 Jack Mellors
Jack Mellors Tom Tipton1
Tom Tipton1 Stephanie Longet
Stephanie Longet Miles Carroll
Miles Carroll
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 July 2020
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.01450
The complement system is a key component of innate immunity which readily responds to invading microorganisms. Activation of the complement system typically occurs via three main pathways and can induce various antimicrobial effects, including: neutralization of pathogens, regulation of inflammatory responses, promotion of chemotaxis, and enhancement of the adaptive immune response. These can be vital host responses to protect against acute, chronic, and recurrent viral infections. Consequently, many viruses (including dengue virus, West Nile virus and Nipah virus) have evolved mechanisms for evasion or dysregulation of the complement system to enhance viral infectivity and even exacerbate disease symptoms. The complement system has multifaceted roles in both innate and adaptive immunity, with both intracellular and extracellular functions, that can be relevant to all stages of viral infection. A better understanding of this virus-host interplay and its contribution to pathogenesis has previously led to: the identification of genetic factors which influence viral infection and disease outcome, the development of novel antivirals, and the production of safer, more effective vaccines. This review will discuss the antiviral effects of the complement system against numerous viruses, the mechanisms employed by these viruses to then evade or manipulate this system, and how these interactions have informed vaccine/therapeutic development. Where relevant, conflicting findings and current research gaps are highlighted to aid future developments in virology and immunology, with potential applications to the current COVID-19 pandemic.
The complement system is a heat-labile component of native plasma involving both extracellular and cell surface membrane-associated proteins which form a major constituent of the innate immune response. The whole system is comprised of over 30 proteins which have the potential to react via an enzymatic cascade in response to recognition of various stimuli, such as pathogen-associated molecular patterns (PAMPs) and abnormal or damaged host cells. Activation of the complement system typically occurs via three distinct target recognition pathways (the classical, lectin, and alternative pathways) which converge at a single point; the cleavage of complement component 3 (C3). Each pathway has its own unique protease zymogens to recognize and respond to different antigens, but all pathways primarily work to: opsonise pathogens, lyse pathogens and infected cells, regulate the inflammatory response, and enhance the clearance of immune complexes and cell debris (1, 2).
In the context of viral infections, the complement system has been shown to exhibit numerous antiviral mechanisms via direct neutralization of both enveloped and non-enveloped viruses, and/or the promotion of other immune responses. The complement system can directly neutralize virus particles through opsonisation (3), membrane attack complex (MAC) formation on the virion (4), MAC formation on virus-infected cells (5), or targeting of intracellular viral components for proteasomal degradation (6). Other immune responses may also be modulated by the complement system to promote viral clearance, including: the regulation of inflammation/chemotaxis (7), the induction of the antiviral state (6), and the enhancement of adaptive immune responses specific to viral antigens (8, 9). The effectiveness and outcome of this response can vary depending on the infectious agent and host genetic variability.
The classical complement pathway is typically activated when hexameric C1q proteins bind to the fragment crystallisable (FC) CH2-domains of antigen-bound IgM and/or IgG immune complexes (10–12). The binding affinity of C1q to IgG is dependent on the IgG isotype with the greatest affinity for IgG-3, then IgG-1, a weak association with IgG-2, and no interaction with IgG-4 (13). However, for downstream activation and complement lysis activity, the response is more efficient following IgG1-C1q interactions rather than IgG3-C1q interactions (14). C1q is also a versatile pattern recognition molecule. In absence of antibodies, C1q can directly bind to apoptotic cells (15) or proteins on the cell-surface membrane of some pathogens, such as human immunodeficiency virus (HIV) (16) and dengue virus (DENV) (17). C1q can also bind other host plasma proteins such as C-reactive protein (18), fibronectin (19, 20), decorin (21), lactoferrin (22), pentraxin-3 (23), and serum amyloid P component (24).
The C1q molecule is an assembly of six heterotrimers, each containing three chains (C1qA, C1qB, and C1qC) with a central collagenous stem and a globular head at the C-terminus. In native plasma, the C1q molecule forms a calcium-dependent complex with two C1r and two C1s serine proteases to form the C1 complex (25). Ligand recognition and binding via the C1q molecule in the C1 complex induces a conformational change and autoactivation of the C1r2s2 tetramer to activate the classical complement pathway (11, 12). Activated C1s cleaves complement proteins C4 and C2 into active fragments C4b and C2a, along with an inactive fragment (C2b), and a protease-activated receptor (PAR)1/PAR4 ligand (C4a) which increases endothelial cell permeability (26). Non-covalent binding of C4b and C2a forms the classical pathway C3 convertase, C4bC2a, responsible for cleavage of C3 into C3a (anaphylatoxin) and C3b (active component of the convertase). The newly formed C4bC2aC3b complex is a C5 convertase formed from either the classical or lectin complement pathway activation, which cleaves the C5 molecule into C5a (anaphylatoxin) and C5b. C5 proteolysis and the successive steps are then the same for each of the three complement pathways - C5b is deposited onto the activating surface and subsequent, irreversible binding of C6, C7, C8, and multiple copies of C9 to permeate the cell surface membrane and form the MAC (11, 12, 27). All three complement pathways are summarized in Figure 1.
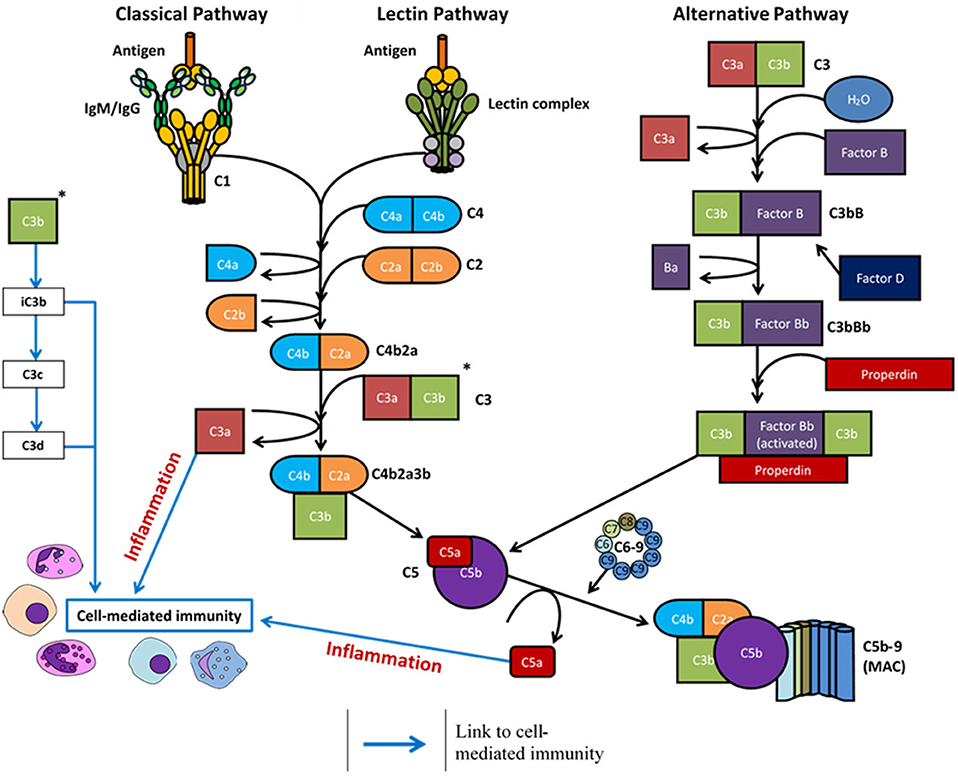
Figure 1. Overview of the complement system following activation via antigen (classical and lectin pathways) or spontaneous hydrolysis (alternative pathway). Complement activation eventuates in formation of the membrane attack complex (MAC) and the cleavage products regulate inflammation (C3a and C5a) and cell-mediated immunity (C3a, C3c, C3d, C5a).
The lectin complement pathway follows the same enzymatic cascade as the classical pathway but is distinct in the antigens and proteases required for its activation. The lectin pathway is activated in response to invading pathogens via direct binding of PAMPs by various C1q-like lectins, complexed with mannose-binding lectin (MBL)-associated serine proteases (MASPs)-1/2/3. These C1q-like activators are MBL, ficolin-1 (M-ficolin), ficolin-2 (L-ficolin), ficolin-3 (H-ficolin), and collectin-11 (CL-11) (28–30). In humans, MBL is typically present as a trimer, tetramer, pentamer, or hexamer and these oligomeric structures influence its carbohydrate binding properties (31, 32). Each monomeric subunit in the complex is a homotrimer with each polypeptide containing a cysteine-rich region at the N-terminus, followed by a collagen-like domain, a neck region, and a carbohydrate recognition domain capable of binding specific sugars present on pathogenic surfaces i.e., N-acetyl-D-glucosamine and D-mannose (33, 34).
Similar to MBL, multimeric ficolin complexes are assembled through homotrimer subunits with cysteine-rich N-terminal segments which form interchain disulphide bonds, followed by collagen-like regions, but they are unique in their ability to bind distinct carbohydrates via their C-terminal fibrinogen-like domains (35–37). Ficolin-1 is predominantly synthesized in monocytes and granulocytes where it can be found present on their surface or extracellularly in native human plasma. Ficolin-2 is synthesized in the liver and secreted into the bloodstream where it can bind to various acetylated structures and sugars via three inner binding sites (38). Ficolin-3 is synthesized in the liver and lungs and is present in native plasma at a higher concentration than ficolins-1/2, although less is known about its functional capabilities (39). Collectin-11 (CL-11) can form heterotrimeric complexes with collectin liver 1 (CL-10) in serum and can also associate with MASPs to activate the lectin complement pathway (40).
Once one or more of these lectins have complexed with MASP-2 and bound their specified target, MASP-2 then cleaves C4 and C2 to form the C3 convertase (C4bC2a complex). Following the proteolytic cleavage of C3, the lectin complement pathway follows the same catalytic process as the classical pathway (Figure 1) (2). The roles of MASP-1 and MASP-3 in this pathway are relatively ambiguous (29, 41). MASP-1 is capable of cleaving complement component C2 and, to a much lower extent, components C3 and C4 (29, 42), whilst MASP-3 may have a negative regulatory role of the lectin pathway through downregulation of MASP-2 cleavage activity (43).
The alternative pathway does not require the specific protein-protein or protein-carbohydrate interactions seen with the other two pathways. Under normal physiological conditions, ~1% of C3 per hour undergoes spontaneous hydrolysis as the internal thioester bond is cleaved to produce C3(H2O). This process is augmented in the presence of various surfaces which lack complement regulatory proteins, as electrostatic and/or hydrophobic interactions adsorb C3 to the surface to induce conformational changes (44, 45). C3(H2O) can then bind factor B to induce another conformational change, as factor B is then cleaved into two components by factor D: Ba (which dissociates from the complex) and Bb (which remains bound in the complex). The protein complex C3bBb is the alternative pathway C3 convertase, which is stabilized through the binding of properdin to produce C3bBbP and can cleave further C3 molecules through the serine protease activity of fragment Bb. The alternative pathway therefore has the potential to both activate and enhance complement activity through an amplification loop; cleaved C3 components produce C3 convertases which cleave further C3 molecules (46, 47). Cleavage of C3 also yields C3a and C3b, where C3b remains bound in the complex to form the alternative pathway C5 convertase, C3bBbPC5b, and C3a acts as an anaphylatoxin. The rest of the complement cascade is then identical for all pathways (Figure 1) (48, 49).
Although complement activity typically occurs via the three pathways described, less conventional mechanisms of activation and immune modulation can occur, and have been discussed in a recent review (50). Typically, properdin is described as a positive regulator of the alternative pathway through stabilization of the C3 and C5 convertases. But its functions extend beyond this, including complement activation via direct antigen recognition of invading pathogens and apoptotic/necrotic cells (51–53), and as a potential ligand for NKp46-mediated natural killer cell activation and subsequent secretion of X-chemokine ligand 1 (54). Similarly, C3 and its cleavage products are often described as extracellular components, yet they can have intracellular signaling roles to eliminate pathogens, alter cytokine signaling profiles and induce Th1 responses (6, 55–57).
Most complement proteins are primarily synthesized in the liver and secreted into the bloodstream; this process is rapidly upregulated during infection (58). Complement proteins can also be produced by epithelial cells (59), endothelial cells (60), and circulating immune cells such as dendritic cells, granulocytes, macrophages, and monocytes (61, 62). Local production of complement also occurs in immune privileged sites including the brain (63), eyes (64), and testis (65). To regulate this activity and prevent damage to healthy cells and tissues, many regulatory proteins are primarily expressed as either soluble plasma proteins or cell-surface receptors (Figure 2 and Table 1).
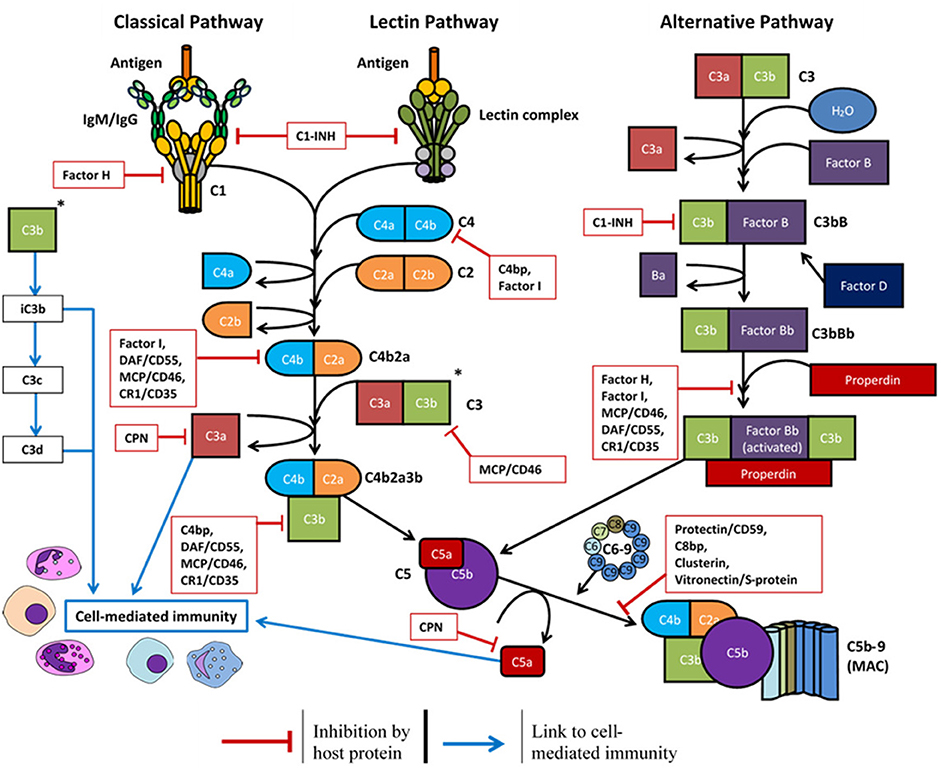
Figure 2. Overview of the complement system and the host soluble/membrane-bound regulatory proteins (red boxes): C1-INH, C1-inhibitor; C4bp, C4-binding protein; C8bp, C8-binding protein; CPN, carboxypeptidase-N; CR1, complement receptor 1; DAF, decay-accelerating factor; MCP, membrane cofactor protein.
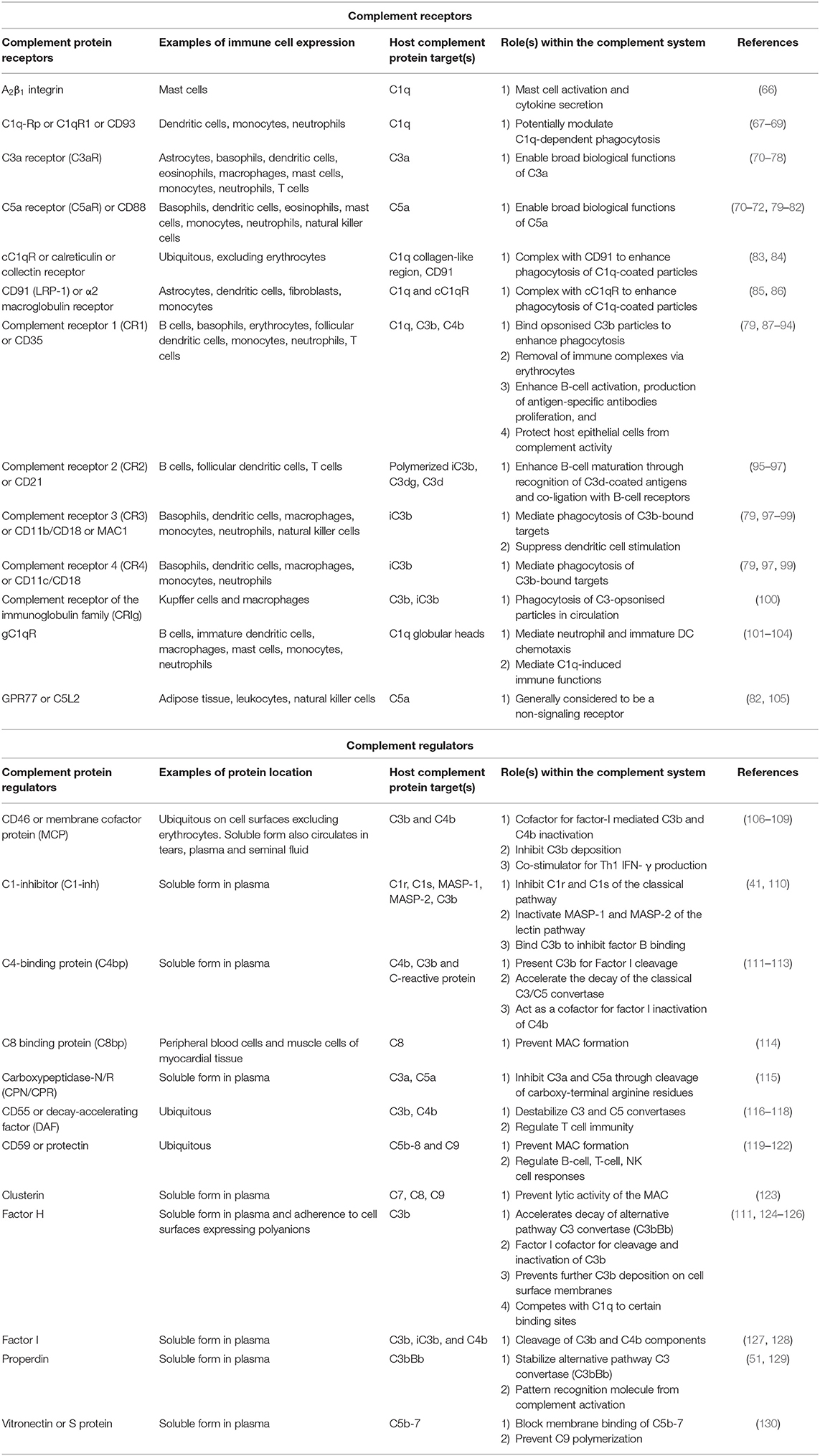
Table 1. Overview of key complement regulatory proteins and receptors, their location within plasma and on circulating immune cells, and their roles within the complement system of humans.
Complement regulatory proteins may be unique to a certain pathway or influence the downstream activity of all three pathways. Factor H, factor I, and properdin are unique to the alternative pathway. Factor H is both a soluble and cell-surface membrane regulator (124) which accelerates the decay of the C3 convertase (C3bBb) to reduce complement deposition (125), and it functions as a Factor I cofactor to cleave C3b and C4b components (111). Properdin is a positive regulator of the alternative pathway which stabilizes the C3 convertase (C3bBb) and promotes its association with further C3b molecules (129). The C1-inhibitor (C1-inh) is a negative regulator of all three pathways. C1-inh competes with factor B to limit activation of the alternative pathway (110), inhibits C1r and C1s to prevent classical pathway activation (41), and inactivates MASP-1 and MASP-2 to prevent lectin pathway activation (41).
Further proteins are required to regulate the downstream complement activity. Carboxypeptidase-N/R regulates the anaphylatoxin activity of C3a and C5a via cleavage of their arginine residues (115). C8 binding protein (114), clusterin (123), and vitronectin/S protein (130) are all soluble proteins which prevent the complete assembly of the MAC. CD46/membrane cofactor protein (106, 107), CD55/decay-accelerating factor (116, 117), and CD59/protectin (119) are ubiquitously expressed on the surface of host cell surface membranes to protect the cell from complement deposition.
One of the key functions of the complement system is to assist in the killing and containment of invading pathogens, including bacteria (131), fungi (132), protozoa (133), and viruses (134). Previous reviews have discussed various evasion mechanisms adopted by viruses to dysregulate or evade this complement activity (135–138). This knowledge may then be exploited and has previously identified novel methods for vaccine and therapeutic intervention—an area which has not been extensively discussed in the previous reviews.
The antiviral mechanisms of complement have been divided into four main sections in this discussion; physiologically however, each section is not exclusive as they work together to form a complete system. Briefly, complement deposition on a virion can block interactions with host cell receptors, aggregate virus particles, signal intracellularly to induce an antiviral state, and enhance phagocytosis (3, 6, 139). This can lead to formation of the MAC and lyse lipid membranes of enveloped viruses (140) or lyse infected host cells expressing viral antigens (141). Activation of the complement system also produces pro-inflammatory anaphylatoxins (C3a, C5a, and putatively C4a) which can enhance phagocytosis and in some cases, worsen disease symptoms (142). Lastly, these processes can enhance the adaptive immune response to viral antigens, induce a Th1 response (56), modulate Treg and Th17 responses (143), prolong B-cell memory and significantly increase antigen-specific antibody titres (144). Many of these functions may be evaded or manipulated by different viruses (shown in Figure 3) and such examples are provided throughout.
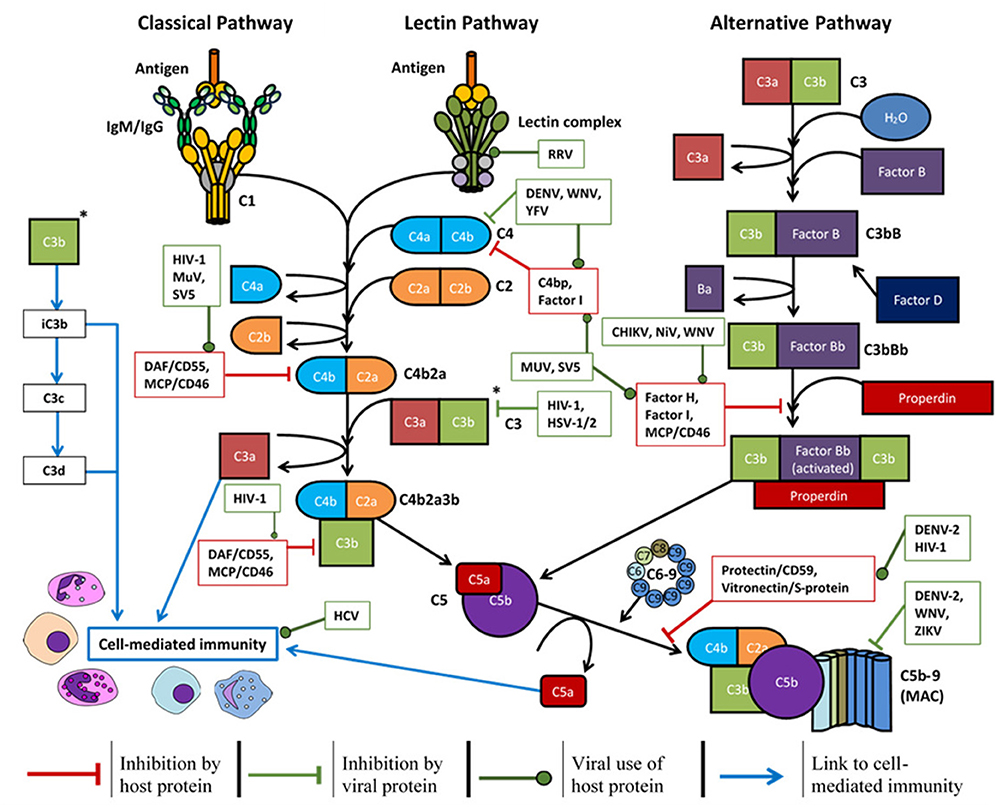
Figure 3. Overview of the complement system, host soluble/membrane-bound regulatory proteins influenced by the viruses mentioned (red boxes), and the regulation exerted by certain viruses (green boxes) to promote survival: CHIKV, chikungunya virus; DENV, dengue virus; HCV, hepatitis C virus; HIV-1, human immunodeficiency virus-1; HSV-1/2, herpes simplex virus-1/2; MuV, mumps virus; NiV, Nipah virus; RRV, Ross River virus; SV5, simian virus 5; WNV, West Nile virus; YFV, yellow fever virus; ZIKV, Zika virus.
All three complement pathways can lead to virus opsonisation and complement deposition following activation. The outcome of this response largely depends on the infectious agent and could enhance viral infection, suppress viral infection, or be dysregulated by the expression of some viral proteins.
The MBL protein of the lectin pathway can interact with numerous viral antigens and have varying effects on neutralization or viral enhancement. MBL can directly bind the Ebola virus (EBOV) glycoprotein (GP). High doses of MBL, relative to other complement proteins, can enhance EBOV-GP pseudotyped virus infection into primary human macrophages and human monocyte-derived macrophage cell lines (145). However, MBL opsonisation of the EBOV-GP can neutralize EBOV-GP-pseudotyped virus by preventing cell interactions via DC-SIGN (3). MBL has also been successfully used as a rescue therapy in 40% of mice when administered at supra-physiological levels, 24 h post-lethal challenge with a mouse adapted EBOV strain (146). So, in the context of EBOV infection, the effects of MBL appear to be dependent on the cellular target and the relative concentrations of other complement protein components.
MBL has also been shown to bind the HIV-1 protein, gp120. This interaction was sufficient to neutralize cell-line adapted HIV infection of CD4+ H9 lymphoblasts (134). A later study reported a similar finding, although much higher concentrations of MBL were required to achieve the same level of neutralization (50 μg/mL rather than 1 μg/mL of MBL), and these findings were not replicated when using HIV primary isolates or other cell lines for infection. In the later study, MBL was shown to be sufficient for virus opsonisation but not neutralization (147). This highlights an important consideration for in vitro studies when investigating complement and pseudovirus interactions, as small method variations can yield significantly different results. Where possible, in vivo experiments can help validate this work and address possible discrepancies. Further possible implications of MBL during HIV infection have been shown in a study of single nucleotide polymorphisms (SNPs). SNPs in the MBL gene which result in low serum concentrations of MBL were associated with increased risk of HIV infection and poorer prognosis following AIDS diagnosis (148).
Downstream from MBL binding, complement components are deposited on HIV virions which increase viral uptake and internalization into dendritic cells (DCs). Both complement-opsonised and complement-free HIV binding was reduced through the blockage of C-type lectins, integrins and CD4. However, the use of individual blockers showed that complement-opsonised HIV utilized β1- and β2-integrin for binding and uptake, whereas complement-free HIV utilized β2- and β7-integrin (149). A similar observation has been reported for herpes simplex virus (HSV)-2 during the infection of human DCs. Complement deposition and interactions with complement receptor 3 (CR3) enhanced HSV-2 infection of immature DCs and increased the production of new virus particles, whereas complement with the use of neutralizing antibodies significantly reduced infection (150). This highlights another important point with regards to in vitro investigations of complement and viral infection. Plasma is often heat-inactivated for use in cell culture to overcome concerns of complement-mediated cytotoxicity. Consequently, investigations of virus-host cell interactions may overlook important complement-mediated interactions that would normally be present during infection.
The varied effects of MBL opsonisation during viral infection have also been described for severe acute respiratory syndrome coronavirus (SARS-CoV). Multiple studies have shown the potential for MBL to bind immobilized SARS-CoV or the SARS-CoV spike protein (151, 152). This interaction was shown to be dependent on a single N-linked glycosylation site of the spike protein and this binding could prevent spike protein interactions with DC-SIGN but not the angiotensin-converting enzyme 2 (ACE2) receptor or cathepsin-L (152). Ip et al. showed that MBL binding to immobilized SARS-CoV could also inhibit SARS infection into fetal rhesus kidney cells and enhance deposition of C4 (151). However, Leth-Larsen et al. did not observe any interactions between MBL and SARS-CoV spike protein in their study (153). Similar to HIV, several studies have found a significant difference of MBL SNPs associated with lower or deficient MBL serum levels in SARS patients compared to healthy Chinese population control groups (151, 154), and a reduction of MBL protein concentrations in SARS patient sera (151). However, one other study observed no significant correlation of MBL-deficient SNPs in SARS patients compared to healthy Chinese population control groups (155). The role of MBL in SARS-CoV infections appears conflicted but could be significant. As later discussed, the downstream effects of complement activation do significantly influence symptoms of coronavirus infections.
Other complement proteins and downstream products of its activation can opsonise virus particles. For DENV and West Nile virus (WNV), neutralization of the virions occurs in a C3 and C4 dependent manner following MBL binding. For WNV, neutralization was achieved independent of downstream C5 and therefore did not require formation of the MAC (156). For Simian virus 5 (SV5), complement-mediated neutralization is predominantly achieved through C3 deposition and the formation of virion aggregates, rather than virion lysis. For the closely related Mumps virus (MuV) however, the opposite effect is observed with few aggregates formed but greater susceptibility to complement lysis (157). Similarly, complement activation in the presence of influenza A virus causes virion aggregation and opsonisation of the hemagglutinin receptor. Although to achieve neutralization, IgM antibodies and activation of the classical pathway is required (139).
For Chandipura virus (CHPV), the alternative pathway and factors C3, C5, and factor B were required for complement-mediated virus neutralization in absence of C8 or antibodies (158). A different study utilized antibodies to observe classical pathway activation and reported that C1q, C3, and C4 were essential components for neutralization, but this was independent of factor B and C8 (159). The discrepancy of the importance of Factor B for CHPV neutralization could depend on the presence of antibodies and the classical pathway.
As mentioned previously, complement opsonisation of virions can enhance infection through interactions with complement receptors on host phagocytic cells (149, 160). However, some complement proteins can have a protective intracellular function as well, which is independent of cell-type (6). Enveloped viruses may naturally evade the intracellular functions of complement, as the protein deposition would occur on the lipid membrane. So, for viral entry via membrane fusion or endocytosis, it is expected that the complement-opsonised viral envelope would be left on the host cell surface membrane or endosome plasma membrane. This has been demonstrated in vitro using respiratory syncytial virus (RSV), an enveloped virus which enters the cell via membrane fusion, where complement intracellular signaling was absent following infection (6).
The intracellular immune function of complement has a better-defined role for non-enveloped viruses, although the area of intracellular complement immunity is still relatively new (161). In a C1-dependent manner and independent of downstream complement activity, C4 deposition on the capsid of non-enveloped human adenovirus 5 has been shown to contain the virus within the endosome, by blocking the fiber shedding and protein VI exposure mechanisms required for capsid disassembly (162). The use of an adenovirus type 5 vector (AdV) also showed that intracellular sensing of complement could inhibit infection and degrade the virus particle (6). A comparison of complement-coated AdV to AdV only, showed that intracellular C3 signaling induced the activation of pro-inflammatory cytokines (IFN-β, IL-6, IL-1β) through NF-κB, interferon-regulatory factor (IRF), and activating protein-1 (AP-1) transcription factor activation. Intracellular C3 sensing was shown to be mitochondrial antiviral-signaling protein (MAVS)-dependent, and independent of PAMPs and pattern recognition receptors. Sensing of complement-coated AdV also targeted the virion for degradation by valosin-containing protein (VCP) and the proteasome. C3-mediated signaling could induce an antiviral state in previously uninfected cells, as the supernatant from complement-coated AdV infected cells was able to protect uninfected HeLa cells from infection with interferon-sensitive Sindbis virus. Lastly, some viruses have evolved evasion mechanisms to overcome the complement-mediated intracellular immune response. Rhinoviruses and polioviruses were shown to inhibit the intracellular C3 complement signaling mechanism through the expression of a cytosolic 3C protease to degrade C3 (6).
Discussed in more detail below, some viruses encode proteases which enhance degradation of the C3 convertase to prevent further complement deposition or MAC formation. This can protect the virion from complement opsonisation and viral lysis.
Following complement deposition and opsonisation, the complement cascade can progress to assembly of the MAC. MAC formation can perturb and lyse lipid membranes of enveloped viruses or destroy infected cells expressing viral antigens to reduce viral load (4, 5, 140, 141). Again, viral proteins can be expressed to dysregulate and evade this response.
Zika virus (ZIKV) can lead to classical pathway activation via formation of antigen-antibody complexes or through direct binding of C1q. For ZIKV derived from insect cell lines, this interaction resulted in MAC formation and a reduction of viral titres in vitro. However, ZIKV derived from human cell lines were more resistant to complement mediated neutralization (4). ZIKV and other Flaviviruses (including yellow fever virus (YFV), DENV, and WNV) express and secrete the non-structural protein 1 (NS-1) to regulate complement activity. The NS1 protein has a wide variety of functions in complement regulation which include: antagonism of C4 (163), recruitment of host C4 binding protein (164), recruitment of host factor H (165), recruitment of host vitronectin and inhibition of C9 polymerisation (166). However, the DENV NS1 protein is also capable of complement activation and the resulting soluble C5b-9 complexes have been found to correlate with disease severity in patients with dengue shock syndrome (167). This discrepancy was addressed with the possibility that relative IgM, C4 and soluble NS1 concentrations in plasma, at different sites of infection, could influence the extent of inhibition and therefore have varied effects on complement activity (163).
Similarly, Nipah virus (NiV) exhibits factor I-like activity, either through acquisition of factor I host protein or through inherent protease activity. Unlike soluble factor-I, NiV exhibits no capacity for C4b cleavage and showed no significant cleavage of C3b with a CD46 cofactor, despite its integration in the NiV lipid membrane. However, NiV is capable of C3b cleavage into iC3b with factor I cofactors (factor H and soluble CR1) to protect against virus neutralization (168). Chikungunya virus (CHIKV) also exhibits factor I-like activity, likely of viral origin and dependent on host factor H concentrations, to cleave C3b into inactive iC3b and resist complement-mediated neutralization (169). MuV, SV5, and HIV-1 can all recruit host cell CD46 into the viral lipid membranes during the budding process to protect from complement deposition and neutralization (170, 171). HIV-1 also incorporates glycosyl phosphatidylinositol-anchored CD55 and CD59 for further protection from complement mediated neutralization (170). Conversely, complement deposition has been shown to enhance HIV-1 infectivity into peripheral blood mononuclear cells through interactions with complement receptors (160, 172). This highlights the complexity of complement and viral interactions with dualistic mechanisms, which has previously been reviewed in the context of HIV-1 infection (173).
Infected host cells which present viral antigens on the cell surface membrane can activate the classical pathway, as the antigens bind IgM/IgG to induce complement dependent cytotoxicity (CDC). The infected cell is then lysed via the MAC in an attempt to reduce viral load. For Influenza A virus infection, complement-dependent lysis (CDL) monoclonal antibodies can cross-react with H1 and H2 hemagglutinin subtypes for broader protection than neutralizing monoclonal antibodies (141). Similarly, broadly neutralizing anti-HIV-1 antibodies can bind the viral envelope protein expressed on infected primary lymphocytes to initiate complement deposition. The deposition does not result in a rapid lytic effect but neutralizes viral spread to further cells (174). For HSV-1 and HSV-2, the glycoprotein C (gC)-1 is expressed to protect virions and infected cells from complement mediated neutralization. The gC-1 protein binds C3, C3b, and C3c to prevent subsequent binding of C5 or properdin. Modification of gC-1 on HSV infected cells can therefore increase their susceptibility to antibody neutralization and CDC (5, 175).
Some of the cleavage products from complement activation can function as anaphylatoxins and have broader immune regulatory functions. Primarily, cleavage products C3a and C5a can be generated via all three pathways and act as potent immune regulators, whilst C4a is generated via the classical and lectin pathways only (7). The role of C4a as an anaphylatoxin is disputed as it currently has no known anaphylatoxin receptor associated with its activity (176). However, it does function as an effector protein that is derived from complement activation, which enhances endothelial cell permeability and increases stress fiber formation via PAR1 and PAR2 (26). The roles of C3a and C5a are better described as anaphylatoxins, with the latter demonstrating higher stability and broader biological activity. C5a recruits neutrophils to the site of inflammation and both C3a and C5a can recruit: eosinophils, fibroblasts, macrophages, mast cells, and monocytes (70–72, 177–179). These two anaphylatoxins demonstrate a large functional overlap but each have their own discrete functions. To varying degrees, both are capable of stimulating the production of pro-inflammatory mediators from monocytes and macrophages via inflammasome-caspase-1 activation (180, 181). Both can induce the degranulation of mast cells (182–185), basophils (186–188), and eosinophils (189). Both induce respiratory bursts in eosinophils (190) and neutrophils, although only C5a shows chemotactic activity for neutrophils whereas C3a may actually prevent neutrophil mobilization from the bone marrow (191). Further, only C5a can stimulate respiratory bursts in macrophages (192).
The activity of C3a and C5a is mediated via binding to two main G-protein coupled receptors; C3aR or C5aR, respectively (193). A secondary, non-G-protein coupled receptor (C5L2) has been shown to bind C5a and potentially regulate its biological functions in vitro, although its primary functions are not yet clear (105). These receptors are widespread across different cell types including both myeloid cells and non-myeloid cells (e.g., astrocytes, microglia, hepatocytes, endothelial and epithelial cells) to produce various biological functions dependent on the cell type (194, 195). C3a and C5a activity is further regulated by the enzyme carboxypeptidase-N. Carboxypeptidase-N cleaves the carboxy terminal arginine amino acid of these anaphylatoxins to generate products with greatly reduced (C5a-desArg) or absent (C3a-desArg) anaphylatoxin activities (196). The cleavage product C5a-desArg retains some chemotactic activity to recruit distant immune cells (188, 197) whilst C3a-desArg can function as a hormone for lipid metabolism (198). Beyond their roles in chemotaxis, C3a and C5a have been associated with: the induction of smooth muscle contraction (199), regulation of vasodilation (200), an increase in vascular permeability (201), and the production of various cytokines including IL-1β, IL-8/CXCL-8, CCL5, IL-6, TNFα (180, 193).
During viral infection, excessive complement activation leading to a strong pro-inflammatory response is often associated with more severe disease symptoms. The negative impact of complement activation has been associated with more severe symptoms during SARS-CoV and MERS-CoV infections. Infection of C3 deficient mice with SARS-CoV revealed that the loss of complement activity resulted in milder disease outcomes (202). Compared to the wild-type, the C3 deficient mice showed: no significant weight loss, improved respiratory function, reduced lung pathology, and lower levels of inflammatory cytokines and chemokines (202). Proteomic analysis has shown that a product of complement activation, C3c α chain, was significantly higher in SARS-patient sera compared to non-SARS patient sera (203). Similarly, increased concentrations of C5a and C5b-9 were observed in sera lung tissues of hDPP4-transgenic mice challenged with MERS-CoV. The subsequent use of a C5aR antibody to prevent C5a functional activity resulted in reduced tissue damage and a lower viral load (204). Cytotoxic effects of complement may also occur post-SARS-CoV infection. Autoantibodies elicited 1-month after infection against epithelial and endothelial cells can mediate complement-dependent cytotoxicity and enhance lysis against A549 cells and human placenta endothelial cells (205).
In patients with severe DENV infection and dengue shock syndrome, overactivity of the alternative pathway has been reported with increased levels of NS1, C5a, and sC5b-9 in pleural fluids, which likely contribute to the symptoms of increased vascular permeability (167, 206). In DENV infected cells, indicators of the alternative complement pathway are upregulated, with a relatively higher concentration of Factor B to factor H proteins and increased cell surface C3b deposition (206).
In mice infected with Ross River virus (RRV), complement activation products have been identified in serum and inflamed tissues. Similar observations have been made in the synovial fluid of RVV-infected patients. In C3 knockout mice, the signs of severe disease and tissue damage from RVV infection were diminished compared to wild-type, which suggests complement promotes RRV-induced inflammation (207). RRV infected cells express the viral E2 protein which is glycosylated with N-linked glycans. E2 N-linked glycans are antigens for MBL and can activate complement via the lectin pathway, which results in greater inflammation and tissue damage during RVV infection (208, 209).
Complement activation also plays an important role in linking the innate and adaptive immune responses. This interaction can enhance the production of antigen-specific antibody titres and shape the T-cell response to target viral pathogens more efficiently. The importance of complement in the regulation of T-cell immunity has previously been reviewed (50, 210).
Cognate and co-stimulatory interactions (CD80-, and CD86-CD28, and CD40-CD40 ligand) between antigen presenting cells (APCs) and T-cells results in the local production of C3, factor B, factor D, and C5. Receptors C3aR and C5aR are also upregulated on the T-cell surface whilst production of decay accelerating factor (DAF) is down-regulated. The local production of complement components from immune cells enables signaling via C3aR and C5aR in an autocrine and paracrine manner. Complement C3 can also be processed intracellularly, or internalized as C3 (H2O) from the alternative pathway, to increase pro-inflammatory cytokine expression from T-cells and recycled back to the T-cell surface (55, 57). A major component of C3 cleavage on the T-cell surface is iC3b. T-cell membrane bound iC3b binds to CR3 (and possibly CR4) on monocyte derived DCs to enhance T-cell proliferation (9). Absence of C3aR and C5aR leads to: reduced complement protein and receptor regulation, lack of co-stimulatory molecule expression, impaired cytokine production (IL-1, IL-23, and IL-12), an induction of an iTreg cell response, and suppression of T-cell proliferation (211–213).
Activation of both C3aR and C5aR on DCs by their respective anaphylatoxins (C3a and C5a) can mediate the production of IL-6, IL-23, the IL-12 receptor, and TGF-β1 to promote T-cell differentiation into antiviral Th1 and Th17 subsets (143). Induction of the Th1 response also depends on C3aR and CD46 activation on T-cells via their T-cell derived ligands (56). In mice infected with Influenza A virus, inhibition of the C5aR lead to a reduction in influenza-specific cytotoxic CD8+ T-cells (214) and C3 deficiency lead to increased viral titres and delayed viral clearance (215). C3 is also required for the production of antigen-specific CD8+ T-cell responses during lymphocytic choriomeningitis virus infection in mice (216). During HCV infection, the HCV core protein can interact with gC1qR on host immune cells and suppress the T-cell response. This interaction inhibits T-cell proliferation in a dose-dependent manner to downregulate CD69 activation and reduce the production of IFN-γ and IL-2 from T-cells (217–219). HCV core protein interaction with gC1qR on monocyte-derived DCs inhibits IL-12 production and promotes Th2 cytokine production to limit differentiation into Th1 cells (218). HCV core protein interaction with gC1qR on B-cells has a differential response to the one observed on T-cells and DCs, as it increases cell surface costimulatory and chemokine receptor expression and enhances B-cell proliferation (219). Furthermore, the HCV core protein exhibits intracellular functions, as it can suppress the T-cell factor-4 transcription factor required for C9 promoter activity regulation. This reduces C9 mRNA and protein levels which are required for complete MAC assembly (220).
As discussed previously, complement activation can result in C3 deposition on the surface of virions. C3 and its cleavage products can interact with the B-cell receptor and B-cell co-receptor complex (CR2/CD21 ligated with CD19 and CD81) to lower the B-cell activation threshold by several orders of magnitude. This can dramatically increase antibody titres, modulate the proliferation of mature B cells, and protect the B-cells from CD95-mediated elimination (8, 144). Immune complexes coated in C3 and C3 cleavage products covalently interact with complement receptors on follicular DCs (FDCs). The C3-coated immune complexes on FDCs are then presented to B-cells in the germinal center for optimal B-cell responses, including: antibody production, somatic hypermutation, class switching, and affinity maturation (87, 221). FDCs can then retain the C3-coated complexes within the lymphoid for extended periods of time to generate memory B-cells and promote survival (222).
Alternatively, some aspects of the complement system can suppress certain responses of adaptive immunity: stimulation of CR3 on DCs can suppress the release of inflammatory cytokines (98) and C1q-differentiated DCs demonstrate an increased phagocytic capacity but reduced expression of CD80, CD83, and CD86 required for T cell activation (223).
It is apparent that the complement system has important implications for virus neutralization and development of the adaptive immune response. As our knowledge of virus and complement interactions improves, this can inform novel approaches for intervention and the development of therapeutics and vaccines. One such example is the use of rupintrivir against rhinoviral infections. Rhinoviruses encode a cytosolic 3C protease which cleaves intracellular C3 to avoid the intracellular mechanisms of complement, mentioned previously. Rupintrivir inhibits the viral cytosolic 3C protease to increase susceptibility of the virus to intracellular complement immunity (6). Similarly, the use of Fab fragments could prevent the C4 inhibition of human adenovirus 5 vector for its use in adenoviral gene therapy to promote efficient transgene delivery (162).
Due to the multifaceted and complex immune functions of the complement system, direct manipulation of complement would need to be carefully considered. Inhibition of the complement system could increase susceptibility to other diseases, whilst over-stimulation could result in autoimmunity and damage to host cells. A method of complement stimulation through inhibition of the CD59 regulator has been proposed for the treatment of latent HIV-1 infection in cells. The use of provirus stimulants and a CD59 inhibitor showed a dose-response effect of cell sensitization to antibody-dependent cell-mediated lysis and reduced viral load. Aside from the target cells, no significant non-specific cytolytic effects were observed in vitro. CD59 protects host cells from complement activity, is ubiquitously expressed, and so its inhibition has the potential to damage host cells (224, 225). Deletion of CD59 in mice did not have a lethal outcome, however absence of the complement regulatory protein did lead to intravascular haemolysis and thrombosis (226). Treatment in the context of HIV-1 infection would be short-term however (224) and could be an exception for an otherwise incurable disease. Similar approaches have been considered for other life-threatening diseases such as cancerous conditions (227, 228).
Methods of complement inhibition have also demonstrated therapeutic benefit. Mentioned previously, excessive complement activation is associated with more severe outcomes of MERS-CoV and SARS-CoV infections. Use of a C5aR antibody to block the pro-inflammatory effects of C5a in MERS-CoV infected hDPP4-transgenic mice resulted in: lower concentrations of pro-inflammatory cytokines, reduced viral replication in lung tissues, reduced lung and spleen tissue damage, and a reduction of viral antigen and microglia activation in the brain (204). Excessive complement activation and similar lung pathology during SARS-CoV infection has also been observed in H5N1 influenza cases, where the use of C3aR and C5aR antagonists reduced signs of acute lung injury and viral load in H5N1-infected mice (229).
A novel coronavirus, SARS-CoV-2, has recently emerged and is the causative agent of COVID-19—an acute self-limiting disease which has the potential to progress to severe disease and death. Symptoms of severe disease involve major alveolar damage, wide-spread lung inflammation, and progressive respiratory failure (230, 231). The pathological features of lung, liver, and heart tissue in a severe case of COVID-19 greatly resembled those seen in SARS-CoV and MERS-CoV infections which are complement-mediated (202–205, 231). MBL has been shown to activate complement via binding to SARS-CoV spike protein in some studies and this could translate to the SARS-CoV-2 spike protein, which contains N-linked glycosylation sites that are targets for MBL (151, 232). Thus, the widespread lung inflammation observed in severe cases of COVID-19 could be exacerbated by excessive complement activation. Furthermore, viral infections with similar lung pathology to COVID-19 have demonstrated therapeutic benefit with the administration of complement inhibitors targeting C3a/C3aR or C5a/C5aR. This has been shown for H5N1 (229), H7N9 (233), and MERS-CoV (204) infections. So, it seems plausible that the lung inflammation in severe cases of COVID-19 is exacerbated by excessive complement activation and this pathologic inflammation could be attenuated through use of complement inhibitors.
Clinical trials are currently being conducted with the use of a C5a inhibitor, the monoclonal antibody IFX-1, which has proven to be well tolerated in 300 clinical trial participants and aims to reduce inflammation whilst preserving MAC formation (234). As SARS-CoV-2 is an enveloped virus (235), it is possible that MAC formation could have some beneficial antiviral effects. Therefore, a C5a inhibitor such as IFX-1 (InflaRX) may be favorable mechanistically over a C5 inhibitor such as eculizumab (Soliris), which is also considered for use in clinical trials (NCT04288713) (236). The IFX-1 monoclonal antibody targets a specific conformational epitope of the C5a molecule to block its anaphylatoxin activity, whilst C5b and downstream complement activity are preserved (237). Eculizumab is a monoclonal antibody which targets the C5 molecule to prevent cleavage into C5a and C5b, and therefore inhibits all downstream complement activity (238). The distinction between the two antibodies is the preservation of MAC activity which could be relevant against the enveloped SARS-CoV-2, although the effects of complement lysis and whether it occurs on SARS-CoV-2 is not yet known.
Because the efficacy and safety of eculizumab is already well characterized (239), it is logical that this would take precedence over lesser-known options for urgent clinical trials. The use of eculizumab has already proven beneficial for treatment of severe cases of COVID-19, which shows that complement is partly responsible for the symptoms in severe cases (240). It would be interesting to compare the effects of preserving the MAC during infection with the enveloped SARS-CoV-2, as it may offer an antiviral, as well as an anti-inflammatory, effect. But it is also possible that Coronaviruses have an intrinsic evasion mechanism, perhaps similar to the ones described in this review, to avoid the lytic activity of the MAC. Another important consideration could be the stage of infection for implementing complement inhibitors: maintaining complement activity may have a beneficial impact early on in infection for virus neutralization and the development of adaptive immunity, and intervention may only be required to treat excessive inflammation in severe cases.
The complement system has several important considerations for vaccine development, one example being its involvement in antibody dependent enhancement (ADE). ADE is commonly observed when non-neutralizing antibodies are present following initial priming of the immune system. Non-neutralizing antibodies can still bind the viral target with the potential to cross-link with Fc receptors, or activate complement and interact with complement receptors, to enhance viral infection of host cells (241). ADE is more commonly observed to be Fc receptor-mediated, however complement-mediated ADE has been reported for HIV-1 (242), MERS-CoV (243), and EBOV (244).
But complement activation can have a positive effect against viral infections in the presence of some non-neutralizing antibodies. Use of the non-neutralizing influenza virus M2 extracellular vaccine in mice required functional C3 to confer protection and induce effective humoral and cell-mediated immune responses (245). A similar effect has been reported for monoclonal antibodies against human cytomegalovirus (HCMV). Following the use of gB/MF59 HCMV vaccination in humans, the immune sera had enhanced neutralization potency toward HCMV in the presence of complement. Certain HCMV monoclonal antibodies rely on complement for viral neutralization, which appears distinct from CDC or virolysis, and is likely the result of blocking virus-host interactions (246). Complement activity has also been implicated for optimal protection with non-neutralizing antibody mAB-13G8 against Crimean-Congo haemorrhagic fever virus infection in adult mice (247).
In Flavivirus infections, the mechanism of ADE is predominantly shown to be Fc mediated (248). Complement has been shown to augment antibody-mediated neutralization of WNV in vitro (249) and the addition of C1q has been shown to lower the antibody concentrations required for WNV neutralization in vitro, which correlated with protective effects observed in vivo (250). C1q was also shown to mediate effects of ADE from Flavivirus infections in a subclass specific manner, whilst MBL, factor B, or C5 depletion had no significant effect (251). Although IgG subclasses are known to bind C1q with varying avidities, the mechanism to explain this effect on ADE has not been identified. This could highlight the importance of selecting the right antibody subclass when considering monoclonal antibody therapies.
In general, vaccines which effectively engage the complement system may gave rise to a more potent, virolytic serological response. For HIV vaccination in macaques, the presence of complement augmented virus neutralization and complement-mediated neutralizing antibody titres correlated with vaccine-mediated protection (252). Other approaches have modified vaccines to utilize aspects of the complement system for increased antigen immunogenicity, such as complement component C3d. C3d is an end-stage cleavage product from C3 activation which interacts with CR2 on B-cells, T-cells, and FDCs. When bound to an antigen, C3d can dramatically reduce the B-cell activation threshold for a stronger, more antigen-specific antibody response (8, 144, 253). CR2 on FDCs interacts with iC3b, C3d, and C3dg to enhance antibody titres and promote long-term B-cell memory development (254). C3d also bears T-cell epitopes so even with a lack of CR2 expression, the peptide can be internalized and presented on HLA II molecules to autoreactive T-helper cells and enhance antibody responses (255, 256). C3d does not interact with other components of the complement system and so the associated risks are reduced, however a large enough reduction in the B-cell activation threshold could potentially lead to antibody-mediated autoimmune responses.
C3d has been used as a vaccine adjuvant against several different viruses. DNA vaccines encoding the envelope glycoprotein of porcine reproductive and respiratory syndrome virus were more effective at increasing antigen specific neutralizing antibody titres, IFN-γ levels, and IL-4 levels when engineered with gene copies encoding the CR2 binding site of C3d in the same plasmid construct (257). Similarly, use of hepatitis E virus peptide (HEV-p179) for DNA vaccination in mice had enhanced anti-HEV-p179 antibody titres and avidity when fused with three tandem C3d copies as genetic adjuvants (258). C3d has also been used as genetic adjuvant for DNA vaccines against Newcastle disease virus and HIV-1 for increased efficacy and higher, longer-lasting antibody titres (259, 260). Fusion of C3d to target antigens is another approach for the development of safer, more immunogenic DNA vaccines. Coupling of C3d to the secretory form of Influenza virus haemagglutinin in mice provided an effective and safer mechanism for mucosal vaccination compared to the use of other adjuvants i.e., cholera toxin B subunits and Escherichia coli labile toxin (261). So, the use of C3d as an adjuvant can help to overcome the low immunogenicity associated with DNA vaccines, whilst maintaining their safety.
Many of the viruses discussed can activate complement, resulting in beneficial and/or detrimental effects on its survival. In the examples where viral-mediated complement activation has been more extensively studied, a viral mechanism is often identified which protects the virus from certain antiviral functions, such as the acquisition of CD46, CD55, CD59 to protect from MAC formation or the expression of a regulatory protein to inhibit the complement cascade at various points.
For the viruses which have been shown to activate complement but do not have a clear evasion/regulatory mechanism, such as MERS-CoV (204), SARS-CoV (202, 205), EBOV (3), and possibly SARS-CoV-2, it is plausible that the mechanism simply has not yet been identified. The viruses which activate complement would consequently trigger the downstream antiviral effects, both intracellularly and extracellularly. Therefore, it seems plausible that these viruses would utilize a mechanism, similar to the ones described in this review, to evade this antiviral activity and promote their survival. If such a regulatory protein or process is identified, then these may present as possible antiviral targets, similar to the targeting of the rhinovirus 3C protease with rupintrivir (6).
The complex interplay between viruses and the complement system can have profound implications for protection via innate immunity and the development of effective adaptive immunity. The effects of the complement system can vary between viral infections, and even during the different stages of the same viral infection, so a clear understanding of these mechanisms is important to improve efficiency of vaccine/therapeutic development whilst mitigating risk. Such developments can also be applied for non-viral pathogens (including bacteria, fungi, protozoa) and to broader, more systemic functions of the complement system including: interferon signaling (262, 263), metabolism (264), brain development (265), and the coagulation system (266).
Components of the complement system form an ancient aspect of innate immunity in vertebrates (267) and even some invertebrates (268, 269). Therefore, many animals which act as viral hosts or reservoirs for zoonoses also have an active complement system for targeting pathogens i.e., bats (270), cows (271), deer (272), pigs (273), rabbits (274), and rats (274), which the virus may have to overcome to avoid possible antiviral activity. Further viral mechanisms of complement regulation may therefore exist which have not yet been identified and the plasticity of viral genomes could result in the emergence of novel protein regulatory functions. Identifying these novel interactions could be important for the development and augmentation of vaccines and therapeutics or even the possibility of utilizing viral-derived regulatory proteins as therapeutic complement inhibitors in other diseases (137).
The benefits from understanding complement mechanisms in viral diseases may have relevance for the current SARS-CoV-2 outbreak. Previous research has demonstrated the impact of the complement system in coronavirus infections and other diseases, and this knowledge has led to the consideration of several complement inhibitors as therapeutics for severe cases of COVID-19.
JM wrote the manuscript and designed the tables and figures. TT, SL, and MC provided guidance and revised the manuscript. All authors contributed to the article and approved the submitted version.
JM was funded via PHE PhD studentship programme.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Sarma JV, Ward PA. The complement system. Cell Tissue Res. (2011) 343:227–35. doi: 10.1007/s00441-010-1034-0
2. Nesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol. (2012) 2:103–11. doi: 10.1556/EuJMI.2.2012.2.2
3. Ji X, Olinger GG, Aris S, Chen Y, Gewurz H, Spear GT. Mannose-binding lectin binds to Ebola and Marburg envelope glycoproteins, resulting in blocking of virus interaction with DC-SIGN and complement-mediated virus neutralization. J Gen Virol. (2005) 86(Pt 9):2535–42. doi: 10.1099/vir.0.81199-0
4. Schiela B, Bernklau S, Malekshahi Z, Deutschmann D, Koske I, Banki Z, et al. Active human complement reduces the Zika virus load via formation of the membrane-attack complex. Front Immunol. (2018) 9:2177. doi: 10.3389/fimmu.2018.02177
5. Harris SL, Frank I, Yee A, Cohen GH, Eisenberg RJ, Friedman HM. Glycoprotein C of herpes simplex virus type 1 prevents complement-mediated cell lysis and virus neutralization. J Infect Dis. (1990) 162:331–7. doi: 10.1093/infdis/162.2.331
6. Tam JCH, Bidgood SR, McEwan WA, James LC. Intracellular sensing of complement C3 activates cell autonomous immunity. Science. (2014) 345:1256070. doi: 10.1126/science.1256070
7. Wetsel RA, Kildsgaard J, Haviland DL. Complement anaphylatoxins (C3a, C4a, C5a) and their receptors (C3aR, C5aR/CD88) as therapeutic targets in inflammation. In: Lambris JD, Holers VM, editors. Therapeutic Interventions in the Complement System. Totowa, NJ: Contemporary Immunology, Humana Press (2000). p. 113–53.
8. Fingeroth JD, Heath ME, Ambrosino DM. Proliferation of resting B cells is modulated by CR2 and CR1. Immunol Lett. (1989) 21:291–301. doi: 10.1016/0165-2478(89)90022-9
9. Török K, Kremlitzka M, Sándor N, Tóth EA, Bajtay Z, Erdei A. Human T cell derived, cell-bound complement iC3b is integrally involved in T cell activation. Immunol Lett. (2012) 143:131–6. doi: 10.1016/j.imlet.2012.02.003
10. Duncan AR, Winter G. The binding site for C1q on IgG. Nature. (1988) 332:738–40. doi: 10.1038/332738a0
11. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. (2013) 33:479–92. doi: 10.1016/j.semnephrol.2013.08.001
12. Garcia BL, Zwarthoff SA, Rooijakkers SHM, Geisbrecht BV. Novel evasion mechanisms of the classical complement pathway. J Immunol. (2016) 197:2051. doi: 10.4049/jimmunol.1600863
13. Kaul M, Loos M. Dissection of C1q capability of interacting with IgG time-dependent formation of a tight and only partly reversible association. J Biol Chem. (1997) 272:33234–44. doi: 10.1074/jbc.272.52.33234
14. Bindon CI, Hale G, Brüggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med. (1988) 168:127–42. doi: 10.1084/jem.168.1.127
15. Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, et al. Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol. (2002) 32:1726–36. doi: 10.1002/1521-4141(200206)32:6<1726::AID-IMMU1726>3.0.CO;2-R
16. Ebenbichler CF, Thielens NM, Vornhagen R, Marschang P, Arlaud GJ, Dierich MP. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J Exp Med. (1991) 174:1417–24. doi: 10.1084/jem.174.6.1417
17. Douradinha B, McBurney SP, de Melo KMS, Smith AP, Krishna NK, Barratt-Boyes SM, et al. C1q binding to Dengue Virus inhibits infection of THP-1 and cellular inflammatory responses. Virus Res. (2014) 179:231–4. doi: 10.1016/j.virusres.2013.11.007
18. McGrath FDG, Brouwer MC, Arlaud GJ, Daha MR, Hack CE, Roos A. Evidence that complement protein C1q interacts with C-reactive protein through its globular head region. J Immunol. (2006) 176:2950–7. doi: 10.4049/jimmunol.176.5.2950
19. Bing DH, Almeda S, Isliker H, Lahav J, Hynes RO. Fibronectin binds to the C1q component of complement. Proc Natl Acad Sci USA. (1982) 79:4198–201. doi: 10.1073/pnas.79.13.4198
20. Pearlstein E, Sorvillo J, Gigli I. The interaction of human plasma fibronectin with a subunit of the first component of complement, C1q. J Immunol. (1982) 128:2036–9.
21. Krumdieck R, Höök M, Rosenberg LC, Volanakis JE. The proteoglycan decorin binds C1q and inhibits the activity of the C1 complex. J Immunol. (1992) 149:3695–701.
22. Rainard P. Activation of the classical pathway of complement by binding of bovine lactoferrin to unencapsulated Streptococcus agalactiae. Immunology. (1993) 79:648–52.
23. Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ, et al. Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur J Immunol. (2003) 33:465–73. doi: 10.1002/immu.200310022
24. Sørensen IJ, Nielsen EH, Andersen O, Danielsen B, Svehag SE. Binding of complement proteins C1q and C4bp to serum amyloid P component (SAP) in solid contra liquid phase. Scand J Immunol. (1996) 44:401–7. doi: 10.1046/j.1365-3083.1996.d01-326.x
25. Mortensen SA, Sander B, Jensen RK, Pedersen JS, Golas MM, Jensenius JC, et al. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc Natl Acad Sci USA. (2017) 114:986–91. doi: 10.1073/pnas.1616998114
26. Wang H, Ricklin D, Lambris JD. Complement-activation fragment C4a mediates effector functions by binding as untethered agonist to protease-activated receptors 1 and 4. Proc Natl Acad Sci USA. (2017) 114:10948–53. doi: 10.1073/pnas.1707364114
27. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun. (2016) 7:10587. doi: 10.1038/ncomms10587
28. Liu Y, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I, et al. Human M-ficolin is a secretory protein that activates the lectin complement pathway. J Immunol. (2005) 175:3150–6. doi: 10.4049/jimmunol.175.5.3150
29. Takahashi M, Iwaki D, Kanno K, Ishida Y, Xiong J, Matsushita M, et al. Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J Immunol. (2008) 180:6132–8. doi: 10.4049/jimmunol.180.9.6132
30. Hansen S, Selman L, Palaniyar N, Ziegler K, Brandt J, Kliem A, et al. Collectin 11 (CL-11, CL-K1) Is a MASP-1/3-associated plasma collectin with microbial-binding activity. J Immunol. (2010) 185:6096–104. doi: 10.4049/jimmunol.1002185
31. Teillet F, Dublet B, Andrieu J-P, Gaboriaud C, Arlaud GJ, Thielens NM. The two major oligomeric forms of human mannan-binding lectin: chemical characterization, carbohydrate-binding properties, and interaction with MBL-associated serine proteases. J Immunol. (2005) 174:2870–7. doi: 10.4049/jimmunol.174.5.2870
32. Tateishi K, Kanemoto T, Fujita T, Matsushita M. Characterization of the complex between mannose-binding lectin trimer and mannose-binding lectin-associated serine proteases. Microbiol Immunol. (2011) 55:427–33. doi: 10.1111/j.1348-0421.2011.00330.x
33. Weis WI, Drickamer K, Hendrickson WA. Structure of a C-type mannose-binding protein complexed with an oligosaccharide. Nature. (1992) 360:127–34. doi: 10.1038/360127a0
34. Jensen PH, Weilguny D, Matthiesen F, McGuire KA, Shi L, Højrup P. Characterization of the oligomer structure of recombinant human mannan-binding lectin. J Biol Chem. (2005) 280:11043–51. doi: 10.1074/jbc.M412472200
35. Lu J, Le Y, Kon OL, Chan J, Lee SH. Biosynthesis of human ficolin, an Escherichia coli-binding protein, by monocytes: comparison with the synthesis of two macrophage-specific proteins, C1q and the mannose receptor. Immunology. (1996) 89:289–94. doi: 10.1046/j.1365-2567.1996.d01-732.x
36. Runza VL, Schwaeble W, Männel DN. Ficolins: novel pattern recognition molecules of the innate immune response. Immunobiology. (2008) 213:297–306. doi: 10.1016/j.imbio.2007.10.009
37. Garlatti V, Martin L, Lacroix M, Gout E, Arlaud GJ, Thielens NM, et al. Structural insights into the recognition properties of human ficolins. J Innate Immun. (2010) 2:17–23. doi: 10.1159/000233475
38. Kilpatrick DC, Chalmers JD. Human L-Ficolin (Ficolin-2) and its clinical significance. Biomed Res Int. (2012) 2012:138797. doi: 10.1155/2012/138797
39. Swierzko A, Lukasiewicz J, Cedzynski M, Maciejewska A, Jachymek W, Niedziela T, et al. New functional ligands for ficolin-3 among lipopolysaccharides of Hafnia alvei. Glycobiology. (2012) 22:267–80. doi: 10.1093/glycob/cwr119
40. Henriksen ML, Brandt J, Andrieu J-P, Nielsen C, Jensen PH, Holmskov U, et al. Heteromeric Complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol. (2013) 191:6117–27. doi: 10.4049/jimmunol.1302121
41. Matsushita M, Thiel S, Jensenius JC, Terai I, Fujita T. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J Immunol. (2000) 165:2637–42. doi: 10.4049/jimmunol.165.5.2637
42. Ambrus G, Gál P, Kojima M, Szilágyi K, Balczer J, Antal J, et al. Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and−2: a study on recombinant catalytic fragments. J Immunol. (2003) 170:1374–82. doi: 10.4049/jimmunol.170.3.1374
43. Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, et al. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. (2001) 15:127–35. doi: 10.1016/S1074-7613(01)00161-3
44. Andersson J, Ekdahl KN, Larsson R, Nilsson UR, Nilsson B. C3 adsorbed to a polymer surface can form an initiating alternative pathway convertase. J Immunol. (2002) 168:5786–91. doi: 10.4049/jimmunol.168.11.5786
45. Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: is C3 a contact-activated protein? (2012) 217:1106–10. doi: 10.1016/j.imbio.2012.07.008
46. Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. (2009) 104:115–49. doi: 10.1016/S0065-2776(08)04004-2
47. Zewde N Jr, Gorham RD, Dorado A, Morikis D. Quantitative modeling of the alternative pathway of the complement system. PLoS ONE. (2016) 11:e0152337. doi: 10.1371/journal.pone.0152337
48. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. (2006) 176:1305–10. doi: 10.4049/jimmunol.176.3.1305
49. Tegla CA, Cudrici C, Patel S, Trippe R, Rus V, Niculescu F, et al. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res. (2011) 51:45–60. doi: 10.1007/s12026-011-8239-5
50. Reis ES, Mastellos DC, Hajishengallis G, Lambris JD. New insights into the immune functions of complement. Nat Rev Immunol. (2019) 19:503–16. doi: 10.1038/s41577-019-0168-x
51. Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. (2007) 179:2600–8. doi: 10.4049/jimmunol.179.4.2600
52. Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA. (2008) 105:9023–8. doi: 10.1073/pnas.0801015105
53. Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, et al. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol. (2008) 180:7613–21. doi: 10.4049/jimmunol.180.11.7613
54. Narni-Mancinelli E, Gauthier L, Baratin M, Guia S, Fenis A, Deghmane A-E, et al. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci Immunol. (2017) 2. doi: 10.1126/sciimmunol.aam9628
55. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
56. Ghannam A, Fauquert J-L, Thomas C, Kemper C, Drouet C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol Immunol. (2014) 58:98–107. doi: 10.1016/j.molimm.2013.11.010
57. Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP. A C3(H20) recycling pathway is a component of the intracellular complement system. J Clin Investig. (2017) 127:970–81. doi: 10.1172/JCI89412
58. Zimmer J, Hobkirk J, Mohamed F, Browning MJ, Stover CM. On the functional overlap between complement and anti-microbial peptides. Front Immunol. (2015) 5:689. doi: 10.3389/fimmu.2014.00689
59. Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Investig. (1988) 81:1419–26. doi: 10.1172/JCI113472
60. Langeggen H, Pausa M, Johnson E, Casarsa C, Tedesco F. The endothelium is an extrahepatic site of synthesis of the seventh component of the complement system. Clin Exp Immunol. (2000) 121:69–76. doi: 10.1046/j.1365-2249.2000.01238.x
61. Wittenborn T, Thiel S, Jensen L, Nielsen HJ, Jensenius JC. Characteristics and biological variations of M-Ficolin, a pattern recognition molecule, in plasma. J Innate Immun. (2010) 2:167–80. doi: 10.1159/000218324
62. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. (2017) 188:183–94. doi: 10.1111/cei.12952
63. Rozovsky I, Morgan TE, Willoughby DA, Dugichi-Djordjevich MM, Pasinetti GM, Johnson SA, et al. Selective expression of clusterin (SGP-2) and complement C1qB and C4 during responses to neurotoxins in vivo and in vitro. Neuroscience. (1994) 62:741–58. doi: 10.1016/0306-4522(94)90473-1
64. Sohn J-H, Kaplan HJ, Suk H-J, Bora PS, Bora NS. Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Investig Ophthalmol Vis Sci. (2000) 41:3492–502.
65. Simpson KL, Holmes CH. Differential expression of complement regulatory proteins decay-accelerating factor (CD55), membrane cofactor protein (CD46) and CD59 during human spermatogenesis. Immunology. (1994) 81:452–61.
66. Edelson BT, Stricker TP, Li Z, Dickeson SK, Shepherd VL, Santoro SA, et al. Novel collectin/C1q receptor mediates mast cell activation and innate immunity. Blood. (2006) 107:143–50. doi: 10.1182/blood-2005-06-2218
67. Steinberger P, Szekeres A, Wille S, Stöckl J, Selenko N, Prager E, et al. Identification of human CD93 as the phagocytic C1q receptor (C1qRp) by expression cloning. J Leukoc Biol. (2002) 71:133–40.
68. Norsworthy PJ, Fossati-Jimack L, Cortes-Hernandez J, Taylor PR, Bygrave AE, Thompson RD, et al. Murine CD93 (C1qRp) contributes to the removal of apoptotic cells in vivo but is not required for C1q-mediated enhancement of phagocytosis. J Immunol. (2004) 172:3406–14. doi: 10.4049/jimmunol.172.6.3406
69. Bohlson SS, Silva R, Fonseca MI, Tenner AJ. CD93 is rapidly shed from the surface of human myeloid cells and the soluble form is detected in human plasma. J Immunol. (2005) 175:1239–47. doi: 10.4049/jimmunol.175.2.1239
70. Ehrengruber MU, Geiser T, Deranleau DA. Activation of human neutrophils by C3a and C5A. (1994) 346:181–4. doi: 10.1016/0014-5793(94)00463-3
71. Nilsson G, Johnell M, Hammer CH, Tiffany HL, Nilsson K, Metcalfe DD, et al. C3a and C5a are chemotaxins for human mast cells and act through distinct receptors via a pertussis toxin-sensitive signal transduction pathway. J Immunol. (1996) 157:1693–8.
72. Hartmann K, Henz BM, Krüger-Krasagakes S, Köhl J, Burger R, Guhl S, et al. C3a and C5a stimulate chemotaxis of human mast cells. Blood. (1997) 89:2863–70. doi: 10.1182/blood.V89.8.2863
73. Bischoff SC, de Weck AL, Dahinden CA. Interleukin 3 and granulocyte/macrophage-colony-stimulating factor render human basophils responsive to low concentrations of complement component C3a. Proc Natl Acad Sci USA. (1990) 87:6813–7. doi: 10.1073/pnas.87.17.6813
74. Martin U, Bock D, Arseniev L, Tornetta MA, Ames RS, Bautsch W, et al. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J Exp Med. (1997) 186:199–207. doi: 10.1084/jem.186.2.199
75. Ischenko A, Sayah S, Patte C, Andreev S, Gasque P, Schouft MT, et al. Expression of a functional anaphylatoxin C3a receptor by astrocytes. J Neurochem. (1998) 71:2487–96. doi: 10.1046/j.1471-4159.1998.71062487.x
76. Werfel T, Kirchhoff K, Wittmann M, Begemann G, Kapp A, Heidenreich F, et al. Activated human T lymphocytes express a functional C3a receptor. J Immunol. (2000) 165:6599–605. doi: 10.4049/jimmunol.165.11.6599
77. Peng Q, Li K, Anderson K, Farrar CA, Lu B, Smith RAG, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood. (2008) 111:2452–61. doi: 10.1182/blood-2007-06-095018
78. Mommert S, Aslan D, Ratz L, Stark H, Gutzmer R, Werfel T. The Anaphylatoxin C3a receptor expression on human M2 macrophages is down-regulated by stimulating the histamine H4 receptor and the IL-4 receptor. J Innate Immun. (2018) 10:349–62. doi: 10.1159/000490426
79. Füreder W, Agis H, Willheim M, Bankl HC, Maier U, Kishi K, et al. Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J Immunol. (1995) 155:3152–60.
80. Nataf S, Davoust N, Ames RS, Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. J Immunol. (1999) 162:4018–23.
81. Weinmann O, Gutzmer R, Zwirner J, Wittmann M, Langer K, Lisewski M, et al. Up-regulation of C5a receptor expression and function on human monocyte derived dendritic cells by prostaglandin E2. Immunology. (2003) 110:458–65. doi: 10.1111/j.1365-2567.2003.01764.x
82. Fusakio ME, Mohammed JP, Laumonnier Y, Hoebe K, Köhl J, Mattner J. C5a regulates NKT and NK cell functions in sepsis. J Immunol. (2011) 187:5805–12. doi: 10.4049/jimmunol.1100338
83. Stuart GR, Lynch NJ, Day AJ, Schwaeble WJ, Sim RB. The C1q and collectin binding site within C1 q receptor (cell surface calreticulin). Immunopharmacology. (1997) 38:73–80. doi: 10.1016/S0162-3109(97)00076-3
84. Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol. (2002) 169:3978–86. doi: 10.4049/jimmunol.169.7.3978
85. Moestrup SK, Gliemann J, Pallesen G. Distribution of the α2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. (1992) 269:375–82. doi: 10.1007/BF00353892
86. Duus K, Hansen EW, Tacnet P, Frachet P, Arlaud GJ, Thielens NM, et al. Direct interaction between CD91 and C1q. FEBS J. (2010) 277:3526–37. doi: 10.1111/j.1742-4658.2010.07762.x
87. Fang Y, Xu C, Fu Y-X, Holers VM, Molina H. Expression of Complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol. (1998) 160:5273–9.
88. Wright SD, Silverstein SC. Tumor-promoting phorbol esters stimulate C3b and C3b' receptor-mediated phagocytosis in cultured human monocytes. J Exp Med. (1982) 156:1149–64. doi: 10.1084/jem.156.4.1149
89. Berger M, Sorensen RU, Tosi MF, Dearborn DG, Döring G. Complement receptor expression on neutrophils at an inflammatory site, the Pseudomonas-infected lung in cystic fibrosis. J Clin Investig. (1989) 84:1302–13. doi: 10.1172/JCI114298
90. Hivroz C, Fischer E, Kazatchkine MD, Grillot-Courvalin C. Differential effects of the stimulation of complement receptors CR1 (CD35) and CR2 (CD21) on cell proliferation and intracellular Ca2+ mobilization of chronic lymphocytic leukemia B cells. J Immunol. (1991) 146:1766–72.
91. Rødgaard A, Christensen LD, Thomsen BS, Wiik A, Bendixen G. Complement receptor type 1 (CR1, CD35) expression on peripheral T lymphocytes: both CD4- and CD8-positive cells express CR1. Comp Inflamm. (1991) 8:303–9. doi: 10.1159/000463200
92. Pascual M, Schifferli JA. The binding of immune complexes by the erythrocyte complement receptor 1 (CR1). Immunopharmacology. (1992) 24:101–6. doi: 10.1016/0162-3109(92)90016-6
93. Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement Receptor Type 1 (CR1, CD35) Is a Receptor for C1q. Immunity. (1997) 7:345–55. doi: 10.1016/S1074-7613(00)80356-8
94. Java A, Liszewski MK, Hourcade DE, Zhang F, Atkinson JP. Role of complement receptor 1 (CR1; CD35) on epithelial cells: a model for understanding complement-mediated damage in the kidney. Mol Immunol. (2015) 67:584–95. doi: 10.1016/j.molimm.2015.07.016
95. Iida K, Nadler L, Nussenzweig V. Identification of the membrane receptor for the complement fragment C3d by means of a monoclonal antibody. J Exp Med. (1983) 158:1021–33. doi: 10.1084/jem.158.4.1021
96. Levy E, Ambrus J, Kahl L, Molina H, Tung K, Holers VM. T lymphocyte expression of complement receptor 2 (CR2/CD21): a role in adhesive cell-cell interactions and dysregulation in a patient with systemic lupus erythematosus (SLE). Clin Exp Immunol. (1992) 90:235–44. doi: 10.1111/j.1365-2249.1992.tb07935.x
97. Vorup-Jensen T, Jensen RK. Structural Immunology of Complement Receptors 3 and 4. Front Immunol. (2018) 9:2716. doi: 10.3389/fimmu.2018.02716
98. Behrens EM, Sriram U, Shivers DK, Gallucci M, Ma Z, Finkel TH, et al. Complement receptor 3 ligation of dendritic cells suppresses their stimulatory capacity. J Immunol. (2007) 178:6268–79. doi: 10.4049/jimmunol.178.10.6268
99. Sándor N, Kristóf K, Paréj K, Pap D, Erdei A, Bajtay Z. CR3 is the dominant phagocytotic complement receptor on human dendritic cells. Immunobiology. (2013) 218:652–63. doi: 10.1016/j.imbio.2012.07.031
100. Helmy KY, Katschke KJ, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. (2006) 124:915–27. doi: 10.1016/j.cell.2005.12.039
101. Dedio J, Jahnen-Dechent W, Bachmann M, Müller-Esterl W. The multiligand-binding protein gC1qR, Putative C1q receptor, is a mitochondrial protein. J Immunol. (1998) 160:3534–42. doi: 10.1016/S0162-3109(99)00082-X
102. Leigh EAL, Ghebrehiwet B, Perera PST, Bird NI, Strong P, Kishore U, et al. C1q-mediated chemotaxis by human neutrophils: involvement of gClqR and G-protein signalling mechanisms. Biochem J. (1998) 330:247–54. doi: 10.1042/bj3300247
103. Vegh Z, Kew RR, Gruber BL, Ghebrehiwet B. Chemotaxis of human monocyte-derived dendritic cells to complement component C1q is mediated by the receptors gC1qR and cC1qR. Mol Immunol. (2006) 43:1402–7. doi: 10.1016/j.molimm.2005.07.030
104. Pednekar L, Pathan AA, Paudyal B, Tsolaki AG, Kaur A, Abozaid SM, et al. Analysis of the interaction between globular head modules of human C1q and its candidate receptor gC1qR. Front Immunol. (2016) 7:567. doi: 10.3389/fimmu.2016.00567
105. Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry. (2003) 42:9406–15. doi: 10.1021/bi034489v
106. McNearney T, Ballard L, Seya T, Atkinson JP. Membrane cofactor protein of complement is present on human fibroblast, epithelial, and endothelial cells. J Clin Investig. (1989) 84:538–45. doi: 10.1172/JCI114196
107. Oglesby TJ, Allen CJ, Liszewski MK, White DJG, Atkinson JP. Membrane cofactor protein (CD46) protects cells from complement- mediated attack by an intrinsic mechanism. J Exp Med. (1992) 175:1547–51. doi: 10.1084/jem.175.6.1547
108. Hara T, Kuriyama S, Kiyohara H, Nagase Y, Matsumoto M, Seya T. Soluble forms of membrane cofactor protein (CD46, MCP) are present in plasma, tears, and seminal fluid in normal subjects. Clin Exp Immunol. (1992) 89:490–4. doi: 10.1111/j.1365-2249.1992.tb06986.x
109. Friec GL, Sheppard D, Whiteman P, Karsten CM, Shamoun SA-T, Laing A, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol. (2012) 13:1213–21. doi: 10.1038/ni.2454
110. Jiang H, Wagner E, Zhang H, Frank MM. Complement 1 inhibitor is a regulator of the alternative complement pathway. J Exp Med. (2001) 194:1609–16. doi: 10.1084/jem.194.11.1609
111. Seya T, Nakamura K, Masaki T, Ichihara-Itoh C, Matsumoto M, Nagasawa S. Human factor H and C4b-binding protein serve as factor I-cofactors both encompassing inactivation of C3b and C4b. Mol Immunol. (1995) 32:355–60. doi: 10.1016/0161-5890(94)00157-V
112. Sjöberg AP, Trouw LA, McGrath FDG, Hack CE, Blom AM. Regulation of complement activation by C-reactive protein: targeting of the inhibitory activity of C4b-binding protein. J Immunol. (2006) 176:7612–20. doi: 10.4049/jimmunol.176.12.7612
113. Wenderfer SE, Soimo K, Wetsel RA, Braun MC. Analysis of C4 and the C4 binding protein in the MRL/lpr mouse. Arthr Res Ther. (2007) 9:R114. doi: 10.1186/ar2320
114. Blaas-Mautner P, Filsinger S, Berger B, Roelcke D, Hänsch GM. C8 binding protein bears I antigenic determinants. Ann Hematol. (1991) 62:64–7. doi: 10.1007/BF01714902
115. Campbell WD, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. (2002) 46:131–4. doi: 10.1111/j.1348-0421.2002.tb02669.x
116. Medof ME, Walter EI, Rutgers JL, Knowles DM, Nussenzweig V. Identification of the complement decay-accelerating factor (DAF) on epithelium and glandular cells and in body fluids. J Exp Med. (1987) 165:848–64. doi: 10.1084/jem.165.3.848
117. Harris CL, Pettigrew DM, Lea SM, Morgan BP. Decay-accelerating factor must bind both components of the complement alternative pathway C3 convertase to mediate efficient decay. J Immunol. (2007) 178:352–9. doi: 10.4049/jimmunol.178.1.352
118. Fang C, Miwa T, Song W-C. Decay-accelerating factor regulates T-cell immunity in the context of inflammation by influencing costimulatory molecule expression on antigen-presenting cells. Blood. (2011) 118:1008–14. doi: 10.1182/blood-2011-04-348474
119. Meri S, Morgan BP, Davies A, Daniels RH, Olavesen MG, Waldmann H, et al. Human protectin (CD59), an 18,000-20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. (1990) 71:1–9.
120. Korty PE, Brando C, Shevach EM. CD59 functions as a signal-transducing molecule for human T cell activation. J Immunol. (1991) 146:4092–8.
121. Kimberley FC, Sivasankar B, Morgan P. Alternative roles for CD59. Mol Immunol. (2007) 44:73–81. doi: 10.1016/j.molimm.2006.06.019
122. Xie X-H, Gao M-H, Zhang B, Wang M-J, Wang J. Post-transcriptional CD59 gene silencing by siRNAs induces enhanced human T lymphocyte response to tumor cell lysate-loaded DCs. Cell Immunol. (2012) 274:1–11. doi: 10.1016/j.cellimm.2012.02.013
123. Tschopp J, Chonn A, Hertig S, French LE. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement C7, C8 beta, and the b domain of C9. J Immunol. (1993) 151:2159–65.
124. Barlow PN, Hageman GS, Lea SM. Complement factor H: using atomic resolution structure to illuminate disease mechanisms. Adv Exp Med Biol. (2008) 632:117–42. doi: 10.1007/978-0-387-78952-1_10
125. Bettoni S, Bresin E, Remuzzi G, Noris M, Donadelli R. Insights into the effects of complement factor H on the assembly and decay of the alternative pathway C3 proconvertase and C3 convertase. J Biol Chem. (2016) 291:8214–30. doi: 10.1074/jbc.M115.693119
126. Kishore U, Sim RB. Factor H as a regulator of the classical pathway activation. Immunobiology. (2012) 217:162–8. doi: 10.1016/j.imbio.2011.07.024
127. Medicus RG, Melamed J, Arnaout MA. Role of human factor I and C3b receptor in the cleavage of surface-bound C3bi molecules. Eur J Immunol. (1983) 13:465–70. doi: 10.1002/eji.1830130607
128. Masaki T, Matsumoto M, Nakanishi I, Yasuda R, Seya T. Factor I-dependent inactivation of human complement C4b of the classical pathway by C3b/C4b receptor (CR1, CD35) and membrane cofactor protein (MCP, CD46). J Biochem. (1992) 111:573–8. doi: 10.1093/oxfordjournals.jbchem.a123799
129. Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. (2006) 281:2128–32. doi: 10.1074/jbc.M508928200
130. Sheehan M, Morris CA, Pussell BA, Charlesworth JA. Complement inhibition by human vitronectin involves non-heparin binding domains. Clin Exp Immunol. (1995) 101:136–41. doi: 10.1111/j.1365-2249.1995.tb02289.x
131. Heesterbeek DAC, Angelier ML, Harrison RA, Rooijakkers SHM. Complement and bacterial infections: from molecular mechanisms to therapeutic applications. J Innate Immun. (2018) 10:455–64. doi: 10.1159/000491439
132. Kozel TR, Pfrommer GS. Activation of the complement system by Cryptococcus neoformans leads to binding of iC3b to the yeast. Infect Immun. (1986) 52:1–5. doi: 10.1128/IAI.52.1.1-5.1986
133. Mejia P, Diez-Silva M, Kamena F, Lu F, Fernandes SM, Seeberger PH, et al. Human C1-inhibitor suppresses malaria parasite invasion and cytoadhesion via binding to parasite glycosylphosphatidylinositol and host cell receptors. J Infect Dis. (2016) 213:80–9. doi: 10.1093/infdis/jiv439
134. Ezekowitz RA, Kuhlman M, Groopman JE, Byrn RA. A human serum mannose-binding protein inhibits in vitro infection by the human immunodeficiency virus. J Exp Med. (1989) 169:185–96. doi: 10.1084/jem.169.1.185
135. Favoreel HW, Van de Walle GR, Nauwynck HJ, Pensaert MB. Virus complement evasion strategies. J Gen Virol. (2003) 84:1–15. doi: 10.1099/vir.0.18709-0
136. Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology. (2011) 411:362–73. doi: 10.1016/j.virol.2010.12.045
137. Abou-El-Hassan H, Zaraket H. Viral-derived complement inhibitors: current status and potential role in immunomodulation. Exp Biol Med. (2017) 242:397–410. doi: 10.1177/1535370216675772
138. Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement evasion strategies of viruses: an overview. Front Microbiol. (2017) 8:1117. doi: 10.3389/fmicb.2017.01117
139. Jayasekera JP, Moseman EA, Carroll MC. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J Virol. (2007) 81:3487–94. doi: 10.1128/JVI.02128-06
140. Huber M, Fischer M, Misselwitz B, Manrique A, Kuster H, Niederöst B, et al. Complement lysis activity in autologous plasma is associated with lower viral loads during the acute phase of HIV-1 Infection. PLoS Med. (2006) 3:e441. doi: 10.1371/journal.pmed.0030441
141. Terajima M, Cruz J, Co MDT, Lee J-H, Kaur K, Wilson PC, et al. Complement-dependent lysis of influenza A virus-infected cells by broadly cross-reactive human monoclonal antibodies. J Virol. (2011) 85:13463. doi: 10.1128/JVI.05193-11
142. Peng Q, Li K, Sacks SH, Zhou W. The role of anaphylatoxins C3a and C5a in regulating innate and adaptive immune responses. Inflamm Aller Drug Targets. (2009) 8:236–46. doi: 10.2174/187152809788681038
143. Weaver DJ, Reis ES, Pandey MK, Köhl G, Harris N, Gerard C, et al. C5a receptor-deficient dendritic cells promote induction of Treg and Th17. Eur J Immunol. (2010) 40:710–21. doi: 10.1002/eji.200939333
144. Barrington RA, Schneider TJ, Pitcher LA, Mempel TR, Ma M, Barteneva NS, et al. Uncoupling CD21 and CD19 of the B-cell coreceptor. Proc Natl Acad Sci USA. (2009) 106:14490–5. doi: 10.1073/pnas.0903477106
145. Brudner M, Karpel M, Lear C, Chen L, Yantosca LM, Scully C, et al. Lectin-dependent enhancement of ebola virus infection via soluble and transmembrane C-type lectin receptors. PLoS ONE. (2013) 8:60838. doi: 10.1371/journal.pone.0060838
146. Michelow IC, Lear C, Scully C, Prugar LI, Longley CB, Yantosca LM, et al. High-dose mannose-binding lectin therapy for Ebola virus infection. J Infect Dis. (2011) 203:175–9. doi: 10.1093/infdis/jiq025
147. Ying H, Ji X, Hart ML, Gupta K, Saifuddin M, Zariffard MR, et al. Interaction of mannose-binding lectin with HIV type 1 is sufficient for virus opsonization but not neutralization. AIDS Res Human Retroviruses. (2004) 20:327–35. doi: 10.1089/088922204322996563
148. Garred P, Madsen HO, Balslev U, Hofmann B, Pedersen C, Gerstoft J, et al. Susceptibility to HIV infection and progression of AIDS in relation to variant alleles of mannose-binding lectin. Lancet. (1997) 349:236–40. doi: 10.1016/S0140-6736(96)08440-1
149. Tjomsland V, Ellegård R, Che K, Hinkula J, Lifson JD, Larsson M. Complement opsonization of HIV-1 enhances the uptake by dendritic cells and involves the endocytic lectin and integrin receptor families. PLoS ONE. (2011) 6:e23542. doi: 10.1371/journal.pone.0023542
150. Crisci E, Ellegård R, Nyström S, Rondahl E, Serrander L, Bergström T, et al. Complement opsonization promotes herpes simplex virus 2 infection of human dendritic cells. J Virol. (2016) 90:4939–50. doi: 10.1128/JVI.00224-16
151. Ip WKE, Chan KH, Law HKW, Tso GHW, Kong EKP, Wong WHS, et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis. (2005) 191:1697–704. doi: 10.1086/429631
152. Zhou Y, Lu K, Pfefferle S, Bertram S, Glowacka I, Drosten C, et al. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J Virol. (2010) 84:8753–64. doi: 10.1128/JVI.00554-10
153. Leth-Larsen R, Zhong F, Chow VTK, Holmskov U, Lu J. The SARS coronavirus spike glycoprotein is selectively recognized by lung surfactant protein D and activates macrophages. Immunobiology. (2007) 212:201–11. doi: 10.1016/j.imbio.2006.12.001
154. Zhang H, Zhou G, Zhi L, Yang H, Zhai Y, Dong X, et al. Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect Dis. (2005) 192:1355–61. doi: 10.1086/491479
155. Yuan FF, Tanner J, Chan PKS, Biffin S, Dyer WB, Geczy AF, et al. Influence of FcgammaRIIA and MBL polymorphisms on severe acute respiratory syndrome. Tissue Antigens. (2005) 66:291–6. doi: 10.1111/j.1399-0039.2005.00476.x
156. Fuchs A, Lin T-Y, Beasley DW, Stover CM, Schwaeble WJ, Pierson TC, et al. Direct complement restriction of flavivirus infection requires glycan recognition by mannose-binding lectin. Cell Host Microbe. (2010) 8:186–95. doi: 10.1016/j.chom.2010.07.007
157. Johnson JB, Capraro GA, Parks GD. Differential mechanisms of complement-mediated neutralization of the closely related paramyxoviruses simian virus 5 and mumps virus. Virology. (2008) 376:112–23. doi: 10.1016/j.virol.2008.03.022
158. Gupta P, Tripathy AS. Alternative pathway of complement activation has a beneficial role against Chandipura virus infection. Med Microbiol Immunol. (2019). doi: 10.1007/s00430-019-00648-z
159. Kunnakkadan U, Nag J, Kumar NA, Mukesh RK, Suma SM, Johnson JB. Complement-mediated neutralization of a potent neurotropic human pathogen, Chandipura virus, is dependent on C1q. J Virol. (2019) 93:e00994-19. doi: 10.1128/JVI.00994-19
160. Bajtay Z, Speth C, Erdei A, Dierich MP. Cutting edge: productive HIV-1 infection of dendritic cells via complement receptor type 3 (CR3, CD11b/CD18). J Immunol. (2004) 173:4775–8. doi: 10.4049/jimmunol.173.8.4775
161. Arbore G, Kemper C, Kolev M. Intracellular complement – the complosome – in immune cell regulation. Mol Immunol. (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
162. Bottermann M, Foss S, Caddy SL, Clift D, van Tienen LM, Vaysburd M, et al. Complement C4 prevents viral infection through capsid inactivation. Cell Host Microbe. (2019) 25:617–29.e7. doi: 10.1016/j.chom.2019.02.016
163. Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, et al. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med. (2010) 207:793–806. doi: 10.1084/jem.20092545
164. Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP. Binding of Flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol. (2011) 187:424–33. doi: 10.4049/jimmunol.1100750
165. Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, et al. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci USA. (2006) 103:19111–6. doi: 10.1073/pnas.0605668103
166. Conde JN, da Silva EM, Allonso D, Coelho DR, da Andrade IS, et al. Inhibition of the membrane attack complex by Dengue Virus NS1 through interaction with vitronectin and terminal complement proteins. J Virol. (2016) 90:9570–81. doi: 10.1128/JVI.00912-16
167. Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, et al. Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J Infect Dis. (2006) 193:1078–88. doi: 10.1086/500949
168. Johnson JB, Borisevich V, Rockx B, Parks GD. A novel factor I activity in Nipah Virus inhibits human complement pathways through cleavage of C3b. J Virol. (2015) 89:989–98. doi: 10.1128/JVI.02427-14
169. Nag J, Mukesh RK, Suma SM, Kunnakkadan U, Kumar NA, Johnson JB. A factor I-like activity associated with chikungunya virus contributes to its resistance to the human complement system. J Virol. (2020) 94:e02062-19. doi: 10.1128/JVI.02062-19
170. Saifuddin M, Hedayati T, Atkinson JP, Holguin MH, Parker CJ, Spear GT. Human immunodeficiency virus type 1 incorporates both glycosyl phosphatidylinositol-anchored CD55 and CD59 and integral membrane CD46 at levels that protect from complement-mediated destruction. J Gen Virol. (1997) 78:1907–11. doi: 10.1099/0022-1317-78-8-1907
171. Johnson JB, Grant K, Parks GD. The paramyxoviruses simian virus 5 and mumps virus recruit host cell CD46 to evade complement-mediated neutralization. J Virol. (2009) 83:7602–11. doi: 10.1128/JVI.00713-09
172. Nielsen SD, Sørensen AM, Schønning K, Lund O, Nielsen JO, Hansen JE. Complement-mediated enhancement of HIV-1 infection in peripheral blood mononuclear cells. Scand J Infect Dis. (1997) 29:447–52. doi: 10.3109/00365549709011852
173. Yu Q, Yu R, Qin X. The good and evil of complement activation in HIV-1 infection. Cell Mol Immunol. (2010) 7:334–40. doi: 10.1038/cmi.2010.8
174. Dufloo J, Guivel-Benhassine F, Buchrieser J, Lorin V, Grzelak L, Dupouy E, et al. Anti-HIV-1 antibodies trigger non-lytic complement deposition on infected cells. EMBO Rep. (2019) 21:e49351. doi: 10.15252/embr.201949351
175. Kostavasili I, Sahu A, Friedman HM, Eisenberg RJ, Cohen GH, Lambris JD. Mechanism of complement inactivation by glycoprotein C of herpes simplex virus. J Immunol. (1997) 158:1763–71.
176. Barnum SR. C4a: an anaphylatoxin in name only. J Innate Immun. (2015) 7:333–9. doi: 10.1159/000371423
177. Marder SR, Chenoweth DE, Goldstein IM, Perez HD. Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des Arg. J Immunol. (1985) 134:3325–31.
178. Daffern PJ, Pfeifer PH, Ember JA, Hugli TE. C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activation. J Exp Med. (1995) 181:2119–27. doi: 10.1084/jem.181.6.2119
179. Kalant D, Cain SA, Maslowska M, Sniderman AD, Cianflone K, Monk PN. The chemoattractant receptor-like protein C5L2 binds the C3a des-Arg77/acylation-stimulating protein. J Biol Chem. (2003) 278:11123–9. doi: 10.1074/jbc.M206169200
180. Takabayashi T, Vannier E, Clark BD, Margolis NH, Dinarello CA, Burke JF, et al. A new biologic role for C3a and C3a desArg: regulation of TNF-alpha and IL-1 beta synthesis. J Immunol. (1996) 156:3455–60.
181. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. (2014) 192:2837–45. doi: 10.4049/jimmunol.1302484
182. Schulman ES, Post TJ, Henson PM, Giclas PC. Differential effects of the complement peptides, C5a and C5a des Arg on human basophil and lung mast cell histamine release. J Clin Investig. (1988) 81:918–23. doi: 10.1172/JCI113403
183. Kubota Y. The effect of human anaphylatoxins and neutrophils on histamine release from isolated human skin mast cells. J Dermatol. (1992) 19:19–26. doi: 10.1111/j.1346-8138.1992.tb03174.x
184. El-Lati SG, Dahinden CA, Church MK. Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J Investig Dermatol. (1994) 102:803–6. doi: 10.1111/1523-1747.ep12378589
185. Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem. (2011) 286:15003–9. doi: 10.1074/jbc.M111.221036
186. Glovsky MM, Hugli TE, Ishizaka T, Lichtenstein LM, Erickson BW. Anaphylatoxin-induced histamine release with human leukocytes: studies of C3a leukocyte binding and histamine release. J Clin Investig. (1979) 64:804–11. doi: 10.1172/JCI109527
187. Kretzschmar T, Jeromin A, Gietz C, Bautsch W, Klos A, Köhl J, et al. Chronic myelogenous leukemia-derived basophilic granulocytes express a functional active receptor for the anaphylatoxin C3a. Eur J Immunol. (1993) 23:558–61. doi: 10.1002/eji.1830230239
188. Bürgi B, Brunner T, Dahinden CA. The degradation product of the C5a anaphylatoxin C5adesarg retains basophil-activating properties. Eur J Immunol. (1994) 24:1583–9. doi: 10.1002/eji.1830240720
189. Takafuji S, Tadokoro K, Ito K, Dahinden CA. Degranulation from human eosinophils stimulated with C3a and C5a. Int Arch Aller Immunol. (1994) 104 (Suppl. 1):27–9. doi: 10.1159/000236743
190. Elsner J, Oppermann M, Czech W, Dobos G, Schöpf E, Norgauer J, et al. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur J Immunol. (1994) 24:518–22. doi: 10.1002/eji.1830240304
191. Wu MCL, Brennan FH, Lynch JPL, Mantovani S, Phipps S, Wetsel RA, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci USA. (2013) 110:9439–44. doi: 10.1073/pnas.1218815110
192. Iles KE, Dickinson DA, Watanabe N, Iwamoto T, Forman HJ. AP-1 activation through endogenous H2O2 generation by alveolar macrophages. Free Radic Biol Med. (2002) 32:1304–13. doi: 10.1016/S0891-5849(02)00840-7
193. Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine M. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. (2003) 17:1003–14. doi: 10.1096/fj.02-0737com
194. Gasque P, Singhrao SK, Neal JW, Götze O, Morgan BP. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol. (1997) 150:31–41.
195. Gasque P, Singhrao SK, Neal JW, Wang P, Sayah S, Fontaine M, et al. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J Immunol. (1998) 160:3543–54.
196. Bajic G, Yatime L, Klos A, Andersen GR. Human C3a and C3a desArg anaphylatoxins have conserved structures, in contrast to C5a and C5a desArg. Protein Sci. (2013) 22:204–12. doi: 10.1002/pro.2200
197. Swerlick RA, Yancey KB, Lawley TJ. Inflammatory properties of human C5a and C5a des Arg/ in mast cell-depleted human skin. J Investig Dermatol. (1989) 93:417–22. doi: 10.1016/0022-202X(89)90069-9
198. Barbu A, Hamad OA, Lind L, Ekdahl KN, Nilsson B. The role of complement factor C3 in lipid metabolism. Mol Immunol. (2015) 67:101–7. doi: 10.1016/j.molimm.2015.02.027
199. Drouin SM, Kildsgaard J, Haviland J, Zabner J, Jia HP, McCray PB, et al. Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol. (2001) 166:2025–32. doi: 10.4049/jimmunol.166.3.2025
200. Schumacher WA, Fantone JC, Kunkel SE, Webb RC, Lucchesi BR. The anaphylatoxins C3a and C5a are vasodilators in the canine coronary vasculature in vitro and in vivo. Agents Actions. (1991) 34:345–9. doi: 10.1007/BF01988727
201. Williams TJ. Vascular permeability changes induced by complement-derived peptides. Agents Actions. (1983) 13:451–5. doi: 10.1007/BF02176415
202. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio. (2018) 9, e01753–18. doi: 10.1128/mBio.01753-18
203. Pang RT, Poon TC, Chan KA, Lee NL, Chiu RW, Tong Y-K, et al. Serum proteomic fingerprints of adult patients with severe acute respiratory syndrome. Clin Chem. (2006) 52:421–9. doi: 10.1373/clinchem.2005.061689
204. Jiang Y, Zhao G, Song N, Li P, Chen Y, Guo Y, et al. Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg Microbes Infect. (2018) 7:1–12. doi: 10.1038/s41426-018-0063-8
205. Yang Y-H, Huang Y-H, Chuang Y-H, Peng C-M, Wang L-C, Lin Y-T, et al. Autoantibodies against human epithelial cells and endothelial cells after severe acute respiratory syndrome (SARS)-associated coronavirus infection. J Med Virol. (2005) 77:1–7. doi: 10.1002/jmv.20407
206. Cabezas S, Bracho G, Aloia AL, Adamson PJ, Bonder CS, Smith JR, et al. Dengue virus induces increased activity of the complement alternative pathway in infected cells. J Virolo. (2018) 92:e00633–18. doi: 10.1128/JVI.00633-18
207. Morrison TE, Fraser RJ, Smith PN, Mahalingam S, Heise MT. Complement contributes to inflammatory tissue destruction in a mouse model of ross river virus-induced disease. J Virol. (2007) 81:5132–43. doi: 10.1128/JVI.02799-06
208. Gunn BM, Morrison TE, Whitmore AC, Blevins LK, Hueston L, Fraser RJ, et al. Mannose binding lectin is required for Alphavirus-induced arthritis/myositis. PLOS Pathog. (2012) 8:e1002586. doi: 10.1371/journal.ppat.1002586
209. Gunn BM, Jones JE, Shabman RS, Whitmore AC, Sarkar S, Blevins LK, et al. Ross River virus envelope glycans contribute to disease through activation of the host complement system. Virology. (2018) 515:250–60. doi: 10.1016/j.virol.2017.12.022
210. Kwan W, van der Touw W, Heeger PS. Complement regulation of T cell immunity. Immunol Res. (2012) 54:247–53. doi: 10.1007/s12026-012-8327-1
211. Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. (2005) 201:1523–30. doi: 10.1084/jem.20041967
212. Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. (2008) 28:425–35. doi: 10.1016/j.immuni.2008.02.001
213. Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3+ regulatory T cells. Nat Immunol. (2013) 14:162–71. doi: 10.1038/ni.2499
214. Kim AHJ, Dimitriou ID, Holland MCH, Mastellos D, Mueller YM, Altman JD, et al. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses. J Immunol. (2004) 173:2524–9. doi: 10.4049/jimmunol.173.4.2524
215. Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat Med. (2002) 8:373–8. doi: 10.1038/nm0402-373
216. Suresh M, Molina H, Salvato MS, Mastellos D, Lambris JD, Sandor M. Complement component 3 is required for optimal expansion of CD8 T cells during a systemic viral infection. J Immunol. (2003) 170:788–94. doi: 10.4049/jimmunol.170.2.788
217. Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Investig. (2000) 106:1239. doi: 10.1172/JCI10323
218. Waggoner SN, Hall CHT, Hahn YS. HCV core protein interaction with gC1q receptor inhibits Th1 differentiation of CD4+ T cells via suppression of dendritic cell IL-12 production. J Leukoc Biol. (2007) 82:1407–19. doi: 10.1189/jlb.0507268
219. Yao ZQ, Prayther D, Trabue C, Dong ZP, Moorman J. Differential regulation of SOCS-1 signalling in B and T lymphocytes by hepatitis C virus core protein. Immunology. (2008) 125:197–207. doi: 10.1111/j.1365-2567.2008.02829.x
220. Kim H, Meyer K, Bisceglie AMD, Ray R. Hepatitis C virus suppresses C9 complement synthesis and impairs membrane attack complex function. J Virol. (2013) 87:5858–67. doi: 10.1128/JVI.00174-13
221. Aydar Y, Sukumar S, Szakal AK, Tew JG. The influence of immune complex-bearing follicular dendritic cells on the IgM response, Ig class switching, and production of high affinity IgG. J Immunol. (2005) 174:5358–66. doi: 10.4049/jimmunol.174.9.5358
222. Klaus GG, Humphrey JH. The generation of memory cells. I. The role of C3 in the generation of B memory cells. Immunology. (1977) 33:31–40.
223. Castellano G, Woltman AM, Schlagwein N, Xu W, Schena FP, Daha MR, et al. Immune modulation of human dendritic cells by complement. Eur J Immunol. (2007) 37:2803–11. doi: 10.1002/eji.200636845
224. Lan J, Yang K, Byrd D, Hu N, Amet T, Shepherd N, et al. Provirus activation plus CD59 blockage triggers antibody-dependent complement-mediated lysis of latently HIV-1–infected cells. J Immunol. (2014) 193:3577–89. doi: 10.4049/jimmunol.1303030
225. Yang K, Lan J, Shepherd N, Hu N, Xing Y, Byrd D, et al. Blockage of CD59 function restores activities of neutralizing and nonneutralizing antibodies in triggering antibody-dependent complement-mediated lysis of HIV-1 virions and provirus-activated latently infected cells. J Virol. (2015) 89:9393–406. doi: 10.1128/JVI.01614-15
226. Holt DS, Botto M, Bygrave AE, Hanna SM, Walport MJ, Morgan BP. Targeted deletion of the CD59 gene causes spontaneous intravascular hemolysis and hemoglobinuria. Blood. (2001) 98:442–9. doi: 10.1182/blood.V98.2.442
227. Macor P, Tripodo C, Zorzet S, Piovan E, Bossi F, Marzari R, et al. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res. (2007) 67:10556–63. doi: 10.1158/0008-5472.CAN-07-1811
228. Zhao W-P, Zhu B, Duan Y-Z, Chen Z-T. Neutralization of complement regulatory proteins CD55 and CD59 augments therapeutic effect of herceptin against lung carcinoma cells. Oncol Rep. (2009) 21:1405–11. doi: 10.3892/or_00000368
229. Sun S, Zhao G, Liu C, Wu X, Guo Y, Yu H, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol. (2013) 49:221–30. doi: 10.1165/rcmb.2012-0428OC
230. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Different. (2020) 27:1451–4. doi: 10.1038/s41418-020-0530-3
231. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020). 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
232. Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–292.e6. doi: 10.1101/2020.02.19.956581
233. Sun S, Zhao G, Liu C, Fan W, Zhou X, Zeng L, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African Green Monkeys. Clin Infect Dis. (2015) 60:586–95. doi: 10.1093/cid/ciu887
234. Clinical Trials Arena. InflaRx Starts Dosing Covid-19 Coronavirus Patients in Europe. (2020). Available online at: https://www.clinicaltrialsarena.com/news/inflarx-covid-19-dosing-europe/ (accessed April 7, 2020).
235. Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, et al. The species Severe acute respiratory syndrome-related coronavirus : classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. (2020) 5:536–44. doi: 10.1038/s41564-020-0695-z
236. U.S. National Library of Medicine. Eculizumab (Soliris) in Covid-19 Infected Patients - Full Text View - ClinicalTrials.gov. (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT04288713 (accessed April 7, 2020).
237. InflaRx. The InflaRx Technology. (2017). Available online at: https://www.inflarx.de/Home/Research---Development/Technology.html (accessed May 15, 2020).
238. Dubois EA, Cohen AF. Eculizumab. Br J Clin Pharmacol. (2009) 68:318–9. doi: 10.1111/j.1365-2125.2009.03491.x
239. Rondeau E, Cataland SR, Al-Dakkak I, Miller B, Webb NJA, Landau D. Eculizumab safety: five-year experience from the global atypical hemolytic uremic syndrome registry. Kidney Int Rep. (2019) 4:1568–76. doi: 10.1016/j.ekir.2019.07.016
240. Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. (2020) 24:4040–7. doi: 10.26355/eurrev_202004_20875
241. Kulkarni R. Antibody-dependent enhancement of viral infections. Dyn Immune Activ Viral Dis. (2019) 9–41. doi: 10.1007/978-981-15-1045-8_2
242. Robinson WE. Mechanism for complement-mediated, antibody-dependent enhancement of human immunodeficiency virus type 1 infection in MT2 cells is enhanced entry through CD4, CD21, and CXCR4 chemokine receptors. Viral Immunol. (2006) 19:434–47. doi: 10.1089/vim.2006.19.434
243. Houser KV, Broadbent AJ, Gretebeck L, Vogel L, Lamirande EW, Sutton T, et al. Enhanced inflammation in New Zealand white rabbits when MERS-CoV reinfection occurs in the absence of neutralizing antibody. PLoS Pathog. (2017) 13:6565. doi: 10.1371/journal.ppat.1006565
244. Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-dependent enhancement of Ebola virus infection. J Virol. (2003) 77:7539–44. doi: 10.1128/JVI.77.13.7539-7544.2003
245. Kim Y-J, Kim K-H, Ko E-J, Kim M-C, Lee Y-N, Jung Y-J, et al. Complement C3 plays a key role in inducing humoral and cellular immune responses to influenza virus strain-specific hemagglutinin-based or cross-protective M2 extracellular domain-based vaccination. J Virol. (2018) 92. doi: 10.1128/JVI.00969-18
246. Li F, Freed DC, Tang A, Rustandi RR, Troutman MC, Espeseth AS, et al. Complement enhances in vitro neutralizing potency of antibodies to human cytomegalovirus glycoprotein B (gB) and immune sera induced by gB/MF59 vaccination. NPJ VaccIN. (2017) 2:855–864. doi: 10.1038/s41541-017-0038-0
247. Golden JW, Shoemaker CJ, Lindquist ME, Zeng X, Daye SP, Williams JA, et al. GP38-targeting monoclonal antibodies protect adult mice against lethal Crimean-Congo hemorrhagic fever virus infection. Sci Adv. (2019) 5:eaaw9535. doi: 10.1126/sciadv.aaw9535
248. Pierson TC, Fremont DH, Kuhn RJ, Diamond MS. Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: implications for vaccine development. Cell Host Microbe. (2008) 4:229–38. doi: 10.1016/j.chom.2008.08.004
249. Mehlhop E, Whitby K, Oliphant T, Marri A, Engle M, Diamond MS. Complement activation is required for induction of a protective antibody response against West Nile virus infection. J Virol. (2005) 79:7466–77. doi: 10.1128/JVI.79.12.7466-7477.2005
250. Mehlhop E, Nelson S, Jost CA, Gorlatov S, Johnson S, Fremont DH, et al. C1q reduces the stoichiometric threshold for antibody-mediated neutralization of West Nile Virus. Cell Host Microbe. (2009) 6:381–91. doi: 10.1016/j.chom.2009.09.003
251. Mehlhop E, Ansarah-Sobrinho C, Johnson S, Engle M, Fremont DH, Pierson TC, et al. C1q inhibits antibody-dependent enhancement of flavivirus infection in vitro and in vivo in an IgG subclass specific manner. Cell Host Microbe. (2007) 2:417–26. doi: 10.1016/j.chom.2007.09.015
252. Page M, Quartey-Papafio R, Robinson M, Hassall M, Cranage M, Stott J, et al. Complement-mediated virus infectivity neutralisation by HLA antibodies is associated with sterilising immunity to SIV challenge in the Macaque Model for HIV/AIDS. PLoS ONE. (2014) 9:e88735. doi: 10.1371/journal.pone.0088735
253. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. (1996) 271:348–50. doi: 10.1126/science.271.5247.348
254. Mattsson J, Yrlid U, Stensson A, Schön K, Karlsson MCI, Ravetch JV, et al. Complement activation and complement receptors on follicular dendritic cells are critical for the function of a targeted adjuvant. J Immunol. (2011) 187:3641–52. doi: 10.4049/jimmunol.1101107
255. Knopf PM, Rivera DS, Hai S-H, McMurry J, Martin W, De Groot AS. Novel function of complement C3d as an autologous helper T-cell target. Immunol Cell Biol. (2008) 86:221–5. doi: 10.1038/sj.icb.7100147
256. De Groot AS, Ross TM, Levitz L, Messitt TJ, Tassone R, Boyle CM, et al. C3d adjuvant effects are mediated through the activation of C3d-specific autoreactive T cells. Immunol Cell Biol. (2015) 93:189–97. doi: 10.1038/icb.2014.89
257. Zhang D, Xia Q, Wu J, Liu D, Wang X, Niu Z. Construction and immunogenicity of DNA vaccines encoding fusion protein of murine complement C3d-p28 and GP5 gene of porcine reproductive and respiratory syndrome virus. Vaccine. (2011) 29:629–35. doi: 10.1016/j.vaccine.2010.11.046
258. Yang S, Wang C, Fang X, Zhai L, Dong C, Ding L, et al. Fusion of C3d molecule with neutralization epitope(s) of hepatitis E virus enhances antibody avidity maturation and neutralizing activity following DNA immunization. Virus Res. (2010) 151:162–9. doi: 10.1016/j.virusres.2010.04.011
259. Green TD, Montefiori DC, Ross TM. Enhancement of antibodies to the human immunodeficiency virus type 1 envelope by using the molecular adjuvant C3d. J Virol. (2003) 77:2046–55. doi: 10.1128/JVI.77.3.2046-2055.2003
260. Zhao K, Duan X, Hao L, Wang X, Wang Y. Immune effect of Newcastle Disease Virus DNA vaccine with C3d as a molecular adjuvant. J Microbiol Biotechnol. (2017) 27:2060–9. doi: 10.4014/jmb.1708.08017
261. Watanabe I, Ross TM, Tamura S, Ichinohe T, Ito S, Takahashi H, et al. Protection against influenza virus infection by intranasal administration of C3d-fused hemagglutinin. Vaccine. (2003) 21:4532–8. doi: 10.1016/S0264-410X(03)00510-3
262. Elkon KB, Santer DM. Complement, interferon and lupus. Curr Opin Immunol. (2012) 24:665–70. doi: 10.1016/j.coi.2012.08.004
263. Lebel M-È, Langlois M-P, Daudelin J-F, Tarrab E, Savard P, Leclerc D, et al. Complement component 3 regulates IFN-α production by plasmacytoid dendritic cells following TLR7 activation by a plant Virus–like nanoparticle. J Immunol. (2017) 198:292–9. doi: 10.4049/jimmunol.1601271
264. Hess C, Kemper C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity. (2016) 45:240–54. doi: 10.1016/j.immuni.2016.08.003
265. Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. (2011) 48:1592–603. doi: 10.1016/j.molimm.2011.04.003
266. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. (2008) 632:71–9. doi: 10.1007/978-0-387-78952-1_6
267. Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev. (2016) 274:9–15. doi: 10.1111/imr.12474
268. Al-Sharif WZ, Sunyer JO, Lambris JD, Smith LC. Sea urchin coelomocytes specifically express a homologue of the complement component C3. J Immunol. (1998) 160:2983–97.
269. Poole AZ, Kitchen SA, Weis VM. The role of complement in Cnidarian-Dinoflagellate symbiosis and immune challenge in the sea Anemone Aiptasia pallida. Front Microbiol. (2016) 7:519. doi: 10.3389/fmicb.2016.00519
270. Moore MS, Reichard JD, Murtha TD, Zahedi B, Fallier RM, Kunz TH. Specific alterations in complement protein activity of little brown myotis (Myotis lucifugus) hibernating in white-nose syndrome affected sites. PLoS ONE. (2011) 6:e27430. doi: 10.1371/journal.pone.0027430
271. Rainard P. The complement in milk and defense of the bovine mammary gland against infections. Vet Res. (2003) 34:647–70. doi: 10.1051/vetres:2003025
272. Isogai E, Kamewaka Y, Isogai H, Kimura K, Fujii N, Nishikawa T. Complement-mediated killing of Borrelia garinii–bactericidal activity of wild deer serum. Microbiol Immunol. (1994) 38:753–6. doi: 10.1111/j.1348-0421.1994.tb01852.x
273. Khoa DVA, Wimmers K. Genetic association of the Porcine C9 complement component with hemolytic complement activity. Asian Austr J Anim Sci. (2015) 28:1354–61. doi: 10.5713/ajas.14.0734
Keywords: virology, immunology, complement system, innate immunity, vaccines, therapeutics
Citation: Mellors J, Tipton T, Longet S and Carroll M (2020) Viral Evasion of the Complement System and Its Importance for Vaccines and Therapeutics. Front. Immunol. 11:1450. doi: 10.3389/fimmu.2020.01450
Received: 07 April 2020; Accepted: 04 June 2020;
Published: 09 July 2020.
Edited by:
Aurelio Cafaro, Istituto Superiore di Sanità (ISS), ItalyReviewed by:
François J. M. A. Meurens, UMR INRAE-Oniris 1300 Oniris - Nantes Atlantic National College of Veterinary Medicine, FranceCopyright © 2020 Mellors, Tipton, Longet and Carroll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jack Mellors, amFjay5tZWxsb3JzQHBoZS5nb3YudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.