. Immunol Lett. (2019) 209:28–35. doi: 10.1016/j.imlet.2019.03.011
132. Al-Nedawi K, Szemraj J, Cierniewski CS. Mast cell-derived exosomes activate endothelial cells to secrete plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol. (2005) 25:1744–9. doi: 10.1161/01.ATV.0000172007.86541.76
133. Carroll-Portillo A, Surviladze Z, Cambi A, Lidke DS, Wilson BS. Mast cell synapses and exosomes: membrane contacts for information exchange. Front Immunol. (2012) 3:46. doi: 10.3389/fimmu.2012.00046
134. Raposo G, Tenza D, Mecheri S, Peronet R, Bonnerot C, Desaymard C. Accumulation of major histocompatibility complex class II molecules in mast cell secretory granules and their release upon degranulation. Mol Biol Cell. (1997) 8:2631–45. doi: 10.1091/mbc.8.12.2631
135. Vincent-Schneider H, Thery C, Mazzeo D, Tenza D, Raposo G, Bonnerot C. Secretory granules of mast cells accumulate mature and immature MHC class II molecules. J Cell Sci. (2001) 114:323–34.
136. Eldh M, Ekstrom K, Valadi H, Sjostrand M, Olsson B, Jernas M, et al. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS ONE. (2010) 5:e15353. doi: 10.1371/journal.pone.0015353
137. Lotvall J, Valadi H. Cell to cell signalling via exosomes through esRNA. Cell Adh Migr. (2007) 1:156–8. doi: 10.4161/cam.1.3.5114
138. Xiao H, He M, Xie G, Liu Y, Zhao Y, Ye X, et al. The release of tryptase from mast cells promote tumor cell metastasis via exosomes. BMC Cancer. (2019) 19:1015. doi: 10.1186/s12885-019-6203-2
139. Liang Y, Qiao L, Peng X, Cui Z, Yin Y, Liao H, et al. The chemokine receptor CCR1 is identified in mast cell-derived exosomes. Am J Transl Res. (2018) 10:352–67.
140. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
141. Chesney JA, Mitchell RA, Yaddanapudi K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol. (2017) 102:727–40. doi: 10.1189/jlb.5VMR1116-458RRR
142. Zoller M. Janus-faced myeloid-derived suppressor cell exosomes for the good and the bad in cancer and autoimmune disease. Front Immunol. (2018) 9:137. doi: 10.3389/fimmu.2018.00137
143. Burke M, Choksawangkarn W, Edwards N, Ostrand-Rosenberg S, Fenselau C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J Proteome Res. (2014) 13:836–43. doi: 10.1021/pr400879c
144. Adams KR, Chauhan S, Patel DB, Clements VK, Wang Y, Jay SM, et al. Ubiquitin conjugation probed by inflammation in myeloid-derived suppressor cell extracellular vesicles. J Proteome Res. (2018) 17:315–24. doi: 10.1021/acs.jproteome.7b00585
145. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. (2014) 159:499–513. doi: 10.1016/j.cell.2014.09.051
146. Challagundla KB, Wise PM, Neviani P, Chava H, Murtadha M, Xu T, et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst. (2015) 107:djv135. doi: 10.1093/jnci/djv135
147. Koch R, Aung T, Vogel D, Chapuy B, Wenzel D, Becker S, et al. Nuclear trapping through inhibition of exosomal export by indomethacin increases cytostatic efficacy of doxorubicin and pixantrone. Clin Cancer Res. (2016) 22:395–404. doi: 10.1158/1078-0432.CCR-15-0577
148. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. (2013) 19:57–64. doi: 10.1038/nm.2999
149. Ghiringhelli F, Apetoh L. The interplay between the immune system and chemotherapy: emerging methods for optimizing therapy. Expert Rev Clin Immunol. (2014) 10:19–30. doi: 10.1586/1744666X.2014.865520
150. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
151. Deng Z, Rong Y, Teng Y, Zhuang X, Samykutty A, Mu J, et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene. (2017) 36:639–51. doi: 10.1038/onc.2016.229
152. van Ravenswaay Claasen HH, Kluin PM, Fleuren GJ. Tumor infiltrating cells in human cancer. On the possible role of CD16+ macrophages in antitumor cytotoxicity. Lab Invest. (1992) 67:166–74.
153. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. (2006) 124:263–6. doi: 10.1016/j.cell.2006.01.007
154. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
155. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. (2017) 17:457–74. doi: 10.1038/nrc.2017.51
156. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
157. Zhu Y, Chen X, Pan Q, Wang Y, Su S, Jiang C, et al. A comprehensive proteomics analysis reveals a secretory path- and status-dependent signature of exosomes released from tumor-associated macrophages. J Proteome Res. (2015) 14:4319–31. doi: 10.1021/acs.jproteome.5b00770
158. Zheng P, Luo Q, Wang W, Li J, Wang T, Wang P, et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional apolipoprotein E. Cell Death Dis. (2018) 9:434. doi: 10.1038/s41419-018-0465-5
159. Lan J, Sun L, Xu F, Liu L, Hu F, Song D, et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. (2019) 79:146–58. doi: 10.1158/0008-5472.CAN-18-0014
160. Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, et al. Exosomes released from tumor-associated macrophages transfer miRNAs that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol Res. (2018) 6:1578–92. doi: 10.1158/2326-6066.CIR-17-0479
161. Zhu X, Shen H, Yin X, Yang M, Wei H, Chen Q, et al. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exp Clin Cancer Res. (2019) 38:81. doi: 10.1186/s13046-019-1095-1
162. Cianciaruso C, Beltraminelli T, Duval F, Nassiri S, Hamelin R, Mozes A, et al. Molecular profiling and functional analysis of macrophage-derived tumor extracellular vesicles. Cell Rep. (2019) 27:3062–80.e11. doi: 10.1016/j.celrep.2019.05.008
163. Born W, Miles C, White J, O'Brien R, Freed JH, Marrack P, et al. Peptide sequences of T-cell receptor delta and gamma chains are identical to predicted X and gamma proteins. Nature. (1987) 330:572–4. doi: 10.1038/330572a0
164. Hayday AC, Saito H, Gillies SD, Kranz DM, Tanigawa G, Eisen HN, et al. Structure, organization, and somatic rearrangement of T cell gamma genes. Cell. (1985) 40:259–69. doi: 10.1016/0092-8674(85)90140-0
165. Wu D, Wu P, Wu X, Ye J, Wang Z, Zhao S, et al. Ex vivo expanded human circulating Vdelta1 gammadeltaT cells exhibit favorable therapeutic potential for colon cancer. Oncoimmunology. (2015) 4:e992749. doi: 10.4161/2162402X.2014.992749
166. Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. (2013) 13:88–100. doi: 10.1038/nri3384
167. Ismaili J, Olislagers V, Poupot R, Fournie JJ, Goldman M. Human gamma delta T cells induce dendritic cell maturation. Clin Immunol. (2002) 103:296–302. doi: 10.1006/clim.2002.5218
168. Conti L, Casetti R, Cardone M, Varano B, Martino A, Belardelli F, et al. Reciprocal activating interaction between dendritic cells and pamidronate-stimulated gammadelta T cells: role of CD86 and inflammatory cytokines. J Immunol. (2005) 174:252–60. doi: 10.4049/jimmunol.174.1.252
169. Devilder MC, Maillet S, Bouyge-Moreau I, Donnadieu E, Bonneville M, Scotet E. Potentiation of antigen-stimulated V gamma 9V delta 2 T cell cytokine production by immature dendritic cells (DC) and reciprocal effect on DC maturation. J Immunol. (2006) 176:1386–93. doi: 10.4049/jimmunol.176.3.1386
170. Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human gammadelta T Cells. Science. (2005) 309:264–8. doi: 10.1126/science.1110267
171. Wakita D, Sumida K, Iwakura Y, Nishikawa H, Ohkuri T, Chamoto K, et al. Tumor-infiltrating IL-17-producing gammadelta T cells support the progression of tumor by promoting angiogenesis. Eur J Immunol. (2010) 40:1927–37. doi: 10.1002/eji.200940157
172. Carmi Y, Rinott G, Dotan S, Elkabets M, Rider P, Voronov E, et al. Microenvironment-derived IL-1 and IL-17 interact in the control of lung metastasis. J Immunol. (2011) 186:3462–71. doi: 10.4049/jimmunol.1002901
173. Benevides L, da Fonseca DM, Donate PB, Tiezzi DG, De Carvalho DD, de Andrade JM, et al. IL17 Promotes mammary tumor progression by changing the behavior of tumor cells and eliciting tumorigenic neutrophils recruitment. Cancer Res. (2015) 75:3788–99. doi: 10.1158/0008-5472.CAN-15-0054
174. Ma S, Cheng Q, Cai Y, Gong H, Wu Y, Yu X, et al. IL-17A produced by gammadelta T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. (2014) 74:1969–82. doi: 10.1158/0008-5472.CAN-13-2534
175. Rei M, Goncalves-Sousa N, Lanca T, Thompson RG, Mensurado S, Balkwill FR, et al. Murine CD27− Vgamma6+ gammadelta T cells producing IL-17A promote ovarian cancer growth via mobilization of protumor small peritoneal macrophages. Proc Natl Acad Sci USA. (2014) 111:E3562–70. doi: 10.1073/pnas.1403424111
176. Patin EC, Soulard D, Fleury S, Hassane M, Dombrowicz D, Faveeuw C, et al. Type I IFN receptor signaling controls IL7-dependent accumulation and activity of protumoral IL17A-producing gammadeltaT cells in breast cancer. Cancer Res. (2018) 78:195–204. doi: 10.1158/0008-5472.CAN-17-1416
177. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell. (2014) 25:621–37. doi: 10.1016/j.ccr.2014.03.014
178. Yan J, Huang J. Innate gammadeltaT17 cells convert cancer-elicited inflammation into immunosuppression through myeloid-derived suppressor cells. Oncoimmunology. (2014) 3:e953423. doi: 10.4161/21624011.2014.953423
179. Patil RS, Shah SU, Shrikhande SV, Goel M, Dikshit RP, Chiplunkar SV. IL17 producing gammadeltaT cells induce angiogenesis and are associated with poor survival in gallbladder cancer patients. Int J Cancer. (2016) 139:869–81. doi: 10.1002/ijc.30134
180. Rutkowski MR, Stephen TL, Svoronos N, Allegrezza MJ, Tesone AJ, Perales-Puchalt A, et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell. (2015) 27:27–40. doi: 10.1016/j.ccell.2014.11.009
181. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. (2014) 40:785–800. doi: 10.1016/j.immuni.2014.03.013
182. Ventimiglia LN, Alonso MA. Biogenesis and function of T cell-derived exosomes. Front Cell Dev Biol. (2016) 4:84. doi: 10.3389/fcell.2016.00084
183. Pitt JM, Kroemer G, Zitvogel L. Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J Clin Invest. (2016) 126:1139–43. doi: 10.1172/JCI87316
184. Li L, Lu S, Liang X, Cao B, Wang S, Jiang J, et al. γδTDEs: an efficient delivery system for miR-138 with Anti-tumoral and immunostimulatory roles on oral squamous cell carcinoma. Mol Ther Nucleic Acids. (2019) 14:101–13. doi: 10.1016/j.omtn.2018.11.009
185. Ness-Schwickerath KJ, Jin C, Morita CT. Cytokine requirements for the differentiation and expansion of IL-17A- and IL-22-producing human Vgamma2Vdelta2 T cells. J Immunol. (2010) 184:7268–80. doi: 10.4049/jimmunol.1000600
186. Caccamo N, La Mendola C, Orlando V, Meraviglia S, Todaro M, Stassi G, et al. Differentiation, phenotype, and function of interleukin-17-producing human Vgamma9Vdelta2 T cells. Blood. (2011) 118:129–38. doi: 10.1182/blood-2011-01-331298
187. Jacquin-Porretaz C, Cordonnier M, Nardin C, Boullerot L, Chanteloup G, Vautrot V, et al. Increased levels of interleukin-17A exosomes in psoriasis. Acta Derm Venereol. (2019) 99:1143–7. doi: 10.2340/00015555-3300
188. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
189. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
190. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
191. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
192. Togashi Y, Nishikawa H. Regulatory T cells: molecular and cellular basis for immunoregulation. Curr Top Microbiol Immunol. (2017) 410:3–27. doi: 10.1007/82_2017_58
193. Paluskievicz CM, Cao X, Abdi R, Zheng P, Liu Y, Bromberg JS. T regulatory cells and priming the suppressive tumor microenvironment. Front Immunol. (2019) 10:2453. doi: 10.3389/fimmu.2019.02453
194. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
195. Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. (1998) 188:287–96. doi: 10.1084/jem.188.2.287
196. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
197. Perez VL, Van Parijs L, Biuckians A, Zheng XX, Strom TB, Abbas AK. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. (1997) 6:411–7. doi: 10.1016/S1074-76130080284-8
198. Steinbrink K, Wolfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. (1997) 159:4772–80.
199. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
200. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 limits anti-tumor immunity. Immunity. (2016) 44:316–29. doi: 10.1016/j.immuni.2016.01.013
201. Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. (2006) 177:896–904. doi: 10.4049/jimmunol.177.2.896
202. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
203. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
204. Zhang H, Nestor CE, Zhao S, Lentini A, Bohle B, Benson M, et al. Profiling of human CD4+ T-cell subsets identifies the TH2-specific noncoding RNA GATA3-AS1. J Allergy Clin Immunol. (2013) 132:1005–8. doi: 10.1016/j.jaci.2013.05.033
205. Bryniarski K, Ptak W, Jayakumar A, Pullmann K, Caplan MJ, Chairoungdua A, et al. Antigen-specific, antibody-coated, exosome-like nanovesicles deliver suppressor T-cell microRNA-150 to effector T cells to inhibit contact sensitivity. J Allergy Clin Immunol. (2013) 132:170–81. doi: 10.1016/j.jaci.2013.04.048
206. Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, et al. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. (2013) 43:2430–40. doi: 10.1002/eji.201242909
207. Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, et al. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity. (2014) 41:89–103. doi: 10.1016/j.immuni.2014.05.019
208. Tung SL, Boardman DA, Sen M, Letizia M, Peng Q, Cianci N, et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci Rep. (2018) 8:6065. doi: 10.1038/s41598-018-24531-8
209. Aiello S, Rocchetta F, Longaretti L, Faravelli S, Todeschini M, Cassis L, et al. Extracellular vesicles derived from T regulatory cells suppress T cell proliferation and prolong allograft survival. Sci Rep. (2017) 7:11518. doi: 10.1038/s41598-017-08617-3
210. Monleon I, Martinez-Lorenzo MJ, Monteagudo L, Lasierra P, Taules M, Iturralde M, et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J Immunol. (2001) 167:6736–44. doi: 10.4049/jimmunol.167.12.6736
211. Yin W, Ouyang S, Li Y, Xiao B, Yang H. Immature dendritic cell-derived exosomes: a promise subcellular vaccine for autoimmunity. Inflammation. (2013) 36:232–40. doi: 10.1007/s10753-012-9539-1
212. Lindenbergh MFS, Stoorvogel W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu Rev Immunol. (2018) 36:435–59. doi: 10.1146/annurev-immunol-041015-055700
213. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. (2013) 39:38–48. doi: 10.1016/j.immuni.2013.07.004
214. Yang Y, Hong Y, Nam GH, Chung JH, Koh E, Kim IS. Virus-mimetic fusogenic exosomes for direct delivery of integral membrane proteins to target cell membranes. Adv Mater. (2017) 29:1605604. doi: 10.1002/adma.201605604
215. Won YW, Patel AN, Bull DA. Cell surface engineering to enhance mesenchymal stem cell migration toward an SDF-1 gradient. Biomaterials. (2014) 35:5627–35. doi: 10.1016/j.biomaterials.2014.03.070
216. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. (2011) 29:341–5. doi: 10.1038/nbt.1807
217. Hung ME, Leonard JN. Stabilization of exosome-targeting peptides via engineered glycosylation. J Biol Chem. (2015) 290:8166–72. doi: 10.1074/jbc.M114.621383
218. Delcayre A, Estelles A, Sperinde J, Roulon T, Paz P, Aguilar B, et al. Exosome display technology: applications to the development of new diagnostics and therapeutics. Blood Cells Mol Dis. (2005) 35:158–68. doi: 10.1016/j.bcmd.2005.07.003
219. Veron P, Segura E, Sugano G, Amigorena S, Thery C. Accumulation of MFG-E8/lactadherin on exosomes from immature dendritic cells. Blood Cells Mol Dis. (2005) 35:81–8. doi: 10.1016/j.bcmd.2005.05.001
Keywords: extracellular vesicles, exosomes, nanovesicles, immune cells, cancer, immunotherapy, immune suppression
Citation: Li C, Donninger H, Eaton J and Yaddanapudi K (2020) Regulatory Role of Immune Cell-Derived Extracellular Vesicles in Cancer: The Message Is in the Envelope. Front. Immunol. 11:1525. doi: 10.3389/fimmu.2020.01525
Received: 12 March 2020; Accepted: 09 June 2020;
Published: 16 July 2020.
Edited by:
Fabio Malavasi, University of Turin, ItalyReviewed by:
Theresa L. Whiteside, University of Pittsburgh School of Medicine, United StatesEmily R. Levy, National Institutes of Health (NIH), United States
Copyright © 2020 Li, Donninger, Eaton and Yaddanapudi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kavitha Yaddanapudi, a2F2aXRoYS55YWRkYW5hcHVkaUBsb3Vpc3ZpbGxlLmVkdQ==
†These authors have contributed equally to this work