 Adonis Sfera
Adonis Sfera Carolina Osorio
Carolina Osorio Nyla Jafri1†
Nyla Jafri1† Eddie Lee Diaz
Eddie Lee Diaz- 1Patton State Hospital, San Bernardino, CA, United States
- 2Department of Psychiatry, Loma Linda University, Loma Linda, CA, United States
- 3Department of Medicine, The University of Texas Rio Grande Valley, Edinburg, TX, United States
Severe acute respiratory syndrome coronavirus 2 has spread rapidly around the globe. However, despite its high pathogenicity and transmissibility, the severity of the associated disease, COVID-19, varies widely. While the prognosis is favorable in most patients, critical illness, manifested by respiratory distress, thromboembolism, shock, and multi-organ failure, has been reported in about 5% of cases. Several studies have associated poor COVID-19 outcomes with the exhaustion of natural killer cells and cytotoxic T cells, lymphopenia, and elevated serum levels of D-dimer. In this article, we propose a common pathophysiological denominator for these negative prognostic markers, endogenous, angiotensin II toxicity. We hypothesize that, like in avian influenza, the outlook of COVID-19 is negatively correlated with the intracellular accumulation of angiotensin II promoted by the viral blockade of its degrading enzyme receptors. In this model, upregulated angiotensin II causes premature vascular senescence, leading to dysfunctional coagulation, and immunity. We further hypothesize that angiotensin II blockers and immune checkpoint inhibitors may be salutary for COVID-19 patients with critical illness by reversing both the clotting and immune defects (Graphical Abstract).
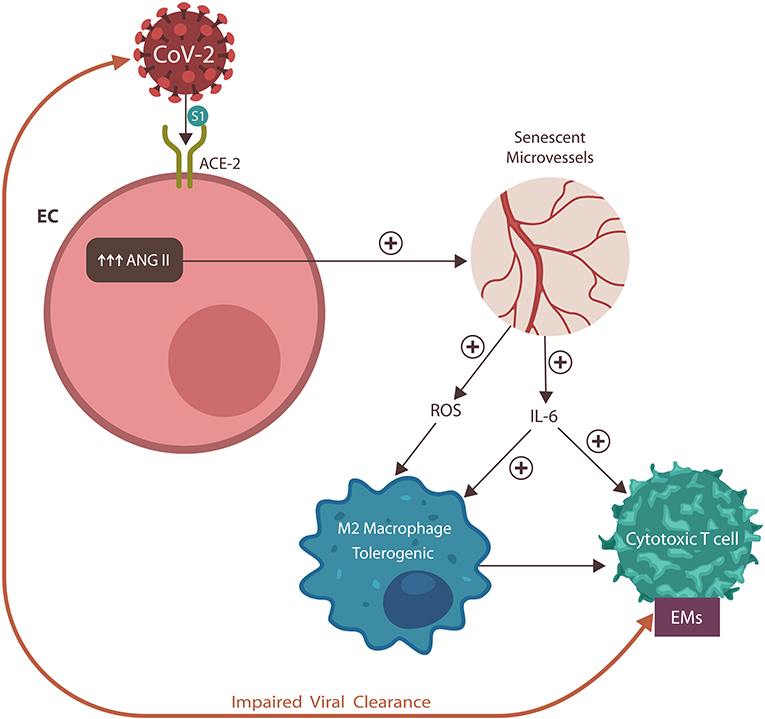
Graphical Abstract. The SARS-CoV-2 virus engages the angiotensin-converting enzyme-2 (ACE-2) protein, displacing its physiological ligand. As a result, angiotensin II (ANG II) accumulates in endothelial cells (ECs), inducing vascular senescence with upregulation of interleukin-6 (IL-6) and reactive oxygen species (ROS), impairing both innate and adaptive immunity. These changes engender dysfunctional coagulation (not shown) and the expression of exhausting markers (EM). In return, these immune defects disrupt viral clearance, engendering a vicious cycle and poor COVID-19 prognosis.
Introduction
High transmissibility, asymptomatic carriers, and the absence of herd immunity have contributed to the rapid worldwide spread of COVID-19 disease (1, 2). Although up to 50% of the affected individuals are free of clinical manifestations, about 5% of patients display serious complications, consisting of acute respiratory distress syndrome (ARDS), thromboembolism, sepsis, and multi-organ failure, often leading to death (3, 4).
COVID-19 disease is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is genetically related to SARS-CoV-1, known for engendering the 2002–2003 SARS epidemic. Several studies at the time have connected this virus to severe lymphopenia, involving cytotoxic T-cells (CTCs), and natural killer (NK) cells, which are indispensable for antiviral immunity (5, 6). In addition, faulty coagulation, associated with deep venous thrombosis (DVT) and pulmonary embolism (PE), has further complicated the management of this syndrome (7). These prior findings have been replicated in relation to SARS-CoV-2 and seem to precede the development of critical illness, suggesting that defective immunity may play a major role in this disease (8–10). Indeed, as in avian influenza, the upregulation of NK cell, and CTC exhaustion markers (EMs) has been observed (11). This is somewhat surprising, as these molecules are uncommon in acute viral infections and characterize cancer and viruses associated with chronic illness, such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), or cytomegalovirus (CMV) (12). In oncology, lowering EMs with immune checkpoint inhibitors (ICIs) is an established anti-tumor therapy aimed at reinvigorating host immunity, a modality with potential benefits in COVID-19 (13).
Under normal circumstances, EMs lower immune reactions to prevent autoimmunity. However, chronic inflammation can also elicit this response by prolonged stimulation of T cell receptors (TCRs) (14). Many viruses, likely including SARS-CoV-2, exploit EM pathways to avert detection. For example, SARS-CoV-2 gains access to host cells via angiotensin-converting enzyme-2 (ACE-2) associated with the renin-angiotensin system (RAS), which, aside from regulating arterial blood pressure, plays a major role in immunity (15). In this respect, SARS-CoV-2 appears to act like avian influenza viruses H5N1 and H7N9, elevating the serum levels of angiotensin II (ANG II), interleukin-6 (IL-6), and EMs (16–20).
As viral replication is more efficient in senescent cells, many viruses, including CMV and probably SARS-CoV-2, promote this phenotype in host cells to facilitate invasion (19, 21, 22). Senescent cells are characterized by proliferation arrest and a specific secretome, senescence-associated secretory phenotype (SASP). This is marked by upregulated IL-6 and reactive oxygen species (ROS), which were also detected in COVID-19 disease (23). Indeed, SARS-CoV-2 has been associated with upregulation of ANG II, a molecule previously shown to promote senescence in vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) (24–26).
We hypothesize that vascular senescence-mediated upregulation of IL-6 and ROS is responsible for both coagulation and immune dysfunction. Furthermore, this pathology, evidenced by the elevated plasma levels of EMs and D-dimer, heralds a poor COVID-19 prognosis (27). We further hypothesize that ICIs and angiotensin II blockers may help critically ill COVID-19 patients by reversing the virus-induced premature vascular senescence.
A Brief Pathophysiology of COVID-19 Disease
The SARS-CoV-2 virus gains access to host cells by engaging ACE-2 proteins, which are abundantly expressed in many tissues, including alveolar epithelial cells type II (AEC II), intestinal epithelial cells (IECs), and ECs (26, 28, 29). Interestingly, these cells function as “non-professional” antigen-presenting cells (APCs), so viral invasion directly affects their immune function. It has been established that viruses often evade detection by exploiting immunity-related host receptors. For example, the human poliovirus enters host cells via CD155, which is a receptor for T-cell immunoglobulin and ITIM domains (TIGIT) and an EM associated with functional downregulation of the CTCs and NK cells (30). Human immunodeficiency virus (HIV) upregulates EMs by maintaining a constant low-grade inflammation that repeatedly stimulates TCRs, “desensitizing” them (31). Other examples of virus-induced cellular senescence or EM upregulation are hepatitis C virus (HCV) and cytomegalovirus (CMV) (21, 32).
ACE-2 Downregulation and Critical Illness
In SARS-CoV-1 or SARS-CoV-2 infection, unfavorable prognosis has been associated with ACE-2 downregulation (33). This is a surprising and counterintuitive finding, as fewer viral entry portals should improve the clinical outcome. However, novel studies have shown that decreased levels of ACE-2 proteins cause higher illness severity and more end-organ damage (34) (Figure 1).
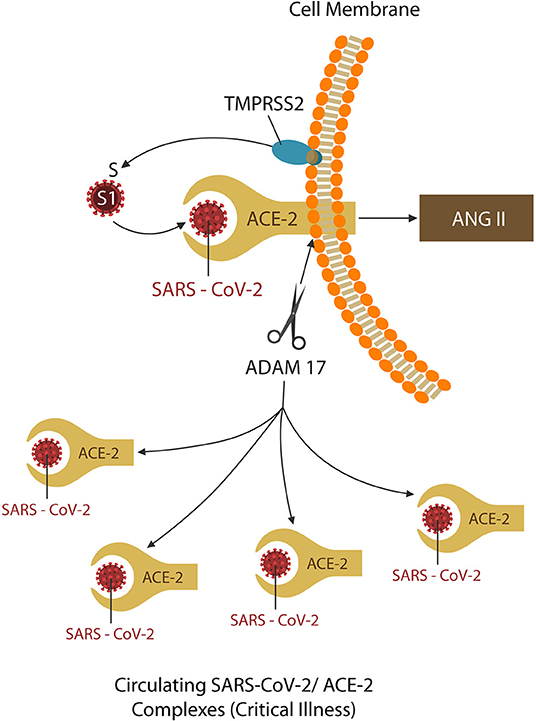
Figure 1. TMPRSS2 and ADAM17 are two virus-usurped host proteases. The former primes the spike (S) protein into S1, the active receptor binding site, promoting viral ingress. The latter, ADAM17, sheds the ACE-2 ectodomain, downregulating these proteins. The shed virus-ACE-2 complexes are soluble and readily spread by the circulation, causing end-organ damage, and critical illness. Some protease inhibitors may downregulate both TMPRSS2 and ADAM17, providing added therapeutic benefit for COVID-19 patients.
On closer scrutiny, ACE-2 downregulation takes place as these proteins are shed (along with the attached virus) from the cell membranes and are spread by circulation throughout the body. This occurs as SARS-CoV-2 spike (S) protein engages ACE-2 by usurping two host proteases: type II transmembrane serine protease (TMPRSS2), which facilitates viral ingress (by cleaving the S antigen into S1, the active binding site), and ADAM17, which downregulates ACE-2 proteins (by shedding them together with the attached virus) (33–37). For this reason, the latter, responsible for COVID-19 complications and end-organ damage, may be more harmful to the host (Figure 1). Indeed, since the origination of this pandemic, the research focus has been on blocking TMPRSS2 to prevent viral entry, rather than ADAM17 inhibition to avert critical illness (26).
SARS-CoV-2 and Cellular Senescence
Under normal circumstances, ACE-2 terminates the action of angiotensin (ANG I), and ANG II by cleaving these peptides into ANG 1-9 and ANG 1-7, respectively (Figure 2). In the absence of ACE-2 (due to viral blockade and downregulation), both ANG I and ANG II accumulate. However, as ACE-1 is not engaged by the virus, the conversion of ANG I to ANG II continues unabated, leading to the unopposed accumulation of ANG II. Excess ANG II has been associated with mitochondrial oxidative damage and ROS and IL-6 upregulation, impairing both coagulation and immunity (38) (Figure 2). SARS-CoV-2 may induce vascular aging and EC senescence by two mechanisms: ADAM-17 activation and NO depletion (27, 39) (Figure 2). Indeed, preclinical studies have shown that ANG II-infused rodents demonstrated mitochondrial loss and muscle atrophy, suggesting that ANG II acts as a mitochondrial toxin (40). Taken together, SARS-CoV-2 triggers premature cellular senescence and possibly organismal aging by damaging mitochondria (41, 42).
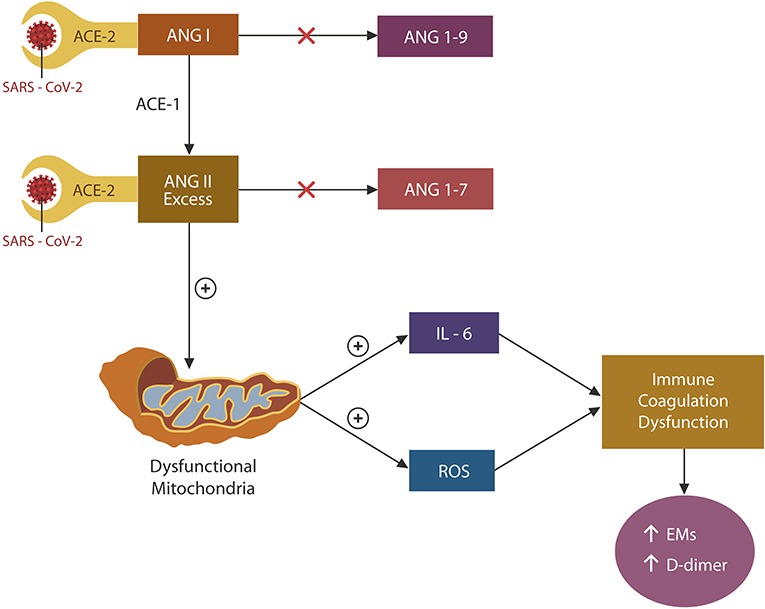
Figure 2. Engagement of ACE-2 by the SARS-CoV-2 virus blocks and downregulates these proteins, impairing the degradation of both ANG I and ANG II. However, since ACE-1 is not affected by the virus, ANG I conversion to ANG II continues unabated, contributing to its accumulation. ANG II excess damages mitochondria, upregulating both IL-6 and ROSs. These molecules induce EC senescence, dysfunctional immunity, and coagulation by upregulating both the exhaustion markers (EM) and D-dimer.
To ARB or Not To ARB?
A controversy involving two antihypertensive drug categories, angiotensin receptor blockers (ARBs), and angiotensin converting enzyme inhibitors (ACEi), arose as a recent paper opined that these agents might upregulate ACE-2, increasing the likelihood of viral infiltration (43). However, others have found these agents not to be harmful to COVID-19 patients and possibly to be beneficial, supporting the hypothesis presented here (44–46).
Taken together, the S1/ACE-2 attachment occupies and downregulates ACE-2 proteins, rendering them incapable of cleaving ANG II, contributing to its accumulation (Figure 2).
Endothelial Senescence: Angiotensin II and SARS-CoV-2 Critical Illness
Under normal circumstances, ECs are facultative APCs that synthesize tissue factors and thrombin inhibitors, maintaining both coagulation and immune homeostasis (27, 47). Although SARS-CoV-2 primarily targets AEC II in the lower respiratory tract, these cells are in close proximity to the underlying endothelium, which is likely to be infected (48). Indeed, body-wide EC damage has been reported in COVID-19, suggesting that the spread of this disease outside the respiratory system is a common occurrence (49). In addition, in COVID-19, like in HIV infection, the elevated serum D-dimer levels were found to herald a higher mortality rate, linking disease severity to impaired endothelia and coagulation (50, 51). Moreover, a recent COVID-19 study found a negative correlation between D-dimer and the number of CTCs and NK cells, connecting dysfunctional coagulation with lymphopenia (52–54).
SARS-CoV-2 and Mitochondrial Damage
Viral replication is more effective in senescent cells, and many viruses, including influenza, have been shown to promote this phenotype in their hosts (19, 22). Indeed, the H7N9 Influenza virus induces host vascular senescence by upregulating ANG II and its signaling via AT-1Rs, causing NO depletion (19, 35, 55–59) (Figure 3). As SARS-CoV-2 is believed to utilize the same mechanism, AT-1R blockers, including losartan, are currently in COVID-19 clinical trials (NCT04335123, NCT04312009, and NCT04311177) (Figure 3).
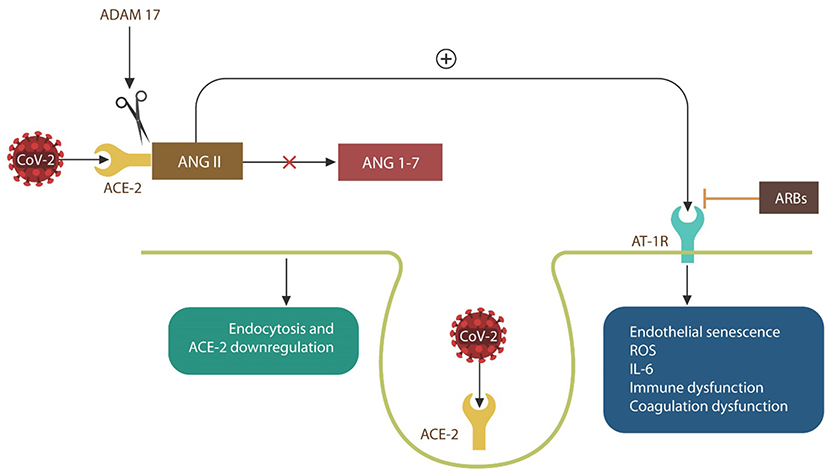
Figure 3. SARS-CoV-2 engagement of ACE-2 blocks ANG II breakdown into ANG 1-7, increasing intracellular ANG II. ANG II signaling via angiotensin 1 receptors (AT-1Rs) (inhibited by ARBs), induces EC senescence and upregulates IL-6 and ROS, causing immune, and coagulation dysfunction. When ACE-2 is bound by the virus, the SARS-CoV-2/ACE-2 complexes enter host cells by endocytosis. Complexes that are not endocytosed are shed by ADAM17, contributing to critical illness.
Several viruses, including polio, HIV, and SARS-CoV-1, induce senescence in host cells by inflicting mitochondrial damage (60–62). For example, the avian influenza H5N1 virus was demonstrated to impair mitochondrial antiviral signaling (MAVS) protein, inhibiting interferon release (63, 64). Since MAVS is indispensable for NK cell and CTC maturation and metabolism, disabling these proteins translates into impaired immunity (65, 66). Aside from altering MAVS, viruses can also lower host immunity by interfering with mitochondrial metabolism. Because NK and CTCs undergo metabolic rewiring to support clonal expansion and effector function upon antigen contact, viral interference with this process impairs immune responses (67). Moreover, ROSs released by the virus-damaged mitochondria not only impair NO synthesis but also activate ADAM 17, causing EC senescence by two distinct mechanisms (27, 39, 68, 69). For this reason, ADAM17 inhibitors, deemed effective against SARS-CoV-1, should be investigated against SARS-CoV-2 (35).
Angiotensin II, a Mitochondrial Toxin
In COVID-19 patients, elevated serum levels of ANG II were found to be directly correlated with viral load and the severity of lung injuries (70, 71). Moreover, ACE-2 downregulation has been directly linked to the critical pulmonary pathology, suggesting that unopposed ANG II acts as an endogenous toxin (72). On the other hand, a recombinant human ACE-2 (rhACE-2) was found beneficial in a small cohort of SARS-CoV-1 patients and is currently in COVID-19 clinical trials (clinical trial identifier NCT04335136) (73, 74).
Taking this evidence together, intracellular ANG II is an endogenous mitochondrial poison, causing premature endothelial senescence that damages end organs, impairing COVID-19 prognosis.
Hypothesis- Putting It All Together
In light of the above discussion, we hypothesize the following:
1. ANG II is a mitochondrial toxin that, under normal circumstances, is rapidly removed by ACE-2, which converts it into ANG 1-7.
In favor of this statement, we point to several studies showing that in the absence of hydrolyzing enzyme, ACE-2, ANG II accumulates intracellularly, inducing mitochondrial elimination or damage throughout the body endothelia (36, 75–77).
2. ACE-2 proteins are both occupied and downregulated by the SARS-CoV-2 virus and are therefore incapable of hydrolyzing ANG II.
In favor of this assertion, we cite studies reporting that the SARS-CoV-2 receptor-binding domain (RBD) exhibits significantly higher affinity for ACE-2 and a higher degree of ACE-2 downregulation compared to the related SARS-CoV-1 (78, 79).
3. The attachment of SARS-CoV-2 to ACE-2 is positively correlated with ANG II accumulation and negatively correlated with ACE-2 levels.
In favor of this statement are novel findings showing that ANG II serum levels are positively correlated with both the SARS-CoV-2 viral load and lung injuries (70, 71). In addition, the density of ACE-2 protein has been found to be negatively correlated with COVID-19 critical illness (72).
4. Excess ANG II promotes premature EC senescence along with dysfunctional coagulation and immunity.
Several COVID-19 studies have associated poor disease prognosis with ANG II-induced endothelial dysfunction, impaired coagulation, and the overexpression of EMs (8, 27, 78, 80).
5. SARS-CoV-2-mediated ANG II accumulation causes IL-6 and ROS upregulation, damaging the endothelia.
Novel studies have associated SARS-CoV-2 infection with elevation of IL-6, a cytokine that inhibits endothelial NO synthesis, causing senescence (81, 82). On the other hand, IL-6-blocking antibodies are currently in clinical trials for COVID-19 (clinical trial identifier NCT04322773). Moreover, ROSs upregulate ADAM17 and lower NO, triggering vascular aging (27, 39, 68, 69). Conversely, ROS scavengers, including camostat mesylate and anti-inflammatory/antioxidant supplements, are currently in COVID-19 clinical trials (clinical trial identifiers NCT04321096 and NCT04323228).
6. Immune checkpoint inhibitors and ANG II blockers may help critically ill COVID-19 patients by reversing premature vascular senescence, restoring immune homeostasis.
We base this assertion on novel studies showing beneficial effects of rhACE-2 and ARBs, including losartan, in SARS-CoV-2 patients. Losartan and rhACE-2 clinical trials are listed above (83, 84). Moreover, cancer patients with SARS-CoV-2 who were undergoing immunotherapy were found to have a better COVID-19 prognosis than those on chemotherapy, suggesting that ICIs may be helpful against SARS-CoV-2 (85). Furthermore, the clinical trial “Personalized Immunotherapy for SARS-CoV-2 (COVID-19) Associated with Organ Dysfunction (ESCAPE)” (clinical trial identifier NCT04339712) is currently assessing the potential benefit of these agents against COVID-19.
In the remaining sections of this article, we look through the prism of this pathophysiological hypothesis, attempting to identify new target molecules, or pathways that might emerge from this model (Table 1). We also point to the neuropsychiatric manifestations of COVID-19 that, as demonstrated by prior pandemics, are often delayed and involve both movement and neurodegenerative disorders.
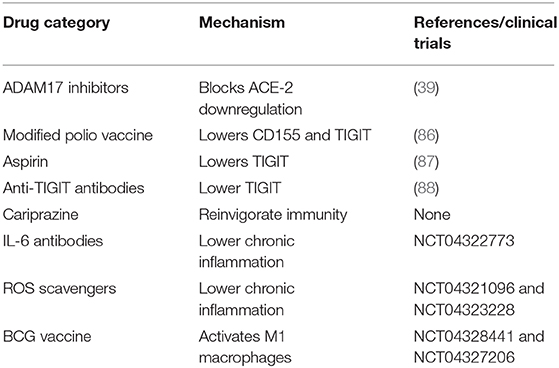
Table 1. Potential COVID-19 therapies based on the presented hypothesis.
TIGIT: In the Eye of the “Cytokine Storms”
COVID-19 patients may present with a wide variety of immune and inflammatory responses, ranging from hyperinflammation or “cytokine storms” to immune suppression or exhaustion (89, 90). This has raised a clinical dilemma: should immunity be augmented or lowered in COVID-19 patients? Indeed, it appears that some individuals require anti-inflammatory drugs, while others are in need of immune activators (91, 92). Along these lines, both NK cells and anti-inflammatory agents are currently in COVID-19 clinical trials, indicating that both categories may be called upon due to the fact that individual immune responses to this virus can be highly variable (NCT04375176, NCT04329650). On the one hand, the SARS-CoV-2 virus likely averts detection by inducing immune disruption, while on the other, the host may unleash excessive inflammation to limit viral infection. Since human CTCs and NK cells possess a functional RAS, the virus-induced immune impairments may be mediated by this system (93). Indeed, preclinical studies have found that ARBs, including losartan, can prevent COVID-19 pulmonary injuries, suggesting that ANG II/AT-1R signaling drives the immune defects associated with SARS-CoV-2 (70). Moreover, as the TIGIT pathway has been found to promote immune dysregulation in response to many viral infections, it is likely that SARS-CoV-2 may manipulate this EM to evade detection (10). Indeed, elevated levels of IL-10, a TIGIT-signaling cytokine, have been documented in COVID-19 patients, suggesting that SARS-CoV-2 exploits these proteins to cover its molecular signatures (Figure 4) (94).
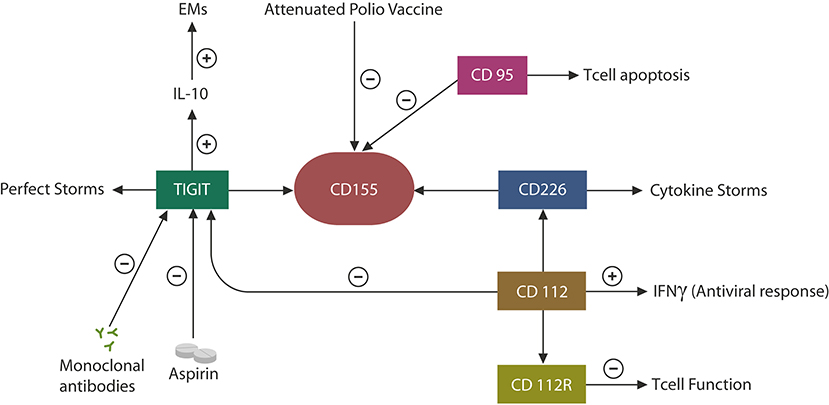
Figure 4. CD155 can be engaged by TIGIT, leading to immune exhaustion or by the competing molecule, CD 226, augmenting immunity. In individuals with a degree of immune senescence, TIGIT may be more likely to engage CD155, while in persons more prone to autoimmunity, CD226 may bind CD155, generating hyperinflammation, or “cytokine storms.” CD112 expression in CTCs and NK cells triggers antiviral responses by interferon release. CD112R upregulation lowers immune function as this protein, like TIGIT, functions as an EM. Viruses also exploit the CD95 pathway to induce CTC apoptosis (mimicking infection resolution). Aspirin and anti-TIGIT antibodies may decrease TIGIT, IL-10, and EMs, potentially benefiting COVID-19 patients. Attenuated polio vaccine may have similar effects by inhibiting the CD155–TIGIT axis.
Viruses often bind to cell membrane receptors associated with immune suppression or senescence to achieve both host cell entry and a progeny-permissive microenvironment. For example, the human poliovirus attaches to CD155, the TIGIT receptor, to upregulate this EM and inhibit host immunity (95) (Figure 4). As CD155 is associated with other immune-suppressing proteins, including CD95 and CD112 and its receptor (CD112R), it is likely that an immune inhibitory network exists around CD155 that can be exploited by viral agents to avert detection (Figure 4). For example, alpha herpesvirus targets CD112, which controls the expression of interferon gamma (IFNγ), while influenza virus induces CTC apoptosis via CD95 (96, 97) (Figure 4). Moreover, recent studies have reported that CD112R functions as a human EMs, suggesting that along with TIGIT, it may be responsible for many viral infections, including SARS-CoV-2 (98). On the other hand, TIGIT competes for CD155 binding with CD226, a receptor associated with the hyperinflammation of autoimmune disorders, suggesting the existence of a host-driven “cytokine storms” axis opposed to the virally induced immune suppressant, TIGIT (30, 99) (Figure 4). Indeed, it was recently reported that individuals expressing the CD226 G allele (which binds to CD155 with higher affinity) exhibited severe influenza symptoms, linking this gene to critical COVID-19 illness (100).
Taken together, the TIGIT–CD155–CD226 axis likely comprises a major immune switch usurped by many viruses, likely including SARS-CoV-2, to avert host detection (30, 99). As elevated serum levels of TIGIT and IL-10 have been documented in SARS-CoV-2 infection, the attenuated polio vaccine may be beneficial against COVID-19, as it inhibits CD155 and its immunosuppressive network (Figure 4).
Older Individuals and COVID-19 Critical Illness
The COVID-19 pandemic appears to affect the elderly more than children or younger adults, suggesting that immune senescence may play a role in its pathogenesis (101–103). Since ANG II/AT-1R signaling triggers immune exhaustion, older COVID-19 patients may present with more complex immune defects engendered by the simultaneous expression of exhaustion and senescence markers (104). Indeed, novel preclinical studies have demonstrated that TIGIT knockdown can reverse premature cellular and immune aging, suggesting that downregulation of this molecule may benefit COVID-19 patients (105).
Aside from older individuals, persons with higher levels of pro-inflammatory cytokines, including those with obesity and diabetes, may be at higher risk of TIGIT overexpression and COVID-19 complications. Indeed, SARS-CoV-2 critical illness is more prevalent in individuals with these conditions, as reported by the Louisiana Department of Health Update from 3/27/2020 (106) (http://ldh.la.gov/index.cfm/newsroom/detail/5517).
It is therefore possible that in individuals predisposed to autoimmunity, such as those expressing the CD226 G allele, SARS-CoV-2 may tilt the immune balance toward CD155–CD226 interaction, generating “cytokine storms.” On the other hand, in persons with preexisting immune defects, such as immune senescence, the CD155–TIGIT interaction may be enabled, engendering more profound immune deficits (by adding immune exhaustion to the previously aged CTCs and NK cells) (107, 108). Indeed, immune senescence appears to be the likely cause of the lower prevalence of autoimmune diseases and poorer response to vaccines in the elderly population (109, 110). For this reason, we surmise that the unfavorable COVID-19 prognosis is directly correlated with plasma TIGIT levels and that anti-TIGIT monoclonal antibodies could be salutary for COVID-19 patients (Figures 4, 5). Furthermore, as recombinant polio vaccines were reported to provide suitable vector systems for antigen attachment, connecting viral S protein to this vector may expedite the development of a SARS-CoV-2 vaccine (111).
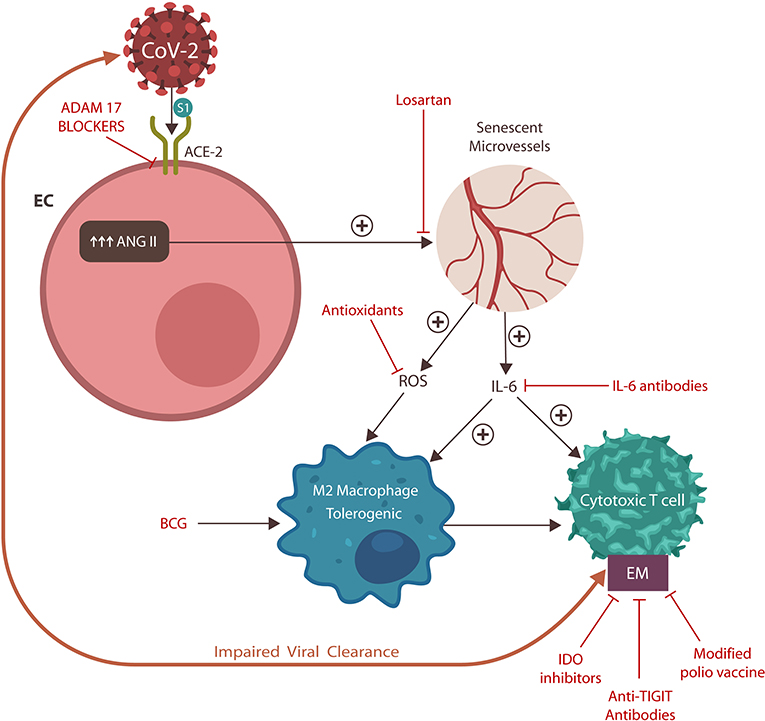
Figure 5. Potential anti-SARS-CoV-2 therapeutics and their action sites. ADAM17 blockers prevent ACE-2 downregulation and critical illness. Losartan blocks AT-1Rs, counteracting vascular senescence. Antioxidants and IL-6 antibodies work downstream, downregulating EM, and reinvigorating immunity. BCG activates innate immunity, improving antigen presentation to CTCs. EMs may also be lowered by cariprazine (not shown) and IDO inhibitors. Modified polio vaccine and immune checkpoint inhibitors, including anti-TIGIT antibodies, may be helpful by lowering EMs.
The Virus, THE RAS, and The DAS
Aside from expressing an RAS, immune cells, including dendritic cells (DC), CTCs, and NK cells, also possess a viable dopaminergic system (DAS) that plays a major role in the crosstalk between immunity and the brain (112). While the central nervous system (CNS) DAS is adequately elucidated, the role of dopamine (DA) in peripheral immunity has been less emphasized. Moreover, although a local RAS with a role in aging and cognition has previously been described in the brain, its interaction with DAS is an emerging topic in neurodegeneration, especially Parkinson's disease (PD) (113).
Nearly 40% of COVID-19 patients present with neuropsychiatric symptoms, suggesting that this virus, like many previous pandemic-related viruses, may be neurotropic (114). Indeed, delirium, seizures, impaired consciousness, and acute cerebrovascular disease have already been described in COVID-19 patients, suggesting that SARS-CoV-2 possesses the capability of altering brain functions (114, 115). Interestingly, previous studies have associated elevated D-dimer levels with strokes and delirium, indicating that, aside from the peripheral involvement, this molecule may be the herald of unfavorable neuropsychiatric outcome in COVID-19 patients (116, 118).
Aside from entering the CNS via ANG II/AT-1Rs-related senescent endothelia, SARS-CoV-2 may access the brain directly via the cribriform plate, possibly explaining the anosmia symptom described by many COVID-19 patients (119). In addition, as influenza A virus utilizes the same entry portal and lowers local immunity by inducing the nasal expression of indoleamine 2,3-dioxygenase (IDO), SARS-CoV-2 may employ a similar mechanism (120). Furthermore, IDO inhibitors, an emerging cancer therapy, may be beneficial for the neuropsychiatric manifestations of SARS-CoV-2.
Upon CNS arrival, the virus likely blocks astrocytic and neuronal ACE-2, elevating ANG II levels. In this regard, several studies have linked excessive brain ANG II to premature neuronal aging and Alzheimer's disease (AD) (120). Conversely, longevity was associated with the suppression of this molecule (121). Indeed, ARBs and ACEi were found to be protective against both PD and AD, indicating that RAS may play a key role in neurodegeneration (122–124). Moreover, in 2017, the US Food and Drug Administration approved a synthetic form of human angiotensin II, Giapreza, for the treatment of septic shock. The listed adverse effects of this compound include delirium, thrombotic events, and infection, resembling the central SARS-CoV-2 manifestations. Since ANG II accumulation may be essential for COVID-19 pathogenesis, Giapreza should probably be avoided in SARS-CoV-2-associated septic shock (113, 117, 125).
A growing body of evidence has demonstrated that DA mediates the crosstalk between immunity and the CNS, suggesting that RAS, and DAS signaling may be responsible for both peripheral and central COVID-19 manifestations. Indeed, since, at the body periphery, DA alters the activation of CTCs and NK cells, it is not surprising that DA blockers are capable of inhibiting the replication of several viruses (126–129). For example, the antiviral properties of chlorpromazine have been well-documented, as this compound protects against viral entry, preventing the exploitation of immune cells (130, 131). For these reasons, we believe that patients taking antipsychotic medications may be, at least partially, protected against COVID-19, as suggested by the emerging data on forensic inpatients (unpublished research). Moreover, as reinvigoration of CTCs can be achieved by blocking dopamine D3 receptors in DCs, selective D3 partial agonists, such as cariprazine, should be assessed for COVID-19 efficacy (132). Finally, the link between excessive DA and immune defects is further substantiated by the fact that methamphetamine (METH) users with chronically elevated DA levels often present with lymphopenia as well as CTC and NK cell dysfunction (133–135). For these reasons, METH users are probably at high risk of developing SARS-CoV-2 complications. Moreover, METH was found to augment brain ANG II/AT-1R signaling, promoting neuronal senescence, and neurocognitive deficits, further emphasizing the connection between RAS and DAS in both neurodegenerative and addictive disorders (136, 137). Conversely, ARBs are currently being tested for METH addiction, as preclinical studies have reported decreased self-administration of this stimulant in candesartan-treated rodents (138). This points to the fact that a better understanding of COVID-19 may have unintended consequences: improved treatment of addictions.
Taken together, the synergistic actions of ANG II and METH illustrate the intertwined role of RAS and DAS in both COVID-19 and substance use disorders, suggesting that candesartan may be the treatment of choice for COVID-19 in METH users.
Conclusions
SARS-CoV-2 infection has spread around the world in a short time interval, but its prognosis is variable. Since the onset of this pandemic, there has been an overemphasis on the virus itself and less attention on host immunity.
It has been said that Nature plays a cruel game of chess in which the host and pathogen can only thrive by outmaneuvering each other. Like influenza viruses, cancer, and chronic viral infections, SARS-CoV-2 evades detection by disguising itself as an ACE-2 ligand. The host responds by mobilizing its innate and adaptive immunity to eliminate the virus, but the latter proceeds to downregulate host immune defenses by augmenting EMs. In a desperate move, the host unleashes “cytokine storms” to reinvigorate its suppressed immune cells and overcome the virus. However, this extreme maneuver sacrifices the vulnerable individuals, such as those with chronic inflammation, damaged endothelia, and defective immunity. But Nature has rarely been fair to the weak, as their demise contributes to herd immunity. And the life-death cycles go on and on, moves and countermoves, hosts and pathogens. Indeed, it has been said that man can come up with better and better mousetraps, but Nature can always build better mice.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. (2020) 109:102433. doi: 10.1016/j.jaut.2020.102433
2. Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, Yang J, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. (2020) 395:514–23. doi: 10.1016/S0140-6736(20)30154-9
3. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019. pneumonia in Wuhan, China. JAMA Internal Med. (2020). doi: 10.1001/jamainternmed.2020.0994. [Epub ahead of print].
4. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019. (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA. (2020). 323:1239–42 doi: 10.1001/jama.2020.2648
5. Wong RS, Wu A, To KF, Nelson L, Lam CEK, Wong CK, et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ. (2003) 326:1358–62. doi: 10.1136/bmj.326.7403.1358
6. Oh HL, Chia A, Chang CX, Leong HN, Ling LL, Gijsbert M, et al. Engineering T cells specific for a dominant severe acute respiratory syndrome coronavirus CD8 T cell epitope. J Virol. (2011) 85:10464–71. doi: 10.1128/JVI.05039-11
7. Xiang-Hua Y, Le-Min W, Ai-Bin L, Zhu G, Riquan L, Xu-You Z, et al. Severe acute respiratory syndrome and venous thromboembolism in multiple organs. Am J Respir Crit Care Med. (2010) 182:436–7. doi: 10.1164/ajrccm.182.3.436
8. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019. (COVID-19). medRxiv. (2020). doi: 10.1101/2020.02.18.20024364
9. Channappanavar R, Zhao J, Perlman S. T cell-mediated immune response to respiratory coronaviruses. Immunol Res. (2014) 59:118–28. doi: 10.1007/s12026-014-8534-z
10. Schorer M, Rakebrandt N, Lambert K, Hunziker A, Pallmer K, Oxenius A, et al. TIGIT limits immune pathology during viral infections. Nat Commun. (2020) 11:1288. doi: 10.1038/s41467-020-15025-1
11. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
12. Saeidi A, Zandi K, Cheok YY, Saeidi H, Wong WF, Yie Qin Lee C, et al. T-cell exhaustion in chronic infections: reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front Immunol. (2018) 9:2569. doi: 10.3389/fimmu.2018.02569
13. Liu M, Guo F. Recent updates on cancer immunotherapy. Precis Clin Med. (2018) 1:65–74. doi: 10.1093/pcmedi/pby011
14. Jackson SR, Berrien-Elliott MM, Meyer JM, Wherry EJ, Teague RM. CD8+ T cell exhaustion during persistent viral infection is regulated independently of the virus-specific T cell receptor. Immunol Invest. (2013) 42:204–20. doi: 10.3109/08820139.2012.751397
15. South AM, Diz D, Chappell MC. COVID-19 ACE2 and the cardiovascular consequences. Am J Physiol Heart Circ Physiol. (2020) 318:H1084–90. doi: 10.1152/ajpheart.00217.2020
16. Zou Z, Yan Y, Shu Y, Gao R, Sun X, Ju X, et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun. (2014) 5:3594. doi: 10.1038/ncomms4594
17. Paquette SG, Banner D, Zhao Z, Fang Y, Huang SSH, et al. Interleukin-6 is a potential biomarker for severe pandemic H1N1 influenza A infection. PLoS ONE. (2012) 7:e38214. doi: 10.1371/journal.pone.0038214
18. Rutigliano JA, Sharma S, Morris MY, John A, Oguin TH, McClaren JL, et al. Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J Virol. (2014) 88:1636–51. doi: 10.1128/JVI.02851-13
19. Yan Y, Du Y, Zheng H, Wang G, Rui Li, Chen J, et al. NS1 of H7N9 influenza A virus induces NO-mediated cellular senescence in Neuro2a cells. Cell Physiol Biochem. (2017) 43:1369–80. doi: 10.1159/000481848
20. Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol. (2012) 10:4–18. doi: 10.2174/157016112798829760
21. Solana R, Tarazona R, Aiello AE, Akbar AN, Appay V, Beswick M, et al. CMV and Immunosenescence: from basics to clinics. Immun Ageing. (2012) 9:23. doi: 10.1186/1742-4933-9-23
22. Kim JA, Seong RK, Shin OS. Enhanced viral replication by cellular replicative senescence. Immune Netw. (2016) 16:286–95. doi: 10.4110/in.2016.16.5.286
23. Qudrat A, Wong J, Truong K. Engineering mammalian cells to seek senescence-associated secretory phenotypes. J Cell Sci. (2017) 130:3116–23. doi: 10.1242/jcs.206979
24. Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. (2006) 114:953–60. doi: 10.1161/CIRCULATIONAHA.106.626606
25. Shan H, Bai X, Chen X. Angiotensin II induces endothelial cell senescence via the activation of mitogen-activated protein kinases. Cell Biochem Funct. (2008) 26:459–66. doi: 10.1002/cbf.1467
26. Luther JM, Gainer JV, Murphey LJ, James Luther M, Gainer JV, Murphey LJ, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension. (2006) 48:1050–7. doi: 10.1161/01.HYP.0000248135.97380.76
27. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. (2020) 18:844–7. doi: 10.1111/jth.14768
28. Debbabi H, Ghosh S, Kamath AB, Debbabi H, Kamath AB, Alt J, et al. Primary type II alveolar epithelial cells present microbial antigens to antigen-specific CD4+ T cells. Am J Physiol Lung Cell Mol Physiol. (2005) 289:L274–9. doi: 10.1152/ajplung.00004.2005
29. Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol. (2018) 9:2144. doi: 10.3389/fimmu.2018.02144
30. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. (2012) 188:3869–75. doi: 10.4049/jimmunol.1103627
31. Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narváez AB, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. (2004) 104:942–7. doi: 10.1182/blood-2003-09-3333
32. Naggie S. Hepatitis C virus, inflammation, and cellular aging: turning back time. Top Antivir Med. (2017) 25:3–6.
33. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S. TMPRSS2 S and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. (2014) 88:1293–307. doi: 10.1128/JVI.02202-13
34. AlGhatrif M, Cingolani O, Lakatta EG. The dilemma of coronavirus disease 2019. aging cardiovascular disease: insights from cardiovascular aging science. JAMA Cardiol. (2020). doi: 10.1001/jamacardio.2020.1329
35. Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci USA. (2008) 105:7809–14. doi: 10.1073/pnas.0711241105
36. Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res. (2009) 84:137–44. doi: 10.1093/cvr/cvp176
37. Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L84–96. doi: 10.1152/ajplung.00071.2009
38. Moss ML, Minond D. Recent advances in ADAM17 research: a promising target for cancer and inflammation. Mediators Inflamm. (2017) 2017:9673537. doi: 10.1155/2017/9673537
39. Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. (2013) 19:1085–94. doi: 10.1089/ars.2012.4604
40. Sukhanov S, Semprun-Prieto L, Yoshida T, Higashi Y, Galvez S, Delafontaine P, et al. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci. (2011) 342:143–7. doi: 10.1097/MAJ.0b013e318222e620
41. Sargiacomo C, Sotgia F, Lisanti MP. COVID-19 and chronological aging: senolytics and other anti-aging drugs for the treatment or prevention of corona virus infection? Aging. (2020) 12:6511–7. doi: 10.18632/aging.103001
42. Oudit GY, Kassiri Z, Jiang C, Liu PP, Poutanen SM, Penninger JM, et al. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. (2009) 39:618–25. doi: 10.1111/j.1365-2362.2009.02153.x
43. Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. (2020) 17:259–60. doi: 10.1038/s41569-020-0360-5
44. Mehra MR, Sapan, Desai S, Kuy SR, Henry TD, Patel AN. Cardiovascular disease, drug therapy, and mortality in covid-19. NEJM. (2020). doi: 10.1056/NEJMoa2007621. [Epub ahead of print].
45. Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, et al. Renin-angiotensin-aldosterone system inhibitors and risk of covid-19. N Engl J Med. (2020). doi: 10.1056/NEJMoa2008975. [Epub ahead of print].
46. Mancia GMD, Rea F, Ludergnani M, Apolone GMD, Corrao G. enin–angiotensin–aldosterone system blockers and the risk of covid-19. NEJM. (2020). doi: 10.1056/NEJMoa2006923. [Epub ahead of print].
47. van Hinsbergh VW. Endothelium–role in regulation of coagulation and inflammation. Semin Immunopathol. (2012) 34:93–106. doi: 10.1007/s00281-011-0285-5
48. Armstrong SM, Wang C, Tigdi J, Si X, Dumpit C, Charles S, et al. Influenza infects lung microvascular endothelium leading to microvascular leak: role of apoptosis and claudin-5. PLoS ONE. (2012) 7:e47323. doi: 10.1371/journal.pone.0047323
49. Zhou F, Yu T, Du R, Fan G, Du R, Fan G, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
50. Hileman CO, Longenecker CT, Carman TL, Milne GL, Labbato DE, Storer NJ, et al. Elevated D-dimer is independently associated with endothelial dysfunction: a cross-sectional study in HIV-infected adults on antiretroviral therapy. Antivir Ther. (2012) 17:1345–9. doi: 10.3851/IMP2297
51. Wan S, Xiang Y, Fang W, Zheng Y, Boqun L, Hu Y, et al. Clinical features and treatment of COVID-19 patients in northeast Chongqing. J Med Virol. (2020) 92:797–806. doi: 10.1002/jmv.25783
52. Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L, et al. Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J Infect Dis. (2020) 221:1762–9. doi: 10.1093/infdis/jiaa150
53. Zheng M, Gao Y, Wang, Song GG, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
54. Pantsulaia Ia, Ciszewski WM, Niewiarowska J. Senescent endothelial cells: potential modulators of immunosenescence and ageing. Ageing Res Rev. (2016) 29:13–25. doi: 10.1016/j.arr.2016.05.011
55. Yang P, Gu H, Zhao Z, Wang W, Cao B, Chengcai Lai C, et al. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci Rep. (2014) 4:7027. doi: 10.1038/srep07027
56. Ye S, Lowther S, Stambas J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J Virol. (2015) 89:2672–83. doi: 10.1128/JVI.03529-14
57. Otani A, Takagi H, Oh H, Oh H, Suzuma K. Angiotensin II-stimulated vascular endothelial growth factor expression in bovine retinal pericytes. Invest Ophthalmol Vis Sci. (2000) 41:1192–9.
58. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol. (2019) 4:eaay0555. doi: 10.1126/sciimmunol.aay0555
59. Li YL, Zhao H, Ren XB. Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med. (2016) 13:206–14. doi: 10.20892/j.issn.2095-3941.2015.0070
60. Sun J, Longchamps RJ, Piggott DA, Castellani CA, Sumpter JA, Brown TT, et al. Association between HIV infection and mitochondrial DNA copy number in peripheral blood: a population-based, prospective cohort study. J Infect Dis. (2019) 219:1285–93. doi: 10.1093/infdis/jiy658
61. Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. (2014) 193:3080–9. doi: 10.4049/jimmunol.1303196
62. Yarosz EL, Chang CH. The role of reactive oxygen species in regulating T cell-mediated immunity and disease. Immune Netw. (2018) 18:e14. doi: 10.4110/in.2018.18.e14
63. Bian Q, Lu J, Zhang L, Chi Y, Li Y, Guo H. Highly pathogenic avian influenza A virus H5N1 non-structural protein 1 is associated with apoptotic activation of the intrinsic mitochondrial pathway. Exp Ther Med. (2017) 14:4041–6. doi: 10.3892/etm.2017.5056
64. Long JC, Fodor E. The PB2 subunit of the influenza A virus RNA polymerase is imported into the mitochondrial matrix. J Virol. (2016) 90:8729–38. doi: 10.1128/JVI.01384-16
65. Haynes LD, Verma S, McDonald B, Wu R, Tacke R, Nowyhed HN, et al. Cardif (MAVS) regulates the maturation of NK cells. J Immunol. (2015) 195:2157–67. doi: 10.4049/jimmunol.1402060
66. Angajala A, Lim S, Phillips JB, Kim J-H, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol. (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
67. Previte DM, O'Connor EC, Novak EA, Martins CP, Mollen KP, Piganelli JD. Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS ONE. (2017) 12:e0175549. doi: 10.1371/journal.pone.0175549
68. Fisicaro P, Barili V, Montanini B, Acerbi G, Ferracin M, Guerrieri F, et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med. (2017) 23:327–36. doi: 10.1038/nm.4275
69. Morancho B, Martínez-Barriocanal Á, Villanueva J, Arribas J. Role of ADAM17 in the non-cell autonomous effects of oncogene-induced senescence. Breast Cancer Res. (2015) 17:106. doi: 10.1186/s13058-015-0619-7
70. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. (2020) 63:364–74. doi: 10.1007/s11427-020-1643-8
71. Reddy R, Asante I, Liu S, Parikh P, Liebler J, Borok Z, et al. Circulating angiotensin peptides levels in acute respiratory distress syndrome correlate with clinical outcomes: a pilot study. PLoS ONE. (2019) 14:e0213096. doi: 10.1371/journal.pone.0213096
72. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. (2005) 11:875–9. doi: 10.1038/nm1267
73. Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Jason D, et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care. (2017) 21:234. doi: 10.1186/s13054-017-1823-x
74. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. (2020) 46:586–90. doi: 10.1007/s00134-020-05985-9
75. Zhao M, Bai M, Ding G, Zhang Y, Huang S, Jia Z, et al. Angiotensin II stimulates the NLRP3 inflammasome to induce podocyte injury and mitochondrial dysfunction. Kidney Dis. (2018) 4:83–94. doi: 10.1159/000488242
76. Kadoguchi T, Kinugawa S, Takada S, Fukushima A, Furihata T, Homma T, et al. Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol. (2015) 100:312–22. doi: 10.1113/expphysiol.2014.084095
77. van Kats JP, de Lannoy LM, Jan Danser AH, van Meegen JR, Verdouw PD, Schalekamp MA. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension. (1997) 30:42–9. doi: 10.1161/01.HYP.30.1.42
78. Guo J, Huang Z, Lin L, Lv J. Coronavirus Disease (2019). (COVID-19) and cardiovascular disease: a viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 infection. J Am Heart Assoc. (2020) 9:e016219. doi: 10.1161/JAHA.120.016219
79. Tai W, He L, Zhang, Pu JX, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. (2020) 17:613–20. doi: 10.1038/s41423-020-0400-4
80. Han H, Yang L, Liu R, Liu F, Wu KL, Li J, et al. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. (2020). doi: 10.1515/cclm-2020-0188. [Epub ahead of print].
81. Hung MJ, Cherng WJ, Hung MY, Wu HT, Pang JH. Interleukin-6 inhibits endothelial nitric oxide synthase activation and increases endothelial nitric oxide synthase binding to stabilized caveolin-1 in human vascular endothelial cells. J Hypertens. (2010) 28:940–51. doi: 10.1097/HJH.0b013e32833992ef
82. Chen X, Zhao B, Qu Y, Chen Y, Xiong, J Feng JY, et al. Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely associated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. medRxiv. (2020). doi: 10.1101/2020.02.29.20029520
83. Gurwitz D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev Res. (2020). doi: 10.1002/ddr.21656. [Epub ahead of print].
84. Meng J, Xiao G, Zhang J, He X, Ou M, Jing Bi J, et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. (2020) 9:757–60. doi: 10.1080/22221751.2020.1746200
85. Bersanelli M, Scala S, Affanni P, Veronesi L, Colucci ME, Banna GL, et al. Immunological insights on influenza infection and vaccination during immune checkpoint blockade in cancer patients. Immunotherapy. (2020) 12:105–10. doi: 10.2217/imt-2019-0200
86. Desjardins A, Gromeier M, Herndon JE II, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
87. Bode-Böger SM, Martens-Lobenhoffer J, Täger M, Schröder H, Scalera F. Aspirin reduces endothelial cell senescence. Biochem Biophy Res Commun. (2005) 334:1226–32. doi: 10.1016/j.bbrc.2005.07.014
88. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
89. Zhao M. Cytokine storm and immunomodulatory therapy in COVID-19: role of chloroquine and anti-IL-6 monoclonal antibodies. Int J Antimicrob Agents. (2020) 55:105982. doi: 10.1016/j.ijantimicag.2020.105982
90. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, Jammal TE, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev. (2020) 19:102567. doi: 10.1016/j.autrev.2020.102567
91. Zha L, Li S, Pan L, Yeshan Li, French N, Liyun C, et al. Corticosteroid treatment of patients with coronavirus disease 2019. Med J Aust. (2020) 212:416–20. doi: 10.5694/mja2.50577
92. He R, Lu Z, Zhang L, Fan T, Xiong R, Shen X, et al. The clinical course and its correlated immune status in COVID-19 pneumonia. J Clin Virol. (2020) 127:104361. doi: 10.1016/j.jcv.2020.104361
93. Jurewicz M, McDermott DH, Sechler JM, Tinckam K, Takakura A, Carpenter CB, et al. Human T and natural killer cells possess a functional renin-angiotensin system: further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol. (2007) 18:1093–102. doi: 10.1681/ASN.2006070707
94. Chiappelli F, Khakshooy A, Greenberg G. CoViD-19 immunopathology and immunotherapy. Bioinformation. (2020) 16:219–22. doi: 10.6026/97320630016219
95. Knowlson S, Burlison J, Giles E, Fox H, Macadam AJ, Minor PD. New strains intended for the production of inactivated polio vaccine at low-containment after eradication. PLoS Pathog. (2015) 11:e1005316. doi: 10.1371/journal.ppat.1005316
96. Fujimoto I, Takizawa T, Ohba Y, Nakanishi Y. Co-expression of Fas and Fas-ligand on the surface of influenza virus-infected cells. Cell Death Differ. (1998) 5:426–31. doi: 10.1038/sj.cdd.4400362
97. Grauwet K, Cantoni C, Parodi M, De Maria A, Devriendt B, Pendeet D, et al. Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc Natl Acad Sci USA. (2014). 111:16118–23. doi: 10.1073/pnas.1409485111
98. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med. (2016) 213:167–76. doi: 10.1084/jem.20150785
99. Ayano M, Tsukamoto H, Kohno K, Ueda N, Tanaka A, Mitoma H, et al. Increased CD226 expression on CD8+ T cells is associated with upregulated cytokine production and endothelial cell injury in patients with systemic sclerosis. J Immunol. (2015) 195:892–900. doi: 10.4049/jimmunol.1403046
100. Redlberger-Fritz M, Vietzen H, Puchhammer-Stöckl E. Association of severe influenza virus infections with CD226 (DNAM-1) variants. J Infect Dis. (2019) 220:1162–5. doi: 10.1093/infdis/jiz270
101. Cristiani L, Mancino E, Matera L, Nenna R, Pierangeli A, Scagnolari C, et al. Will children reveal their secret? The coronavirus dilemma. Eur Respir J. (2020). doi: 10.1183/13993003.00749-2020. [Epub ahead of print].
102. Nikolich-Zugich J, Knox KS, Rios CT, Natt B, Bhattacharya D, Fain MJ. SARS-CoV-2 and COVID-19 in older adults: what we may expect regarding pathogenesis, immune responses, and outcomes. GeroScience. (2020) 42:1013. doi: 10.1007/s11357-020-00193-1
103. Liu K, Chen Y, Lin R, Han K. Clinical features of COVID-19 in elderly patients: a comparison with young and middle-aged patients. J Infect. (2020) 80:e14–8. doi: 10.1016/j.jinf.2020.03.005
104. Crowley SD, Rudemiller NP. Immunologic Effects of the Renin-Angiotensin System. J Am Soc Nephrol. (2017) 28:1350-1361. doi: 10.1681/ASN.2016101066
105. Song Y, Wang B, Song R, Hao Y, Wang D, Li Y, et al. T-cell immunoglobulin and ITIM domain contributes to CD8+T-cell immunosenescence. Aging Cell. (2018) 17. doi: 10.1111/acel.12716
106. Aguilar EG, Murphy WJ. Obesity induced T cell dysfunction and implications for cancer immunotherapy. Curr Opin Immunol. (2018) 51:181–6. doi: 10.1016/j.coi.2018.03.012
107. Pan XX, Wu F, Chen XH, Chen D-R, Chen H-J, Kong L-R, et al. T cell senescence accelerates Angiotensin II-induced target organ damage. Cardiovasc Res. (2020) cvaa032. doi: 10.1093/cvr/cvaa032
108. Nakamura K, Yaguchi T, Ohmura G, Kobayashi A, Kawamura N, Iwata T, et al. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci. (2018) 109:54–64. doi: 10.1111/cas.13423
109. Vadasz Z, Haj T, Kessel A, Toubi E. Age-related autoimmunity. BMC Med. (2013) 11:94. doi: 10.1186/1741-7015-11-94
110. Aspinall R, Del Giudice G, Effros, Grubeck-Loebenstein B RB, Sambhara S. Challenges for vaccination in the elderly. Immun Ageing. (2007) 4:9. doi: 10.1186/1742-4933-4-9
111. Mandl S, Sigal LJ, Rock KL, Andino R. Poliovirus vaccine vectors elicit antigen-specific cytotoxic T cells and protect mice against lethal challenge with malignant melanoma cells expressing a model antigen. Proc Natl Acad Sci USA. (1998) 95:8216–21. doi: 10.1073/pnas.95.14.8216
112. Beck GCh, Brinkkoetter P, Hanusch C, Schulte J, van Ackern K, van der Woude FK, et al. Clinical review: immunomodulatory effects of dopamine in general inflammation. Crit Care. (2004) 8:485–91. doi: 10.1186/cc2879
113. Labandeira-Garcia JL, Rodríguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ. Brain renin-angiotensin system and microglial polarization: implications for aging and neurodegeneration. Front Aging Neurosci. (2017) 9:129. doi: 10.3389/fnagi.2017.00129
114. Guo YJ, Chang MH, Chen PL, Lee YS, Chang YC, Liao YC. Predictive value of plasma (D)-dimer levels for cancer-related stroke: a 3-year retrospective study. J Stroke Cerebrovasc Dis. (2014) 23:e249–54. doi: 10.1016/j.jstrokecerebrovasdis.2013.10.022
115. Wang T, Du Z, Zhu F, Cao Z, An Y, Gao Y, et al. Comorbidities and multi-organ injuries in the treatment of COVID-19. Lancet. (2020) 395:e52. doi: 10.1016/S0140-6736(20)30558-4
116. Khalique SC, Ferguson N. Angiotensin II (Giapreza): a distinct mechanism for the treatment of vasodilatory shock. Cardiol Rev. (2019) 27:167–9. doi: 10.1097/CRD.0000000000000247
117. Farina N, Bixby A, Alaniz C. Angiotensin II brings more questions than answers. P T. (2018) 43:685–7.
118. Girard TD, Ware LB, Bernard GR, Pandharipande PP, Thompson JL, Shintani AK, et al. Associations of markers of inflammation and coagulation with delirium during critical illness. Intensive Care Med. (2012) 38:1965–73. doi: 10.1007/s00134-012-2678-x
119. Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. (2020) 11:995–8. doi: 10.1021/acschemneuro.0c00122
120. Cooper HA, Scalia R, Rizzo V, Eguchi S. Angiotensin II- and alzheimer-type cardiovascular aging. Circ Res. (2018) 123:651–3. doi: 10.1161/CIRCRESAHA.118.313477
121. Cassis P, Conti S, Remuzzi G, Benigni A. Angiotensin receptors as determinants of life span. Pflugers Arch. (2010) 459:325–32. doi: 10.1007/s00424-009-0725-4
122. Perez-Lloret S, Otero-Losada M, Toblli JE, Capani F. Renin-angiotensin system as a potential target for new therapeutic approaches in Parkinson's disease. Expert Opin Investig Drugs. (2017) 26:1163–73. doi: 10.1080/13543784.2017.1371133
123. Ho JK, Nation DA. Memory is preserved in older adults taking AT1 receptor blockers. Alz Res Therapy. (2017) 9:33. doi: 10.1186/s13195-017-0255-9
124. Lee YC, Lin CH, Wu RM, Lin JW, Chang CH, Lai MS. Antihypertensive agents and risk of Parkinson's disease: a nationwide cohort study. PLoS ONE. (2014) 9:e98961. doi: 10.1371/journal.pone.0098961
125. Biancardi VC, Stern JE. Compromised blood-brain barrier permeability: novel mechanism by which circulating angiotensin II signals to sympathoexcitatory centres during hypertension. J Physiol. (2016) 594:1591–600. doi: 10.1113/JP271584
126. Simanjuntak Y, Liang JJ, Lee YL, Lin YL. Japanese encephalitis virus exploits dopamine D2 receptor-phospholipase C to target dopaminergic human neuronal cells. Front Microbiol. (2017) 8:651. doi: 10.3389/fmicb.2017.00651
127. de Oliveira LC, Ribeiro AM, Albarnaz JD, Torres AA, Guimarães LFZ Pinto AK, et al. The small molecule AZD6244 inhibits dengue virus replication in vitro and protects against lethal challenge in a mouse model. Arch Virol. (2020) 165:671–81. doi: 10.1007/s00705-020-04524-7
128. Smith JL, Stein DA, Shum D, Fischer MA, Radu C, Bhinder B, et al. Inhibition of dengue virus replication by a class of small-molecule compounds that antagonize dopamine receptor d4 and downstream mitogen-activated protein kinase signaling. J Virol. (2014) 88:5533–42. doi: 10.1128/JVI.00365-14
129. Bain KA, Milling S. T cell addiction: can pathogenic T cells be controlled using dopamine receptors? Immunology. (2019) 158:151–2. doi: 10.1111/imm.13127
130. Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. (2002) 76:10455–64. doi: 10.1128/JVI.76.20.10455-10464.2002
131. Bhattacharyya S, Warfield KL, Ruthel G, Bavari S, Aman MJ, Hope TJ. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology. (2010) 401:18–28. doi: 10.1016/j.virol.2010.02.015
132. Figueroa C, Gálvez-Cancino F, Oyarce C, Contreras F, Prado C, Valeria C, et al. Inhibition of dopamine receptor D3 signaling in dendritic cells increases antigen cross-presentation to CD8+ T-cells favoring anti-tumor immunity. J Neuroimmunol. (2017) 303:99–107. doi: 10.1016/j.jneuroim.2016.12.014
133. Loftis JM, Janowsky A. Neuroimmune basis of methamphetamine toxicity. Int Rev Neurobiol. (2014) 118:165–97. doi: 10.1016/B978-0-12-801284-0.00007-5
134. Sriram U, Haldar B, Cenna JM, Gofman L, Potula R. Methamphetamine mediates immune dysregulation in a murine model of chronic viral infection. Front Microbiol. (2015) 6:793. doi: 10.3389/fmicb.2015.00793
135. Potula R, Haldar B, Cenna JM, Sriram U, Fant S. Methamphetamine alters T cell cycle entry and progression: role in immune dysfunction. Cell Death Discov. (2018) 4:44. doi: 10.1038/s41420-018-0045-6
136. Jiang L, Zhu R, Bu Q, Li Y, Shao X, Gu H, et al. Brain renin-angiotensin system blockade attenuates methamphetamine-induced hyperlocomotion and neurotoxicity. Neurotherapeutics. (2018) 15:500–10. doi: 10.1007/s13311-018-0613-8
137. Jan RK, Kydd RR, Russell BR. Functional and structural brain changes associated with methamphetamine abuse. Brain Sci. (2012). 2:434–82. doi: 10.3390/brainsci2040434
Keywords: SARS-CoV-2, cellular senescence, angiotensin II, prognosis, critical illness, immune checkpoint inhibitors
Citation: Sfera A, Osorio C, Jafri N, Diaz EL and Campo Maldonado JE (2020) Intoxication With Endogenous Angiotensin II: A COVID-19 Hypothesis. Front. Immunol. 11:1472. doi: 10.3389/fimmu.2020.01472
Received: 17 April 2020; Accepted: 05 June 2020;
Published: 19 June 2020.
Edited by:
Zisis Kozlakidis, International Agency for Research on Cancer (IARC), FranceReviewed by:
Ana Acacia Sá Pinheiro, Federal University of Rio de Janeiro, BrazilAshraf Siddig Yousif, Ragon Institute of MGH, MIT and Harvard, United States
Copyright © 2020 Sfera, Osorio, Jafri, Diaz and Campo Maldonado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adonis Sfera, ZHIuc2ZlcmFAZ21haWwuY29t
†These authors have contributed equally to this work